 Albert Qin
Albert Qin- Medical Research & Clinical Operations, PharmaEssentia Corporation, Taipei, Taiwan
Interferon-beta (IFN-β), an extracellular cytokine that initiates signaling pathways for gene regulation, has been demonstrated to function as a tumor suppressor protein through lentiviral gene transduction. In this article, I review the relevant previous works and propose a cell cycle-based, tumor suppressor protein-mediated mechanism of anti-cancer surveillance. IFN-β induces a tumor cell cycle alteration that leads to S phase accumulation, senescence entry, and a loss of tumorigenicity in solid tumor cells. IFN-β does not show a significant cell cycle effect in their normal counterparts. Retinoblastoma protein RB1, another tumor suppressor protein, tightly controls the cell cycle and differentiation of normal cells, preventing them from being significantly impacted by the IFN-β effect. The interplay between IFN-β and RB1 acts as a mechanism of cell cycle-based, tumor suppressor protein-mediated anti-cancer surveillance that can selectively suppress solid tumor or proliferating transformed cells from the loss of control leading to cancer. This mechanism has important implications for the treatment of solid tumors.
Introduction
Cancer occurrence and growth involve the activation of oncogenes and loss or inactivation of tumor suppressor genes or proteins. During this process, events such as gene mutations, loss of tumor suppressor functions, and changes in intracellular and extracellular signaling networks accumulate and aggregate, leading to cell transformation or abnormal cell proliferation, cancer, and metastasis. The initial cell transformation or cancer cell formation may not necessarily be sustained to cause established cancer (1, 2). As cancer cells can often escape immune systems or immune surveillance may not always be adequate, it is not clear whether there are other mechanisms of surveillance and control against cancer development in vivo, and if so, how they exist.
Type I interferons alpha and beta
Type I interferons (IFNs) including alpha (IFN-α) and beta (IFN-β), are extracellular cytokines. IFNs function by binding to receptors and activating the Janus tyrosine kinase signal transducer and activator of transcription (JAK-STAT) pathway or other signaling pathways to regulate various genes (3–6). IFN-β exists in a single form and is expressed in virtually all tissues and cell types. IFN-α has 13 subtypes and is noted to be produced in many types of cells including plasmacytoid dendritic and other immune cells (7–9). IFN-α and IFN-β bind to the type I IFN receptor (IFNAR), which is expressed on almost every cell type and is composed of the transmembrane subunits IFNAR1 and IFNAR2 (10–12). They induce similar anti-proliferative, immune-stimulatory, and anti-angiogenic activities (6, 12–15). The anti-proliferative effects include cell cycle inhibition and apoptosis (16–20).
IFNs were reported to cause a cell cycle inhibition, including G1/G0 arrest, S phase prolongation, or both (16–18, 21). These effects appeared to be dependent on the cell type examined. In hematopoietic cancer cells, IFN-α and IFN-β cause G1/G0 cell cycle arrest. The G1/G0 arrest was the most characterized and considered to be the most common IFN effect (18, 22). Data mainly with IFN-α suggest that they induce the expression of the retinoblastoma protein (RB1) or activate its function by reducing its phosphorylation (23–30). RB1 is phosphorylated during the G1 phase and becomes hyper-phosphorylated at the G1 to S phase transition by cyclin-dependent kinases (CDKs) including cyclin D-CDK4/6 and cyclin E-CDK2 (31–38). IFN signaling could suppress CDK2, 4 and 6 activities or their regulatory cyclin subunits and induce gene expression of CDK inhibitors including p19Ink4D and p21WAF1/CIP1, and phosphatase CDC25A (26–30). However, subsequent systematic analyses with various cell types indicated that in human solid tumor cells, the predominant IFN cell cycle effect might differ. IFN-β, and presumably IFN-α induce S phase accumulation and entry into senescence (19, 39). A diagram indicating the differential IFN-induced cell cycle alterations between human hematopoietic cancer and solid tumor cells is shown in Figure 1. Despite the notable similarity and redundancy in IFN-α and IFN-β activities, a recent study suggests that IFN-α subtypes may function mainly to complement, prolong, and amplify the IFN-β effects (40). Therefore, IFN-β represents a prototype of type I IFNs and meticulously mediates its activities potentially with the cooperative and mutually supplemental involvement of IFN-α subtypes.
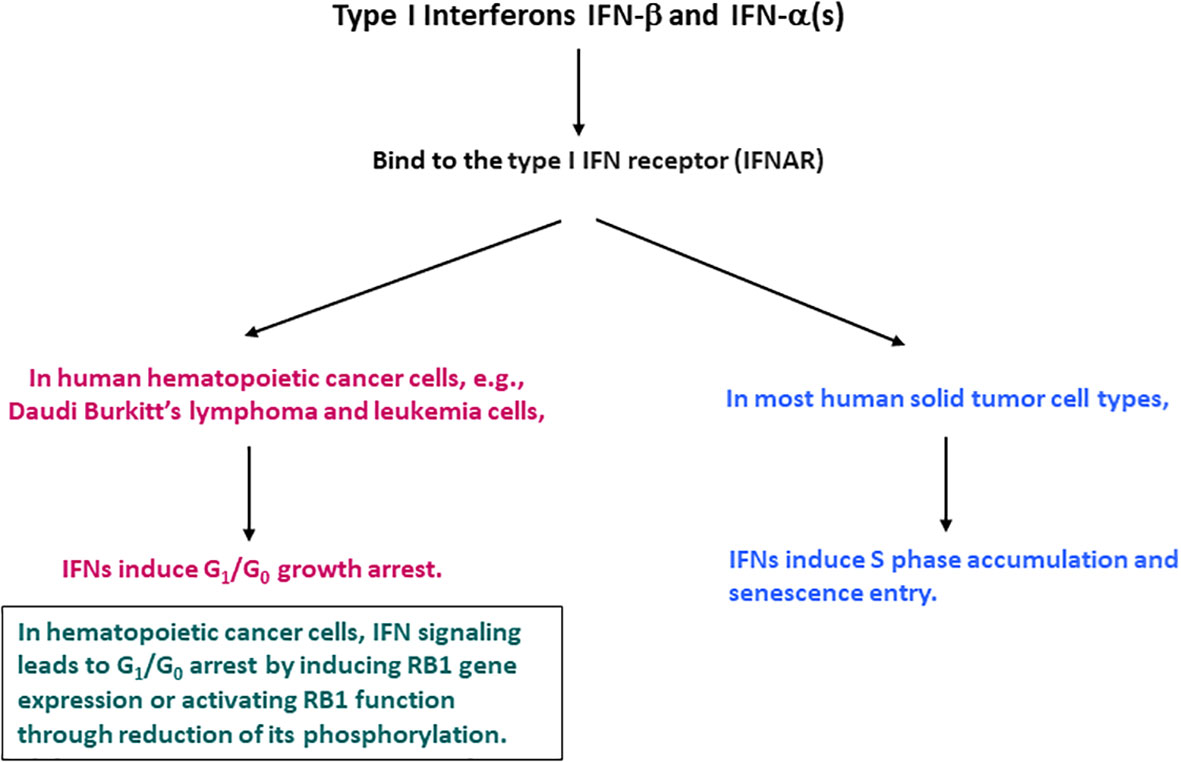
Figure 1 Cell cycle alterations induced by IFN-β and IFN-α in human hematopoietic cancer and solid tumor cells.
Anti-viral effect of IFNs and virus-induced carcinogenesis
Type I IFNs exhibit anti-viral activities (41, 42). The anti-viral activities can suppress virus-induced carcinogenesis. IFN signaling induces many antiviral effector proteins. These proteins can inhibit viral replication or augment the IFN-induced anti-viral response and include the anti-myxovirus-related (Mx) protein family with GTPase activity, 2′,5′-oligoadenylate synthetase 1, protein kinase R, and IFN-stimulated protein of 15 kDa (43). Viruses often antagonize IFN-induced antiviral responses to prevent their removal from cells. For example, hepatitis B virus downregulates MxA gene expression by its precore or core proteins (44) and SARS-CoV-2 virus inhibits endogenous IFN synthesis (45). In this respect, conjugation of polyethylene-glycol (PEG) on IFN molecule has significantly improved the pharmacokinetic profile of IFNs in vivo and allowed more convenient dose schemas than un-PEGylated recombinant IFNs. The PEGylation technology made IFN-based therapies possible with higher in vivo IFN exposures or longer half-lives that can endure the virus-mediated antagonizing effect. This improvement has led to clinically meaningful, therapeutic anti-viral responses (46–52).
Viruses, regardless of being genomic DNA or RNA-based, can impose cancer risk by introduction of viral oncogenes, activation of cellular oncogenes, or inactivation of tumor suppressor genes (53). This can disrupt normal growth and differentiation pathways leading to uncontrolled cellular proliferation and neoplastic transformation. Inflammation and other changes during viral infection can also elicit a virus-induced neoplastic process, potentially leading to cancer (54, 55). Therefore, anti-viral responses by IFNs may inhibit virus-induced tumorigenesis. In this respect, using an anti-viral regimen containing an anti-cancer component such as IFN-based therapy throughout the treatment course of onco-viral infections including hepatitis B or D may potentially minimize cancer occurrence (56).
Immune surveillance
Immune surveillance is an important, well-characterized mechanism for detection and removal of cancer cells (57–59). Cancer progression occurs when cancer cells escape the immune surveillance as part of a process termed as cancer immunoediting (60, 61). IFNs play important roles in immune surveillance against cancer and the cancer immunoediting process during the interaction of cancer cells and the immune system (60, 61). IFN-α and IFN-β are strong immunostimulants (7–9). However, an immune activation by IFNs per se may not be directly linked to an antitumor effect in some cancer models. We explored the IFN-β-induced immune-based, antitumor effect using IFN-β gene therapy in tumor mouse models (62, 63). IFN-β gene therapy showed potent antitumor responses in various immune-deficient and -competent mouse models. The involvement of immune cells was subsequently defined by using depleting antibodies against immune cells (62, 63). The results indicated that the antitumor effect of IFN-β was dependent upon natural killer (NK) cells with a suspected macrophage involvement. Furthermore, IFN-β gene therapy significantly inhibited tumor growth and metastasis via cytotoxic CD8+ T-cells, even with the depletion of CD4+ T helper cells in immune-competent mice (63). In recent years, cancer cell-intrinsic signaling was found to significantly impact the tumor immune landscape (64). Animal modeling and research have provided further insights into our understanding of the effect of IFN-α and IFN-β signaling on the immunity against cancer. The interaction between type I IFN signaling and cellular oncoproteins such as c-MYC and KRAS were implicated in affecting the immune microenvironment including NK cell-mediated immunity (65–67), underscoring the importance of type I IFNs in immune surveillance against cancer.
A mechanism of cell cycle-based, anti-cancer surveillance mediated by the interplay between IFN-β and RB1
An important and intriguing aspect of IFN-β, perhaps somewhat overlooked, is its direct tumor suppressor function, which is different from its general cytotoxic effects. In 2002, we reported our results on IFN-β gene delivery into tumor cells using a lentiviral vector, providing evidence that IFN-β can function as a direct or cancer cell-intrinsic, tumor suppressor protein (39). Abnormalities or deletions in chromosome 9p containing IFN-α, IFN-β, and other genes were previously observed to be frequent in cancer. However, IFN-β gene was not specifically identified to be relevant in cancer occurrence (68–70). We noted that human IFN-β induced overt apoptosis or cytotoxicity in human cancer cells when overexpressed by an adenovirus vector or in combination with chemotherapeutic agents (20, 62, 71). Therefore, we performed lentiviral vector-mediated IFN-β gene transduction with the initial aim of introducing the gene at a low copy number into human tumor cells and characterizing the IFN-β-induced cell cycle effect by separating it from cytotoxicity. Tumor cell clones stably expressing IFN-β were acquired after the gene transduction with the lentivirus. Despite stable IFN-β expression, the cells continuously divided and grew in vitro. All cell clones transduced by the IFN-β gene had a cell cycle alteration showing S phase accumulation and an entry into senescence. Importantly, all the cells lost their ability to form tumors in vivo when implanted back into animals. Therefore, IFN-β functioned as a tumor suppressor protein (39).
The cell cycle profile of the IFN-β-expressing clones was consistent with our earlier observations (19). Given the apparent lack of comprehensive understanding of the IFN-induced cell cycle effect in all cancer types, we used IFN-β in a systematic analysis of various types of human cancer cells and their normal cell counterparts (19). IFN-β did not significantly alter the cell cycle profiles of normal cells grown under normal conditions without growth factor stimulation (19). However, various solid tumor cell types altered their cycle, with more cells detected in the S phase. After further examination, the S phase accumulation, not the G1/G0 growth arrest that was observed in hematopoietic cancer cells, was found to be the cell cycle effect of IFN-β in various types of solid tumor and transformed cells (19). The S phase accumulation appears to be due to an inefficient S phase progression without a G2/M phase accumulation (19, 39), suggesting that an intra-S phase checkpoint is activated. Catastrophic cell death was observed in IFN-β expressing tumor cells (39), indicating that some cells might have moved to the next cell division without the S phase completion. In solid tumor and transformed cells that exhibited the IFN-β-induced S phase accumulation, IFN-β activated the JAK-STAT pathway, as revealed by tyrosine phosphorylation of STAT proteins and activation of DNA-binding complexes including the IFN-stimulated gene factor-3 (ISGF3) (19). Consistent with our work with lentiviral IFN-β gene transduction, we observed a portion of tumor cells exhibiting senescence entry after IFN-β treatment. In solid tumor and cells that showed slow S phase and senescent entry, there was a lack of functional RB1. RB1 function was lost due to gene mutations, inactivation by hyperphosphorylation, or binding of viral oncoproteins. The cell cycle alteration induced by IFN-β was not significant in non-transformed cell counterparts, nor in RB1+/+ tumor cells with an abundant presence of underphosphorylated RB1. In the latter, IFN-β signaling was clearly detected using electrophoretic mobility shift assays for active STAT transcriptional complexes including the transcription factor ISGF3 (19). Therefore, active RB1 in abundance prevented the RB1+/+ tumor cells from developing the IFN-β-induced cell cycle alterations despite IFN-β signaling.
RB1 is a tumor suppressor protein that provides key cell cycle regulation (72). It is phosphorylated at different phases of the cell cycle by cyclin D-CDK4/6 and cyclin E-CDK2 and becomes inactivated via hyperphosphorylation at the G1/S phase boundary possibly by cyclin E-CDK2, in a process regulated by CDK inhibitors (31–38). RB1 interacts with various cellular factors, including transcription factors E2F1-3 and histone deacetylase, to modulate gene expression and maintain normal cell cycle and differentiation (73–82). The loss of RB1, or disruption of its interaction with other cellular factors, is associated with a variety of human cancers (73). Our results indicate that in cells of solid tissue origin, the cell cycle regulatory machinery, hallmarked by functional RB1 interacting appropriately with cellular factors, is a determining factor for whether or not IFN-β induces cell cycle alterations (19).
While the findings initially appeared perplexing, here it is proposed that a cancer cell-intrinsic, cell cycle-based mechanism of anti-cancer surveillance exists for the selective suppression of solid tumor and transformed cells. The extracellular and intracellular interplay between the tumor suppressor proteins, IFN-β and RB1, are central to this mechanism. Specifically, IFN-β signaling provides surveillance and removal of solid tumor and transformed cells by altering their cycle and causing more cells to accumulate in S phase and enter senescence. This effect leads to the loss of a tumorigenesis. Meanwhile, functional RB1 with its interacting cellular factors in normal cells to engage them in the normal cycle and promote differentiation, preventing them from being impacted by the IFN-β-induced cell cycle alterations. When RB1 function is lost due to gene mutation or protein inactivation, cells become susceptible to surveillance and inhibition by IFN-β. This tumor suppressor protein-mediated surveillance serves as a mechanism to selectively suppress solid tumor and transformed cells, including those that are occasionally derived from sporadic gene mutations and changes in epigenetic regulation. Furthermore, cell death in tumor cells that exhibited a very enlarged senescent cell phenotype was observed (39). Therefore, tumor cell senescence can ultimately lead to cell death and result in the removal of tumor cells. A schematic elucidating the mechanism of the tumor suppressor protein-mediated, cell cycle-based surveillance for the selective inhibition of solid tumor and transformed cells by IFN-β and RB1 is shown in Figure 2.
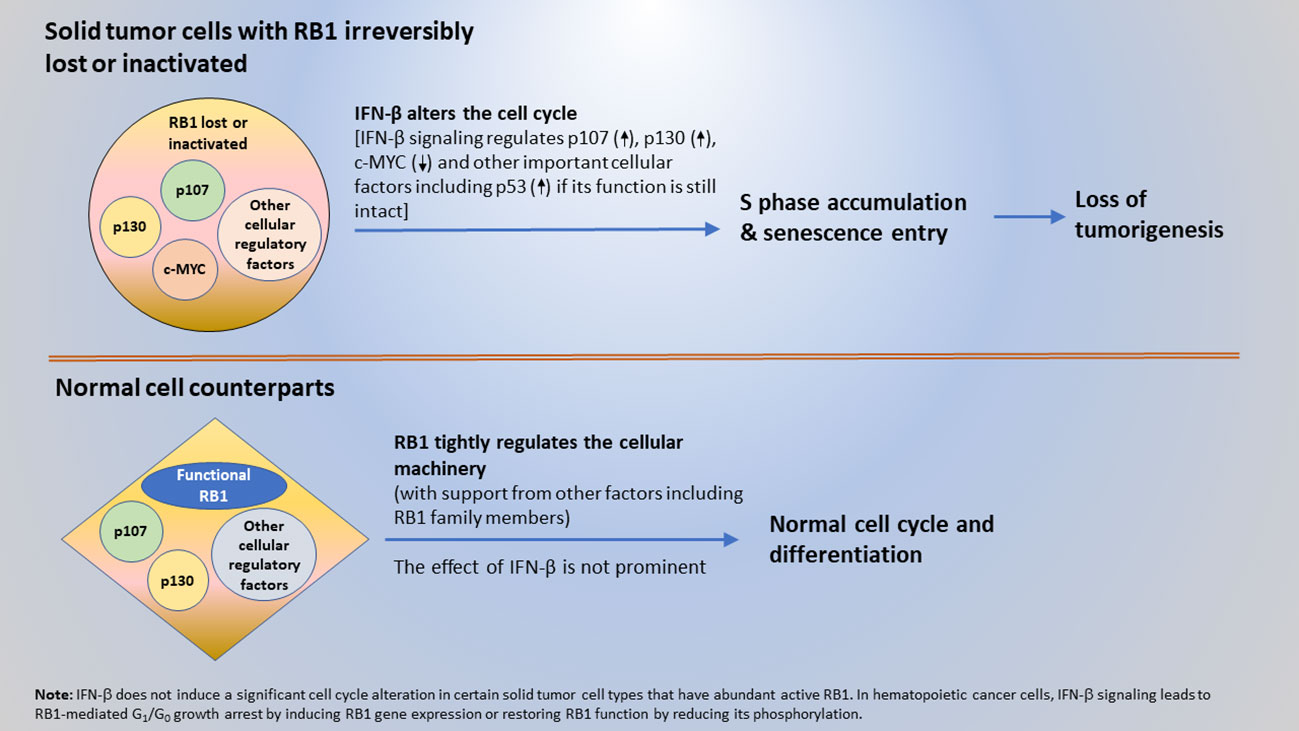
Figure 2 A mechanism of anti-cancer surveillance for selective suppression of tumor or transformed cells mediated by the interplay between tumor suppressor proteins IFN-β and RB1.
Most solid tumors have irreversibly lost RB1 function. Therefore, IFN-β signaling induces cell cycle alterations by interacting with cell cycle regulators other than RB1. However, the incidence of hematopoietic cancer varies. In Burkitt’s lymphoma (Daudi) and leukemia cells, loss of RB1 function appears to be reversible, and IFN-β signaling induces RB1 gene expression or restores RB1 function by reducing its phosphorylation (23–30). In these hematopoietic cancer cells, IFN-β exerts its tumor suppressor function via RB1-mediated G1/G0 growth arrest. The different IFN-β cell cycle effects illustrate that the cell cycle regulatory machinery differs between hematopoietic cancer and solid tumor cells.
Discussion
IFN-β was revealed to possess a tumor suppressor protein function with lentiviral gene transduction (39). It alters the cell cycle of various solid tumor and transformed cell types by inducing S phase accumulation and senescence entry (19, 39). This article elucidates a new mechanism of anti-cancer surveillance mediated by the interplay between tumor suppressor proteins IFN-β and RB1. IFN-β in tissues maintains a presence for detecting tumor and transformed cells. Once a tumor cell is identified, IFN-β alters its cell cycle and causes an inefficient or slow S phase progression accompanied by senescence entry, rendering it no longer cancerous. RB1 interacts in this process in normal cells to engage them in their regular cycle and differentiation. The selective suppression of tumor and transformed cells without significantly affecting their normal counterparts, coordinated by IFN-β and RB1, is an important surveillance and control mechanism against cancer. As IFN-β is often significantly induced in response to viral infections that may impose a potential cancer risk, this cancer cell-intrinsic, cell cycle-based, tumor suppressor protein-mediated surveillance can suppress carcinogenesis or transformation at the cellular level during viral infections.
Differential cell cycle regulatory machinery between hematopoietic cancer and solid tumor cells
In hematopoietic cancer cells, IFN-β signaling can directly restore RB1 function by increasing its gene expression or protein activation with reduction of its phosphorylation to induce an RB1-mediated G1/G0 growth arrest. As a result, there is a direct anti-cancer interaction between IFN-β signaling and RB1 function in the same cells. This differs from the cell cycle effects observed in various types of solid tumor cells, indicating an alternative landscape of the cell cycle regulatory machinery inside solid tumor cells compared with hematopoietic cancer cells. Indeed, the function of RB1 or its complexes that regulate the normal cell cycle are irreversibly lost or disrupted in most solid tumors. In addition, inactivation of the tumor suppressor protein p53 is very frequent in human cancer and could inhibit RB1 function as p53 can upregulate the CDK inhibitor p21WAF1/CIP1 from upstream (83, 84). Therefore, RB1-mediated growth arrest in G1/G0 by IFN-β does not occur in these solid tumor cells. Additional gene mutations or epigenetic changes present in solid tumor cells may help prevent proliferating cells from stopping at G1 and/or entering G0. Ultimately, these solid tumor cells undergo cell cycle alterations with an S phase accumulation due to a slow S phase progression possibly caused by an activation of intra-S phase checkpoint, and senescence entry in response to IFN-β signaling.
Cellular proteins that IFN-β potentially agonizes or antagonizes for suppression of solid tumor cells
With slow S phase progression accompanied by senescence entry, not G1/G0 arrest, being the cell cycle effect of IFN-β in various types of solid tumor cells (19, 39), then how does IFN-β induce the effect in these cells in which RB1 function is irreversibly lost or inactivated? This article postulates that IFN-β does so in the solid tumor cells with irreversibly lost RB function by modulating the RB1 family members p107 and p130, and cellular factors, including c-MYC and other regulatory proteins. p107 and p130 can be associated with promoters in a similar manner as RB1 and cooperate with RB1 in cell cycle regulation, although with functional distinctions (85–88). In normal cells, p107 and p130 may complement or assist RB1 in regulating cell growth and differentiation. However, in tumor cells with irreversibly lost RB1 function, the involvement of p107 or p130 in cell cycle regulation becomes prominent. All RB1 family members are regulated by phosphorylation and bind to E2Fs with differential preferences to regulate gene expression (87, 89, 90). IFNs are suggested to be able to decrease all their phosphorylation (28). Notably different from RB1, which is phosphorylated as cells enter G1 from G0 and becomes hyperphosphorylated at the G1/S boundary, p107 is phosphorylated later in the cell cycle, in the late G1 and S phase onward (28, 91). Additionally, p107 was cloned as an RB1-related protein and is implicated in S phase regulation (92, 93). The expression of p107 in RB1-deficient osteosarcoma cells suppresses the progression of the S phase in addition to G1 (94). Therefore, one possibility is that in tumor cells with loss of RB1 function, IFN-β signaling reduces the phosphorylation of p107 to form p107/E2F or other p107 complexes to induce a p107-mediated suppression of the S phase progression. These complexes may suppress the genes promoting DNA synthesis and S phase progression (e.g., encoding cyclins, CDK1, DNA polymerase subunits, c-MYC, and B-MYB), resulting in the inhibition of S phase progression and more cells accumulating in the S phase. IFN-β also downregulates the growth-promoting gene c-myc in an RB1 family member-independent manner (95, 96), indicating that IFN-β activates different pathways to elicit its cell cycle effect. Newer data indicate that both p107 and p130 are involved in the senescence entry of tumor cells that have lost RB1 function (97–99). Downregulation of c-myc has been suggested to trigger tumor cell senescence (100–102). In addition, IFN-α and IFN-β signaling was shown to induce the transcription of the p53 gene by ISGF3 (103), suggesting that IFN-β signaling involves p53 in inducing the senescent entry of tumor cells retaining a functional p53 and an intact p53-responsive pathway. Moreover, IFN-α induces senescence-promoting CDK inhibitors including p19Ink4D and p21WAF1/CIP1 (29, 30, 104, 105), which suggests that IFN-β signaling may also involve p19Ink4D and p21WAF1/CIP1 in promoting senescence entry. Taken together, these data are consistent with our previous finding that in various types of solid tumor cells with the lost RB1 function, the prominent cell cycle effect of IFN-β is slow S phase progression and senescence entry accompanied by a loss of tumorigenicity. Additionally, a notion raised in this article is that IFN-β elicits its inhibitory effect on cell cycle and tumorigenicity in RB-defective solid tumor cells by inducing a p107-mediated suppression of S phase progression and regulating multiple cellular factors including p107, p130, c-MYC, and other important regulatory proteins.
Clinical implications
The difference in the cell cycle machinery and effects induced by IFN-β between hematologic cancer and solid tumor cells has clinical implications. In hematologic cancers, IFN-β and IFN-α can induce a potent RB1-mediated G1/G0 growth arrest, implying that they are more sensitive to an IFN-based therapy. For solid tumors, however, adequate IFN-β or -α concentrations at the tumor sites to induce a significant cancer cell-intrinsic effect or immunity is a key for therapy success. Previously, clinical treatment for a broad range of solid tumors with IFN proteins, e.g., subcutaneous or intramuscular administration of un-PEGylated IFNs at millions of units/m2 multiple times per week, was generally not successful. This was likely due to an insufficient IFN level at the tumor site to induce an antitumor effect due to the rapid protein clearance after the treatment (20). Intratumor gene therapy with a replication-defective adenoviral vector encoding human IFN-β gene overcame the issue and led to a remarkable antitumor effect (20, 106). The recent approval of a non-replicating adenovirus encoding IFN-α 2b for high-risk Bacillus Calmette-Guérin-unresponsive non-muscle invasive bladder cancer by the US Food and Drug Administration (FDA) highlights the importance of sufficient local IFN levels in solid tumor treatment (107). Recent advances in PEGylation technologies yields IFN-based products with improved pharmacokinetic properties. The new advances may expand the opportunity to use IFN-based therapies in broader cancer indications including advanced metastasis. Ropeginterferon alfa-2b (also known as BESREMi) is a new-generation PEGylated IFN alfa-2b that is administered subcutaneously once every two or more weeks and currently in clinical development for several indications (108–112). In patients with polycythemia vera (PV), a myeloproliferative neoplasm, ropeginterferon alfa-2b provides clinically significant effects at the level of the patient’s bone marrow to selectively inhibit mutation-carrying malignant progenitor cells and increase the ratio of normal versus malignant cells (113–117). The FDA has approved ropeginterferon alfa-2b for the treatment of PV (118). Emerging data indicate that administration of ropeginterferon alfa-2b at a higher starting dose may lead to a quicker and greater level of complete hematological remission and reduction in the mutant variant allele frequency with manageable toxicities (119). Therefore, it is possible that a new generation IFN-based agent, such as ropeginterferon alfa-2b, may provide promising new treatment options for patients with a metastatic cancer at a favorable benefit-risk balance.
Concluding remarks
Slowed S phase progression and senescence entry is the prominent cell cycle effect of IFN-β in various types of human solid tumor cells, not the G1/G0 growth arrest observed in hematopoietic cancer cells (19, 39). The effect is associated with a loss of tumorigenicity in vivo (39). This article elucidates a mechanism of cell cycle-based anti-cancer surveillance. The interplay between tumor suppressor proteins IFN-β and RB1 is central in this new mechanism of surveillance for selective suppression of tumor and transformed cells, while engaging their normal cell counterparts in regular cell cycle and differentiation. The IFN-β effect in various types of tumor cells with irreversibly lost RB1 function is possibly due to its signaling inducing a p107-mediated S phase inhibition and regulating p107, p130, c-MYC and other cellular factors including p53 if its function is still intact. For the future perspectives, the development of new therapeutic means to provide sufficient concentrations of IFN-β or α at tumor sites to achieve an antitumor effect is a key for the success of using an IFN-based therapy in cancer treatment.
Author contributions
The author is the sole contributor of this work and has approved it for publication.
Acknowledgments
The author wrote the manuscript and would like to thank his colleagues for their wonderful support.
Conflict of interest
The author currently works for PharmaEssentia Corporation and serves as the Chief Medical Officer of the company.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ehrlich P. Ueber den jetzigen stand der karzinomforschung. Ned Tijdschr Geneeskd (1909) 5:273–90.
2. Ribatti D. The concept of immune surveillance against tumors: the first theories. Oncotarget (2017) 8:7175–80. doi: 10.18632/oncotarget.12739
3. Darnell JE Jr, Kerr IM, Stark GR. Jak–STAT pathways and transcriptional activation in response to IFNs and other extracellular proteins. Science (1994) 264:1415–20. doi: 10.1126/science.8197455
4. Ihle JN. The janus protein tyrosine kinase family and its role in cytokine signaling. Adv Immunol (1995) 60:1–35. doi: 10.1016/S0065-2776(08)60582-9
5. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem (1998) 67:227–64. doi: 10.1146/annurev.biochem.67.1.227
6. Mazewsky C, Perez RE, Fish EN, Platanias LC. Type I interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front Immunol (2020) 11:606456. doi: 10.3389/fimmu.2020.606456
7. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol (2005) 23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843
8. Vremec D, O’Keeffe M, Hochrein H, Fuchsberger M, Caminschi I, Lahoud M, et al. Production of interferons by dendritic cells, plasmacytoid cells, natural killer cells, and interferon-producing killer dendritic cells. Blood (2007) 109:1165–73. doi: 10.1182/blood-2006-05-015354
9. Santini SM, Di Pucchio T, Lapenta C, Parlato S, Logozzi M, Belardelli F. The natural alliance between type I interferon and dendritic cells and its role in linking innate and adaptive immunity. J Interf Cytokine Res (2002) 22:1071–80. doi: 10.1089/10799900260442494
10. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Droemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15:405–14. doi: 10.1038/nri3845
11. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol (2013) 34:67–73. doi: 10.1016/j.it.2012.10.004
12. Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev (2012) 250(1):317–34. doi: 10.1111/imr.12001
13. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16:131–44. doi: 10.1038/nrc.2016.14
14. Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D’Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst (1995) 87:581–6. doi: 10.1093/jnci/87.8.581
15. Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA (1995) 92:4562–6. doi: 10.1073/pnas.92.10.4562
16. Roos G, Leandersson T, Lundgren E. Interferon-induced cell cycle changes in human hemapoietic cell lines and fresh leukemic cells. Cancer Res (1984) 44:5358–62.
17. Lundblad D, Lundgren E. Block of a glioma cell line in s by interferon. Int J Cancer (1981) 27:749–54. doi: 10.1002/ijc.2910270604
18. Grandér D, Sangfelt O, Erickson S. How does interferon exert its cell growth inhibitory effect? Eur J Haematol (1997) 59:129–35. doi: 10.1111/j.1600-0609.1997.tb00965.x
19. Qin XQ, Runkel L, Deck C, DeDios C, Barsoum J. Interferon-beta induces s phase accumulation selectively in human transformed cells. J Interf Cytokine Res (1997) 17:355–67. doi: 10.1089/jir.1997.17.355
20. Qin XQ, Tao N, Dergay A, Moy P, Fawell S, Davis A, et al. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci USA (1998) 95:14411–6. doi: 10.1073/pnas.95.24.14411
21. Balkwill F, Taylor-Papadimitriou J. Interferons affect both G1 and S+G2 in cells stimulated from quiecence to growth. Nature (1978) 274:798–800. doi: 10.1038/274798a0
22. Einat M, Resnitzky D, Kimchi A. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature (1985) 313:597–600. doi: 10.1038/313597a0
23. Kumar R, Atlas I. Interferon a induces the expression of retinoblastoma gene product in human burkitt lymphoma daudi cells: role in growth regulation. Proc Natl Acad Sci USA (1992) 89:6599–603. doi: 10.1073/pnas.89.14.6599
24. Thomas NS, Burke LC, Bybee A, Linch DC. The phosphorylation state of the retinoblastoma (RB) protein in G0/G1 is dependent on growth status. Oncogene (1991) 6:317–22.
25. Kimchi A. Cytokine triggered molecular pathways that control cell cycle arrest. J Cell Biochem (1992) 50:1–9. doi: 10.1002/jcb.240500102
26. Tiefenbrun N, Melamed D, Levy N, Resnitzky D, Hoffman I, Reed SI, et al. Alpha interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest. Mol Cell Biol (1996) 16(7):3934–44. doi: 10.1128/MCB.16.7.3934
27. Sangfelt O, Erickson S, Einhorn S, Grandér D. Induction of Cip/Kip and Ink4 cyclin dependent kinase inhibitors by interferon-α in hematopoietic cell lines. Oncogene (1997) 14:415–23. doi: 10.1038/sj.onc.1200832
28. Thomas NSB, Pizzey AR, Tiwari S, CD W, Yang J. p130, p107, and pRb are differentially regulated in proliferating cells and during the cell cycle arrest by alpha interferon. J Biol Chem (1998) 273:23659–67. doi: 10.1074/jbc.273.37.23659
29. Matsuoka M, Tani K, Asano S. Interferon-α-induced G1 phase arrest through up-regulated expression of CDK inhibitors, p19Ink4D and p21Cip1 in mouse macrophages. Oncogene (1998) 16:2075–86. doi: 10.1038/sj.onc.1201745
30. Sangfelt O, Erickson S, Castro J, Heiden T, Gustafsson A, Einhorn S, et al. Molecular mechanisms underlying interferon-alpha –induced G0/G1 arrest: CKI-mediated regulation of G1 cdk-complex and activation of pocket proteins. Oncogene (1999) 18:2798–810. doi: 10.1038/sj.onc.1202609
31. DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, et al. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell (1989) 22:1085–95. doi: 10.1016/0092-8674(89)90507-2
32. Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell (1989) 58:1097–105. doi: 10.1016/0092-8674(89)90508-4
33. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin d to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin d-dependent kinase CDK4. Genes Dev (1993) 7(3):331–42.
34. Sherr CJ. D-type cyclins. Trends Biochem Sci (1995) 20:187–90. doi: 10.1016/S0968-0004(00)89005-2
35. Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclin D1 and e with an inducible system. Mol Cell Biol (1994) 14:1669–79. doi: 10.1128/MCB.14.3.1669
36. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol (1998) 18:753–61. doi: 10.1128/MCB.18.2.753
37. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell (1999) 98:859–69. doi: 10.1016/S0092-8674(00)81519-6
38. Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin d activates the Rb tumor suppressor by mono-phosphorylation. eLife (2014) 3:e02872. doi: 10.7554/eLife.02872
39. Kaynor C, Xin M, Wakefield J, Barsoum J, Qin XQ. Direct evidence that IFN-beta functions as a tumor-suppressor protein. J Interf Cytokine Res (2002) 22:1089–98. doi: 10.1089/10799900260442511
40. Wittling MC, Cahalan SR, Levenson EA, Rabin RL. Shared and unique features of human interferon-beta and interferon-alfa subtypes. Front Immunol (2021) 11:605673. doi: 10.3389/fimmu.2020.605673
41. Isaacs A, Lindenmann J. Virus interferon I. Interferon Proc R Soc Lond B Biol Sci (1957) 147:258–67.
42. McNab F, Mayer-Barber K, Sher A. O’Garra a. Type I interferons Infect disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
43. Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi: 10.1038/nri2314
44. Fernandez M, Quiroga JA, Carreno V. Hepatitis b virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins.J Gen Virol (2003) 84:2073–82. doi: 10.1099/vir.0.18966-0
45. King C, Sprent J. Dual nature of type I interferons in SARS-CoV-2-Induced inflammation. Trends Immunol (2021) 42:312–22. doi: 10.1016/j.it.2021.02.003
46. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis c: a randomized trial. Lancet (2001) 358:958–65. doi: 10.1016/S0140-6736(01)06102-5
47. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis c virus infection. N Engl J Med (2002) 347:975–82. doi: 10.1056/NEJMoa020047
48. Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon Alfa-2a HBeAg-positive chronic hepatitis b study group: peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis b. N Engl J Med (2005) 352:2682–95. doi: 10.1056/NEJMoa043470
49. Bonino F, Marcellin P, Lau GK, Hadziyannis S, Jin R, Piratvisuth T, et al. Peginterferon Alfa-2a HBeAg-negative chronic hepatitis b study group: predicting response to peginterferon alpha-2a, lamivudine and the two combined for HBeAg-negative chronic hepatitis b. Gut (2007) 56:699–705. doi: 10.1136/gut.2005.089722
50. Huang YW, Hsu CW, Lu SN, Yu ML, Su CW, Su WW, et al. Ropeginterferon alfa-2b every 2 weeks as a novel pegylated interferon for patients with chronic hepatitis b. Hepatol Int (2020) 14:997–1008. doi: 10.1007/s12072-020-10098-y
51. Lin HH, Hsu SJ, Lu SN, Chuang WL, Hsu CW, Chien RN, et al. Ropeginterferon alfa-2b in patients with genotype 1 chronic hepatitis c: pharmacokinetics, safety, and preliminary efficacy. JGH Open (2021) 5:929–40. doi: 10.1002/jgh3.12613
52. Miyachi N, Zagrijtschuk O, Kang L, Yonezu K, Qin A. Pharmacokinetics and pharmacodynamics of ropeginterferon alfa-2b in healthy Japanese and Caucasian subjects after single subcutaneous administration. Clin Drug Investig (2021) 41:391–404. doi: 10.1007/s40261-021-01026-5
53. Varmus H. How tumor virology evolved into cancer biology and transformed oncology. Annu Rev Cancer Biol (2017) 1:1–18. doi: 10.1146/annurev-cancerbio-050216-034315
54. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol Engl (2016) 16:112–23. doi: 10.1038/nri.2015.9
55. Gosain R, Abdou Y, Singh A, Rana N, Puzanov I, Ernstoff MS. COVID-19 and cancer: a comprehensive review. Curr Oncol Rep (2022) 22:53. doi: 10.1007/s11912-020-00934-7
56. Qin A, Huang YW, Validated CPJA. New-generation pegylated interferon therapy for chronic hepatitis B and possibly D. CTGH (2022) 4(1):343–5.
58. Thomas L. Delayed hypersensitivity in health and disease. In: Lawrence HS, editor. Cellular and humoral aspects of the hypersensitive states (1959) vol. 1959 . New York: Hoeber-Harper. p. 529–32.
59. von Locquenghien M, Rozalén C, Celià-Terrassa T. Interferons in cancer immunoediting: sculpting metastasis and immunotherapy response. J Clin Invest. (2021) 131(1):e143296. doi: 10.1172/JCI143296
60. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
61. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi: 10.1038/ni1102-991
62. Qin XQ, Beckham C, Brown JL, Lukashev M, Barsoum J. Human and mouse IFN-β gene therapy exhibits different anti-tumor mechanisms in mouse models. Mol Ther (2001) 4:356–64. doi: 10.1006/mthe.2001.0464
63. Brown JL, Barsoum J, Qin XQ. CD4+ T helper cell-independent antitumor response mediated by murine IFN-beta gene delivery in immunocompetent mice. J Interferon Cytokine Res (2002) 22:719–28. doi: 10.1089/10799900260100222
64. Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity (2018) 48:399–416. doi: 10.1016/j.immuni.2018.03.004
65. Swaminathan S, Hansen AS, Heftdal LD, Dhanasekaran R, Deutzmann A, Fernandez WDM, et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat Commun (2020) 11(1):2860. doi: 10.1038/s41467-020-16447-7
66. Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, et al. Repression of the type I interferon pathway underlies MYC- and KRAS-dependent evasion of NK and b cells in pancreatic ductal adenocarcinoma. Cancer Discovery (2020) 10(6):872–87. doi: 10.1158/2159-8290.CD-19-0620
67. Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell (2017) 171(6):1284–1300.e21. doi: 10.1016/j.cell.2017.10.022
68. Manuel O, Diaz MO, Rubin CM, Harden A, Ziemin S, Larson RA, et al. Deletions of interferon genes in acute lymphoblastic leukemia. N Engl J Med (1990) 322:77–82. doi: 10.1056/NEJM199001113220202
69. Cowan JM, Halaban R, Francke U. Cytogenetic analysis of melanocytes from premalignant nevi and melanomas. J Natl Cancer Inst (1988) 80:115964. doi: 10.1093/jnci/80.14.1159
70. Henn W, Werner M, Fischer H, Zang KD. Evolution of a 9p- marker in human glioblastoma cell lines. Cytogenet Cell Genet (1987) 46:628.
71. Brickelmaier M, Carmillo A, Goelz S, Barsoum J, Qin XQ. Cytotoxicity of combinations of IFN-beta and chemotherapeutic drugs. J Interf Cytokine Res (2022) 22:873–80. doi: 10.1089/107999002760274881
72. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell (1995) 81:323–30. doi: 10.1016/0092-8674(95)90385-2
73. Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet (2001) 10:699–703. doi: 10.1093/hmg/10.7.699
74. Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell (1991) 65:1053–61. doi: 10.1016/0092-8674(91)90557-F
75. Qin XQ, Livingston DM, Kaelin WG Jr, Adams PD. Deregulated E2F-1 transcription factor expression leads to s-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA (1994) 91:10918–22. doi: 10.1073/pnas.91.23.10918
76. Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin WG Jr. The transcription factor E2F1 is a downstream target of Rb action. Mol Biol Cell (1995) 15:742–55. doi: 10.1128/MCB.15.2.742
77. Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, et al. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Gens Dev (1998) 12:95–106. doi: 10.1101/gad.12.1.95
78. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol (2002) 3:11–20. doi: 10.1038/nrm714
79. Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene (1999) 18:7873–82. doi: 10.1038/sj.onc.1203244
80. Jacks T, Fazeli A, Schmitt E, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature (1992) 359:295–300. doi: 10.1038/359295a0
81. Lee EY, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, et al. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev (1994) 8:2008–21. doi: 10.1101/gad.8.17.2008
82. Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature (1998) 391(6667):597–601. doi: 10.1038/35404
83. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer (2011) 2(4):466–74. doi: 10.1177/1947601911408889
84. Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ (2022) 29:946–60. doi: 10.1038/s41418-022-00988-z
85. Stengel KR, Thangavel C, Solomon DA, Angus SP, Zheng Y, Knudsen ES. Retinoblastoma/107/p130 pocket proteins. J Biol Chem (2009) 284:19265–71. doi: 10.1074/jbc.M808740200
86. Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet (1998) 14:223–9. doi: 10.1016/S0168-9525(98)01470-X
87. Hurford RK Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev (1997) 11:1447–63. doi: 10.1101/gad.11.11.1447
88. Paggi MG, Giordano A. Who is the boss in the retinoblastoma family? the point of view of The little brother. Cancer Res (2001) 61:4651–4.
89. Ginsberg D, Vairo G, Chittenden T, Xiao ZX, Xu G, Wydner KL, et al. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev (1994) 8:2665–79. doi: 10.1101/gad.8.22.2665
90. Beijersbergen Ri, Kerkhoven R, Zhu L, Carlde L, PM V, Bemards R. E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes Dev (1994) 8:2680–90. doi: 10.1101/gad.8.22.2680
91. Ewen M, Faha B, Harlow E, Livingston D. Interaction of p107 with cyclin a independent of complex formation with viral oncoproteins. Science (1992) 255:85–7. doi: 10.1126/science.1532457
92. Ewen ME, Xing YG, Lawrence JB, Livingston DM. Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell (1991) 66:1155–64. doi: 10.1016/0092-8674(91)90038-Z
93. Wirt SE, Sage J. p107 in the public eye: an Rb understudy and more. Cell Div (2010) 5:9. doi: 10.1186/1747-1028-5-9
94. Jiang H, Karnezis AN, Tao M, Guida PM, Zhu L. pRB and p107 have distinct effects when expressed in pRB-deficient tumor cells at physiologically relevant levels. Oncogene (2000) 19:3878–87. doi: 10.1038/sj.onc.1203722
95. Jonak GJ, Knight E Jr. Selective reduction of c-myc mRNA in daudi cells by human beta interferon. Proc Natl Acad Sci USA (1984) 81:1747–50. doi: 10.1073/pnas.81.6.1747
96. Sarkar D, Park E, Fisher P. Defining the mechanism by which IFN-β dowregulates c-myc expression in human melanoma cells: pivotal role for human polynucleotide phosphorylase (hPNPaseold-35). Cell Death Differ (2006) 13:1541–53. doi: 10.1038/sj.cdd.4401829
97. Lehmann B, Brooks AM, Paine MS, Chappell WH, McCubrey JA, Terrian DM. Distinct roles for p107 and p130 in Rb-independent cellular senescence. Cell Cycle (2008) 7:1262–8. doi: 10.4161/cc.7.9.5945
98. Kapic A, Helmbold H, Reimer R, Klotzsche O, Deppert W, Bohn W. Cooperation between p53 and p130(Rb2) in induction of cellular senescence. Cell Death Differ (2006) 13:324–34. doi: 10.1038/sj.cdd.4401756
99. Jackson JG, Pereira-Smith OM. Primary and compensatory roles for RB family members at cell cycle gene promoters that are deacetylated and downregulated in doxorubicin-induced senescence of breast cancer cells. Mol Cell Biol (2006) 26:2501–10. doi: 10.1128/MCB.26.7.2501-2510.2006
100. Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-myc inactivation. Proc Natl Acad Sci USA (2007) 104:13028–33. doi: 10.1073/pnas.0701953104
101. Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene (2008) 27:6623–34. doi: 10.1038/onc.2008.258
102. Li B, Zhang G, Wang Z, Yang Y, Wang C, Fang D, et al. C-myc-activated USP2-AS1 suppresses senescence and promotes tumor progression via stabilization of E2F1 mRNA. Cell Death Dis (2021) 12:1006. doi: 10.1038/s41419-021-04330-2
103. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature (2003) 424(6948):516–23. doi: 10.1038/nature01850
104. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer (2022) 22:340–55. doi: 10.1038/s41568-022-00450-9
105. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle (2012) 11(19):3599–610. doi: 10.4161/cc.21884
106. Qin A. A favorable benefit-risk balance maybe expected with replication-defective adenovirus-mediated interferon gene therapy for cancer treatment. Hum Gene Ther (2023) 34:339–40. doi: 10.1089/hum.2023.028
107. FDA Approves first gene therapy for the treatment of high-risk, non-muscle-invasive bladder cancer. Washington, DC: FDA. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-high-risk-non-muscle-invasive-bladder-cancer (Accessed 19Jan2023).
108. Verstovsek S, Komatsu N, Gill H, Jin J, Lee S-E, Hou H-A, et al. SURPASS-ET: phase III study of ropeginterferon alfa-2b versus anagrelide as second-line therapy in essential thrombocythemia. Future Oncol (2022) 18(27):2999–3009. doi: 10.2217/fon-2022-0596
109. Huang YW, Qin A, Tsai CY, Chen PJ. Novel pegylated interferon for the treatment of chronic viral hepatitis. Viruses (2022) 14:1128. doi: 10.3390/v14061128
110. Chen CY, Chuang WL, Qin A, Zhang WH, Zhu LY, Zhang GQ, et al. A phase 3 clinical trial validating the potency and safety of an innovative, extra-long-acting interferon in chronic hepatitis c. JGH Open (2022) 6:11. doi: 10.1002/jgh3.12825
111. Chen KY, Lee KY, Qin A, Luo CS, Yeh YK, Zheng JQ, et al. Clinical experience with ropeginterferon alfa-2b in the off-label use for the treatment of COVID-19 patients in Taiwan. Adv Ther (2022) 39:2. doi: 10.1007/s12325-021-01998-y
112. Jin J, Qin A, Zhang L, Shen W, Wang W, Zhang J, et al. A phase 2 trial to assess the efficacy and safety of ropeginterferon alfa-2b in Chinese patients with polycythemia Vera. Future Oncol (2023). doi: 10.2217/FON-2022-1141
113. Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, et al. Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood (2015) 126:1762–9. doi: 10.1182/blood-2015-04-637280
114. Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol (2020) 7:e196–208. doi: 10.1016/S2352-3026(19)30236-4
115. Kiladjian JJ, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b. Leukemia (2022) 36(5):1408–11. doi: 10.1038/s41375-022-01528-x
116. Edahiro Y, Ohishi K, Gotoh A, Takenaka K, Shibayama H, Shimizu T, et al. Efficacy and safety of ropeginterferon alfa-2b in Japanese patients with polycythemia vera: an open-label, single-arm, phase 2 study. Int J Hematol (2022) 116:215–27. doi: 10.1007/s12185-022-03341-9
117. Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, et al. Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells. Vitro vivo. Blood Cancer J (2018) 8:94. doi: 10.1038/s41408-018-0133-0
118. FDA US. FDA Approves treatment for rare blood disease [Internet] (2021). Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease (Accessed 2021 Nov 12).
119. Qin A, Urbansky RW, Yu L, Ahmed T, Mascrenhas J. An alternative dosing strategy for ropeginterferon alfa-2b may help improve outcomes in myeloproliferative neoplasms: an overview of previous and ongoing studies with perspectives on the future. Front Oncol (2023) 13:1109866. doi: 10.3389/fonc.2023.1109866
Keywords: tumorigenicity, cell cycle-based anti-cancer surveillance, tumor suppressor protein-mediated mechanism, interferon-beta (IFN-β), retinoblastoma protein RB1
Citation: Qin A (2023) An anti-cancer surveillance by the interplay between interferon-beta and retinoblastoma protein RB1. Front. Oncol. 13:1173467. doi: 10.3389/fonc.2023.1173467
Received: 02 March 2023; Accepted: 05 April 2023;
Published: 27 April 2023.
Edited by:
Daitoku Sakamuro, Augusta University, United StatesReviewed by:
Srividya Swaminathan, City of Hope National Medical Center, United StatesCopyright © 2023 Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert Qin, albert_qin@pharmaessentia.com