 Ezgi Elmas1,2
Ezgi Elmas1,2 Noushin Saljoughian2,3
Noushin Saljoughian2,3 Marcelo de Souza Fernandes Pereira2
Marcelo de Souza Fernandes Pereira2 Brian P. Tullius4
Brian P. Tullius4 Kinnari Sorathia2
Kinnari Sorathia2 Robin J. Nakkula2
Robin J. Nakkula2 Dean A. Lee2,5*
Dean A. Lee2,5* Meisam Naeimi Kararoudi2,3,5*
Meisam Naeimi Kararoudi2,3,5*- 1Molecular, Cellular and Developmental Biology Graduate Program, The Ohio State University, Columbus, OH, United States
- 2Center for Childhood Cancer and Blood Diseases, Abigail Wexner Research Institute at Nationwide Children’s Hospital, Columbus, OH, United States
- 3CRISPR/Gene Editing Core, Abigail Wexner Research Institute at Nationwide Children’s Hospital, Columbus, OH, United States
- 4Pediatric Cellular Therapy, AdventHealth for Children, Orlando, FL, United States
- 5Department of Pediatrics, The Ohio State University, Columbus, OH, United States
Antitumor activity of immune cells such as T cells and NK cells has made them auspicious therapeutic regimens for adaptive cancer immunotherapy. Enhancing their cytotoxic effects against malignancies and overcoming their suppression in tumor microenvironment (TME) may improve their efficacy to treat cancers. Clustered, regularly interspaced short palindromic repeats (CRISPR) genome editing has become one of the most popular tools to enhance immune cell antitumor activity. In this review we highlight applications and practicability of CRISPR/Cas9 gene editing and engineering strategies for cancer immunotherapy. In addition, we have reviewed several approaches to study CRISPR off-target effects.
Introduction
In recent years, adoptive T cell and NK cell therapies and immune checkpoint blockades have been successfully used in the clinic to improve immunotherapy for cancer. Immunotherapies with T and NK cells aim to overcome tumor-mediated immunosuppression and augment immunity against cancer (1–3). Adoptive T cell cancer immunotherapies comprehend tumor-infiltrating lymphocytes (TILs), transgenic T cell receptor (TCR)- T cell and chimeric antigen receptors (CAR)-T cell therapies (1). NK cell immunotherapies with cytokine stimulation, antibodies, and gene CAR-NK cells have been studied to overcome immunosuppression in cancers (2, 4). Although advancement in immunotherapy has been significant and durable, most cancer patients fail to respond to immunotherapy due to resistant tumor nature. Thus, we urgently need to find novel immunotherapies for cancer patients.
CRISPR/Cas9 gene-editing technology application has been widely studied and used in cancer immunotherapy research (5, 6). CRISPR method offers precise and powerful gene-editing efficiency in cancer and immunotherapy research. It has been used to identify essential genes as immune checkpoint targets, generate CAR-T and CAR-NK cells, construct TCR, understand signaling pathways, and screen for new druggable targets in immunotherapy (1, 7–10).
In this review, we describe the fundamentals of CRISPR gene editing in primary human T cells and NK cells. In addition, we highlight the applications of CRISPR/Cas9 technology in engineered T cells and NK cells and how it improves the immune cell function against cancers. Furthermore, several approaches to study off-target effects of CRISPR has been discussed.
CRISPR Gene Editing
CRISPR are classes of repeated DNA sequences that act in coordination with CRISPR-associated (Cas) genes to devote bacterial and archaeal immunity against foreign raider phages and plasmid DNA (11). This system has been tested in several human cells including primary immune cells such as T-cells and NK cells. CRISPR consists of three elements: tracer-RNA, crispr-RNA (complementary to the target gene) and the Cas nuclease protein (12). Recognition of the target gene by guideRNA (Tracer-RNA + crispr-RNA) bound to Cas protein results in double stranded break (DSB) (5, 13, 14). DSBs can be repaired by one of the two highly conserved competing repair mechanisms, named as nonhomologous end-joining (NHEJ) or homology-directed repair (HDR) pathways (15). NHEJ results in insertion/deletion (indel) of nucleotides at the Cas9 targeting site and causes a frame shift in coding region and introduces gene knock-out (15). On the other hand, HDR is essential for insertion of a transgene such as a DNA template encoding a CAR into the Cas9 targeting site through homology repairs when homologous arms for the flanking region of Cas9 targeting site are provided in the DNA template (5). The best approach to deliver CRISPR elements and the DNA template depends on the target tissue or cell, packaging capacity, immunotoxicity, tropism, and integration site (5). Viral delivery has been widely used for human cells. Some of them are non-integrative, like the adeno-associated viruses (AAV) and adenoviruses (AdV), while some are integrative, such as Retroviridae family (MLV; murine leukemia virus or HIV; human immunodeficiency virus) (16, 17). Stable expression of the CRISPR in human primary cells is challenging due to the activation of anti-viral activity of the cells especially in NK cells and expressing a big protein like Cas9 results in low efficiency (18, 19). Therefore, delivery of pre-transcribed gRNA and pre-translated Cas9 as Cas9/Ribonucleoprotein (Cas9/RNP) has been favorable in immune cells (20, 21). Generation CAR expressing immune cells by site-directed gene insertion has been shown to be successful in both NK and T-cells. In this approach the DNA encoding a CAR is delivered as an HDR template by AAV vectors following electroporation of Cas9/RNP (22, 23). Providing optimal homology arms for Cas9-targeting site in the HDR template would be challenging as AAV has a small packaging capacity (less than 5 kb) (24). We have shown that a minimum of 300bp homology arms is required for high efficiency of the transgene integration into the Cas9 targeting site (23).
Introduction to T Cells and Their Role in Cancer Immunotherapy
T cells are one of the most prominent components of the adaptive immune response. They can be distinguished from other lymphocytes by possessing TCR on their cell surface. T cells are developed in the thymus, and they recognize the antigen peptides presented by major histocompatibility complexes (MHC) class I and class II. T cells have two major CD8+ and CD4+ subtypes. CD8+ T cell refers to killer T cells, and CD4+ T cell refers to helper T cells. CD8+ killer T cells are involved in directly eradicating the virally infected cells as well as cancer cells. Even though T cells incredibly work and eliminate the most frustrating cancers, cancer remains one of the most devastating diseases globally and the leading cause of death. Conventional treatment options such as chemotherapy, radiotherapy and surgery have not been very effective in treating cancers. Recently, cell-based therapies, checkpoint blockades, cancer vaccines, oncolytic viruses and other forms of immunotherapies have shown promising clinical outcomes. T cell-based therapies are among the most efficient immunotherapies for cancer patients due to their eminent clinical efficacy (25). These new immunotherapies rely on the ability of T cells to eradicate tumors (26, 27). To enhance their antitumor activity and specificity, great interest in CAR- T cells has been evolved and have been used to treat hematologic malignancies and solid tumors. In autologous CAR-T cell-based therapies, the patient’s own T cells are genetically engineered to express a single-chain CAR which includes an antibody extracellular binding domain that recognizes a tumor cell surface antigen. Tumor antigen is recognized by extracellular domain of the CAR. Signaling activation is achieved by both costimulatory molecule such as CD27, CD28, 41BB and CD3zeta which contains ITAM motives (28). Thus, the engineered CAR-T cells can bind to tumor antigens and lyse the tumor cells independently from MHC, whereas normal T cells require TCR binding to an MHC class peptide antigen for their activation (19). Although CAR-T cell immunotherapies have been shown to be the most promising FDA approved cell based treatments, several challenges remain to be tackled (29). There has been some severe adverse events associated with CAR T cell toxicities (30–37). For example, most of the clinical trials use autologous T cells isolated from patients’ blood. This results in cell manufacturing failures from the early phase of the trial, due to low T cell quality and lymphocyte counts in some of the heavily treated patients (38). Manufacturing of autologous CAR T cell is a time-consuming process, therefore delaying the treatment in patients (33, 34). Additionally, when apheresis product is used for CAR-T cell production, sometimes failure in the process causes unsuccessful CAR-T cell manufacturing and poor response to treatment (30, 39–41). To overcome the problems related to autologous CAR-T cells, allogeneic CAR-T cell therapies has become alternative to autologous CAR-T cells (42–44). However, allogeneic CAR-T cell recognize and attack the recipient’s tissues causing graft-versus-host disease (GvHD) therefore limiting their use in the clinic (45–48). In addition to that, in both autologous and allogeneic CAR-T cells, side effects such as cytokine release syndrome (CRS) and neurologic toxicity in patients remains a challenge to overcome (34–37, 49–55). Efforts in gene-editing technologies such as CRISPR gene editing aid as a potential tool for overcoming the barriers in CAR-T immunotherapies (Figure 1) (27, 38, 56–62).
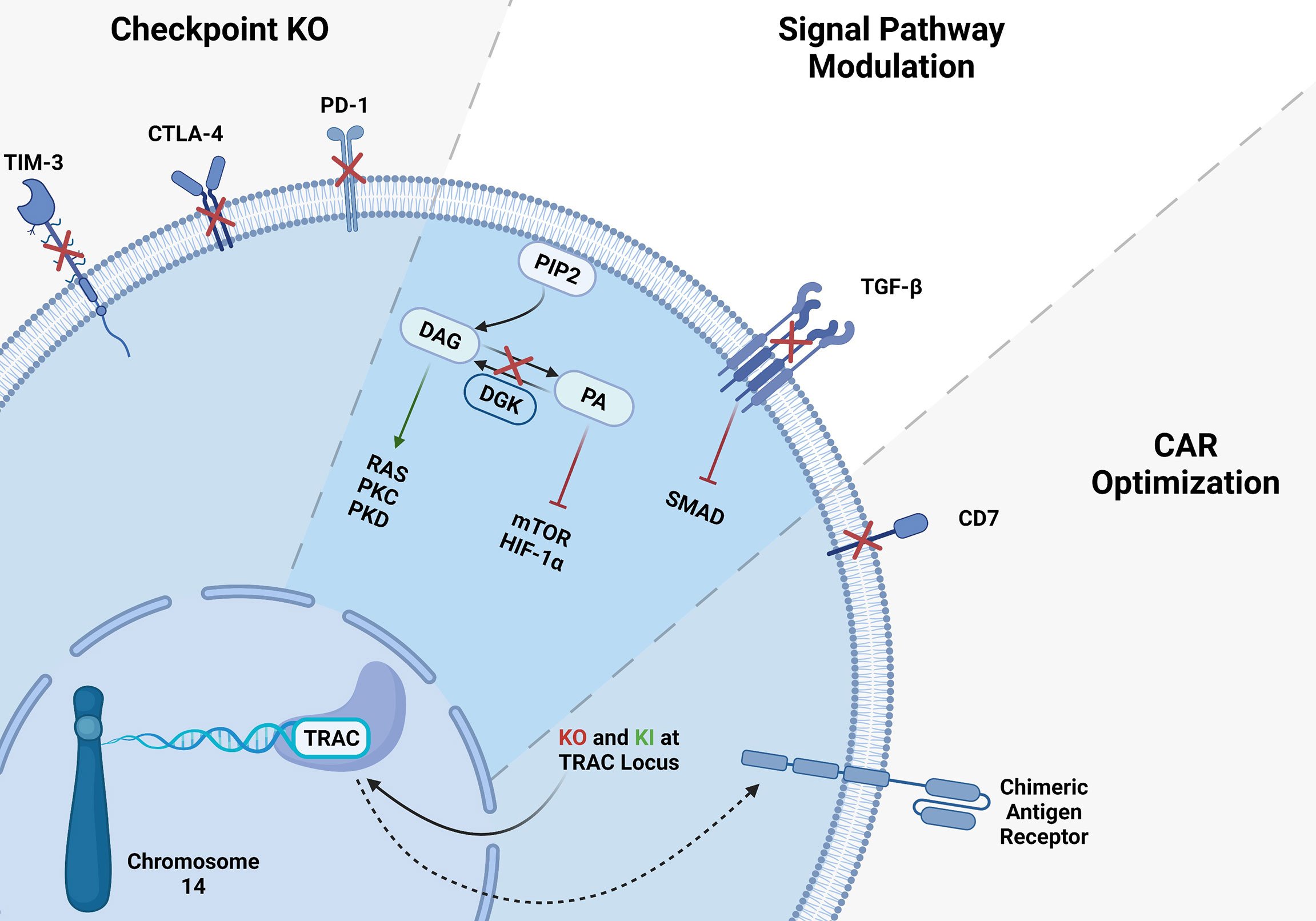
Figure 1 CRISPR gene editing in T-cells. Several gene KO and KI have been tested in T-cells, here we summarized the targeted genes. T cell checkpoint inhibitory receptor KO such as TIM3, CTLA-4 and PD-1 KO resulted in higher antitumor activity of T-cells. CAR-T cell signaling modulation via inhibition of immunosuppressive TGF-β signaling showed significant improvement of CAR-T cells. Integration of CAR-T in TRAC locus may solve the mentioned problems with allogeneic CAR-T therapies.
Examples of CRISPR Edited T Cells
Genome editing technologies facilitate remarkable, highly efficient, and specifically targeted genomic modifications. CRISPR/Cas9 technology has been the most practical and efficient gene-editing method among other strategies for editing the T cells (63–66). Producing off-the-shelf universal CAR- T cells, overcoming T cell exhaustion, and suppressive TME become significant obstacles which CRISPR can be a suitable tool to tackle those issues (Figure 1) (44, 63). Several groups have reported successful gene editing of T-cells using Cas9/RNP (66, 67). Electroporation of Cas9/RNP to edit T-cells has been very efficient and been successfully used in the clinic to treat cancers (68). To solve the limitations of antigen-specific and HLA-matched T cells and generate universal allogeneic CAR-T cells, genetically engineered TCR complexes were developed for immune therapy. Targeted gene editing in T cells has major advantages over lentiviral transduction platforms. For example, lentiviral transduction of TCR leads to variable transgene copy numbers and untargeted transgene integration and therefore initiates variable TCR expression and functionality. Oppositely, TCR editing with CRISPR/Cas9 allows high-efficient gene targeting and avoids random integration (63, 64). CRISPR/Cas9 strategy has also been used to target PD-1, CTLA-4, LAG-3, and TIM-3 inhibitory molecules to overcome tumor mediated immune suppression and enhance CAR-T cell function (22, 69, 70). It also has been shown that diacylglycerol kinase (DGK) CRISPR-Cas9 KO improves the anti-tumor activity of CAR-T cells (71). TGF-β receptor II (TGFBR2) KO with CRISPR/Cas9 was also shown to reduce CAR-T exhaustion and increase the anti-tumor activity of CAR-T cells (72). Inhibition of CD7 and TRAC using CRISPR/Cas9 enhances CAR-T cell-killing activity and prevents fratricide against T-ALL. Sterner et al. (73) showed that CRISPR/cas9 KO of granulocyte-macrophage colony-stimulating factor (GM-CSF) decreased the side effects like cytokine release syndrome and neuroinflammation of CAR-T cell therapies and also improved the CAR-T cell anti-tumor activity in-vivo (73). CRISPR/Cas9 gene not only used for KO, it has been also utilized for gene insertion of exogenous DNAs. Site directed gene knock-in (KI) has improved CAR-T cell antitumor efficiency (74, 75). Several approaches have been developed to deliver the DNA template encoding CARs. Schumann et al. introduced a HDR template into the CXCR4 gene locus by electroporation of a plasmid DNA and Cas9/RNP, and demonstrated successful site directed KI (75). Moreover, insertion of CD19 specific CAR expressing DNA into the TRAC locus has been achieved with the CRISPR/Cas9 method and improved CAR-T cell efficiency. To generate these cells, T cells were electroporated with Cas9 mRNA and gRNA. Next, the HDR template encoding CD19 CAR was delivered to the cells via AAV6 transduction (60). In the T cell engineering era, insertions or deletions of short sequences with CRISPR/Cas9 technology have been very effective, precise, and routinely used. However, it has also been possible to KI longer sequences using ssDNA inserts called the Easi-CRISPR method with high efficiency (74). Cas9 is the most used endonuclease protein in CRISPR systems, but other Cas proteins such as Cas12 or Cpf1 is also used to generated CAR-T cells when combined with AAV gene delivery (22). To generate CAR-T cells with simultaneous KO of checkpoints and knock-in of double CARs, a method called KIKO has been developed. This method uses AAV-Cpf1 to generate KO and double knock-in KIKO-CAR-T cells (22, 76).
Introduction to NK Cells
Natural Killer Cells (NK cells) are type of innate lymphocytes mediates anti-viral and anti-tumor activity. NK cells develop in the bone marrow (BM) and secondary lymphoid tissues such as, tonsils, spleen and lymph nodes (LNs) and they represent 5-20% of circulating lymphocytes in humans (77, 78). NK cells are distinguished from the other immune cells by possessing CD3- and CD56+ phenotype. Human NK cell subsets express also CD16 molecule, which is involved in antibody dependent cellular cytotoxicity (ADCC). NK cells are effector cytotoxic cells, they recognize and destroy their target without prior sensitization. Unlike T cells, they do not need MHC class presentation to enact their cytotoxic properties. Unlike T cells, NK cells recognize and kill tumor in an HLA-independent manner which result in being known as a great candidate for allogeneic anti-tumor cell-based therapies, as they do not cause acute GvHD (79–81). NK cells use KIR receptor and ligand mismatch to recognize cancer cells from self-cells, therefore mediating enhanced engraftment, anti-tumor response, and safe clinical outcomes (79, 81–85). NK cell killing of target cells accomplished with a balance of activating and inhibitory signals engaged around the cell. NK cell activating receptors includes, killer cell’s immunoglobulin-like receptors (KIRs), KIR2DS2, KIR2DS5 KIR3DS1, CD94/NKG2C, NKG2D, NKp30 NKp40, NKp44 and NKp46 recognize ligands present on target cells. NK cells have the ability of recognize non-self by NKp80, SLAM, CD18, CD2 and TLR3/9 receptors. Some of the NK cell inhibitory ligands are PD-1, TIGIT, TIM-3 and LAG-3. Inhibitory KIR ligands, KIR2DL1, 2DL2, and 2DL3 interact with highly polymorphic human leukocyte antigen (HLA). There are three HLA groups, group 1, 2 and HLA-Bw4, which usually bind inhibitory KIR and have long extracellular immunoglobulin structure. It has been shown that patients who receive NK cell immunotherapy containing haplo-mismatched NK cells they have anti-leukemic effects without the risk of GVHD. In hematopoietic stem cell transplant (HSCT) patients, infusions of haplo-mismatched KIR and HLA NK cells has shown benefits of survival and lower relapse rates. If the infused NK cells are identical, they only show benefit if the KIR receptors are activating (86, 87). NK cells can be isolated from peripheral blood, umbilical cord, and induced pluripotent stem cells (iPSCs) (88–91). Once isolated from their primary source, feeder cells, such membrane-bound IL-21 K562s, used to expand NK cells ex-vivo (92). They can be cultured anywhere from 14-21 days in most protocols and can proliferate remarkably over hundreds of folds (92). Cytokines such as IL-2, IL-12, IL-15, IL-18, and IL-21 are also added in NK cell cultures to enhance NK cell proliferation and activation (86, 87). NK cells have several mechanisms to eradicate their targets. One of the main mechanisms is perforin and granzyme induced apoptosis. Granzymes which are serine/proteases, packaged along with perforin and when they release by NK cells, they initiate target apoptosis via caspase-3 pathway. In addition to that, NK cells via Fas ligand and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) pathways can destroy their targets (93–95).
Examples of CRISPR Edited NK Cells
CRISPR editing of NK cells has been challenging, however we and others have shown that using electroporation of Cas9/RNP can solve the issue of low viral transduction efficiency of NK cells (18, 23, 96–103). Gene editing in NK cells in a short period since its invention has been used for serval applications such as to improve their metabolic function, knocking-out checkpoint molecules, improving antibody therapies and generation of CAR-NK (96). One great example of gene engineered NK cells is CD38 knock-out NK cells. NK cells highly express CD38 on their surface. Patients treated with daratumumab (Dara, hereafter), a monoclonal antibody targeting CD38 on multiple myeloma, showed a decrease in NK cells number. This is a result of NK-NK recognition through CD16 biding to Dara coated CD38+ NK cells, referred to as “fratricide.” Beyond the role of the structural marker, CD38 is well described to be associated with a large diversity of physiological and pathological conditions. Our group and others successfully developed NK cells lacking CD38 by introducing the CRISPR/Cas9 as Cas9/RNP via electroporation (96, 101). In particular, CD38 is an NAD-degradation enzyme in mammalian tissues (104–110). Our data demonstrated that CD38KO NK cells have more prominent metabolic profile, increased killing mediated by ADCC against CD38+ multiple myeloma cell lines and patient derived samples and are protected from fratricide mediated by daratumumab (96, 101).
Another important target to improve the NK cell’s function is CISH encoded by CIS gene. CISH has a critical impact on NK cells, and its activation is known to disable JAK-STAT downstream signaling pathways including a decline in NK cell ability to kill malignant cells (111, 112). Different groups have shown that CISH is overexpressed in the presence of IL-2 and IL-15 (113–115). IL15 was previously described as an important factor potentiating NK cells cytokine production and cytotoxicity activity (116–118). Felices et al. have demonstrated that prolonged administration of IL15 can unleash NK cells exhaustion via metabolic failure (119). Delconte et al. showed that CISH was quickly activated after IL15 stimulation in a mouse model, supporting that using gene-editing in NK cells to delete CISH seems to be advantageous (120). Using CRISPR/Cas9 on human iPSC to generate iPSC-CISH knockout NK cells displayed prolonged persistence in vivo and enhanced antitumor activity for acute myeloid leukemia (121, 122). NK cell checkpoint blockade has been used as a promising therapy for liquid and solid tumors. Other candidate for gene editing in NK cells is NKG2A which is an immune checkpoint in CD8+ αβ T cells, natural killer T cells (NKT) and CD56hi NK cells. Upon activation of immune cells, NKG2A leads to decreased effector function (123, 124). Data from the literature have shown that NKG2A drives NK cells to fatigue when highly expressed, and it can be predictive of poor prognosis in liver cancer patients (125). Thus, the blockage of the NKG2A receptor enhances NK cell’s effector function for immunotherapy (126–128). Similarly, Berrien-Elliot et al., have shown that gene-editing using CRISPR/CAS9 to delete NKG2A from human NK cells was able to increase NK cell ability to control HLA-E+ K562 leukemia when compared to control NK cells demonstrating a substantial inhibitory function for NKG2A (129). Additionally, NKG2AKO NK cells did not affect their persistence in NSG mouse model (129), however, the role of NKG2A in NK cells licensing may cause development of unlicensed NK cells with lower cytotoxic activity (130). It is very well established that the PD1/PD-L1 axis has an inhibitory function that can impair many T cells’ functions. This fact has been validated in preclinical models where the inhibition of this signaling cascade is used for cancer treatment (131). Indeed, high expression of PD1 ligand I or II in cancer cell lines impairs cytotoxic function on CD8+ T cells. On the other hand, the absence of a functional PD1 was responsible for tumors rejection in the murine model (132, 133). The blockage of the PD1/PD-L1 axis with monoclonal antibodies repair these effects and unleash T cells to effectively kill tumor cells (132–134). Recently it has been shown that in different malignancies, human NK cells also express PD-1 (135–139). Like T cells, blockade of the PD1/PD-L1 axis was able to activate NK response (140). However, such strategies present limitations, especially regarding off-target toxicity (102). Pomeroy et al. could generate PD1KO NK cells by electroporating mRNA Cas9 and gRNA (102). They demonstrated that PD1KO NK cells showed notably enhanced cytotoxicity and cytokine secretion in vitro and in vivo, decreasing tumor burden that culminated with survival (102). Another promising target for gene editing to boost cancer immunotherapy is the Suppressor of cytokine signaling 3 (SOCS3). The protein SOCS3 is one among eight members of the Suppressor of cytokine signaling family (SOCS1–7 and CIS). Those proteins downregulate cytokine signaling via the JAK/STAT signaling cascade. Murine NK cells upregulated SOCS3 expression after IL-15 stimulation (120). SOCS3 impair inflammation by inhibiting pro-inflammatory signaling pathways, including IL-12 inducing IL-12Rβ2 subunit blockage via the SH2 domain and its signaling pathway mediated by STAT4 (122). The absence of SOCS3 does not impact NK cells function upon IL15 stimulation in murine models. In humans NK cells, our group successfully generated SOCS3KO NK cells using Cas9/RNP and showed higher cell proliferation and enhanced NK cells anti-tumor activity (100). Suggesting SOCS3KO NK cells could be an excellent target for gene-editing to boost cancer immunotherapy. Another novel target is ADAM17, this gene has well described as a membrane-associated protease responsible for cleaving a large variety of membrane molecules, including CD16 (102, 141–144). Blocking ADAM17 activity leads to improvement in cytokine production of human NK cells due to maintaining their CD16 on the cell surface and activating higher ADCC when combined with antibodies (145). Pomeroy et al. have demonstrated that CRISPR-edited ADAM17KO NK cells are prevented against CD16 shedding compared to WT NK cells (102). Additionally, those data are similar to ADAM17 inhibitors where treated groups presented enhanced killing through ADCC. Similarly, Yamamoto et al. showed that ADAM17 gene-edited iPSCs derived NK cells have enhanced ADCC (102, 141, 144–146).
To improve immune cell recognition and killing towards tumor cells, immune cells, including T cells and NK cells are engineered to express chimeric antigen receptors (CARs) (147–149). In one of the first clinical trials using iPSC CD19-CAR NK cells, the patients treated with the CAR-expressing NK cells showed some improvements in their clinical outcomes (150). Generation of CAR-NK cells have been challenging due to the low efficient viral transduction including CAR-NK cells used in the trial mentioned above. Our group recently showed that we could efficiently combine Cas9/RNP approach with self-complementary (sc) Adeno-associated virus (AAV) or single-stranded gene delivery for generating highly efficient human primary CAR-NK cells (98). Using this approach, we developed CD33 CAR-NK cells (98). These CAR-NK were efficiently able to kill AML cells and showed improvement on their activation markers (98). Similar data were obtained when CD33-CARNK cells co-culture with patient samples (97, 98). Recently, Daher et al. showed that CRISPR edited CIS-KO NK cells expressing CAR-IL-15 construct could boost CAR-NK cell function in vitro and xenograft models by increasing aerobic glycolysis (121). This double enhancement of CAR-IL-15/CIS-KO signaling is significantly beneficial in the TME (151). Overall, gene editing of NK cells has been challenging but the recent successes in using CRISRP by electroporating Cas9/RNP helped to improve the outcome of the NK cells therapy (Figure 2) (18, 101, 103, 152, 153). There has been some evidence showing that Polymer-stabilized Cas9 nanoparticles and modified repair templates can increase genome editing efficiency. These modified nanoparticles improved knock-out and knock-in efficiency of the CRISPR gene editing in several primary cells such as NK and T cells (16).
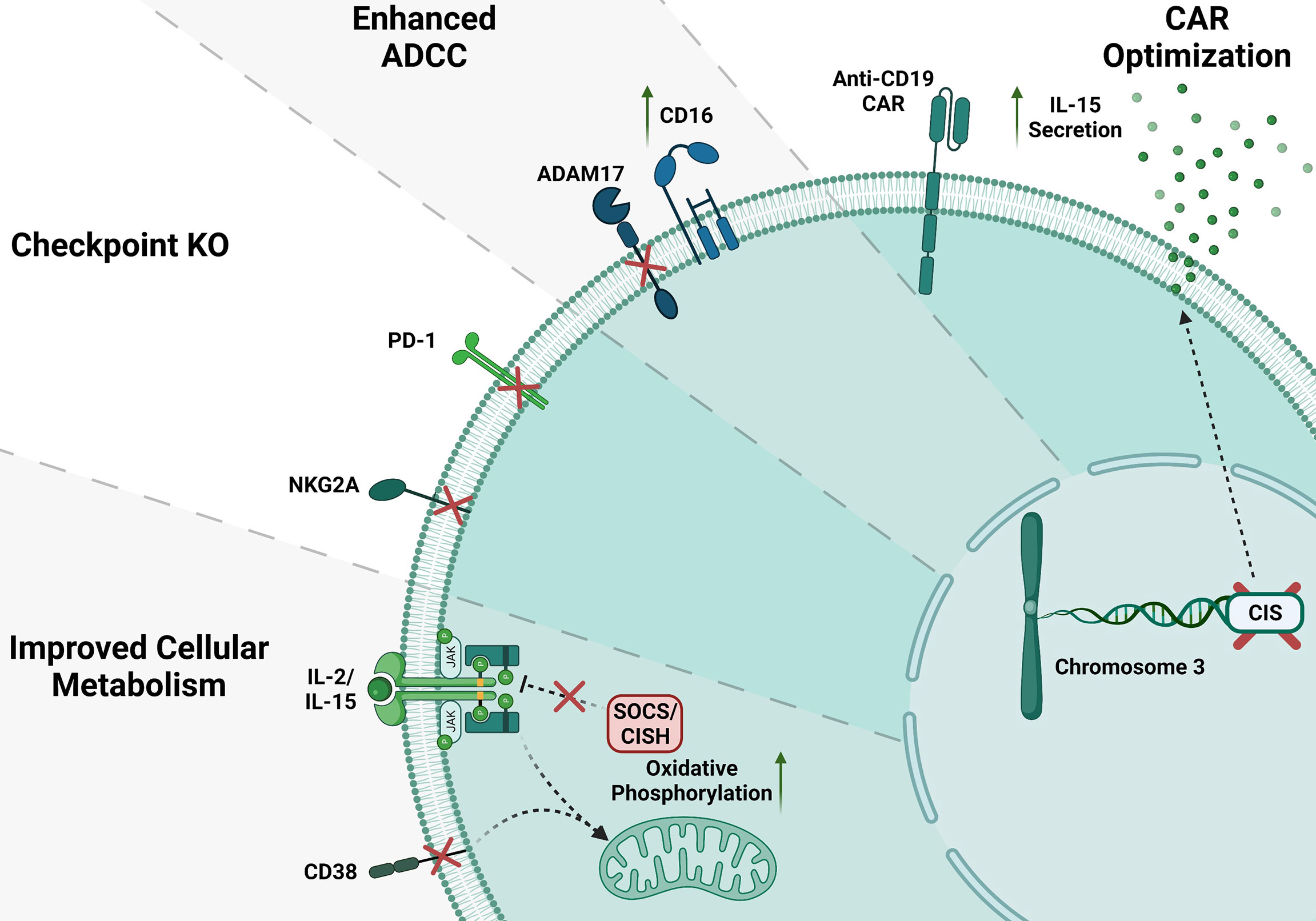
Figure 2 CRISPR gene editing in NK cells. Several gene KO in NK cells have been done to improve NK cell function; here, we show some of the NK cell gene modifications. CD38 and SOCS/CISH KO can improve metabolism in NK cells. Inhibitory checkpoint receptor KO such as NKG2A and PD-1 KO. ADAM17 KO enhance CD16 mediated ADCC. Anti-CD19 CAR NK cells increase IL-15 production and enhance NK cell anti-tumor activity.
Clinical Trials Using CRISPR Edited NK Cells and T Cells
Advancements in immunotherapy and gene therapy opened a new era for clinical trials to treat some hematological malignancies and solid tumors. Along with other platforms, CRISPR/Cas9 technology was adapted and brought up to the clinic to correct some mutations and boost immune responses. CRISPR/Cas9, as a precise gene-editing tool with minimal cytotoxicity and off-target effects, has become a promising approach to treat complex and refractory diseases. However, due to some limitations, including transduction efficiency, off-target mutations, ethical questions, and the deficiency in scientific risk assessment, CRISPR/Cas9 gene-editing clinical trials have not been prevalent, especially for T and NK cells. However, CRISPR has opened its way to the clinic. One of the first in human phase 1 clinical trial of using CRISPR engineered T cell have been used for patients with refractory cancers in the U.S. (clinicaltrials.gov; trial NCT03399448) (68). In this trial, endogenous TCR and immune checkpoint molecule PD-1 were targeted in T cells with CRISPR/Cas9 to improve immunotherapy in several refractory cancers. Two patients with advanced refractory myeloma and one with metastatic sarcoma were treated with these CRISPR-edited cells (68). The results of this trial demonstrated the safety of infusing CRISPR-edited ex-vivo expanded CAR-T cells in patients (151). Examples of some clinical trials with the CRISPR/Cas9 method in T cells are presented in Table 1. However, there are no registered CRISPR/Cas9 transduced CAR-NK cell clinical trials in the United States.
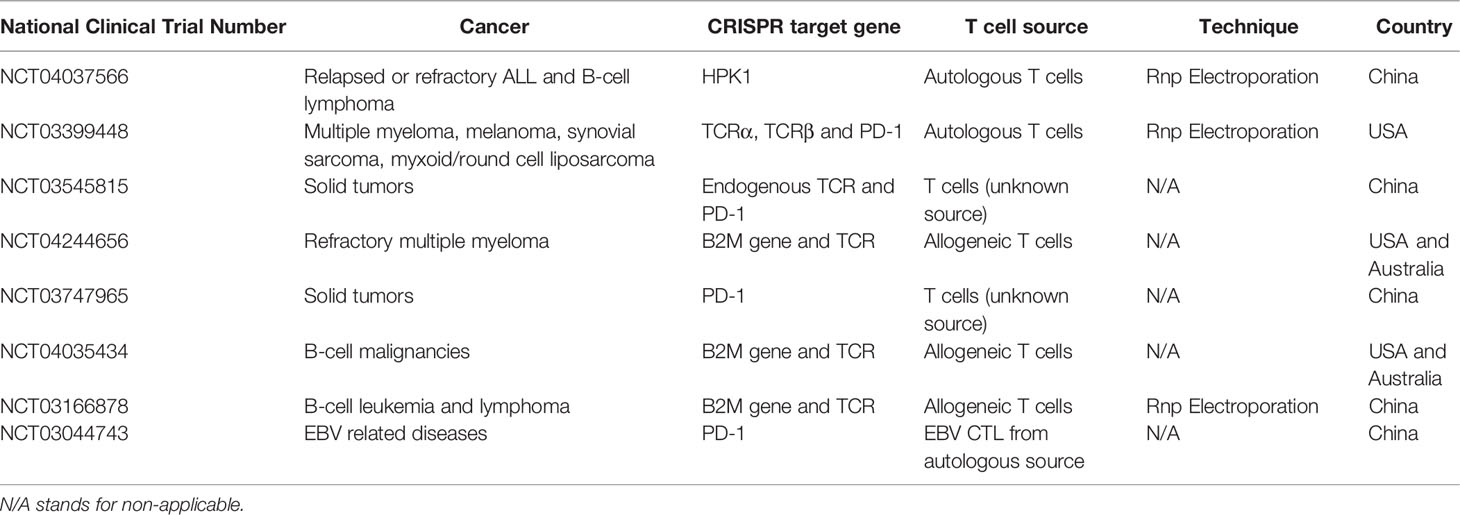
Table 1 Examples of clinical trials with CRISPR/Cas9 gene edited T cells (151).
Off-Target Analysis of CRISPR Edited Immune Cells
Recently by the promises of Cas9 endonuclease, researchers can target multiple genes in immune cells, including T cells and Natural killer (NK), to improve cancer immunotherapy. For these applications that lead to clinical cancer immunotherapy, the induced mutations by CRISPR-Cas9 should be highly precise and specific for the targeted loci with high on-target efficiency and low or no off-target activity. However, rare off-target events are inescapable during the manipulation of the gene of interest. This phenomenon requires scrutiny identification, especially in clinical applications to cure cancers and avoid adverse effects during cancer immunotherapy such as introduction of an oncogene. By developing next-generation sequencing (NGS) a survey of new functional and non-functional variations during gene manipulation became possible (154, 155). NGS has been broadly applied by researchers and employed in clinical trials due to its development in data acquisition with speedy and high-quality recognition (156, 157). Analyzing these NGS-generated data is even more critical to optimize and manage the workflow to fill the gap between massive data and scientific exploration. To date, several methods have been invented to analyze NGS data and off-target effects of CRISPR mediated mutations, such as GUIDE-seq, SITE-Seq, CHANGE-seq, Cas-OFFinder and Churchill (158–162). Some of them like GUIDE-seq, SITE-Seq and CHANGE-seq are based on the PCR amplification of pre-selected potential sites, which predicted by CRISPR/Cas9 design tools, and sequencing the PCR amplicons utilizing Sanger or NGS technologies (158–160, 163). For instance, Schumann et al., used a 2-step PCR method and sequenced with the amplicons with Illumina HiSeq, and identified indel mutations and their spatiality distribution in the target region in primary human T cells (75). In another study the efficiency and indel rates in the created CAR-T cells, using CRISPR-Cas9-mediated multiplex gene editing, was quantified by both surveyor assay and tracking of indels by decomposition (TIDE) analysis (58). Stadtmauer et al. utilized iGUIDE, a modified method of GUIDE-seq, for the Cas9-mediated cleavage specificity analysis in the engineered T cells to cure refractory cancer and found no clinical toxicities (68, 158, 164). Although these methods are simple and available to most molecular biology laboratories, they are not always precise as they are based on the predictions of potential off-target sites by CRISPR/Cas9 design tools in the genome of interest and therefore result in studying limited loci. As a matter of fact, DSBs happened beyond the predicted sites and may be ignored and caused detrimental side effects during the process of clinical cancer immunotherapy (163). This major disadvantage of off-target mutations identification by PCR based methods have been resolved by whole genome sequencing (WGS) which is unbiased and has been used to screen for off-target mutations induced by CRISPR/Cas9 in different cells including human inducible pluripotent stem cells, primary T cells, CAR-T cells (163, 165–167). Using this method, researchers can recognize both small indels and SNPs as well as major deletions, inversions, duplications and, rearrangements (163, 166). The only restriction of whole genome sequencing is missing the most low-frequent off-targets that happens to a small number of clones (163, 168). Cas-OFFinder algorithm have been invented in order to search for potential off-target sites in any sequenced genome regions (161). In a clinical trial, the safety and feasibility of CRISPR–Cas9 PD-1-edited T cells were confirmed after analyzing all the potential off-targets using Cas-OFFinder method in the treatment of lung cancer (169). More recently, as an ultra-fast, definite, highly scalable, and balanced parallelization strategy for discovering human genetic variation in clinical and population-scale genomics, Churchill has been applied for the analysis of next-generation sequencing data (162). We reported the high efficacy of Churchill analysis in verifying off-target events after deletion of CD38 in NK cells via Cas9/RNP and showed low off-target effects of Cas9/RNP (96). It has successfully revealed all the existing mutations and categorized them as missense and non-frameshift and moderate or high impact (96). Overall, WGS can provide more precise landscape of the off-target effects in CRISPR-edited cells. Here, we summarize and compare the current methods in off-target effects analyses of CRIPR edited immune cells (Table 2).

Table 2 Current methods in off-target analyses of CRISPR edited immune cells.
CRISPR Screening in Primary Immune Cells
Genome wide CRISPR screen has been used in several cancer cells to discover novel targets for cancer immunotherapy. CRISPR screening approach has not been extensively used in human primary immune cells due to several technical challenges. However, some studies have shown successful screening approaches in human primary T cells and Cas9-expressing transgenic mice in recent years (19, 170–172).. In general, to perform a CRISPR screen we need to introduce Cas9 and gRNA pool library into the cells (173). These molecules usually delivered to the target cells via lentiviral transduction. However, expressing large proteins such as cas9 using LV vectors in immune cells such as NK cells and T-cells has been challenging and results in low transduction efficiency. Shifrut et al; tested a hybrid approaching which the Cas9 was introduced to the gRNA library expressing cells via electroporation (19). They developed Single guide RNA (sgRNA) lentiviral infection with Cas9 protein electroporation (SLICE) and resulted in discovery of novel genes important in activation and expansion of CD8 T-cells (19). A similar approach was used by other groups to perform CRISPR screening in CAR-T cells (174). To date, there is no publication on CRISPR-screening on NK cells. Our group is investigating some new approaches to overcome issues related to lentiviral transduction of NK cells.
Conclusion
CRISPR gene editing technology has shown to be a very versatile tool for improving anti-tumor activity of NK cells and T-cells. We reviewed here some of the CRISPR edited cells used for cancer immunotherapy. We also reviewed ways to determine the off-target effects of CRISPR and emphasized that Cas9/RNP approach results in low off-target effects. We also mentioned how important information can be discovered by CRISPR screening approach and there are a lot to do the efficiently optimize this method to be used in NK cells and T cells. Overall, CRISPR gene editing shows promising clinical outcome and have potentials to be used more broad Clinical applications such as cancer immunotherapy using NK cells and T cells.
Author Contributions
The corresponding authors MNK and DAL supervised the authors for manuscript completion. The first author EE contributed to manuscript writing and revising. All authors contributed to the article and approved the submitted version.
Conflict of Interest
MN reports personal fees from Kiadis Pharma; in addition, MN has patents US62/825,007; WO2019222503A1; USPTO63/105,722; PCT/US2020/02545; US63/018,108; US62/928,524; US62/987,935; self-driving CAR with royalties paid by Kiadis Pharma. DL reports stock from Courier Therapeutics, personal fees and stock options from Caribou Biosciences, personal fees from Intellia Therapeutics, personal fees from Merck, Sharp, and Dohme, grants, stock, and personal fees from Kiadis Pharma, outside the submitted work; in addition, DL has patents US62/825,007; US63/105,722; US62928,524; PCT-US201/032,670; WO-2019/222,503-A1; PCT-US2020/018,384; US62/805,394; US62/987,935; US62/900,245; US62/815,625; Self-driving CAR with royalties paid to Kiadis Pharma and Membership on the NIH Novel and Exception Therapies and Research Advisory Committee (NExTRAC). MSFP reports stocks from MERCK, Fate Therapeutics, Sorrento Therapeutics, Moderna and received licensing fee from Kiadis Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ou X, Ma Q, Yin W, Ma X, He Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front Cell Dev Biol (2021) 9:674467. doi: 10.3389/fcell.2021.674467
2. Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan X. Genetic Reprogramming for NK Cell Cancer Immunotherapy With CRISPR/Cas9. Immunology (2019) 158:63–9. doi: 10.1111/imm.13094
3. O’Donnell JS, Teng MWL, Smyth MJ. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat Rev Clin Oncol (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
4. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK Cell-Based Immunotherapy for Malignant Diseases. Cell Mol Immunol (2013) 10:230–52. doi: 10.1038/cmi.2013.10
5. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (2013) 339:819–23. doi: 10.1126/science.1231143
6. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell (2014) 159:440–55. doi: 10.1016/j.cell.2014.09.014
7. Evers B, Jastrzebski K, Heijmans JP, Grernrum W, Beijersbergen RL, Bernards R. CRISPR Knockout Screening Outperforms shRNA and CRISPRi in Identifying Essential Genes. Nat Biotechnol (2016) 34:631–3. doi: 10.1038/nbt.3536
8. Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep (2016) 17:1193–205. doi: 10.1016/j.celrep.2016.09.079
9. Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat Commun (2018) 9:1849. doi: 10.1038/s41467-018-03917-2
10. Wei L, Lee D, Law CT, Zhang MS, Shen J, Chin DW, et al. Genome-Wide CRISPR/Cas9 Library Screening Identified PHGDH as a Critical Driver for Sorafenib Resistance in HCC. Nat Commun (2019) 10:4681. doi: 10.1038/s41467-019-12606-7
11. Wiedenheft B, Sternberg SH, Doudna JA. RNA-Guided Genetic Silencing Systems in Bacteria and Archaea. Nature (2012) 482:331–8. doi: 10.1038/nature10886
12. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (2012) 337:816–21. doi: 10.1126/science.1225829
13. Clemmensen OJ, Moll M, Arpi M, de Fine Olivarius N, Nielsen JB. [Bacteriological Autopsy. The Value of Postmortem Heart Blood Culture]. Ugeskr Laeger (1988) 150:101–3.
14. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-Programmed Genome Editing in Human Cells. Elife (2013) 2:e00471. doi: 10.7554/eLife.00471
15. Kass EM, Jasin M. Collaboration and Competition Between DNA Double-Strand Break Repair Pathways. FEBS Lett (2010) 584:3703–8. doi: 10.1016/j.febslet.2010.07.057
16. Nguyen DN, Roth TL, Li PJ, Chen PA, Apathy R, Mamedov MR, et al. Polymer-Stabilized Cas9 Nanoparticles and Modified Repair Templates Increase Genome Editing Efficiency. Nat Biotechnol (2020) 38:44–9. doi: 10.1038/s41587-019-0325-6
17. Verhoeyen E. Advances in Foamy Virus Vector Technology and Disease Correction Could Speed the Path to Clinical Application. Mol Ther (2012) 20:1105–7. doi: 10.1038/mt.2012.97
18. Naeimi Kararoudi M, Tullius BP, Chakravarti N, Pomeroy EJ, Moriarity BS, Beland K, et al. Genetic and Epigenetic Modification of Human Primary NK Cells for Enhanced Antitumor Activity. Semin Hematol (2020) 57:201–12. doi: 10.1053/j.seminhematol.2020.11.006
19. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-Wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell (2018) 175:1958–71.e15. doi: 10.1016/j.cell.2018.10.024
20. Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. Elife (2014) 3:e04766. doi: 10.7554/eLife.04766.010
21. Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, et al. Generation of Knock-Out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J Vis Exp (2018). doi: 10.3791/58237
22. Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-Step Generation of Modular CAR-T Cells With AAV-Cpf1. Nat Methods (2019) 16:247–54. doi: 10.1038/s41592-019-0329-7
23. Kararoudi MN, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. CRISPR-Targeted CAR Gene Insertion Using Cas9/RNP and AAV6 Enhances Anti-AML Activity of Primary NK Cells. bioRxiv (2021). 2021.03.17.435886. doi: 10.1101/2021.03.17.435886
24. Dong JY, Fan PD, Frizzell RA. Quantitative Analysis of the Packaging Capacity of Recombinant Adeno-Associated Virus. Hum Gene Ther (1996) 7:2101–12. doi: 10.1089/hum.1996.7.17-2101
25. Zhi L, Su X, Yin M, Zhang Z, Lu H, Niu Z, et al. Genetical Engineering for NK and T Cell Immunotherapy With CRISPR/Cas9 Technology: Implications and Challenges. Cell Immunol (2021) 369:104436. doi: 10.1016/j.cellimm.2021.104436
26. Taniuchi I. CD4 Helper and CD8 Cytotoxic T Cell Differentiation. Annu Rev Immunol (2018) 36:579–601. doi: 10.1146/annurev-immunol-042617-053411
27. Guedan S, Ruella M, June CH. Emerging Cellular Therapies for Cancer. Annu Rev Immunol (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
28. Terry RL, Meyran D, Fleuren EDG, Mayoh C, Zhu J, Omer N, et al. Chimeric Antigen Receptor T Cell Therapy and the Immunosuppressive Tumor Microenvironment in Pediatric Sarcoma. Cancers (2021) 13:4704. doi: 10.3390/cancers13184704
29. Pavlovic K, Tristán-Manzano M, Maldonado-Pérez N, Cortijo-Gutierrez M, Sánchez-Hernández S, Justicia-Lirio P, et al. Using Gene Editing Approaches to Fine-Tune the Immune System. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.570672
30. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
31. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy - Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
32. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-Up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
33. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
34. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
35. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
36. Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
37. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
38. Singh N, Shi J, June CH, Ruella M. Genome-Editing Technologies in Adoptive T Cell Immunotherapy for Cancer. Curr Hematol Malig Rep (2017) 12:522–9. doi: 10.1007/s11899-017-0417-7
39. Elavia N, Panch SR, McManus A, Bikkani T, Szymanski J, Highfill SL, et al. Effects of Starting Cellular Material Composition on Chimeric Antigen Receptor T-Cell Expansion and Characteristics. Transfusion (2019) 59:1755–64. doi: 10.1111/trf.15287
40. Tötterman TH, Carlsson M, Simonsson B, Bengtsson M, Nilsson K. T-Cell Activation and Subset Patterns are Altered in B-CLL and Correlate With the Stage of the Disease. Blood (1989) 74:786–92. doi: 10.1182/blood.V74.2.786.786
41. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
42. Qasim W. Allogeneic CAR T Cell Therapies for Leukemia. Am J Hematol (2019) 94:S50–4. doi: 10.1002/ajh.25399
43. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T Cells: More Than Ease of Access? Cells (2018) 7:155. doi: 10.3390/cells7100155
44. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-The-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat Rev Drug Discov (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
45. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I Trials Using Sleeping Beauty to Generate CD19-Specific CAR T Cells. J Clin Invest (2016) 126:3363–76. doi: 10.1172/JCI86721
46. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol (2016) 34:1112–21. doi: 10.1200/JCO.2015.64.5929
47. Zhang JP, Zhang R, Tsao ST, Liu YC, Chen X, Lu DP, et al. Sequential Allogeneic and Autologous CAR-T-Cell Therapy to Treat an Immune-Compromised Leukemic Patient. Blood Adv (2018) 2:1691–5. doi: 10.1182/bloodadvances.2018017004
48. Sanber K, Savani B, Jain T. Graft-Versus-Host Disease Risk After Chimeric Antigen Receptor T-Cell Therapy: The Diametric Opposition of T Cells. Br J Haematol (2021) 195:660–8. doi: 10.1111/bjh.17544
49. Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci Trans Med (2015) 7:303ra139–303ra139. doi: 10.1126/scitranslmed.aac5415
50. Turtle CJ, Hay KA, Hanafi L-A, Li D, Cherian S, Chen X, et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol (2017) 35:3010–20. doi: 10.1200/JCO.2017.72.8519
51. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-Cell Depletion and Remissions of Malignancy Along With Cytokine-Associated Toxicity in a Clinical Trial of Anti-CD19 Chimeric-Antigen-Receptor–Transduced T Cells. Blood (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
52. Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's Lymphoma With a Defined Ratio of CD8+ and CD4+ CD19-Specific Chimeric Antigen Receptor–Modified T Cells. Sci Trans Med (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
53. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
54. Neelapu SS. Managing the Toxicities of CAR T-Cell Therapy. Hematol Oncol (2019) 37:48–52. doi: 10.1002/hon.2595
55. Brudno JN, Kochenderfer JN. Recent Advances in CAR T-Cell Toxicity: Mechanisms, Manifestations and Management. Blood Rev (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
56. Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther (2016) 24:570–81. doi: 10.1038/mt.2015.197
57. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci Rep (2017) 7:737. doi: 10.1038/s41598-017-00462-8
58. Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-Mediated Multiplex Gene Editing in CAR-T Cells. Cell Res (2017) 27:154–7. doi: 10.1038/cr.2016.142
59. Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long Terminal Repeat CRISPR-CAR-Coupled "Universal" T Cells Mediate Potent Anti-Leukemic Effects. Mol Ther (2018) 26:1215–27. doi: 10.1016/j.ymthe.2018.02.025
60. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC Locus With CRISPR/Cas9 Enhances Tumour Rejection. Nature (2017) 543:113–7. doi: 10.1038/nature21405
61. Johnson LA, June CH. Driving Gene-Engineered T Cell Immunotherapy of Cancer. Cell Res (2017) 27:38–58. doi: 10.1038/cr.2016.154
62. Salas-Mckee J, Kong W, Gladney WL, Jadlowsky JK, Plesa G, Davis MM, et al. CRISPR/Cas9-Based Genome Editing in the Era of CAR T Cell Immunotherapy. Hum Vaccin Immunother (2019) 15:1126–32. doi: 10.1080/21645515.2019.1571893
63. Morgan MA, Büning H, Sauer M, Schambach A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-The-Shelf” CAR T and CAR NK Cells. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01965
64. Gao Q, Dong X, Xu Q, Zhu L, Wang F, Hou Y, et al. Therapeutic Potential of CRISPR/Cas9 Gene Editing in Engineered T-Cell Therapy. Cancer Med (2019) 8:4254–64. doi: 10.1002/cam4.2257
65. Omori K, Nagata N, Kurata K, Fukushima Y, Sekihachi E, Fujii N, et al. Inhibition of Stromal Cell-Derived Factor-1alpha/CXCR4 Signaling Restores the Blood-Retina Barrier in Pericyte-Deficient Mouse Retinas. JCI Insight (2018) 3. doi: 10.1172/jci.insight.120706
66. Seki A, Rutz S. Optimized RNP Transfection for Highly Efficient CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. J Exp Med (2018) 215:985–97. doi: 10.1084/jem.20171626
67. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol Immunother (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
68. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367:eaba7365. doi: 10.1126/science.aba7365
69. Muller TR, Jarosch S, Hammel M, Leube J, Grassmann S, Bernard B, et al. Targeted T Cell Receptor Gene Editing Provides Predictable T Cell Product Function for Immunotherapy. Cell Rep Med (2021) 2:100374. doi: 10.1016/j.xcrm.2021.100374
70. Morimoto T, Nakazawa T, Matsuda R, Nishimura F, Nakamura M, Yamada S, et al. CRISPR-Cas9-Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22073489
71. Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res (2018) 78:4692–703. doi: 10.1158/0008-5472.CAN-18-0030
72. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-Beta Inhibition via CRISPR Promotes the Long-Term Efficacy of CAR T Cells Against Solid Tumors. JCI Insight (2020) 5. doi: 10.1172/jci.insight.133977
73. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF Inhibition Reduces Cytokine Release Syndrome and Neuroinflammation But Enhances CAR-T Cell Function in Xenografts. Blood (2019) 133:697–709. doi: 10.1182/blood-2018-10-881722
74. Miura H, Quadros RM, Gurumurthy CB, Ohtsuka M. Easi-CRISPR for Creating Knock-in and Conditional Knockout Mouse Models Using Long ssDNA Donors. Nat Protoc (2018) 13:195–215. doi: 10.1038/nprot.2017.153
75. Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of Knock-in Primary Human T Cells Using Cas9 Ribonucleoproteins. Proc Natl Acad Sci USA (2015) 112:10437–42. doi: 10.1073/pnas.1512503112
76. Zhang X, Cheng C, Sun W, Wang H. Engineering T Cells Using CRISPR/Cas9 for Cancer Therapy. In: Sioud M, editor. RNA Interference and CRISPR Technologies: Technical Advances and New Therapeutic Opportunities. New York, NY: Springer US (2020). p. 419–33.
77. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01869
78. Scoville SD, Freud AG, Caligiuri MA. Modeling Human Natural Killer Cell Development in the Era of Innate Lymphoid Cells. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00360
79. Lupo KB, Matosevic S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers (2019) 11:769. doi: 10.3390/cancers11060769
80. Lee DA. Cellular Therapy: Adoptive Immunotherapy With Expanded Natural Killer Cells. Immunol Rev (2019) 290:85–99. doi: 10.1111/imr.12793
81. Lee DA, Denman CJ, Rondon G, Woodworth G, Chen J, Fisher T, et al. Haploidentical Natural Killer Cells Infused Before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biol Blood Marrow Transplant (2016) 22:1290–8. doi: 10.1016/j.bbmt.2016.04.009
82. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295:2097–100. doi: 10.1126/science.1068440
83. Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-Leukemia Activity of Alloreactive NK Cells in KIR Ligand-Mismatched Haploidentical HSCT for Pediatric Patients: Evaluation of the Functional Role of Activating KIR and Redefinition of Inhibitory KIR Specificity. Blood (2009) 113:3119–29. doi: 10.1182/blood-2008-06-164103
84. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful Adoptive Transfer and In Vivo Expansion of Human Haploidentical NK Cells in Patients With Cancer. Blood (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
85. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of Natural Killer Cell Alloreactivity in HLA-Mismatched Hematopoietic Stem Cell Transplantation. Blood (1999) 94:333–9. doi: 10.1182/blood.V94.1.333.413a31_333_339
86. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
87. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural Killer Cells in Cancer Biology and Therapy. Mol Cancer (2020) 19:120. doi: 10.1186/s12943-020-01238-x
88. Tomchuck S, Leung W, Dallas M. Isolation, Expansion and Function of Cord Blood Natural Killer Cells. (P2226). J Immunol (2013) 190:69.51–1. doi: 10.1016/j.bbmt.2012.11.234
89. Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, Purification, and Functional Assessment of Human Peripheral Blood NK Cells. J Vis Exp (2011) e2540. doi: 10.3791/2540
90. Melsen JE, Themeli M, van Ostaijen-Ten Dam MM, van Beelen E, Lugthart G, Hoeben RC, et al. Protocol for Isolation, Stimulation and Functional Profiling of Primary and iPSC-Derived Human NK Cells. Bio Protoc (2020) 10:e3845. doi: 10.21769/BioProtoc.3845
91. Hermanson DL, Ni Z, Kaufman DS. Human Pluripotent Stem Cells as a Renewable Source of Natural Killer Cells. In: Hematopoietic Differentiation of Human Pluripotent Stem Cells. Netherlands: Springer (2015). p. 69–79.
92. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PloS One (2012) 7:e30264. doi: 10.1371/journal.pone.0030264
93. Zhu Y, Huang B, Shi J. Fas Ligand and Lytic Granule Differentially Control Cytotoxic Dynamics of Natural Killer Cell Against Cancer Target. Oncotarget (2016) 7:47163–72. doi: 10.18632/oncotarget.9980
94. Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, et al. TRAIL Identifies Immature Natural Killer Cells in Newborn Mice and Adult Mouse Liver. Blood (2005) 105:2082–9. doi: 10.1182/blood-2004-08-3262
95. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of Murine NK Cell Cytotoxicity Requires the Translation of a Pre-Existing Pool of Granzyme B and Perforin mRNAs. Immunity (2007) 26:798–811. doi: 10.1016/j.immuni.2007.04.010
96. Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH, et al. CD38 Deletion of Human Primary NK Cells Eliminates Daratumumab-Induced Fratricide and Boosts Their Effector Activity. Blood (2020) 136:2416–27. doi: 10.1182/blood.2020006200
97. Naeimi Kararoudi M, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. Optimization and Validation of CAR Transduction Into Human Primary NK Cells Using CRISPR and AAV. SSRN Electron J (2021). doi: 10.2139/ssrn.3869896
98. Naeimi Kararoudi M, Likhite S, Elmas E, Schwartz M, Sorathia K, Yamamoto K, et al. CD33 Targeting Primary CAR-NK Cells Generated By CRISPR Mediated Gene Insertion Show Enhanced Anti-AML Activity. Blood (2020) 136:3–3. doi: 10.1182/blood-2020-142494
99. Naeimi Kararoudi M, Hejazi SS, Elmas E, Hellström M, Naeimi Kararoudi M, Padma AM, et al. Clustered Regularly Interspaced Short Palindromic Repeats/Cas9 Gene Editing Technique in Xenotransplantation. Front Immunol (2018) 9:1711–1. doi: 10.3389/fimmu.2018.01711
100. Kararoudi MN, Elmas E, Lamb MG, Chakravarti N, Trikha P, Lee DA. Disruption of SOCS3 Promotes the Anti-Cancer Efficacy of Primary NK Cells. Blood (2018). doi: 10.1182/blood-2018-99-116621
101. Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 Knockout Natural Killer Cells Expressing an Affinity Optimized CD38 Chimeric Antigen Receptor Successfully Target Acute Myeloid Leukemia With Reduced Effector Cell Fratricide. Haematologica (2020). doi: 10.3324/haematol.2020.271908
102. Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther (2020) 28:52–63. doi: 10.1016/j.ymthe.2019.10.009
103. Rautela J, Surgenor E, Huntington ND. Drug Target Validation in Primary Human Natural Killer Cells Using CRISPR RNP. J Leukoc Biol (2020) 108:1397–408. doi: 10.1002/JLB.2MA0620-074R
104. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction Through an SIRT3-Dependent Mechanism. Cell Metab (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
105. Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, et al. The Enzyme CD38 (a NAD Glycohydrolase, EC 3.2.2.5) Is Necessary for the Development of Diet-Induced Obesity. FASEB J (2007) 21:3629–39. doi: 10.1096/fj.07-8290com
106. Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, et al. Regulation of SIRT 1 Mediated NAD Dependent Deacetylation: A Novel Role for the Multifunctional Enzyme CD38. Biochem Biophys Res Commun (2006) 349:353–9. doi: 10.1016/j.bbrc.2006.08.066
107. Aksoy P, White TA, Thompson M, Chini EN. Regulation of Intracellular Levels of NAD: A Novel Role for CD38. Biochem Biophys Res Commun (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
108. Chiang SH, Harrington WW, Luo G, Milliken NO, Ulrich JC, Chen J, et al. Genetic Ablation of CD38 Protects Against Western Diet-Induced Exercise Intolerance and Metabolic Inflexibility. PloS One (2015) 10:e0134927. doi: 10.1371/journal.pone.0134927
109. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol Rev (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
110. Chini EN. CD38 as a Regulator of Cellular NAD: A Novel Potential Pharmacological Target for Metabolic Conditions. Curr Pharm Des (2009) 15:57–63. doi: 10.2174/138161209787185788
111. Krebs DL, Hilton DJ. SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells (2001) 19:378–87. doi: 10.1634/stemcells.19-5-378
112. Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of Cytokine Signaling: The SOCS Perspective. Cytokine Growth Factor Rev (2013) 24:241–8. doi: 10.1016/j.cytogfr.2013.03.005
113. Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative Regulation of Cytokine Signaling and Immune Responses by SOCS Proteins. Arthritis Res Ther (2005) 7:100–10. doi: 10.1186/ar1741
114. Hanada T, Kinjyo I, Inagaki-Ohara K, Yoshimura A. Negative Regulation of Cytokine Signaling by CIS/SOCS Family Proteins and Their Roles in Inflammatory Diseases. Rev Physiol Biochem Pharmacol (2003) 149:72–86. doi: 10.1007/s10254-003-0015-z
115. Inagaki-Ohara K, Hanada T, Yoshimura A. Negative Regulation of Cytokine Signaling and Inflammatory Diseases. Curr Opin Pharmacol (2003) 3:435–42. doi: 10.1016/S1471-4892(03)00070-5
116. Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, et al. Interleukin (IL) 15 is a Novel Cytokine That Activates Human Natural Killer Cells via Components of the IL-2 Receptor. J Exp Med (1994) 180:1395–403. doi: 10.1084/jem.180.4.1395
117. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 Trans-Presentation Promotes Human NK Cell Development and Differentiation In Vivo. J Exp Med (2009) 206:25–34. doi: 10.1084/jem.20082013
118. Geller MA, Miller JS. Use of Allogeneic NK Cells for Cancer Immunotherapy. Immunotherapy (2011) 3:1445–59. doi: 10.2217/imt.11.131
119. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous Treatment With IL-15 Exhausts Human NK Cells via a Metabolic Defect. JCI Insight (2018) 3. doi: 10.1172/jci.insight.96219
120. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a Potent Checkpoint in NK Cell-Mediated Tumor Immunity. Nat Immunol (2016) 17:816–24. doi: 10.1038/ni.3470
121. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a Cytokine Checkpoint Enhances the Fitness of Armored Cord Blood CAR-NK Cells. Blood (2021) 137:624–36. doi: 10.1182/blood.2020007748
122. Yamamoto K, Yamaguchi M, Miyasaka N, Miura O. SOCS-3 Inhibits IL-12-Induced STAT4 Activation by Binding Through its SH2 Domain to the STAT4 Docking Site in the IL-12 Receptor Beta2 Subunit. Biochem Biophys Res Commun (2003) 310:1188–93. doi: 10.1016/j.bbrc.2003.09.140
123. Jin S, Deng Y, Hao JW, Li Y, Liu B, Yu Y, et al. NK Cell Phenotypic Modulation in Lung Cancer Environment. PloS One (2014) 9:e109976. doi: 10.1371/journal.pone.0109976
124. Creelan BC, Antonia SJ. The NKG2A Immune Checkpoint - a New Direction in Cancer Immunotherapy. Nat Rev Clin Oncol (2019) 16:277–8. doi: 10.1038/s41571-019-0182-8
125. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A Expression Contributes to NK Cell Exhaustion and Predicts a Poor Prognosis of Patients With Liver Cancer. Oncoimmunology (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
126. Li F, Wei H, Wei H, Gao Y, Xu L, Yin W, et al. Blocking the Natural Killer Cell Inhibitory Receptor NKG2A Increases Activity of Human Natural Killer Cells and Clears Hepatitis B Virus Infection in Mice. Gastroenterology (2013) 144:392–401. doi: 10.1053/j.gastro.2012.10.039
127. Cichocki F, Miller JS. Setting Traps for NKG2A Gives NK Cell Immunotherapy a Fighting Chance. J Clin Invest (2019) 129:1839–41. doi: 10.1172/JCI128480
128. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking Expression of Inhibitory Receptor NKG2A Overcomes Tumor Resistance to NK Cells. J Clin Invest (2019) 129:2094–106. doi: 10.1172/JCI123955
129. Berrien-Elliott MM, Pamela W, Neal C, Wagner JA, Becker-Hapak M, Schappe T, et al. Primary Human NK Cell Gene-Editing Reveals a Critical Role for NKG2A in Cytokine-Induced Memory-Like NK Cell Responses. Blood (2019) 134:3237–7. doi: 10.1182/blood-2019-129162
130. Zhang X, Feng J, Chen S, Yang H, Dong Z. Synergized Regulation of NK Cell Education by NKG2A and Specific Ly49 Family Members. Nat Commun (2019) 10:5010. doi: 10.1038/s41467-019-13032-5
131. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
132. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by Monoclonal Antibodies Potentiates Cancer Therapeutic Immunity. Cancer Res (2005) 65:1089–96. doi: 10.1093/intimm/dxq049
133. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on Tumor Cells in the Escape From Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc Natl Acad Sci USA (2002) 99:12293–7. doi: 10.1073/pnas.192461099
134. Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, et al. B7-H1 Blockade Augments Adoptive T-Cell Immunotherapy for Squamous Cell Carcinoma. Cancer Res (2003) 63:6501–5. doi: 10.1158/0008-5472.1089.65.3
135. Benson DM Jr., Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell Versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti-PD-1 Antibody. Blood (2010) 116:2286–94. doi: 10.1182/blood-2010-02-271874
136. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 Mediates Functional Exhaustion of Activated NK Cells in Patients With Kaposi Sarcoma. Oncotarget (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
137. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a Subset of Human Natural Killer Cells Expressing High Levels of Programmed Death 1: A Phenotypic and Functional Characterization. J Allergy Clin Immunol (2017) 139:335–46.e3. doi: 10.1016/j.jaci.2016.04.025
138. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased Expression of Programmed Cell Death Protein 1 on NK Cells Inhibits NK-Cell-Mediated Anti-Tumor Function and Indicates Poor Prognosis in Digestive Cancers. Oncogene (2017) 36:6143–53. doi: 10.1038/onc.2017.209
139. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages is More Prominent in Hodgkin Lymphoma Than DLBCL. Blood (2018) 131:1809–19. doi: 10.1182/blood-2017-07-796342
140. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128:4654–68. doi: 10.1172/JCI99317
141. Arribas J, Esselens C. ADAM17 as a Therapeutic Target in Multiple Diseases. Curr Pharm Des (2009) 15:2319–35. doi: 10.2174/138161209788682398
142. Mishra HK, Ma J, Walcheck B. Ectodomain Shedding by ADAM17: Its Role in Neutrophil Recruitment and the Impairment of This Process During Sepsis. Front Cell Infect Microbiol (2017) 7:138. doi: 10.3389/fcimb.2017.00138
143. Zunke F, Rose-John S. The Shedding Protease ADAM17: Physiology and Pathophysiology. Biochim Biophys Acta Mol Cell Res (2017) 1864:2059–70. doi: 10.1016/j.bbamcr.2017.07.001
144. Wu J, Mishra HK, Walcheck B. Role of ADAM17 as a Regulatory Checkpoint of CD16A in NK Cells and as a Potential Target for Cancer Immunotherapy. J Leukoc Biol (2019) 105:1297–303. doi: 10.1002/JLB.2MR1218-501R
145. Mishra HK, Pore N, Michelotti EF, Walcheck B. Anti-ADAM17 Monoclonal Antibody MEDI3622 Increases IFNgamma Production by Human NK Cells in the Presence of Antibody-Bound Tumor Cells. Cancer Immunol Immunother (2018) 67:1407–16. doi: 10.1007/s00262-018-2193-1
146. Yamamoto K, Blum R, Kaufman DS. ADAM17-Deficient Pluripotent Stem Cell-Derived Natural Killer Cells Possess Improved Antibody-Dependent Cellular Cytotoxicity and Antitumor Activity. Blood (2020) 136:2–2. doi: 10.1182/blood-2020-137766
147. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-Specific TCR-Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat Med (2015) 21:914–21. doi: 10.1038/nm.3910
148. Stern LA, Jonsson VD, Priceman SJ. CAR T Cell Therapy Progress and Challenges for Solid Tumors. Cancer Treat Res (2020) 180:297–326. doi: 10.1007/978-3-030-38862-1_11
149. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
150. Aherne CM, Collins CB, Rapp CR, Olli KE, Perrenoud L, Jedlicka P, et al. Coordination of ENT2-Dependent Adenosine Transport and Signaling Dampens Mucosal Inflammation. JCI Insight (2018) 3. doi: 10.1172/jci.insight.121521
151. Basar R, Daher M, Rezvani K. Next-Generation Cell Therapies: The Emerging Role of CAR-NK Cells. Hematol Am Soc Hematol Educ Prog (2020) 2020:570–8. doi: 10.1182/hematology.2020002547
152. Robbins GM, Wang M, Pomeroy EJ, Moriarity BS. Nonviral Genome Engineering of Natural Killer Cells. Stem Cell Res Ther (2021) 12:350. doi: 10.1186/s13287-021-02406-6
153. Huang RS, Lai MC, Shih HA, Lin S. A Robust Platform for Expansion and Genome Editing of Primary Human Natural Killer Cells. J Exp Med (2021) 218. doi: 10.1084/jem.20201529
154. Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, et al. Systematic Investigation of Genetic Vulnerabilities Across Cancer Cell Lines Reveals Lineage-Specific Dependencies in Ovarian Cancer. Proc Natl Acad Sci USA (2011) 108:12372–7. doi: 10.1073/pnas.1109363108
155. International Human Genome Sequencing Consortium. Finishing the Euchromatic Sequence of the Human Genome. Nature (2004) 431:931–45. doi: 10.1038/nature03001
156. Mardis ER. A Decade's Perspective on DNA Sequencing Technology. Nature (2011) 470:198–203. doi: 10.1038/nature09796
157. Brownstein CA, Beggs AH, Homer N, Merriman B, Yu TW, Flannery KC, et al. An International Effort Towards Developing Standards for Best Practices in Analysis, Interpretation and Reporting of Clinical Genome Sequencing Results in the CLARITY Challenge. Genome Biol (2014) 15:R53. doi: 10.1186/gb-2014-15-3-r53
158. Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-Seq Enables Genome-Wide Profiling of Off-Target Cleavage by CRISPR-Cas Nucleases. Nat Biotechnol (2015) 33:187–97. doi: 10.1038/nbt.3117
159. Cameron P, Fuller CK, Donohoue PD, Jones BN, Thompson MS, Carter MM, et al. Mapping the Genomic Landscape of CRISPR-Cas9 Cleavage. Nat Methods (2017) 14:600–6. doi: 10.1038/nmeth.4284
160. Lazzarotto CR, Malinin NL, Li Y, Zhang R, Yang Y, Lee G, et al. CHANGE-Seq Reveals Genetic and Epigenetic Effects on CRISPR-Cas9 Genome-Wide Activity. Nat Biotechnol (2020) 38:1317–27. doi: 10.1038/s41587-020-0555-7
161. Bae S, Park J, Kim JS. Cas-OFFinder: A Fast and Versatile Algorithm That Searches for Potential Off-Target Sites of Cas9 RNA-Guided Endonucleases. Bioinformatics (2014) 30:1473–5. doi: 10.1093/bioinformatics/btu048
162. Kelly BJ, Fitch JR, Hu Y, Corsmeier DJ, Zhong H, Wetzel AN, et al. Churchill: An Ultra-Fast, Deterministic, Highly Scalable and Balanced Parallelization Strategy for the Discovery of Human Genetic Variation in Clinical and Population-Scale Genomics. Genome Biol (2015) 16:6. doi: 10.1186/s13059-014-0577-x
163. Zischewski J, Fischer R, Bortesi L. Detection of on-Target and Off-Target Mutations Generated by CRISPR/Cas9 and Other Sequence-Specific Nucleases. Biotechnol Adv (2017) 35:95–104. doi: 10.1016/j.biotechadv.2016.12.003
164. Nobles CL, Reddy S, Salas-McKee J, Liu X, June CH, Melenhorst JJ, et al. iGUIDE: An Improved Pipeline for Analyzing CRISPR Cleavage Specificity. Genome Biol (2019) 20:14. doi: 10.1186/s13059-019-1625-3
165. Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, et al. Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human iPSCs. Cell Stem Cell (2014) 15:12–3. doi: 10.1016/j.stem.2014.06.011
166. Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, et al. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell (2014) 15:27–30. doi: 10.1016/j.stem.2014.04.020
167. Yang L, Grishin D, Wang G, Aach J, Zhang CZ, Chari R, et al. Targeted and Genome-Wide Sequencing Reveal Single Nucleotide Variations Impacting Specificity of Cas9 in Human Stem Cells. Nat Commun (2014) 5:5507. doi: 10.1038/ncomms6507
168. Wu X, Kriz AJ, Sharp PA. Target Specificity of the CRISPR-Cas9 System. Quant Biol (2014) 2:59–70. doi: 10.1007/s40484-014-0030-x
169. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients With Refractory non-Small-Cell Lung Cancer. Nat Med (2020) 26:732–40. doi: 10.1038/s41591-020-0840-5
170. Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell (2019) 178:1189–204.e23. doi: 10.1016/j.cell.2019.07.044
171. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 Programs Long-Lived Effector T Cells for Cancer Therapy. Nature (2019) 576:471–6. doi: 10.1038/s41586-019-1821-z
172. Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, et al. A Genome-Wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell (2015) 162:675–86. doi: 10.1016/j.cell.2015.06.059
173. Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. Genome-Scale CRISPR-Cas9 Knockout and Transcriptional Activation Screening. Nat Protoc (2017) 12:828–63. doi: 10.1038/nprot.2017.016
Keywords: NK cells, CRISPR, T cell, immunotherapy, off-target analysis, CRISPR screening, CAR-T cells, CAR-NK cell
Citation: Elmas E, Saljoughian N, de Souza Fernandes Pereira M, Tullius BP, Sorathia K, Nakkula RJ, Lee DA and Naeimi Kararoudi M (2022) CRISPR Gene Editing of Human Primary NK and T Cells for Cancer Immunotherapy. Front. Oncol. 12:834002. doi: 10.3389/fonc.2022.834002
Received: 12 December 2021; Accepted: 07 March 2022;
Published: 05 April 2022.
Edited by:
Massimo Fantini, Precision Biologics, Inc., United StatesReviewed by:
Ramon Arens, Leiden University Medical Center, NetherlandsHiroki Torikai, University of Texas MD Anderson Cancer Center, United States
Copyright © 2022 Elmas, Saljoughian, de Souza Fernandes Pereira, Tullius, Sorathia, Nakkula, Lee and Naeimi Kararoudi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meisam Naeimi Kararoudi, meisam.naeimikararoudi@nationwidechildrens.org; Dean A. Lee, Dean.Lee@nationwidechildrens.org