 Muchun Li
Muchun Li Xiaozhen Zhang
Xiaozhen Zhang Xueli Bai
Xueli Bai Tingbo Liang
Tingbo Liang- 1Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Zhejiang Provincial Key Laboratory of Pancreatic Disease, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Zhejiang Provincial Innovation Center for the Study of Pancreatic Diseases, Hangzhou, China
- 4Zhejiang Provincial Clinical Research Center for the Study of Hepatobiliary & Pancreatic Diseases, Hangzhou, China
- 5Cancer Center, Zhejiang University, Hangzhou, China
Tumor necrosis factor (TNF) receptor type II (TNFR2) is expressed in various tumor cells and some immune cells, such as regulatory T cells and myeloid-derived suppressing cells. TNFR2 contributes a lot to the tumor microenvironment. For example, it directly promotes the occurrence and growth of some tumor cells, activates immunosuppressive cells, and supports immune escape. Existing studies have proved the importance of TNFR2 in cancer treatment. Here, we reviewed the activation mechanism of TNFR2 and its role in signal transduction in the tumor microenvironment. We summarized the expression and function of TNFR2 within different immune cells and the potential opportunities and challenges of targeting TNFR2 in immunotherapy. Finally, the advantages and limitations of TNFR2 to treat tumor-related diseases are discussed, and the problems that may be encountered in the clinical development and application of targeted anti-TNFR2 agonists and inhibitors are analyzed.
Introduction
TNFR2-Related Signaling Pathways in Cancer
Tumor necrosis factor (TNF) plays a role in many pathophysiological processes, especially in the different periods of cell growth, inflammatory and immune responses, as well as tumor progression and metastasis (1, 2). Studies show that TNF functions through complicated signaling pathways, which affect practically any type of cell, through binding to two kinds of receptors, type I and II (TNFR1, TNFR2) (3).
TNF activates TNFR2 by recruiting a complex composed of the adapter protein. These mainly include TNF receptor-associated factor 2 (TRAF2), TRAF2-associated proteins, and apoptosis-related makers such as cIAP1/2. This process leads to the depletion of these compounds and affects other functions of these molecules in tumor cells (4–6). For example, the depletion of the adapter TRAF2-cIAP1/2 complexes in the cytoplasmic matrix can antagonize TNFR1-mediated the classical NF-κB pathway (7). Interestingly, the depletion of these complexes can lead to the decrease of NF-κB-related expression, causing the increase of NIK kinase expression and activating the alternative NF-κB pathway (8). TNFR2, through PI3K/Akt, can also induce phosphorylation of IKKβ and lead to the stimulation of the canonical NF-κB pathway (2). However, only TNFR2 binding to the cell membrane-bound TNF activates the NF-κB-induced non-canonical pathway (2). Moreover, TNFR2 binds to the non-receptor tyrosine kinase BMX constitutively, resulting in the stimulation of Akt pathways and the regulation of TNFR2-mediated NF-κB signaling (9, 10). Unlike TNFR1, which is TRAF2-dependent, TNFR2 induces BMX activation independent of TRAF2. BMX interacts with TNFR2 not through ligand connection at first, but a direct connection with different BMX domains at the C-terminal domain of TNFR2, which doesn’t overlap with the TRAF2-binding sequence (9).
TNFR2 is not only expressed on many different types of tumors and malignant cells but is also enriched in the tumor microenvironment (11–13). TNF regulates different signaling pathways in the tumor microenvironment through TNFR2 and participates in the occurrence and growth of tumors (Figure 1). Intriguingly, mTNF can not only act as a ligand but also a receptor and can transmit signals in both directions. Transmembrane TNF, in some cells, can combine with sTNFR2 to deliver the reverse signal to the target cell (14). In addition, the transmembrane TNF can also be used as a receptor to deliver the signal back to the cell after binding to its natural receptor (15, 16). TNFR2 can prevent cancerous cells from DNA damage through the Akt signaling pathway in breast cancer. At the same time, it activates NF-κB through MAPK, leading to rapid tumor cell growth (17, 18). mTNF/TNFR2 signaling stimulates reciprocal PI3K/Akt signaling, thereby increasing the phosphorylation of STAT5, which impairs Th17 differentiation (19). In angiogenesis, the PI3K/Akt pathway is activated by TNFR2 and then Etk is recruited to form a complex of TNFR2, Etk, and VEGFR2, which can influence cell growth and proliferation (20, 21). In immune-mediated inflammatory bowel disease models, TNFR2 can lead to tight junction dysregulation through activation of MLCK, which leads to the decrease in cell apoptosis-related defenses and the induction of colitis (22). Moreover, TNFR2 mediates JNK signaling via AIP1 association, an adaptor molecule specific for JNK signaling, independent of TRAF2, regulating vascular endothelial cell function (23). TNFR2 can also induce BIRC3/cIAP2 transcripts dependent on TRAF1 and decrease the transcription and expression of NKp46/NCR1, leading to tumor deterioration in mice and adverse outcomes in patients with gastrointestinal stromal tumors (24). In macrophages, TNFR2 sensitizes pro-inflammatory signals by activating p38/MAPK and NF-κB signaling pathways and triggering TRAF2 degradation signals (25). In gastric lymphoma, miR-17 accelerates tumor development by influencing the HSP60/TNFR2 pathway (26). Meng et al. found that TNFR2 activates YAP signaling by regulating heterogeneous nuclear ribonucleoprotein K (hnRNPK), which promotes primary liver cancer development in hepatic progenitor cells (27).
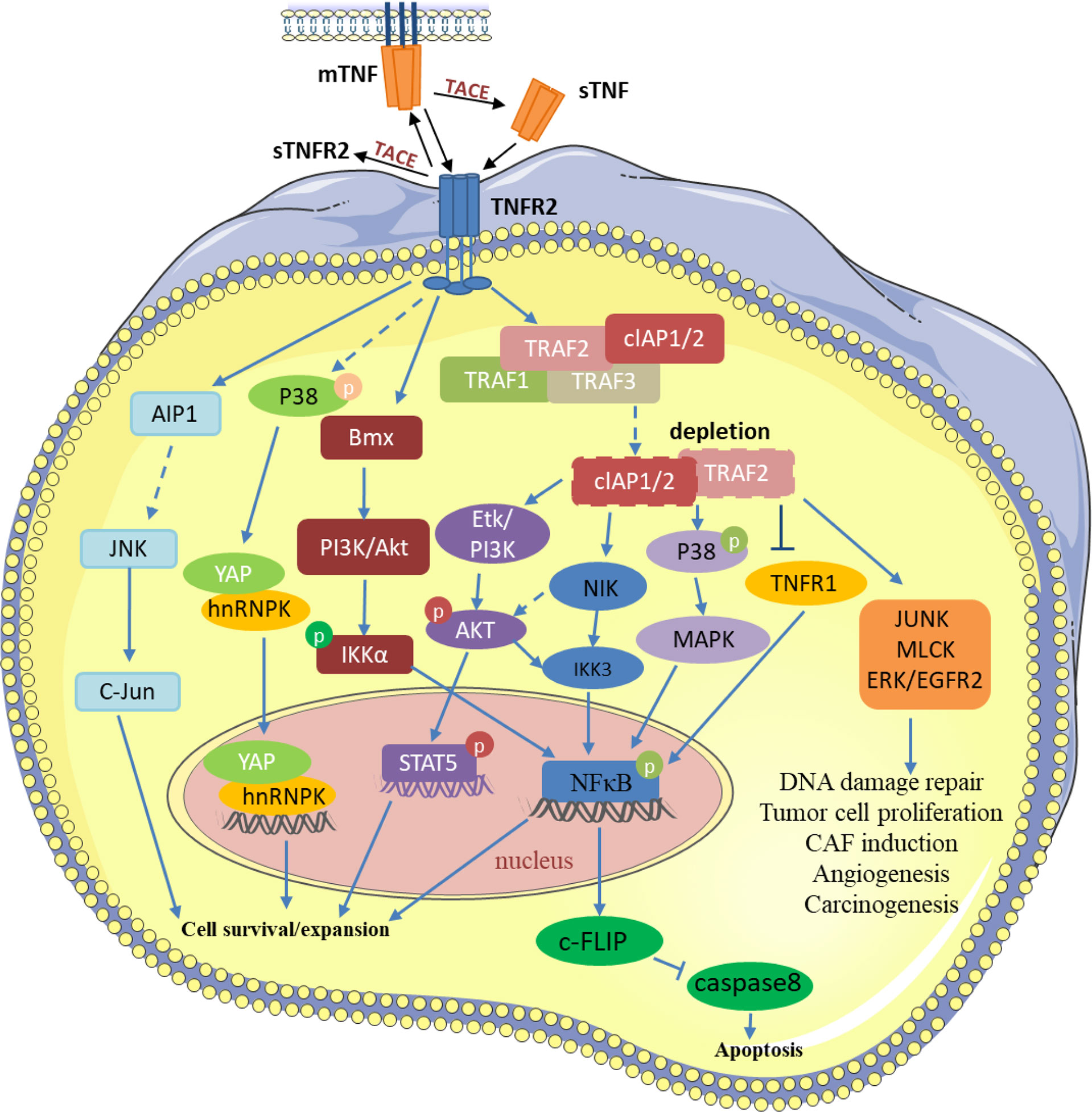
Figure 1 TNF/TNFR2 participates in various processes of tumor development by regulating different signaling pathways in the tumor and tumor microenvironment. TRAF2 and TRAF2-related proteins, such as TRAF1 and cIAP1/2, are recruited to activate TNFR2. Then, TNFR2 activates NF-κ B, STAT5, YAP, and other transcription factors through different pathways to induce the transcription of its target genes, thereby inhibiting tumor cell apoptosis and promoting the development of tumor cells. TNFR2 also participates in various changes in the tumor microenvironment through signal transduction such as JUNK, MLCK, and EGFR2. P, Phosphorylation.
TNF-α mediates distinct signaling pathways through two structurally distinct receptors, TNFR1 and TNFR2, and thus has distinct functions in the tumor environment. Since both TNFR1 and TNFR2 bind cIAP1/2 and TRAF2 and the activation order of TNFR1 and TNFR2 ultimately determines the life and death of tumor cells, the mechanism and complexity of its signaling pathway obviously need to be further explored. Previously, owing to the extensive non-specific effects of TNF, this signaling pathway was abandoned as the main treatment option during clinical anti-tumor therapy. Through the recent increased attention to TNFR2, we found that whereas TNFR1 effectively promotes cancer cell death by activating NF-κB signaling, the activation of TNFR2 on tumor cells and immunosuppressive cells might be detrimental to anticancer therapy. Therefore, we need more specific therapeutic regimens to target TNFR1 and TNFR2, rather than TNF, which can effectively avoid the treatment side effects caused by the non-specific action of TNF and make cancer treatment more efficient.
Mechanisms of TNFR2 Activation
TNF is a type II protein that can be translocated from the membrane (mTNF) and take a soluble form (sTNF) in the cytoplasm after being sheared by the TNF-converting enzyme (TACE) (1). The TNF homology domain (THD) exists in the above-mentioned two forms of TNF to control trimer constitution as well as receptor binding (1). The THD is the key component of the TNF superfamily, while the cysteine-rich domain (CRD) is an important structural feature (28, 29). TNFR1 and TNFR2 are typical members of the TNF receptor superfamily, and they can be activated by mTNF. However, sTNF can selectively activate TNFR1, and not TNFR2, to trigger efficient receptor signaling despite high-affinity binding (30). Therefore, the activation of TNFR2 is largely dependent on the transmembrane TNF expressed on the neighboring cells. TNFR1 has a cytoplasmic death domain (DD) and it binds to the proteins containing a DD, leading to pro-inflammatory signaling, as well as cytotoxic-related signaling pathway activation. However, TNFR2 possesses just one TRAF2 binding site but no DD (31). Thus, TNFR2 recruits the TRAF1/TRAF2-cIAP1/2 complex and activates an alternative NF-κB pathway, as well as various kinases (1).
TNFR2 can auto-associate in the absence of TNF and locates on the first N-terminal CRD position of the molecule that does not bind to the ligand (32). This part of the TNF receptor is called the pre-ligand binding assembly domain, which may play a role during ligand binding. It also initiates the formation of the active receptor (32). Studies have shown that TNFR2 dimers can be formed closer to TNF rather than monomeric TNFR2 (33). There are three molecules of TNFR2 that interact with a TNF trimer in a parallel way (34). Notably, the TNF3-TNFR23 complex cannot independently and accurately activate TNFR2. Therefore, more than one TNF3-TNFR23 complex interacts to stimulate intracellular signaling cascades. Three homologous TRAF2 adaptor proteins form a polymer, and each TRAF2 interacts with the C-terminus of TNFR2 (35). Because the TRAF2 trimer only interacts with a single cIAP1 or cIAP2 molecule, it is necessary to form multiple (TNF-TNFR2-TRAF2)3-cIAP1/2 complexes to ensure the activation of cIAP1/2 molecules. It is important to the first step for TNFR2 to perform its function (36). In addition to the highly complex binding to TRAF2, TNFR2 can also bind to other proteins, such as adaptor proteins like BAX and AIP (9, 23). As the expression of mTNF on adjacent cell membranes increases, the mTNF-TNFR2 interaction strengthens, which further activates TNFR2. Instead, sTNF can also stimulate TNFR2 activation when physically linked sTNF trimers are bound by antibodies or co-expressed with an oligomerizing domain (7, 37), although the mechanism remains to be explored.
Compared with mTNF, sTNF can also interact with TNFR2 but fails to trigger effective receptor signaling. Therefore, how TNF effectively activates TNFR2 or how TNF-based TNFR2-stimulating drugs accurately distinguish between TNFR1 and TNFR2 should be clarified in vitro. Rauert et al. found that bacterially produced sTNF mutants contain large amounts of integrated trimers of ligands that can activate TNFR2. However, the corresponding eukaryotic trimeric variant of sTNF is unable to activate TNFR2. Notably, they found that the monomeric TNF variant, flag-TNC-scTNF(143N/145R), could stimulate TNFR2 specifically in the absence of oligomerization (7). In addition, Rauert et al. introduced specific mutations into the binding site of TNFR2 and mTNF with an intracellular YFP domain fusion expression plasmid that can specifically activate TNFR2 (7). Moreover, previous studies have demonstrated that mTNF-containing exosomes are capable of stimulating TNFR2 in vitro (38–40). Although the mechanism is not fully clear, TNFR2 might stimulate cells that are not in direct contact with TNF-expressing cells. Therefore, future research should focus on improving the activity of sTNF towards TNFR2 and evaluate the potential of TNF-based TNFR2-stimulating antibodies.
Soluble TNFR2
Membrane-bound TNFR2 can be cleaved to soluble TNFR2 (sTNFR2) by TACE enzymes when TNFR2 trimerizes with TNF and forms a tightly clustered complex (41). Membrane-bound TNFR2 is not only immunosuppressive on Tregs but is also immunostimulatory on T effector cells (Teffs), which depends on the cell type (42). However, the function of sTNFR2 is consistently immunosuppressive (43).
Soluble TNFR2 is an indicator in the serum of patients with cancer, and it also represents the level of active TNFR2 in the TNF-stimulated cell culture medium (41). Studies have shown that IL-2, TNF, or TNFR2 agonists can quickly stimulate CD4+ T cells to produce abundant sTNFR2 in vitro (44). Furthermore, activated Tregs can release high amounts of sTNFR2 (43). It has also been reported that some pathogens can stimulate the shedding of TNFR2 mediated by IL-10, thereby inhibiting the secretion of TNF (45). At present, the neutralizing effect of soluble TNFR2 ectodomain on TNF promotes TNFR2 to have a shedding-protective function (46). In contrast, a TNFR2 antagonist can block TNF-TNFR2 binding, which may maintain or decrease the expression of mTNFR2 on Tregs, and also affect the expression of sTNFR2 cleaved from Tregs (47). In addition, Torrey et al. found that pre-diagnosis plasma sTNFR2 levels are significantly related to increased overall mortality in colorectal cancer (48). In malignant ovarian tumors, sTNFR2 affects tumor grade and differentiation (49). Thus, we can speculate that TNFR2 antagonistic antibody therapy can be applied to patients with cancer with bad survival and a high level of serum sTNFR2.
The Function of TNFR2 in the Tumor Microenvironment
Expression and Clinical Features of TNFR2 in Various Cancers
To elucidate the potential functions and the clinical relevance of TNFR2 in various cancers better, we investigate the TNFR2 expression profiles in 30 kinds of human cancers. The research methods included Gene Expression Profiling Interactive Analysis (GEPIA: http://gepia.cancer-pku.cn/) and Tumor and Immune System Interaction Database (TISIDB: http://cis.hku.hk/TISIDB/index.php) (Figures 2, 3). As shown in Figure 2A, compared to normal tissues, TNFR2 is expressed at a higher level in pancreatic adenocarcinoma (PAAD), glioblastoma multiforme (GBM), brain lower-grade glioma (LGG), kidney renal clear cell carcinoma (KIRC), stomach adenocarcinoma, and testicular germ cell tumors. Meanwhile, the expression of TNFR2 is decreased in other tumors, including breast invasive carcinoma, lung adenocarcinoma (LUAD), and lymphoid neoplasm diffuse large B-cell lymphoma.
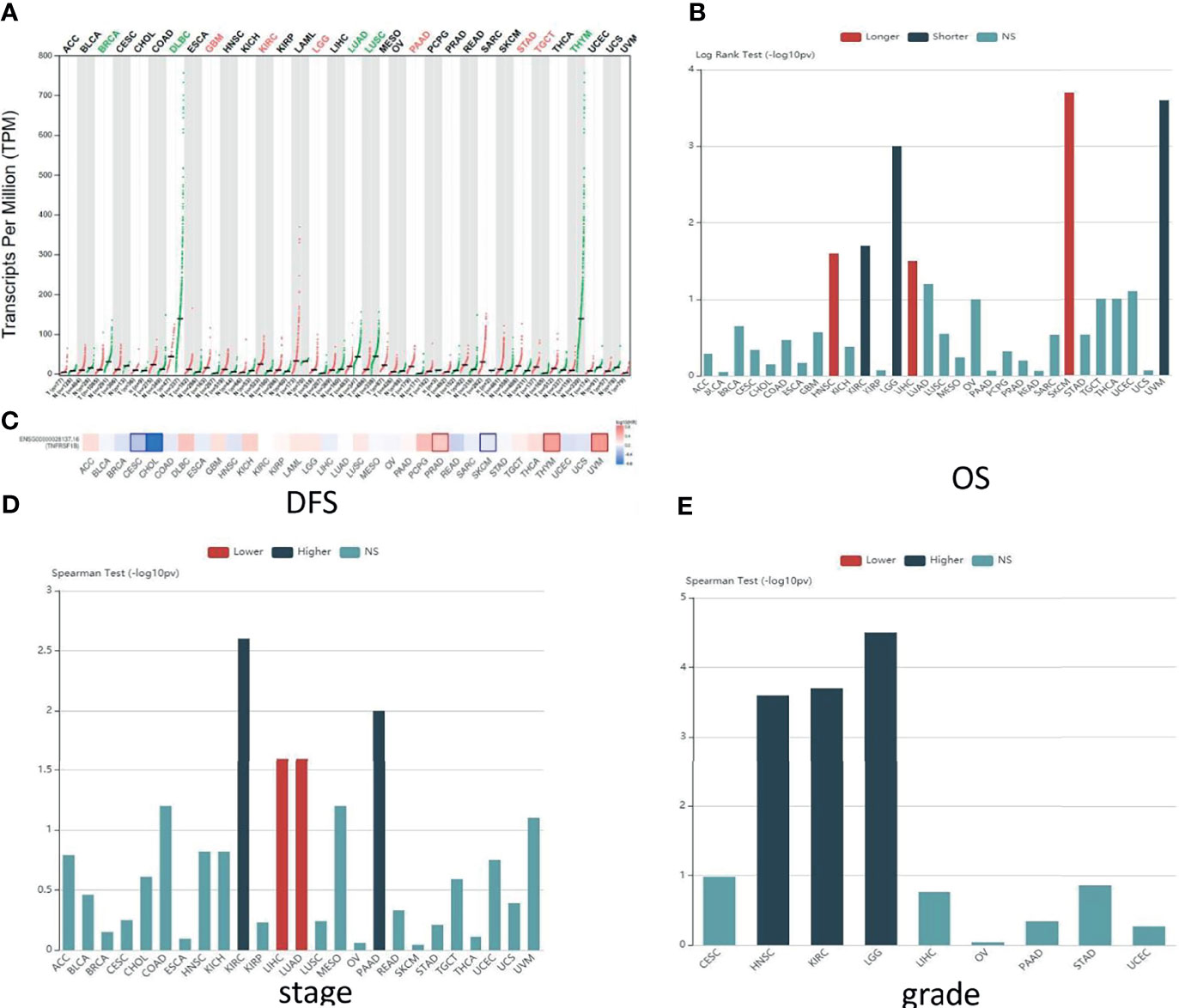
Figure 2 Predictions of TNFR2 function in various cancers. (A) The TNFR2 expression profile across all tumor samples and paired normal tissues from the TCGA database through GEPIA. Each dot represents expression in samples. The red font represents the significantly high expression of TNFR2 in the tumor, and the green font represents the significantly low expression (P < 0.05). (B) Analysis of the relationship between the expression of TNFR2 and the overall survival (OS) of various cancer patients from the TCGA database through TISIDB. The red bar (longer) indicates a correlation between higher TNFR2 expression and better overall survival rates for cancer patients; the green bar (shorter) indicates a correlation between higher TNFR2 expression and decreased cancer patient overall survival rates; the blue bar (NS) indicates that the TNFR2 expression level is not correlated with the overall survival rate of cancer patients. (C) TNFR2 expression is related to patient disease-free survival (DFS) in various cancers. Data were obtained from the TCGA database through GEPIA (P < 0.05). The red represents a positive correlation between TNFR2 expression and disease-free survival in cancer patients, and the blue represents a negative correlation. (D, E) Using large-scale RNA-Seq data sets of multiple cancer types from the TCGA database, we analyzed the relationship between TNFR2 expression and tumor stage and grade through TIBIS prediction. The red bar (lower) indicates a correlation between higher TNFR2 expression and a lower stage or grade of cancer; the green bar (higher) indicates a correlation between higher TNFR2 expression and an increased cancer stage or grade; the blue bar (NS) indicates that the TNFR2 expression level is not correlated with the stage or grade of cancer.
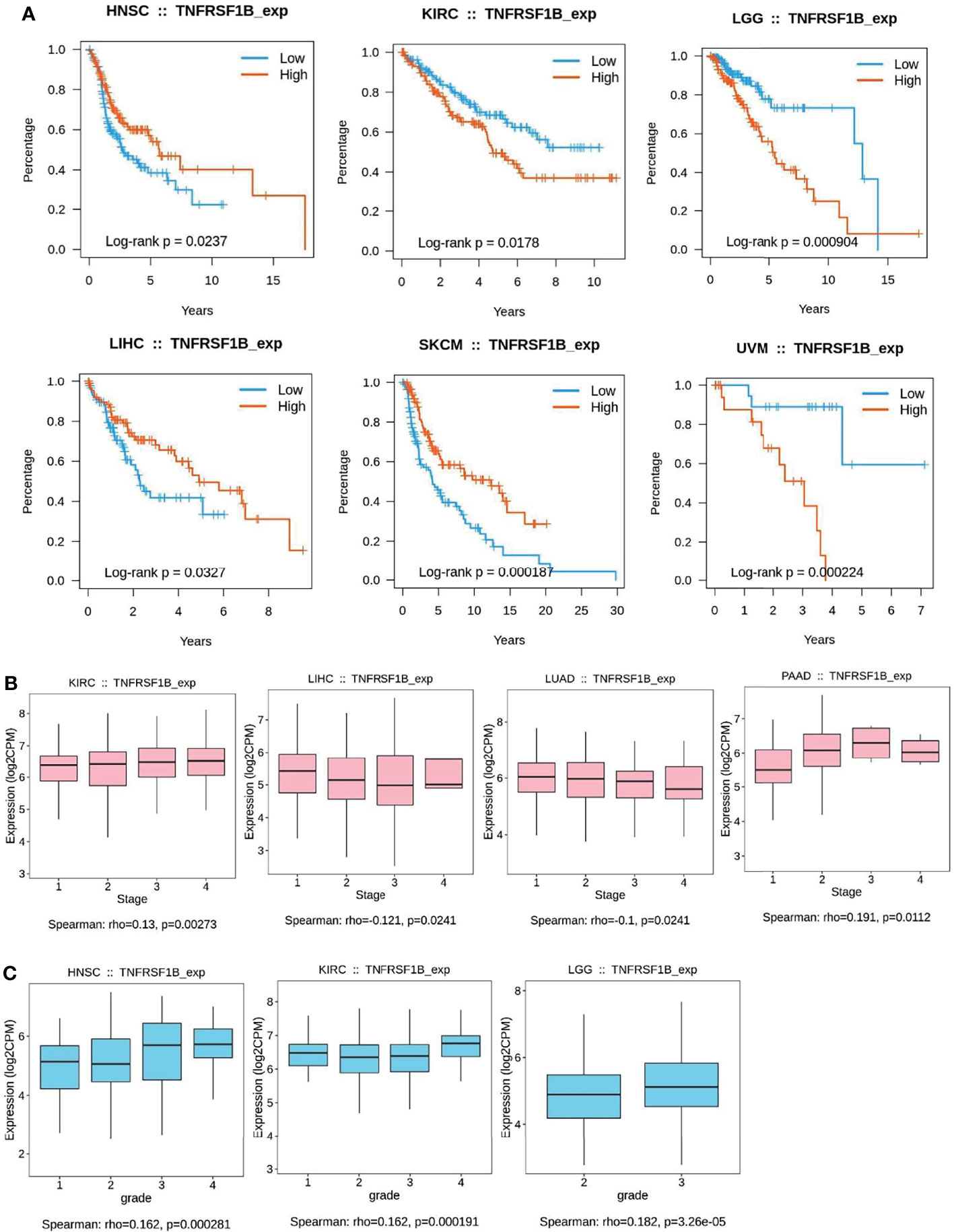
Figure 3 Kaplan–Meier curves to demonstrate the clinic pathological significance of TNFR2. (A) Analysis of the relationship between the expression of TNFR2 and the overall survival (OS) of various cancer patients from TCGA database through TISIDB (P < 0.05). (B, C) Analysis of the relationship between TNFR2 expression and tumor stage and grade through TIBIS prediction (P < 0.05).
To further investigate the clinic correlation between TNFR2 and the terms of prognostic and pathological features and also analyze the connection between TNFR2 level and overall survival (OS), TNM stage, disease-free survival (DFS), and tumor grade, TISIDB was used (Figures 2B–E, 3A–C). Intriguingly, the results revealed a significant association between TNFR2 expression in tumor tissues and prognostic outcome. For example, high expression of TNFR2 is associated with a worse prognostic outcome for UVM, LGG, and KIRC, whereas it leads to the opposite result in HNSC, LIHC, and SKCM (Figures 2B, 3A). Furthermore, TNFR2 expression was positively associated with the TNM stage in KIRC and PAAD, but this relationship was negatively associated with LIHC and LUAD (Figures 2D, 3B). As for tumor grade, an increased level of TNFR2 is usually related to a worse grade in HNSC, KIRC, and LGG (Figures 2E, 3C). In conclusion, the clinical results of the TCGA database indicate that TNFR2 has a crucial function during the development and progression of various cancers.
The Role of TNFR2 in Immune Cells
TNFR1 is widely expressed in almost all kinds of cells, but TNFR2 expression has limitations. TNFR2 is only expressed in subgroups of the lymphatic system, such as Tregs, endothelial cells, and myeloid-derived suppressor cells (MDSCs) (50).
TNFR2 was originally thought to be a stimulator of T cells, like other receptors of TNFRSF (51). T cells have always been a crucial target for cancer immunotherapy. Immunosuppressive tumor-infiltrating regulatory T cells (Tregs) play a major role in the stabilization of the immunosuppressive tumor microenvironment (52, 53). Tregs can not only directly help tumor cells escape the fate of apoptosis but can also make tumor cells survive by inhibiting a subset of CD8+ Teffs (54). Reportedly, the TNFR2 expression on Treg cells is superlatively suppressive (55, 56) and is related to the poor prognosis of patients (57). Moreover, activated Tregs can release a large amount of sTNFR2, which enriches the immunosuppressive mechanism of Tregs from another perspective (43). Meanwhile, TNFR2 can increase the activities and phenotypic stability of Treg cells (58). Several studies have shown that TNFR2+ Tregs promote the growth of primary tumors and tumor metastasis (58, 59). Further, in the intracellular pathway of human Tregs, TNFR2 enhances IL-2-induced proliferation of Tregs and expansion of cell numbers through the non-canonical NF-κB pathway (60). It has been reported that CD8+ Tregs can also express TNFR2 and are involved in the phenotypic stability, proliferation, activation, and inhibitory activities of CD8+ Tregs (42, 61). Although CD8+ Tregs contribute to tumor immune evasion in the tumor microenvironment (62), the mechanism by which TNFR2 mediates the function of CD8+ Tregs in cancer immune evasion remains to be further investigated. Interestingly, there more TNFR2 is expressed on Treg cells under the tumor microenvironment than that under healthy and normal conditions (63). This also provides a favorable condition for TNFR2 as a new tumor therapy target. Dadiani et al. showed that TNFR2+ tumor-infiltrating lymphocytes (TILs) are closely related to improvements in patient prognosis in triple-negative breast cancer (TNBC). This might be due to the sensitivity of Tregs to chemotherapy, leading to them being preferentially reduced during treatment (64, 65). Moreover, TNFR2+ Tregs can restrain pro-inflammatory processes in many malignancies, which is closely related to increased tumor progression (66, 67). Jiang et al. found that TNF-α can accelerate naive CD4+ T cell differentiation into Th9 cells. Moreover, TNFR2 can enhance Th9 cell growth and survival through STAT5/NF-κB pathways and increase the tumor-infiltrating capability in a mouse tumor model (68). In addition, more immunosuppressive markers are expressed in these TNFR2+ Tregs, including CTLA-4 and CD73. TNFR2+ Tregs can also express an increased amount of inhibitory immune cytokines, such as IL-10 or TGF-β, which helps them exert a stronger immunosuppressive effect (69). Therefore, we speculate that targeting this group of highly suppressive TNFR2+ Treg cells might result in the destruction of multiple immune regulatory circuits in the tumor microenvironment (70).
TNFR2 is also present on other conventional T cells, where it mostly acts as a costimulatory molecule (71, 72). Increased expression of TNFR2 on Teffs following T-cell receptor stimulation is critical not only for Teff proliferation and activation but also for the induction of activation-induced cell death (AICD) (42, 73). AICD can terminate the Teff proliferative response, which is mainly dependent on TRAF2, a downstream mediator of TNFR2 (74). Similarly, knockdown of TNFR2 impairs the proliferative capacity of conventional CD4+ T and CD8+ T cells and reduces their stimulated production of IL-2, IFN-γ, and TNFα (51, 75, 76). Furthermore, the increased release of sTNFR2 can also inhibit the anti-tumor function of Teffs (43). However, the proper chemotherapy-driven exposure of neo-antigens, such as TNFR2, on Teffs may activate them against the tumor cells (57, 77). Here, we speculate that TNFR2 may play opposite roles in Tregs and Teffs, thereby regulating the immune response in the tumor microenvironment. Therefore, we can formulate an appropriate treatment plan based on its double-sided properties to eliminate harmful immunosuppressive cells, especially TNFR2+ Treg cells, and increase the number of immune-stimulatory cells, such as TNFR2+ CD8+ T cells, thereby activating anti-tumor reactions.
In addition to T cells, it has been reported that TNFR2 could exert a suppressive or stimulatory effect in the tumor microenvironment by influencing various immune cells (Table 1), although published studies have mainly focused on the immunological co-suppressive effect of TNFR2 through immune cells. A previous study has found that TNFR2 suppresses the NK cell growth by activating the BIRC3/TRAF1 signaling pathway and promoting the immunosuppressive function of NK cells in the tumor microenvironment (24). Recent studies have shown that TNFR2 promotes MDSC generation and accumulation via increasing the level of c-FLIP and decreasing caspase-8 activity (79). Moreover, TNFR2 signaling can also affect the immunosuppressive function of mesenchymal stem cells (MSCs) (81, 82). Hu et al. also found that mTNF-α, but not sTNF-α, activates MDSCs through TNFR2, increases the production and release of immunosuppressive factors including NO, ROS, IL-10, and TGF-β, and reverses the inhibitory effect of T cell proliferation (84). TNF/TNFR2 is also a key signaling pathway that regulates the immunosuppressive function of endothelial progenitor cells (EPCs) (83). Furthermore, the activation of TNFR2 induces the p38MAPK-NF-κB pathway and induces TRAF2 protein degradation in macrophages (25). TNFR2 expressed on tumor-associated macrophages is related to the malignancy of human TNBC and participates in its metastasis (85). It is reported that TNFR2 expression coincides with the expression of IL-10, which is produced by regulatory B cells. More importantly, selective TNFR2 stimulation enhances the expression of IL-10 (80). T cells play a central role in regulating tumor-specific immune responses. Nevertheless, macrophages, MDSCs, MSCs, NK cells, EPCs, and B cells also contribute to immune regulation. Interestingly, the suppression of these immune cells is dependent on TNFR2. To fully clarify the association between TNFR2 and TILs, we further analyzed the association between TILs and the expression of TNFR2 in human cancers using the TCGA database via TISIDB across 30 cancer types. We also found that the expression of TNFR2 was significantly positively correlated with the levels of many immune cells, including NK cells, Tregs, CD8+ T cells, and MDSCs, in 30 types of cancers (Figure 4A). Thus, TNFR2 plays an important role in the tumor microenvironment through these cells. However, how TNFR2 affects the biological functions of these cells in the tumor microenvironment and the specific regulatory mechanisms remain elusive and require further exploration.

Table 1 TNFR2-mediated signaling and TNFR2 function in immune cells.
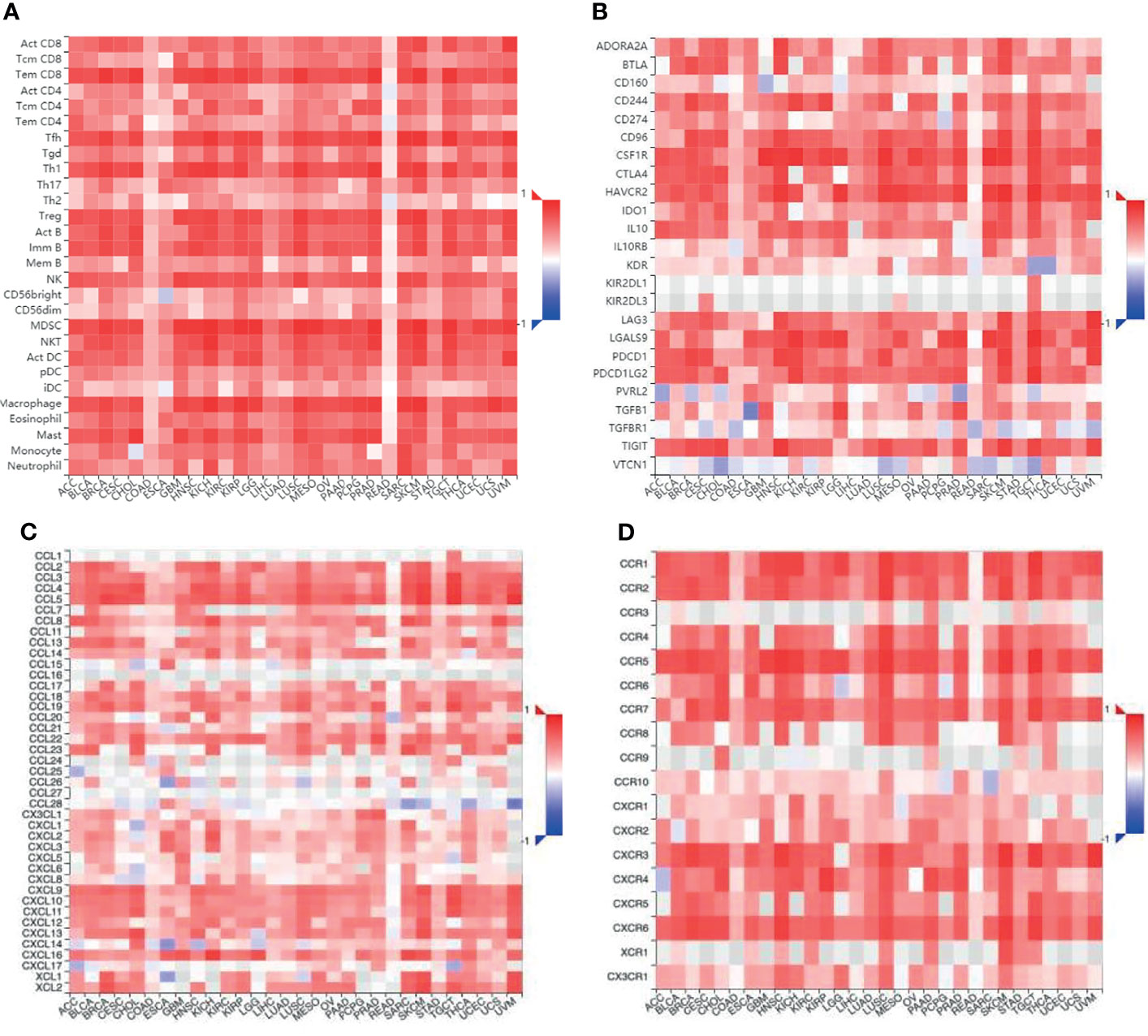
Figure 4 The relationship between different immune cells and immunomodulators and the expression of TNFR2 in various cancers. (A) Bioinformatics analysis of the correlation of TNFR2 expression and immune cell numbers. (B) The relationship between TNFR2 expression and immune inhibitors. (C, D) The correlation between TNFR2 and the expression of chemokines and their receptors. All data were obtained from the TCGA database through TISIDB.
Exploration of the Mechanism of TNFR2 in the Tumor Immune Microenvironment
In tumor cells, TNFR2 promotes tumor progression directly or indirectly by maintaining a favorable immune microenvironment for tumors and via different signaling pathways. Moreover, TNFR2 is expressed in some immune cells and various tumor cells. It has been reported that TNFR2 is abnormally expressed on various tumor cells such as those of breast cancer, ovarian cancer, skin cancer, renal cell carcinoma, colon cancer, and multiple myeloma (77, 86–91). How TNFR2 functions in the complex tumor microenvironment has also been explored. In renal carcinoma, TNFR2 on endothelial cells and renal tubular epithelial cells, upon injury-inducing stimuli, activates endothelial/epithelial tyrosine kinases, which in turn activate vascular endothelial growth factor receptor 2 to promote cell division and proliferation (89, 91). In a mouse model of lung cancer, the knockdown of TNFR2 on tumor cells promotes apoptosis and downregulates pro-angiogenic factors in endothelial progenitor cells (92). A recent study showed that TNF-α, produced by macrophages, can stabilize PD-L1 via activation of p65/CSN5 and enhance its interaction with PD-1 to elude T cell immune surveillance (93). However, in this process, whether TNF-α stabilizes PD-L1 through TNFR1 or TNFR2 remains to be verified. Recently, our group found that TNF-α regulates the transcriptional level of PD-L1 in pancreatic cancer cells through TNFR2-p65 NF-κB signaling, promoting its interaction with PD-1, thereby leading to CD8+ T cell immune surveillance evasion. Meanwhile, anti-TNFR2 and PD-L1 antibody combination therapy inhibits tumor growth, reduces Treg and tumor-associated macrophage infiltration, and induces the activation of CD8+ T cells in the pancreatic cancer microenvironment (94). Furthermore, in colon cancer (CT26) model, TNFR2 overexpression on cancer cells promotes increased TNFR2+ Tregs in draining lymph nodes and abundant sTNFR2 expression in peripheral blood (95). These studies suggest that TNFR2 on tumor cells in the tumor microenvironment can affect tumor growth by directly or indirectly regulating surrounding cells. In turn, TNFR2 on other cells in the tumor microenvironment also affects the expression of TNFR2 on tumor cells. It has been reported that soluble TNFR2, which is highly secreted by Tregs in the tumor microenvironment, can bind to membrane TNF on tumor cells to form a reverse transduction signaling pathway to induce the NF-κB pathway, thereby promoting the survival of lymphoma cells (14). These findings further enrich our understanding of the intricate roles of TNFR2 in regulating the tumor microenvironment. However, the key signaling events associated with TNFR2 in the tumor immune microenvironment and the mechanisms of TNFR2 interactions between different cells remain elusive. Therefore, we still need a more in-depth exploration of the characteristics and regulatory mechanisms of TNFR2 in various cells to more accurately treat TNFR2-related tumor diseases.
TNFR2 and Immune Checkpoint/Immune-Modulatory Factors
Some changes that may occur in the treatment enhance the tumor immunosuppressive effect and ultimately lead to treatment failure. Therefore, immunosuppressive cells and factors need to be taken into account during tumor treatment. Tumor-infiltrating Tregs are considered one of the main immunosuppressive cells regulating the tumor immune response (53, 96, 97). However, finding a specific way to diminish the host Tregs has remained particularly challenging, particularly within the tumor microenvironment (98–100).
Immune checkpoint inhibitors are providing new ideas for cancer immunotherapy, but their therapeutic effects are uneven. Some autoimmune side effects or immune dysregulation may be caused by anti-CTLA-4 or anti-PD-(L)1 antibody-targeted treatment (101). TNFR2 is becoming a new immune checkpoint molecule. It has better prospects than other immune checkpoint molecules because its expression is limited to a small group of effective Tregs and some immune cells. For example, the restricted expression of TNFR2 may explain why no serious autoimmune response was observed in Tnfr2-/- mice (102). Previous studies have shown that antagonistic antibodies against TNFR2 restrain the NF-κB pathway and inhibit Treg cell function, leading to tumor cell death (47). Furthermore, these anti-TNFR2 antibodies mostly affect tumor-infiltrating Treg cells because they exhibit higher TNFR2 expression levels than normal Treg cells. Targeting TNFR2 on Treg cells is well tolerated and clinically more promising. The tumor microenvironment is altered to a huge extent upon anti-TNFR2 therapy through the specific depletion of Tregs and activation of Teffs, thus inducing immune responses (103). Therefore, we believe that TNFR2 could be a promising marker in tumor immunotherapy.
We examined that the expression of TNFR2 is frequently and positively correlated with that of most immuno-inhibitors, such as PD-L1, CTLA-4, and LAG3, using the TCGA database through TISIDB (Figure 4B). Moreover, the expression of TNFR2, as well as some chemokines and their receptors also showed a positive correlation (Figures 4C, D). Therefore, we speculate a possibility that the efficacy of some checkpoint inhibitors may be enhanced upon combination therapies with anti-TNFR2 antibodies, for example, anti-PD-1, anti-CTLA-4, and CXCR4 inhibitors. Indications of this are also present in recent reports. Katherine et al. found that the combination of anti-TNFR2 and anti-PD-1 could be helpful in the development of a new immunotherapy method for the model of colon cancer (103). The combination of anti-PD-1 and anti-TNFR2 will lead to the death of most suppressive Tregs in the tumor microenvironment. It also increases the ratio of CD8+ T cells to Tregs compared with the single therapy. Furthermore, if anti-TNFR2 therapy was used in combination with anti-PD-1 therapy, or if anti-TNFR2 therapy is used after anti-PD-1 therapy, the therapeutic effect could be optimal. It is known that blocking the PD-1 checkpoint re-activates specific markers on Teffs and repairs the cell viability (78). Therefore, we speculate that PD-1 blockade might enhance TNFR2 expression in Teffs. Interestingly, researchers have found that anti-TNFR2 antibodies can notably decrease PD-1 expression in CD8+ T cells (104). This is the reason behind the proposed unique combination of anti-TNFR2 therapy and anti-PD-1 therapy. Dadiani reported that the appearance of TNFR2+ TILs is beneficial for the prognosis of patients with TNBC (105). However, there is no stable correlation between PD-1+ TILs and survival rate. The active state of PD-L1+ TILs increases the beneficial effect of TNFR2+ TILs. However, low or high levels of PD-1+ TILs in tumors do not promote the beneficial effect of TNFR2+ TILs. For the relationship between the subtype of immune infiltration and prognosis, TNFR2+ TILs could be a more stable immune target than PD-1+ TILs in TNBC. Therefore, it may be better not to block TNFR2+ TILs during TNBC treatment, which may enhance the immunotherapy efficiency of anti-PD-1 regimens. It is believed that the anti-TNFR2 antibody could be very helpful in a breast cancer mouse model. However, this model could also achieve a better result through combination with therapies like CpG or anti-CD25 (106). The expression of CXCR4 is related to tumor progression (107). Interestingly, the expression of CXCR4 on Tregs has a significant positive correlation with the expression of TNFR2 in acute myeloid leukemia (AML). Furthermore, the interaction and expression of CXCR4/CXCL12 promote an increase in TNFR2+ Tregs in patients with AML (69). Therefore, we conclude that blocking the TNFR2 checkpoint could be an attractive immunotherapy method, the effects of which may increase if combined with other checkpoint inhibitors.
TNFR2 and Cancer Immunotherapy
At present, the common methods of cancer immunotherapy include blocking immunosuppressive Tregs and thereby promoting the survival of tumor cells, as well as methods related to immune response, such as T cell activation and complement activation (108). Preventing the expansion of Tregs is currently considered to be the primary means of many cancer treatments (53).
It has been reported that TNFR2 can be triggered by agonists or antagonists to bidirectionally regulate Treg activity in adult CD4+ T cells. Antagonism causes Treg contraction, while agonism leads to Tregs expansion in vitro (109). Consequently, therapeutic targeting of TNFR2 may enable the decrease in the Treg activity and eliminate the immune-related suppressing cells. This would help the immune system to defend against the tumors and improve the cancer treatment effect. Another benefit of choosing TNFR2 as a novel target for tumor therapy is that TNFR2 can be found on some malignant cells. Increased levels of TNFR2 will improve the development of tumor cells (110). Thus, blocking TNFR2 not only enhances the anti-tumor immune response but may also directly kill tumor cells.
TNFR2 agonism and antagonism play essential roles in autoimmune and tumor microenvironments. Several anti-TNFR2 agonist antibodies that can enhance the activity of effector T cells have been reported previously (77), as well as some antagonist antibodies that can block the binding of TNF to TNFR2 and inhibit the cleavage of TNFR2 from mTNFR2 to sTNFR2 (111, 112). Blocking the TNF-TNFR2 interaction probably weakens TNFR2 surface expression on inhibitory Tregs and then destabilizes Tregs because TNF can accelerate TNFR2 expression on T cells. Torrey et al. found that the Tregs in ovarian cancer were more susceptible to TNFR2 antagonist treatment compared to Tregs in healthy tissues. The reason may be the relatively high expression of TNFR2 on tumor-infiltrating Tregs (113, 114). Thus, it is possible that TNFR2 antagonists selectively inhibit the activity of Tregs in tumors. However, they may not affect the function of regular Tregs around the tissues. This is the key to maintaining a stable immune environment. It is well known that highly suppressive Tregs and Teffs can express TNFR2. Although elevated TNFR2 expression on Teffs can promote Teffs development and enhance their ability to suppress Treg-mediated inhibition, TNFR2 expression was much higher on the tumor-invasive Tregs than that on Teffs (58, 113). Thus, in immunotherapy with TNFR2 antagonists, the lethality to Tregs may be greater than that to Teffs. The TNFR2 antagonist also inhibits TNFR2 cleavage from mTNFR2 to sTNFR2 in Tregs (111). Overall, treatment with TNFR2 antagonists would favor the activation and amplification of Teffs for a more potent antitumor immune response.
Recently, agonistic antibodies against TNFR2 have also been studied. Tam et al. constructed a new type of anti-TNFR2 antibody in mice, named Y9, which can recognize the receptor outside the TNF-binding domain (104). Y9 antibody treatment, mediated by CD8+ T cells and NK cells expands population and enhances the functionality of CD8+ T cells while not altering the suppressive function of Tregs and changing the ratio of CD8+ T cells to Tregs in vitro. Interestingly, Y9 antibody treatment not only contributes to short-term anti-tumor activity but also maintains long-term immune memory in many tumor models (104). A combination of the Y9 antibody with anti-PD-1 or -PD-L1 antibodies could further improve the anti-tumor efficacy. Moreover, this combination therapy results in a better effect than the combination of anti-PD-1 with anti-CTLA-4 theraphy (104). They also constructed anti-human TNFR2 antibodies Ab1 and Ab2, which exhibit properties similar to the Y9 antibody (104). These results show that the effect of the TNFR2 agonist antibody Y9 is very encouraging in anti-tumor immunotherapy, justifying the clinical development of human anti-TNFR2 antibodies. At present, domestic and foreign biopharmaceutical companies have begun to develop anti-TNFR2 antibodies, but most related research is still in the early preclinical stage, and the fastest progress has been the advancement of research to phase I clinical trials (Table 2).
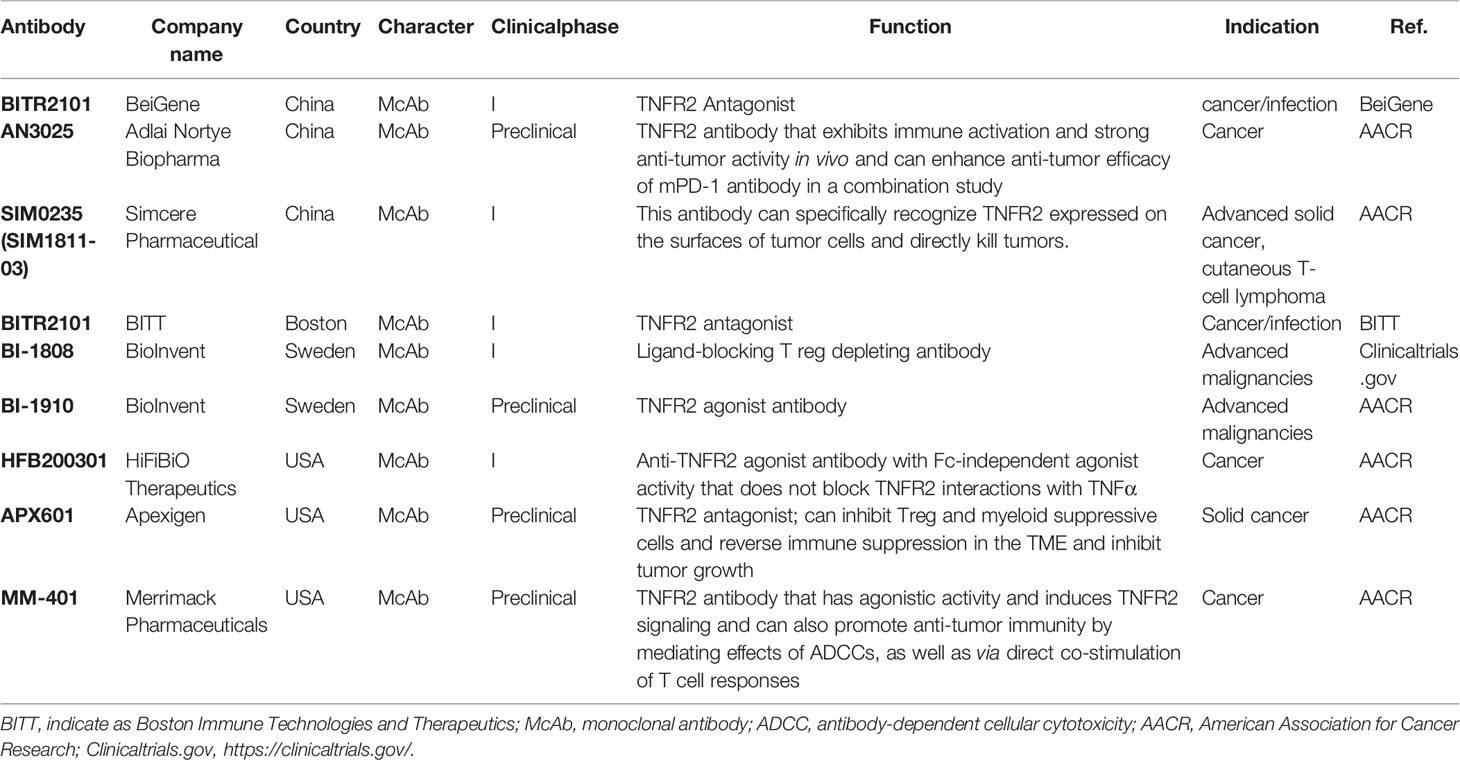
Table 2 The clinical progress of TNFR2-Targeting Treatment antibody research and development.
In current immunotherapies for cancer, the TNF/TNFR2 pathway is critical for the suppression of Tregs. Interestingly, specific inhibition of IL-6, instead of TNF, downregulates the population of TNFR2+ Tregs in advanced ovarian tumor ascites (63), which indicates that IL-6 is involved in the accumulation of TNFR2+ Tregs. During the treatment of acute myelocytic leukemia, the decrease in the number of TNFR2+ Tregs and the increase in the expression of IL-2 and IFN-γ can explain the combination of azacitidine and pabirestat can improve the therapeutic effect (69). In colon cancer, a new murine monoclonal anti-TNFR2 antibody (TY101) therapy combined with R848 (a synthetic TLR7/8 agonist) and HMGN1 (N1, a dendritic cell-activating TLR4 agonist) synergistically inhibits murine colon cancer and is more effective when compared with the single treatment with any of the above-mentioned drugs (115). Treatment of patients with advanced lymphoma with TNFR2 antagonists cause increased death of TNFR2+ Tregs and tumor cells and maintains the normal level of CD26- lymphocyte population (111). Additionally, immunotherapy with TNFR2 antagonists promotes the rapid expansion of Teff cells and stabilizes the normal ratio of Tregs to Teffs (111). Besides, small molecules from natural products can also specifically bind to TNFR2 and disrupt TNF-TNFR2 interactions (116). According to reports, Treg cells prevent glycolysis by inhibiting the mTOR pathway (117–120), while TNFR2 co-stimulation can allow thymus-derived Treg (tTreg) cells to undergo glycolysis (121). Therefore, in addition to antagonists, TNFR2 co-stimulation also induces metabolic remodeling of human Treg cells, which may broaden the applications of immunotherapy. In summary, TNFR2 targeted therapy may be a new approach to improve the efficacy of anti-tumor immunotherapy, as well as an adjuvant to improve the efficacy of other immune checkpoint inhibitors.
Concluding Remarks and Perspective
In tumor cells, TNFR2 promotes tumor progression directly or indirectly by maintaining a favorfavorable immune microenvironment for tumors and via different signaling pathways. Unlike TNFR1, which induces cell apoptosis, TNFR2 mainly promotes the growth and malignant transformation of cancer cells. TNFR2 expression is restricted to certain tumor cells and subpopulations of the lymphoid system, especially immunosuppressive cells. These properties make TNFR2 an ideal target for precise cancer treatment. Existing studies have confirmed that TNFR2 has excellent potential in tumor immunotherapy. Moreover, some antibody-based TNFR2 agonists and TNF antagonists have been proposed and have strong clinical practice potential. However, there are still many unanswered questions that require extensive preclinical verification. The development of the TNFR2 antibody, clinical development strategy, and selection of indications are also facing severe challenges.
The key to Treg-related anti-tumor treatment strategies is whether they can effectively and accurately regulate Tregs. Remarkably, TNFR2 can selectively regulate Tregs, which are more specific and safer than other immune checkpoints. Although breakthroughs have been made in tumor immune checkpoint therapy, relying on combination therapies has become a trend to improve the therapeutic effect. We predict that the therapeutic effect of TNFR2-treatment combined with other targets has the potential to match the effect of PD-(L)1-targeting therapies in the future. TNFR2 antibody has shown good anti-tumor activity in a single administration test in an animal model, and the combined effect with PD-(L)1 antibody was more significant. There is evidence that blocking TNF-TNFR2 reduces the expression of PD-L1 by monocytes (122). Moreover, PD-1 blockade can restore the expression of Teffs activation markers, including TNFR2. These results may explain why the combination of TNFR2 and PD-(L)1 antibody treatment affects salience, but it is still necessary to continue to explore the mechanism. As a new immunotherapy model, TNFR2 targeting may be combined with well-established immune checkpoint targets, including CTLA-4 and Tim3, in order to achieve the best effect in tumor immunotherapy. This plan may be a more effective and safer treatment and will be extensively investigated in future studies. In addition, whether the combination of anti-TNFR2 antibodies and TNF blockade will significantly improve the therapeutic effect remains to be explored.
In the treatment of various tumors, targeted therapy based on monoclonal antibodies shows significantly improved therapeutic effects on patients. Nevertheless, the long-term efficacy of this treatment is limited by its resistance mechanisms and other conditions. It is well known that PD-(L)1 or CTLA-4 have immunosuppressive functions in the tumor microenvironment, but when the antibodies against CTLA-4 or PD-(L)1 regulate Tregs, they can cause immune disorders and even serious autoimmune diseases and other side effects. Therefore, the success of clinical studies of anti-CTLA-4 or PD-L1 drugs has been limited. The design of bifunctional or multifunctional antibodies as a single agent to target multiple antigens has become a new immunotherapy strategy. The bifunctional PD-L1/TGF-βRII antibody (bintrafusp alfa) can direct the anti-TGF-β antibody to the tumor microenvironment via its anti-PD-L1 component, thereby achieving simultaneous inhibition of TGF-β and PD-L1 (123). The bifunctional PD-L1/TGF-βRII antibody (bintrafusp alfa) can use the anti-PD-L1 antibody to direct anti-TGF-β antibody to the tumor microenvironment, thereby achieving simultaneous inhibition of TGF-β and PD-L1. The bifunctional antibody-mediated inhibition of the immunosuppressive TGF-β and PD-1/PD-L1 pathways can improve the effect of tumor immunotherapy, which is a characteristic immunotherapy regimen. TNFR2 antibody is more specific and safer than other immunotherapies because it specifically recognizes the tumor microenvironment. Therefore, the development of bispecific functional antibodies or multifunctional specific antibodies that simultaneously target TNFR2 and other immune checkpoints will be more beneficial to tumor immunotherapy. The TNFR2 antibody can carry other immune target antibodies to directly target tumors or immunosuppressive cells, which dramatically reduces drug resistance and severe adverse reactions. These novel multifunctional antibodies demonstrate a powerful potential in immunotherapy for different cancer types.
There has been accumulating evidence showing that TNFR2 is expressed and plays a crucial role in immune cells. Especially, TNFR2+ Tregs, which are associated with elevated disease progression, suggest that TNFR2 could be used as a potential therapeutic target for cancer therapies (47, 52, 68, 124, 125). However, understanding the relationship between TNF/TNFR2 and immune cell responses is elusive and controversial. For example, the TNF/TNFR2 signaling pathway potentially activates CD8+ Tregs and CD8+ Teffs simultaneously, which have antagonistic relationships. Therefore, blocking the TNF/TNFR2 pathway may suppress the protective Tregs or Teffs and impair the treatment (42). Interestingly, another study demonstrated that chemotherapy could reduce the content of CD4+ TNFR2+ Tregs and increase the ratio of protective CD8+ TNFR2+ TILs in TNBC (115). Furthermore, another study showed that TNFR2+ TILs have a strong association with improved survival in patients with TNBC (105). Therefore, therapeutics against TNFR2 may negatively affect patients with TNBC, which is different from other tumors. Consequently, targeting TNFR2 alone may not yield good results. To provide a safer and more precise treatment approach for tumor immunotherapy, we need to explore the specific mechanism of TNFR2 in the tumor microenvironment and accurately understand the expression level of TNFR2 and the ratio of TNFR2+ T cells in various T cell subgroups.
Finally, TNFR2 has attracted attention for its ability to promote tumor cell survival and proliferation, and this property provides a strong rationale for TNFR2 as a potentially effective therapeutic target. Since TNFR2 promotes tumorigenesis and progression, most studies have also focused on cancers that express high levels of TNFR2. As shown in Figure 2A, most tumor tissues show higher levels of TNFR2 expression than normal tissues. However, some tumor tissues express lower levels of TNFR2 than normal tissues. In future clinical studies, appropriate protocols should be designed according to the level of TNFR2 expressed in different tumors. In addition, we can also see from Figure 2 that the expression level of TNFR2 is not positively correlated with the poor prognosis and disease-free survival of patients in all tumors. Moreover, the clinical stage and pathological grade of different tumors were not completely consistent with the expression level of TNFR2. This inconsistency may be related to the tumor type and the complexity of tumor progression. For example, in breast cancer, the prognosis could be affected by different factors, including age, menopausal status, clinical stage, pathological grade, and receptors on the surface of tumor cells (126–128). Studies have found that TNFR2 expression and menopausal status might significantly affect the DFS rate of breast cancer patients (129). This difference could also be related to cell types and proportions in different tumor environments. Although TNFR2+ Tregs represent the largest immunosuppressive subset in the tumor immune microenvironment, anti-tumor cells in the TME benefit from the activation of the TNFα/TNFR2 pathway signaling, such as TNFR2+ Teff cells. In the intricate tumor microenvironment, this dual function of TNFR2 can be out of balance due to certain factors. Dadiani et al. demonstrated that large numbers of TNFR2+ TILs can significantly improve survival in TNBC patients, whereas unfavorable PD-1+ TIL levels counteract the favorable effect of TNFR2+ TILs on disease outcomes (105). Interestingly, PD-1 expression itself might result from a dynamic process during T cell activation (130, 131), and thus, if we provide appropriate conditions, the effect of PD-1+ TIL levels on TNFR2+ TILs could be improved. Therefore, different tumor types and different disease stages must be considered when targeting TNFR2 in therapy. A more comprehensive assessment of the function of TNFR2 in different tumors is required in future studies.
Author Contributions
XB and TL provided direction and guidance throughout the preparation of this manuscript. ML wrote and edited the manuscript. XZ discussed and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Key Research and Development Program [2019YFC1316000 to TBL]; the National Natural Science Foundation of China [U20A20378 and 81830089 to TBL, 81871925 and 82071867 to XLB]; the Key Research and Development Program of ZheThe page range of references has been revisedjiang Province [2019C03019 to TBL, 2020C03117 to XLB].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Wajant H, Pfizenmaier K, Scheurich P. Tumor Necrosis Factor Signaling. Cell Death Differ (2003) 10:45–65. doi: 10.1038/sj.cdd.4401189
2. Brenner D, Blaser H, Mak TW. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat Rev Immunol (2015) 15:362–74. doi: 10.1038/nri3834
3. Wajant H, Scheurich P. TNFR1-Induced Activation of the Classical NF-kappaB Pathway. FEBS J (2011) 278:862–76. doi: 10.1111/j.1742-4658.2011.08015.x
4. Duckett CS, Thompson CB. CD30-Dependent Degradation of TRAF2: Implications for Negative Regulation of TRAF Signaling and the Control of Cell Survival. Genes Dev (1997) 11:2810–21. doi: 10.1101/gad.11.21.2810
5. Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic Crosstalk of TNF Receptors: TNF-R2-Induces Depletion of TRAF2 and IAP Proteins and Accelerates TNF-R1-Dependent Activation of Caspase-8. J Cell Sci (2002) 115:2757–70. doi: 10.1242/jcs.115.13.2757
6. Li X, Yang Y, Ashwell JD. TNF-RII and C-IAP1 Mediate Ubiquitination and Degradation of TRAF2. Nature (2002) 416:345–7. doi: 10.1038/416345a
7. Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane Tumor Necrosis Factor (TNF) Induces P100 Processing via TNF Receptor-2 (TNFR2). J Biol Chem (2010) 285:7394–404. doi: 10.1074/jbc.M109.037341
8. Sun SC. The non-Canonical NF-kappaB Pathway in Immunity and Inflammation. Nat Rev Immunol (2017) 17:545–58. doi: 10.1038/nri.2017.52
9. Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a Tumor Necrosis Factor Receptor Type 2-Specific Kinase: Role in Endothelial Cell Migration and Angiogenesis. Mol Cell Biol (2002) 22:7512–23. doi: 10.1128/MCB.22.21.7512-7523.2002
10. So T, Croft M. Regulation of PI-3-Kinase and Akt Signaling in T Lymphocytes and Other Cells by TNFR Family Molecules. Front Immunol (2013) 4:139. doi: 10.3389/fimmu.2013.00139
11. Vanamee ES, Faustman DL. TNFR2: A Novel Target for Cancer Immunotherapy. Trends Mol Med (2017) 23:1037–46. doi: 10.1016/j.molmed.2017.09.007
12. Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, Pietryga-Krieger A, et al. Myeloid Suppressor Cells Require Membrane TNFR2 Expression for Suppressive Activity. Immun Inflamm Dis (2014) 2:121–30. doi: 10.1002/iid3.19
13. Shamdani S, Uzan G, Naserian S. TNFalpha-TNFR2 Signaling Pathway in Control of the Neural Stem/Progenitor Cell Immunosuppressive Effect: Different Experimental Approaches to Assess This Hypothetical Mechanism Behind Their Immunological Function. Stem Cell Res Ther (2020) 11:307. doi: 10.1186/s13287-020-01816-2
14. Zhang H, Yan D, Shi X, Liang H, Pang Y, Qin N, et al. Transmembrane TNF-Alpha Mediates "Forward" and "Reverse" Signaling, Inducing Cell Death or Survival via the NF-kappaB Pathway in Raji Burkitt Lymphoma Cells. J Leukoc Biol (2008) 84:789–97. doi: 10.1189/jlb.0208078
15. Tartaglia LA, Pennica D, Goeddel DV. Ligand Passing: The 75-kDa Tumor Necrosis Factor (TNF) Receptor Recruits TNF for Signaling by the 55-kDa TNF Receptor. J Biol Chem (1993) 268:18542–8. doi: 10.1016/S0021-9258(17)46661-0
16. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-Alpha: Structure, Function and Interaction With Anti-TNF Agents. Rheumatol (Oxford) (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
17. Yang F, Zhao N, Wu N. TNFR2 Promotes Adriamycin Resistance in Breast Cancer Cells by Repairing DNA Damage. Mol Med Rep (2017) 16:2962–8. doi: 10.3892/mmr.2017.6898
18. Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W, Salatino M, et al. TNF Alpha Acting on TNFR1 Promotes Breast Cancer Growth via P42/P44 MAPK, JNK, Akt and NF-Kappa B-Dependent Pathways. Exp Cell Res (2008) 314:509–29. doi: 10.1016/j.yexcr.2007.10.005
19. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front Immunol (2018) 9:784. doi: 10.3389/fimmu.2018.00784
20. Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, et al. Etk/Bmx Transactivates Vascular Endothelial Growth Factor 2 and Recruits Phosphatidylinositol 3-Kinase to Mediate the Tumor Necrosis Factor-Induced Angiogenic Pathway. J Biol Chem (2003) 278:51267–76. doi: 10.1074/jbc.M310678200
21. Zhou Z, Gengaro P, Wang W, Wang XQ, Li C, Faubel S, et al. Role of NF-kappaB and PI 3-Kinase/Akt in TNF-Alpha-Induced Cytotoxicity in Microvascular Endothelial Cells. Am J Physiol Renal Physiol (2008) 295:F932–41. doi: 10.1152/ajprenal.00066.2008
22. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 Activates MLCK-Dependent Tight Junction Dysregulation to Cause Apoptosis-Mediated Barrier Loss and Experimental Colitis. Gastroenterology (2013) 145:407–15. doi: 10.1053/j.gastro.2013.04.011
23. Ji W, Li Y, Wan T, Wang J, Zhang H, Chen H, et al. Both Internalization and AIP1 Association are Required for Tumor Necrosis Factor Receptor 2-Mediated JNK Signaling. Arterioscler Thromb Vasc Biol (2012) 32:2271–9. doi: 10.1161/ATVBAHA.112.253666
24. Ivagnes A, Messaoudene M, Stoll G, Routy B, Fluckiger A, Yamazaki T, et al. TNFR2/BIRC3-TRAF1 Signaling Pathway as a Novel NK Cell Immune Checkpoint in Cancer. Oncoimmunology (2018) 7:e1386826. doi: 10.1080/2162402X.2017.1386826
25. Ruspi G, Schmidt EM, McCann F, Feldmann M, Williams RO, Stoop AA, et al. TNFR2 Increases the Sensitivity of Ligand-Induced Activation of the P38 MAPK and NF-kappaB Pathways and Signals TRAF2 Protein Degradation in Macrophages. Cell Signal (2014) 26:683–90. doi: 10.1016/j.cellsig.2013.12.009
26. Wang LL, Dong JJ, An BZ, Liang J, Cai KR, Jin ZS, et al. Has-miR-17 Increases the Malignancy of Gastric Lymphoma by HSP60/TNFR2 Pathway. J Biol Regul Homeost Agents (2020) 34:1317–24. doi: 10.23812/20-60-A
27. Meng Y, Zhao Q, An L, Jiao S, Li R, Sang Y, et al. A TNFR2-hnRNPK Axis Promotes Primary Liver Cancer Development via Activation of YAP Signaling in Hepatic Progenitor Cells. Cancer Res (2021) 81:3036–50. doi: 10.1158/0008-5472.CAN-20-3175
28. Bodmer JL, Schneider P, Tschopp J. The Molecular Architecture of the TNF Superfamily. Trends Biochem Sci (2002) 27:19–26. doi: 10.1016/s0968-0004(01)01995-8
29. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell (2001) 104:487–501. doi: 10.1016/s0092-8674(01)00237-9
30. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The Transmembrane Form of Tumor Necrosis Factor is the Prime Activating Ligand of the 80 kDa Tumor Necrosis Factor Receptor. Cell (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
31. Xie P. TRAF Molecules in Cell Signaling and in Human Diseases. J Mol Signal (2013) 8:7. doi: 10.1186/1750-2187-8-7
32. Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A Domain in TNF Receptors That Mediates Ligand-Independent Receptor Assembly and Signaling. Science (2000) 288:2351–4. doi: 10.1126/science.288.5475.2351
33. Moosmayer D, Wajant H, Gerlach E, Schmidt M, Brocks B, Pfizenmaier K. Characterization of Different Soluble TNF Receptor (TNFR80) Derivatives: Positive Influence of the Intracellular Domain on Receptor/Ligand Interaction and TNF Neutralization Capacity. J Interferon Cytokine Res (1996) 16:471–7. doi: 10.1089/jir.1996.16.471
34. Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, et al. Solution of the Structure of the TNF-TNFR2 Complex. Sci Signal (2010) 3:ra83. doi: 10.1126/scisignal.2000954
35. Park YC, Burkitt V, Villa AR, Tong L, Wu H. Structural Basis for Self-Association and Receptor Recognition of Human TRAF2. Nature (1999) 398:533–8. doi: 10.1038/19110
36. Mace PD, Smits C, Vaux DL, Silke J, Day CL. Asymmetric Recruitment of cIAPs by TRAF2. J Mol Biol (2010) 400:8–15. doi: 10.1016/j.jmb.2010.04.055
37. Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, et al. Conversion of Membrane-Bound Fas(CD95) Ligand to its Soluble Form is Associated With Downregulation of its Proapoptotic Activity and Loss of Liver Toxicity. J Exp Med (1998) 187:1205–13. doi: 10.1084/jem.187.8.1205
38. Chee J, Angstetra E, Mariana L, Graham KL, Carrington EM, Bluethmann H, et al. TNF Receptor 1 Deficiency Increases Regulatory T Cell Function in Nonobese Diabetic Mice. J Immunol (2011) 187:1702–12. doi: 10.4049/jimmunol.1100511
39. Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor Necrosis Factor-Alpha Regulation of CD4+CD25+ T Cell Levels in NOD Mice. Proc Natl Acad Sci USA (2002) 99:12287–92. doi: 10.1073/pnas.172382999
40. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF With TNF Receptor Type 2 Promotes Expansion and Function of Mouse CD4+CD25+ T Regulatory Cells. J Immunol (2007) 179:154–61. doi: 10.4049/jimmunol.179.1.154
41. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A Metalloproteinase Disintegrin That Releases Tumour-Necrosis Factor-Alpha From Cells. Nature (1997) 385:729–33. doi: 10.1038/385729a0
42. Ye LL, Wei XS, Zhang M, Niu YR, Zhou Q. The Significance of Tumor Necrosis Factor Receptor Type II in CD8(+) Regulatory T Cells and CD8(+) Effector T Cells. Front Immunol (2018) 9:583. doi: 10.3389/fimmu.2018.00583
43. van Mierlo GJ, Scherer HU, Hameetman M, Morgan ME, Flierman R, Huizinga TW, et al. Cutting Edge: TNFR-Shedding by CD4+CD25+ Regulatory T Cells Inhibits the Induction of Inflammatory Mediators. J Immunol (2008) 180:2747–51. doi: 10.4049/jimmunol.180.5.2747
44. Dri P, Gasparini C, Menegazzi R, Cramer R, Alberi L, Presani G, et al. TNF-Induced Shedding of TNF Receptors in Human Polymorphonuclear Leukocytes: Role of the 55-kDa TNF Receptor and Involvement of a Membrane-Bound and non-Matrix Metalloproteinase. J Immunol (2000) 165:2165–72. doi: 10.4049/jimmunol.165.4.2165
45. Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium Tuberculosis Evades Apoptosis of Host Macrophages by Release of TNF-R2, Resulting in Inactivation of TNF-Alpha. J Immunol (1998) 161:2636–41.
46. Balcewicz-Sablinska MK, Gan H, Remold HG. Interleukin 10 Produced by Macrophages Inoculated With Mycobacterium Avium Attenuates Mycobacteria-Induced Apoptosis by Reduction of TNF-Alpha Activity. J Infect Dis (1999) 180:1230–7. doi: 10.1086/315011
47. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 With Antagonistic Antibodies Inhibits Proliferation of Ovarian Cancer Cells and Tumor-Associated Tregs. Sci Signal (2017) 10:eaaf8608. doi: 10.1126/scisignal.aaf8608
48. Babic A, Shah SM, Song M, Wu K, Meyerhardt JA, Ogino S, et al. Soluble Tumour Necrosis Factor Receptor Type II and Survival in Colorectal Cancer. Br J Cancer (2016) 114:995–1002. doi: 10.1038/bjc.2016.85
49. Nomelini RS, Borges Junior LE, de Lima CA, Chiovato AFC, Micheli DC, Tavares-Murta BM, et al. TNF-R2 in Tumor Microenvironment as Prognostic Factor in Epithelial Ovarian Cancer. Clin Exp Med (2018) 18:547–54. doi: 10.1007/s10238-018-0508-3
50. Kalliolias GD, Ivashkiv LB. TNF Biology, Pathogenic Mechanisms and Emerging Therapeutic Strategies. Nat Rev Rheumatol (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
51. Kim EY, Teh HS. TNF Type 2 Receptor (P75) Lowers the Threshold of T Cell Activation. J Immunol (2001) 167:6812–20. doi: 10.4049/jimmunol.167.12.6812
52. Speiser DE, Ho PC, Verdeil G. Regulatory Circuits of T Cell Function in Cancer. Nat Rev Immunol (2016) 16:599–611. doi: 10.1038/nri.2016.80
53. Facciabene A, Motz GT, Coukos G. T-Regulatory Cells: Key Players in Tumor Immune Escape and Angiogenesis. Cancer Res (2012) 72:2162–71. doi: 10.1158/0008-5472.CAN-11-3687
54. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T Cells in Tumors From Patients With Early-Stage non-Small Cell Lung Cancer and Late-Stage Ovarian Cancer. Cancer Res (2001) 61:4766–72.
55. Nguyen DX, Ehrenstein MR. Anti-TNF Drives Regulatory T Cell Expansion by Paradoxically Promoting Membrane TNF-TNF-RII Binding in Rheumatoid Arthritis. J Exp Med (2016) 213:1241–53. doi: 10.1084/jem.20151255
56. He T, Liu S, Chen S, Ye J, Wu X, Bian Z, et al. The P38 MAPK Inhibitor SB203580 Abrogates Tumor Necrosis Factor-Induced Proliferative Expansion of Mouse CD4(+)Foxp3(+) Regulatory T Cells. Front Immunol (2018) 9:1556. doi: 10.3389/fimmu.2018.01556
57. Yan F, Du R, Wei F, Zhao H, Yu J, Wang C, et al. Expression of TNFR2 by Regulatory T Cells in Peripheral Blood is Correlated With Clinical Pathology of Lung Cancer Patients. Cancer Immunol Immunother (2015) 64:1475–85. doi: 10.1007/s00262-015-1751-z
58. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, et al. Tumor Necrosis Factor Receptor 2-Dependent Homeostasis of Regulatory T Cells as a Player in TNF-Induced Experimental Metastasis. Carcinogenesis (2013) 34:1296–303. doi: 10.1093/carcin/bgt038
59. Ham B, Wang N, D'Costa Z, Fernandez MC, Bourdeau F, Auguste P, et al. TNF Receptor-2 Facilitates an Immunosuppressive Microenvironment in the Liver to Promote the Colonization and Growth of Hepatic Metastases. Cancer Res (2015) 75:5235–47. doi: 10.1158/0008-5472.CAN-14-3173
60. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 Ligation in Human T Regulatory Cells Enhances IL2-Induced Cell Proliferation Through the non-Canonical NF-kappaB Pathway. Sci Rep (2018) 8:12079. doi: 10.1038/s41598-018-30621-4
61. Horwitz DA, Pan S, Ou JN, Wang J, Chen M, Gray JD, et al. Therapeutic Polyclonal Human CD8+ CD25+ Fox3+ TNFR2+ PD-L1+ Regulatory Cells Induced Ex-Vivo. Clin Immunol (2013) 149:450–63. doi: 10.1016/j.clim.2013.08.007
62. Filaci G, Fenoglio D, Fravega M, Ansaldo G, Borgonovo G, Traverso P, et al. CD8+ CD28- T Regulatory Lymphocytes Inhibiting T Cell Proliferative and Cytotoxic Functions Infiltrate Human Cancers. J Immunol (2007) 179:4323–34. doi: 10.4049/jimmunol.179.7.4323
63. Kampan NC, Madondo MT, McNally OM, Stephens AN, Quinn MA, Plebanski M. Interleukin 6 Present in Inflammatory Ascites From Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front Immunol (2017) 8:1482. doi: 10.3389/fimmu.2017.01482
64. Emens LA, Middleton G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol Res (2015) 3:436–43. doi: 10.1158/2326-6066.CIR-15-0064
65. Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, et al. Pathologic Complete Response to Neoadjuvant Chemotherapy of Breast Carcinoma is Associated With the Disappearance of Tumor-Infiltrating Foxp3+ Regulatory T Cells. Clin Cancer Res (2008) 14:2413–20. doi: 10.1158/1078-0432.CCR-07-4491
66. Naserian S, Shamdani S, Arouche N, Uzan G. Regulatory T Cell Induction by Mesenchymal Stem Cells Depends on the Expression of TNFR2 by T Cells. Stem Cell Res Ther (2020) 11:534. doi: 10.1186/s13287-020-02057-z
67. Beldi G, Bahiraii S, Lezin C, Nouri Barkestani M, Abdelgawad ME, Uzan G, et al. TNFR2 Is a Crucial Hub Controlling Mesenchymal Stem Cell Biological and Functional Properties. Front Cell Dev Biol (2020) 8:596831. doi: 10.3389/fcell.2020.596831
68. Jiang Y, Chen J, Bi E, Zhao Y, Qin T, Wang Y, et al. TNF-Alpha Enhances Th9 Cell Differentiation and Antitumor Immunity via TNFR2-Dependent Pathways. J Immunother Cancer (2019) 7:28. doi: 10.1186/s40425-018-0494-8
69. Govindaraj C, Tan P, Walker P, Wei A, Spencer A, Plebanski M. Reducing TNF Receptor 2+ Regulatory T Cells via the Combined Action of Azacitidine and the HDAC Inhibitor, Panobinostat for Clinical Benefit in Acute Myeloid Leukemia Patients. Clin Cancer Res (2014) 20:724–35. doi: 10.1158/1078-0432.CCR-13-1576
70. He J, Li R, Chen Y, Hu Y, Chen X. TNFR2-Expressing CD4(+)Foxp3(+) Regulatory T Cells in Cancer Immunology and Immunotherapy. Prog Mol Biol Transl Sci (2019) 164:101–17. doi: 10.1016/bs.pmbts.2019.03.010
71. Aspalter RM, Eibl MM, Wolf HM. Regulation of TCR-Mediated T Cell Activation by TNF-RII. J Leukoc Biol (2003) 74:572–82. doi: 10.1189/jlb.0303112
72. Reiner SL. Development in Motion: Helper T Cells at Work. Cell (2007) 129:33–6. doi: 10.1016/j.cell.2007.03.019
73. Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OM, Oppenheim JJ. Expression of Costimulatory TNFR2 Induces Resistance of CD4+FoxP3- Conventional T Cells to Suppression by CD4+FoxP3+ Regulatory T Cells. J Immunol (2010) 185:174–82. doi: 10.4049/jimmunol.0903548
74. Twu YC, Gold MR, Teh HS. TNFR1 Delivers Pro-Survival Signals That are Required for Limiting TNFR2-Dependent Activation-Induced Cell Death (AICD) in CD8+ T Cells. Eur J Immunol (2011) 41:335–44. doi: 10.1002/eji.201040639
75. Kim EY, Chi HH, Rajaiah R, Moudgil KD. Exogenous Tumour Necrosis Factor Alpha Induces Suppression of Autoimmune Arthritis. Arthritis Res Ther (2008) 10:R38. doi: 10.1186/ar2393
76. Kim EY, Priatel JJ, Teh SJ, Teh HS. TNF Receptor Type 2 (P75) Functions as a Costimulator for Antigen-Driven T Cell Responses In Vivo. J Immunol (2006) 176:1026–35. doi: 10.4049/jimmunol.176.2.1026
77. Williams GS, Mistry B, Guillard S, Ulrichsen JC, Sandercock AM, Wang J, et al. Phenotypic Screening Reveals TNFR2 as a Promising Target for Cancer Immunotherapy. Oncotarget (2016) 7:68278–91. doi: 10.18632/oncotarget.11943
78. Buchan S, Manzo T, Flutter B, Rogel A, Edwards N, Zhang L, et al. OX40- and CD27-Mediated Costimulation Synergizes With Anti-PD-L1 Blockade by Forcing Exhausted CD8+ T Cells to Exit Quiescence. J Immunol (2015) 194:125–33. doi: 10.4049/jimmunol.1401644
79. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF Signaling Drives Myeloid-Derived Suppressor Cell Accumulation. J Clin Invest (2012) 122:4094–104. doi: 10.1172/JCI64115
80. Ticha O, Moos L, Wajant H, Bekeredjian-Ding I. Expression of Tumor Necrosis Factor Receptor 2 Characterizes TLR9-Driven Formation of Interleukin-10-Producing B Cells. Front Immunol (2017) 8:1951. doi: 10.3389/fimmu.2017.01951
81. Djouad F, Fritz V, Apparailly F, Louis-Plence P, Bony C, Sany J, et al. Reversal of the Immunosuppressive Properties of Mesenchymal Stem Cells by Tumor Necrosis Factor Alpha in Collagen-Induced Arthritis. Arthritis Rheum (2005) 52:1595–603. doi: 10.1002/art.21012
82. Miettinen JA, Pietila M, Salonen RJ, Ohlmeier S, Ylitalo K, Huikuri HV, et al. Tumor Necrosis Factor Alpha Promotes the Expression of Immunosuppressive Proteins and Enhances the Cell Growth in a Human Bone Marrow-Derived Stem Cell Culture. Exp Cell Res (2011) 317:791–801. doi: 10.1016/j.yexcr.2010.12.010
83. Naserian S, Abdelgawad ME, Afshar Bakshloo M, Ha G, Arouche N, Cohen JL, et al. The TNF/TNFR2 Signaling Pathway is a Key Regulatory Factor in Endothelial Progenitor Cell Immunosuppressive Effect. Cell Commun Signal (2020) 18:94. doi: 10.1186/s12964-020-00564-3
84. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-Alpha Promotes Suppressive Activities of Myeloid-Derived Suppressor Cells via TNFR2. J Immunol (2014) 192:1320–31. doi: 10.4049/jimmunol.1203195
85. Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T, et al. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res (2015) 75:4063–73. doi: 10.1158/0008-5472.CAN-14-3394
86. Arnott CH, Scott KA, Moore RJ, Robinson SC, Thompson RG, Balkwill FR. Expression of Both TNF-Alpha Receptor Subtypes is Essential for Optimal Skin Tumour Development. Oncogene (2004) 23:1902–10. doi: 10.1038/sj.onc.1207317
87. Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, et al. A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol Cell Proteomics (2005) 4:1920–32. doi: 10.1074/mcp.M500279-MCP200
88. Rauert H, Stuhmer T, Bargou R, Wajant H, Siegmund D. TNFR1 and TNFR2 Regulate the Extrinsic Apoptotic Pathway in Myeloma Cells by Multiple Mechanisms. Cell Death Dis (2011) 2:e194. doi: 10.1038/cddis.2011.78
89. Al-Lamki RS, Wang J, Yang J, Burrows N, Maxwell PH, Eisen T, et al. Tumor Necrosis Factor Receptor 2-Signaling in CD133-Expressing Cells in Renal Clear Cell Carcinoma. Oncotarget (2016) 7:24111–24. doi: 10.18632/oncotarget.8125
90. Hamilton KE, Simmons JG, Ding S, Van Landeghem L, Lund PK. Cytokine Induction of Tumor Necrosis Factor Receptor 2 is Mediated by STAT3 in Colon Cancer Cells. Mol Cancer Res (2011) 9:1718–31. doi: 10.1158/1541-7786.MCR-10-0210
91. Al-Lamki RS, Sadler TJ, Wang J, Reid MJ, Warren AY, Movassagh M, et al. Tumor Necrosis Factor Receptor Expression and Signaling in Renal Cell Carcinoma. Am J Pathol (2010) 177:943–54. doi: 10.2353/ajpath.2010.091218
92. Sasi SP, Yan X, Enderling H, Park D, Gilbert HY, Curry C, et al. Breaking the 'Harmony' of TNF-Alpha Signaling for Cancer Treatment. Oncogene (2012) 31:4117–27. doi: 10.1038/onc.2011.567
93. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
94. Zhang X, Lao M, Xu J, Duan Y, Yang H, Li M, et al. Combination Cancer Immunotherapy Targeting TNFR2 and PD-1/PD-L1 Signaling Reduces Immunosuppressive Effects in the Microenvironment of Pancreatic Tumors. J Immunother Cancer (2022) 10:e003982. doi: 10.1136/jitc-2021-003982
95. Chen X LP, Yang X, Miao X. ( 2018) Tumor Necrosis Factor Receptor II (TNFR2) Promotes the Growth of Mouse CT26 Colon Cancer. J Immunol (2018) 178:200–7.
96. Cari L, Nocentini G, Migliorati G, Riccardi C. Potential Effect of Tumor-Specific Treg-Targeted Antibodies in the Treatment of Human Cancers: A Bioinformatics Analysis. Oncoimmunology (2018) 7:e1387705. doi: 10.1080/2162402X.2017.1387705
97. Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting Regulatory T Cells in Cancer. Cancer Res (2011) 71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156
98. Shimizu J, Yamazaki S, Sakaguchi S. Induction of Tumor Immunity by Removing CD25+CD4+ T Cells: A Common Basis Between Tumor Immunity and Autoimmunity. J Immunol (1999) 163:5211–8.
99. Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade and Depletion of CD25(+) Regulatory T Cells in Antitumor Therapy Reveals Alternative Pathways for Suppression of Autoreactive Cytotoxic T Lymphocyte Responses. J Exp Med (2001) 194:823–32. doi: 10.1084/jem.194.6.823
100. Smyth MJ, Ngiow SF, Teng MW. Targeting Regulatory T Cells in Tumor Immunotherapy. Immunol Cell Biol (2014) 92:473–4. doi: 10.1038/icb.2014.33
101. Kong YC, Flynn JC. Opportunistic Autoimmune Disorders Potentiated by Immune-Checkpoint Inhibitors Anti-CTLA-4 and Anti-PD-1. Front Immunol (2014) 5:206. doi: 10.3389/fimmu.2014.00206
102. Erickson SL, de Sauvage FJ, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, et al. Decreased Sensitivity to Tumour-Necrosis Factor But Normal T-Cell Development in TNF Receptor-2-Deficient Mice. Nature (1994) 372:560–3. doi: 10.1038/372560a0
103. Case K, Tran L, Yang M, Zheng H, Kuhtreiber WM, Faustman DL. TNFR2 Blockade Alone or in Combination With PD-1 Blockade Shows Therapeutic Efficacy in Murine Cancer Models. J Leukoc Biol (2020) 107:981–91. doi: 10.1002/JLB.5MA0420-375RRRRR
104. Tam EM, Fulton RB, Sampson JF, Muda M, Camblin A, Richards J, et al. Antibody-Mediated Targeting of TNFR2 Activates CD8(+) T Cells in Mice and Promotes Antitumor Immunity. Sci Transl Med (2019) 11:eaax0720. doi: 10.1126/scitranslmed.aax0720
105. Dadiani M, Necula D, Kahana-Edwin S, Oren N, Baram T, Marin I, et al. TNFR2+ TILs are Significantly Associated With Improved Survival in Triple-Negative Breast Cancer Patients. Cancer Immunol Immunother (2020) 69:1315–26. doi: 10.1007/s00262-020-02549-0
106. Nie Y, He J, Shirota H, Trivett AL, Yang, Klinman DM, et al. Blockade of TNFR2 Signaling Enhances the Immunotherapeutic Effect of CpG ODN in a Mouse Model of Colon Cancer. Sci Signal (2018) 11:eaan0790. doi: 10.1126/scisignal.aan0790
107. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a Prognostic Marker in Acute Myelogenous Leukemia. Blood (2007) 109:786–91. doi: 10.1182/blood-2006-05-024844
108. Dhodapkar M, Zhang AD, Puthumana J, Downing NS, Shah ND, Ross JS. Characteristics of Clinical Studies Used for US Food and Drug Administration Supplemental Indication Approvals of Drugs and Biologics, 2017 to 2019. JAMA Netw Open (2021) 4:e2113224. doi: 10.1001/jamanetworkopen.2021.13224
109. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous Expansion of Human T-Regulatory Cells via Tumor Necrosis Factor Receptor 2. Sci Rep (2013) 3:3153. doi: 10.1038/srep03153
110. Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic Analysis of Mycosis Fungoides and Sezary Syndrome Identifies Recurrent Alterations in TNFR2. Nat Genet (2015) 47:1056–60. doi: 10.1038/ng.3370
111. Torrey H, Khodadoust M, Tran L, Baum D, Defusco A, Kim YH, et al. Targeted Killing of TNFR2-Expressing Tumor Cells and Tregs by TNFR2 Antagonistic Antibodies in Advanced Sezary Syndrome. Leukemia (2019) 33:1206–18. doi: 10.1038/s41375-018-0292-9
112. Chen X, Oppenheim JJ. Targeting TNFR2, an Immune Checkpoint Stimulator and Oncoprotein, is a Promising Treatment for Cancer. Sci Signal (2017) 10:eaal2328. doi: 10.1126/scisignal.aal2328
113. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting Edge: Expression of TNFR2 Defines a Maximally Suppressive Subset of Mouse CD4+CD25+FoxP3+ T Regulatory Cells: Applicability to Tumor-Infiltrating T Regulatory Cells. J Immunol (2008) 180:6467–71. doi: 10.4049/jimmunol.180.10.6467
114. Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, et al. Impaired Th1 Immunity in Ovarian Cancer Patients is Mediated by TNFR2+ Tregs Within the Tumor Microenvironment. Clin Immunol (2013) 149:97–110. doi: 10.1016/j.clim.2013.07.003
115. Jiang M, Liu J, Yang, Tross D, Li P, Chen F, et al. A TNFR2 Antibody by Countering Immunosuppression Cooperates With HMGN1 and R848 Immune Stimulants to Inhibit Murine Colon Cancer. Int Immunopharmacol (2021) 101:108345. doi: 10.1016/j.intimp.2021.108345
116. Shaikh F, He J, Bhadra P, Chen X, Siu SWI. TNF Receptor Type II as an Emerging Drug Target for the Treatment of Cancer, Autoimmune Diseases, and Graft-Versus-Host Disease: Current Perspectives and In Silico Search for Small Molecule Binders. Front Immunol (2018) 9:1382. doi: 10.3389/fimmu.2018.01382
117. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T Cell Receptor Signaling Controls Foxp3 Expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA (2008) 105:7797–802. doi: 10.1073/pnas.0800928105
118. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25:1282–1293 e7. doi: 10.1016/j.cmet.2016.12.018
119. Chapman NM, Zeng H, Nguyen TM, Wang Y, Vogel P, Dhungana Y, et al. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis. Nat Commun (2018) 9:2095. doi: 10.1038/s41467-018-04392-5
120. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16:1174–84. doi: 10.1038/ni.3269
121. de Kivit S, Mensink M, Hoekstra AT, Berlin I, Derks RJE, Both D, et al. Stable Human Regulatory T Cells Switch to Glycolysis Following TNF Receptor 2 Costimulation. Nat Metab (2020) 2:1046–61. doi: 10.1038/s42255-020-00271-w
122. Ou JN, Wiedeman AE, Stevens AM. TNF-Alpha and TGF-Beta Counter-Regulate PD-L1 Expression on Monocytes in Systemic Lupus Erythematosus. Sci Rep (2012) 2:295. doi: 10.1038/srep00295
123. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JM, et al. Dual Targeting of TGF-Beta and PD-L1 via a Bifunctional Anti-PD-L1/TGF-betaRII Agent: Status of Preclinical and Clinical Advances. J Immunother Cancer (2020) 8:e000433. doi: 10.1136/jitc-2019-000433
124. Ablamunits V, Bisikirska B, Herold KC. Acquisition of Regulatory Function by Human CD8(+) T Cells Treated With Anti-CD3 Antibody Requires TNF. Eur J Immunol (2010) 40:2891–901. doi: 10.1002/eji.201040485
125. Govindaraj C, Scalzo-Inguanti K, Scholzen A, Li S, Plebanski M. TNFR2 Expression on CD25(hi)FOXP3(+) T Cells Induced Upon TCR Stimulation of CD4 T Cells Identifies Maximal Cytokine-Producing Effectors. Front Immunol (2013) 4:233. doi: 10.3389/fimmu.2013.00233
126. Mouttet D, Lae M, Caly M, Gentien D, Carpentier S, Peyro-Saint-Paul H, et al. Estrogen-Receptor, Progesterone-Receptor and HER2 Status Determination in Invasive Breast Cancer. Concordance Between Immuno-Histochemistry and MapQuant Microarray Based Assay. PloS One (2016) 11:e0146474. doi: 10.1371/journal.pone.0146474
127. Pizot C, Boniol M, Mullie P, Koechlin A, Boniol M, Boyle P, et al. Physical Activity, Hormone Replacement Therapy and Breast Cancer Risk: A Meta-Analysis of Prospective Studies. Eur J Cancer (2016) 52:138–54. doi: 10.1016/j.ejca.2015.10.063
128. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer Statistics. CA Cancer J Clin (2011) 61:69–90. doi: 10.3322/caac.20107
129. Yang F, Zhao Z, Zhao N. Clinical Implications of Tumor Necrosis Factor Receptor 2 in Breast Cancer. Oncol Lett (2017) 14:2393–8. doi: 10.3892/ol.2017.6410
130. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 Pathway From Discovery to Clinical Implementation. Front Immunol (2016) 7:550. doi: 10.3389/fimmu.2016.00550
Keywords: TNFR2, signaling pathway, immune response, immune checkpoint, cancer treatment, tumor immune microenvironment
Citation: Li M, Zhang X, Bai X and Liang T (2022) Targeting TNFR2: A Novel Breakthrough in the Treatment of Cancer. Front. Oncol. 12:862154. doi: 10.3389/fonc.2022.862154
Received: 25 January 2022; Accepted: 22 March 2022;
Published: 14 April 2022.
Edited by:
Afshin Derakhshani, University of Calgary, CanadaReviewed by:
Chien-Feng Li, National Health Research Institutes, TaiwanBehzad Mansoori, Molecular & Cellular Oncogenesis Program, United States
Copyright © 2022 Li, Zhang, Bai and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingbo Liang, liangtingbo@zju.edu.cn; Xueli Bai, shirleybai@zju.edu.cn