Corrigendum: The NaV1.7 Channel Subtype as an Antinociceptive Target for Spider Toxins in Adult Dorsal Root Ganglia Neurons
 Tânia C. Gonçalves
Tânia C. Gonçalves Evelyne Benoit
Evelyne Benoit Michel Partiseti1
Michel Partiseti1 Denis Servent
Denis Servent- 1Sanofi R&D, Integrated Drug Discovery – High Content Biology, Paris, France
- 2Service d’Ingénierie Moléculaire des Protéines, CEA de Saclay, Université Paris-Saclay, Gif-sur-Yvette, France
- 3Institut des Neurosciences Paris-Saclay, UMR CNRS/Université Paris-Sud 9197, Gif-sur-Yvette, France
Although necessary for human survival, pain may sometimes become pathologic if long-lasting and associated with alterations in its signaling pathway. Opioid painkillers are officially used to treat moderate to severe, and even mild, pain. However, the consequent strong and not so rare complications that occur, including addiction and overdose, combined with pain management costs, remain an important societal and economic concern. In this context, animal venom toxins represent an original source of antinociceptive peptides that mainly target ion channels (such as ASICs as well as TRP, CaV, KV and NaV channels) involved in pain transmission. The present review aims to highlight the NaV1.7 channel subtype as an antinociceptive target for spider toxins in adult dorsal root ganglia neurons. It will detail (i) the characteristics of these primary sensory neurons, the first ones in contact with pain stimulus and conveying the nociceptive message, (ii) the electrophysiological properties of the different NaV channel subtypes expressed in these neurons, with a particular attention on the NaV1.7 subtype, an antinociceptive target of choice that has been validated by human genetic evidence, and (iii) the features of spider venom toxins, shaped of inhibitory cysteine knot motif, that present high affinity for the NaV1.7 subtype associated with evidenced analgesic efficacy in animal models.
Introduction
According to the International Association for the Study of Pain, at least 10% of the world’s population suffer from pain since 1 over 10 adults has experienced or had (acute, chronic, intermittent or combined) pain with a median of suffering time around 7 years (Goldberg and McGee, 2011). The unpleasant sensation of pain is necessary to maintain the body integrity. However, it is often accompanied by long-term complications not only limited to comorbidities, as depression, but also including social and economic concerns as inability to work, social isolation and intrusive thoughts, leading to costs of more than 600 billion US dollars annually (Holmes, 2016). Pain care is thus a global public health priority whose management must be regulated in its totality by policies.
Nowadays, mild to moderate pain may be treated effectively with a combination of physical modalities (e.g., ice, rest and splints) and non-opioid analgesics (e.g., non-steroidal anti-inflammatory drugs, acetaminophen or other adjuvant medications). In contrast, the health system is pushed into its limits to treat debilitating chronic pain because the therapy is ineffective and/or associated with devastating effects. Indeed, management of chronic and severe pain, especially related to cancers or neuropathies, often requires opioids (Savage et al., 2008). Unfortunately, the opioid abuse and overdose often lead to death, which stimulates industries and academics to find an alternative with acceptable undesired effects (Negus, 2018).
In this context, the likely promising target for therapeutic treatment to fight pain and avoid central side-effects is the neuron located in the periphery dorsal root ganglia (DRG) which conveys pain from the skin and tendons to the central nervous system (CNS). The DRG neurons are well-known to express various families of transmembrane proteins, including ion channels, G-protein-coupled receptors (GPCRs) and gap junctions/pannexins (Pan et al., 2008; Spray and Hanani, 2017; Yekkirala et al., 2017). Among the ion channel family, the most extensively studied targets for pain treatment are voltage-gated calcium (CaV) and sodium (NaV) channels. In particular, it is well established that small molecules that target NaV channels attenuate chronic and debilitating pain in humans, as exemplified by tetrodotoxin (TTX). However, due to a lack of selectivity, pronounced side-effects have been described, such as nausea, dizziness, oral numbness and tingling, limiting thus the therapeutic development of this molecule (Hagen et al., 2017). During the last decade, the attraction of scientists for the NaV1.7 channel subtype has greatly increased, due to its validation by human genetic diseases as a pain target. Many studies have been reported in the literature to describe gating modulators or pore blockers that affect the functional properties of this subtype (Vetter et al., 2017). Therefore, the present review will focus on the fascinating spider venom toxins which represent an original source of proteins possessing complex structures associated with specific electrophysiological effects and prone to be more selective for the NaV1.7 channel subtype mainly expressed in DRG neurons.
Primary Sensory Neurons as Front Door for Pain
The cellular elements involved in pain transmission from the peripheral to the CNS are detailed in Figure 1. The noxious information is first detected by the nociceptors of peripheral and visceral tissue, and then conveyed by the dendrites of primary sensory neurons (PSNs). The nociceptors are located at the level of free nerve endings of Aδ and C fibers of PSNs that respond to noxious stimuli and are widely found throughout skin and internal tissue. Three main types of pain receptors exist: the thermal, the mechanical and the polymodal receptors, activated by temperature, high pressure and mechanical, thermal or/and chemical stimuli, respectively (Figure 1, Box 1). The PSNs are pseudo-bipolar neurons which send their axons, components of dorsal roots, to the laminas I, II and V of the dorsal horn of spinal cord and establish synapses with the dendrites of secondary sensory neurons (SSNs) (Figure 1, Box 2). The SSNs, in turn, bring the noxious information to the hypothalamus and connect to tertiary sensory neurons (TSNs) whose cell bodies constitute, in part, the brain cortex (Figure 1, Box 3). At each CNS level, the information is integrated and modulated by different ascending/descending control systems such as the medullary control, named “gate control,” and the diffuse inhibitory control including the noradrenergic and serotoninergic pathways induced by nociception from the higher centers to the dorsal horn, giving the affective, sensory and cognitive dimensions to the human experience of pain (Porreca and Navratilova, 2017).
FIGURE 1
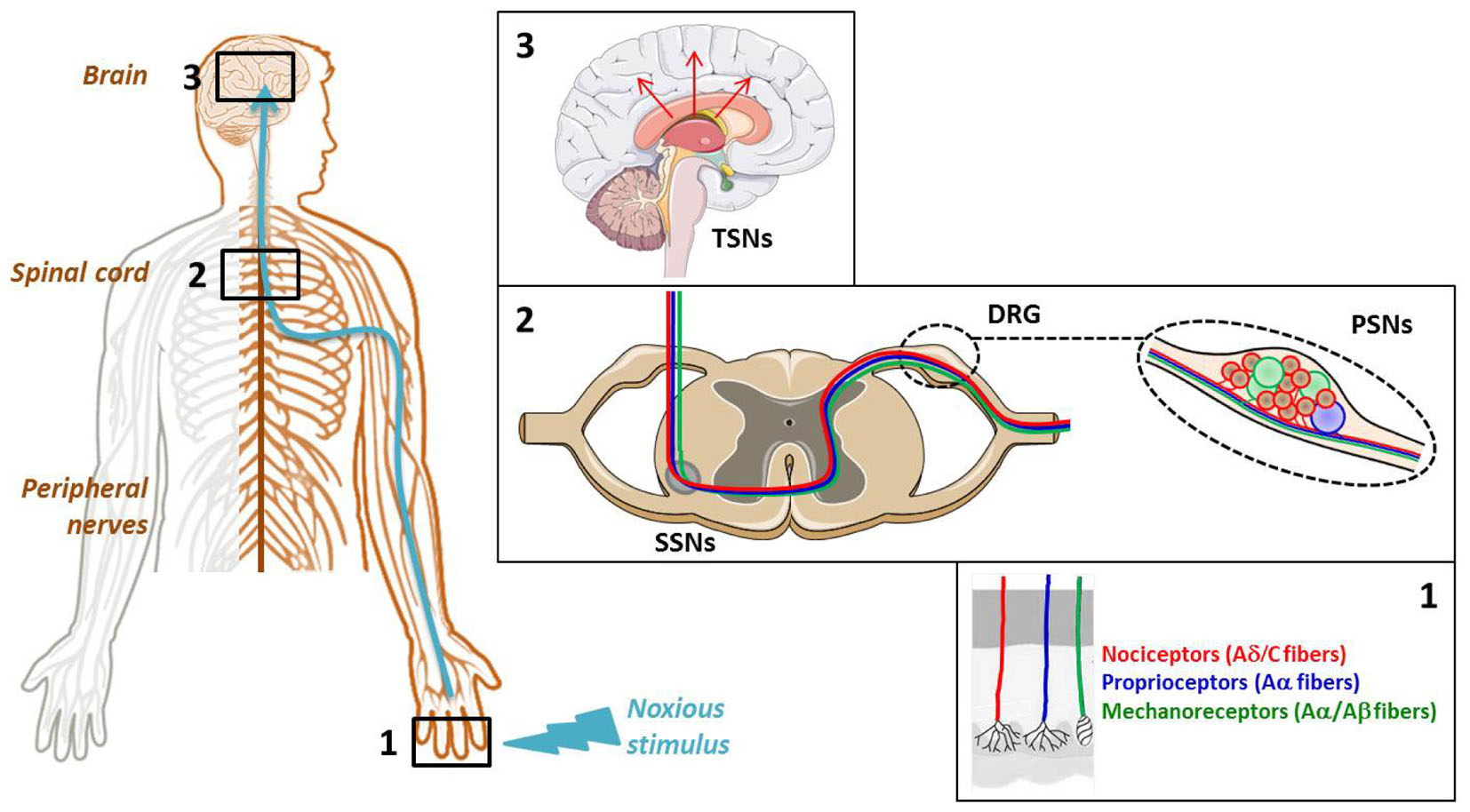
FIGURE 1. Cellular elements involved in pain transmission from the peripheral to the central nervous system (CNS). (Box 1) The pain (thermal, high pressure, mechanical, chemical) information is first detected by the receptors located at the level of free nerve endings of primary sensory neuron (PSN) fibers. (Box 2) Then, it is conveyed by the dendrites of these neurons, components of dorsal root ganglia (DRG), to the dorsal horn of spinal cord where it is transmitted to the dendrites of secondary sensory neurons (SSNs). (Box 3) Finally, it is brought to the hypothalamus via the tertiary sensory neurons (TSNs) whose cell bodies constitute, in part, the brain cortex.
The neuron bodies of PSNs constitute the 31 pairs of DRG, coming out all along the spinal marrow: 8 cervical (C1-C7, note that the first cervical spinal nerve is born above C1 and the eighth one below C7), 12 thoracic (T1–T12), 5 lumbar (L1–L5), 5 sacred (S1–S5), and 1 coccygeal (Co) which is vestigial. The cranial sensory (trigeminal or Gasser’s) ganglion (nerve V) conveys facial skin sensitivity, the spiral (or cochlear) and vestibular (or Scarpa’s) ganglia (nerve VIII) serve the hearing and balance senses, respectively, and the geniculate ganglion (nerve VII) transfers facial sensations, with the contribution of the superior and inferior (or petrous) ganglia of glossopharyngeal nerve (nerve IX) and the superior (or jugular) and inferior (or nodose) ganglia of vagus nerve (nerve X).
Dorsal root ganglia present a rich capillary bed in cell body area (Figure 2), with the particularity of high fenestrations between two endothelial cells being permeable to low and high molecular weight compounds (Petterson and Olsson, 1989; Parke and Whalen, 2002; Jimenez-Andrade et al., 2008; Berta et al., 2017). In contrast to the cell body area, the nerve fiber area wrapped by the epineural sheath, i.e., the dura mater continuum in peripheral nervous system (PNS), presents a blood-nerve barrier similar to the CNS blood-brain barrier (BNB), with a lot of tight junctions between cells that prevent the passage of unwanted drugs (Jimenez-Andrade et al., 2008; Liu et al., 2018).
FIGURE 2
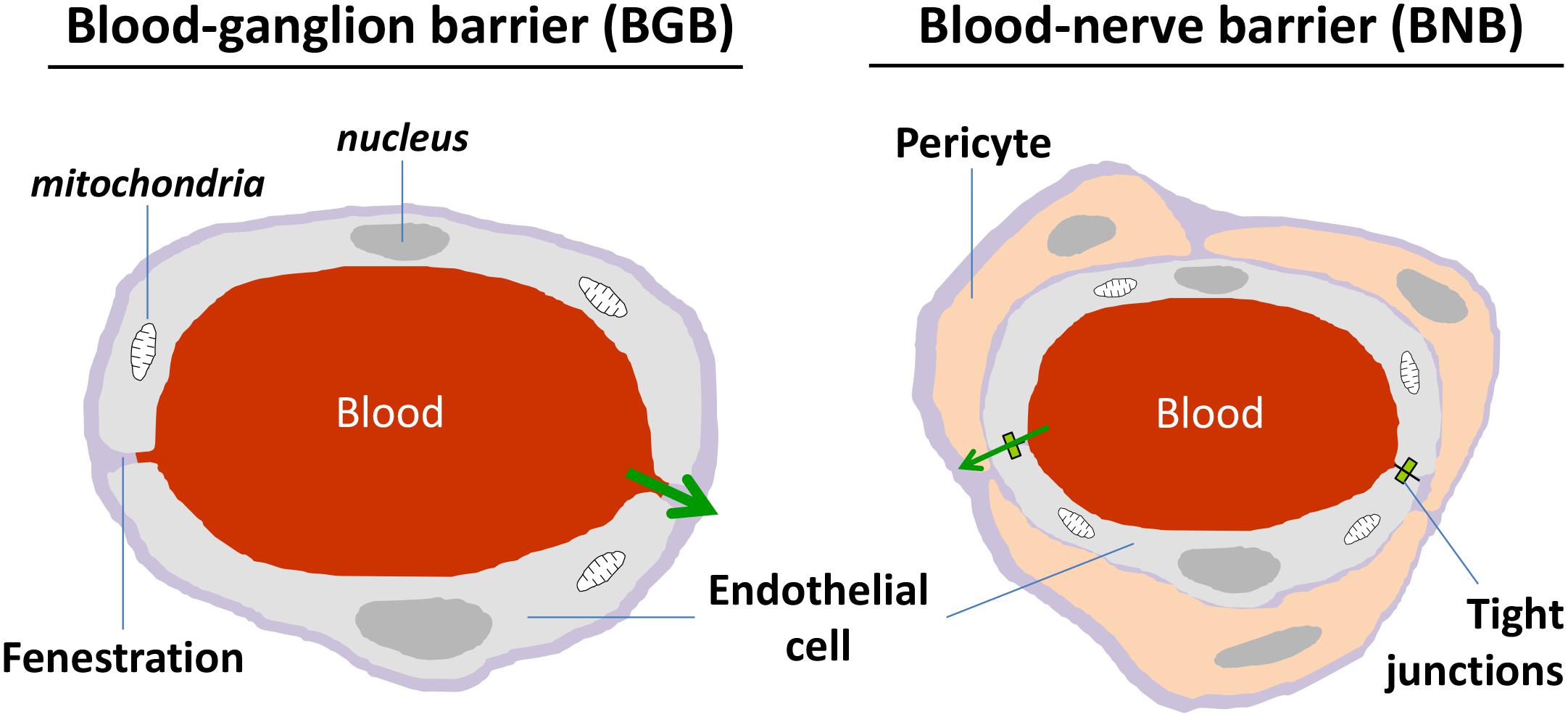
FIGURE 2. Schematic representation of morphological characteristics of ganglion and nerve capillaries. Ganglion capillaries differ from nerve ones by the presence of fenestration and absence of narrow tight junctions. Nerve endothelial cells are surrounded by pericytes.
Soma of PSNs relaying the sensory information are part of the DRG which also contain other different cell types such as glial cells, endothelial cells and macrophages. Two groups of DRG neurons may be distinguished using light and electronic microscopy: the small dark neurons (cross-sectional area ≤ 800 μm2 and diameter ≤ 30 μm) composed of high threshold, slowly-conducting unmyelinated (C) and/or thinly myelinated (Aδ) nerve fibers, and the large light neurons (cross-sectional area > 800 μm2 and diameter > 30 μm) constituted of low threshold, fast-conducting thickly-myelinated (Aα and Aβ) nerve fibers (Elliott and Elliott, 1993; Taddese et al., 1995; Ho and O’Leary, 2011). The small DRG neurons that convey mainly pain message are subdivided into two groups: the non-peptidergic and the peptidergic neurons, depending on isolectin-IB4 labeling (Table 1). This subdivision of small neurons results from the expression level of runt-related transcription factor 1 (RUNX1), responsible for neuropeptide expression, regulated by the nerve growth factor (NGF) signaling during cell growth and differentiation (Luo et al., 2007). In adult DRG neurons, RUNX and neurogenin transcription factors regulate the expression of (i) glial cell line-derivated neurotrophic factor (GDNF) and tyrosine kinase c-Ret co-receptors (allowing the GDNF-ligand expression required for cell post-natal survival and indicative of non-peptidergic neurons), and (ii) the tropomyosin receptor-kinase receptors (TrkA, B and C which bind NGF or brain-derived neurotrophic factor, neurotrophin-4 and neurotrophin-3, respectively). The expression of growth factor receptors is therefore of great help to better characterizing adult DRG neurons (Ernsberger, 2009). Although only the small DRG neurons which are not labeled by isolectin-IB4 are peptidergic, the high dense-core vesicles of large neurons may also contain peptides, depending on both the vesicle size and the nerve condition, i.e., normal or injured (Wiesenfeld-Hallin and Xu, 2001). The peptidergic neurons deliver not only substance P and calcitonin gene-related peptide, but also somatostatin, vasoactive intestinal peptide and cholecystokinin. When released in the CNS areas associated with pain transmission, these neuropeptides affect the expression pattern of SSNs, PSNs and peripheral organs (Moraes et al., 2014). The type of cytoskeleton neurofilaments present in DRG neurons is correlated with both the axonal diameter and the conduction velocity of action potential: intermediate neurofilament peripherin (57 kDa) is expressed in slowly-conducting unmyelinated (C) and/or thinly myelinated (Aδ) nerve fibers whereas the heavy neurofilament NF200 (200 kDa) is expressed in fast-conducting thickly-myelinated (Aα and Aβ) nerve fibers. The expression of the cell adhesion nectin-like molecule 1, interacting with the cytoskeleton, reflects the myelination level of nerve fibers (Ho and O’Leary, 2011).
TABLE 1
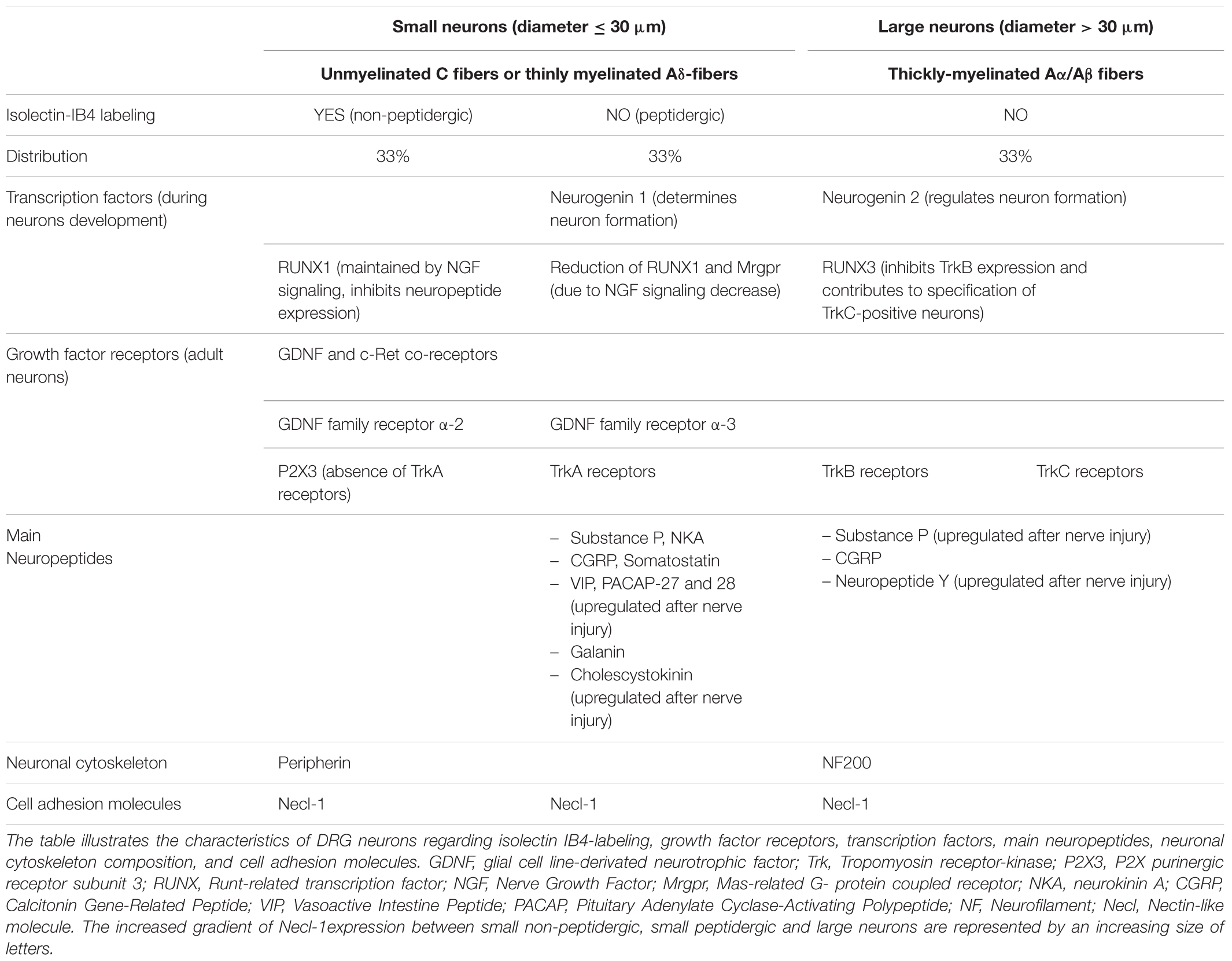
TABLE 1. Characteristics of DRG neurons.
Because of sequencing advances, a large scaled and more precise genetic characterization of DRG is now possible to better identifying the function and underlying mechanisms of each neuron. Therefore, an innovative approach to get rid of pain sensation, without affecting other physiological pain (or itch) pathways, would be to inhibit/remove only the population of DRG neurons that are responsible for the noxious disturbance (Liem et al., 2016; Li et al., 2018).
Electrophysiological Studies of Drg Neurons in vitro
Different types of tissues or individual cells can be used to perform electrophysiological studies of DRG neurons in vitro, each of them offering advantages and disadvantages. Hence, the primary cell cultures of rodent models (rats or mice) provide freshly isolated DRG neurons, however dissociated using enzymatic treatments which may disturb, in some extent, their functioning and thus their electrophysiological recordings. However, the two enzymatic procedures needed to replate the cells (i.e., detach and again deposit them on glass-slides) 24 h after their dissociation, in order to slow down extensive neurite growth that could limit adequate electrophysiological recordings, represent an aggressive cell treatment but were reported to have no marked effect on the neuronal action potential (Caviedes et al., 1990). In any case, a delay of 4–7 days between cell dissociation and recordings is primordial to obtain adequate membrane conditions for experiments. A more physiological alternative to avoid cell dissociation and thus enzymatic procedures is to use DRG explants, i.e., slices of DRG previously inserted in 2% agar. Under these conditions, neurons are kept in their native environment and their plasma membrane is not altered (Scholz et al., 1998; Scholz and Vogel, 2000). However, the maximal life-time of DRG explants, as that of primary cell cultures, is of about 2 weeks.
Another possibility is thus the use of immortalized DRG neurons which offer the advantage of being maintained in cultures for long periods of time by changing freshly-made medium daily. The principle consists in immortalizing DRG neurons from human fetuses or rodents by using a tetracycline-responsive v-myc oncogene (Sah et al., 1997; Raymon et al., 1999), a medium previously conditioned with the rat thyroid cell line UCHT1 (Allen et al., 2002), or telomerase reverse transcriptase expression vectors added in the medium (Chen et al., 2007). Immortalized DRG neurons may also be directly obtained from transgenic rats harboring the temperature-sensitive large T-antigen gene (Nishiya et al., 2011). Immortalized human DRG neurons became an advance 30 years ago because of human tissue short supply. This type of more homogeneous cell lines is of great interest for high throughput screening of antinociceptive compounds.
Recently, the development of the induced pluripotent stem cell (iPSC) technology opens up new perspectives in personalized medicine, drug discovery or cell therapy. In the context of pain studies, iPSCs, derived for example from mesenchymal cells of a patient with inherited pain disease, are dedifferentiated to acquire the neuronal phenotype (bipolar cells) with the appropriate external medium containing neural growth factors. Then, the cell cultures will allow performing electrophysiological studies and pharmacological validation of a drug directly on targets presenting the mutation responsible for the patient pain phenotype (Cao et al., 2016; Sommer, 2016; Yang Y. et al., 2018).
Voltage-Gated Sodium Channels Expressed in Drg Neurons
NaV channels are crucial transmembrane proteins for the communication of excitable cells in vertebrate and invertebrate organisms, due to their important role in action potential genesis and propagation. In terms of discovery, these channels are the founding members of a superfamily comprising more than 140 members grouped into eight families (voltage-gated Na, K and Ca channels, Ca-activated K channels, cyclic nucleotide-modulated ion channels, transient receptor potential (TRP) channel, inward-rectifying K channels and two-pore K channels) which, after the GPCRs, constitute the second largest group of signaling molecules encoded by the human genome (Yu and Catterall, 2004).
The fundamental functional features that allow NaV channels to perform their role in cellular electrical signaling include a high selective permeation of Na ions and a gating system whose opening and closing are controlled by both the time and the membrane potential. Currently available data indicate that these channels consist of a pore-forming α-subunit (glycoprotein of 220–260 kDa) which is formed by four homologous domains (designated DI to DIV), each comprising six hydrophobic transmembrane α helices segments (designated S1–S6) connected by extra- and intra-cellular loops (Figure 3). The channel pore formation is attributed to the hairpin-like P loops connecting S5 and S6 segments (extracellular part of the pore) and to the S6 segments (intracellular part of the pore) of each domain. The channel activation (opening) is associated with the S4 segments of each domain, containing repeated motifs of positively charged amino acid residues (arginine) followed by two hydrophobic residues, which lead to the opening of the pore by moving outward under the influence of the membrane electric field to initiate protein conformational change. The channel inactivation (closing), meanwhile, is associated with the intracellular loop connecting DIII and DIV domains, including the isoleucine, phenylalanine and methionine (IFM) motif (Catterall, 2000; Goldin et al., 2000; Payandeh et al., 2011). Ten α-subunits of the mammalian NaV channel, referred as NaV1.1-1.10 (the first and second numbers indicating the gene subfamily and the specific channel isoform, respectively), have been identified so far. These subunits, which exhibit 40–70% sequence homology and closely related structures, can be distinguished according to their specific expression in tissues and their sensitivity to TTX, a well-known blocker of NaV channels (Table 2). The structure, functional characteristics and phylogenetic relationships of the various NaV channel subtypes have been largely detailed in the literature (Catterall, 2000; Goldin et al., 2000; Goldin, 2001, 2002; Catterall et al., 2005b, 2007).
TABLE 2
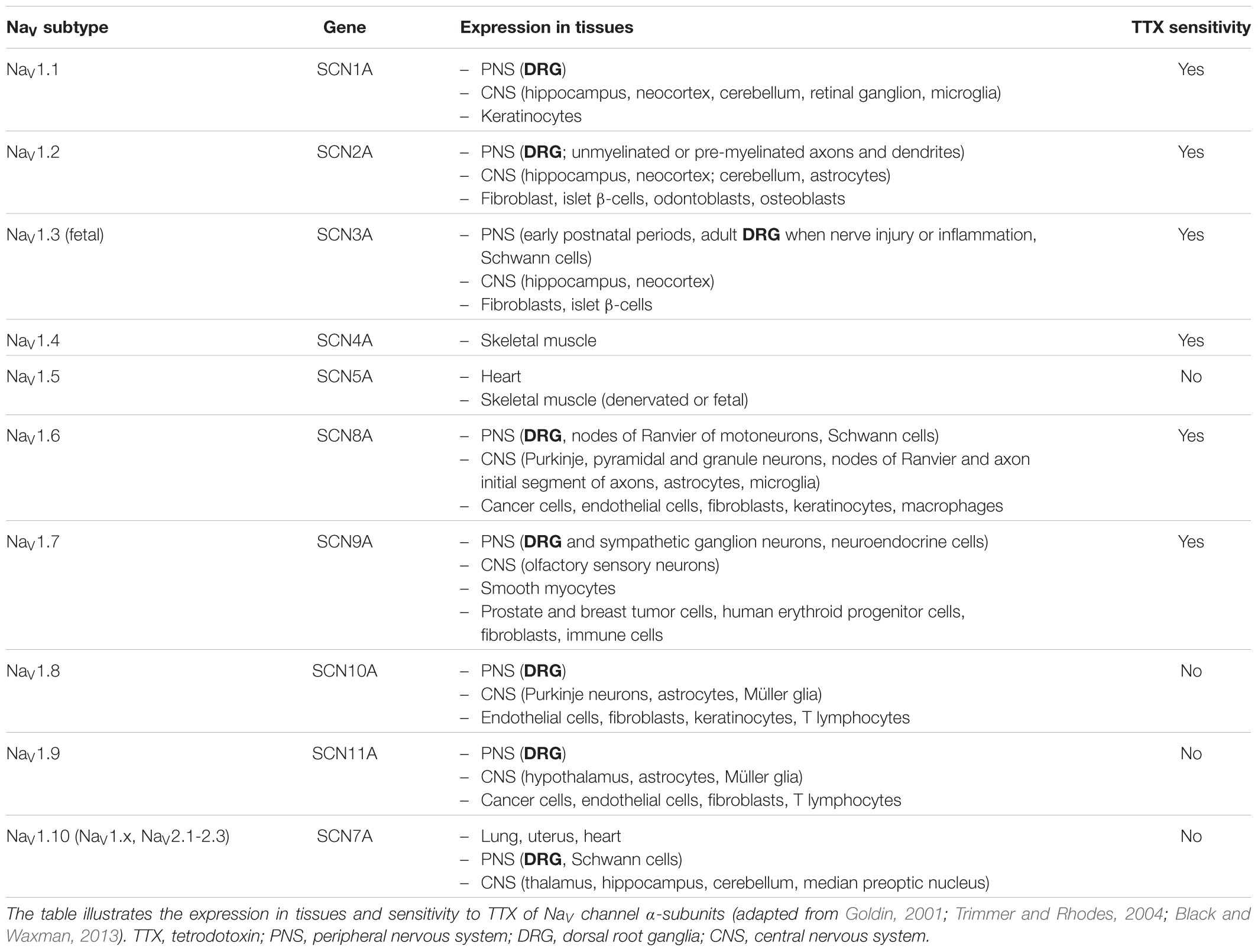
TABLE 2. Expression in tissues and TTX sensitivity of NaV channel subtypes.
FIGURE 3
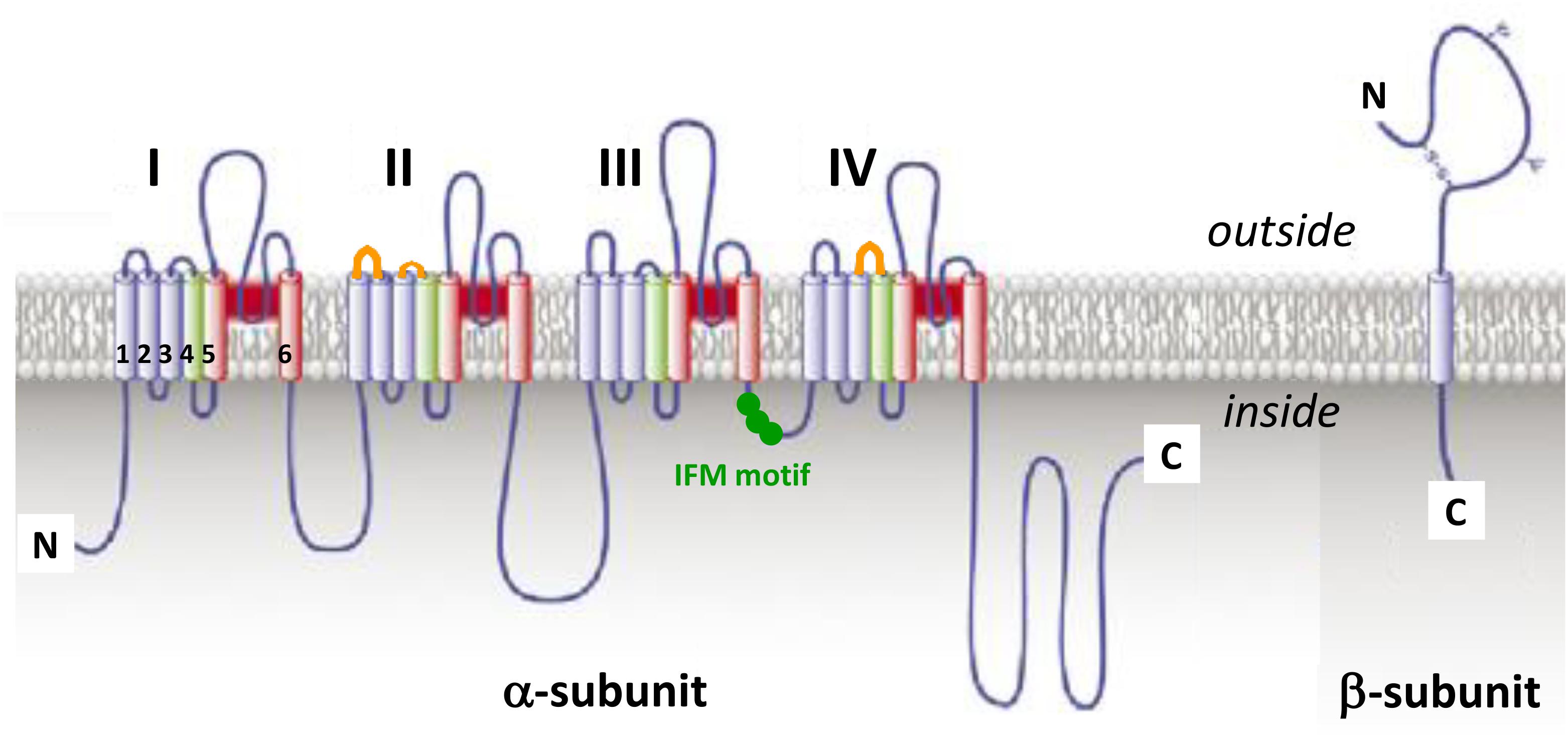
FIGURE 3. The voltage-gated sodium channel. Schematic representations of α-subunit and auxiliary β-subunit of NaV channels, in which cylinders are transmembrane α helices. In red: S5 and S6 pore-forming segments, in green: S4 voltage-sensor segment, and in blue: S1, S2, and S3 segments. IFM, isoleucine, phenylalanine and methionine residues. The orange loops in DII and DIV domains correspond to spider toxins binding sites (adapted from Catterall et al., 2007).
The expression, pharmacology and functioning of NaV channels can be altered by post-translational modifications (PTMs) of α-subunits, such as acetylation, phosphorylation, glycosylation and palmitoylation that occur after translation of mRNAs into peptidic chains or during secretory pathways. These PTMs greatly contribute to the development of chronic pain syndromes and may also modulate the toxin sensitivity of NaV channels (Liu et al., 2012). In acquired, but not in inherited, pain syndromes, various signaling pathway activations may alter expression and functioning of NaV channels (Laedermann et al., 2015). In mammals, the α-subunit is associated with an auxiliary β-subunit (glycoprotein of 30–40 kDa), consisting of a single transmembrane α helices segment, a long N-terminal extracellular immunoglobulin type V domain and a short C-terminal intracellular domain (Figure 3), which may in particular modulate the channel functioning, regulate its trafficking and expression at the membrane surface and/or link it to cytoskeleton proteins (Catterall, 2000; Xie et al., 2016). Therefore, it is likely that the β-subunit type and presence or absence in overexpression systems, or even in native tissues, will have big impact on NaV channel readout during molecule screening experiments, in particular. Hence, the functional behavior of NaV1.8 subtype has been reported to be highly dependent on the type of β-subunit expressed under normal and disease conditions (Vijayaragavan et al., 2004). Among the four auxiliary β-subunits (β1-β4) identified so far, only β2- and β4-subunit have been reported to be covalently linked, by their N-terminal domain, to NaV channel α-subunits (Namadurai et al., 2015). Recently, β4-subunit has been highlighted as a painkiller target because of its action of regulating fast resurgent Na currents in sensory neurons associated with pain disorders (Xie et al., 2013; Lewis and Raman, 2014; Barbosa et al., 2015).
Seven over the ten NaV channel subtypes (NaV1.1, 1.3, 1.6–1.10) which are expressed in DRG neurons are detailed in Table 3. All these subtypes are thus potentially involved in conveying noxious stimuli and may represent a target for pain treatment. Indeed, the NaV1.6–1.9 subtypes, as main actors of pain anatomical and physiological integrities, have been genetically proved to be linked to human pain disorders. However, the high expression of Nav1.7 subtype in DRG neurons (see Figure 4) and its multiple reported mutations inducing genetic-painful and painless disorders, largely documented in the literature, make this subtype one of the most promising target to alleviate pain. The contribution of NaV1.1 and 1.10 subtypes to pain message was evidenced by pharmacological approaches, and the NaV1.3 (fetal) subtype was reported to be overexpressed during injury-induced pain. It is worth noting that NaV1.2 is the only subtype that does not transmit pain message in the PNS, although present in DRG neurons. In the CNS, mutations in the sequence coding for this subtype have been reported to induce epileptogenic and/or neurodevelopmental disorders (Liao et al., 2010; Hackenberg et al., 2014; Ben-Shalom et al., 2017; Wolff et al., 2017).
TABLE 3
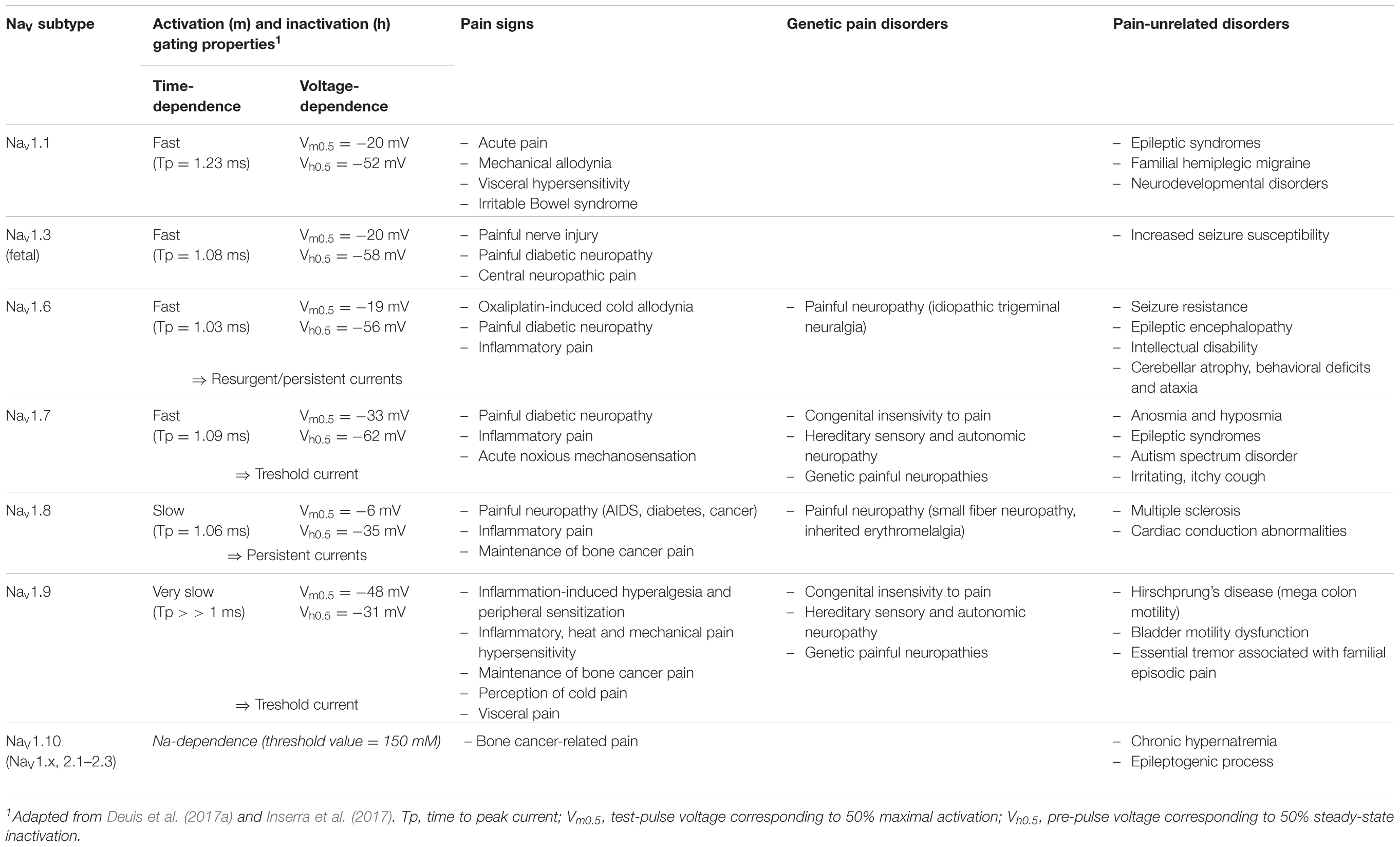
TABLE 3. Electrophysiological properties and disorders associated with NaV channel subtypes expressed in DRG neurons and involved in pain.
FIGURE 4
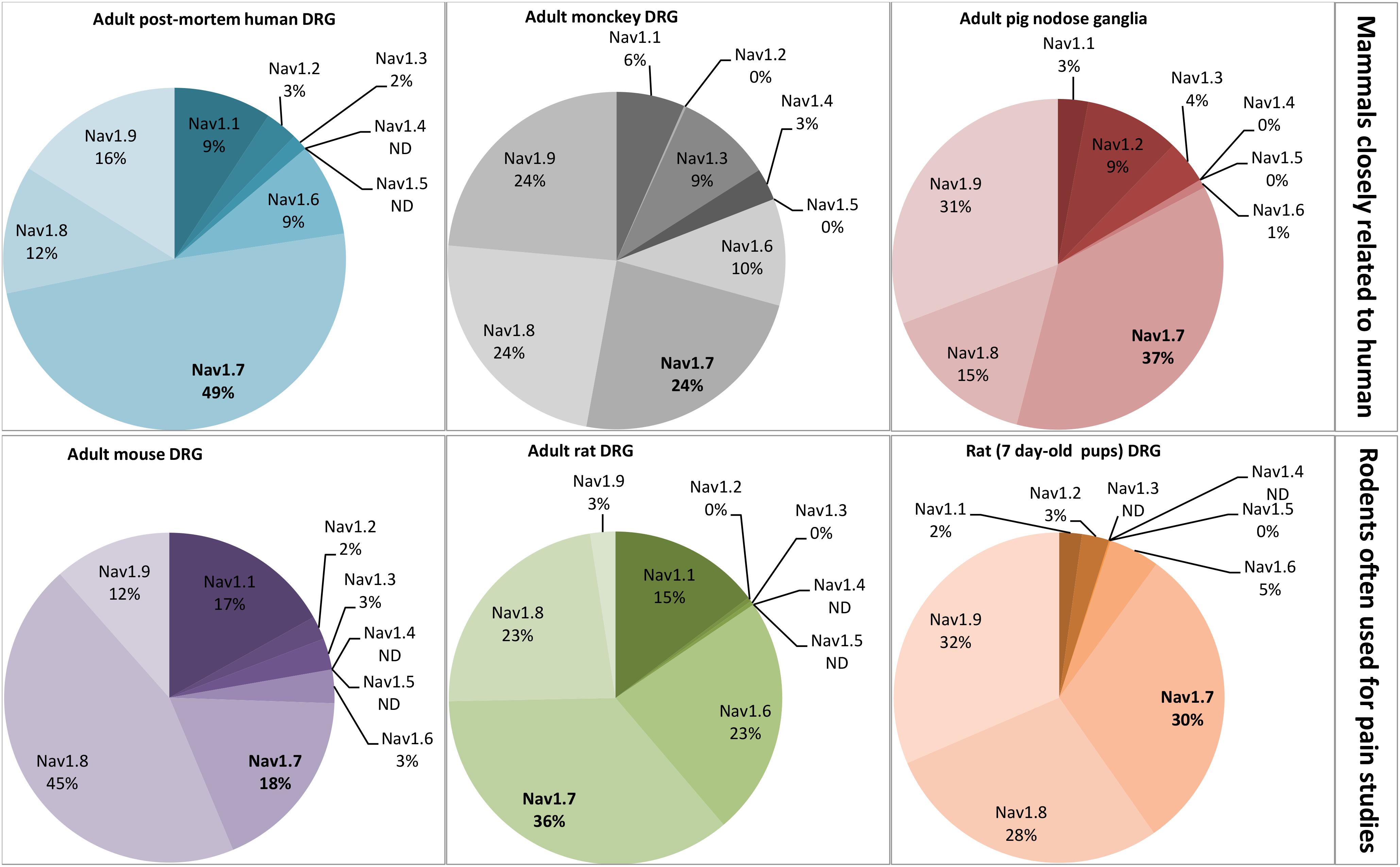
FIGURE 4. Relative proportion of NaV channel α-subunits detected in mammalian dorsal root ganglia (DRG) neurons by RT-PCR. DRG neurons are from some representative mammals of different orders reported in the literature: primates, artyodactyla and rodents. The first order, including human and monkey, is closely related to the pig, belonging to the second order. The rodents, more distant from the human, represent the model often used for pain studies. The adult post-mortem human DRG neurons were obtained from healthy donors. All the data are from DRG neurons of adult mammals except those from 7 day-old rats. ND, non-determined. Adapted from Raymond et al. (2004), Berta et al. (2008), Ho and O’Leary (2011), Muroi and Undem (2014), and Chang et al. (2018).
NaV1.1, encoded by the SCN1A gene, is a TTX-sensitive, fast-activating and inactivating NaV channel. Its expression is located in the CNS, PNS (more precisely in DRG neurons) and keratinocytes (Trimmer and Rhodes, 2004; Black and Waxman, 2013). This subtype was recently reported as a potential pain target involved in neuropathic pain (Irritable Bowel Syndrome, visceral hypersensitivity) and in acute pain and mechanical allodynia, due to the correlation between its activity and pain behaviors in rodent models using the activating spider toxin Hm1a and the selective inhibitory small molecule ICA-121431 (Osteen et al., 2016, 2017; Salvatierra et al., 2018). The important function of NaV1.1 in the CNS is highlighted by more than 500 mutations in its coding sequence that cause epileptic syndromes (Febrile Seizure, Generalized Epilepsy with Febrile Seizures +, and Severe Myoclonic Epilepsy of Infancy also known as Dravet syndrome) (Catterall et al., 2010). Moreover, three of these mutations are also correlated to familial hemiplegic migraine, and several copy number variants have been linked to neurodevelopmental disorders such as intellectual disability, autism and psychiatric disease (Dichgans et al., 2005; Fry et al., 2016; Xiong et al., 2016).
NaV1.3, encoded by the SCN3A gene, is also a TTX-sensitive, fast-activating and inactivating NaV channel (Cummins et al., 2001). This fetal subtype is normally expressed in early postnatal periods. However, it is also expressed at very low levels in adult sensory primary afferents, and is rapidly upregulated in DRG after peripheral axotomy by sciatic nerve transection or chronic constriction (Waxman et al., 1994; Black et al., 1999; Dib-Hajj et al., 1999) or by tight ligation of the spinal nerve (Boucher et al., 2000; Kim et al., 2001), and in painful diabetic neuropathy (Tan et al., 2015; Yang et al., 2016). NaV1.3 promotes the spontaneous ectopic discharge observed during nerve injury. In particular, its over-expression after spinal cord injury leads to rhythmic oscillatory burst firing, alternating with single spikes and silent periods, in second order dorsal horn sensory neurons, and to spindle wave firing mode in thalamus (ventral posterior lateral) neurons with identifiable peripheral receptive fields (Hains et al., 2003; Lai et al., 2003). The central neuropathic pain is also explained by NaV1.3 upregulation which induces neuronal hyperexcitability and alters the process of somatosensory information (Hains et al., 2003; Hains et al., 2005, 2006). Recently, loss-of-function of the SCN3A gene, resulting in reduced expression or deficient trafficking to the plasma membrane of the protein, was reported to contribute to increased seizure susceptibility (Lamar et al., 2017).
NaV1.6, encoded by the SCN8A gene, is a TTX-sensitive fast-activating and inactivating NaV channel expressed in the PNS (DRG neurons, nodes of Ranvier of motoneurons, Schwann cells), in the CNS (Purkinje, pyramidal and granule neurons, nodes of Ranvier and initial segment of axons, astrocytes, microglia) and in non-neuronal tissues such as cancer cells, endothelial cells, fibroblasts, keratinocytes and macrophages (Trimmer and Rhodes, 2004; Black and Waxman, 2013; Israel et al., 2017). This subtype is upregulated in various peripheral pain pathways including oxaliplatin-induced cold allodynia (Deuis et al., 2013), type-2 diabetic neuropathic pain (Ren et al., 2012) and inflammatory pain (Xie et al., 2013). The NaV1.6 α-subunit, covalently linked to the β4-subunit, can underlie excitatory, persistent and resurgent currents which induce repetitive firing and abnormal spontaneous activity of sensory neurons (Lewis and Raman, 2014; Barbosa et al., 2015; Xie et al., 2016). A NaV1.6-gene mutation resulting in gain-of-function has been reported to potentiate transient and resurgent Na currents, leading to increased excitability in trigeminal neurons, exacerbating thus the pathophysiology of vascular compression and contributing to idiopathic trigeminal neuralgia (Grasso et al., 2016; Tanaka et al., 2016). In contrast to NaV1.1 and 1.2, NaV1.6 is involved in seizure resistance (Makinson et al., 2014). The knock-down of NaV1.6 in the brain was shown to compensate the NaV1.1-gene mutation-induced imbalance of excitation over inhibition involved in epileptogenic disorders, which motivates the necessity to find specific NaV1.6 inhibitors to treat debilitating or fatal form of epilepsy such as the Dravet syndrome (Catterall, 2012; Anderson et al., 2017). Finally, more than ten human de novo mutations of NaV1.6 gene have been identified in patients with two types of CNS disorders, epileptic encephalopathy and intellectual disability (O’Brien and Meisler, 2013).
NaV1.7, encoded by the SCN9A gene, is a TTX-sensitive fast-activating and inactivating NaV channel. It is expressed in the somatosensory system (mainly in C- and Aβ-type DRG neurons) and in the sympathetic ganglion neurons (myenteric and visceral sensory neurons) of PNS, but only in the olfactory sensory neurons of CNS. This subtype is also present in smooth myocytes (Jo et al., 2004; Israel et al., 2017; Vetter et al., 2017), and in non-excitable cells such as prostate and breast tumor cells, human erythroid progenitor cells, fibroblasts and immune cells (Black and Waxman, 2013; Israel et al., 2017). This subtype is a threshold channel since it is involved in the action potential (i.e., pain message) triggering by regulating the resting membrane potential of DRG. The implication of NaV1.7 in neuropathic (diabetes) and inflammatory pain, as well as in acute noxious mechanosensation, has been explained by gene upregulation or variants (Dib-Hajj et al., 2013; Blesneac et al., 2018). In addition, multiple NaV1.7 genetic mutations have been linked to painless or painful phenotypes. Hence, congenital SCN9A loss-of-function mutations, such as congenital insensivity to pain and type IID of hereditary sensory and autonomic neuropathy, can induce genetic diseases with a complete absence of pain. In contrast, the SCN9A gain-of-function mutations cause genetic painful neuropathies such as small fiber neuropathy, primary erythromelalgia and paroxysmal extreme pain disorder (de Lera Ruiz and Kraus, 2015; Vetter et al., 2017). The NaV1.7 expression in the CNS is responsible for anosmia and hyposmia, always linked to painless phenotypes, and epilepsy (presence of different variants in patients showing seizures and Dravet syndrome, and of two SCN9A mutations related to epilepsy phenotype), as well as to autism spectrum disorder (Dib-Hajj et al., 2013; Mulley et al., 2013; Rubinstein et al., 2018; Yang C. et al., 2018). NaV1.7 has also been reported to be the major NaV subtype in irritating, itchy cough conveyed by DRG neurons (Muroi and Undem, 2014; Sun et al., 2017).
NaV1.8, encoded by the SCN10A gene, is a TTX-resistant NaV channel that exhibits slow activation and inactivation, as well as rapid repriming kinetics in C- and Aβ-type DRG neurons. With its slow kinetics and high activation threshold, this subtype corresponds to 80–90% of the inward current necessary to the rising phase of action potentials (Renganathan et al., 2001; Patrick Harty and Waxman, 2007). It is ectopically expressed in the CNS Purkinje neurons during multiple sclerosis disorder, and becomes thus a target of choice to develop a treatment for this disorder (Han et al., 2016). NaV1.8 mRNAs were also detected and quantified in astrocytes, Müller glia, endothelial cells, fibroblasts, keratinocytes and T lymphocytes (Black and Waxman, 2013). This subtype has been reported to contribute to neuropathic pain, notably associated with acquired immunodeficiency syndrome, diabetes and cancer, as well as to inflammatory pain (Thakor et al., 2009; Qiu et al., 2012; Belkouch et al., 2014; Liu X. D. et al., 2014). Moreover, SCN10A gain-of-function mutations are associated, not only with the above mentioned neuropathic pain, but also with small fiber neuropathy and inherited erythromelalgia (Faber et al., 2012; Huang et al., 2016; Kist et al., 2016). Finally, genetic variations of SCN10A have been reported to correlate with cardiac conduction abnormalities in patients suffering from hypertrophic cardiomyopathy-like atrial fibrillation and Brugada syndrome (Zimmer et al., 2014; Behr et al., 2015; Iio et al., 2015).
NaV1.9, encoded by the SCN11A gene, is a TTX-resistant NaV channel with very slow activation and inactivation kinetics. This subtype is also a threshold channel but exhibits different biophysical properties, compared with NaV1.7 subtype. Roughly 80% of small-diameter sensory DRG neurons but only a few large-diameter ones and trigeminal ganglia (including C-type nociceptive cells) were reported to express NaV1.9 mRNAs (Dib-Hajj et al., 1998). The expression pattern of this subtype is merely limited to the PNS, despite spots of expression in the CNS (hypothalamus, astrocytes, Müller glia), endothelial cells, fibroblasts, and T lymphocytes. It was also detected in some cancers such as lymphoma and small cell lung cancer (Black and Waxman, 2013; Israel et al., 2017). NaV1.9 plays a major role (i) in inflammatory, heat and mechanical pain hypersensitivity, as revealed in both (sub) acute and chronic inflammatory pain models, (ii) in the maintenance of bone cancer pain (with the NaV1.8 subtype), (iii) in the perception of cold pain under normal and pathological conditions, and (iv) in visceral pain (Lolignier et al., 2011; Qiu et al., 2012; Dib-Hajj et al., 2015; Lolignier et al., 2015; Hockley et al., 2016; Lolignier et al., 2016). More recently, multiple NaV1.9 genetic mutations were linked to painless or painful phenotypes, making this subtype the second target of interest (after the NaV1.7 subtype) to treat pain. Hence, on one hand, congenital SCN11A loss-of-function mutations, such as congenital insensitivity to pain and type VII of hereditary sensory and autonomic neuropathy, were reported to result in genetic diseases with a complete absence of pain (Leipold et al., 2013; Woods et al., 2015; Phatarakijnirund et al., 2016; Huang et al., 2017; King et al., 2017). On the other hand, the SCN11A gain-of-function mutations lead to genetic painful neuropathies such as small fiber neuropathy and rare inheritable pain disorders (Zhang et al., 2013; Huang et al., 2014; Han et al., 2015; Leipold et al., 2015; Kleggetveit et al., 2016; Okuda et al., 2016; Han et al., 2017). Finally, NaV1.9 expression has also been linked to the Hirschprung’s disease (affecting the mega colon motility), and implicated in the development of inflammation-based bladder motility dysfunction and in essential tremor associated with familial episodic pain (Ritter et al., 2009; O’Donnell et al., 2016; Leng et al., 2017).
NaV1.10, encoded by the SCN7A gene and also named NaV1.x or NaV2.12.3 (according to the species), is an atypical subtype associated with leak currents and considered as descendant of NaV channel α1-subunits despite, notably, a less than 50% sequence homology and marked discrepancies in S4 segments and the intracellular loop connecting DIII and DIV domains (Goldin et al., 2000; Yu and Catterall, 2003; Nehme et al., 2012). In addition and in contrast to other NaV channel subtypes, NaV1.10 is not activated by the membrane potential but is sensitive to extracellular concentration of Na ions with a threshold value of 150 mM (Hiyama et al., 2002). It is expressed in the lung, uterus and heart, in the PNS neurons (e.g., medium to large-sized DRG neurons, non-myelinating Schwann cells) and in the CNS (e.g., thalamus, hippocampus, cerebellum, median preoptic nucleus) (Fukuoka et al., 2008; Garcia-Villegas et al., 2009) In particular, this subtype is clearly present in the primary regions implicated in hydromineral homeostasis, such as the subfornical organ, the vascular organ of the lamina terminalis and the median eminence which control the Na-intake behavior by changing neuronal excitability (Watanabe et al., 2006; Xing et al., 2015; Kinsman et al., 2017). It is involved in autoimmunity process causing chronic hypernatremia (Hiyama et al., 2010) and in epilectogenic process (Gorter et al., 2010). Recently, the inhibition or suppression of NaV1.10 was reported to reduce pain behaviors in a bone cancer-induced model by decreasing the excitability of DRG neurons (Ke et al., 2012).
Using electrophysiological studies of DRG neurons in vitro for drug-discovery research may be limited by the relative proportions of targeted NaV channel subtypes, as exemplified by the plant alkaloid paclitaxel whose effects differ between the models used (Chang et al., 2018). Indeed, the relative proportion of NaV channel subtypes varies between small- and large-diameter DRG neurons, the first one expressing more TTX-resistant and less TTX-sensitive subtypes than the second one in both rodent and human neurons (Djouhri et al., 2003; Zhang et al., 2017). In addition, the relative proportion of NaV subtypes varies according to the species studied. This is illustrated in Figure 4 by the relative quantification of each NaV channel subtype mRNA in small-diameter DRG neurons, the most documented in the literature because of their interest to treat pain, in various mammalian species. As expected from their importance in pain process, NaV1.6–1.9 subtypes are relatively more expressed than NaV1.1–1.3 subtypes, and the expression of the pain-unrelated NaV1.4 and 1.5 subtypes, when detected, is extremely low and their function unknown (Ho et al., 2013).
The DRG neurons from rodent models are preferentially used for pain studies, compared with those from human, because they are rapidly available, easy to manipulate, cheap and exhibit well-conserved anatomical and physiological properties. However, adult mice and rat differ in their relative proportions of NaV subtypes: more than 50% of mouse DRG NaV subtypes are TTX-resistant (i.e., 45% of NaV1.8 and 12% of NaV1.9) whereas it is the opposite in rat DRG neurons (i.e., 15% of NaV1.1, 23% of NaV1.6 and 36% of NaV1.7) (Berta et al., 2008; Chang et al., 2018). Interestingly, the level of expression of NaV subtypes is greatly influenced by the age of mammal, i.e., the neuron maturation, as exemplified by the high expression of NaV1.9 subtype in pup rat DRG neurons which is replaced by NaV1.1 and 1.6 subtype expression in adult rat DRG neurons (Ho and O’Leary, 2011). PCR analysis of the seven NaV subtypes expressed in DRG neurons reveals that post-mortem human DRG neurons from healthy donors show relatively high expression of NaV1.7 (49%) and low expression of NaV1.8 (12%), whereas the mouse DRG neurons present high expression of NaV1.8 (45%) and low expression of NaV1.7 (18%), the adult rat DRG neurons having an intermediate expression of NaV1.7 (36%) and NaV1.8 (23%) (Chang et al., 2018).
The mammals closely related to human (i.e., adult monkey and pig) roughly conserve the NaV subtype expression pattern of post-mortem human DRG neurons, i.e., NaV1.7 ≥ NaV1.9 ≥ NaV1.8 (Raymond et al., 2004; Muroi and Undem, 2014). Although the adult post-mortem human DRG neurons obtained from healthy donors are valuable in terms of physiology to estimate the relative proportions of NaV subtypes in living humans (Zhang et al., 2017; Chang et al., 2018), and even if some mammalian models seem closed to human, the message needs to be always shaded when extrapolated to human. Moreover, RT-PCR consists in averaging NaV subtype mRNAs present in nucleus of cell population, and does not represent strictly the level of functional NaV subtypes located in cell plasma membranes.
In several mammals DRG neurons, alternative splicing of NaV α-subunit genes has been detected, resulting in the expression of multiple proteins. However, the functional significance of this process has not been completely elucidated (Dietrich et al., 1998; Schirmeyer et al., 2014). Some variants seem to lead to subunits showing redundant or no obvious pharmacological and/or functional differences, compared with the wild-type subunit (Schirmeyer et al., 2014). However, different pharmacological and functional properties between variant and wild-type subunits are evidenced in the literature, such as their sensitivity to drugs/toxins (Dietrich et al., 1998; Tan et al., 2002; Thompson et al., 2011; Boullot et al., 2017), their functional specificity regarding tissue/cell localization (Song et al., 2004), and their involvement in membrane excitability via the regulation of translational repression (Lin and Baines, 2015). Some alternative splice events are unique to DRG neurons. Hence, significant changes in the splicing patterns of Scn8a and Scn9a genes were observed in a rat model of neuropathic pain, leading to down-regulation of all transcripts (Raymond et al., 2004). Moreover, four alternative splice variants of SCN9A gene were reported to be expressed in human DRG neurons. The difference between two of them at the exon 5 level (exons 5A and 5N) results in two different amino acid residues, located in the S3 segment of DI domain acid. One of them, negatively charged, may be involved in modifications of NaV channel activation and de-activation, impacting thus the paroxysmal extreme pain disorder disease phenotype (mutation I1461T). The two other alternative splice variants differ at the exon 11 level, leading to the presence (11L) or absence (11S) of an 11-amino acid sequence in the intracellular loop connecting DI and DII domains of NaV channels, an important region for protein kinase A regulation which will thus influence neuronal excitability and pain sensation (Chatelier et al., 2008; Jarecki et al., 2009). Recently, a (NAT) was reported to be a potential candidate gene for patients with inherited (primary erythromelalgia, paroxysmal extreme pain disorder, and painful small fiber neuropathy) or acquired chronic pain disorders linked to the SCN9A locus, taking into account that the sense gene must not contain mutations which lead to sense gene-NAT pairing. This is the first example of a new therapy based on increased native antisense mRNAs to treat chronic pain in humans (Koenig et al., 2015).
Analgesic Spider Toxins Targeting the NaV1.7 Channel Subtype
Arachnids (araneae order) are the most diverse group of venomous animals with more than 46,000 extant species subdivided in araneomorph (crossing fangs) and mygalomorph (parallel fangs) suborders. Theraphosidae, the most studied and represented family in Arachnoserver 3.0 database, belongs to the latter suborder, with approximatively 470 species, a bit more than one quarter of all species (Pineda et al., 2018). Each spider venom contains from 100s to 1000s peptides (Escoubas, 2006), meaning that more than 10 million spider-venom peptides with an original sequence remain to be discovered since only approximatively 0.01% of these toxins have been explored until now (Klint et al., 2012, 2015b). The major components of most spider venoms are small disulfide-rich peptide toxins (Saez et al., 2010).
Because of their major role in action potential genesis and propagation in CNS, PNS, heart, smooth and skeletal muscles, NaV channels are crucial for vital functions and are thus targeted by various groups of toxins that interact with at least six specific channel receptor-sites (Cestele and Catterall, 2000; Catterall et al., 2007; Gilchrist et al., 2014; Israel et al., 2017). Toxins that alter these channels may affect one or more of their three essential properties: activation, inactivation and ion selectivity. In that regard, toxins that have been isolated from different venomous animals (such as spiders, scorpions, cone snails, sea anemones and centipedes) may be classified as pore blockers and/or gating modifiers (Israel et al., 2017). The main source of the approximately 20 analgesic peptide toxins targeting the NaV1.7 subtype is the venoms of tarantula constitutive of the theraphosidae family (Figure 5) (Klint et al., 2015b; Vetter et al., 2017). It is worth nothing that this family also contains many NaV channel activators (Deuis et al., 2017b), such as Hm1a toxin which has been reported to induce a painful behavior when injected in rodents (Jami et al., 2017). A small amount of these toxins also target other ion channel types located at the level of DRG neurons and, thus, taking part into pain processing such as TRP channels A1 antagonized by Protoxin (ProTx)-I and Phα1β, acid-sensitive ionic channel (ASIC)1a inhibited with high affinity by psalmotoxin (PcTx)-1, and N-type CaV channels targeted by Phα1β, although with less potency than for TRPA1 (de Souza et al., 2013; Gui et al., 2014; Osmakov et al., 2014; Tonello et al., 2017).
FIGURE 5

FIGURE 5. Sequence alignment obtained by Multalin (version 5.4.1) of different potential analgesic toxins sorted by NaV spider toxins families, using their UniProtKB identifiers. The consensus sequence is shown above each alignment, with the disulfide bond connectivity. In dark blue, highly conserved amino acid residues (100%), and in light blue, poorly conserved amino acid residues (>50%). The Greek letter(s) before the toxin name is associated to its type of action: μ for NaV channel inhibition, β for shift in the voltage-dependence of NaV channel activation, ω for CaV channel inhibition, and κ for KV channel inhibition. ProTx, protoxin; HnTx, hainantoxin; CcoTx, ceratotoxin; HwTx, huwentoxin; JzTx, jingzhaotoxin; aa, amino acid residues; NaSpTx, spider NaV channel toxin.
Spider toxins targeting the NaV1.7 subtype with an IC50 less than 500 nM are considered as analgesic toxin inhibitors (Klint et al., 2015a), and belong to the three first classes of spider NaV channel toxins (NaSpTx), based on their primary structure and disulfide framework (Figure 5). It is worth noting that this classification also includes spider toxins which target not only the NaV1.7 channel subtype but also other subtypes of ionic channels, as exemplified by the ω-TRTX-Gr2a toxin (GpTx-1) which was initially reported as a CaV3.1 subtype blocker after isolation from the Chilean tarantula, Grammostola rosea, venom (Ono et al., 2011). The NaSpTx peptides are gating modifier toxins (GMTs) because they alter channel gating by stabilizing voltage-sensors (mainly S3–S4 segments of DII domain) in a closed, or resting, configuration state (Table 4) (Klint et al., 2012). The NaV1.7 analgesic spider toxin inhibitors are shaped by inhibitory cystine knot (ICK) scaffold due to 6 cysteines, arranged into a ring composed of two disulfide-bridges crossed by a third one (Saez et al., 2010). These peptides share a conserved amphipathic surface profile characterized by a high proportion of hydrophobic/aromatic amino acid residues, such as Trp, Tyr and Phe, surrounded by charged amino acids which constitute a dipole potential with negative (Asp and Glu) and positive (Lys and Arg) zones (Jung et al., 2005; Cai et al., 2015). Finding more selective GMTs than pore-blockers of NaV1.7 subtypes is likely because the voltage-sensors are more variable in terms of amino acid sequence than the pore region of NaV channels (Catterall et al., 2005a; Payandeh et al., 2011).
TABLE 4
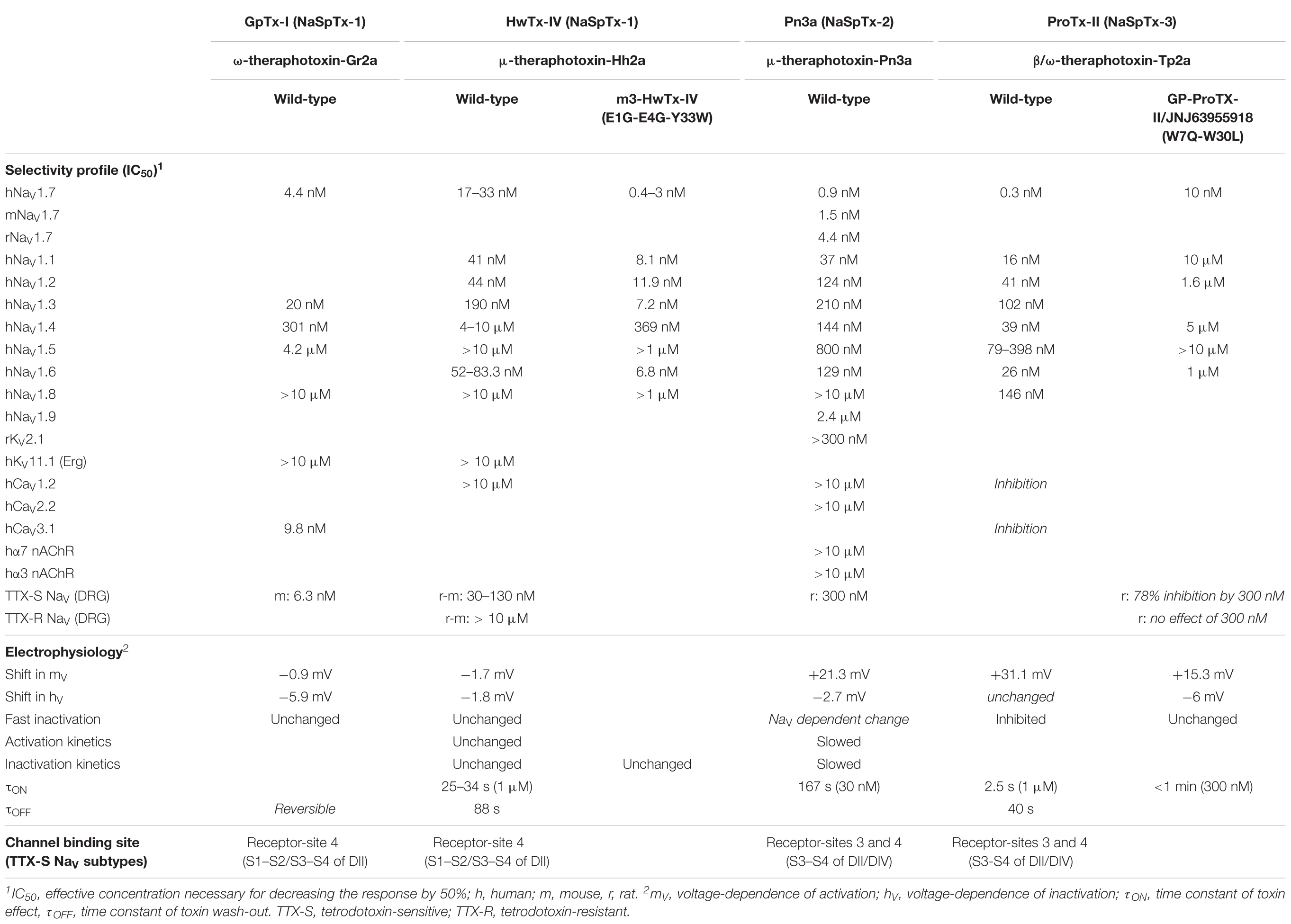
TABLE 4. Selectivity profile, electrophysiological characteristics and channel binding site of NaV1.7 potential analgesic peptide toxins representative of NaV spider toxins (NaSpTx) families.
ProTx-III, ceratotoxin-1, GpTx-1, Cd1a, huwentoxin (HwTx)-IV, hainantoxin (HnTx)-IV, Hd1a, HnTx-III and HnTx-I are NaV1.7 potential analgesic peptide toxins, composed of 33–35 amino acid residues, that belong to NaSpTx-1 family, with nanomolar affinities (IC50 between 2.1 and 440 nM) for this NaV subtype. According to their electrophysiological properties, these toxins act as pore blockers of the NaV1.7 subtype and, except for ProTx-III and ceratotoxin-1, induce minor alterations (less than 5 mV) in the voltage-dependence of its activation and steady-state inactivation (Figure 5 and Table 4). Various mutants of ProTx-III and ceratotoxin-1were produced, showing a 10–20-mV shift in the voltage-dependence of NaV1.7 activation without any change in its fast and steady-state inactivation (Bosmans et al., 2006; Cardoso et al., 2015), in agreement with their interaction with the receptor-site 4 of NaV channels (i.e., S3–S4 segments of DII domain). Models of docking toxins on NaV channels have been reported, placing toxin peptides in the cleft between the channel S1–S2 and S3–S4 transmembrane α-helices (Minassian et al., 2013; Cai et al., 2015; Murray et al., 2016). Even the main channel amino acid residues involved in toxin-channel interactions were located in the extracellular loop connecting S3 and S4 segments of DII domain, some residues of the extracellular loop connecting S1 and S2 segments of DII domain helping to stabilize the toxin binding to the channel (Figure 3).
Pn3a, Df1a, ProTx-I, Phlo1b and jingzhaotoxin (JzTx)-34 are NaV1.7 potential analgesic peptide toxins, composed of 34–35 amino acid residues, belonging to the NaSpTx-2 family and having also nanomolar affinities (IC50 between 0.9 and 610 nM) for this NaV subtype (Figure 5 and Table 4). This toxin family produces important alterations in the voltage-dependence of both activation (10–37-mV positive shifts) and steady-state inactivation (2.7–17.5-mV negative shifts). In addition, the fast inactivation of some TTX-sensitive NaV channels is also affected by some of these toxins. This is in agreement with the known toxin receptor-sites 3 (i.e., S3-S4 segments of DIV domain) and 4 of NaV channels (Deuis et al., 2017b).
ProTx-II, JzTx-V, GsAF1, GrTx-1 and Phlo2a are NaV1.7 potential analgesic peptide toxins from the NaSpTx-3 family, composed of 26–30 amino acid residues, and showing nanomolar affinities (IC50 between 0.3 and 333 nM) for this NaV subtype (Figure 5 and Table 4). The toxin action consists in major alterations in the voltage-dependence of activation (10–31-mV positive shifts) with only minor modifications of the voltage-dependence of steady-state inactivation (up to 5-mV positive shifts). The channel binding sites of these toxins are the receptor-site 4 alone for JzTx-V or in addition with the receptor-site 3 for ProTx-II, those for the other toxins having not been reported (Smith et al., 2007; Moyer et al., 2018). As a consequence, the channel fast inactivation is altered by ProTx-II but not by JzTx-V.
Pn3a is the toxin that has been studied on the biggest panel of ionic channels and receptors reported so far (Deuis et al., 2017a), including all NaV channel subunits, some cardiosafety targets (such as KV11.1 and CaV1.2 channel subtypes) and other transmembrane proteins expressed at the membrane of DRG neurons (KV2.1, α3-α7 nAChR, CaV2.2 subtypes). In particular, this toxin was tested on human and rodent (mouse and rat) NaV1.7 subtypes, showing minor loss of potency (between 2 and 5 fold) for mouse and rat. These results are in agreement with the 93.0% (human versus mouse) and 92.8% (human versus rat) sequence identities between species [data obtained from high quality protein multiple sequence alignments using CLUSTAL Oméga version 1.2.3 (web version)]. The other toxins have not been screened exhaustively: mainly human NaV subtypes and cardiovascular targets, such as KV11.1, CaV1.2 and CaV3.1 subtypes. Big efforts were engaged to decrease toxin potency for NaV1.4 and 1.5 subtypes to avoid neuromuscular and cardiac side-effects (Murray et al., 2016) or to directly find toxin possessing these characteristics (Xiao et al., 2008). Moreover, the interest to find analgesic toxins highly selective for NaV1.1 and 1.6 subtypes, or to improve the toxin selectivity for these two subtypes, decreased during the past years because of the consequent inhibition of action potential transmission via axonal nodes of Ranvier which leads to central and peripheral (at the level of neuromuscular junctions) side-effects. Hence, the HwTx-IV mutant m3-HwTx-IV, presenting an additional hydrophobic patch (Gly1-Gly4-Trp33) has a reinforced inhibitory potency for the NaV1.7 subtype while improving NaV1.1, 1.2 and 1.6 subtype selectivity (Rahnama et al., 2017). Moreover, the ProTx-II mutant JNJ63955918/GP-ProTX-II (W7Q-W30L) presents a 14-fold decreased potency for the NaV1.7 subtype but an improved selectivity against NaV1.1, 1.2, 1.4, and 1.6 subtypes, thus avoiding side-effects such as seizures, arrhythmias and impaired motor functioning (Flinspach et al., 2017; Goncalves et al., 2018). A new strategy was recently proposed to increase the selectivity among the off-target panel, consisting in finding antagonist antibodies specific of NaV1.7 subtype. The results obtained are controversial and need to be further confirmed (Lee et al., 2014; Liu et al., 2016). More recently, another approach was reported, using an antibody-drug conjugated: a potent NaV1.7 toxin inhibitor (a GpTx-1 analog), connected by a PEG-linker to an antibody, showed greater stability in plasma and a biodistribution restricted to the regions expressing NaV1.7 subtype, decreasing thus possible side-effect occurrence (Biswas et al., 2017).
Bilayer membranes that surrounded channel proteins seem to be important to stabilize the interactions between amphipathic GMTs and NaV channels. Toxins and their mutants brought a better understanding of the so-called trimolecular complex relations. Hence, the affinity of GMTs for bilayer membrane lipids highlighted the type of amino acid residues implicated in these interactions that could also impact the toxin selectivity for NaV channels (Deplazes et al., 2016; Henriques et al., 2016; Agwa et al., 2017, 2018; Zhang et al., 2018). Moreover, the pharmacological sensitivity of NaV channels for toxins may be modulated by PTMs on the NaV channel protein itself (Liu et al., 2012). Indeed, palmitoylation of rat NaV1.2 subtype was reported to modify the subtype sensitivity to phrixotoxin (PaurTx)-3 and ProTx-II, producing a 10-fold increased affinity by binding to simultaneously the voltage-sensor domain and the surrounding membrane, without affecting ProTx-I binding (Bosmans et al., 2011; Henriques et al., 2016). PTMs could thus be of major interest and have to be also considered as potential therapeutic targets.
GpTx-1, HwTx-IV, HnTx-IV, HnTx-III, Pn3a, JzTx-34, ProTx-II and JzTx-V were tested on rodent DRG neurons, revealing high affinity for TTX-sensitive NaV channels associated with high selectivity against TTX-resistant NaV channels in mouse and rat DRG neurons (Liu et al., 2002, 2013; Peng et al., 2002; Deuis et al., 2016, 2017a; Flinspach et al., 2017; Goncalves et al., 2018; Moyer et al., 2018; Zeng et al., 2018). The action of HwTx-IV, meanwhile, is not equivalent on TTX-sensitive NaV channels of rat and mouse DRG neurons, the IC50 being 4-fold lower in rat neurons likely due to the presence of high sensitive NaV subtypes that are poorly or not expressed in mouse DRG neurons (Peng et al., 2002; Goncalves et al., 2018). Electrophysiological recordings under physiological conditions or quantification of altered expression of some proteins relevant in pain processing, in DRG neurons after toxin application, allowed characterizing the toxin potential analgesic effect at the cellular level. Hence, in DRG neurons, a JzTx-V mutant [CyA-JzTx-V (M6J-E17X-I28G), i.e., AM-0422] and ProTx-II were reported to inhibit or diminish action potential firing induced by chemical (capsaicin) and mechanical stimulation, or to alter spinal nociceptive processing induced by burn injury, respectively (Moyer et al., 2018; Torres-Perez et al., 2018).
Acute pain is a physiological function associated with injury that is essential for human survival. This kind of pain is normally short-lasting (<3 months). Beyond this period of time and without real injury, pathologic chronic pain is considered to result from damage in the transmission pain system itself. Several bioassays are available to appraise acute or chronic pain using standard or specific rodent models. Hence, mechanical and thermal stimulation assays or global gait analysis are classical to evaluate acute pain. The manual or electronic von Frey filament (or paw pressure) test is commonly used to assess to mechanical pain, while tail flick or water immersion and hot/cold plate tests are dedicated to assess to thermal pain. In these tests, the pain is provoked by pressure or extreme temperature on healthy rodent models. The inflammatory (caused by subcutaneously injected formalin, carrageenan or Freund’s adjuvant compound, or intraperitoneally injected acid acetic) and neuropathic (caused by nerve constriction or ligation injury, chemotherapy-induced neuropathy) chronic pain are usually evaluated with the above mentioned mechanical and thermal stimulation assays, associated to a global evaluation of animal behavior. However, under these conditions, the pathological pain transmission system induced by inflammatory proteins or nerve injury is evaluated (Bridges et al., 2001; Mogil, 2009).
Various analgesic toxins have been proposed as candidates to replace opioids, because of their well-known side-effects. Hence, HwTx-IV, HnTx-IV, Pn3a and ProTx-II were shown to decrease pain at the level of morphine relief, in a dose-depending manner, in neuropathic (mainly spared nerve injury and diabetic neuropathy) and all inflammatory pain models, revealing a real evidence of their analgesic potential as drugs (Liu et al., 2014a,b, Tanaka et al., 2015; Deuis et al., 2017a; Flinspach et al., 2017). Pn3a and the ProTx-II mutant JNJ63955918 were also described as being effective on acute thermal pain tests (Deuis et al., 2017a; Flinspach et al., 2017). Despite the large number of pain tests, a new and original pharmacological one was recently proposed. It consists in specifically inhibiting fast inactivation and increasing peak current associated to NaV1.7 subtype, by the local or systematical injection of OD1 scorpion toxin whose (EC50) being in the nanomolar range (Deuis et al., 2016). This test has the advantages of being less invasive and more sensitive than neuropathic and inflammatory tests. Indeed, only a low amount of toxin candidate to be evaluated is necessary to relieve pain, as exemplified by GpTx-I, Cd1a, m3-HwTx-IV and Pn3a (Deuis et al., 2016; Cardoso et al., 2017; Rahnama et al., 2017; Sousa et al., 2017). However, the question of lack of physiological relevance of this test may be raised.
The best galenic form to make the patients compliant with their treatment is the pills for oral administration. However, in the pain tests, NaV1.7 potential analgesic peptide toxins are often administrated by peripheral routes, such as subcutaneous (intraplantar), intraperitoneal or intramuscular injection) or intrathecal route. ProTx-II and its mutant, despite being the best candidate toxins with the highest affinity and selectivity, are unable to pass through the BNB (see Figure 2) and inhibit action potential transmission along nerves, except if a disruption of the perineurial barrier occurs (Schmalhofer et al., 2008; Hackel et al., 2012). After 24 h of intravenous infusion in vivo, ProTx-II can access DRG neurons but not sciatic nerves and CNS tissues (Liu et al., 2018). Thus, the fenestrations in Blood-Glangia-Barrier (BGB, see Figure 2) are the entry doors for large peptide toxins but their spreading to dorsal root of spinal cord and to distal nerve endings will depend on BNB. The intrathecal route is one possibility to bypass both the BBB and BNB, showing analgesic effects of ProTx-II in rodent pain tests with the risk of the post-lumbar puncture syndrome (headache down to the shoulders, nausea, vertigo and tinnitus) or the effects of the chemical compound itself if the injection is failed. The other possibility consists of toxin co-injection with hypertonic saline solution to disturb BNB (decrease claudin-1 mRNA, one protein responsible for tight-junction) and lead to toxin penetration (Hackel et al., 2012; Tanaka et al., 2015; Flinspach et al., 2017).
Conclusion
Venoms are usually associated with a lethal effect due to the presence in this complex mixture of toxins that have been selected during the evolution process to target crucial physiological systems of the preys. Nevertheless, due to their high affinity and selectivity profiles for specific receptors and ion channels involved in various pathophysiological processes, peptide toxins may be exploited as pharmacological tools and/or therapeutic drugs. Currently, six venom-derived drugs are used for the treatment of hypertension, acute coronary syndromes or diabetes, but the most promising therapeutic area is probably the pain and more precisely, the chronic pain. One peptide, isolated from cone snail venom, has been approved by FDA more than 14 years ago for the treatment of severe chronic pain (ziconotide), and several drug-leads, mainly issue from spider venoms, are actually in development. Among the various receptors and ion channels involved in pain transmission and which are targeted by venom peptides, the NaV1.7 subtype is one of the most promising due to its peripheral location in DRG neurons which in addition present facilitated permeability to high molecular weight drugs. Furthermore, human genetic diseases, associated with NaV1.7 mutations and leading to painless/painful phenotypes, validate this subtype as a pain target. Several spider toxins have been recently identified and characterized for their analgesic property due to their interactions with NaV1.7. Furthermore, their engineering was associated with the optimization of their pharmacological (affinity for NaV1.7 and selectivity profile) and biodistribution properties, reinforcing the potential of these venom-derived peptides as leads for therapeutic development. Finally, new paradigm used in the venom-peptide discovery, based on transcriptomic/proteomic technologies and on a toxin-driven approach, should increase the diversity of toxins identified and the rate of new drug lead discovery, more particularly for the treatment of chronic pain.
Author Contributions
TG performed the major part of the bibliographic research and of the review writing. EB, MP, and DS critically read the review.
Funding
This research was funded by a collaborative grant (#153114) between Sanofi Research & Development (Chilly-Mazarin, France) and the French Alternative Energies and Atomic Energy Commission (CEA, Gif sur Yvette, France). TG was supported by a doctoral CIFRE fellowship from Sanofi.
Conflict of Interest Statement
TG and MP are current or former employees of Sanofi.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ASIC, acid-sensitive ionic channel; BGB, blood-ganglia-barrier; BNB, blood-nerve-barrier; CaV channel, voltage-gated calcium channel; CNS, central nervous system; DRG, dorsal root ganglia; EC50, effective concentration necessary for increasing the response by 50%; GDNF, glial cell line-derivated neurotrophic factor; GMT, gating modifier toxin; GPCR, G-protein-coupled receptor; HnTx, hainantoxin; HwTx, huwentoxin; IC50, effective concentration necessary for decreasing the response by 50%; iPSCs, induced pluripotent stem cells; JzTx, jingzhaotoxin; KV channel, voltage-gated potassium channels; NaSpTx, spider NaV channel toxins; NAT, natural antisense transcript; NaV channel, voltage-gated sodium channel; NGF, nerve growth factor; PaurTx, Phrixotoxin; PcTx-1, psalmotoxin-1; PNS, peripheral nervous system; ProTx, protoxin; PSN, primary sensory neuron; PTM, post-translational modification; Ret, “rearranged during transfection" proto-oncogene; RUNX, Runt-related transcription factor; SSN, secondary sensory neuron; TRP channel, transient receptor potential channel; TSN, tertiary sensory neuron; TTX, tetrodotoxin.
References
Agwa, A. J., Lawrence, N., Deplazes, E., Cheneval, O., Chen, R. M., Craik, D. J., et al. (2017). Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochim. Biophys. Acta 1859, 835–844. doi: 10.1016/j.bbamem.2017.01.020
Agwa, A. J., Peigneur, S., Chow, C. Y., Lawrence, N., Craik, D. J., Tytgat, J., et al. (2018). Gating modifier toxins isolated from spider venom: modulation of voltage-gated sodium channels and the role of lipid membranes. J. Biol. Chem. 293, 9041–9052. doi: 10.1074/jbc.RA118.002553
Allen, D. D., Cardenas, A. M., Arriagada, C., Bennett, L. B., Garcia, C. J., Caviedes, R., et al. (2002). A dorsal root ganglia cell line derived from trisomy 16 fetal mice, a model for down syndrome. Neuroreport 13, 491–496. doi: 10.1097/00001756-200203250-00027
Anderson, L. L., Hawkins, N. A., Thompson, C. H., Kearney, J. A., and George, A. L. Jr. (2017). Unexpected efficacy of a novel sodium channel modulator in dravet syndrome. Sci. Rep. 7:1682. doi: 10.1038/s41598-017-01851-9
Barbosa, C., Tan, Z. Y., Wang, R., Xie, W., Strong, J. A., Patel, R. R., et al. (2015). Navbeta4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol. Pain 11:60. doi: 10.1186/s12990-015-0063-9
Behr, E. R., Savio-Galimberti, E., Barc, J., Holst, A. G., Petropoulou, E., Prins, B. P., et al. (2015). Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 106, 520–529. doi: 10.1093/cvr/cvv042
Belkouch, M., Dansereau, M. A., Tetreault, P., Biet, M., Beaudet, N., Dumaine, R., et al. (2014). Functional up-regulation of Nav1.8 sodium channel in Abeta afferent fibers subjected to chronic peripheral inflammation. J. Neuroinflammation 11:45. doi: 10.1186/1742-2094-11-45
Ben-Shalom, R., Keeshen, C. M., Berrios, K. N., An, J. Y., Sanders, S. J., and Bender, K. J. (2017). Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82, 224–232. doi: 10.1016/j.biopsych.2017.01.009
Berta, T., Poirot, O., Pertin, M., Ji, R. R., Kellenberger, S., and Decosterd, I. (2008). Transcriptional and functional profiles of voltage-gated Na(+) channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 37, 196–208. doi: 10.1016/j.mcn.2007.09.007
Berta, T., Qadri, Y., Tan, P. H., and Ji, R. R. (2017). Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin. Ther. Targets 21, 695–703. doi: 10.1080/14728222.2017.1328057
Biswas, K., Nixey, T. E., Murray, J. K., Falsey, J. R., Yin, L., Liu, H., et al. (2017). Engineering antibody reactivity for efficient derivatization to generate NaV1.7 inhibitory GpTx-1 peptide-antibody conjugates. ACS Chem. Biol. 12, 2427–2435. doi: 10.1021/acschembio.7b00542
Black, J. A., Cummins, T. R., Plumpton, C., Chen, Y. H., Hormuzdiar, W., Clare, J. J., et al. (1999). Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J. Neurophysiol. 82, 2776–2785. doi: 10.1152/jn.1999.82.5.2776
Black, J. A., and Waxman, S. G. (2013). Noncanonical roles of voltage-gated sodium channels. Neuron 80, 280–291. doi: 10.1016/j.neuron.2013.09.012
Blesneac, I., Themistocleous, A. C., Fratter, C., Conrad, L. J., Ramirez, J. D., Cox, J. J., et al. (2018). Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain 159, 469–480. doi: 10.1097/j.pain.0000000000001116
Bosmans, F., Milescu, M., and Swartz, K. J. (2011). Palmitoylation influences the function and pharmacology of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 108, 20213–20218. doi: 10.1073/pnas.1108497108
Bosmans, F., Rash, L., Zhu, S., Diochot, S., Lazdunski, M., Escoubas, P., et al. (2006). Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 69, 419–429. doi: 10.1124/mol.105.015941
Boucher, T. J., Okuse, K., Bennett, D. L., Munson, J. B., Wood, J. N., and Mcmahon, S. B. (2000). Potent analgesic effects of GDNF in neuropathic pain states. Science 290, 124–127. doi: 10.1126/science.290.5489.124
Boullot, F., Castrec, J., Bidault, A., Dantas, N., Payton, L., Perrigault, M., et al. (2017). Molecular characterization of voltage-gated sodium channels and their relations with paralytic shellfish toxin bioaccumulation in the pacific oyster crassostrea gigas. Mar. Drugs 15:E21. doi: 10.3390/md15010021
Bridges, D., Thompson, S. W., and Rice, A. S. (2001). Mechanisms of neuropathic pain. Br. J. Anaesth. 87, 12–26. doi: 10.1093/bja/87.1.12
Cai, T., Luo, J., Meng, E., Ding, J., Liang, S., Wang, S., et al. (2015). Mapping the interaction site for the tarantula toxin hainantoxin-IV (beta-TRTX-Hn2a) in the voltage sensor module of domain II of voltage-gated sodium channels. Peptides 68, 148–156. doi: 10.1016/j.peptides.2014.09.005
Cao, L., Mcdonnell, A., Nitzsche, A., Alexandrou, A., Saintot, P. P., Loucif, A. J., et al. (2016). Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci. Transl. Med. 8:335ra356. doi: 10.1126/scitranslmed.aad7653
Cardoso, F. C., Dekan, Z., Rosengren, K. J., Erickson, A., Vetter, I., Deuis, J. R., et al. (2015). Identification and characterization of ProTx-III [mu-TRTX-Tp1a], a new voltage-gated sodium channel inhibitor from venom of the tarantula Thrixopelma pruriens. Mol. Pharmacol. 88, 291–303. doi: 10.1124/mol.115.098178
Cardoso, F. C., Dekan, Z., Smith, J. J., Deuis, J. R., Vetter, I., Herzig, V., et al. (2017). Modulatory features of the novel spider toxin mu-TRTX-Df1a isolated from the venom of the spider Davus fasciatus. Br. J. Pharmacol. 174, 2528–2544. doi: 10.1111/bph.13865
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi: 10.1016/S0896-6273(00)81133-2
Catterall, W. A., Cestele, S., Yarov-Yarovoy, V., Yu, F. H., Konoki, K., and Scheuer, T. (2007). Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141. doi: 10.1016/j.toxicon.2006.09.022
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005a). International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005b). International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425.
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859. doi: 10.1113/jphysiol.2010.187484
Catterall, W.A. (2012). “Sodium channel mutations and epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. Noebels, M. Avoli, M. Rogawski, R. Olsen, and A. Delgado-Escueta (Rockville, MD: Bethesda). doi: 10.1093/med/9780199746545.003.0052
Caviedes, P., Ault, B., and Rapoport, S. I. (1990). Replating improves whole cell voltage clamp recording of human fetal dorsal root ganglion neurons. J. Neurosci. Methods 35, 57–61. doi: 10.1016/0165-0270(90)90094-V
Cestele, S., and Catterall, W. A. (2000). Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82, 883–892. doi: 10.1016/S0300-9084(00)01174-3
Chang, W., Berta, T., Kim, Y. H., Lee, S., Lee, S. Y., and Ji, R. R. (2018). Expression and role of voltage-gated sodium channels in human dorsal root ganglion neurons with special focus on Nav1.7, Species Differences, and Regulation by Paclitaxel. Neurosci. Bull. 34, 4–12. doi: 10.1007/s12264-017-0132-3
Chatelier, A., Dahllund, L., Eriksson, A., Krupp, J., and Chahine, M. (2008). Biophysical properties of human Na v1.7 splice variants and their regulation by protein kinase A. J. Neurophysiol. 99, 2241–2250. doi: 10.1152/jn.01350.2007
Chen, W., Mi, R., Haughey, N., Oz, M., and Hoke, A. (2007). Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. J. Peripher. Nerv. Syst. 12, 121–130. doi: 10.1111/j.1529-8027.2007.00131.x
Cummins, T. R., Aglieco, F., Renganathan, M., Herzog, R. I., Dib-Hajj, S. D., and Waxman, S. G. (2001). Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001
de Lera Ruiz, M., and Kraus, R. L. (2015). Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem. 58, 7093–7118. doi: 10.1021/jm501981g
de Souza, A. H., Castro, C. J. Jr., Rigo, F. K., De Oliveira, S. M., Gomez, R. S., et al. (2013). An evaluation of the antinociceptive effects of Phalpha1beta, a neurotoxin from the spider Phoneutria nigriventer, and omega-conotoxin MVIIA, a cone snail Conus magus toxin, in rat model of inflammatory and neuropathic pain. Cell. Mol. Neurobiol. 33, 59–67. doi: 10.1007/s10571-012-9871-x
Deplazes, E., Henriques, S. T., Smith, J. J., King, G. F., Craik, D. J., Mark, A. E., et al. (2016). Membrane-binding properties of gating modifier and pore-blocking toxins: membrane interaction is not a prerequisite for modification of channel gating. Biochim. Biophys. Acta 1858, 872–882. doi: 10.1016/j.bbamem.2016.02.002
Deuis, J. R., Dekan, Z., Wingerd, J. S., Smith, J. J., Munasinghe, N. R., Bhola, R. F., et al. (2017a). Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 7:40883. doi: 10.1038/srep40883
Deuis, J. R., Mueller, A., Israel, M. R., and Vetter, I. (2017b). The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 127, 87–108. doi: 10.1016/j.neuropharm.2017.04.014
Deuis, J. R., Wingerd, J. S., Winter, Z., Durek, T., Dekan, Z., Sousa, S. R., et al. (2016). Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of NaV1.7-Mediated Pain. Toxins 8:E78. doi: 10.3390/toxins8030078
Deuis, J. R., Zimmermann, K., Romanovsky, A. A., Possani, L. D., Cabot, P. J., Lewis, R. J., et al. (2013). An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain 154, 1749–1757. doi: 10.1016/j.pain.2013.05.032
Dib-Hajj, S. D., Black, J. A., and Waxman, S. G. (2015). NaV1.9: a sodium channel linked to human pain. Nat. Rev. Neurosci. 16, 511–519. doi: 10.1038/nrn3977
Dib-Hajj, S. D., Fjell, J., Cummins, T. R., Zheng, Z., Fried, K., Lamotte, R., et al. (1999). Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 83, 591–600. doi: 10.1016/S0304-3959(99)00169-4
Dib-Hajj, S. D., Tyrrell, L., Black, J. A., and Waxman, S. G. (1998). NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U.S.A. 95, 8963–8968. doi: 10.1073/pnas.95.15.8963
Dib-Hajj, S. D., Yang, Y., Black, J. A., and Waxman, S. G. (2013). The Na(V)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62. doi: 10.1038/nrn3404
Dichgans, M., Freilinger, T., Eckstein, G., Babini, E., Lorenz-Depiereux, B., Biskup, S., et al. (2005). Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377. doi: 10.1016/S0140-6736(05)66786-4
Dietrich, P. S., Mcgivern, J. G., Delgado, S. G., Koch, B. D., Eglen, R. M., Hunter, J. C., et al. (1998). Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J. Neurochem. 70, 2262–2272. doi: 10.1046/j.1471-4159.1998.70062262.x
Djouhri, L., Newton, R., Levinson, S. R., Berry, C. M., Carruthers, B., and Lawson, S. N. (2003). Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J. Physiol. 546, 565–576. doi: 10.1113/jphysiol.2002.026559
Elliott, A. A., and Elliott, J. R. (1993). Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J. Physiol. 463, 39–56. doi: 10.1113/jphysiol.1993.sp019583
Ernsberger, U. (2009). Role of neurotrophin signalling in the differentiation of neurons from dorsal root ganglia and sympathetic ganglia. Cell Tissue Res. 336, 349–384. doi: 10.1007/s00441-009-0784-z
Escoubas, P. (2006). Molecular diversification in spider venoms: a web of combinatorial peptide libraries. Mol. Divers. 10, 545–554. doi: 10.1007/s11030-006-9050-4
Faber, C. G., Lauria, G., Merkies, I. S., Cheng, X., Han, C., Ahn, H. S., et al. (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 109, 19444–19449. doi: 10.1073/pnas.1216080109
Flinspach, M., Xu, Q., Piekarz, A. D., Fellows, R., Hagan, R., Gibbs, A., et al. (2017). Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep. 7:39662. doi: 10.1038/srep39662
Fry, A. E., Rees, E., Thompson, R., Mantripragada, K., Blake, P., Jones, G., et al. (2016). Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Med. Genet. 17:34. doi: 10.1186/s12881-016-0294-2
Fukuoka, T., Kobayashi, K., Yamanaka, H., Obata, K., Dai, Y., and Noguchi, K. (2008). Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 510, 188–206. doi: 10.1002/cne.21786
Garcia-Villegas, R., Lopez-Alvarez, L. E., Arni, S., Rosenbaum, T., and Morales, M. A. (2009). Identification and functional characterization of the promoter of the mouse sodium-activated sodium channel Na(x) gene (Scn7a). J. Neurosci. Res. 87, 2509–2519. doi: 10.1002/jnr.22069
Gilchrist, J., Olivera, B. M., and Bosmans, F. (2014). Animal toxins influence voltage-gated sodium channel function. Handb. Exp. Pharmacol. 221, 203–229. doi: 10.1007/978-3-642-41588-3_10
Goldberg, D. S., and McGee, S. J. (2011). Pain as a global public health priority. BMC Public Health 11:770. doi: 10.1186/1471-2458-11-770
Goldin, A. L. (2001). Resurgence of sodium channel research. Annu. Rev. Physiol. 63, 871–894. doi: 10.1146/annurev.physiol.63.1.871
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. doi: 10.1016/S0896-6273(00)00116-1
Goncalves, T. C., Boukaiba, R., Molgo, J., Amar, M., Partiseti, M., Servent, D., et al. (2018). Direct evidence for high affinity blockade of NaV1.6 channel subtype by huwentoxin-IV spider peptide, using multiscale functional approaches. Neuropharmacology 133, 404–414. doi: 10.1016/j.neuropharm.2018.02.016
Gorter, J. A., Zurolo, E., Iyer, A., Fluiter, K., Van Vliet, E. A., Baayen, J. C., et al. (2010). Induction of sodium channel Na(x) (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy. Epilepsia 51, 1791–1800. doi: 10.1111/j.1528-1167.2010.02678.x
Grasso, G., Landi, A., and Alafaci, C. (2016). A novel pathophysiological mechanism contributing to trigeminal neuralgia. Mol. Med. doi: 10.2119/molmed.2016.00172 [Epub ahead of print].
Gui, J., Liu, B., Cao, G., Lipchik, A. M., Perez, M., Dekan, Z., et al. (2014). A tarantula-venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1-S4 gating domain. Curr. Biol. 24, 473–483. doi: 10.1016/j.cub.2014.01.013
Hackel, D., Krug, S. M., Sauer, R. S., Mousa, S. A., Bocker, A., Pflucke, D., et al. (2012). Transient opening of the perineurial barrier for analgesic drug delivery. Proc. Natl. Acad. Sci. U.S.A. 109, E2018-E2027. doi: 10.1073/pnas.1120800109
Hackenberg, A., Baumer, A., Sticht, H., Schmitt, B., Kroell-Seger, J., Wille, D., et al. (2014). Infantile epileptic encephalopathy, transient choreoathetotic movements, and hypersomnia due to a De Novo missense mutation in the SCN2A gene. Neuropediatrics 45, 261–264. doi: 10.1055/s-0034-1372302
Hagen, N. A., Cantin, L., Constant, J., Haller, T., Blaise, G., Ong-Lam, M., du Souich, P., et al. (2017). Tetrodotoxin for moderate to severe cancer-related pain: a multicentre, randomized, double-blind, placebo-controlled, parallel-design trial. Pain Res. Manag. 2017:7212713. doi: 10.1155/2017/7212713
Hains, B. C., Klein, J. P., Saab, C. Y., Craner, M. J., Black, J. A., and Waxman, S. G. (2003). Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003
Hains, B. C., Saab, C. Y., and Waxman, S. G. (2005). Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain 128, 2359–2371. doi: 10.1093/brain/awh623
Hains, B. C., Saab, C. Y., and Waxman, S. G. (2006). Alterations in burst firing of thalamic VPL neurons and reversal by Na(v)1.3 antisense after spinal cord injury. J. Neurophysiol. 95, 3343–3352. doi: 10.1152/jn.01009.2005
Han, C., Huang, J., and Waxman, S. G. (2016). Sodium channel Nav1.8: emerging links to human disease. Neurology 86, 473–483. doi: 10.1212/WNL.0000000000002333
Han, C., Yang, Y., De Greef, B. T., Hoeijmakers, J. G., Gerrits, M. M., Verhamme, C., et al. (2015). The domain II S4-S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med. 17, 158–169. doi: 10.1007/s12017-015-8347-9
Han, C., Yang, Y., Te Morsche, R. H., Drenth, J. P., Politei, J. M., Waxman, S. G., et al. (2017). Familial gain-of-function Nav1.9 mutation in a painful channelopathy. J. Neurol. Neurosurg. Psychiatry 88, 233–240. doi: 10.1136/jnnp-2016-313804
Henriques, S. T., Deplazes, E., Lawrence, N., Cheneval, O., Chaousis, S., Inserra, M., et al. (2016). Interaction of tarantula venom peptide proTx-II with lipid membranes is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem. 291, 17049–17065. doi: 10.1074/jbc.M116.729095
Hiyama, T. Y., Matsuda, S., Fujikawa, A., Matsumoto, M., Watanabe, E., Kajiwara, H., et al. (2010). Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron 66, 508–522. doi: 10.1016/j.neuron.2010.04.017
Hiyama, T. Y., Watanabe, E., Ono, K., Inenaga, K., Tamkun, M. M., Yoshida, S., et al. (2002). Na(x) channel involved in CNS sodium-level sensing. Nat. Neurosci. 5, 511–512. doi: 10.1038/nn0602-856
Ho, C., and O’Leary, M. E. (2011). Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 46, 159–166. doi: 10.1016/j.mcn.2010.08.017
Ho, W. S., Davis, A. J., Chadha, P. S., and Greenwood, I. A. (2013). Effective contractile response to voltage-gated Na+ channels revealed by a channel activator. Am. J. Physiol. Cell Physiol. 304, C739–C747. doi: 10.1152/ajpcell.00164.2012
Hockley, J. R., Winchester, W. J., and Bulmer, D. C. (2016). The voltage-gated sodium channel NaV 1.9 in visceral pain. Neurogastroenterol. Motil. 28, 316–326. doi: 10.1111/nmo.12698
Huang, J., Han, C., Estacion, M., Vasylyev, D., Hoeijmakers, J. G., Gerrits, M. M., et al. (2014). Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137, 1627–1642. doi: 10.1093/brain/awu079
Huang, J., Vanoye, C. G., Cutts, A., Goldberg, Y. P., Dib-Hajj, S. D., Cohen, C. J., et al. (2017). Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J. Clin. Invest. 127, 2805–2814. doi: 10.1172/JCI92373
Huang, Q., Chen, Y., Gong, N., and Wang, Y. X. (2016). Methylglyoxal mediates streptozotocin-induced diabetic neuropathic pain via activation of the peripheral TRPA1 and Nav1.8 channels. Metabolism 65, 463–474. doi: 10.1016/j.metabol.2015.12.002
Iio, C., Ogimoto, A., Nagai, T., Suzuki, J., Inoue, K., Nishimura, K., et al. (2015). Association between genetic variation in the SCN10A gene and cardiac conduction abnormalities in patients with hypertrophic cardiomyopathy. Int. Heart 56, 421–427. doi: 10.1536/ihj.14-411
Inserra, M. C., Israel, M. R., Caldwell, A., Castro, J., Deuis, J. R., Harrington, A. M., et al. (2017). Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin-1. Sci. Rep. 7:42810. doi: 10.1038/srep42810
Israel, M. R., Tay, B., Deuis, J. R., and Vetter, I. (2017). Sodium channels and venom peptide pharmacology. Adv. Pharmacol. 79, 67–116. doi: 10.1016/bs.apha.2017.01.004
Jami, S., Erickson, A., Brierley, S. M., and Vetter, I. (2017). Pain-causing venom peptides: insights into sensory neuron pharmacology. Toxins 10:E15 doi: 10.3390/toxins10010015
Jarecki, B. W., Sheets, P. L., Xiao, Y., Jackson, J. O. II, and Cummins, T. R. (2009). Alternative splicing of Na(V)1.7 exon 5 increases the impact of the painful PEPD mutant channel I1461T. Channels 3, 259–267. doi: 10.4161/chan.3.4.9341
Jimenez-Andrade, J. M., Herrera, M. B., Ghilardi, J. R., Vardanyan, M., Melemedjian, O. K., and Mantyh, P. W. (2008). Vascularization of the dorsal root ganglia and peripheral nerve of the mouse: implications for chemical-induced peripheral sensory neuropathies. Mol. Pain 4:10. doi: 10.1186/1744-8069-4-10
Jo, T., Nagata, T., Iida, H., Imuta, H., Iwasawa, K., Ma, J., et al. (2004). Voltage-gated sodium channel expressed in cultured human smooth muscle cells: involvement of SCN9A. FEBS Lett. 567, 339–343. doi: 10.1016/j.febslet.2004.04.092
Jung, H. J., Lee, J. Y., Kim, S. H., Eu, Y. J., Shin, S. Y., Milescu, M., et al. (2005). Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 44, 6015–6023. doi: 10.1021/bi0477034
Ke, C. B., He, W. S., Li, C. J., Shi, D., Gao, F., and Tian, Y. K. (2012). Enhanced SCN7A/Nax expression contributes to bone cancer pain by increasing excitability of neurons in dorsal root ganglion. Neuroscience 227, 80–89. doi: 10.1016/j.neuroscience.2012.09.046
Kim, C. H., Oh, Y., Chung, J. M., and Chung, K. (2001). The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res. 95, 153–161. doi: 10.1016/S0169-328X(01)00226-1
King, M. K., Leipold, E., Goehringer, J. M., Kurth, I., and Challman, T. D. (2017). Pain insensitivity: distal S6-segment mutations in NaV1.9 emerge as critical hotspot. Neurogenetics 18, 179–181. doi: 10.1007/s10048-017-0513-9
Kinsman, B. J., Nation, H. N., and Stocker, S. D. (2017). Hypothalamic signaling in body fluid homeostasis and hypertension. Curr. Hypertens. Rep. 19:50. doi: 10.1007/s11906-017-0749-7
Kist, A. M., Sagafos, D., Rush, A. M., Neacsu, C., Eberhardt, E., Schmidt, R., et al. (2016). SCN10A mutation in a patient with erythromelalgia enhances c-fiber activity dependent slowing. PLoS One 11:e0161789. doi: 10.1371/journal.pone.0161789
Kleggetveit, I. P., Schmidt, R., Namer, B., Salter, H., Helas, T., Schmelz, M., et al. (2016). Pathological nociceptors in two patients with erythromelalgia-like symptoms and rare genetic Nav 1.9 variants. Brain Behav. 6:e00528. doi: 10.1002/brb3.528
Klint, J. K., Chin, Y. K., and Mobli, M. (2015a). Rational engineering defines a molecular switch that is essential for activity of spider-venom peptides against the analgesics target NaV1.7. Mol. Pharmacol. 88, 1002–1010. doi: 10.1124/mol.115.100784
Klint, J. K., Smith, J. J., Vetter, I., Rupasinghe, D. B., Er, S. Y., Senff, S., et al. (2015b). Seven novel modulators of the analgesic target NaV 1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 172, 2445–2458. doi: 10.1111/bph.13081
Klint, J. K., Senff, S., Rupasinghe, D. B., Er, S. Y., Herzig, V., Nicholson, G. M., et al. (2012). Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 60, 478–491. doi: 10.1016/j.toxicon.2012.04.337
Koenig, J., Werdehausen, R., Linley, J. E., Habib, A. M., Vernon, J., Lolignier, S., et al. (2015). Regulation of Nav1.7: a conserved SCN9A natural antisense transcript expressed in dorsal root ganglia. PLoS One 10:e0128830. doi: 10.1371/journal.pone.0128830
Laedermann, C. J., Abriel, H., and Decosterd, I. (2015). Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front. Pharmacol. 6:263. doi: 10.3389/fphar.2015.00263
Lai, J., Hunter, J. C., and Porreca, F. (2003). The role of voltage-gated sodium channels in neuropathic pain. Curr. Opin. Neurobiol. 13, 291–297. doi: 10.1016/S0959-4388(03)00074-6
Lamar, T., Vanoye, C. G., Calhoun, J., Wong, J. C., Dutton, S. B. B., Jorge, B. S., et al. (2017). SCN3A deficiency associated with increased seizure susceptibility. Neurobiol. Dis. 102, 38–48. doi: 10.1016/j.nbd.2017.02.006
Lee, J. H., Park, C. K., Chen, G., Han, Q., Xie, R. G., Liu, T., et al. (2014). A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell 157, 1393–1404. doi: 10.1016/j.cell.2014.03.064
Leipold, E., Hanson-Kahn, A., Frick, M., Gong, P., Bernstein, J. A., Voigt, M., et al. (2015). Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat. Commun. 6:10049. doi: 10.1038/ncomms10049
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Giesselmann, S., Baets, J., et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404. doi: 10.1038/ng.2767
Leng, X. R., Qi, X. H., Zhou, Y. T., and Wang, Y. P. (2017). Gain-of-function mutation p.Arg225Cys in SCN11A causes familial episodic pain and contributes to essential tremor. J. Hum. Genet. 62, 641–646. doi: 10.1038/jhg.2017.21
Lewis, A. H., and Raman, I. M. (2014). Resurgent current of voltage-gated Na(+) channels. J. Physiol. 592, 4825–4838. doi: 10.1113/jphysiol.2014.277582
Li, C., Wang, S., Chen, Y., and Zhang, X. (2018). Somatosensory neuron typing with high-coverage single-Cell RNA sequencing and functional analysis. Neurosci. Bull. 34, 200–207. doi: 10.1007/s12264-017-0147-9
Liao, Y., Anttonen, A. K., Liukkonen, E., Gaily, E., Maljevic, S., Schubert, S., et al. (2010). SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 75, 1454–1458. doi: 10.1212/WNL.0b013e3181f8812e
Liem, L., Van Dongen, E., Huygen, F. J., Staats, P., and Kramer, J. (2016). The dorsal root ganglion as a therapeutic target for chronic pain. Reg. Anesth. Pain Med. 41, 511–519. doi: 10.1097/AAP.0000000000000408
Lin, W. H., and Baines, R. A. (2015). Regulation of membrane excitability: a convergence on voltage-gated sodium conductance. Mol. Neurobiol. 51, 57–67. doi: 10.1007/s12035-014-8674-0
Liu, D., Tseng, M., Epstein, L. F., Green, L., Chan, B., Soriano, B., Lim, D., et al. (2016). Evaluation of recombinant monoclonal antibody SVmab1 binding to Na V1.7 target sequences and block of human Na V1.7 currents. F1000Res. 5:2764.
Liu, H., Chen, Y., Huang, L., Sun, X., Fu, T., Wu, S., et al. (2018). Drug distribution into peripheral nerve. J. Pharmacol. Exp. Ther. 365, 336–345. doi: 10.1124/jpet.117.245613
Liu, X. D., Yang, J. J., Fang, D., Cai, J., Wan, Y., and Xing, G. G. (2014). Functional upregulation of nav1.8 sodium channels on the membrane of dorsal root Ganglia neurons contributes to the development of cancer-induced bone pain. PLoS One 9:e114623. doi: 10.1371/journal.pone.0114623
Liu, Y., Tang, J., Zhang, Y., Xun, X., Tang, D., Peng, D., et al. (2014a). Synthesis and analgesic effects of mu-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins 6, 2363–2378. doi: 10.3390/toxins6082363
Liu, Y., Wu, Z., Tang, D., Xun, X., Liu, L., Li, X., et al. (2014b). Analgesic effects of Huwentoxin-IV on animal models of inflammatory and neuropathic pain. Protein Pept. Lett. 21, 153–158. doi: 10.2174/09298665113206660119
Liu, Z., Cai, T., Zhu, Q., Deng, M., Li, J., Zhou, X., et al. (2013). Structure and function of hainantoxin-III, a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels isolated from the Chinese bird spider Ornithoctonus hainana. J. Biol. Chem. 288, 20392–20403. doi: 10.1074/jbc.M112.426627
Liu, Z., Tao, J., Ye, P., and Ji, Y. (2012). Mining the virgin land of neurotoxicology: a novel paradigm of neurotoxic peptides action on glycosylated voltage-gated sodium channels. J. Toxicol. 2012:843787. doi: 10.1155/2012/843787
Liu, Z. H., Chen, P., and Liang, S. P. (2002). Synthesis and oxidative refolding of hainantoxin-IV. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao 34, 516–519.
Lolignier, S., Amsalem, M., Maingret, F., Padilla, F., Gabriac, M., Chapuy, E., et al. (2011). Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One 6:e23083. doi: 10.1371/journal.pone.0023083
Lolignier, S., Eijkelkamp, N., and Wood, J. N. (2015). Mechanical allodynia. Pflugers Arch. 467, 133–139. doi: 10.1007/s00424-014-1532-0
Lolignier, S., Gkika, D., Andersson, D., Leipold, E., Vetter, I., Viana, F., et al. (2016). New insight in cold pain: role of ion channels, modulation, and clinical perspectives. J. Neurosci. 36, 11435–11439. doi: 10.1523/JNEUROSCI.2327-16.2016
Luo, W., Wickramasinghe, S. R., Savitt, J. M., Griffin, J. W., Dawson, T. M., and Ginty, D. D. (2007). A hierarchical NGF signaling cascade controls Ret-dependent and Ret-independent events during development of nonpeptidergic DRG neurons. Neuron 54, 739–754. doi: 10.1016/j.neuron.2007.04.027
Makinson, C. D., Tanaka, B. S., Lamar, T., Goldin, A. L., and Escayg, A. (2014). Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiol. Dis. 68, 16–25. doi: 10.1016/j.nbd.2014.03.014
Minassian, N. A., Gibbs, A., Shih, A. Y., Liu, Y., Neff, R. A., Sutton, S. W., et al. (2013). Analysis of the structural and molecular basis of voltage-sensitive sodium channel inhibition by the spider toxin huwentoxin-IV (mu-TRTX-Hh2a). J. Biol. Chem. 288, 22707–22720. doi: 10.1074/jbc.M113.461392
Mogil, J. S. (2009). Animal models of pain: progress and challenges. Nat. Rev. Neurosci. 10, 283–294. doi: 10.1038/nrn2606
Moraes, E. R., Kushmerick, C., and Naves, L. A. (2014). Characteristics of dorsal root ganglia neurons sensitive to Substance P. Mol. Pain 10:73. doi: 10.1186/1744-8069-10-73
Moyer, B. D., Murray, J. K., Ligutti, J., Andrews, K., Favreau, P., Jordan, J. B., et al. (2018). Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS One 13:e0196791. doi: 10.1371/journal.pone.0196791
Mulley, J. C., Hodgson, B., Mcmahon, J. M., Iona, X., Bellows, S., Mullen, S. A., et al. (2013). Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia 54, e122–e126. doi: 10.1111/epi.12323
Muroi, Y., and Undem, B. J. (2014). Targeting voltage gated sodium channels NaV1.7, Na V1.8, and Na V1.9 for treatment of pathological cough. Lung 192, 15–20. doi: 10.1007/s00408-013-9533-x
Murray, J. K., Long, J., Zou, A., Ligutti, J., Andrews, K. L., Poppe, L., et al. (2016). Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the NaV1.7 inhibitory peptide GpTx-1. J. Med. Chem. 59, 2704–2717. doi: 10.1021/acs.jmedchem.5b01947
Namadurai, S., Yereddi, N. R., Cusdin, F. S., Huang, C. L., Chirgadze, D. Y., and Jackson, A. P. (2015). A new look at sodium channel beta subunits. Open Biol. 5:140192. doi: 10.1098/rsob.140192
Negus, S. S. (2018). Addressing the opioid crisis: the importance of choosing translational endpoints in analgesic drug discovery. Trends Pharmacol. Sci. 39, 327–330. doi: 10.1016/j.tips.2018.02.002
Nehme, B., Henry, M., Mouginot, D., and Drolet, G. (2012). The expression pattern of the Na(+) Sensor, Na(X) in the hydromineral homeostatic network: a comparative study between the rat and mouse. Front. Neuroanat. 6:26. doi: 10.3389/fnana.2012.00026
Nishiya, Y., Yokokawa, S., Fukuda, A., Yamagata, T., Inayoshi, A., Obinata, M., et al. (2011). The generation of rat dorsal root ganglion cell lines to identify the target of KW-7158, a novel treatment for overactive bladder. Neurosci. Res. 71, 278–288. doi: 10.1016/j.neures.2011.07.1823
O’Brien, J. E., and Meisler, M. H. (2013). Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 4:213. doi: 10.3389/fgene.2013.00213
O’Donnell, A. M., Coyle, D., and Puri, P. (2016). Decreased Nav1.9 channel expression in Hirschsprung’s disease. J. Pediatr. Surg. 51, 1458–1461. doi: 10.1016/j.jpedsurg.2016.05.007
Okuda, H., Noguchi, A., Kobayashi, H., Kondo, D., Harada, K. H., Youssefian, S., et al. (2016). Infantile pain episodes associated with novel Nav1.9 mutations in familial episodic pain syndrome in japanese families. PLoS One 11:e0154827. doi: 10.1371/journal.pone.0154827
Ono, S., Kimura, T., and Kubo, T. (2011). Characterization of voltage-dependent calcium channel blocking peptides from the venom of the tarantula Grammostola rosea. Toxicon 58, 265–276. doi: 10.1016/j.toxicon.2011.06.006
Osmakov, D. I., Andreev, Y. A., and Kozlov, S. A. (2014). Acid-sensing ion channels and their modulators. Biochemistry 79, 1528–1545. doi: 10.1134/S0006297914130069
Osteen, J. D., Herzig, V., Gilchrist, J., Emrick, J. J., Zhang, C., Wang, X., et al. (2016). Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499. doi: 10.1038/nature17976
Osteen, J. D., Sampson, K., Iyer, V., Julius, D., and Bosmans, F. (2017). Pharmacology of the Nav1.1 domain IV voltage sensor reveals coupling between inactivation gating processes. Proc. Natl. Acad. Sci. U.S.A. 114, 6836–6841. doi: 10.1073/pnas.1621263114
Pan, H. L., Wu, Z. Z., Zhou, H. Y., Chen, S. R., Zhang, H. M., and Li, D. P. (2008). Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 117, 141–161. doi: 10.1016/j.pharmthera.2007.09.003
Parke, W. W., and Whalen, J. L. (2002). The vascular pattern of the human dorsal root ganglion and its probable bearing on a compartment syndrome. Spine 27, 347–352. doi: 10.1097/00007632-200202150-00004
Patrick Harty, T., and Waxman, S. G. (2007). Inactivation properties of sodium channel Nav1.8 maintain action potential amplitude in small DRG neurons in the context of depolarization. Mol. Pain 3:12.
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi: 10.1038/nature10238
Peng, K., Shu, Q., Liu, Z., and Liang, S. (2002). Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol. Chem. 277, 47564–47571. doi: 10.1074/jbc.M204063200
Petterson, C. A., and Olsson, Y. (1989). Blood supply of spinal nerve roots. An experimental study in the rat. Acta Neuropathol. 78, 455–461. doi: 10.1007/BF00687706
Phatarakijnirund, V., Mumm, S., Mcalister, W. H., Novack, D. V., Wenkert, D., Clements, K. L., et al. (2016). Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9. Bone 84, 289–298. doi: 10.1016/j.bone.2015.11.022
Pineda, S. S., Chaumeil, P. A., Kunert, A., Kaas, Q., Thang, M. W. C., Le, L., et al. (2018). ArachnoServer 3.0: an online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 34, 1074–1076. doi: 10.1093/bioinformatics/btx661
Porreca, F., and Navratilova, E. (2017). Reward, motivation, and emotion of pain and its relief. Pain 158(Suppl. 1), S43–S49. doi: 10.1097/j.pain.0000000000000798
Qiu, F., Jiang, Y., Zhang, H., Liu, Y., and Mi, W. (2012). Increased expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 within dorsal root ganglia in a rat model of bone cancer pain. Neurosci. Lett. 512, 61–66. doi: 10.1016/j.neulet.2012.01.069
Rahnama, S., Deuis, J. R., Cardoso, F. C., Ramanujam, V., Lewis, R. J., Rash, L. D., et al. (2017). The structure, dynamics and selectivity profile of a NaV1.7 potency-optimised huwentoxin-IV variant. PLoS One 12:e0173551. doi: 10.1371/journal.pone.0173551
Raymon, H. K., Thode, S., Zhou, J., Friedman, G. C., Pardinas, J. R., Barrere, C., et al. (1999). Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J. Neurosci. 19, 5420–5428. doi: 10.1523/JNEUROSCI.19-13-05420.1999
Raymond, C. K., Castle, J., Garrett-Engele, P., Armour, C. D., Kan, Z., Tsinoremas, N., et al. (2004). Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. J. Biol. Chem. 279, 46234–46241. doi: 10.1074/jbc.M406387200
Ren, Y. S., Qian, N. S., Tang, Y., Liao, Y. H., Yang, Y. L., Dou, K. F., et al. (2012). Sodium channel Nav1.6 is up-regulated in the dorsal root ganglia in a mouse model of type 2 diabetes. Brain Res. Bull. 87, 244–249. doi: 10.1016/j.brainresbull.2011.10.015
Renganathan, M., Cummins, T. R., and Waxman, S. G. (2001). Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640. doi: 10.1152/jn.2001.86.2.629
Ritter, A. M., Martin, W. J., and Thorneloe, K. S. (2009). The voltage-gated sodium channel Nav1.9 is required for inflammation-based urinary bladder dysfunction. Neurosci. Lett. 452, 28–32. doi: 10.1016/j.neulet.2008.12.051
Rubinstein, M., Patowary, A., Stanaway, I. B., Mccord, E., Nesbitt, R. R., Archer, M., et al. (2018). Association of rare missense variants in the second intracellular loop of NaV1.7 sodium channels with familial autism. Mol. Psychiatry 23, 231–239. doi: 10.1038/mp.2016.222
Saez, N. J., Senff, S., Jensen, J. E., Er, S. Y., Herzig, V., Rash, L. D., et al. (2010). Spider-venom peptides as therapeutics. Toxins 2, 2851–2871. doi: 10.3390/toxins2122851
Sah, D. W., Ray, J., and Gage, F. H. (1997). Bipotent progenitor cell lines from the human CNS. Nat. Biotechnol. 15, 574–580. doi: 10.1038/nbt0697-574
Salvatierra, J., Castro, J., Erickson, A., Li, Q., Braz, J., Gilchrist, J., et al. (2018). NaV1.1 inhibition can reduce visceral hypersensitivity. JCI Insight 3:121000. doi: 10.1172/jci.insight.121000
Savage, S. R., Kirsh, K. L., and Passik, S. D. (2008). Challenges in using opioids to treat pain in persons with substance use disorders. Addict. Sci. Clin. Pract. 4, 4–25. doi: 10.1151/ascp08424
Schirmeyer, J., Szafranski, K., Leipold, E., Mawrin, C., Platzer, M., and Heinemann, S. H. (2014). Exon 11 skipping of SCN10A coding for voltage-gated sodium channels in dorsal root ganglia. Channels 8, 210–215. doi: 10.4161/chan.28146
Schmalhofer, W. A., Calhoun, J., Burrows, R., Bailey, T., Kohler, M. G., Weinglass, A. B., et al. (2008). ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 74, 1476–1484. doi: 10.1124/mol.108.047670
Scholz, A., Gruss, M., and Vogel, W. (1998). Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J. Physiol. 513(Pt 1), 55–69. doi: 10.1111/j.1469-7793.1998.055by.x
Scholz, A., and Vogel, W. (2000). Tetrodotoxin-resistant action potentials in dorsal root ganglion neurons are blocked by local anesthetics. Pain 89, 47–52. doi: 10.1016/S0304-3959(00)00345-6
Smith, J. J., Cummins, T. R., Alphy, S., and Blumenthal, K. M. (2007). Molecular interactions of the gating modifier toxin ProTx-II with NaV 1.5: implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 282, 12687–12697. doi: 10.1074/jbc.M610462200
Sommer, C. (2016). Exploring pain pathophysiology in patients. Science 354, 588–592. doi: 10.1126/science.aaf8935
Song, W., Liu, Z., Tan, J., Nomura, Y., and Dong, K. (2004). RNA editing generates tissue-specific sodium channels with distinct gating properties. J. Biol. Chem. 279, 32554–32561. doi: 10.1074/jbc.M402392200
Sousa, S. R., Wingerd, J. S., Brust, A., Bladen, C., Ragnarsson, L., Herzig, V., et al. (2017). Discovery and mode of action of a novel analgesic β-toxin from the African spider Ceratogyrus darlingi. PLoS One 12:e0182848. doi: 10.1371/journal.pone.0182848
Spray, D. C., and Hanani, M. (2017). Gap junctions, pannexins and pain. Neurosci. Lett. doi: 10.1016/j.neulet.2017.06.035 [Epub ahead of print].
Sun, H., Kollarik, M., and Undem, B. J. (2017). Blocking voltage-gated sodium channels as a strategy to suppress pathological cough. Pulm. Pharmacol. Ther. 47, 38–41. doi: 10.1016/j.pupt.2017.05.010
Taddese, A., Nah, S. Y., and Mccleskey, E. W. (1995). Selective opioid inhibition of small nociceptive neurons. Science 270, 1366–1369. doi: 10.1126/science.270.5240.1366
Tan, A. M., Samad, O. A., Dib-Hajj, S. D., and Waxman, S. G. (2015). Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol. Med. 21, 544–552. doi: 10.2119/molmed.2015.00063
Tan, J., Liu, Z., Nomura, Y., Goldin, A. L., and Dong, K. (2002). Alternative splicing of an insect sodium channel gene generates pharmacologically distinct sodium channels. J. Neurosci. 22, 5300–5309. doi: 10.1523/JNEUROSCI.22-13-05300.2002
Tanaka, B. S., Zhao, P., Dib-Hajj, F. B., Morisset, V., Tate, S., Waxman, S. G., and Dib-Hajj, S. D. (2016). A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol. Med. 22, 338–348. doi: 10.2119/molmed.2016.00131
Tanaka, K., Sekino, S., Ikegami, M., Ikeda, H., and Kamei, J. (2015). Antihyperalgesic effects of ProTx-II, a Nav1.7 antagonist, and A803467, a Nav1.8 antagonist, in diabetic mice. J. Exp. Pharmacol. 7, 11–16. doi: 10.2147/JEP.S79973
Thakor, D. K., Lin, A., Matsuka, Y., Meyer, E. M., Ruangsri, S., Nishimura, I., and Spigelman, I. (2009). Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol. Pain 5:14. doi: 10.1186/1744-8069-5-14
Thompson, C. H., Kahlig, K. M., and George, A. L. Jr. (2011). SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52, 1000–1009. doi: 10.1111/j.1528-1167.2011.03040.x
Tonello, R., Fusi, C., Materazzi, S., Marone, I. M., De Logu, F., Benemei, S., et al. (2017). The peptide phalpha1beta, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice. Br. J. Pharmacol. 174, 57–69. doi: 10.1111/bph.13652
Torres-Perez, J. V., Adamek, P., Palecek, J., Vizcaychipi, M., Nagy, I., and Varga, A. (2018). The NAv1.7 blocker protoxin II reduces burn injury-induced spinal nociceptive processing. J. Mol. Med. 96, 75–84. doi: 10.1007/s00109-017-1599-0
Trimmer, J. S., and Rhodes, K. J. (2004). Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 66, 477–519. doi: 10.1146/annurev.physiol.66.032102.113328
Vetter, I., Deuis, J. R., Mueller, A., Israel, M. R., Starobova, H., Zhang, A., et al. (2017). NaV1.7 as a pain target - from gene to pharmacology. Pharmacol. Ther. 172, 73–100. doi: 10.1016/j.pharmthera.2016.11.015
Vijayaragavan, K., Powell, A. J., Kinghorn, I. J., and Chahine, M. (2004). Role of auxiliary beta1-, beta2-, and beta3-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem. Biophys. Res. Commun. 319, 531–540. doi: 10.1016/j.bbrc.2004.05.026
Watanabe, E., Hiyama, T. Y., Shimizu, H., Kodama, R., Hayashi, N., Miyata, S., et al. (2006). Sodium-level-sensitive sodium channel Na(x) is expressed in glial laminate processes in the sensory circumventricular organs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R568–R576. doi: 10.1152/ajpregu.00618.2005
Waxman, S. G., Kocsis, J. D., and Black, J. A. (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 72, 466–470. doi: 10.1152/jn.1994.72.1.466
Wiesenfeld-Hallin, Z., and Xu, X. J. (2001). Neuropeptides in neuropathic and inflammatory pain with special emphasis on cholecystokinin and galanin. Eur. J. Pharmacol. 429, 49–59. doi: 10.1016/S0014-2999(01)01305-X
Wolff, M., Johannesen, K. M., Hedrich, U. B. S., Masnada, S., Rubboli, G., Gardella, E., et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336. doi: 10.1093/brain/awx054
Woods, C. G., Babiker, M. O., Horrocks, I., Tolmie, J., and Kurth, I. (2015). The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur. J. Hum. Genet. 23, 561–563. doi: 10.1038/ejhg.2014.166
Xiao, Y., Luo, X., Kuang, F., Deng, M., Wang, M., Zeng, X., and Liang, S. (2008). Synthesis and characterization of huwentoxin-IV, a neurotoxin inhibiting central neuronal sodium channels. Toxicon 51, 230–239. doi: 10.1016/j.toxicon.2007.09.008
Xie, W., Strong, J. A., Ye, L., Mao, J. X., and Zhang, J. M. (2013). Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 154, 1170–1180. doi: 10.1016/j.pain.2013.02.027
Xie, W., Tan, Z. Y., Barbosa, C., Strong, J. A., Cummins, T. R., and Zhang, J. M. (2016). Upregulation of the sodium channel NaVbeta4 subunit and its contributions to mechanical hypersensitivity and neuronal hyperexcitability in a rat model of radicular pain induced by local dorsal root ganglion inflammation. Pain 157, 879–891. doi: 10.1097/j.pain.0000000000000453
Xing, D., Wu, Y., Li, G., Song, S., Liu, Y., Liu, H., et al. (2015). Role of cerebrospinal fluid-contacting nucleus in sodium sensing and sodium appetite. Physiol. Behav. 147, 291–299. doi: 10.1016/j.physbeh.2015.04.034
Xiong, Z., Yi, L., Cao, D., He, W., Chen, J., Gao, S., and Sun, X. (2016). Dravet syndrome with autism inherited from a paternal mosaic heterozygous mutation on SCN1A. J. Neurol. Sci. 369, 53–56. doi: 10.1016/j.jns.2016.07.038
Yang, C., Hua, Y., Zhang, W., Xu, J., Xu, L., Gao, F., and Jiang, P. (2018). Variable epilepsy phenotypes associated with heterozygous mutation in the SCN9A gene: report of two cases. Neurol. Sci. 39, 1113–1115. doi: 10.1007/s10072-018-3300-y
Yang, Y., Mis, M. A., Estacion, M., Dib-Hajj, S. D., and Waxman, S. G. (2018). NaV1.7 as a pharmacogenomic target for pain: moving toward precision medicine. Trends Pharmacol. Sci. 39, 258–275. doi: 10.1016/j.tips.2017.11.010
Yang, L., Li, Q., Liu, X., and Liu, S. (2016). Roles of voltage-gated tetrodotoxin-sensitive sodium channels NaV1.3 and NaV1.7 in diabetes and painful diabetic neuropathy. Int. J. Mol. Sci. 17:E1479 doi: 10.3390/ijms17091479
Yekkirala, A. S., Roberson, D. P., Bean, B. P., and Woolf, C. J. (2017). Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 16, 545–564. doi: 10.1038/nrd.2017.87
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4:207.
Yu, F. H., and Catterall, W. A. (2004). The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004:re15. doi: 10.1126/stke.2532004re15
Zeng, X., Li, P., Chen, B., Huang, J., Lai, R., Liu, J., and Rong, M. (2018). Selective closed-State Nav1.7 blocker JZTX-34 exhibits analgesic effects against pain. Toxins 10:E64. doi: 10.3390/toxins10020064
Zhang, A. H., Sharma, G., Undheim, E. A. B., Jia, X., and Mobli, M. (2018). A complicated complex: ion channels, voltage sensing, cell membranes and peptide inhibitors. Neurosci. Lett. 679, 35–47 doi: 10.1016/j.neulet.2018.04.030
Zhang, X., Priest, B. T., Belfer, I., and Gold, M. S. (2017). Voltage-gated Na(+) currents in human dorsal root ganglion neurons. eLife 6:e23235 doi: 10.7554/eLife.23235
Zhang, X. Y., Wen, J., Yang, W., Wang, C., Gao, L., Zheng, L. H., et al. (2013). Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 93, 957–966. doi: 10.1016/j.ajhg.2013.09.016
Keywords: voltage-gated sodium channels, NaV1.7 channel subtype, spider toxins, pain, dorsal root ganglia neurons, electrophysiology
Citation: Gonçalves TC, Benoit E, Partiseti M and Servent D (2018) The NaV1.7 Channel Subtype as an Antinociceptive Target for Spider Toxins in Adult Dorsal Root Ganglia Neurons. Front. Pharmacol. 9:1000. doi: 10.3389/fphar.2018.01000
Received: 22 June 2018; Accepted: 14 August 2018;
Published: 04 September 2018.
Edited by:
Yuri N. Utkin, Institute of Bioorganic Chemistry (RAS), RussiaReviewed by:
Sandrine Cestèle, UMR7275 Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), FranceMaria Elena De Lima, Universidade Federal de Minas Gerais, Brazil
Copyright © 2018 Gonçalves, Benoit, Partiseti and Servent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denis Servent, denis.servent@cea.fr