Whole Exome Sequencing in a Population With Severe Congenital Anomalies of Kidney and Urinary Tract
 Meredith Harris
Meredith Harris Meredith P. Schuh
Meredith P. Schuh David McKinney3
David McKinney3  Kenneth Kaufman
Kenneth Kaufman Elif Erkan
Elif Erkan- 1Division of Nephrology and Hypertension, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 2Division of Nephrology, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, United States
- 3University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 4Center for Autoimmune Genomics and Etiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
Fetal and neonatal interventions (e.g., amnioinfusions, amniotic shunting, and infant dialysis) have increased survival of infants with severe Congenital Anomalies of the Kidney and Urinary Tract (CAKUT), however, outcomes vary dramatically. Our aim was to perform Whole Exome Sequencing (WES) in a unique severe CAKUT population with the goal to identify new variants that will enhance prediction of postnatal outcomes. We performed trio WES on five infants with severe CAKUT (undergoing fetal interventions and/or those who initiated renal replacement therapy (RRT) within 1 month of life) and their parents as well as three singletons. We identified three potential candidate gene variants (NSUN7, MTMR3, CEP162) and validated two variants in known CAKUT genes (GATA3 and FRAS1) showing strong enrichment in this severe phenotype population. Based on our small pilot study of a unique severe CAKUT population, WES appears to be a potential tool to help predict the course of infants with severe CAKUT prenatally.
Introduction
Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) are the leading cause of End Stage Kidney Disease (ESKD) in the pediatric population, representing 30–60% of chronic kidney disease (1, 2). Clinical presentation of these patients varies greatly from asymptomatic patients with normal renal function to ESKD at birth (3). With improved prenatal ultrasound testing, about two-thirds of CAKUT is diagnosed prenatally, with about 50% detected between 18 and 22 weeks of gestation (4).
Infants with the most severe presentations have oligohydramnios or anhydramnios during gestation. Etiologies include primary structural abnormalities, renal agenesis, or bladder outlet obstruction (BOO). The Center for Fetal Care (CFC) at Cincinnati Children’s Hospital Medical Center is currently one of the few centers in the United States offering amnioinfusions and amniotic shunting for pregnancies with oligo/anhydramnios. Fetal MRI and ultrasound are performed to diagnose the etiology of low amniotic fluid and to aid in parental guidance during a multidisciplinary meeting including neonatology, nephrology, urology, maternal fetal medicine, fetal surgery, palliative care, and neonatology (Figures 1, 2). After this meeting, parents may elect to terminate the pregnancy, not undergo any invasive interventions, or undergo fetal interventions. During amnioinfusions, fluid (Lactated Ringers at our institute) is administered into the uterus at serial intervals. During amniotic shunting, a shunt is placed between the bladder and the uterus to overcome the obstruction in patients with BOO. Both interventions increase fluid availability to the lungs to enhance pulmonary development during the canalicular stage (5). In total, 108 mothers have undergone fetal interventions from 2014 to 2018 in our center, increasing survival to discharge of these CAKUT infants from 17 to ∼50%.
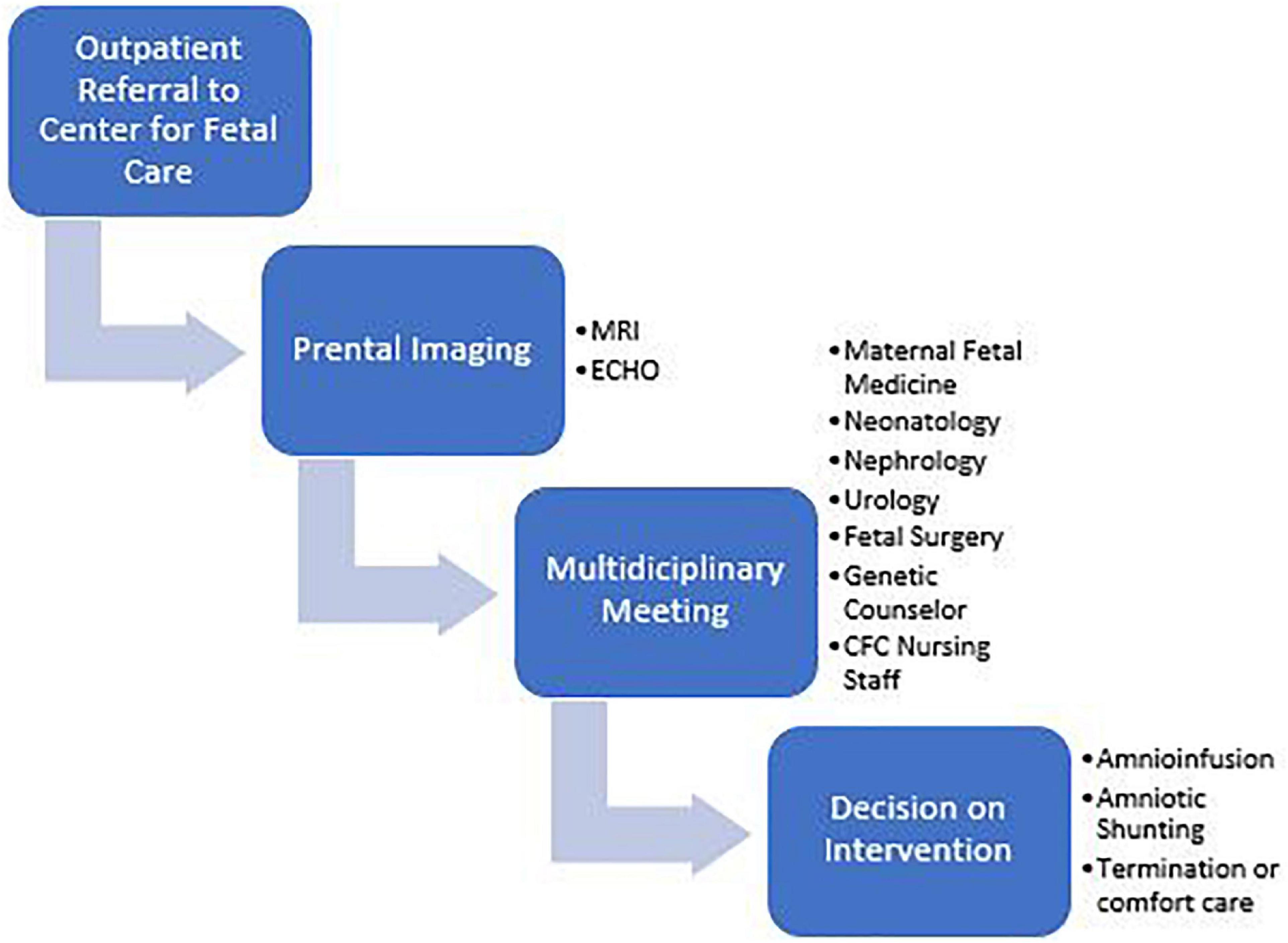
Figure 1. Process diagram for families referred to the center for fetal Care.

Figure 2. MRI of a singleton fetus in utero at 25 weeks of gestation with bilateral multicystic dysplastic kidneys and absence of amniotic fluid.
While fetal interventions improve pulmonary survivorship, they do not improve renal outcomes. Our CFC is one of very few centers that offers dialysis for infants as small as 1.8 kg. As a bridge to peritoneal dialysis, we perform prolonged intermittent renal replacement therapy (PIRRT) using the Aquadex® Smart Flow Ultrafiltration Device (6). As a result of successes with fetal interventions and infant dialysis, our center has a unique surviving CAKUT population with a severe phenotype that previously would be incompatible with life. Namely, we have the largest living population of patients with bilateral Multicystic dysplastic kidney (bMCDK), a condition that was previously fatal before fetal interventions as these patients have no amniotic fluid, and subsequently, poor lung development. To date, there is minimal research in living bMCDK populations.
The long-term implications of this previously lethal anomaly are unknown but are imperative for accurate and informed prenatal counseling. CAKUT is known to cover a wide variety of phenotypes with a complex molecular basis with both environmental and genetic contributors. Traditional genetic approaches, such as targeted investigation of single genes, provided the foundation of knowledge of the genetics of CAKUT, covering 5–10% of CAKUT cases (7–10). Genome-wide genotyping has dramatically expanded the list of genes known to cause the disease. More recently, various genetic testing including Whole Exome Sequencing (WES) has identified more than 40 monogenic genes, attributing to up to 20% of CAKUT diagnoses (11, 12). Even with these advances, genetic etiology of the disease is unknown in most cases. Previous work supports that the detection rate of genetic etiologies is higher for bilateral renal anomalies (13, 14).
With a new emerging severe CAKUT population, there is a critical need to provide targeted prenatal counseling via identification of genotype-phenotype correlations associated with severe CAKUT prior to aggressive fetal interventions. Based on previous studies, we hypothesize that our phenotypically severe CAKUT population will contain higher numbers of variants in novel kidney disease-causing genes. Here, we establish the framework for WES testing in severe CAKUT during the early postnatal period. As outcomes in our severe CAKUT population remain variable from intrauterine demise to successful kidney transplantation, our goal is to develop new metrics based on genetic testing that will enhance the ability to predict the postnatal outcomes of these infants.
Materials and Methods
Inclusion criteria included subjects whose mothers underwent fetal interventions (amnioinfusions or amniotic shunting) for severe CAKUT or who started dialysis within 1 month of life. The phenotype of these patients included bMCDK as well as BOO severe enough to warrant fetal interventions for oligo/anhydramnios. Biological parents were included for the purpose of trio WES testing. Infants with minor or moderate CAKUT not requiring fetal interventions as well as infants with bilateral renal agenesis (BRA) were excluded from this study. BRA was excluded as our center does not currently offer intervention to this population (15). All study methods were IRB approved at our center and the delivery hospital centers.
Sample Collection
For infants, 1 ml of whole blood was obtained for WES. Whole blood samples were collected in an EDTA microtainer, mixed on a nutator, and centrifuged for 5 min at 3 665 G at 4°C. Parental blood (gold standard) or saliva was collected and stored at the internal biorepository to use for WES according to the above methods.
DNA was extracted from whole blood samples using an AutoGen Flex STAR machine via precipitation using Qiagen chemistry. A positive and negative control were used to validate the run. Extracted DNA was placed into a cryovial (0.175 ml). This vial was rotated overnight and placed in an incubator for 30 min to ensure even distribution of DNA in the sample. The sample then underwent DNA quantification and a gel quality check. The sample was then stored at −20°C.
Identification of Genetic Variants by Genetic Analyses
For WES analysis, sequencing was performed on genomic DNA libraries created by fragmentation and annealing of linkers according to the Agilent TruSeq (Illumina) protocol. Exome capture was performed with IDTERPv1 capture kits. Sequencing was performed on the Illumina NovaSeq platforms. Fastq files were aligned to the NCBI human reference genome (build 37) using the Burrows-Wheeler Aligner. Multi-sample variant call format (vcf) files were generated using the Broad Institutes GATK software following their best practices. Variants were filtered based on a 1% minor allele frequency in the general population using both public and internal data sets. Variants underwent quality control filtering to remove sequencing artifacts, were annotated to identify amino acid altering variants, were scored using multiple functional prediction algorithms and were fit to homozygous recessive, compound heterozygous, and de Novo genetic models of inheritance. We required the read depth to be >15, genotype quality score >20, and used zygosity based filtering based on the ratio of alternate alleles to reference alleles. Reference alleles required the following: Homozygous reference alleles-ratio less than 0.15, homozygous alternate alleles-ratio greater than 0.85 and heterozygous alleles-ratio between 0.3 and 0.7. In our experience these filters remove ∼99% of the sequencing artifacts.
Variants were prioritized using a multifaceted approach using the GeneCards, NCBI and OMIM databases to assess gene function, gene expression, gene enrichment, and severity of predicted biological impact on gene function using SIFT (16), Polyphen (17), Mutation Taster (18), Mutation Assessor (19), and FATHMM (20). ToppFun was used to further detect functional enrichment of candidate genes (21). We used Franklin by Genoox (22) to classify variants according to the 2015 American College of Genetics and Genomics (ACMG) criteria. These classifications are based on population data, computational and predictive data, functional data, segregation data, de novo data, allelic data as well as other additional data (23).
Results
Patient Demographics
We enrolled 18 patients along with their parents (Figure 3). From the enrolled patients, we obtained blood from eight liveborn patients for WES. Six patients underwent fetal demise, and 2 patients did not consent for WES. Demographics of this population are in Table 1. Six of these families have the rare diagnosis of bMCDK.
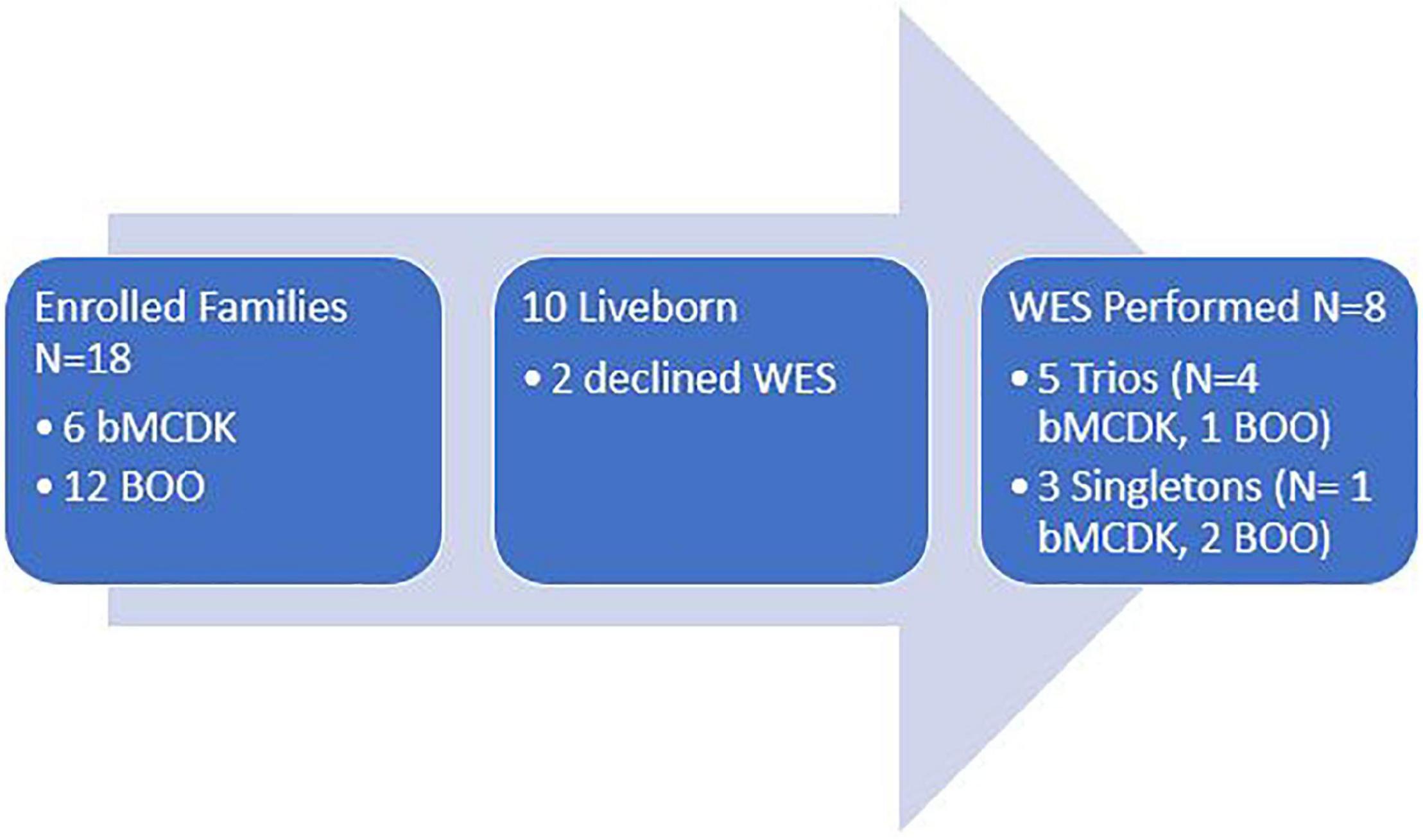
Figure 3. Study enrollment population based on diagnosis and study performed. WES, whole exome sequencing. bMCDK, bilateral multicystic dysplastic kidney. BOO, bladder outlet obstruction.

Table 1. Demographics of the patient population with severe congenital anomalies of the kidney and urinary tract (CAKUT) by diagnosis.
Whole Exome Sequencing
We performed variant calling on 5 trios (4 bMCDK) and 3 singleton samples (1 bMCDK). We identified 3 novel gene candidates (NSUN7, MTMR3, CEP162) and validated known variants of 2 CAKUT genes GATA3 and FRAS1. The bMCDK families are designated by F1, F2, F3, and F4. In one bMCDK family, we did not detect any candidate genes. The three candidate gene variants are described in Table 2. In bMCDK family F2, we identified a de novo frameshift deletion of five base pairs of NSUN7 near the terminal end of chromosome 4, a protein coding gene involved in the RNA methylation transferase pathway. NSUN7 is moderately expressed in the kidney (∼4 RPKM). In bMCDK family F3, we identified a de novo SNV of MTMR3 on chromosome 22 which 3/5 analyses predict to be damaging (C to T exchange). MTMR3 mRNA is expressed in the renal cortex and medulla at a level of ∼5 RPKM. Additionally, two bMCDK patients shared a non-synonymous SNV in CEP162 (F1, F2-A to G exchange). This mutation, found in 0.0009% of the population, is predicted to be damaging in 4/5 analyses. All 3 novel gene candidates were classified as variants of unknown significance (VUS) using ACMG guidelines (22).
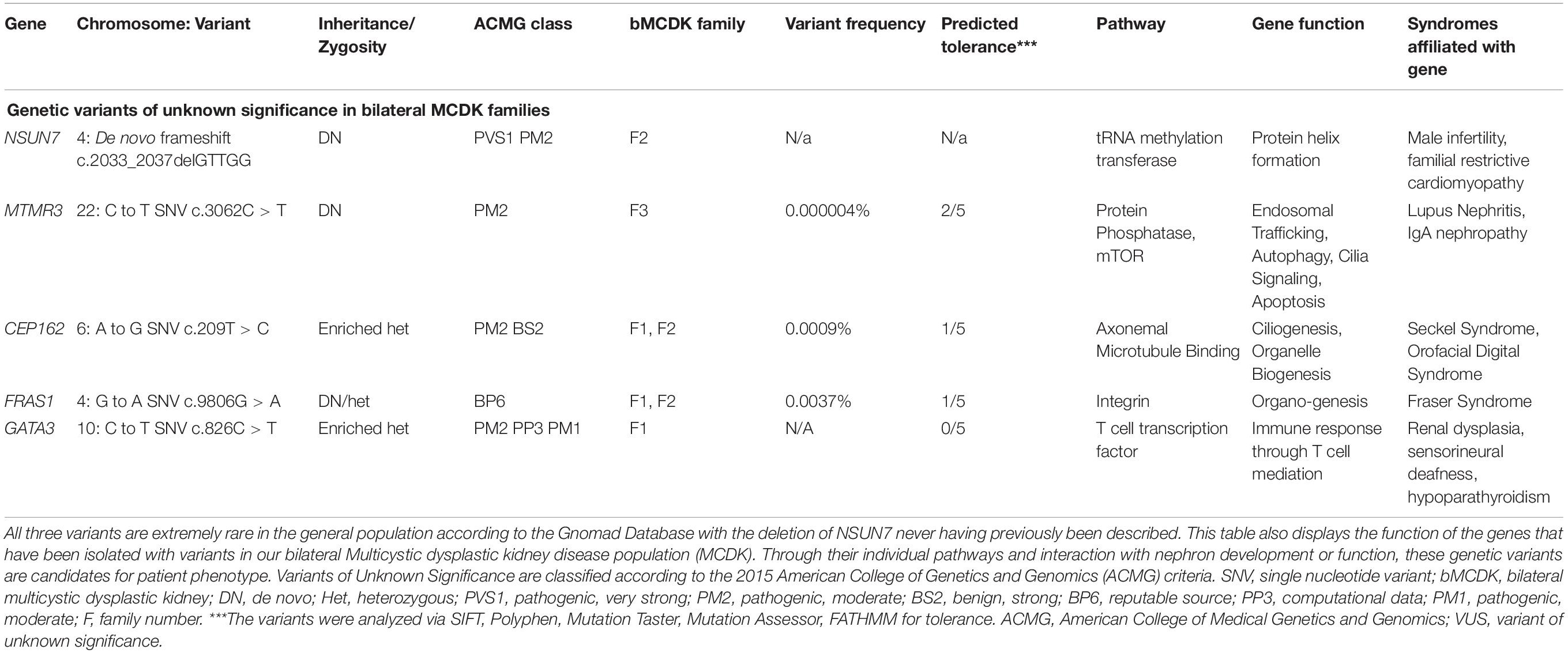
Table 2. Three candidate genes and two previously described congenital anomalies of the kidney and urinary tract genes in the bilateral Multicystic dysplastic kidney disease (MCKD) population.
In 2 bMCDK families, we identified a rare variant in FRAS1 not associated with Fraser Syndrome. FRAS1 RNA is expressed extensively in the kidney at a level of ∼4 Reads per Kilobase of Transcript (RPMK) (Figure 4) (24). We identified a rare SNV in FRAS1 on chromosome 4 in 2 individual bMCDK families (F1 and F2-G to A exchange, present in 0.0037% of the population). This mutation is predicted to be damaging in 4/5 analyses (SIFT, Polyphen, Mutation Taster, Mutation Assessor, FATHMM). Additionally, MCDK family F1 had a de novo SNV in GATA3, which is a known variant for CAKUT(C to T exchange) (11).
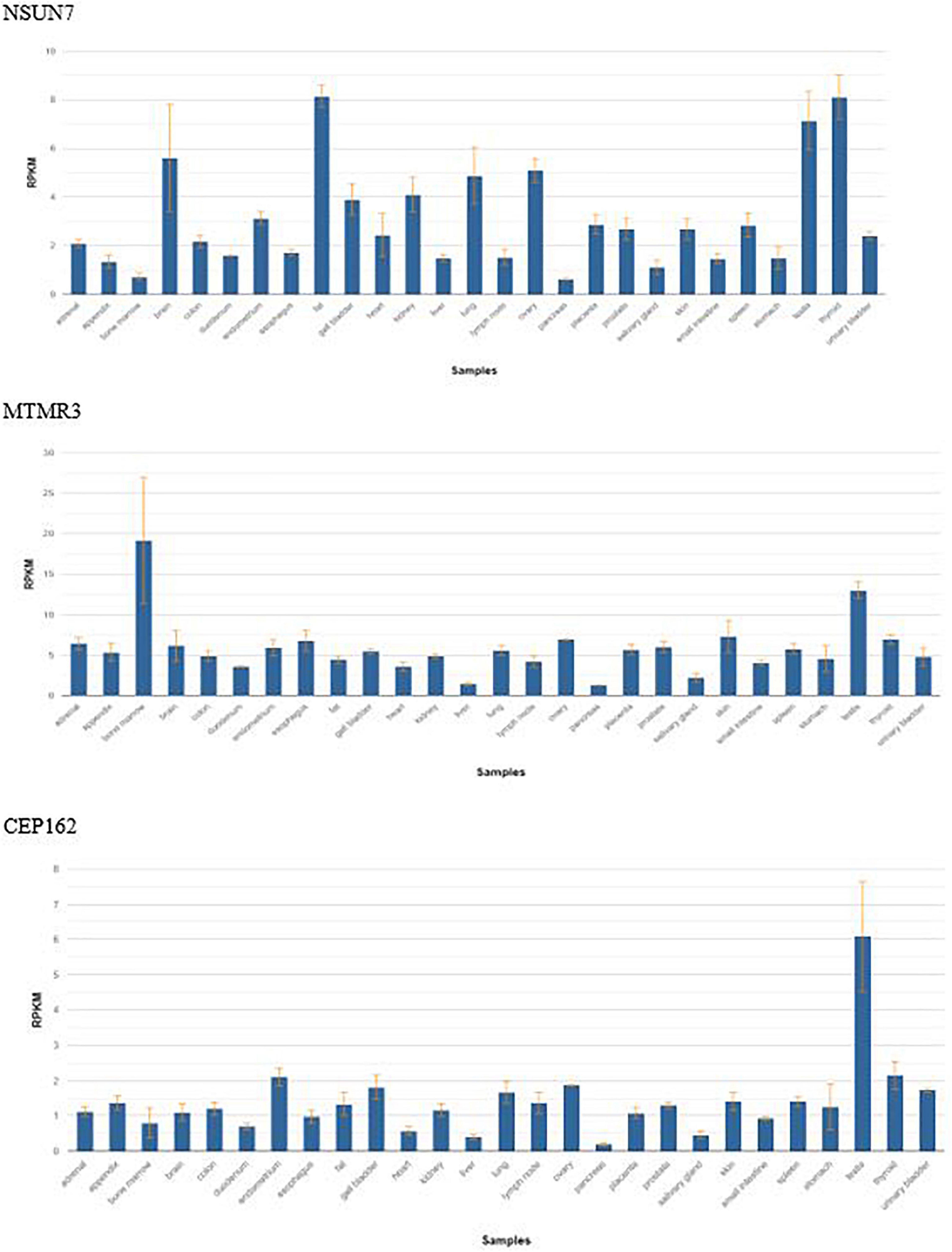
Figure 4. Frequency of gene expression by tissue of the three candidate genes found in bilateral Multicystic dysplastic kidney disease. RPKM, reads per kilobase of transcript.
In addition to these 5 genetic variants, we also found 6 genes with rare variants enriched in the GO annotation GO:0001701:in utero embryonic development of 526 genes (P = 1.3 × 10–4 TopGene). These genes include GATA3 (as previously mentioned), TTN, TGBR1, CDKN1C, HSD17B2, NCAPG2.
Discussion
Previously incompatible with life, infants with severe CAKUT may develop unknown manifestations of their underlying genetic variants. In our pilot study of 18 patients in a unique severe CAKUT population requiring fetal interventions and/or early infant dialysis, we have discovered three novel CAKUT candidate genes and validated two previously established CAKUT genes in our study population as well as identified 6 rare variants implicated in embryonic development. As one of the few centers in North America that performs fetal interventions on infants with severe CAKUT as well as performs infant dialysis via PIRRT, we have a unique population of severely affected patients that has not previously been studied. Targeted prenatal counseling and postnatal care is imperative for this growing population.
Little is known about the genetic etiology or clinical outcomes of bMCDK given its high rate of fetal demise without intervention. Ishiwa et al. found that patients with a unilateral MCDK with a contralateral hypoplastic kidney (less severe phenotype than bMCDK) have an increased likelihood of genetic mutations (25). Previous studies also support that the detection rate of a genetic etiology for CAKUT is higher for bilateral renal anomalies (12, 13). This is reinforced in our study, where despite a small patient population, we identified strong genetic variant enrichment. Additionally, identifying variants in FRAS1 and GATA3 establishes feasibility of our project through identification of known genes associated with CAKUT, while MTMR3, CEP162, and NSUN7 represent novel gene unique to this severe patient phenotype. The clinical implications of these variants are shown in Table 2 and are further discussed below.
GATA3 known to be implicated in CAKUT, is a critical regulator of ureteric bud positioning upstream of RET signaling (26). GATA3 mutations are known to cause HDR syndrome, characterized by hypoparathyroidism, deafness and renal dysplasia (27, 28). The proband in this study did not have hearing loss or hypoparathyroidism (although this is likely masked by the bone mineral disease of ESKD). Importantly, validation of GATA3 in one bMCDK family validates our study design and supports strong enrichment in a small sample size.
In our study, we identified several rare variants in FRAS1 not associated with Fraser Syndrome. Of note, one rare variant was present in 2 different bMCDK families, further pointing to enrichment. FRAS1 encodes the protein FRAS1, an extracellular matrix protein. Mutations in this gene can be associated with Fraser syndrome, an autosomal recessive mutation associated with cryptophthalmos, cutaneous syndactyly, and renal agenesis (29–31), as well as isolated CAKUT (12, 32, 33). Saisawat et al. described four heterozygous truncating mutations in FRAS1 as a potential cause of non-syndromic CAKUT in a pooled exome analysis of 40 CAKUT patients (32). Notably, the phenotype in this study was less severe than ours with 29 patients having unilateral MCDK only. However, one patient in this pooled exome study with a compound heterozygous mutation in FRAS1 had a sibling with previous intrauterine demise secondary to bilateral renal agenesis, suggesting various FRAS1 mutations may cause more severe renal phenotypes. This is further supported by a recent publication identifying biallelic FRAS1 variants in four cases of bilateral renal agenesis (34).
This study introduced three candidate genes implicated in severe CAKUT. In one bMCDK family, we detected a de novo frameshift deletion in NSUN7. NSUN7 is part of a 6 gene family of the NOL1/NOP2/SUN domain that catalyzes the methylation of cytosine to 5-methycytosine (m5c) (35, 36). NSUN7 encodes a methyltransferase protein by the same name and has been implicated in familial restrictive cardiomyopathy as well as male infertility through the mechanism of sperm dysmotility (37). NSUN7 encodes for an RNA methyltransferase and is currently not described in renal development literature or associated with the CAKUT phenotype. However, single-cell RNA seq expression data of the mid-gestation human kidney demonstrates NSUN7 sharing expression patterns of known progenitor markers, including CITED1 and EYA1 (38). NSUN7 is also expressed in the distal portion of the developing human (39) and murine kidney (40). NSUN2-NSUN7 has been implicated in aberrant murine embryogenesis in multiple organ systems including the brain and liver, often through inappropriate protein helix formation and decreased expression of genes that require methylation by NSUN7 (36, 41). In studies on NSUN7 mutations in asthenospermic men, a single SNV lead to a change in the structure of the helix, coil and strand of the protein with impairment of protein function (41). Inappropriately folded proteins induce the Unfolded Protein Response which triggers a cytotoxic pathway, leading to apoptosis and impairment in cell function and structure (42). Additionally, NOP2/NSUN1 and NSUN2 are implicated in epigenetic modifications to create transcriptionally active chromatin (43). Due to the role of NSUN7 in epigenetic methylation of proteins, depletion of NSUN leads to diminished expression of the enhancer RNA of these proteins, especially ones that rely on cytosine modification (36). Unfortunately, studies on the role of the NOL1/NOP2 SUN domain in renal embryogenesis are lacking (35). However, impaired cystine methylation during renal development has been implicated in renal pathologies including the SIX2, PAX2 gene and the TGF-β pathway leading to podocyte loss, fibrosis, mesangial proliferation, and tubular dysgenesis (44–47). Further research in the CAKUT patient population will be critical to understand how NSUN7 is involved in kidney development.
Additionally, we identified a rare SNV in MTMR3 in one bMCDK family. MTMR3 belongs to the protein-phosphatase family and is structurally similar to myotubularin. It is highly expressed in the ureteric lineage of the developing mouse kidney (40) and the proximal s-shaped body of the human nephron (39). The myotubularin family regulates endosomal trafficking, apoptosis, and autophagy. MTMR3 inhibits the activity of mTORC1, regulating autophagy (48–51). Mtor deletion in mice have severe paucity of glomeruli which lead to death after birth as Mtor regulates cell growth, proliferation and protein synthesis (52). Variants of MTMR3 have also recently been implicated in lupus nephritis and IgA nephropathy, implicating MTMR3 as important for renal function and immune regulation (53).
Similar to MTMR3, CEP162 is also moderately expressed throughout the renal tubules (54). In the developing kidney, CEP162 is highly expressed from nephron progenitor to differentiated structures (55), implicating its role early in development. The protein CEP162 is an axoneme-associated protein that promotes transition zone assembly of the cilia (56). Loss of CEP162 arrests ciliogenesis at the transition zone of the cilia in animal models (56). Dysfunctional cilia, leading to “ciliopathies” have been linked to more than 180 proteins (57–59). Primary cilia are located on the polarized epithelial cells of the renal tubule and are involved in sensation rather than motility (60). Dysregulated cilia-dependent processes have been attributed to cyst formation and progression of chronic kidney disease (61). MCDK has not previously been considered a ciliopathy, but the strong enrichment of these genes supports an early developmental spectrum of ciliopathy presentation resulting in a severe phenotype.
Limitations of this study include a small sample size. However, this pilot data supports strong enrichment of candidate genes in a novel severe phenotype population. This study is limited by exclusion of patients with BRA. In a study by Riddle et al. completed at our institution, all 8 patients who underwent serial amnioinfusions for BRA died either in utero (n = 6) or within 30 days of delivery (n = 2) (15). As a result of this study, fetal interventions and infant dialysis are no longer offered to mothers of infants with BRA at our institution. It is promising that with even the small number of samples sequenced, we are identifying new variants in CAKUT genes as well as suggestions of gene enrichment in pathways involved in CAKUT.
Even with these limitations, this study lays the groundwork for identifying additional genetic causes for severe CAKUT. The goal of this study is to provide individualized and accurate prenatal and postnatal counseling for families with anomalies previously incompatible with life. Clinically, there is a wide range of variability in outcomes in this population including survival to delivery, pulmonary survivorship, degree of respiratory support, need for urological interventions, length of hospitalization, survival to hospital discharge and degree of developmental delay, which have been difficult to predict. We aim to enhance our genetic data with biomarker data currently being investigated to develop a polygenic risk score (PRS) to predict and screen for disease severity early in pregnancy, as well as counsel families on other associated findings with these genetic mutations. Polygenic risk scores have been used successfully in other conditions such as diabetes mellitus and urinary tract stones (62, 63). In our model, weight will be placed differently based on (1) known CAKUT risk variants, (2) variants in known CAKUT genes of unknown significance, and (3) variants in plausible genes.
Based on our small pilot study of a unique severe CAKUT population, WES testing appears to be a promising diagnostic tool to help predict the course of infants with severe CAKUT prenatally. We believe this data, together with clinical outcomes, will improve our ability to counsel parents and improve clinical outcomes as well as help us further understand the role of the intricate genetic network in renal embryogenesis.
Data Availability Statement
The datasets presented in this study are not readily available because it may be possible for this specific de-identified genetic data to be re-identified. Requests to access the datasets should be directed to MH.
Ethics Statement
The studies involving human participants were reviewed and approved by the Cincinnati Children’s Hospital IRB. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
MH and EE: study conception and design. MH, EE, and DM: data collection. MH, MS, KK, and EE: analysis and interpretation of results and draft manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Funding
This study was funded by an NIH T-32 grant through the Division of Nephrology at Cincinnati Children’s Hospital Medical Center.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the Maternal Fetal Care Physicians at Cincinnati Children’s Hospital Medical Center and the University of Cincinnati.
References
1. Soares CM, Diniz JS, Lima EM, Oliveira GR, Canhestro MR, Colosimo EA, et al. Predictive factors of progression to chronic kidney disease stage 5 in a predialysis interdisciplinary programme. Nephrol Dial Transplant. (2009) 24:848–55. doi: 10.1093/ndt/gfn547
2. Warady BA, Abraham AG, Schwartz GJ, Wong CS, Muñoz A, Betoko A, et al. Predictors of rapid progression of glomerular and nonglomerular kidney disease in children and adolescents: the chronic kidney disease in children (CKiD) cohort. Am J Kidney Dis. (2015) 65:878–88. doi: 10.1053/j.ajkd.2015.01.008
3. Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. (2012) 27:363–73.
4. Malin G, Tonks AM, Morris RK, Gardosi J, Kilby MD. Congenital lower urinary tract obstruction: a population-based epidemiological study. BJOG. (2012) 119:1455–64.
5. Haeri S, Simon DH, Pillutla K. Serial amnioinfusions for fetal pulmonary palliation in fetuses with renal failure. J Matern Fetal Neonatal Med. (2017) 30:174–6.
6. Askenazi D, Ingram D, White S, Cramer M, Borasino S, Coghill C, et al. Smaller circuits for smaller patients: improving renal support therapy with aquadex. Pediatr Nephrol. (2016) 31:853–60.
7. Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. (1997) 15:157–64. doi: 10.1038/ng0297-157
8. Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. (1999) 8:2001–8. doi: 10.1093/hmg/8.11.2001
9. Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. (1995) 9:358–64. doi: 10.1038/ng0495-358
10. Vervoort VS, Smith RJ, O’Brien J, Schroer R, Abbott A, Stevenson RE, et al. Genomic rearrangements of EYA1 account for a large fraction of families with BOR syndrome. Eur J Hum Genet. (2002) 10:757–66. doi: 10.1038/sj.ejhg.5200877
11. Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest. (2018) 128:4–15.
12. van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. (2018) 29:2348–61.
13. Zhou X, Wang Y, Shao B, Wang C, Hu P, Qiao F, et al. Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole-exome sequencing. J Clin Lab Anal. (2020) 34:e23480.
14. Rasmussen M, Sunde L, Nielsen ML, Ramsing M, Petersen A, Hjortshøj TD, et al. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin Genet. (2018) 93:860–9. doi: 10.1111/cge.13185
15. Riddle S, Habli M, Tabbah S, Lim FY, Minges M, Kingma P, et al. Contemporary outcomes of patients with isolated bilateral renal agenesis with and without fetal intervention. Fetal Diagn Ther. (2020) 47:675–81.
16. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4.
17. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9.
18. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6.
19. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118.
20. Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. (2015) 31:1536–43.
21. ToppGene Suite. Division of Biomedical Informatics. Cincinnati, OH: Cincinnati Children’s Hospital Medical Center (2007-2022).
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24.
24. National Institutes of Health. Gene. Bethesda, MD: National Center for Biotechnology Information (2022).
25. Ishiwa S, Sato M, Morisada N, Nishi K, Kanamori T, Okutsu M, et al. Association between the clinical presentation of congenital anomalies of the kidney and urinary tract (CAKUT) and gene mutations: an analysis of 66 patients at a single institution. Pediatr Nephrol. (2019) 34:1457–64. doi: 10.1007/s00467-019-04230-w
26. Grote D, Boualia SK, Souabni A, Merkel C, Chi X, Costantini F, et al. Gata3 acts downstream of beta-catenin signaling to prevent ectopic metanephric kidney induction. PLoS Genet. (2008) 4:e1000316. doi: 10.1371/journal.pgen.1000316
27. Ali A, Christie PT, Grigorieva IV, Harding B, Van Esch H, Ahmed SF, et al. Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet. (2007) 16:265–75. doi: 10.1093/hmg/ddl454
28. Chenouard A, Isidor B, Allain-Launay E, Moreau A, Le Bideau M, Roussey G. Renal phenotypic variability in HDR syndrome: glomerular nephropathy as a novel finding. Eur J Pediatr. (2013) 172:107–10. doi: 10.1007/s00431-012-1845-y
29. McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. (2003) 34:203–8. doi: 10.1038/ng1142
30. Vrontou S, Petrou P, Meyer BI, Galanopoulos VK, Imai K, Yanagi M, et al. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat Genet. (2003) 34:209–14.
31. van Haelst MM, Scambler PJ, Hennekam RC. Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am J Med Genet A. (2007) 143a:3194–203. doi: 10.1002/ajmg.a.31951
32. Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Gunther B, Airik R, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. (2012) 81:196–200. doi: 10.1038/ki.2011.315
33. Kohl S, Hwang DY, Dworschak GC, Hilger AC, Saisawat P, Vivante A, et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. (2014) 25:1917–22. doi: 10.1681/ASN.2013101103
34. Jordan P, Dorval G, Arrondel C, Morinière V, Tournant C, Audrezet MP, et al. Targeted next-generation sequencing in a large series of fetuses with severe renal diseases. Hum Mutat. (2022) 43:347–61. doi: 10.1002/humu.24324
35. Chi L, Delgado-Olguin P. Expression of NOL1/NOP2/sun domain (Nsun) RNA methyltransferase family genes in early mouse embryogenesis. Gene Expr Patterns. (2013) 13:319–27. doi: 10.1016/j.gep.2013.06.003
36. Aguilo F, Li S, Balasubramaniyan N, Sancho A, Benko S, Zhang F, et al. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1alpha. Cell Rep. (2016) 14:479–92. doi: 10.1016/j.celrep.2015.12.043
37. Khosronezhad N, Hosseinzadeh Colagar A, Mortazavi SM. The Nsun7 (A11337)-deletion mutation, causes reduction of its protein rate and associated with sperm motility defect in infertile men. J Assist Reprod Genet. (2015) 32:807–15. doi: 10.1007/s10815-015-0443-0
38. Schuh MP, Alkhudairy L, Potter A, Potter SS, Chetal K, Thakkar K, et al. The rhesus macaque serves as a model for human lateral branch nephrogenesis. J Am Soc Nephrol. (2021) 32:1097–112. doi: 10.1681/ASN.2020101459
39. Lindström NO, Sealfon R, Chen X, Parvez RK, Ransick A, De Sena Brandine G, et al. Spatial transcriptional mapping of the human nephrogenic program. Dev Cell. (2021) 56:2381–98.e6. doi: 10.1016/j.devcel.2021.07.017
40. Ransick A, Lindstrom NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, et al. Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell. (2019) 51:399–413.e7. doi: 10.1016/j.devcel.2019.10.005
41. Khosronezhad N, Colagar AH, Jorsarayi SG. T26248G-transversion mutation in exon7 of the putative methyltransferase Nsun7 gene causes a change in protein folding associated with reduced sperm motility in asthenospermic men. Reprod Fertil Dev. (2015) 27:471–80. doi: 10.1071/RD13371
42. Tavasolian F, Hosseini AZ, Mirzaei A, Abdollahi E, Jandaghi P, Soudi S, et al. Unfolded protein response-mediated modulation of mesenchymal stem cells. IUBMB Life. (2020) 72:187–97. doi: 10.1002/iub.2154
43. Willbanks A, Wood S, Cheng JX. RNA epigenetics: fine-tuning chromatin plasticity and transcriptional regulation, and the implications in human diseases. Genes (Basel). (2021) 12:627. doi: 10.3390/genes12050627
44. Nicolaou N, Renkema KY, Bongers EM, Giles RH, Knoers NV. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. (2015) 11:720–31.
45. Ko YA, Mohtat D, Suzuki M, Park AS, Izquierdo MC, Han SY, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. (2013) 14:R108. doi: 10.1186/gb-2013-14-10-r108
46. Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. (2007) 13:580–92. doi: 10.1016/j.devcel.2007.09.004
47. Lefevre GM, Patel SR, Kim D, Tessarollo L, Dressler GR. Altering a histone H3K4 methylation pathway in glomerular podocytes promotes a chronic disease phenotype. PLoS Genet. (2010) 6:e1001142. doi: 10.1371/journal.pgen.1001142
48. Naughtin MJ, Sheffield DA, Rahman P, Hughes WE, Gurung R, Stow JL, et al. The myotubularin phosphatase MTMR4 regulates sorting from early endosomes. J Cell Sci. (2010) 123(Pt 18):3071–83.
49. Zou J, Chang SC, Marjanovic J, Majerus PW. MTMR9 increases MTMR6 enzyme activity, stability, and role in apoptosis. J Biol Chem. (2009) 284:2064–71. doi: 10.1074/jbc.M804292200
50. Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, et al. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. (2010) 11:468–78. doi: 10.1111/j.1600-0854.2010.01034.x
51. Hao F, Itoh T, Morita E, Shirahama-Noda K, Yoshimori T, Noda T. The PtdIns3-phosphatase MTMR3 interacts with mTORC1 and suppresses its activity. FEBS Lett. (2016) 590:161–73. doi: 10.1002/1873-3468.12048
52. Volovelsky O, Nguyen T, Jarmas AE, Combes AN, Wilson SB, Little MH, et al. Hamartin regulates cessation of mouse nephrogenesis independently of Mtor. Proc Natl Acad Sci USA. (2018) 115:5998–6003. doi: 10.1073/pnas.1712955115
53. Zhou XJ, Nath SK, Qi YY, Cheng FJ, Yang HZ, Zhang Y, et al. Brief report: identification of MTMR3 as a novel susceptibility gene for lupus nephritis in northern Han Chinese by shared-gene analysis with IgA nephropathy. Arthritis Rheumatol. (2014) 66:2842–8. doi: 10.1002/art.38749
54. The Human Protein Atlas. CEP162. (2022). Available online at: https://www.proteinatlas.org/ENSG00000135315-CEP162/tissue/kidney (accessed 2022).
55. Lindström NO, Sealfon R, Chen X, Parvez R, Ransick A, De Sena Brandine G, et al. Spatial transcriptional mapping of the human nephrogenic program. bioRxiv[Preprint]. (2020). 2020.04.27.060749 doi: 10.1016/j.devcel.2021.07.017
56. Wang WJ, Tay HG, Soni R, Perumal GS, Goll MG, Macaluso FP, et al. CEP162 is an axoneme-recognition protein promoting ciliary transition zone assembly at the cilia base. Nat Cell Biol. (2013) 15:591–601. doi: 10.1038/ncb2739
57. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. (2007) 8:880–93. doi: 10.1038/nrm2278
58. Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TT, Racher H, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol. (2015) 17:1074–87. doi: 10.1038/ncb3201
59. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. (2017) 18:533–47.
60. Bacallao RL, McNeill H. Cystic kidney diseases and planar cell polarity signaling. Clin Genet. (2009) 75:107–17.
61. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. (2011) 26:1039–56.
62. Paranjpe I, Tsao N, Judy R, Paranjpe M, Chaudhary K, Klarin D, et al. Derivation and validation of genome-wide polygenic score for urinary tract stone diagnosis. Kidney Int. (2020) 98:1323–30.
Keywords: CAKUT, genetics, amnioinfusion, whole exome sequencing, bladder outlet obstruction
Citation: Harris M, Schuh MP, McKinney D, Kaufman K and Erkan E (2022) Whole Exome Sequencing in a Population With Severe Congenital Anomalies of Kidney and Urinary Tract. Front. Pediatr. 10:898773. doi: 10.3389/fped.2022.898773
Received: 17 March 2022; Accepted: 01 June 2022;
Published: 04 August 2022.
Edited by:
Bassam R. Ali, United Arab Emirates University, United Arab EmiratesReviewed by:
Andrew Mallett, Townsville University Hospital, AustraliaLaurel Willig, Children’s Mercy Hospital, United States
Copyright © 2022 Harris, Schuh, McKinney, Kaufman and Erkan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meredith Harris, Merharris@luriechildrens.org