Neonatal sclerosing cholangitis with novel mutations in DCDC2 (doublecortin domain-containing protein 2) in Chinese children
 Xia Wei1,†
Xia Wei1,†  Yuan Fang1,†
Yuan Fang1,†  Jian-She Wang2,‡
Jian-She Wang2,‡  Yi-Zhen Wang1,‡
Yi-Zhen Wang1,‡  Yuan Zhang3,‡
Yuan Zhang3,‡  Kuerbanjiang Abuduxikuer2*
Kuerbanjiang Abuduxikuer2*  Lian Chen4*
Lian Chen4*
- 1Department of Pathology, Anhui Provincial Children's Hospital, Hefei, China
- 2Department of Hepatology, Children's Hospital of Fudan University, Shanghai, China
- 3Department of Ultrasound, Children's Hospital of Fudan University, Shanghai, China
- 4Department of Pathology, Children's Hospital of Fudan University, Shanghai, China
Background: Neonatal sclerosing cholangitis (NSC) is a rare and severe autosomal recessive inherited liver disease with mutations in DCDC2, commonly requiring liver transplantation (LT) for decompensated biliary cirrhosis in childhood.
Methods: The information of four Chinese patients with NSC caused by mutations in DCDC2 from Children's Hospital of Fudan University were gathered. The four patients' clinicopathological and molecular features were summarized by clinical data, liver biopsy, immunohistochemical, and molecular genetic analysis.
Results: All patients presented with jaundice, hepatosplenomegaly, hyperbilirubinemia and bile embolism, and high serum γ-glutamyl transferase activity (GGT). Liver biopsies revealed varying degrees of bile duct hyperplasia, portal-tract inflammation, and/or fibrosis. Whole-exome sequencing (WES) found novel heterozygous variants of c.1024-1G > T /p.? and c.544G > A /p. Gly182Arg in the DCDC2.
Conclusion: This study expands the genetic spectrum of DCDC2 in NSC.
Author Summary: Neonatal sclerosing cholangitis (NSC) is an uncommon and severe inherited liver disease, which mutations in DCDC2 are responsible for, often leading to liver transplantation (LT) for end-stage liver disease in childhood. Herein, we collected the information of four Chinese patients with NSC caused by mutations in DCDC2 and analysed these patients’ clinical, pathological, and molecular features. We identified novel mutations (c.1024-1G > T and c.544G > A) in DCDC2, which have not been reported. This study adds to the genotype spectrum of DCDC2 causing NSC and expands the understanding of this rare disease.
Introduction
Neonatal sclerosing cholangitis (NSC, OMIM 617394) is a rare and severe neonatal-onset cholestatic liver disease, which ultimately develops into end-stage liver disease and has to undergo liver transplant in childhood (1). The main clinical manifestations include jaundice, hepatosplenomegaly, hyperbilirubinemia and high serum gamma glutamyl transferase activity (GGT), which was firstly reported in 1987 (2). It is difficult to distinguish from biliary atresia and other neonatal cholestatic liver disease in early NSC (3). Typical imaging and liver biopsy pathology can assist in establishing diagnosis. At the early stage of the disease, the typical cholangiogram features of NSC are as follows: unobstructed bile ducts, thin and irregular intrahepatic duct with or without extrahepatic bile ducts change (4). Liver biopsies often revealed bile duct hyperplasia, portal-tract inflammation, and/or fibrosis. In 2004, Hadj-Rabia et al. firstly reported NSC caused by claudin 1 (CLDN1) gene associated with ichthyosis, which encodes a tight-junction protein (5). In 2016, Girard et al. (4)and Grammatikopoulos et al. (6) verified that doublecortin domain containing 2 (DCDC2) gene mutations can lead to NSC. Apart from that, DCDC2 pathogenic variants were reported to be associated with renal-hepatic ciliopathy with nephronophthisis, non-syndromic recessive deafness, dyslexia and central nervous system impairment (7–10). With the development of whole-exome sequencing (WES), more and more DCDC2 mutations have been identified. To our knowledge, seventeen cases of NSC caused by DCDC2 mutation with 12 variants have been reported worldwide (4, 6, 10–14).
In this study, we summarized the clinical and laboratory features, presentation, and disease progression of NSC in four Chinese children caused by DCDC2 mutation, two of whom have been reported in our previous publication (11). The Liver biopsies showed various degrees of bile duct hyperplasia, portal-tract inflammation, and/or fibrosis. WES detected compound heterozygous variants c.1024-1G > T and c.544G > A, and homozygous variant c.529dupA in DCDC2, of which the former had not been previously reported. All patients were treated with Ursodeoxycholic acid capsules, Cholestyramine, Vitamins A, D, E and K1 to relieve symptoms and prevent the rapid progression of the disease. Over time, the symptoms of all patients have improved during follow-up. Our study identified novel mutations in DCDC2 and expanded the molecular spectrum of NSC.
Materials and methods
Patient
The inclusion criteria included patients had the disorder of liver function, cholestasis with elevated GGT during infancy, in whom cholangiopathy was demonstrated on histopathology or imaging. Meanwhile, WES confirmed mutations in only DCDC2. Moreover, the cholangiography showed no atresia of the bile duct. Then the patient can be definitively diagnosed with a DCDC2-related NSC. Exclusion criteria covered biliary atresia, ichthyosis-like skin lesions, Alagille syndrome or immune dysregulation. The information of four Chinese pediatric patients with DCDC2-related NSC in the Children's Hospital of Fudan University were reviewed. Clinical, pathological, and molecular features of all patients were analysed by clinical manifestations, laboratory investigations, liver biopsy, immunohistochemistry, and molecular genetic analysis. This study conformed to the provisions of the institutional ethics committee and the Declaration of Helsinki (as revised in 2013). The patients' parents shared all procedures including treatment and signed the written informed consent. The written informed consents were obtained for publication of any potentially identifiable images or data included in this paper.
Liver biopsy
Archival formalin-fixed, paraffin-embedded liver biopsy samples from patient 1 and 2 were obtained from surgical biopsy. For each patient, tissue sections were cut at 4 µm and stained with haematoxylin-eosin (HE), and special staining for periodic acid–Schiff (PAS), masson, reticulin, copper, and iron.
Immunohistochemistry
4-µm sections were deparaffinized, rehydrated, and pretreated with 3% H2O2 to eliminate endogenous peroxidase activity. Moreover, they were treated with EDTA (pH 9) or citrate buffer (pH 6) for heat-mediated antigen retrieval before commencing with the immunohistochemical staining protocol. The antibodies used included CK7, CK19, CD68, CD163, CD3, and CD8, which were purchased from http://www.maxim.com.cn (Fuzhou, China). The method of application is carried out according to the instruction manual. Finally, the sections were treated with DAB and counterstained with hematoxylin. In addition, a rabbit polyclonal antibody against DCDC2 (purchased from http://www.ptgc.com, product code: 26978-1-AP) with a 1:200 dilution was utilized. The specific experimental procedures were carried out in accordance with the instructions. A normal liver sample (donated by a surgical patient) was prepared as the positive control, simultaneously omitting the first antibody as the negative control.
Molecular genetic analysis
EDTA-anticoagulated whole blood specimens were used for the patients and their parents. DNA was extracted from peripheral blood. DNA libraries were prepared following the manufacturer's instructions, then sequenced on the Illumina platform. The whole exome capture high-throughput sequencing technology was utilized, with average coverage of 90-110X. Variants were detected using Genome Analysis Tool Kit (GATK) software (version: v3.2), including base quality score recalibration, InDels position and variant quality score recalibration, SNVs and InDels variant discovery and typing. And the AD:DP:GQ:PL was 13,5:18:61:61,0,420. The variants were annotated using Variant Effect Predictor (VEP) software, in which the functional coding regions and splicing site variants were called for further analysis, mainly including loss-of-function variants (mutations to obtain stop codons, frameshift mutations, and critical splicing point mutations), missense mutations, and non-frameshift deletions/insertions. Data were aligned against the Human Gene Mutation Database (HGMD) Professional (http://www.hgmd.cf.ac.uk), 1000 Genome Database (www.1000 genomes.org), Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org), dbSNP152 (https://www.ncbi.nlm.nih.gov/snp), and Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org). Damage prediction of the genetic variants was conducted by Mutation Significance Cutoff (MSC) (https://lab.rockefeller.edu/casanova/MSC), which was applied to CADD (https://cadd.gs.washington.edu), PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (https://sift.bii.a-star.edu.sg/www/SIFT_indels2.html), with a confidence interval of 99% and database source of HGMD and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). The pathogenicity of amino acid changes caused by gene mutations was predicted by MutationTaster (http://www.mutationtaster.org) as well. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, genetic variants were classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, and benign. Thereafter, pathogenic variants of DCDC2 were identified with the transcript of NM_001195610, which were validated by Sanger sequencing in patient 1. We conducted protein modeling by SWISS-model (https://www.swissmodel.expasy.org) with an UniProtKB code Q9UHG0, and the mutated structures were analyzed and visualized using PyMol (http://www.pymol.org).
Literature search
In addition, we conducted a retrospective analysis of the literature. We used NSC and DCDC2 as the terms, and retrieved a total of 7 related literatures from Pub Med, Spring Link, CNKI and Wan fang databases. There were 17 DCDC2-related NSC, summing to 21 cases including 4 cases in our paper. Then, we summarized and analyzed their clinical, pathological and molecular characteristics.
Results
Clinical features
There are four patients (2 female and 2 male) from 3 families, among whom patient 3 and 4 are siblings. All parents denied consanguinity. The age distribution is 6 months to 15 years. All patients presented with jaundice as the primary symptom in the first week of birth. Three patients (Patient 1, Patient 3 and Patient 4) suffered from recurrent respiratory infections during the course of the disease. Patient 1 developed sepsis and enteritis due to staphylococcus aureus and enterovirus infections during her first hospitalization. Patient 4 was infected with cytomegalovirus (CMV) and Epstein-Barr virus (EBV) after birth. Moreover, he developed into biliary cirrhosis at 10 months of age. Developmental delay and other medical history in four patients were unremarkable. Physical examination revealed the livers were located 3–5 cm below the right costal and the spleens 1–6 cm below the left costal. Spider nevus of the face in patient 4 was positive. Other physical examinations showed no positive signs. Biochemical findings included liver dysfunction with elevated total bile acid, total hyperbilirubinemia and GGT. Other blood tests except coagulation function showed no obvious abnormalities. Laparoscopic cholangiography was performed in patient 1 and 2, that revealed bile duct patency and dysplasia of the intrahepatic biliary tree. Magnetic resonance and cholangiopancreatography in patient 3 showed multiple and irregular dilatations of intrahepatic bile ducts. Hepatobiliary isotope imaging excluded biliary atresia (BA) (11). Computed tomography scan and angiography in patient 4 showed hydrocephalus and aneurysm of the communicating segment of the left internal carotid artery with vascular malformation (11). The clinical data and major lab investigations of four patients were listed in Table 1. All patients were treated with Ursodeoxycholic acid, Cholestriamine and took vitamin K1/A/E/AD supplements. The symptoms of all patients were relieved and the blood test indicators improved during regular examination.
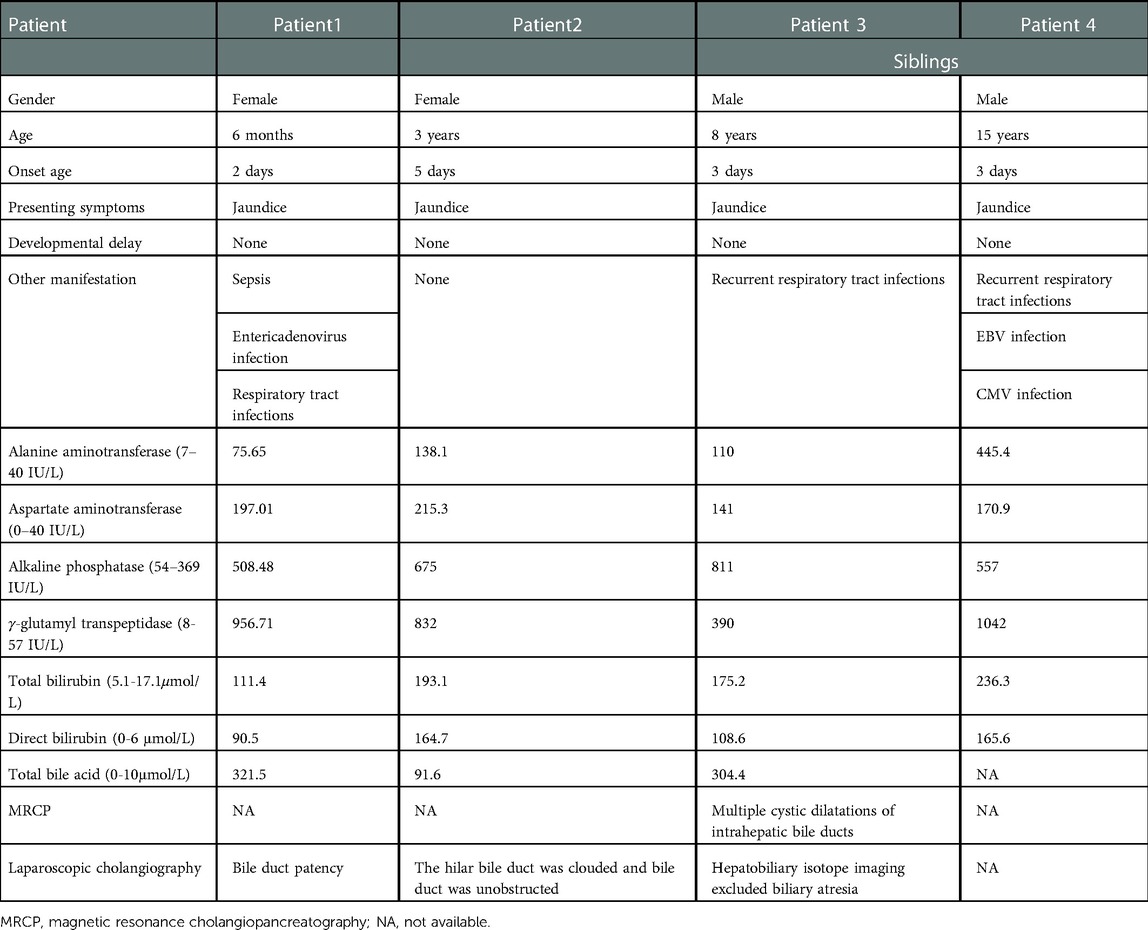
Table 1. The clinical data and predominant laboratory investigations of four Chinese patients.
Liver biopsy
Liver biopsy of patient 1 showed that the structural destruction of liver lobule and the formation of pseudo-lobule (Figure 1A). High magnification exhibited balloon-like degeneration of hepatocytes, giant cell changes of hepatocytes and bile plugs formation in hepatocytes and capillary bile ducts (Figure 1B). Ductular proliferation, the formation of bile thrombus in bile duct, moderate portal-tract inflammation, and fibrosis were found (Figure 1C). PAS staining revealed small vacuoles in some hepatocytes (Figure 1D), while copper staining was negative. Liver lobules were separated by the hyperplastic fibrous tissue in the portal area, formatting bridging-like fibrosis and pseudo-lobules, which was identified by Masson staining (Figure 1E). Reticulin staining indicated the preserved reticular scaffold structure (Figure 1F). Iron staining showed a little iron deposition. Liver histopathology presented cholestatic cirrhosis with stage 4 based on the Scheurer histopathologic scoring system (15). The pathological features of the liver biopsy in patient 2 were similar to those in patient 1, but without pseudo-lobule formation. The degree of inflammation and fibrous tissue hyperplasia in the portal area was less severe. Liver histopathology was assessed with stage 3.
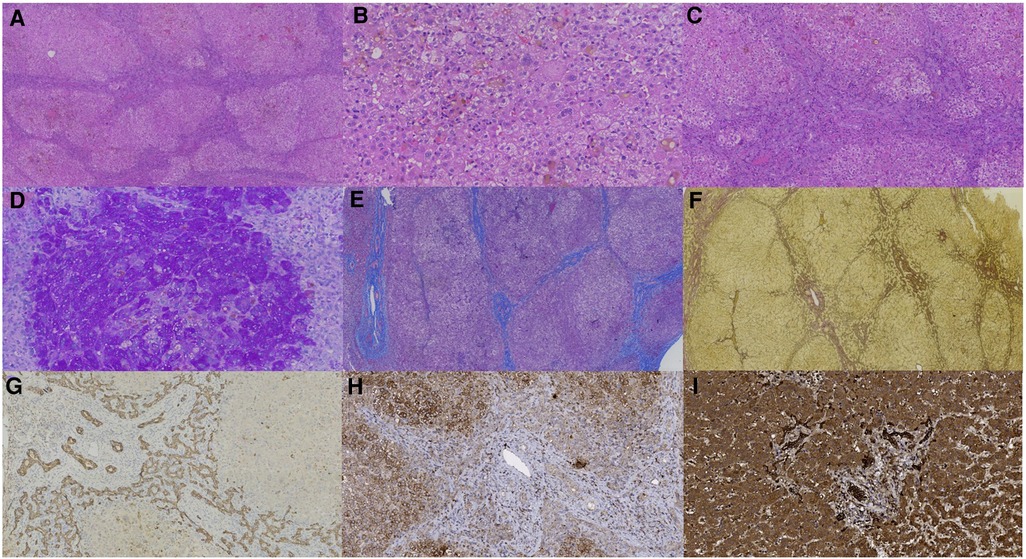
Figure 1. Liver biopsy (all originally magnified principal images). Hematoxylin and eosin staining showed the discorded architecture of liver lobules and the formation of pseudo-lobules (A × 40), balloon-like hepatic cells, giant cell changes of hepatocytes and bile plugs formation in hepatocytes and capillary bile ducts (B × 200), and ductular proliferation, moderate portal-tract inflammation, and fibrosis in the portal area (C × 100). PAS staining identified small vacuoles in partial hepatocytes (D × 200). Formation of bridging-like fibrosis and pseudo-lobule was highlighted by Masson staining (E × 50). The preserved reticular scaffold structure was displayed by reticulin staining (F × 40). Obvious hyperplasia of bile ducts along the margins of pseudo-lobule was showed by immunohistochemical staining of CK7 (G × 100). DCDC2 immunohistochemical staining showed weaker expression in patient 1 (H × 200) than in normal control (I × 200).
Immunohistochemistry
In patient 1, CK7 (Figure 1G) and CK19 staining showed obvious hyperplasia of bile ducts around pseudo-lobules. CD68 and CD163 staining manifested moderate proliferated Kupffer cells in hepatic sinusoids, and lymphocytes infiltrating in the portal area were indicated by CD3 and CD8 staining. It was important to notice that the expression of DCDC2 was significantly reduced in patient sample (Figure 1H), compared with the normal liver sample (Figure 1I), which showed a cytoplasmic positive pattern in hepatic and bile duct cells. The results of immunohistochemical staining in patient 2 and patient 1 were resemble.
Molecular genetic analysis
WES in 4 patients identified compound heterozygous variants c.1024-1G > T and c.544G > A (Figure 2A) in exon 5 and 10, and homozygous variant c.529dupA in exon 4 in DCDC2. In patient 1, The c.1024-1G > T was inherited from the healthy father and the c.544G > A was inherited from the healthy mother, resulting to the alteration of glycine to arginine at amino acid position 182 (p.Gly182Arg), which was conserved across various species (Figure 2C). In patient 3 and 4, the c.529dupA was inherited from their parents who were asymptomatic carriers (Figure 2B), which was reported in our publication. The MutationTaster score of c.1024-1G > T and c.544G > A was 1.0, both predicted as likely pathogenic by MSC. Both mutations of c.1024-1G > T and c.544G > A were assessed to be likely pathogenic in line with the guideline of the ACMG. The nomenclature of variants was on the basis of the recommendations of the Human Genome Variation Society (HGVS, http://www.hgvs.org/varnomen). Wild-type and mutated DCDC2 were modeled by PyMol (http://www.pymol.org) which substantiated the change of Gly182Arg residue had no effect on the polar contact with them around amino acid Val 184 (Figure 2D). Spatial conformation of the frameshift mutation (p. Ile177Asnfs*20) predicted by protein modeling demonstrated that the mutated amino acid at position 177 moved backward 20 positions and then terminated, generating protein truncation (Figure 2E). Figure 2F illustrated DCDC2 protein domains and the location of amino acid changes in our report.
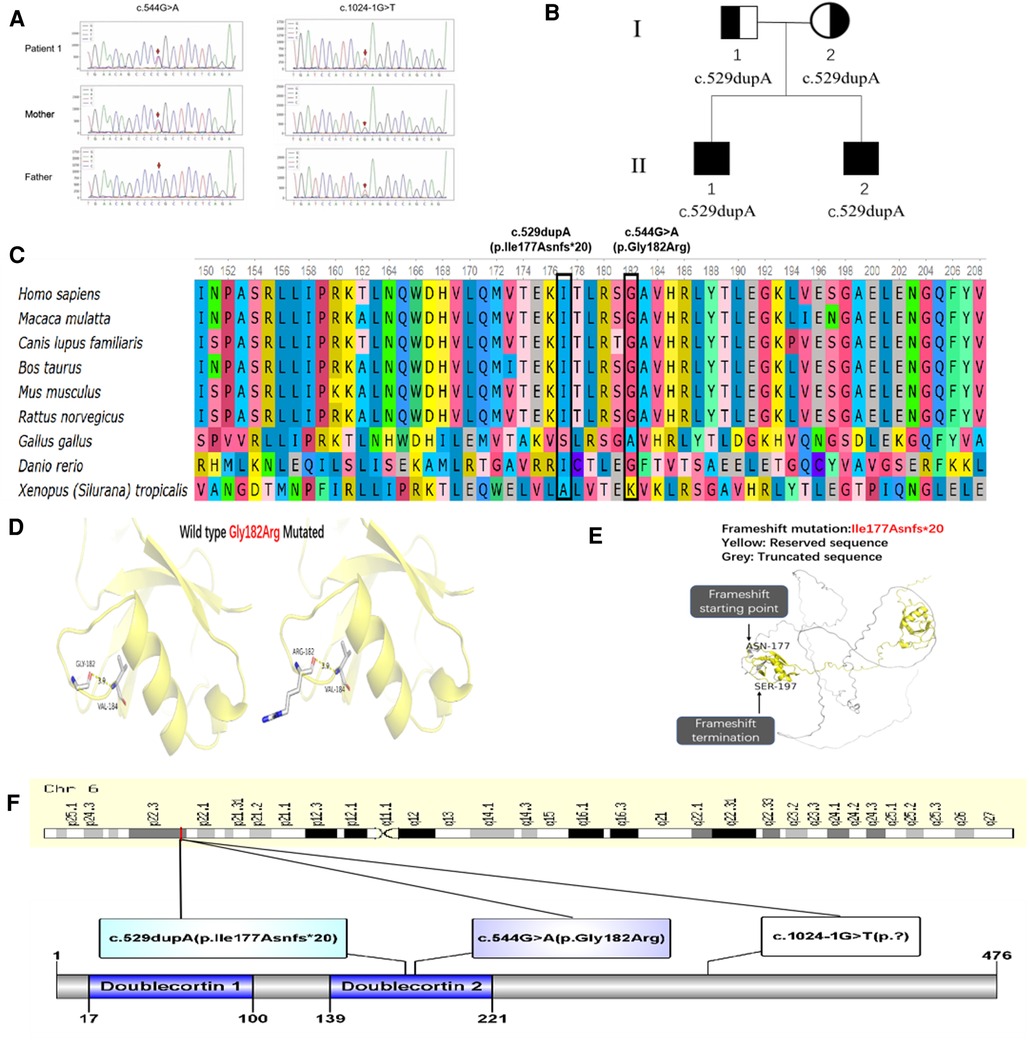
Figure 2. (A) Sanger sequencing confirmation of c.1024-1G > T and c.544G > A mutation of DCDC2 in patient 1 and his parents. (B) Pedigree of patient 3 and 4, I-1 was the father, I-2 was the mother, II-1 was patient 3, II-2 was patient 4. (C) Conservation status of amino acid residues of the two variants across various species in four patients. (D) Wild and mutated types of the p. Gly182Arg variant in patient 1 compared by PyMol. (E) Spatial conformation of the frameshift mutation (p. Ile177Asnfs*20) showed by protein modeling. (F) Illustration of DCDC2 protein domains, location of amino acid changes in four patients.
Retrospective analysis of the literature
The main features of twenty-one cases of DCDC2-related NSC were displayed in Table 2. The twenty-one patients with DCDC2-related NSC included sixteen Asian and Europeans (nine originating from Caucasian, Arabic and Turkish and seven from China). The male to female ratio approximates 3:1. The occurrence of NSC among siblings and high parental consanguinity implies autosomal recessive inheritance (16). Our study demonstrates forty-three percent of patients have parental consanguinity and fifty percent of patients are siblings. Most patients develop the disease within the first six months of life, with jaundice and/or acholic stools as the earliest symptoms. Upper gastrointestinal bleeding, ascites, hepatosplenomegaly and coagulopathy occurred in some patients at different stages of the disease. Biochemical test in all patients showed liver dysfunction with total hyperbilirubinemia and GGT with or without coagulation dysfunction. Five patients suffered from recurrent respiratory tract infections, one of whom had sepsis and enteric adenovirus infection and the other had CMV and EBV infection in our report, which may suggest impaired immune function in NSC patients. The renal involvement was found in four patients. Aneurysms were present in 2 patients (P4 and P9). Developmental delay and hypotonia were reported in 2 patients (P20 and P21). One patient (P6) had mild intellectual disability. These studies attest that DCDC2-related NSC mainly makes impact on the liver and meanwhile, urinary, nervous and muscular systems may be implicated. 13 individuals (62%) in 21 patients suffered from NSC received LT for end-stage liver disease at an average age of 11.9 years old (range 1–25). In all, three patients died among two of whom died of end-stage liver disease when awaiting for LT and one died after LT for complications of severe renal failure. Overall mortality was 14%.
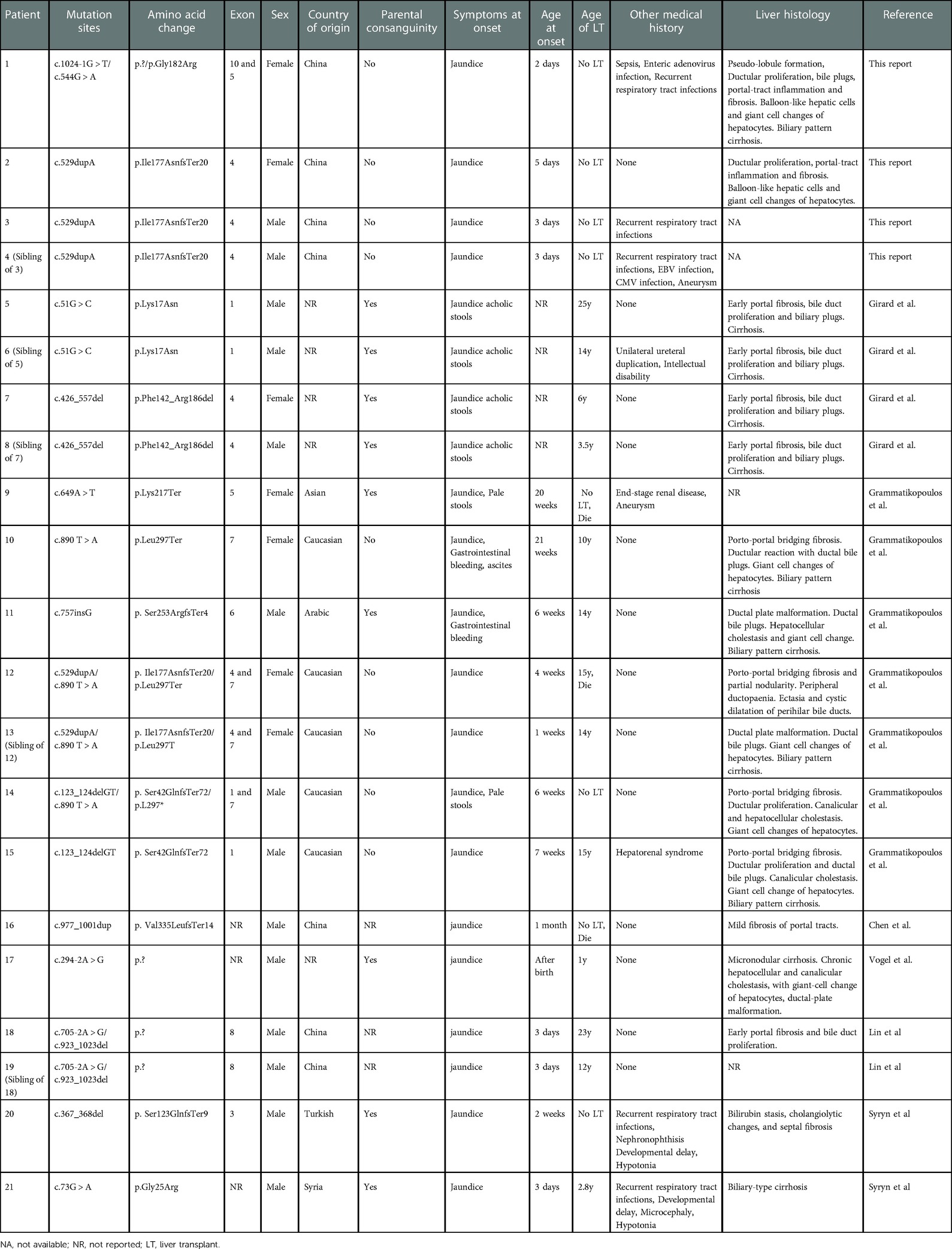
Table 2. Main features of the twenty-one reported patients with NSC caused by mutations in DCDC2.
Liver biopsies were performed in seventeen patients, five of whom developed into biliary cirrhosis and others showed various degrees of liver fibrosis, including portal fibrosis, portal tract inflammation, bile duct proliferation or plate malformation with or without ductal bile plugs, giant cell changes of hepatocyte. It's worth noting that results of the histology on liver biopsies bring to mind the biliary atresia and congenital hepatic fibrosis, which makes it challenging to establish the diagnosis of NSC. DCDC2 immunohistochemical staining was performed in some patients, which revealed DCDC2 staining was heighten in the cytoplasm of cholangiocytes while a lesser extent or absent at the apical margin of cholangiocytes (4, 6, 10). These findings suggest that mutations in DCDC2 perturbed protein expression and localization. The ultrastructural characteristics of hepatocytes were evaluated using a transmission electron microscope (TEM) in individual patient. In liver tissue, lobular cytoplasmic necrosis, dilatation of canalicular lumina with amorphous bile, were seen as well as blunting of microvilli, and cytoplasmic blebbing into the canalicular lumen, while cholangiocellular primary cilia were existent or absent (6, 10).
Discussion
NSC is a rare hereditary liver disease in nurseling, for which DCDC2 mutation was mainly responsible (4, 6). With the development of WES, researchers found biallelic mutations in DCDC2 can cause familial NSC (13). The DCDC2 is located on chromosome 6p22.3, containing 11 exons, which encodes DCDC2 protein known to bind tubulin and enhance microtubule polymerization (17). Grati et al. found the pathogenicity of DCDC2 mutations in the aspects of protein expression and localization as well as their influence on ciliogenesis in cholangiocytes (7). The primary cilia of cholangiocytes are important for the regulation of bile flow and its composition (18). Grammatikopoulos et al. suggested that the absence of DCDC2 may be related to the formation of “cytotoxic” bile or the dysregulation of the cholangiocyte's homeostatic mechanisms, possibly by Wnt signalling pathway (8). DCDC2 has been shown to interact with KIF3A, a subunit of the Kinesin-2 complex that is essential for cilia formation and maintenance, which can modulate ciliary signaling by Wnt signaling pathway (17). DCDC2 negatively regulates Wnt/β-catenin signaling pathway by interacting with dishevelled 3, a key regulator of the pathway (17). The deletion of the second doublecortin domain of DCDC2 also has been shown to hold back the decrease of Wnt/β-catenin signaling in the case of Wnt inhibitors (8). Therefore, the inhibitor of Wnt signaling pathway perhaps be a promising treatment for patients with NSC to prevent the progression of the disease.
It was reflected in our report that NSC are more common among siblings and parental consanguinity. The symptoms of the four Chinese patients at onset we reported were consistent with those documented in the literatures. Three patients had the history of upper respiratory infection, one of whom had aneurysm. Two of our patients had a history of virus infection, including enteric adenovirus, EBV, and CMV. Developmental delay, hypotonia, intellectual disability microcephaly and unilateral ureteral duplication reported in the literatures (7–10) were not found in our patients. At present, it is not very clear whether nerve, muscle, or immune system involvements are related to mutations in DCDC2. Researchers hypothesized that the involvement of these systems may be related to the inactivation of the cilium function or the disorder of Wnt signaling pathways due to DCDC2 mutations (10). More cases and further studies are needed to confirm this hypothesis. None of our four patients either received a liver transplant or died, including one with severe cirrhosis. Our patients were given Ursodeoxycholic acid capsules, Cholestyramine, Vitamins A, D, E and K1. Liver function and symptoms in all patients have improved. However, two patients have the early cirrhosis, which makes the outcome of the disease disappointed. The final outcome still needs further follow-up.
In terms of liver biopsy pathology, the pathological changes of our patients were similar to those with DCDC2-related NSC reported in the literature, showing bile duct proliferation, fibrosis and ductal bile plugs at different degrees with or without giant cell changes of hepatocytes. The histological changes of liver biopsy were similar to those reported in the previous literature.
We also reported a novel mutation (c.1024-1G > T /p.? and c.544G > A /p. Gly182Arg) in DCDC2. What's more, DCDC2 immunohistochemical staining revealed that the expression level of DCDC2 protein in NSC patients was significantly decreased, compared with normal liver tissue. It demonstrated that DCDC2 mutation affected protein expression, which was consistent with published literatures. But its effect on protein function needs to be determined in future study. Besides, we sum up the amounts of patients suffering from NSC with mutations in DCDC2 to 21 from 16 irrelevant families and pathogenic variants to 15. These variants include 4 non-truncating, 8 truncating, and 3 splice site variants (Table 2), which will give a more complete picture of DCDC2-related NSC.
In this paper, we discovered novel mutations in DCDC2 causing NSC in 4 Chinese children, which extended the genetic spectrum of DCDC2. For the moment, there is no specific treatment for patients with NSC, and patients with biliary cirrhosis require liver transplantation for survival.
Data availability statement
The data presented in this study is included in the article/Supplementary Material. The DNA datasets are not readily available due to privacy restrictions, further inquiries should be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Children's Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XW and YF drafted and revised the work and they made the same contribution to this research. J-SW, Y-ZW and YZ analyzed and interpreted the data for the work, and KA and LC designed the work and approved the final version to be published. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patients and their parents for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen HL, Wu SH, Hsu SH, Liou BY, Chen HL, Chang MH. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. (2018) 25(1):75. doi: 10.1186/s12929-018-0475-8
2. Amedee-Manesme O, Bernard O, Brunelle F, Hadchouel M, Polonovski C, Baudon JJ, et al. Sclerosing cholangitis with neonatal onset. J Pediatr. (1987) 111(2):225–9. doi: 10.1016/s0022-3476(87)80072-0
3. Kerkar N, Chan A. Autoimmune hepatitis, sclerosing cholangitis, and autoimmune sclerosing cholangitis or overlap syndrome. Clin Liver Dis. (2018) 22(4):689–702. doi: 10.1016/j.cld.2018.06.005
4. Girard M, Bizet AA, Lachaux A, Gonzales E, Filhol E, Collardeau-Frachon S, et al. DCDC2 Mutations cause neonatal sclerosing cholangitis. Hum Mutat. (2016) 37(10):1025–9. doi: 10.1002/humu.23031
5. Hadj-Rabia S, Baala L, Vabres P, Hamel-Teillac D, Jacquemin E, Fabre M, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology. (2004) 127(5):1386–90. doi: 10.1053/j.gastro.2004.07.022
6. Grammatikopoulos T, Sambrotta M, Strautnieks S, Foskett P, Knisely AS, Wagner B, et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol. (2016) 65(6):1179–87. doi: 10.1016/j.jhep.2016.07.017
7. Grati M, Chakchouk I, Ma Q, Bensaid M, Desmidt A, Turki N, et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet. (2015) 24(9):2482–91. doi: 10.1093/hmg/ddv009
8. Schueler M, Braun DA, Chandrasekar G, Gee HY, Klasson TD, Halbritter J, et al. DCDC2 Mutations cause a renal-hepatic ciliopathy by disrupting wnt signaling. Am J Hum Genet. (2015) 96(1):81–92. doi: 10.1016/j.ajhg.2014.12.002
9. Meng H, Smith SD, Hager K, Held M, Liu J, Olson RK, et al. DCDC2 Is associated with Reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci USA. (2005) 102(47):17053–8. doi: 10.1073/pnas.0508591102
10. Syryn H, Hoorens A, Grammatikopoulos T, Deheragoda M, Symoens S, Vande Velde S, et al. Two cases of DCDC2-related neonatal sclerosing cholangitis with developmental delay and literature review. Clin Genet. (2021) 100(4):447–52. doi: 10.1111/cge.14012
11. Li JQ, Lu Y, Qiu YL, Wang JS. Neonatal sclerosing cholangitis caused by DCDC2 variations in two siblings and literature review. Zhonghua Er Ke Za Zhi. (2018) 56(8):623–7. Chinese. doi: 10.3760/cma.j.issn.0578-1310.2018.08.013
12. Vogel GF, Maurer E, Entenmann A, Straub S, Knisely AS, Janecke AR, et al. Co-existence of ABCB11 and DCDC2 disease: infantile cholestasis requires both next generation sequencing and clinical-histopathologic correlation. Eur J Hum Genet. (2020) 28(6):840–4. doi: 10.1038/s41431-020-0613-0
13. Lin Y, Zhang J, Li X, Zheng D, Yu X, Liu Y, et al. Biallelic mutations in DCDC2 cause neonatal sclerosing cholangitis in a Chinese family. Clin Res Hepatol Gastroenterol. (2020) 44(5):e103–8. doi: 10.1016/j.clinre.2020.02.015
14. Chen J, Zhang XX, Liu HD, Chen X. Neonatal sclerosing cholangitis caused by a novel DCDC2 gene variant: a case report and literature review. Chin Pediatr Emerg Med. (2020) 27(2):158–60. doi: 10.3760/cma.j.issn.1673-4912.2020.02.019
15. Scheuer PJ. Classification of chronic viral hepatitis: a need for reassessment. J Hepatol. (1991) 13(3):372–4. doi: 10.1016/0168-8278(91)90084-o
16. Debray D, Pariente D, Urvoas E, Hadchouel M, Bernard O. Sclerosing cholangitis in children. J Pediatr. (1994) 124(1):49–56. doi: 10.1016/s0022-3476(94)70253-5
17. Massinen S, Hokkanen ME, Matsson H, Tammimies K, Tapia-Páez I, Dahlström-Heuser V, et al. Increased expression of the dyslexia candidate gene DCDC2 affects length and signaling of primary cilia in neurons. PLoS One. (2011) 6(6):e20580. doi: 10.1371/journal.pone.0020580
Keywords: neonatal sclerosing cholangitis, biliary cirrhosis, DCDC2, ciliopathy, novel mutations
Citation: Wei X, Fang Y, Wang J, Wang Y, Zhang Y, Abuduxikuer K and Chen L (2023) Neonatal sclerosing cholangitis with novel mutations in DCDC2 (doublecortin domain-containing protein 2) in Chinese children. Front. Pediatr. 11:1094895. doi: 10.3389/fped.2023.1094895
Received: 10 November 2022; Accepted: 9 January 2023;
Published: 3 February 2023.
Edited by:
Kenneth KY Wong, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Shaotao Tang, Huazhong University of Science and Technology, ChinaClara Sze Man Tang, The University of Hong Kong, Hong Kong SAR, China
© 2023 Wei, Fang, Wang, Wang, Zhang, Abuduxikuer and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuerbanjiang Abuduxikuer kurbanjanmph@sina.com Lian Chen doctchenlian@163.com
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work
Specialty Section This article was submitted to Pediatric Gastroenterology, Hepatology and Nutrition, a section of the journal Frontiers in Pediatrics