 Jonas Goldin Diness1*
Jonas Goldin Diness1* Lea Abildgaard1
Lea Abildgaard1 Sofia Hammami Bomholtz1,2
Sofia Hammami Bomholtz1,2 Mark Alexander Skarsfeldt1,2
Mark Alexander Skarsfeldt1,2 Nils Edvardsson1,3
Nils Edvardsson1,3 Ulrik S. Sørensen1Morten Grunnet1
Ulrik S. Sørensen1Morten Grunnet1 Bo Hjorth Bentzen1,3*
Bo Hjorth Bentzen1,3*- 1Acesion Pharma, Copenhagen, Denmark
- 2Department of Biomedical Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Department of Molecular and Clinical Medicine/Cardiology, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
Background: Hypokalemia reduces the cardiac repolarization reserve. This prolongs the QT-interval and increases the risk of ventricular arrhythmia; a risk that is exacerbated by administration of classical class 3 anti-arrhythmic agents.
Small conductance Ca2+-activated K+-channels (KCa2) are a promising new atrial selective target for treatment of atrial fibrillation. Under physiological conditions KCa2 plays a minor role in ventricular repolarization. However, this might change under hypokalemia because of concomitant increases in ventriculay -60r intracellur Ca2+.
Purpose: To study the effects of pharmacological KCa2 channel inhibition by the compounds AP14145, ICA, or AP30663 under hypokalemic conditions as compared to dofetilide and hypokalemia alone time-matched controls (TMC).
Methods: The current at +10 mV was compared in HEK293 cells stably expressing KCa2.3 perfused first with normo- and then hypokalemic solutions (4 mM K+ and 2.5 mM K+, respectively). Guinea pig hearts were isolated and perfused with normokalemic (4 mM K+) Krebs-Henseleit solution, followed by perfusion with drug or vehicle control. The perfusion was then changed to hypokalemic solution (2.5 mM K+) in presence of drug. 30 animals were randomly assigned to 5 groups: ICA, AP14145, AP30663, dofetilide, or TMC. QT-interval, the interval from the peak to the end of the T wave (Tp–Te), ventricular effective refractory period (VERP), arrhythmia score, and ventricular fibrillation (VF) incidence were recorded.
Results: Hypokalemia slightly increased KCa2.3 current compared to normokalemia. Application of KCa2 channel inhibitors and dofetilide prolonged the QT interval corrected for heart rate. Dofetilide, but none of the KCa2 channel inhibitors increased Tp–Te during hypokalemia. During hypokalemia 4/6 hearts in the TMC group developed VF (two spontaneously, two by S1S2 stimulation) whereas 5/6 hearts developed VF in the dofetilide group (two spontaneously, three by S1S2 stimulation). In comparison, 0/6, 1/6, and 1/6 hearts developed VF when treated with the KCa2 channel inhibitors AP30663, ICA, or AP14145, respectively.
Conclusion: Hypokalemia was associated with an increased incidence of VF, an effect that also seen in the presence of dofetilide. In comparison, the structurally and functionally different KCa2 channel inhibitors, ICA, AP14145, and AP30663 protected the heart from hypokalemia induced VF. These results support that KCa2 inhibition may be associated with a better safety and tolerability profile than dofetilide.
Introduction
Intra- and extra-cellular K+ concentrations are closely regulated physiologically. Hypokalemia, defined as K+ serum levels below 3.5 mM, has been reported to occur in 16% of patients when first admitted to acute medical departments (Jensen et al., 2015). Hypokalemia is an adverse effect of diuretic therapy prescribed to treat hypertension, heart failure and other conditions, but can also develop from e.g. vomiting and diarrhea. Hypokalemia lowers the Na/K-ATPase activity, hyperpolarizes the resting membrane potential, and despite increasing the driving force for outward K+ current it suppresses the cardiac IK1, IKr, and Ito currents. This reduction in the repolarizing reserve in turn prolongs the cardiac action potential, and promotes the development of early-after depolarizations (EAD) and initiation of ventricular tachycardia (VT) (for review see (Weiss et al., 2017)). Moreover, action potential prolongation and hypokalemia increase intracellular Ca2+ loading through increased Ca2+ influx via calcium channels, and by lowered Na+/Ca2+ activity (calcium efflux) secondary to the reduced Na+/K+-ATPase activity and consequent elevated intracellular Na+ concentrations (Aronsen et al., 2015; Weiss et al., 2017). It has been suggested that the increase in intracellular calcium activates ventricular KCa2 channels, functioning as a protective mechanism against ventricular arrhythmia during hypokalemia (Chan et al., 2015). If that is the case, KCa2 channel inhibition should be proarrhythmic under hypokalemic conditions. The KCa2 channel also known as the small conductance calcium activated K+, or SK, channel is a novel drug target for treatment of atrial fibrillation (AF) (Diness et al., 2010; Qi et al., 2014; Skibsbye et al., 2014; Haugaard et al., 2015; Diness et al., 2017). Under physiological conditions KCa2 channels appear to play a minor role in ventricular repolarization. However, this might change under pathophysiological conditions such as heart failure and myocardial infarction or hypokalemia (Chua et al., 2011; Chang et al., 2013a; Chang et al., 2013b; Gui et al., 2013; Bonilla et al., 2014; Chan et al., 2015; Hundahl et al., 2017).
Classical class III anti-arrhythmic drugs such as sotalol and dofetilide inhibit KV11.1 thereby reducing IKr and prolonging the QT-interval (Redfern et al., 2003). The drug-induced impairment of the repolarizing reserve and consequent risk for ventricular arrhythmia is further potentiated by hypokalemia (McKibbin et al., 1984).
Because hypokalemia increases intracellular calcium and compromises the repolarizing reserve, we hypothesized that inhibition of KV11.1 and KCa2 would be pro-arrhythmic in a hypokalemic setting. To study this we investigated the effects of KCa2 channel inhibition under hypokalemic conditions as compared to the class III anti-arrhythmic agent dofetilide. Moreover, we also explored if KCa2.3 channel conductance per se is affected by hypokalemia.
Materials and Methods
Electrophysiology
The experiments were performed on HEK293 cells stably expressing KCa2.3. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM1965, Substrat- og sterilcentralen, University of Copenhagen, Denmark) supplemented with 10% fetal bovine serum (Biowest, France), 100 U/ml of penicillin/streptomycin (Sigma, Germany), and 100 µg/ml geneticin (Gibco, USA).Whole cell patch clamping was performed on an automated whole-cell patch-clamp system (QPatch 16 HT) with single-hole Qplates (Sophion, Denmark). The Qpatch automatically generates giga seals, whole-cell formation, compound application, voltage-clamping, and recording of current. On the day of experiment, HEK293 cells expressing human KCa2.3 were treated with detachin (Genlantis, CA, USA) and resuspended in serum Free medium (C5467 SAFC, Buchs, Switzerland) containing 25 mM HEPES 0.04 mg/ml soy bean trypsin inhibitor (T6522 Sigma) and 100U/ml penicillin/streptomycin.
The extracellular solution consisted of (in mM): NaCl 145; CaCl2 2; MgCl2 1; 10 HEPES and 10 glucose and 4 mM KCl or 2.5 mM KCl for the normo- and hypokalemic conditions. The pH was adjusted to 7.4 with NaOH. The intracellular solution contained (in mM): 154 KCl, 10 mM HEPES and 10 mM EGTA. CaCl2 was added to give calculated free concentrations of Ca2+ of 0.4 µM (K0.4) and MgCl2 was added to give a free concentration of 1 mM. Free concentrations were calculated by the Eqcal software.
Currents were elicited by a voltage ramp from −140 mV to +20 mV (200 ms in duration) applied every fifth second from a holding potential of −80 mV. Data were sampled at 10 kHz, four-order Bessel filter, cutoff frequency 3 kHz, and 80 % Rs compensation. The currents were recorded for 500 s in normokalemic solution before exchanging to hypokalemic extracellular solution for 200 s.
Isolated Perfused Guinea Pig Heart Experiments
Guinea pig experiments were performed under licenses from the Danish Ministry of justice (2018-15-0201-01430 and 2016-15-0201-00850) and in accordance with the Danish guidelines for animal experiments according to the European Commission Directive 86/609/EEC.
30 female Guinea pigs (weighing between 350 and 600 g) from the Dunkin Hartley strain (HsdPoc : DH) (Charles River, Scanbur A/S, Karlslunde, Denmark) were used for the study, and randomized to a hypokalemia alone time-matched control group (TMC) or groups of n = 6 treated with ICA, AP14145, AP30663, or dofetilide. The animals were anaesthetized (200 mg/kg pentobarbital and lidocaine hydrochloride 0.150 ml/100 g body weight i.p., (Glostrup Apotek, Denmark)), and a dose of 1000 IU/kg heparin was injected intravenously. After artificial ventilation was established (volume, 12 ml/kg; rate, 60 strokes/min) a thoracotomy was performed. The perfusion cannula was inserted and fixed in the aorta for retrograde perfusion with (normokalemic) Krebs-Henseleit solution: (in mM L−1: NaCl 120.0, NaHCO3 25.0, KCl 4.0, MgSO4 0.6, NaH2PO4 0.6, CaCl2 2.5, glucose 11.0, saturated with 95% O2 and 5% CO2, 37°C, pH 7.4). Hearts were then mounted in a Langendorff set-up (Hugo Sachs Elektronik, Harvard Apparatus GmbH, Germany) and perfused at a constant perfusion pressure of 60 mmHg. Electrocardiography (ECG) was measured with three ECG electrodes placed close to the heart. A pacing electrode was placed on the left ventricle. All data were acquired at 2 KHz using the 16-channel PowerLab system (ADInstruments, Oxford, UK), and monitoried by LabChart 7 software (ADInstruments).
After stabilization, the heart was subjected to three subsequent 20-min perfusion periods (Figure 1): 1) baseline recording in normokalemia (4 mM K+), 2) drug (ICA, AP14145, AP30663, dofetilide, or DMSO) + normokalemia, and 3) drug + hypokalemia (2.5 mM K+). At the end of each perfusion period ventricular effective refractory period (VERP) was measured by applying electrical stimulation (five times rheobase) with a fixed interval of 200 ms (S1S1 stimulation) and for every 10th beat an extra stimulus (S2) was applied with 10 ms decrements starting from 150 ms. The VERP was defined as the longest S1–S2 interval failing to elicit a ventricular action potential. Between the VERP recordings, the heart was kept unpaced. QT-intervals and the intrinsic heart rate (HR) were analyzed just prior to electrical stimulation (mean of 100 last beats).
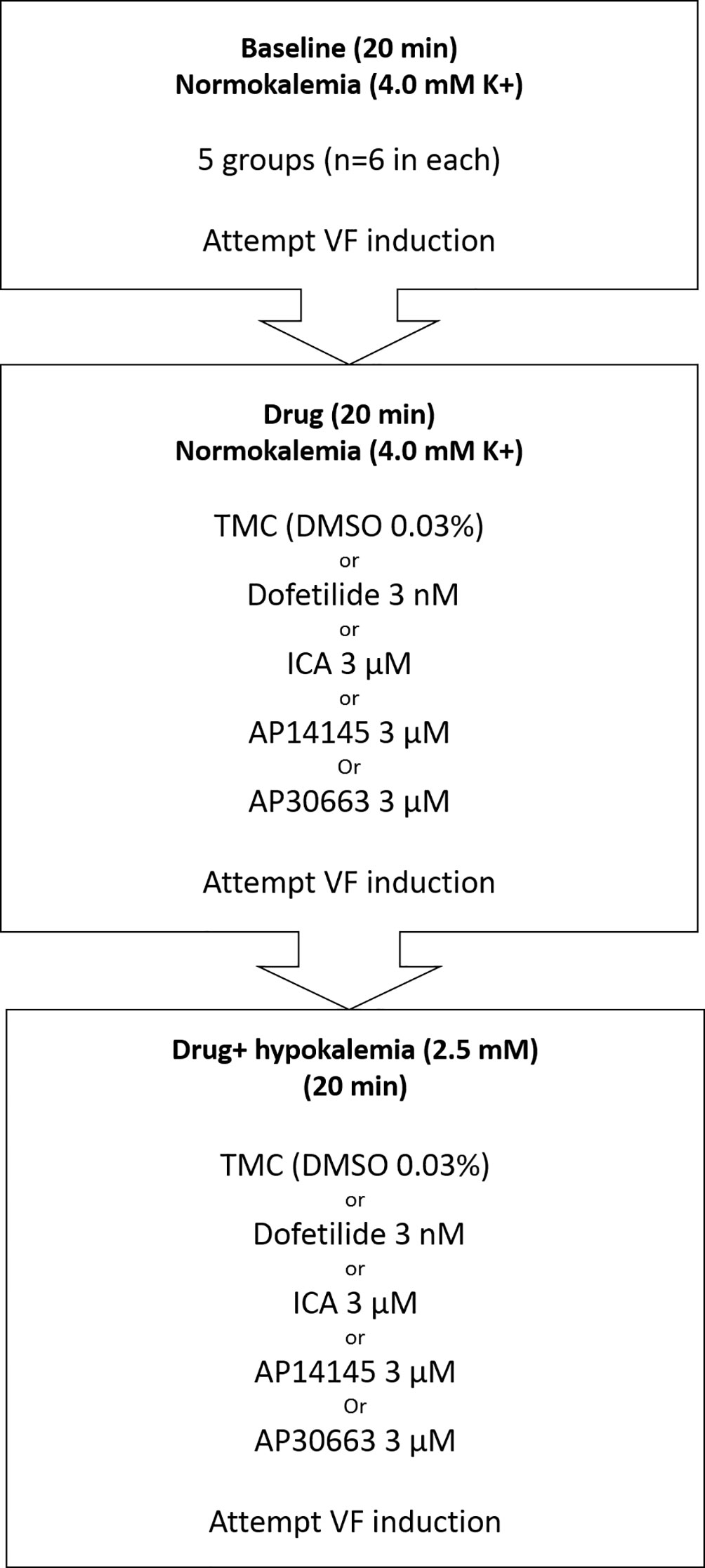
Figure 1 Flowchart of isolated perfused guinea pig experiments and the five experimental groups (TMC, Dofetilide, ICA, AP14145, and AP30663). Three 20-min perfusion periods: baseline, drug, and drug + hypokalemia. VF induction by electrical stimulation (S1–S2) was attempted at the end of each period. TMC, time-matched control; VF, ventricular fibrillation.
Arrhythmia Scoring
The ECG recordings from the three perfusion periods were analyzed for the incidence of ventricular extra systoles (VES) and occurrence of bi-, tri-, and quadrigeminy, VT, and ventricular fibrillation (VF). VT was defined as six or more consecutive VES with an increased HR of >50 beats per minute (bpm). Non-sustained VT (NSVT) < 30 beats; sustained VT (SVT) > 30 beats.
A predefined arrhythmia score system was used to score the severity of the arrhythmia see Table 1. The arrhythmia score was the sum of the individual scores.

Table 1 Arrhythmia scoring.
Data Analysis
All data and figures were analyzed using Prism 8 (GraphPad Software, CA, USA).
In vitro electrophysiology data were analyzed as follows: After removing outliers by ROUT testing (Q = 10%), a paired Wilcoxon test was used to compare the currents during normo- and hypokalemia.
QT-intervals were corrected for HR using the guinea pig specific HR QT correction formula: QTcH = QT/(RR/0.28)0.7861 (Holzgrefe et al., 2014).
Changes from baseline values were compared between each drug group with the TMC group for HR, QTcH, the interval from the peak to the end of the T wave (Tp–Te), VERP, and arrhythmia score by using two-way ANOVA or, in case of missing values, a mixed-effects analysis with Sidak’s multiple comparisons post hoc test. P values are given with three decimals.
Results
Effects of Hypokalemia on outward K+ KCa2.3 Currents
To investigate whether there was a direct effect on KCa2.3 current as a consequence of change in extracellular K+ a number of patch-clamp experiments on heterologously expressed KCa2.3 channels were conducted. As expected from the Nernst equation, when switching from 4 mM to 2.5 mM extracellular K+, the voltage at 0 current was shifted toward more hyperpolarized potentials (from −80.23 ± 1.22 to −87.13 ± 1.75 mV, p < 0.001, n = 10) (Figure 2A), and based on this, a higher driving force for K+-efflux from the cell at +20 mV is achieved. There was slightly more current at +20 mV during hypokalemia than during normokalemia (181 ± 44 pA/pF vs 154 ± 32 pA/pF, Figure 2B).
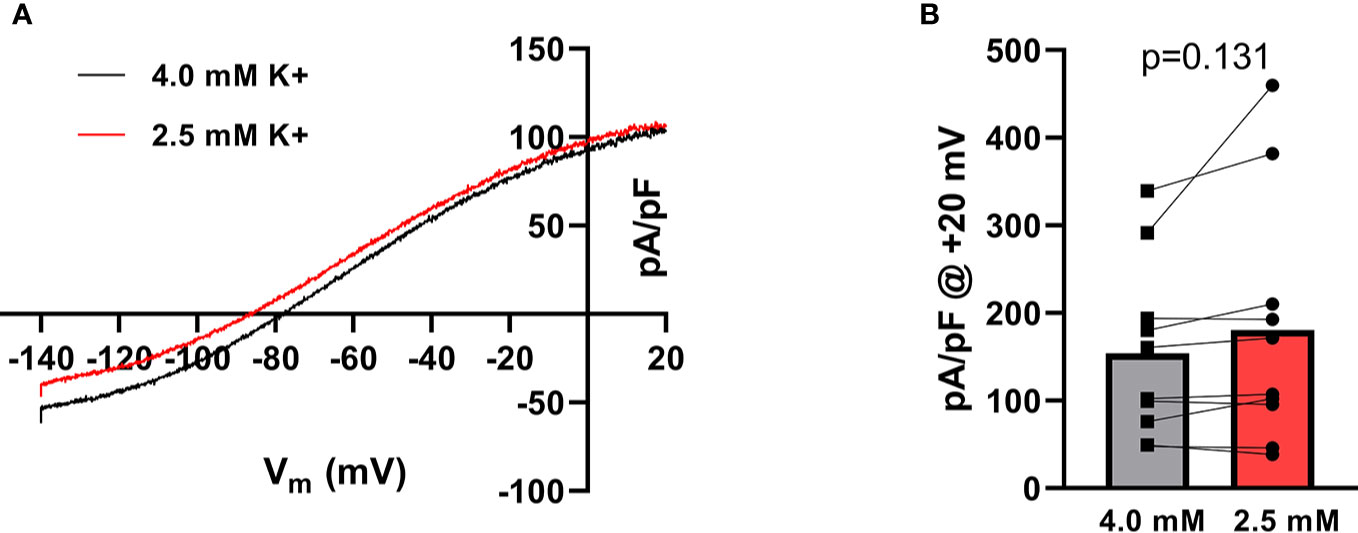
Figure 2 Hypokalemia shifts the reversal potential of K+ in HEK cells expressing KCa2.3. (A) Current–voltage relationship of KCa2.3 current measured at 4.0 mM extracellular K+ and at 2.5 mM K+. Currents were elicited by a voltage-ramp protocol from −140 to +20 mV (200 ms). (B) Summary graph showing the individual current levels at 2.5 mM and 4.0 mM K+, n = 10. Bars represent the mean values.
Effects of Hypokalemia on Repolarization, Heart Rate, and Ventricular Refractoriness
A total of 30 animals were included in the study and randomly assigned to 5 groups: ICA, AP14145, AP30663, dofetilide, or TMC. Despite randomization there were baseline differences in QTcH and HR between the groups (Figure 3). Therefore we evaluated the effect of drugs by comparing changes from baseline (Δ-change = drug-baseline) between TMC and treatment groups for both perfusion periods (drug and drug + hypokalemia) (Table 2).
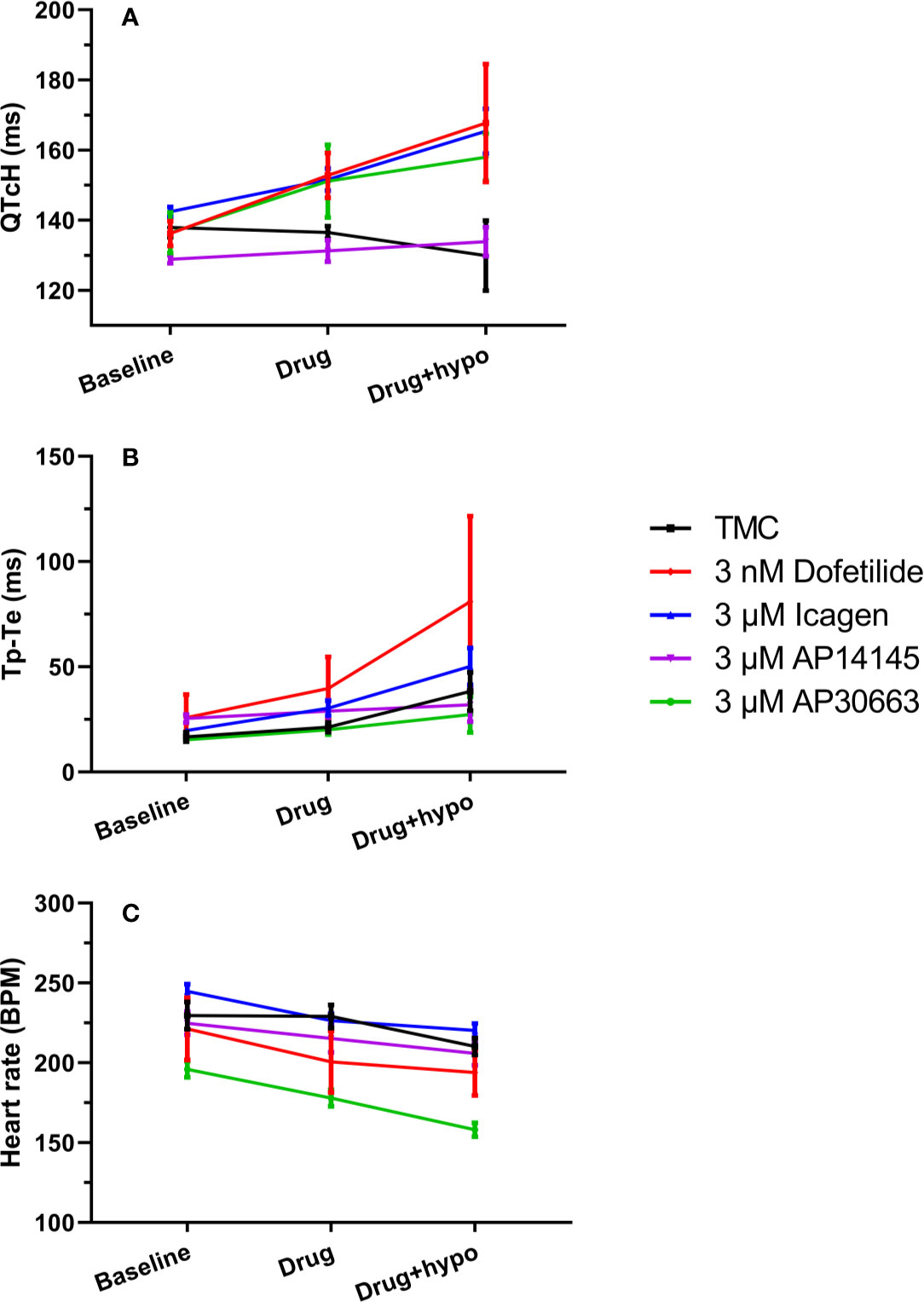
Figure 3 KCa2 channel inhibitors prolong QTc and Tp-Te intervals, and decrease heart rate to a variable degree. (A) Changes in the QT interval corrected for heart rate (QTcH) (B) Changes ins the Tp-Te interval and (C) heart rate in the five groups. Data are presented as mean ± SEM. For QTc and heart rate: N = 6 except for measurement during hypokalemia where n = 5 for ICA and n = 4 for TMC and dofetilide because of spontaneous ventricular fibrillation. TMC, time-matched control.
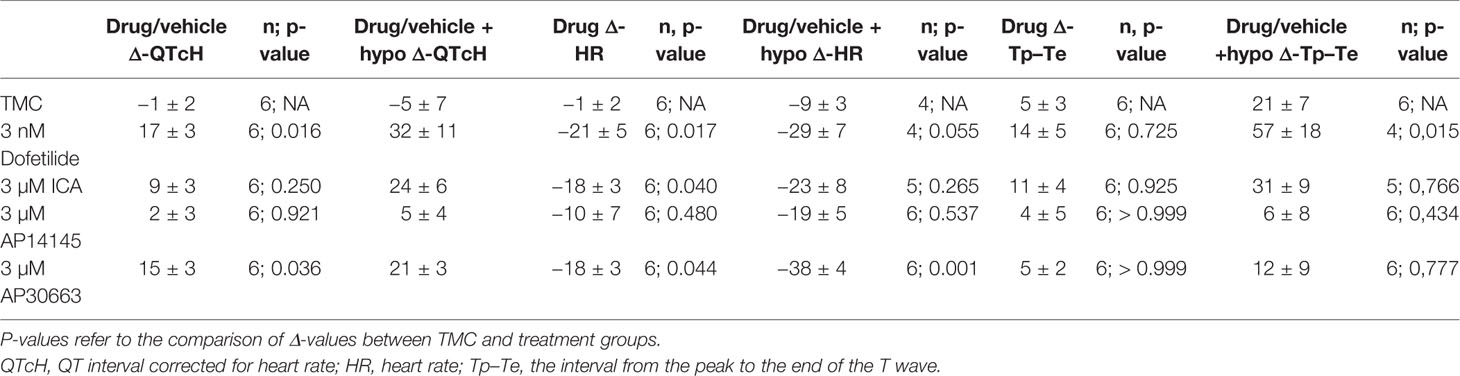
Table 2 Changes (Δdrug − baseline; Δdrug + hypo − baseline) in QTcH, and HR for each group.
The effects of hypokalemia (2.5 mM K+) versus normokalemia (4.0 mM K+) were studied in the TMC group. The QTcH at baseline was 138 ± 3 ms during normokalemia and 130 ± 10 ms during hypokalemia (Figure 3A, Table 2). There was also no effect on HR (Figure 3B, Table 2). While no effects on QT and VERP were observed, the dispersion of ventricular repolarization marked by the Tp–Te interval was increased from 17 ± 2 ms during baseline to 38 ± 9 ms during hypokalemia (Figure 3, Tables 2 and 3) and a ventricular impact of hypokalemia was clearly visible by the increased incidence of arrhythmia (Figure 4). The small impact of hypokalemia on QT and the increased arrhythmia susceptibility reflect what has been previously reported in guinea pigs (Osadchii and Olesen, 2009).
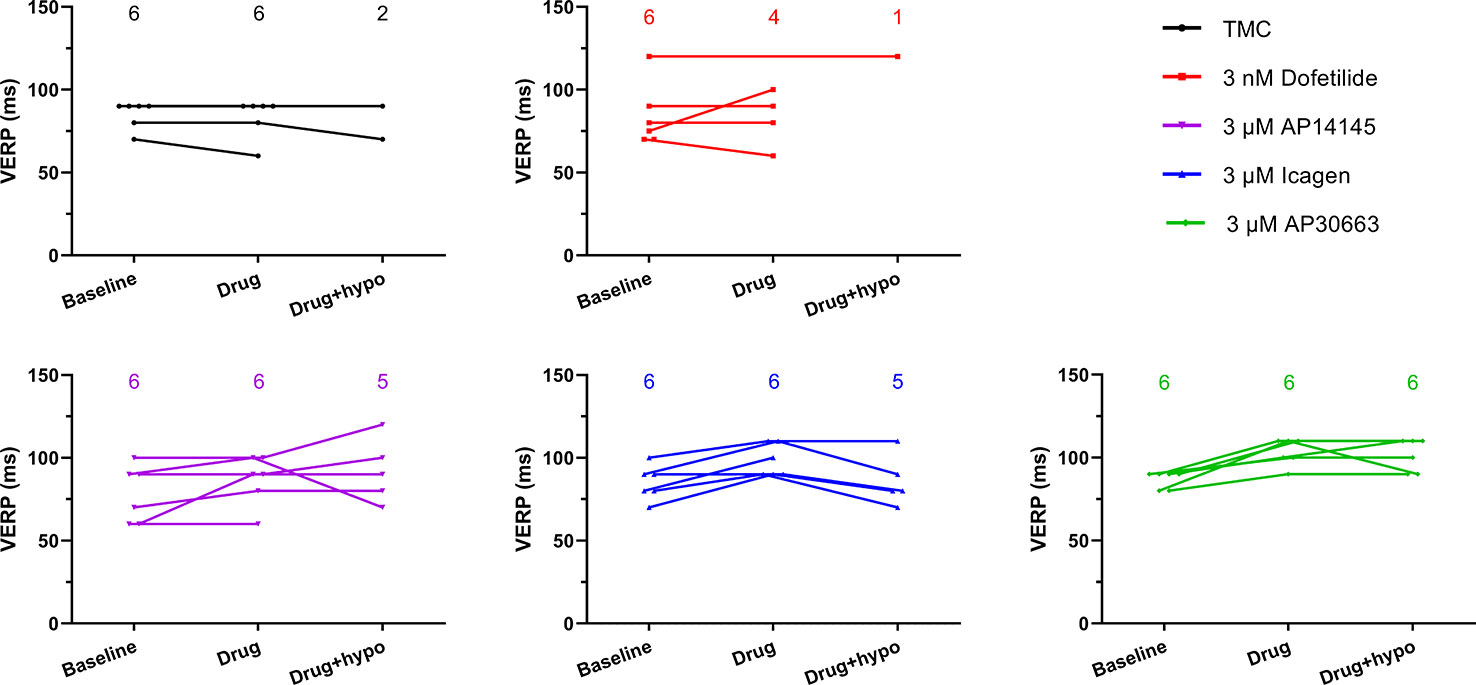
Figure 4 The ventricular effective refractory period (VERP) in the hypokalemia alone time-matched control (TMC) group as well as in the dofetilide group and three KCa2 channel inhibitor groups at three time points. The number of data points for each time point are given in each graph. VERP could not be measured in all cases during hypokalemia due to the presence of ventricular fibrillation.
Effects of Kv11.1 or KCa2 Inhibition on Repolarization, HR, and Ventricular Refractoriness
During normokalemia, dofetilide and AP30663 both increased QTcH versus baseline compared with TMC (Table 2, Figure 3A). The KCa2 channel inhibitor ICA increased QTcH versus baseline, but not in a statistically significant manner compared to TMC. However, none of the KCa2 channel inhibitors nor dofetilide affected the Tp–Te interval during normokalemia compared to the TMC group (Table 2).
Dofetilide, AP30663, and ICA all decreased HR versus baseline compared with TMC (Table 2, Figure 3B).
The VERP at a paced HR of 300 bpm, was increased by the KCa2 channel inhibitors AP30663 and ICA, but not by the Kv11.1 blocker dofetilide. The KCa2 channel inhibitor AP14145 did not significantly change QTcH, VERP, or HR versus baseline when compared to TMC.
Effects of KCa2 or Kv11.1 Inhibition on Ventricular Refractoriness and Repolarization and on HR During Hypokalemia
During hypokalemia dofetilide, AP30663, and ICA all continued to increase QTcH versus baseline compared with TMC (Table 2, Figure 3A). Interestingly, dofetilide increased the dispersion of ventricular repolarization during hypokalemia even more than hypokalemia alone; the Tp–Te increase from baseline was 57 ± 18 ms compared to the increase seen in the TMC group which was increased by 21 ± 7 ms by hypokalemia. None of the KCa2 channel inhibitors affected Tp–Te during hypokalemia compared to the TMC group.
In the TMC group the HR was decreased during hypokalemia, and the decrease in HR versus baseline seen by dofetilide and ICA during hypokalemia was no longer significantly different from the decrease of HR in the TMC group during hypokalemia (Table 2, Figure 3B). The KCa2 channel inhibitor AP30663 decreased the HR to a degree that was greater than the decrease in the TMC group.
The VERP at a paced HR of 300 bpm, was increased by the KCa2 channel inhibitors AP30663 and ICA (Table 3, Figure 4). However, due to induction of VF in the 4/6 hearts during hypokalemia in the TMC group before the VERP could be established, the statistical comparison to the TMC group should be taken with a grain of salt. In the dofetilide group, VF was induced in 5/6 hearts before VERP could be established during hypokalemia. The one dofetilide heart without VF induction had a baseline VERP of 120 which was markedly higher than the mean of the other 5 dofetilide hearts at baseline (76 ± 4 ms). In this particular heart the VERP could not be measured during normokalemia since it could not be paced at a constant rate of 300 bpm. During hypokalemia, however, the VERP remained unchanged from the baseline value of 120 ms.
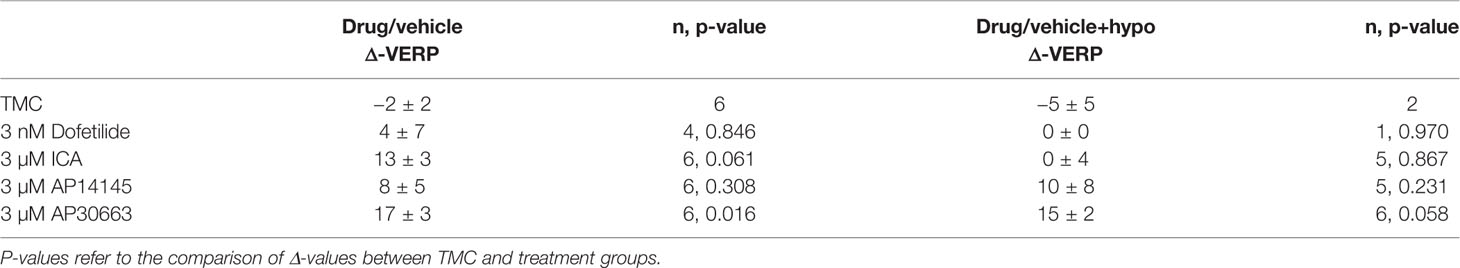
Table 3 Changes (Δdrug − baseline; Δdrug + hypo − baseline) in VERP for each group.
The KCa2 channel inhibitor AP14145 neither changed QTcH, VERP, or HR versus baseline when compared to TMC during hypokalemia as was the case during normokalemia.
KCa2 Channel Inhibition Decreased the Risk of Ventricular Arrhythmia
Hypokalemia increased the incidence of ventricular arrhythmia in the TMC group. Further impairment of the repolarizing IKr current by dofetilide resulted in VF in five out of six animals. Two of them developed spontaneously, one before hypokalemia and one during hypokalemia. In comparison the groups receiving KCa2 inhibitors only presented with VF in 1/6 (ICA and AP14145) or 0/6 (AP30663) (Figure 3).
The presence of VF versus no VF was matched by the arrhythmia scores in the different groups: The TMC and dofetilide were not significantly different, whereas all KCa2 channel inhibitors resulted in a lower arrhythmia score, both before and during hypokalemia as compared to the TMC group (Figure 5, Table 4).
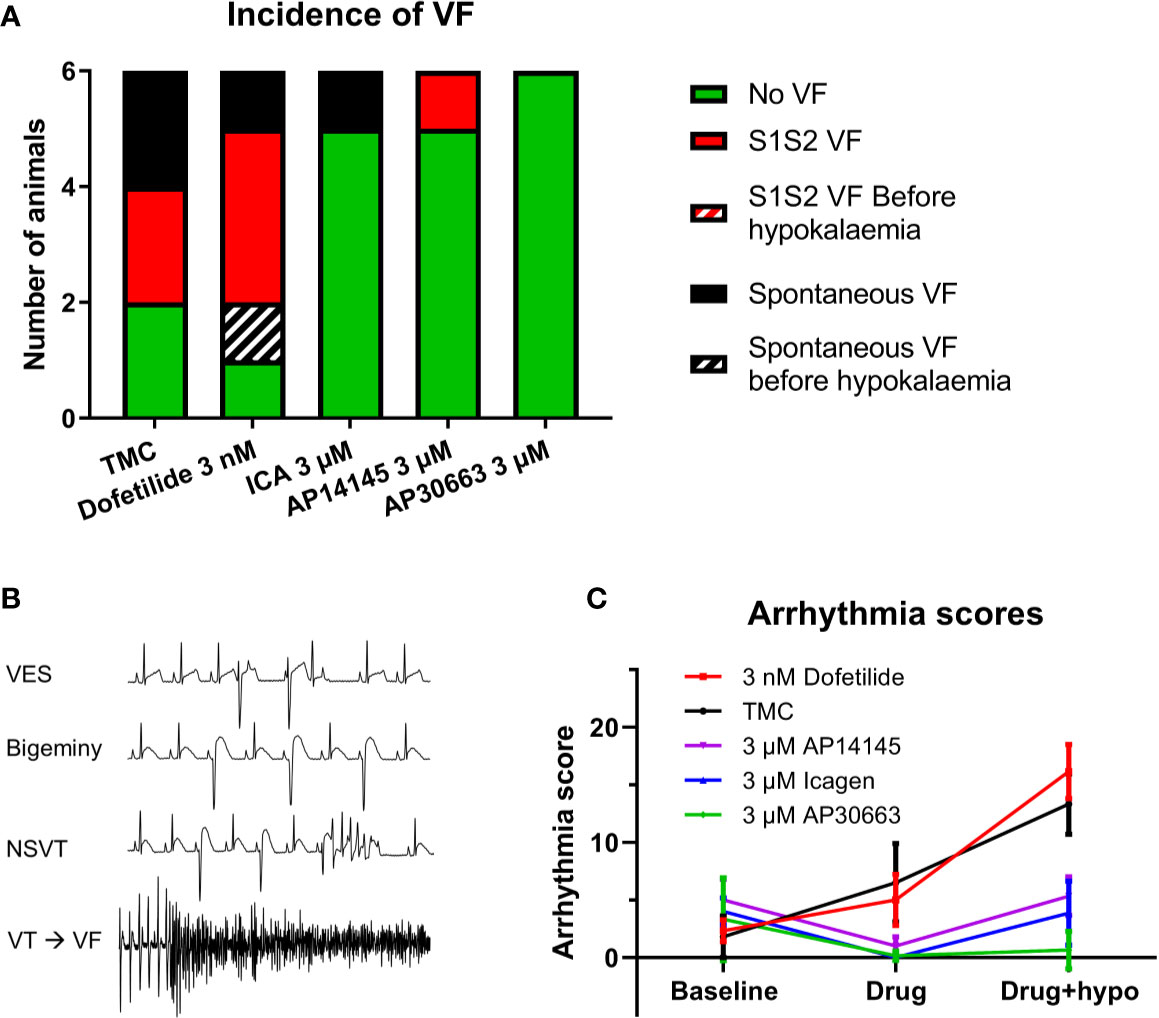
Figure 5 KCa2 channel inhibitors reduces the incidence of ventricular arrhythmia. (A) Incidence of ventricular fibrillation (VF) in the five groups. (B) ECG examples of arrhythmias recorded during hypokalemia + 3 nM Dofetilide. (C) Arrhythmia score for the five groups for each perfusion period (n = 6, data are presented as mean ± SEM). ECG, electrocardiography.
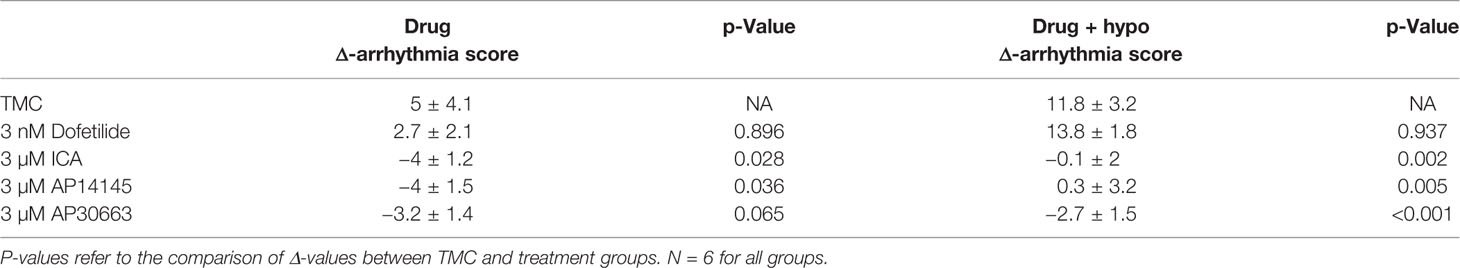
Table 4 Changes (Δdrug − baseline; Δdrug + hypo − baseline) in arrhythmia-score for each group.
Discussion
Hypokalemia was associated with an increased incidence of ventricular arrhythmia. This was also seen in the presence of the class III anti-arrhythmic agent dofetilide. On the contrary, the incidence of ventricular arrhythmias during hypokalemia was reduced in the presence of three structurally and functionally different KCa2 channel inhibitors, ICA, AP14145, and AP30663.
Effects of Induced Hypokalemia at the Cellular Level
Hypokalemia suppresses the cardiac IK1, IKr, and Ito current even though the electrical driving force for K+ efflux is increased. This is because lowered extracellular K+ concentrations speeds up the inactivation of IKr and slows the recovery from inactivation of Ito (Sanguinetti and Jurkiewicz, 1992; Firek and Giles, 1995; Yang et al., 1997) and stabilizes the blocking cations (e.g. Mg2+ and polyamines) in the pore of the Kir2.1 (IK1) channel [for review see (Nichols and Lopatin, 1997)], thereby decreasing the repolarizing outward K+ currents. Whether human KCa2 channels are affected by small changes in extracellular K+ is unknown. Like the Kir2.1 channel, the rat KCa2 channel is also inwardly rectifying partly as a result of voltage-dependent blockade by intracellular divalent cations (Soh and Park, 2001). Moreover, as for the Kir2.1 this block is increased at lower extracellular K+ concentration, effectively lowering the KCa2 current (Soh and Park, 2001). In the present study we were unable to clearly demonstrate that outward human KCa2.3 current was inhibited by lowering the extracellular K+ concentration from 4 mM to 2.5 mM. The discrepancy between our recordings and the study by Soh et al. is likely because we addressed the effects of much smaller changes in extracellular K+, but it could also stem from species or KCa subtype differences. In any case, cardiac KCa2 current will not only be inhibited by hypokalemia via changes in cation block, but also activated by the concomitant increase in intracellular Ca2+ that occurs during hypokalemia. During hypokalemia the prolonged action potential duration and lowered Na+/Ca2+ activity increase intracellular Ca2+ concentrations (Aronsen et al., 2015; Weiss et al., 2017). It has been suggested that this increase in intracellular calcium activates ventricular KCa2 channels, functioning as a protective mechanism against ventricular arrhythmia during hypokalemia (Chan et al., 2015). We addressed this hypothesis in the current study by performing experiments on isolated perfused guinea pig hearts. Lowering the extracellular K+ concentration to 2.5 mM resulted in minor changes of ventricular repolarization but a marked increased risk of ventricular tachycardia, which is similar to what has previously been observed in guinea pigs (Osadchii and Olesen, 2009; Osadchii et al., 2009). That hypokalemia did not produce larger changes in ventricular repolarization is likely explained by the fact that repolarization of ventricular action potential in guinea pigs is more dependent on the IKs (KV7.1/KCNE1) current as compared to IKr (KV11.1) (O’Hara and Rudy, 2012).
Application of the class III anti-arrhythmic agent dofetilide did not prevent ventricular arrhythmia. If anything, dofetilide slightly exacerbated the ventricular arrhythmia, however not to any statistically significant extent. In contrast, the KCa2 channel pore blocker ICA and the negative allosteric modulators of the KCa2 channel AP14145 and AP30663 significantly reduced the ventricular arrhythmia score and the incidence of VF, thereby suggesting that KCa2 channel inhibition protected the ventricles from hypokalemia induced arrhythmia.
In this study we observed QTcH prolongations by AP30663 and ICA, but not with AP14145, and both also seemed to prolong the VERP. None of the KCa2 channel inhibitors affected the Tp–Te interval indicating no effects on the dispersion of ventricular repolarization. In comparison, in the guinea pig heart dofetilide prolonged both QTcH and the Tp–Te interval but did not increase the VERP in the few VERP determinations that were possible to complete. Interestingly, the only heart in the dofetilide group in which VF was not induced had a markedly higher baseline VERP than the rest of the group.
Based on these few observations and the KV11.1 selectivity profile of ICA and AP30663 (IC50 > 32 µM and 4–15 µM respectively (Skibsbye et al., 2014; Bentzen et al., 2020), we speculate that the increase of the QTcH and VERP are mainly but not completely caused by ventricular KCa2 inhibition.
Moreover, one could speculate that the pure KV11.1-driven prolongation of the QTcH and Tp–Te interval observed caused by dofetilide was pro-arrhythmic, during hypokalemia, whereas a KCa2-driven prolongation of the QTcH with no effects on the Tp–Te interval appeared to protect the heart from ventricular arrhythmia. The anti-arrhythmic effects and our findings of small increases in QT and VERP (~5–20 ms) support a role of KCa2 channels in the guinea pig ventricle, albeit minor under physiological conditions, which is in line with our previous studies demonstrating the expression of KCNN2 and KCNN3 in guinea pig ventricular tissue and ventricular anti-arrhythmic effects of KCa2 channel inhibitors (Diness et al., 2015; Kirchhoff et al., 2019).
However, the effect of KCa2 channel inhibition on atrial refractoriness has consistently been found to be greater than the ventricular effects and hence KCa2 channels seem to show functional atrial selectivity (Diness et al., 2010; Qi et al., 2014; Diness et al., 2017; Kirchhoff et al., 2019; Bentzen et al., 2020; Diness et al., 2020). Our data during hypokalemia are different from the findings by Chan et al. who suggested that KCa2 channel inhibition by the toxin apamin facilitated VF induction (Chan et al., 2015) in rabbits. Differences in species and methodology might explain this discrepancy, since they used rabbits, and optical mapping. Another difference between our study and that of Chan that could explain some of the discrepancies is the use of the pore blocking peptide apamin versus our three small molecule KCa2 inhibitors of which two (AP30663 and AP14145) are negative allosteric modulators and one (ICA) is a pore blocker. Furthermore, AP14145 and AP30663 in higher concentrations (10 µM) also inhibit the late NaV1.5 current (Bentzen et al., 2020). Hypokalemia-induced increased intracellular Ca2+ has been proposed to activate Ca2+-calmodulin kinase thereby inducing late Na+ currents and facilitating arrhythmia (Pezhouman et al., 2015). We do not know whether this could add to the anti-arrhythmic effects and/or to a modification of the increase in QTcH observed under hypokalemia of these two drugs.
In a recent study, Tazmini et al. explored the cellular mechanisms behind ventricular and atrial arrhythmias during hypokalemia and found that hypokalemia induced Ca2+ overload, Ca2+ waves, and both early and delayed afterdepolarizations in both ventricular and in atrial cells which contain t-tubules (Tazmini et al., 2020). On the contrary, calcium was not important for development of early afterdepolarizations in atrial cardiomyocytes lacking t-tubules. In the present study we did not observe any hypokalemia induced AF episodes and based on the findings of Tazmini et al. it would be interesting to investigate if KCa2 channel inhibition plays a role in this.
Three Different KCa2 Inhibitors—Similarities and Differences
We studied three structurally and functionally different KCa2 channel inhibitors, the pore blocker ICA, and the two negative allosteric modulators of KCa2 channels AP14145 and AP30663, which inhibit the KCa2 channel by shifting the calcium activation curve to higher intracellular calcium concentrations. They all prolong the QTc (2-15 ms), and VERP but to varying degrees, with AP30663 showing the largest prolongation. Interestingly despite differences in mechanism of inhibition and effects on QTc all significantly reduced and prevented the incidence of VF during hypokalemia.
Conclusions
Induced hypokalemia was associated with an increased risk of spontaneous or induced ventricular arrhythmia and VF, a risk that was also seen after administration of dofetilide. In contrast, the structurally and functionally different KCa2 channel inhibitors, ICA, AP14145, and AP30663 decreased this risk and prevented hypokalemia-induced VF. These results support that the KCa2 inhibitors, particularly AP30663, may be associated with a better safety and tolerability profile than dofetilide.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The animal study was reviewed and approved by The Danish Ministry of Environment and Food (license No. 2014-15-0201-00390).
Author Contributions
LA and SB contributed to the acquisition, analysis, and interpretation of data for the work. MS contributed to analysis of the data. JD and BB made the first draft of the manuscript, and contributed to the conception and design of the work as well as the analysis and interpretation of data. All authors contributed to drafting the work and revising it critically for important intellectual content
Funding
This work was supported by the Innovation Fund Denmark, Wellcome Trust [Grant Number 100406/Z/12/Z].
Conflict of Interest
All authors are fully or partly employed in Acesion Pharma and JD, MG, and BB are inventors of Acesion Pharma patents within the field of KCa2 channels.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aronsen, J. M., Skogestad, J., Lewalle, A., Louch, W. E., Hougen, K., Stokke, M. K., et al. (2015). Hypokalaemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing Na+,K+-ATPase α2 activity. J. Physiol. 15593 (6), 1509–1521. doi: 10.1113/jphysiol.2014.279893
Bentzen, B. H., Bomholtz, S. H., Simó-Vicens, R., Folkersen, L., Abildgaard, L., Speerschneider, T., et al. (2020). Mechanisms of action of the KCa2 negative modulator AP30663, a novel compound in development for treatment of atrial fibrillation in man. Front. Pharmacol. doi: 10.3389/fphar.2020.00610
Bonilla, I. M., Long, V. P., Vargas-Pinto, P., Wright, P., Belevych, A., Lou, Q., et al. (2014). Calcium-activated potassium current modulates ventricular repolarization in chronic heart failure. PloS One 9 (10), e108824. doi: 10.1371/journal.pone.0108824
Chan, Y.-H., Tsai, W.-C., Ko, J.-S., Yin, D., Chang, P.-C., Rubart, M., et al. (2015). Small-Conductance Calcium-Activated Potassium Current Is Activated During Hypokalemia and Masks Short-Term Cardiac Memory Induced by Ventricular Pacing. Circulation 132 (15), 1377–1386. doi: 10.1161/CIRCULATIONAHA.114.015125
Chang, P.-C., Hsieh, Y.-C., Hsueh, C.-H., Weiss, J. N., Lin, S.-F., Chen, P.-S. (2013a). Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart Rhythm. 10 (10), 1516–1524. doi: 10.1016/j.hrthm.2013.07.003
Chang, P.-C., Turker, I., Lopshire, J. C., Masroor, S., Nguyen, B.-L., Tao, W., et al. (2013b). Heterogeneous upregulation of apamin-sensitive potassium currents in failing human ventricles. J. Am. Heart Assoc. 32 (1), e004713. doi: 10.1161/JAHA.112.004713
Chua, S.-K., Chang, P.-C., Maruyama, M., Turker, I., Shinohara, T., Shen, M. J., et al. (2011). Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ. Res. 108 (8), 971–979. doi: 10.1161/CIRCRESAHA.110.238386
Diness, J. G., Sørensen, U. S., Nissen, J. D., Al-Shahib, B., Jespersen, T., Grunnet, M., et al. (2010). Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ. Arrhythm Electrophysiol. 3 (4), 380–390. doi: 10.1161/CIRCEP.110.957407
Diness, J. G., Kirchhoff, J. E., Sheykhzade, M., Jespersen, T., Grunnet, M. (2015). Antiarrhythmic Effect of Either Negative Modulation or Blockade of Small Conductance Ca2+-activated K+ Channels on Ventricular Fibrillation in Guinea Pig Langendorff-perfused Heart. J. Cardiovasc. Pharmacol. 66 (3), 294–299. doi: 10.1097/FJC.0000000000000278
Diness, J. G., Skibsbye, L., Simó-Vicens, R., Santos, J. L., Lundegaard, P., Citerni, C., et al. (2017). Termination of Vernakalant-Resistant Atrial Fibrillation by Inhibition of Small-Conductance Ca2+-Activated K+ Channels in Pigs. Circ. Arrhythm Electrophysiol. 10 (10), e005125. doi: 10.1161/CIRCEP.117.005125
Diness, J. G., Kirchhoff, J. E., Speerschneider, T., Abildgaard, L., Edvardsson, N., Sørensen, U. S., et al. (2020). The KCa2 Channel Inhibitor AP30663 Selectively Increases Atrial Refractoriness, Converts Vernakalant-Resistant Atrial Fibrillation and Prevents Its Reinduction in Conscious Pigs. Front. Pharmacol. 11, 159. doi: 10.3389/fphar.2020.00159
Firek, L., Giles, W. R. (1995). Outward currents underlying repolarization in human atrial myocytes. Cardiovasc. Res. 30 (1), 31–38. doi: 10.1016/S0008-6363(95)00014-3
Gui, L., Bao, Z., Jia, Y., Qin, X., Cheng, Z. J., Zhu, J., et al. (2013). Ventricular tachyarrhythmias in rats with acute myocardial infarction involves activation of small-conductance Ca2+-activated K+ channels. Am. J. Physiol. Heart Circ. Physiol. 304 (1), H118–H130. doi: 10.1152/ajpheart.00820.2011
Haugaard, M. M., Hesselkilde, E. Z., Pehrson, S., Carstensen, H., Flethøj, M., Præstegaard, K. F., et al. (2015). Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm. 12 (4), 825–835. doi: 10.1016/j.hrthm.2014.12.028
Holzgrefe, H., Ferber, G., Champeroux, P., Gill, M., Honda, M., Greiter-Wilke, A., et al. (2014). Preclinical QT safety assessment: cross-species comparisons and human translation from an industry consortium. J. Pharmacol. Toxicol. Methods 69 (1), 61–101. doi: 10.1016/j.vascn.2013.05.004
Hundahl, L. A., Sattler, S. M., Skibsbye, L., Diness, J. G., Tfelt-Hansen, J., Jespersen, T. (2017). Pharmacological blockade of small conductance Ca2+-activated K+ channels by ICA reduces arrhythmic load in rats with acute myocardial infarction. Pflugers Arch. 469 (5–6), 739–750. doi: 10.1007/s00424-017-1962-6
Jensen, H. K., Brabrand, M., Vinholt, P. J., Hallas, J., Lassen, A. T. (2015). Hypokalemia in acute medical patients: risk factors and prognosis. Am. J. Med. 128 (1), 60–67.e1. doi: 10.1016/j.amjmed.2014.07.022
Kirchhoff, J. E., Skarsfeldt, M. A., Muthukumarasamy, K. M., Simó-Vicens, R., Bomholtz, S. H., Abildgaard, L., et al. (2019). The KCa2 Channel Inhibitor AP14145, But Not Dofetilide or Ondansetron, Provides Functional Atrial Selectivity in Guinea Pig Hearts. Front. Pharmacol. 10, 668. doi: 10.3389/fphar.2019.00668
McKibbin, J. K., Pocock, W. A., Barlow, J. B., Millar, R. N., Obel, I. W. (1984). Sotalol, hypokalaemia, syncope, and torsade de pointes. Br. Heart J. 51 (2), 157–162. doi: 10.1136/hrt.51.2.157
Nichols, C. G., Lopatin, A. N. (1997). Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191. doi: 10.1146/annurev.physiol.59.1.171
O’Hara, T., Rudy, Y. (2012). Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 302 (5), H1023–H1030. doi: 10.1152/ajpheart.00785.2011
Osadchii, O. E., Olesen, S. P. (2009). Electrophysiological determinants of hypokalaemia-induced arrhythmogenicity in the guinea-pig heart. Acta Physiol. Oxf Engl. 197 (4), 273–287. doi: 10.1111/j.1748-1716.2009.02028.x
Osadchii, O. E., Bentzen, B. H., Olesen, S. P. (2009). Chamber-specific effects of hypokalaemia on ventricular arrhythmogenicity in isolated, perfused guinea-pig heart. Exp. Physiol. 94 (4), 434–446. doi: 10.1113/expphysiol.2008.045567
Pezhouman, A., Singh, N., Song, Z., Nivala, M., Eskandari, A., Cao, H., et al. (2015). Molecular Basis of Hypokalemia-Induced Ventricular Fibrillation. Circulation 132 (16), 1528–1537. doi: 10.1161/CIRCULATIONAHA.115.016217
Qi, X.-Y., Diness, J. G., Brundel, B. J. J. M., Zhou, X.-B., Naud, P., Wu, C.-T., et al. (2014). Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129 (4), 430–440. doi: 10.1161/CIRCULATIONAHA.113.003019
Redfern, W. S., Carlsson, L., Davis, A. S., Lynch, W. G., MacKenzie, I., Palethorpe, S., et al. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58 (1), 32–45. doi: 10.1016/S0008-6363(02)00846-5
Sanguinetti, M. C., Jurkiewicz, N. K. (1992). Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflugers Arch. 420 (2), 180–186. doi: 10.1007/BF00374988
Skibsbye, L., Poulet, C., Diness, J. G., Bentzen, B. H., Yuan, L., Kappert, U., et al. (2014). Small conductance calcium activated potassium (SK) channels contribute to action potential repolarisation in human atria. Cardiovasc. Res. 103 (1), 156–167. doi: 10.1093/cvr/cvu121
Soh, H., Park, C. S. (2001). Inwardly rectifying current-voltage relationship of small-conductance Ca2+-activated K+ channels rendered by intracellular divalent cation blockade. Biophys. J. 80 (5), 2207–2215. doi: 10.1016/S0006-3495(01)76193-0
Tazmini, K., Frisk, M., Lewalle, A., Laasmaa, M., Morotti, S., Lipsett, D. B., et al. (2020). Hypokalemia Promotes Arrhythmia by Distinct Mechanisms in Atrial and Ventricular Myocytes. Circ. Res. 126 (7), 889–906. doi: 10.1161/CIRCRESAHA.119.315641
Weiss, J. N., Qu, Z., Shivkumar, K. (2017). Electrophysiology of Hypokalemia and Hyperkalemia. Circ. Arrhythm Electrophysiol. 10 (3), e004667. doi: 10.1161/CIRCEP.116.004667
Keywords: KCa2 channel inhibitor, hypokalemia, dofetilide, Kv11.1 blockers, SK channel, SK channel inhibitor, arrhythmia, antiarrhythmic drug
Citation: Diness JG, Abildgaard L, Bomholtz SH, Skarsfeldt MA, Edvardsson N, Sørensen US, Grunnet M and Bentzen BH (2020) Inhibition of KCa2 Channels Decreased the Risk of Ventricular Arrhythmia in the Guinea Pig Heart During Induced Hypokalemia. Front. Pharmacol. 11:749. doi: 10.3389/fphar.2020.00749
Received: 26 February 2020; Accepted: 06 May 2020;
Published: 20 May 2020.
Edited by:
Eleonora Grandi, University of California, Davis, United StatesReviewed by:
Na Li, Baylor College of Medicine, United StatesJordi Heijman, Maastricht University, Netherlands
Copyright © 2020 Diness, Abildgaard, Bomholtz, Skarsfeldt, Edvardsson, Sørensen, Grunnet and Bentzen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonas Goldin Diness, jgd@acesionpharma.com; Bo Hjorth Bentzen, bobe@sund.ku.dk