Case Report: Novel biallelic moderately damaging variants in RTTN in a patient with cerebellar dysplasia
 Ferruccio Romano1
Ferruccio Romano1  Elisabetta Amadori2,3
Elisabetta Amadori2,3  Francesca Madia4
Francesca Madia4  Mariasavina Severino5
Mariasavina Severino5  Valeria Capra1
Valeria Capra1  Renata Rizzo6
Renata Rizzo6  Rita Barone6
Rita Barone6  Beatrice Corradi7,8
Beatrice Corradi7,8  Luca Maragliano8,9 Mohammad Sadegh Shams Nosrati4
Luca Maragliano8,9 Mohammad Sadegh Shams Nosrati4  Antonio Falace4
Antonio Falace4  Pasquale Striano2,10
Pasquale Striano2,10  Federico Zara2,4*
Federico Zara2,4*  Marcello Scala2,4*
Marcello Scala2,4*
- 1Genomics and Clinical Genetics Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 2Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, Università Degli Studi di Genova, Genoa, Italy
- 3Child Neuropsichiatry Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 4Medical Genetics Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 5Neuroradiology Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 6Child Neuropsychiatry Unit, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 7Department of Experimental Medicine, University of Genova, Genova, Italy
- 8Center for Synaptic Neuroscience and Technology, Istituto Italiano di Tecnologia, Genova, Italy
- 9Department of Life and Environmental Sciences, Polytechnic University of Marche, Ancona, Italy
- 10Pediatric Neurology and Muscular Diseases Unit, IRCCS, Genoa, Italy
Rotatin, encoded by the RTTN gene, is a centrosomal protein with multiple, emerging functions, including left-right specification, ciliogenesis, and neuronal migration. Recessive variants in RTTN are associated with a neurodevelopmental disorder with microcephaly and malformations of cortical development known as “Microcephaly, short stature, and polymicrogyria with seizures” (MSSP, MIM #614833). Affected individuals show a wide spectrum of clinical manifestations like intellectual disability, poor/absent speech, short stature, microcephaly, and congenital malformations. Here, we report a subject showing a distinctive neuroradiological phenotype and harboring novel biallelic variants in RTTN: the c.5500A>G, p.(Asn1834Asp), (dbSNP: rs200169343, ClinVar ID:1438510) and c.19A>G, p.(Ile7Val), (dbSNP: rs201165599, ClinVar ID:1905275) variants. In particular brain magnetic resonance imaging (MRI) showed a peculiar pattern, with cerebellar hypo-dysplasia, and multiple arachnoid cysts in the lateral cerebello-medullary cisterns, in addition to left Meckel cave. Thus, we compare his phenotypic features with current literature, speculating a possible role of newly identified RTTN variants in his clinical picture, and supporting a relevant variability in this emerging condition.
Introduction
The RTTN gene (MIM × 610436) is located on chromosome 18 (18q22.2 region) and encodes a brain-enriched protein known as Rotatin, that plays crucial roles in brain development. In particular, Rotatin is involved in the early developmental processes of left-right (L-R) specification and axial rotation (1). More recently, Rotatin was found to be relevant to primary ciliogenesis, sonic hedgehog (SHH) signaling, and neuronal migration (2). Additionally, RTTN was suggested to regulate neuronal differentiation, centrosome biogenesis, and cell growth and duplication (2). Cilia formation and centrosome/centriole biogenesis are in fact considered overlapping and interdependent processes (3, 4). Rotatin is directly implicated in the regulation of different phases of the cell cycle and mutant cells show severe mitotic failure, leading to aneuploidy and apoptosis (5). However, several functional aspects of Rotatin remain poorly understood.
Biallelic variants in RTTN are associated with a neurodevelopmental condition characterized by microcephaly, epilepsy, and brain developmental abnormalities. The variety of cellular processes regulated by Rotatin partly explains the heterogeneity of clinical manifestations observed in RTTN patients, especially cerebral malformations, related to pathological proliferation and migration defects (6). Recently, a thorough revision of literature by Vandervore, including all reported cases by 2019, highlighted the “core features”, consisting of intellectual disability, poor/absent speech, short stature and variable cerebral malformations. These include congenital or secondary microcephaly, lissencephaly, gyration anomalies, periventricular heterotopia, and interhemispheric arachnoid cysts. The cortical malformations are often more evident in the frontal areas, suggesting elective underdevelopment of the frontal lobes (7). Among other clinical manifestations, seizures are described as well as other congenital malformations, especially involving the eye (microphtalmia, orbital abnormalities, and optic nerve hypoplasia), the heart, the kidney (pyelocaliectasis, renal ectopy, or agenesis), and/or the urogenital system (6, 8–10).
In this study we report a patient with novel biallelic RTTN variants with predicted moderate pathogenic effect, presenting with a milder clinical phenotype characterized by cognitive impairment and peculiar neuroimaging abnormalities, including cerebellar dysplasia.
Methods
Patient enrollment and clinical assessment
The study was conducted in accordance with the Declaration of Helsinki and approved by the local Institutional Ethics Committees. The patient was enrolled at Istituto Giannina Gaslini, Genoa, and clinically evaluated by pediatric geneticists and neurologists. Informed consent was obtained by the parents.
Genetic investigation
For genetic testing, trio-WES was performed in the family on genomic DNA extracted from peripheral blood. Agilent Sure Select QXT Clinical Research Exome (Agilent Technologies, Santa Clara, CA, USA) was used and Sequencing data were processed with in-house software for the GATK Best Practices pipeline for WES variant analysis execution. After filtering for allele frequency (≤0.01% in public databases, including GnomAD v2.1.1; https://gnomad.broadinstitute.org/), candidate variants were screened according to family segregation, conservation (GERP score), predicted impact on protein function through in silico tools (including SIFT, PolyPhen-2, Mutation Taster), and presence in clinical databases (ClinVar) (11). The variants were eventually validated through Sanger sequencing and candidate variants were classified according to the American College of Medical Genetics and Genomics (ACMG) criteria (12).
Protein modeling
To better characterize the impact of the identified RTTN variants on protein structure, we introduced them in a 3D model of the WT molecule (Figure 1). The predicted structural model of wild-type (WT) human RTTN was retrieved from the AlphaFold Protein Structure Database (entry AF-Q86VV8-F1-model_v4) (13). The protein comprises 2,226 residues arranged in nearly all-helical domains, with the first 180 residues separated from the rest of the chain (Figure 2A). The model quality score (the predicted local-distance difference test, pLDDT) is higher than 70 (high confidence) for almost all the secondary structure elements and below 50 (low confidence) only for limited disordered regions and interhelical loops.
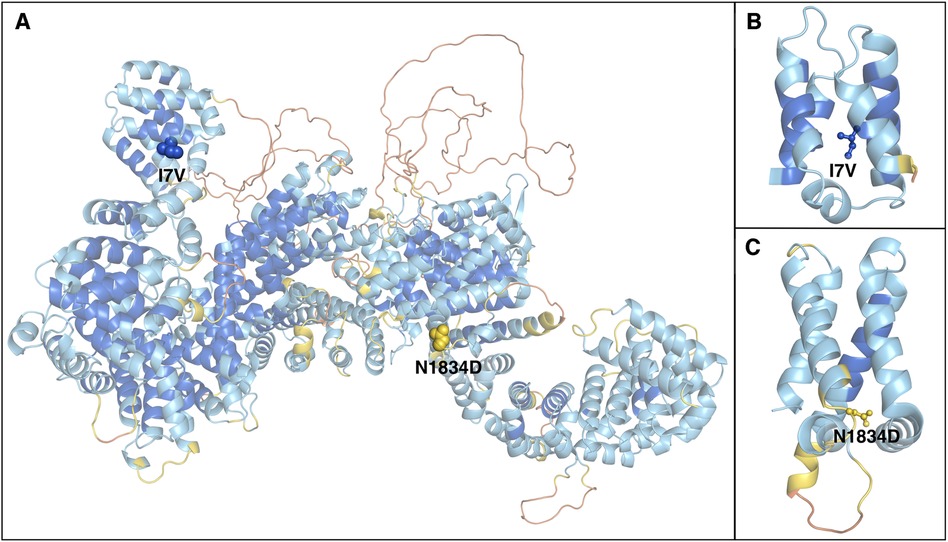
Figure 1. Structural model of the RTTN protein. (A) Alpha Fold predicted model of human rotatin. The protein is represented as cartoons and coloured according to the pLDDT confidence score, from orange (pLDDT < 50) to blue (pLDDT > 90). Mutated residues are represented as spheres and labeled. (B,C) enlarged views of the domains containing the mutations, which are represented as balls and sticks.

Figure 2. Extract from UCSC browser (hg19) showing the conservation of the residues affected by the variants harbored by our patient (highlighted in light blue) in different species and the relative GERP scores.
Results
Clinical description
This is the first 8-year-old boy of non-consanguineous parents of Italian ancestry. His mother is affected by juvenile myoclonic epilepsy since age 17 years and hypothyroidism. His father underwent surgical resection to treat focal epilepsy due to left temporal cortical dysplasia. Pregnancy was characterized by threatened abortion at 4 months. Due to placenta praevia a caesarian section was performed at 37 weeks of gestation. At birth, he only showed mild global hypotonia. Growth parameters at birth are not available but reported within the normal range. Ultrasonography revealed patent ductus arteriosus and enlarged renal pelvis. During the first months of life, the child showed signs of psychomotor delay: head and trunk control was achieved at 8 months, autonomous walking at 17 months, and the first words were pronounced at 18 months, with slow speech progression. Axial hypotonia persisted. At 4 months, the patient began to experience daily absences with eye deviation, lasting a few minutes and with spontaneous resolution. Electroencephalograms (EEGs) showed generalized high voltage epileptic abnormalities (worsened by sleep) in the frontal and left temporal regions. Valproic acid (15 mg/kg/day) was started, with poor clinical response. During the following years, behavioral issues emerged, namely attention deficit hyperactivity disorder (ADHD), severe speech impairment, motor clumsiness, and aggressiveness. Wechsler Preschool and Primary Scale of Intelligence (WPPSI-IV) revealed global psychomotor delay, especially in the verbal/communicative area. Autism Diagnostic Observation Schedule (ADOS-2) led to a diagnosis of autism spectrum disorder (ASD). At last clinical evaluation (age 8 years and 3 months) growth parameters were the following: height 138 cm (75th–90th percentile), weight 42.2 kg (>97th percentile) and head circumference 51 cm (10th percentile).
Neuroimaging analysis
Brain MRI at 3 years and 7 months of age showed cerebellar hypo-dysplasia characterized by abnormal foliar pattern and mildly reduced hemispheric volume, associated with multiple arachnoid cysts in the lateral cerebello-medullary cisterns and left Meckel cave (Figure 3 and Supplementary Figure S1). In addition, there was a developmental venous anomaly in the left posterior temporal region associated with an abnormal cortical infolding and white matter gliosis (Supplementary Figure S2). A small ecchordosis physaliphora was noted at the level of the clivus, while a cystic pineal gland with signs of internal bleeding was detected as incidental finding (Supplementary Figure S2).
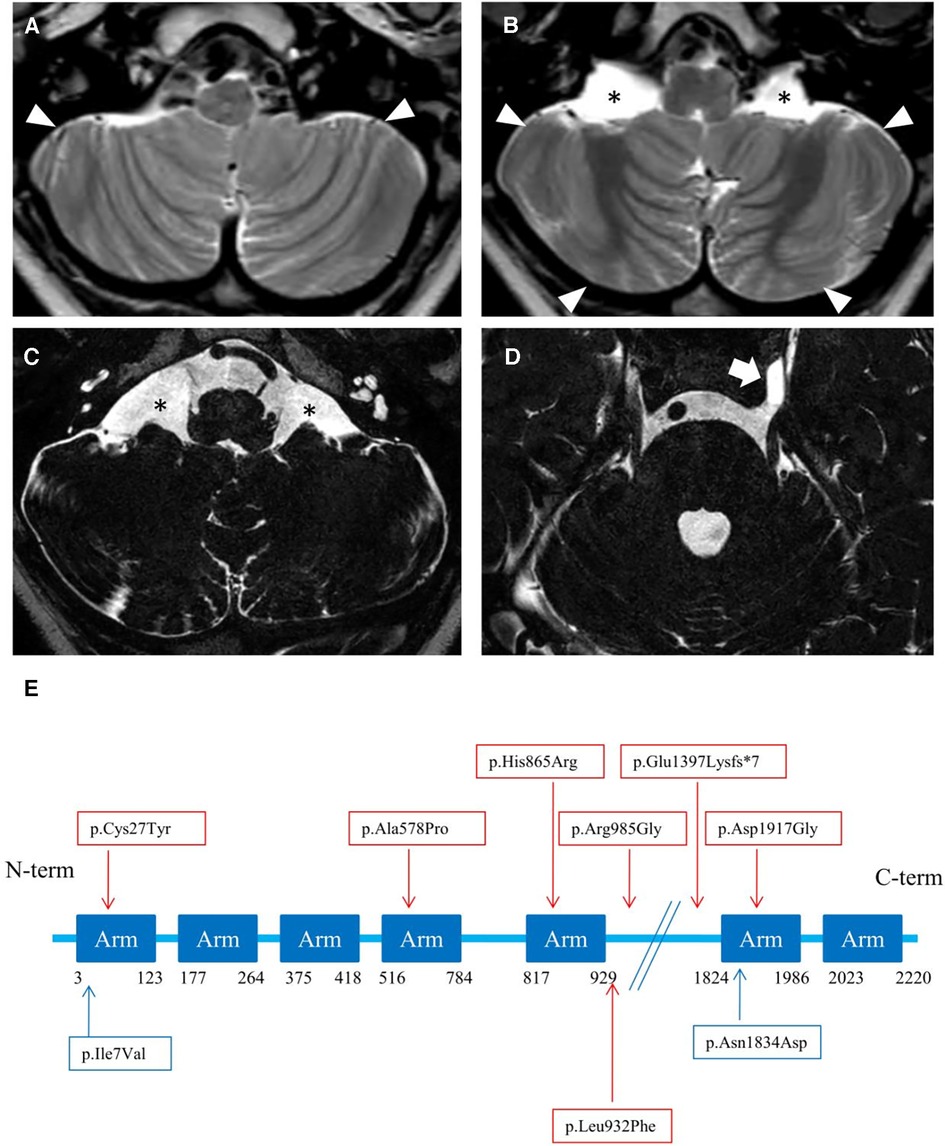
Figure 3. (A–D) neuroimaging of the patient. Brain MRI with axial T2-weighted images (A, B) and high-resolution heavily T2-weighted DRIVE images (C, D) reveal abnormal foliation pattern in the inferior portions of smaller cerebellar hemispheres (arrowheads). There are multiple arachnoid cysts localized in the lateral cerebello-medullary cisterns (asterisks) and left Meckel cave (thick arrow). (E) Linear structure of Rotatin protein showing Armadillo type fold (Arm aminoacids) repeat-rich areas mediating protein-protein interaction, modified from Cavallin et al. (7). The variants identified in the patient of this study are highlighted in blue, others are reported in red.
Genetic results
Genetic investigation included Comparative Genomic Hybridization (CGH)-array, analysis for Fragile-X syndrome and a multi-gene panel for epileptic encephalopathies. All these tests yielded negative results. As a further step of investigation, whole exome sequencing (WES) was performed on DNA from blood's lymphocytes of the proband and his parents. We identified two compound heterozygous variants in the RTTN gene (NM_173630.4) in the patient: the paternally inherited c.5500A>G, p.(Asn1834Asp) variant (rs200169343, ClinVar ID 1438510), and the maternally-inherited c.19A>G, p.(Ile7Val) variant (rs201165599, ClinVar ID 1905275).
Protein modeling
The modelling of the variants on the three-dimensional RTTN protein structure showed that the two mutated residues p.(Ile7Val), (I7V), and p.(Asn1834Asp), (N1834D) are located in the first helix of the outlying N-terminal and on a loop of a tightly packed region, respectively (Figure 1B,C). The impact of mutations on protein stability can be estimated by the difference between the unfolding free energy of the mutated and the WT protein (ΔΔG), with negative values indicating destabilization. We used a machine learning method, ACDC-NN (14), to calculate ΔΔG values for the two substitutions, obtaining −0.60 Kcal/mol and −0.17 Kcal/mol for I7V and N1834D, respectively, thus identifying both as destabilizing.
Discussion
Typical and atypical clinical features
We report a subject harboring compound heterozygous variants in RTTN, presenting with both typical and atypical clinical/neuroradiological features. In addition to the principal neurodevelopmental features of MSSP, our patient suffers from untreatable epilepsy and persistent hypotonia, which are observed in about 17% of cases (6). In line with previously described subjects, our patient presents with behavioral issues, including ADHD, ASD, and aggressiveness. He was also diagnosed with a calico-pyelic dilatation, reported in almost 22% of patients (6). No additional malformation could be detected. Of note, while poor growth and microcephaly are considered seminal features of MSSP, we did not observe them in our patient. The clinical phenotype of MSSP, however, is particularly heterogeneous (Supplementary Table S1) (7) and it is known that microcephaly may be lacking in affected individuals: about 81% of the individuals are microcephalic at birth, and in total 86% have secondary microcephaly, which means that normal head circumference at birth is not an exclusion criterion for RTTN mutations (6).
Typical and atypical neuroradiological features
The cerebral malformations identified in our patient were less severe than other RTTN cases, which are characterized by microcephaly, simplified gyration, lissencephaly/pachygyria, polymicrogyria, nodular heterotopias, midline defects, and cerebellar hypoplasia (6). Indeed, we noticed a peculiar form of cerebellar hypo-dysplasia characterized by bilateral abnormal foliation at the level of the inferior portions of the cerebellar hemispheres with mildly reduced cerebellar volume. The cerebellar cortical pattern has not been described in detail in RTTN subjects with cerebellar hypoplasia. The possible role of RTTN in cerebellar development is not elucidated to date, and cannot be excluded. Vandervore and colleagues reported the presence of (ponto) cerebellar hypoplasia in 30% of patients (2, 3, 6, 8, 9). Thus, the peculiar foliar arrangement observed in our patient might reflect a rotatin defect. No additional malformation of cortical development was detectable, except for a focal cortical infolding associated with a developmental venous anomaly. Based on current knowledge, we can only speculate that isolated cerebellar dysplasia, in absence of other malformative features, may suggest the possibility of a milder spectrum than expected in MSSP.
Arachnoid cysts in subjects with RTTN variants have been described in several locations, including the posterior interhemispheric region, quadrigeminal cistern, and anterior temporal regions. Large interhemispheric cysts were sometimes associated with severe brain disruption, like the hydrocephaly of Rttn-/- knockout mice (1). Finally, two incidental findings were detected, including an ecchordosis physaliphora, a congenital benign hamartomatous lesion originating from nodal cord remnants (15), and a pineal cyst complicated by hemorrhage.
Hypotheses on the phenotypic role of RTTN variants
In our patient, WES analysis led to the identification of two distinct missense variants that the patient harbored in compound heterozygous state. The paternal c.5500A>G, p.(Asn1834Asp) variant is rare in gnomAD (allele frequency 0.0000882) and never reported in homozygous state in healthy individuals. It is classified as a variant of uncertain significance (VUS) with the following parameters: PM2 and BP4. It affects a conserved residue (GERP = 5.78, Figure 2) within the sixth Arm domain of the protein and is predicted to be only moderately damaging according to in silico tools (CADD score = 15.63; SIFT = 0.031; Polyphen-2 = 0.554). Similarly, the maternal c.19A>G, p.(Ile7Val) variant is rare in gnomAD (allele frequency 0.000104) and is never reported in homozygous state in healthy subjects. According to ACMG classification it is considered a VUS (PM2, BP4). This change affects a conserved residue (GERP score = 4.92, Figure 2) within the first Arm domain of the protein and is predicted to have a moderate pathogenic impact (CADD score = 20.9; SIFT = 0.598; Polyphen-2 = 0.023). No additional variants with potential pathogenic impact in other disease-related genes was identifiable and no potentially damaging inherited dominant variant was detected in the proband. Despite the conflicting in silico predictions, both variants are very rare in the general population and only reported in heterozygous state in healthy subjects. Furthermore, they affect conserved residues within functional domains of the RTTN protein and fall in close proximity to previously reported variants associated with MSSP (Figure 3). It is worth noticing that both variants are considered having a destabilizing effect, based on Alphafold protein modeling (Figure 1).
We cannot exclude that the milder phenotype of our patient might reflect a less deleterious effect on protein function of the underlying mutations. Thus, it is possible that the severity and complexity of the phenotype depends on the amount of residual functional rotatin (3). RTTN functions have not yet been fully explored and further studies are required to investigate this hypothesis.
Limitations of the study
A limitation of our study is the unavailability of recent parental brain MRIs. Thus we cannot exclude the presence of subtle brain malformations in the parents. Other possible genetic factors segregating in the family have not been identified, but cannot be excluded. Another limitation is represented by the absence of functional studies, demonstrating a clear (though mild or moderate) effect of the identified variants.
Genotype-phenotype correlations
The phenotypic features in association with biallelic RTTN mutations is heterogeneous, including a vast spectrum of clinical entities. The milder end of the clinical spectrum consists of a neurodevelopmental delay syndrome with mild growth deficiency and a polymicrogyria-like cortical malformation. At the severe end of the spectrum, there is microcephalic primordial dwarfism with different kinds of cortical malformations (6, 8). Extreme phenotypes are reported too, characterized by complex cerebral malformations, heart abnormalities, joint contractures and kidney defects leading, in some cases, to early death (8, 16). The differential diagnosis includes neurodevelopmental conditions with microcephaly and malformations of cortical development, such as the spectrum of complex disorders associated with RAC proteins dysfunction (17–19). Due to the limited number of patients with recessive RTTN mutations it is not clear if some of the sporadically described clinical features are part of the syndrome, or should be considered incidental findings. This is the case of hearing loss, duodenal atresia, and congenital joint contractures (20). More studies are needed to clarify these associations.
We cannot infer clear genotype-phenotype correlations in MSSP. A possible hypothesis is that the protein encoded by RTTN is large (2,226 aminoacids) and has many distinct protein-interacting domains, that act differently during the diverse phases of the cell cycle. Another possible explanation links the amount of RTTN mRNA expression with clinical severity (6). The role of possible modifier genes cannot be excluded. However, the severe phenotypic manifestation associated with several intronic variants suggest the importance of careful analysis of RTTN non-coding regions, especially in case of patients with a severe clinical picture and a single known RTTN variant (6). Moreover, the mechanisms by which RTTN is involved in cell cycle, neuronal migration and ciliogenesis is under debate, still making it difficult to understand how aberrant RTTN gene might result in such a wide range of clinical features. Further studies and clinical description of new cases will be helpful to better understand the complex interplay between mutated RTTN and clinical/neuroradiological issues and to infer a clearer genotype-phenotype association.
Conclusion
We report a patient with novel compound heterozygous variants in RTTN gene presenting with both typical and atypical clinical and neuroradiological features. From a clinical point of view, the patient presents with MSSP signs like ID, ASD, ADHD, aggressiveness, epilepsy and pyelectasis. The neuroradiological findings observed in our patient are milder than the classical pattern reported in MSSP. Furthermore, our patient had normal growth parameters and head circumference, in contrast with most other RTTN individuals. In our case, there were persistent hypotonia and epilepsy, that are only described in a minority of RTTN patients. Overall, we compare our patient's phenotypic features with the ones reported in literature, discussing a possible involvement of newly identified RTTN biallelic variants in his milder clinical and neuroradiological picture. Further studies are needed to better understand the biologic functions of rotatin, and pathological consequences of its genetic mutations.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Comitato Etico Territoriale della Liguria. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
FR: Writing – original draft, Writing – review & editing, Formal analysis, Supervision. EA: Investigation, Writing – review & editing. FM: Data curation, Investigation, Methodology, Software, Writing – review & editing. MS: Data curation, Investigation, Supervision, Writing – review & editing. VC: Supervision, Writing – review & editing. RR: Investigation, Supervision, Writing – review & editing. RB: Investigation, Supervision, Writing – review & editing. BC: Writing – review & editing. LM: Writing – review & editing. MS: Writing – review & editing. AF: Writing – review & editing. PS: Investigation, Supervision, Writing – review & editing. FZ: Investigation, Supervision, Writing – review & editing. MS: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the Italian Ministry of Health, RICERCA CORRENTE 2023. This work was supported by #NEXTGENERATIONEU (NGEU) and funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006)—A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022) to PS, FZ and MS.
Acknowledgments
The authors acknowledge ERN-ITHACA (European Network of Rare Malformation Syndromes) which support a better clinical practice within EU. IRCCS “G. Gaslini” is a member of ERN-Epicare.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1326552/full#supplementary-material
Supplementary Figure S1
Neuroimaging findings of cerebellar hypo-dysplasia. Brain MRI of the patient with axial T2-weighted images at the level of the cerebellum in a normal age-matched subject (A) and in the patient at 7.5 years of age (B). In the patient the volume of the cerebellar hemispheres is reduced and the foliation pattern is abnormal (empty arrows).
Supplementary Figure S2
Brain incidental findings of the patient. Brain MRI of the patient with axial susceptibility weighted (A), T1-weighted (B) and FLAIR (C) images demonstrating a developmental venous anomaly with the typical ‘caput medusae’ (curved arrows) associated with an abnormal cortical infolding (thin arrows) and signal alterations of the surrounding white matter (arrowheads). Sagittal high-resolution heavily T2-weighted DRIVE image (D), post-contrast T1-weighted image (E) and sagittal-reformatted CT scan (F) show a very small non-enhancing retroclival mass (thick arrows) associated with a bony clival defect, visible as a well-demarcated smoothly corticated region, without aggressive features (thin arrow). Note the cystic pineal gland complicated by internal bleeding (arrowheads).
Supplementary Table S1
Clinical Spectrum of phenotypes associated with RTTN variants. Reviewed and modified from Imran Naseer et al. (10). ND, not determined; M, male; F, female.
References
1. Faisst AM, Alvarez-Bolado G, Treichel D, Gruss P. Rotatin is a novel gene required for axial rotation and left-right specification in mouse embryos. Mech Dev. (2002) 113:15–28. doi: 10.1016/S0925-4773(02)00003-5
2. Kheradmand Kia S, Verbeek E, Engelen E, Schot R, Poot RA, de Coo IF, et al. RTTN mutations link primary cilia function to organization of the human cerebral cortex. Am J Hum Genet. (2012) 91(3):533–40. doi: 10.1016/j.ajhg.2012.07.008
3. Grandone A, Torella A, Santoro C, Giugliano T, Del Vecchio Blanco F, Mutarelli M, et al. Expanding the phenotype of RTTN variations: a new family with primary microcephaly, severe growth failure, brain malformations and dermatitis. Clin Genet. (2016) 90(5):445–50. doi: 10.1111/cge.12771
4. Chen HY, Wu CT, Tang CC, Lin YN, Wang WJ, Tang TK. Human microcephaly protein RTTN interacts with STIL and is required to build full-length centrioles. Nat Commun. (2017) 8(1):247. doi: 10.1038/s41467-017-00305-0
5. Chou EJ, Tang TK. Human microcephaly protein RTTN is required for proper mitotic progression and correct spindle position. Cells. (2021) 10(6):1441. doi: 10.3390/cells10061441
6. Vandervore LV, Schot R, Kasteleijn E, Oegema R, Stouffs K, Gheldof A, et al. Heterogeneous clinical phenotypes and cerebral malformations reflected by rotatin cellular dynamics. Brain. (2019) 142(4):867–84. Erratum in: Brain. 2019 Jun 1;142(6):e29. doi: 10.1093/brain/awz045
7. Cavallin M, Bery A, Maillard C, Salomon LJ, Bole C, Reilly ML, et al. Recurrent RTTN mutation leading to severe microcephaly, polymicrogyria and growth restriction. Eur J Med Genet. (2018) 61(12):755–8. doi: 10.1016/j.ejmg.2018.08.001
8. Shamseldin H, Alazami AM, Manning M, Hashem A, Caluseiu O, Tabarki B, et al. RTTN mutations cause primary microcephaly and primordial dwarfism in humans. Am J Hum Genet. (2015) 97(6):862–8. doi: 10.1016/j.ajhg.2015.10.012
9. Wambach JA, Wegner DJ, Yang P, Shinawi M, Baldridge D, Betleja E, et al. Functional characterization of biallelic RTTN variants identified in an infant with microcephaly, simplified gyral pattern, pontocerebellar hypoplasia, and seizures. Pediatr Res. (2018) 84(3):435–41. doi: 10.1038/s41390-018-0083-z
10. Imran Naseer M, AbdulrahmanAbdulkareem A, YousefMuthaffar O, Chaudhary AG. Exome sequencing reveled a compound heterozygous mutations in RTTN gene causing developmental delay and primary microcephaly. Saudi J Biol Sci. (2021) 28(5):2824–9. doi: 10.1016/j.sjbs.2021.02.014
11. Scala M, Accogli A, De Grandis E, Allegri A, Bagowski CP, Shoukier M, et al. A novel pathogenic MYH3 mutation in a child with sheldon-Hall syndrome and vertebral fusions. Am J Med Genet A. (2018) 176(3):663–7. doi: 10.1002/ajmg.a.38593
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
13. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596(7873):583–9. doi: 10.1038/s41586-021-03819-2
14. Benevenuta S, Pancotti C, Fariselli P, Birolo G, Sanavia T. An antisymmetric neural network to predict free energy changes in protein variants. J Phys Appl Phys. (2021) 54:245403. doi: 10.1088/1361-6463/abedfb
15. Lakhani DA, Martin D. Ecchordosis physaliphora: case report and brief review of the literature. Radiol Case Rep. (2021) 16(12):3937–9. Erratum in: Radiol Case Rep. 2022 Nov 25;18(2):730-731. doi: 10.1016/j.radcr.2021.09.049
16. Rump P, Jazayeri O, van Dijk-Bos KK, Johansson LF, van Essen AJ, Verheij JB, et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med Genomics. (2016) 9:7. doi: 10.1186/s12920-016-0167-8
17. Scala M, Nishikawa M, Nagata KI, Striano P. Pathophysiological mechanisms in neurodevelopmental disorders caused by rac GTPases dysregulation: what’s behind neuro-RACopathies. Cells. (2021) 10(12):3395. doi: 10.3390/cells10123395
18. Scala M, Nishikawa M, Ito H, Tabata H, Khan T, Accogli A, et al. Variant-specific changes in RAC3 function disrupt corticogenesis in neurodevelopmental phenotypes. Brain. (2022) 145(9):3308–27. doi: 10.1093/brain/awac106
19. Nishikawa M, Scala M, Umair M, Ito H, Waqas A, Striano P, et al. Gain-of-function p.F28S variant in RAC3 disrupts neuronal differentiation, migration and axonogenesis during cortical development, leading to neurodevelopmental disorder. J Med Genet. (2023) 60(3):223–32. doi: 10.1136/jmedgenet-2022-108483
Keywords: RTTN, microcephaly, cognitive impairment, seizures, cerebellar dysplasia, multiple arachnoid cysts
Citation: Romano F, Amadori E, Madia F, Severino M, Capra V, Rizzo R, Barone R, Corradi B, Maragliano L, Shams Nosrati MS, Falace A, Striano P, Zara F and Scala M (2023) Case Report: Novel biallelic moderately damaging variants in RTTN in a patient with cerebellar dysplasia. Front. Pediatr. 11:1326552. doi: 10.3389/fped.2023.1326552
Received: 23 October 2023; Accepted: 5 December 2023;
Published: 21 December 2023.
Edited by:
Ammar Husami, Cincinnati Children's Hospital Medical Center, United StatesReviewed by:
Louise Bicknell, University of Otago, New ZealandMiguel Angel Alcántara-Ortigoza, National Institute of Pediatrics, Mexico
© 2023 Romano, Amadori, Madia, Severino, Capra, Rizzo, Barone, Corradi, Maragliano, Shams Nosrati, Falace, Striano, Zara and Scala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federico Zara federico.zara@unige.it Marcello Scala mscala.md@gmail.com