 Hongshi Deng1†
Hongshi Deng1† Na Feng
Na Feng Shuzhen Liu
Shuzhen Liu Jiang I. Wu
Jiang I. Wu Xuanming Shi
Xuanming Shi- 1School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui, China
- 2Department of Physiology, University of Texas Southwestern Medical Center, Dallas, TX, United States
Previously we uncovered the epigenetic regulation of medulloblastoma that low levels of H3K27me3 are required for Shh target gene expression and medulloblastoma growth. Since Jmjd3, an H3K27me3 demethylase, is responsible for maintaining low H3K27me3 at Shh target genes, targeting Jmjd3 could be an efficient way to inhibit Shh signaling and medulloblastoma growth. Here we show that the small molecule GSK-J4, an inhibitor of Jmjd3, significantly inhibited the expression of Shh target genes in Shh responsive cell models and primary cerebellar granule neuron precursors. GSK-J4 also significantly reduced the growth of primary Shh medulloblastoma cultures. Treating human medulloblastoma cell line DaoY by GSK-J4 led to cell cycle arrest at G0/G1 phase with decreased cells in S-phase. Tumor cell proliferation was significantly inhibited by GSK-J4 treatment. Gene expression analyses showed that GSK-J4 additionally constrained the expression of key genes in cholesterol biosynthesis. Our results highlight the possibility that targeting H3K27me3 demethylase Jmjd3 with GSK-J4 to inhibit Shh signaling and cholesterol metabolism is a potential application to treat Shh medulloblastoma.
Introduction
Shh ligand covalently modified with N-terminal palmitoylation and C-terminal cholesterol plays essential roles in mammalian embryonic development, cell homeostasis and tumor formation (1). When Shh ligand binds to its receptor Patched (Ptch), Smoothened (Smo) is released from Ptch to activate transcription activator Gli1/2-Act. The translocated Gli1/2-Act in the nucleus activates Shh signaling by increasing the expression of general Shh target genes such as Gli1, Ptch1 and Hhip (2, 3). During cerebellar development, Shh molecules are produced from Purkinje neurons and activate mitogenic target genes such as N-myc and Ccnd1 in cerebellar granule neural precursors (CGNPs). Hence, Shh drives the proliferation of CGNPs and contributes to cerebellar development (4–6). During normal cerebellar development, Shh levels are properly modulated and reach a peak at an early postnatal stage. However, the overactive Shh signaling caused by mutations in Shh pathway genes such as Smo and Ptch1 is the driving force for Shh-type medulloblastoma (7, 8). SmoM2 is a Smo mutant resulting in constitutively active Shh signaling (9). This mutant has been widely used to establish mouse Shh medulloblastoma tumor models (10, 11).
Medulloblastoma is the most common malignant pediatric brain tumor, and accounts for about 20% of all childhood brain tumors (12, 13). Genomic studies classified medulloblastoma into four subgroups: Wnt, Shh, Group 3 and Group 4 (8, 14–17). The Wnt and Shh groups were named according to the principally activated signaling pathways in the tumors. Wnt-type tumors arise from the embryonic brain stem and lower rhombic lip progenitor cells (18). In contrast, Shh-type tumors originate from CGNPs with active Shh signaling (19, 20). This type of tumor is often found in infants and adults and accounts for about 25% of all medulloblastoma (11). Group 3 tumor features large cell/anaplastic phenotype and is driven by oncogenes c-myc and OTX2 (18, 21, 22). The cell origin of Group 4 medulloblastoma is thought to originate from unipolar brush cells (23–25). Current therapies for medulloblastoma consist of surgery, radiotherapy and chemotherapy. The standard radiotherapy includes craniospinal irradiation with a radiation boost to tumor bed. More often, radiotherapy medicated by Linac-based photons is being replaced by proton-beam based radiotherapy. Although prognosis is improved, patients still have severe long-term side effects (8, 26). Having a better understanding of both the active Shh signaling pathway and cell of origin provides possibilities of targeted therapies for medulloblastoma (27).
Epigenetic regulators play important roles in Shh signaling and medulloblastoma development (10, 11, 28–31). Previously we identified histone 3 lysine 27 trimethylation (H3K27me3) demethylase Jmjd3 as a key player in Shh target gene activation (10). We showed that Jmjd3 mediated H3K27me3 demethylation and epigenetic changes are required for the activation of Shh target genes in response to Shh stimulation (11, 31). Genetic knockout of Jmjd3 significantly inhibited Shh medulloblastoma growth in culture and in mouse models (10). Therefore, pharmacological inhibition of Jmjd3 could be a promising treatment option for medulloblastoma. Kruidenier et al. reported that an ethyl ester GSK-J4 inhibits H3K27me3 demethylase activities (32). By targeting Jmjd3, GSK-J4 has been successfully applied to inhibit the growth and proliferation of various tumors including pediatric brainstem glioma, chondrosarcomas, and lung adenocarcinoma in mouse xenograft models or in vivo studies (33–35).
In this report, we demonstrated that GSK-J4 efficiently inhibits Shh signaling and Shh-type medulloblastoma growth. We confirmed the notion in several Shh responsive cell models including immortalized fibroblasts NIH3T3, primary MEFs, normal CGNPs, primary mouse tumor cells, and human medulloblastoma cell line DaoY. In DaoY medulloblastoma cells, GSK-J4 additionally inhibits tumor cholesterol metabolism, which contributes to Shh signaling and cell proliferation. These results highlighted the potential translation to use the small molecule inhibitor GSK-J4 targeting H3K27me demethylase Jmjd3 for Shh-type medulloblastoma treatment.
Materials and methods
Mice
The SmoM2 transgenic mice were purchased from Jackson Laboratory (Strain #:005130). SmoM2 CAG-CreER mice spontaneously develop Shh-type medulloblastoma (9).
Cell lines, primary MEF, CGNP and medulloblastoma cell cultures
NIH3T3 cells, DaoY cells, wild type MEF cells, Gli3-/- and Jmjd3-/- MEF cells were cultured in DMEM containing 10% FBS, sodium pyruvate, penicillin, streptomycin, minimal amino acid, L-glutamine and 2-mercaptoethanol. Jmjd3-/- MEF cells were provided by Dr. T. Akira (36). Primary CGNP cultures were derived from dissociated P4 wild type mouse cerebella and cultured in DMEM/F12 media containing 25 mM KCl, N2, penicillin, streptomycin, and 10% FBS as previously described (31). For Shh induction, Shh conditioned media produced from Shh-CM 293T cells (37) were added to MEF and CGNP cultures at 1:20 dilution. NIH3T3 cells or primary MEF cells were treated with Shh in 0.5% FBS media for 24 hours before harvesting. Primary tumor cells were derived from dissociated SmoM2 medulloblastoma and cultured in DMEM/F12 media containing B27, N2, EGF, and FGF2. For GSK-J4 (XcessBio, M60063-2) treatment, media were mixed with indicated concentrations of the compound, and DMSO was used as a solvent control. The ATP assay for cell viability analysis was carried out as described (10).
Western blot
Western blot was carried out as previously described (38). In detail, cultured cells were washed with ice-cold 1x PBS for three times, and lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.05% SDS, 0.5% DOC, 1% NP-40). Cell lysates were separated on SDS-PAGE gels. Antibodies against Gli1 and Caspase3 were purchased from Cell Signaling and Protein Tech, respectively. HRP-conjugated secondary antibodies were from Jackson Immunology. GAPDH was detected as a loading control.
Reverse-transcription quantitative PCR
RT-qPCR was performed as described (38). RNAs from cells or tissues were extracted with TRIZOL (Invitrogen). The concentration of RNA samples was determined with NanoDrop One (Thermo Scientific). cDNAs were synthesized by reverse transcription using Iscript. iTaq reagents were used for quantitative PCR. Both Iscript and iTaq were purchased from Bio-Rad. A Bio-Rad real-time PCR system (C1000 Thermal Cycler) was used for quantitative PCR. Levels of GAPDH mRNA were used to normalize input RNAs. Relative gene expression was calculated by ΔΔCt as previously described (38). Graphics shown are representative of experiments performed in triplicate. Sequences of PCR-primers are listed in extended data.
Chromatin immunoprecipitation assay
ChIP experiments were performed as previously described (10). Dissociated cells were crosslinked with paraformaldehyde, and DNA was sonicated to fragments (200-1000 bp) in the nuclear lysis buffer using a Sonic Dismembrator 550 (Fisher Scientific) at level 3, output 10%, for 5 cycles of 7 sec on, 30 sec off, in ice water. An antibody against H3K27me3 (#39536, Active Motif) was used in the precipitation step. Precipitated DNA was captured using pre-blocked Protein G agarose beads (Pierce) in the presence of BSA and salmon sperm DNA, followed by purification and subjection to quantitative PCR. The primer sequences are CGCTCACTTCCCTCGTATATCCTTC, GGCAGTATAGGGTCCCTCAAGGG. 10% of input was used for positive control and normalizing results, which were presented as percentages of input.
RNA-seq and bioinformatics analyses
RNA-seq analyses were performed in the Sequencing and Bioinformatics Center in Anhui Medical University. Total RNAs were extracted using TRIzol Reagents (Invitrogen), and mRNAs were separated using magnetic beads (Vazyme, N401). Reverse transcription was performed to obtain cDNA using SuperScript™ II (Invitrogen, # 18064014). cDNA libraries were established using the Tn5 DNA Library Prep Kit from Illumina (TRANS, KP101-11) and sequenced for PE150. Fastq data were analysed with Fastqc and processed using the pipeline described in the Linux system (39). The expression levels of genes were quantified with featureCounts (40), and the differentially expressed genes were called by DESeq2 program with a fold-change > 2 and padj <0.05. Data were analysed and presented using R studio for GO, KEGG, GSEA and STRING analyses.
Immunofluorescence staining
DaoY cells were seeded on cover glasses at a density of 105 cells/well in 24-well plates. Cells were treated with 10 μM of BrdU for 2 hours before staining. For staining, cells on cover glasses were fixed using methanol, then washed with PBS for 3 times. The cells were then treated with 2 M of HCl for 20 min, and blocked with 5% goat serum for 30 min at room temperature. The cells were incubated with mouse anti-BrdU antibodies (ABclonal, A1482) o/n at 4°C and then with GFP-Conjugated goat anti-mouse IgG antibodies (ZGGB-BIO, ZF-0312) for 30 min at 20°C.
Cell cycle assay
The cells were cultured with GSK-J4 for 24 hours in 6 cm dishes at a density of 4 x 105 cells per dish. Cell cycle assay was performed according to the manufacturer’s instructions (Beyotime, C1052). Stained cells were applied to a BD FACSVerse flow cytometer (BD Bioscience), and data were analysed using ModFit software.
Statistical analysis
Each experiment was repeated at least three times except for RNA-seq analyses, data shown were from representative assays with 3 technical repeats. Data are expressed as means plus standard deviation (s.d.). Statistical analyses were performed using a two-tailed, unpaired Student’s t-test provided by Microsoft Office Excel. A p value of <0.05 will be considered significant.
Results
Shh signaling is impaired by GSK-J4 targeting H3K27me3 demethylase Jmjd3
Since Shh medulloblastoma is caused by over-active Shh signaling in cerebellum, modulating Shh signaling could be an effective way to constrain the tumor growth. Previously we reported that Jmjd3 plays an essential role in Shh signaling activation and Shh-medulloblastoma growth (10). Kruidenier et al. developed an H3K27me3 demethylase inhibitor, GSK-J4, to modulate Jmjd3 activity, making it possible to intervene the diseases that depend on Jmjd3 (32). To determine whether GSK-J4 can block Shh signaling by targeting Jmjd3, we chose the immortalized embryonic fibroblast NIH3T3 cells (41), which is responsive to Shh signaling and could activate Shh target genes such as Gli1, Ptch1, Hhip, and Hck (10, 11, 31, 38). Among them, Gli1 encodes a transcription factor forming a positive feedback loop to further activate Shh/Gli1 target genes. The mRNA level of Gli1 faithfully reflects the activities of Shh pathway (31), hence we use Gli1 as a readout of the pathway. Previously 30 µM of GSK-J4 was applied to inhibit the proinflammatory macrophage response through targeting the jumonji domain of histone demethylase (32), we hence tested this concentration of GSK-J4 on NIH3T3 cells, and treated the cells with Shh conditioned media and various concentrations of GSK-J4 at 0.01, 0.05, 0.24, 1.2, 6, and 30 μM (Figures 1A, B). The Treatment of GSK-J4 (Figure 1A, lane 4-8) significantly reduced the Shh induced Gli1 protein levels (Figure 1A, lane 2) when the concentration of GSK-J4 is above 6 μM. The quantifications indicated that 30 μM of GSK-J4 is higher than the IC50 in NIH 3T3 cells for target gene Gli1 expression (Figure 1B).
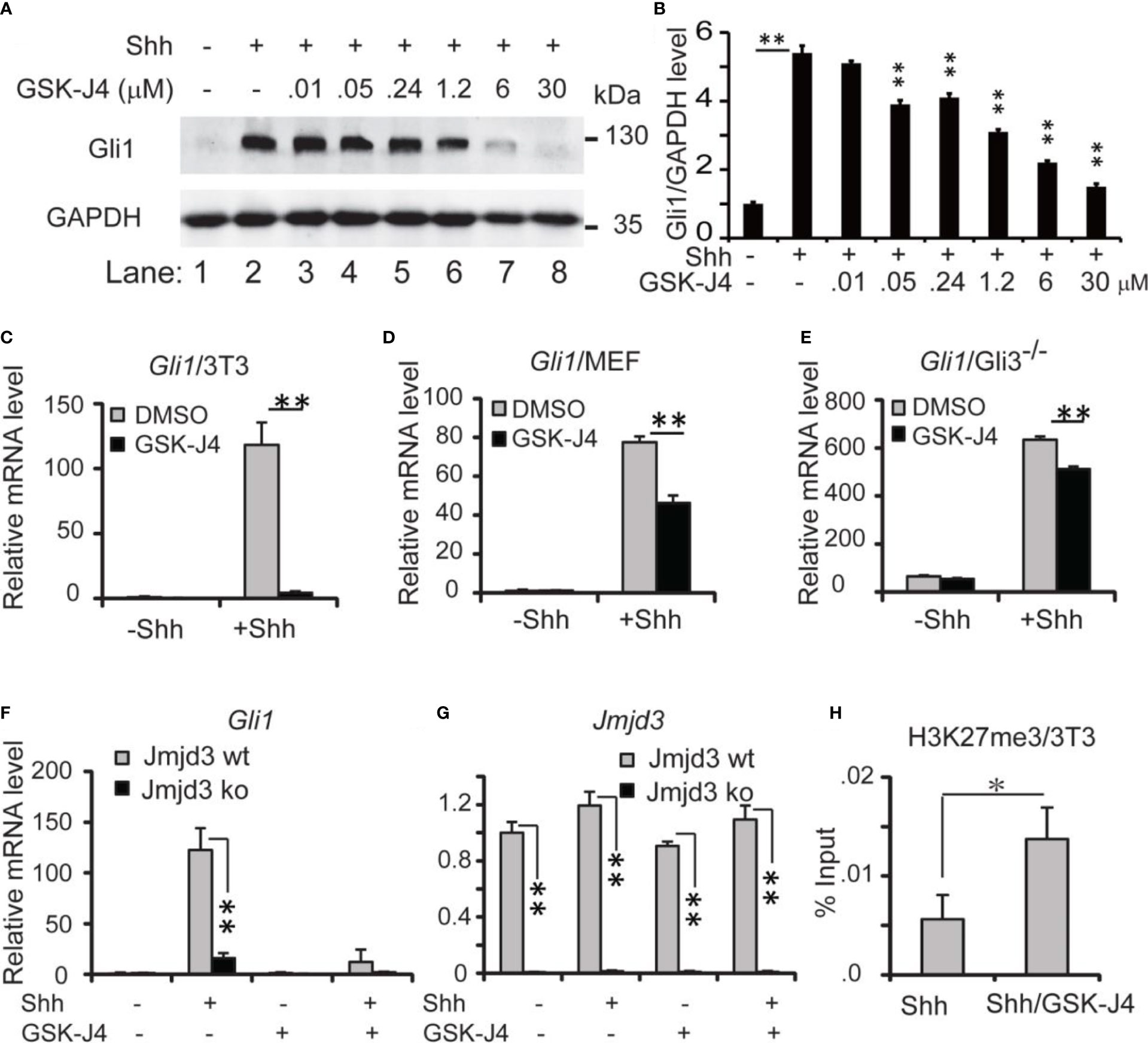
Figure 1 Shh signaling is impaired by GSK-J4 targeting H3K27me3 demethylase Jmjd3. (A) Effects of GSK-J4 at various concentrations on Gli1 protein levels. (B) Ratio of Gli1 protein levels against GAPDH in (A) calculated by Image J Significance is against Shh induced level (rf Figure 1A, lane 2) except for the one between Figure 1A lane 1 and Figure 1A lane 2. (C) GSK-J4 impairs Shh-induced Gli1 mRNA expression in NIH3T3 cells by RT-qPCR analysis. (D) GSK-J4 inhibits Shh-induced Gli1 mRNA expression in primary MEF cells. (E) GSK-J4 significantly inhibits Shh-induced Gli1 mRNA expression in primary Gli3-/- MEF cells. (F) Shh-induced Gli1 mRNA expression is impaired in primary Jmjd3-/- MEF cells, and GSK-J4 does not further decrease the Gli1 mRNA level. (G) Jmjd3 mRNA expression is reduced by Jmjd3 knockout. (H) GSK-J4 raises the local H3K27me3 enrichment at mouse Gli1 promoter region shown by ChIP q-PCR assay. The concentration of GSK-J4 is 30 μM if not indicated for all figures. Significance determined using Student’s t-test. **p < 0.01 and *p < 0.05.
Upon addition of Shh conditioned media to NIH3T3 cells, Gli1 mRNA levels were significantly increased, indicating an activated Shh signaling pathway (Figure 1C). After cells were treated with 30 µM of GSK-J4 for 24 hours, Gli1 expression was significantly decreased to ~10% (Figure 1C). Thus, GSK-J4 is a potential inhibitor of the Shh signaling pathway. Since the immortalized NIH3T3 cells were generated from mouse embryonic fibroblast (MEF), we carried out the GSK-J4 inhibition experiment using primary MEF cells. Similarly, the Shh-induced Gli1 expression was significantly reduced by GSK-J4 treatment, albeit to a lesser extent than that in NIH3T3 cells (Figure 1D vs 1C). The GSK-J4 inhibition of Shh target genes was further confirmed in the primary Gli3-/- MEF cells (Figure 1E). The basal Gli1 mRNA level in Gli3-/- MEFs is higher than that in wildtype MEF since Gli3 is a repressor in the Shh signaling pathway. GSK-J4 treatment also significantly reduced Gli1 basal expression levels in Gli3-/- MEFs. Given our finding that Jmjd3 plays an important role in the Shh signaling pathway (10), we examined whether GSK-J4 inhibition of Shh gene expression is indeed through targeting Jmjd3. When primary Jmjd3-/- MEFs were treated with or without GSK-J4 in the presence or absence of Shh, we observed that Jmjd3 deficiency largely blocked both the Shh-induced target gene expression and GSK-J4 inhibition (Figures 1F, G). To understand the molecular mechanism in which GSK-J4 inhibits the Shh signaling pathway, we performed an H3K27me3 ChIP assay. In NIH3T3 cells treated with GSK-J4, the H3K27me3 levels at the Gli1 gene regulatory region near its promoter were significantly increased (Figure 1H), suggesting impaired demethylation activities of Jmjd3. Taken together, GSK-J4 blocked Shh signaling by inhibiting Jmjd3 demethylase activities.
Growth of CGNP cells is sensitive to GSK-J4
Shh secreted from Purkinje neurons is essential for CGNP proliferation and cerebellar development (1). To determine whether GSK-J4 inhibits CGNP proliferation by inhibiting the Shh signaling pathway, we isolated primary CGNP cells from P4 mouse cerebella, and treated them with Shh conditioned media (37). Shh-induced target gene expression in CGNP cells was significantly impaired by 30 µM of GSK-J4 treatment for 24 hours (Figure 2A). To further investigate inhibition of GSK-J4 in detail, we treated CGNP cells with 30 µM of GSK-J4 for various durations. We observed that Gli mRNA levels were significantly decreased after 2 hours of treatment with GSK-J4, and the decrease reached 50% after 3 hours (Figure 2B). To quantitate the effects of GSK-J4 on CGNP cell growth, we treated the CGNP cells in the presence of Shh with various concentrations of GSK-J4 for 24 hours and measured ATP percentage to indicate cell survival. 6 µM of GSK-J4 treatment reduced 50% of cell growth in CGNP cells (Figure 2C). We further measured CGNP growth at various time points after 30 µM of GSK-J4 treatment. We observed no significant growth change until at 24 h time point (Figure 2D, E). Therefore, the decrease of Gli1 expression after GSK-J4 treatment at earlier time points was not caused by GSK-J4 toxicity. These results confirmed that GSK-J4 inhibits the Shh signaling pathway in CGNP cells. Furthermore, at 24 h time point after GSK-J4 treatment, CGNP cell numbers were significantly reduced in Shh treated group, but not in the untreated one (Figures 2D, E), suggesting that the GSK-J4 inhibition of CGNP cell growth requires Shh signaling. Hence, consistent with our previous observation that Jmjd3 is required for Shh target gene expression and cerebellum development (10), pharmacological inhibition of Jmjd3 by GSK-J4 also efficiently inhibited Shh target gene expression during cerebellar development.
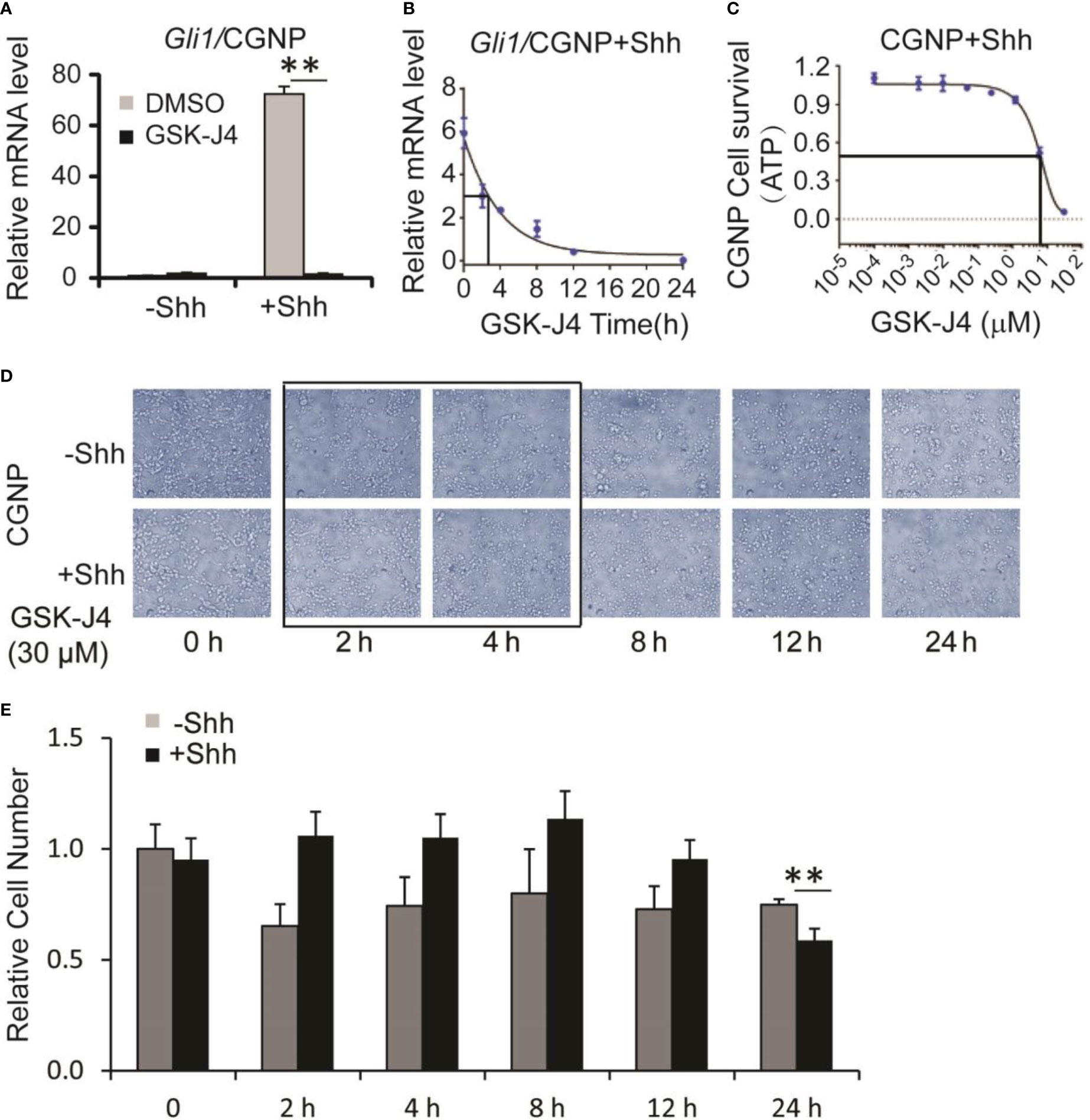
Figure 2 Shh signaling and growth of CGNP cells are inhibited by GSK-J4. (A) GSK-J4 decreases Shh-induced Gli1 expression in P4 CGNP cells. (B) Time course of GSK-J4 effect on Gli1 expression in P4 CGNP cells. (C) Dose dependent survival of CGNP cells after the treatment of GSK-J4 shown by ATP assay. (D, E) Cell performances after the treatment of GSK-J4 for indicated time courses and doses. The cells with frame have significant changes after treatment with GSK-J4. Significance determined using t-test. **p < 0.01.
GSK-J4 shows antitumor activity in medulloblastoma
Next, we determined whether GSK-J4 inhibits Shh-type medulloblastoma. We bred Rosa26-SmoM2, actin-creER transgenic mice, in which SmoM2 mutation causes constitutive Shh signaling and Shh-type medulloblastoma (9). From mouse medulloblastoma tumors, we cultured primary tumor cells and treated them with 30 µM of GSK-J4. Gli1 expression was significantly inhibited as shown by RT-qPCR and the tumor cell growth was completely blocked by GSK-J4 as measured by ATP assay (Figures 3A, C), suggesting that GSK-J4 is more effective in inhibiting the growth of medulloblastoma cells compared to CGNP cells. To find the minimal treatment time of GSK-J4, we treated the tumor cells with 30 µM of GSK-J4 in a time course (Figure 3B) and found that the Gli1 mRNA level decreased 50% after 6-7 hours. Consistently, most of the tumor cells were killed before 8 hours (Figure 3B). We next determined minimal concentration of GSK-J4 to inhibit medulloblastoma growth by treating the tumor cells with various concentrations of GSK-J4 for 24 hours. Our results suggested that 6 µM of GSK-J4 significantly inhibited the tumor cell growth (Figures 3C–E). Since Shh signaling is constitutively elevated in Shh-type medulloblastoma, Shh ligand had no significant effect on Gli1 expression, and GSK-J4 showed similar effects on tumor cell growth in the presence or absence of Shh (Figure 3F).
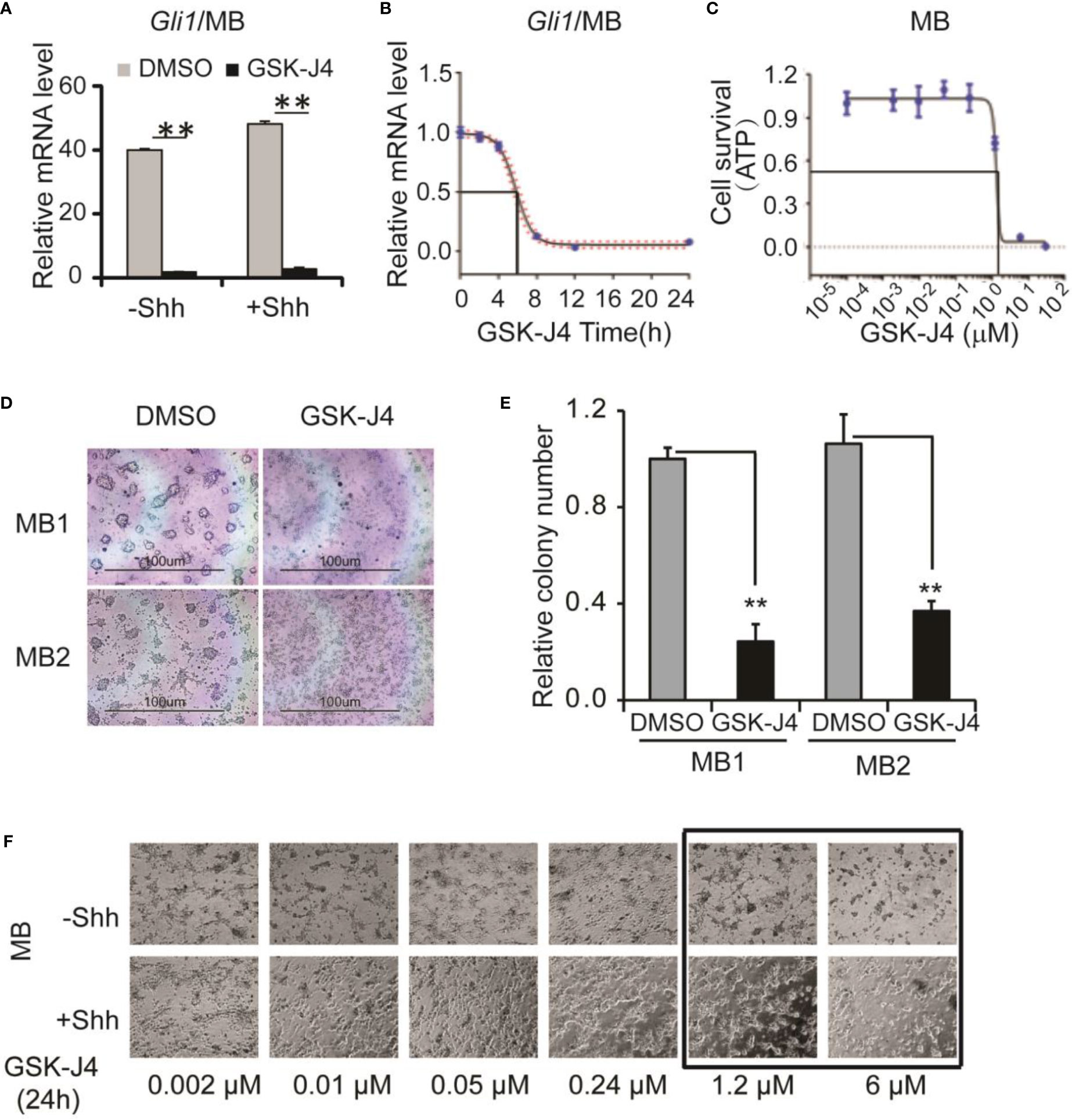
Figure 3 GSK-J4 shows antitumor activity in mouse primary medulloblastoma cells. (A) Shh target gene Gli1 mRNA expression is inhibited by the treatment of GSK-J4 on mouse Shh-type primary medulloblastoma cells (MB). (B) Time course of GSK-J4 effects on Gli1 expression in mouse primary medulloblastoma cells. (C) Dose dependent survival of primary medulloblastoma cells after the treatment of GSK-J4 shown by ATP assay. (D) Survival of primary medulloblastoma cells treated with GSK-J4. (E) Tumor colonies after treatment of GSK-J4. (F) Time course performances of primary medulloblastomas after the treatment of GSK-J4. The cells with frame have significant performance changes after treatment with GSK-J4. Significance determined using t-test. **p < 0.01.
GSK-J4 inhibits the proliferation of medulloblastoma cells
To translate the finding that GSK-J4 is a candidate to treat medulloblastoma, we measured Gli1 mRNA level changes before and after GSK-J4 treatments in a human Shh-type medulloblastoma cell line DaoY. Gli1 was expressed actively in DaoY cells regardless of Shh treatment, and GSK-J4 significantly inhibited Gli1 expression (Figure 4A). To determine the sensitivity of DaoY cells to GSK-J4 treatment, we measured cell survival under 2 µM, 6 µM or 30µM of GSK-J4 treatment, and found that 30 µM GSK-J4 significantly hindered cell viability of DaoY medulloblastoma cells (Figure 4B). To determine whether the cell number reduction was caused by inhibition of proliferation or increase of apoptosis, we performed cell cycle analyses. We found that 30µM of GSK-J4 treatment significantly induced cell cycle arrest at G0/G1 phase, with an extension at G0/G1phase and a reduction of S-phase (Figure 4C). BrdU incorporation assay in DaoY cell cultures further verified our hypothesis that GSK-J4 could inhibit DaoY medulloblastoma cell cycle progression. BrdU positive cells were significantly decreased upon 6 µM of GSK-J4 treatment, and 30 µM of GSK-J4 treatment showed further reduction (Figures 4D, E). After treatment with GSK-J4, Caspase3 levels were slightly increased as shown by western blot (Figure 4F), suggesting minor cell apoptosis caused by GSK-J4 treatment. Taken together, GSK-J4 inhibits the proliferation of human DaoY medulloblastoma cells.
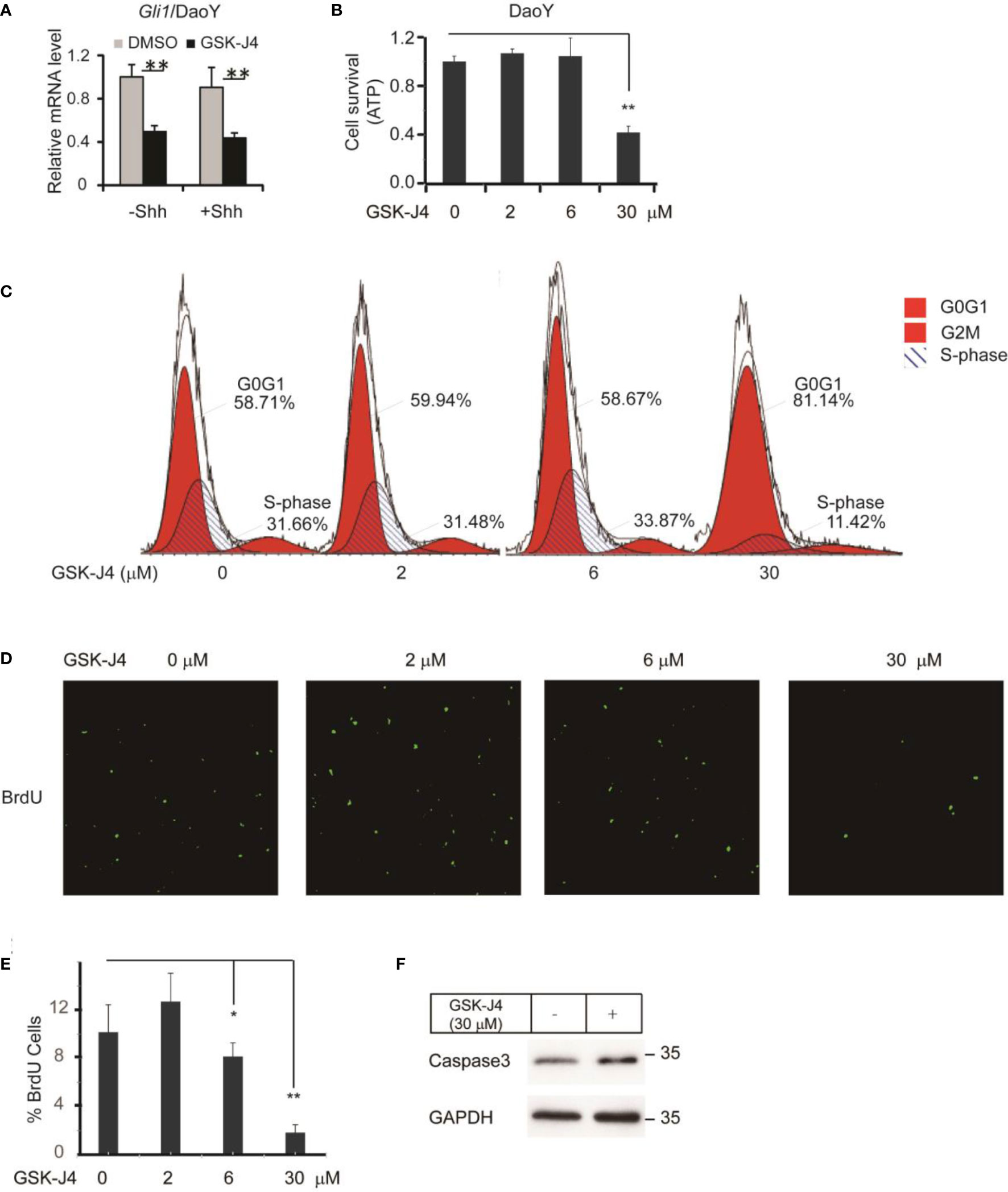
Figure 4 Growth of human DaoY medulloblastoma cells is inhibited and cells are arrested at G0/G1 by treatment of GSK-J4. (A) Shh target gene Gli1 expression is impaired by GSK-J4 in human medulloblastoma cell line DaoY. (B) Growth of DaoY Cells is inhibited by GSK-J4. (C) Growth of DaoY Cells is arrested at G0/G1 phase with decrease of S-phase by GSK-J4. (D, E) The proliferation of DaoY cells is reduced by treatment of GSK-J4. (F) Apoptosis of DaoY tumor cells is not dramatically induced by GSK-J4. Significance determined using Student’s t-test; *p<0.05, **p<0.01.
RNA-seq and bioinformatics analyses show biosynthesis of cholesterol as an additional target of GSK-J4
To understand the global effects of GSK-J4 on medulloblastoma cells, we carried out RNA-seq of DaoY cells treated with or without GSK-J4 (n=2). Heatmap of mRNA levels showed comparable samples used for this assay (Figure 5A). Totally, 1208 genes had higher expression after treatment with GSK-J4, and 1154 genes decreased their expression (Figure 5B). The down-regulation of these genes possibly resulted from the inhibition of H3K27me3 demethylase activities, while the upregulation could be caused by indirect effects. GO analysis showed sterol biosynthesis genes were enriched in the down-regulated gene population (Figure 5D). The heatmap further confirmed the significant down-regulation of cholesterol biosynthesis genes (Figure 5C). Consistently, KEGG assay also showed steroid biosynthesis genes decreased after treatment with GSK-J4 (Figure 5E). Using STRING package, we identified the top 10 genes of sterol biosynthesis pathway as presented in Figure 5F, highlighting MVD, DHCR7, DHCR24, LSS and HSD17B7 in cholesterol biosynthesis. On the other hand, the up-regulated genes mostly were associated with protein degradation and unfolding pathways (Figure 6A), which may result in dys-regulated metabolism and disease (Figure 6B).
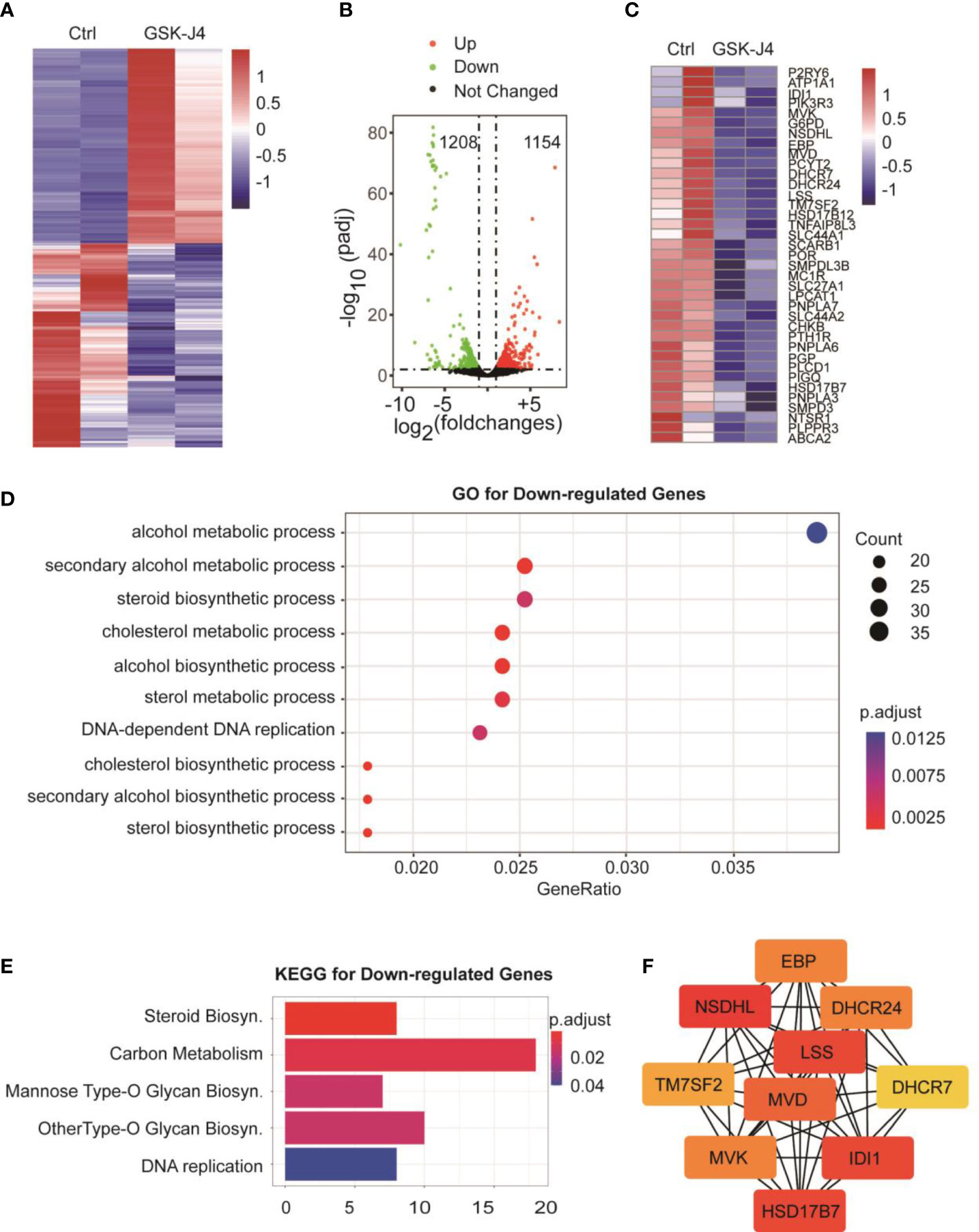
Figure 5 Biosynthesis of cholesterol and phospholipids are impaired by GSK-J4 in DaoY cells. (A) Heatmap of gene mRNA levels in DaoY cells after treatment with GSK-J4. (B) Volcano plot of the differentially expressed genes in DaoY cells after treatment with GSK-J4. (C) Heatmap of the mRNA level of cholesterol biosynthesis genes in DaoY cells after treatment with GSK-J4. (D) GO analysis of the down-regulated genes in DaoY cells treated with GSK-J4. (E) KEGG analysis of the down-regulated genes in DaoY cells treated with GSK-J4. (F) STRING analysis for key genes in cholesterol biosynthesis.
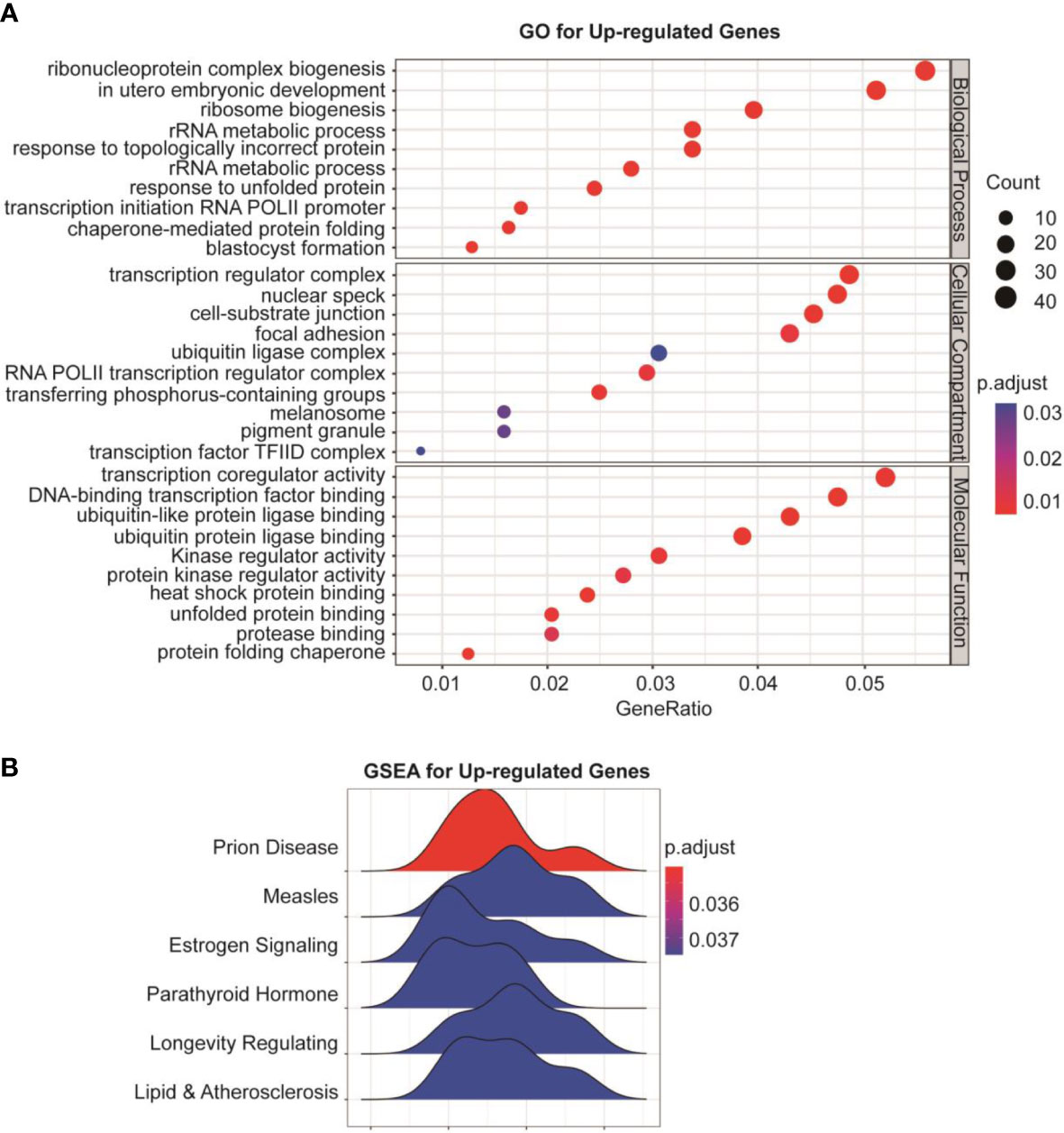
Figure 6 Protein degradation and unfolding are activated by GSK-J4 in DaoY cells. (A) GO analysis of the up-regulated genes in DaoY cells treated with GSK-J4. (B) GSEA analysis of the up-regulated genes in DaoY cells treated with GSK-J4.
Discussion
We broadly tested an epigenetic inhibitor GSK-J4 in Shh-responsive tissue cultures, including Shh responsive cell models NIH3T3, primary MEF cells, primary CGNP cells and medulloblastoma cells. Importantly, we did pilot experiments in human DaoY Shh-medulloblastoma cell line. We provide strong evidences showing decreased mRNA levels of the key Shh target gene Gli1 by GSK-J4 treatment in all settings. Since Gli1 is a positive feedback transcription factor in Shh signaling, it plays important roles to amplify Shh signals in vivo. One strategy for inhibiting Shh signaling in tumor growth is by inhibiting Gli1 levels (27).
Our results together showed that GSK-J4 is a potential inhibitor for Shh-type medulloblastoma growth (Figures 4B–D), and it can be developed for possible targeted therapy of pediatric Shh-type medulloblastoma. Hashizume et al. showed that GSK-J4 is a candidate for brainstem glioma therapy by inhibiting H3K27me3 demethylase (33). The group administered GSK-J4 by intraperitoneal injection, and observed active derivative GSK-J1 in brain tissues, suggesting the small molecule can reach cerebellum for medulloblastoma therapy. In our study, GSK-J4 treatment reduced active tumor cells and BrdU incorporation to genome. Cells were arrested at G0/G1 phase with significant decrease in S-phase after GSK-J4 treatment, together showing inhibited proliferation by GSK-J4.
Surprisingly, we found the expression of cholesterol biosynthesis genes was significantly reduced by GSK-J4 treatment. The key biosynthesis genes included MVD, DHCR7, DHCR24, LSS, HSK17B7 and TM7SF2 (Figure 7A), suggesting that the biosynthesis of cholesterol was impaired in cells treated with GSK-J4. These cholesterol biosynthesis genes were possibly directly regulated by Jmjd3. It was reported that the reduction of cholesterol biosynthesis resulted in S-phase decrease and G0/G1 extension (42), which is consistent with our current finding. Besides the essential role in cell physiological activities, cholesterol is a functional modification of the Shh signaling pathway proteins. Both Shh ligand and membrane protein Smoothed are covalently modified by cholesterol (43, 44). Hence, our study suggested GSK-J4 can also regulate Shh signaling by restraining cholesterol metabolism.
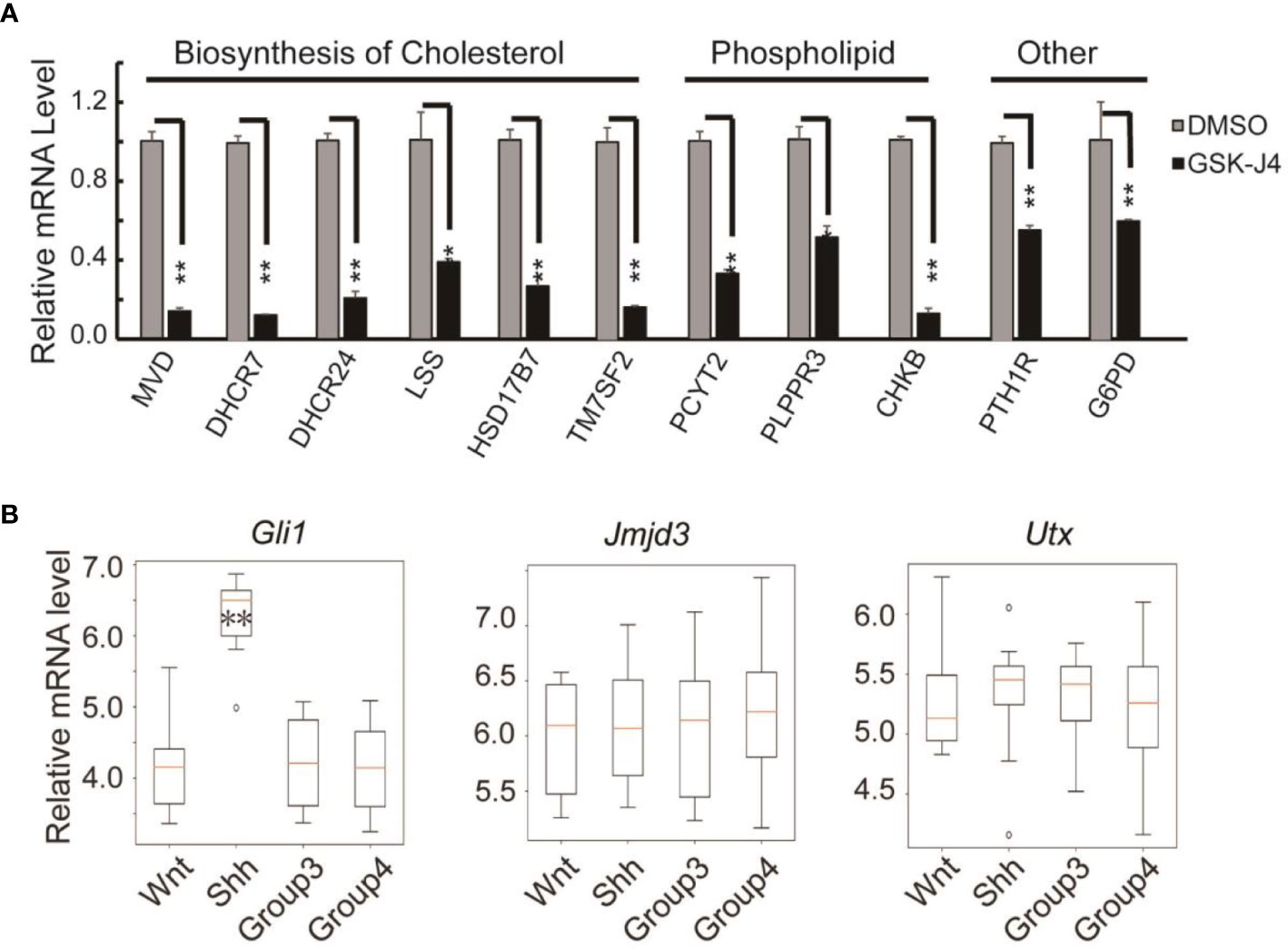
Figure 7 Verification of cholesterol and phospholipids biosynthesis gene expression decreases in DaoY cells treated with GSK-J4. (A) RT-qPCR verification of the expression of cholesterol and phospholipids biosynthesis genes in DaoY cells treated with GSK-J4. (B) Gene expression levels by microarray analysis in four subgroups of human medulloblastoma (GSE37418 (7)). Number of samples in each group, wnt: 8, Shh: 10, Group3: 17, Group4: 41. Significance determined using Student’s t-test. **p< 0.01, *p< 0.05.
GSK-J4 could inhibit the H3K27me3 demethylase activities of both Jmjd3 and Utx. We think the GSK-J4 effects on Shh signaling and medulloblastoma were largely through targeting Jmjd3. The two H3K27me3 demethylases have diverse functions in Shh-type medulloblastoma formation, though they have similar expression levels among different types of medulloblastoma (Figure 7B). Jmjd3 functions as a co-activator for Shh target genes, whereas UTX could inhibit medulloblastoma initiation by promoting tumor differentiation (29). Our study together with others’ showed that GSK-J4 is a promising agent for targeted therapy of human medulloblastoma, and other tumors dependent on Shh signaling and cholesterol metabolism (Figure 8). Hence, our study highlights the possibility of using GSK-J4 that targets H3K27me3 demethylase Jmjd3 to treat Shh medulloblastoma patients.
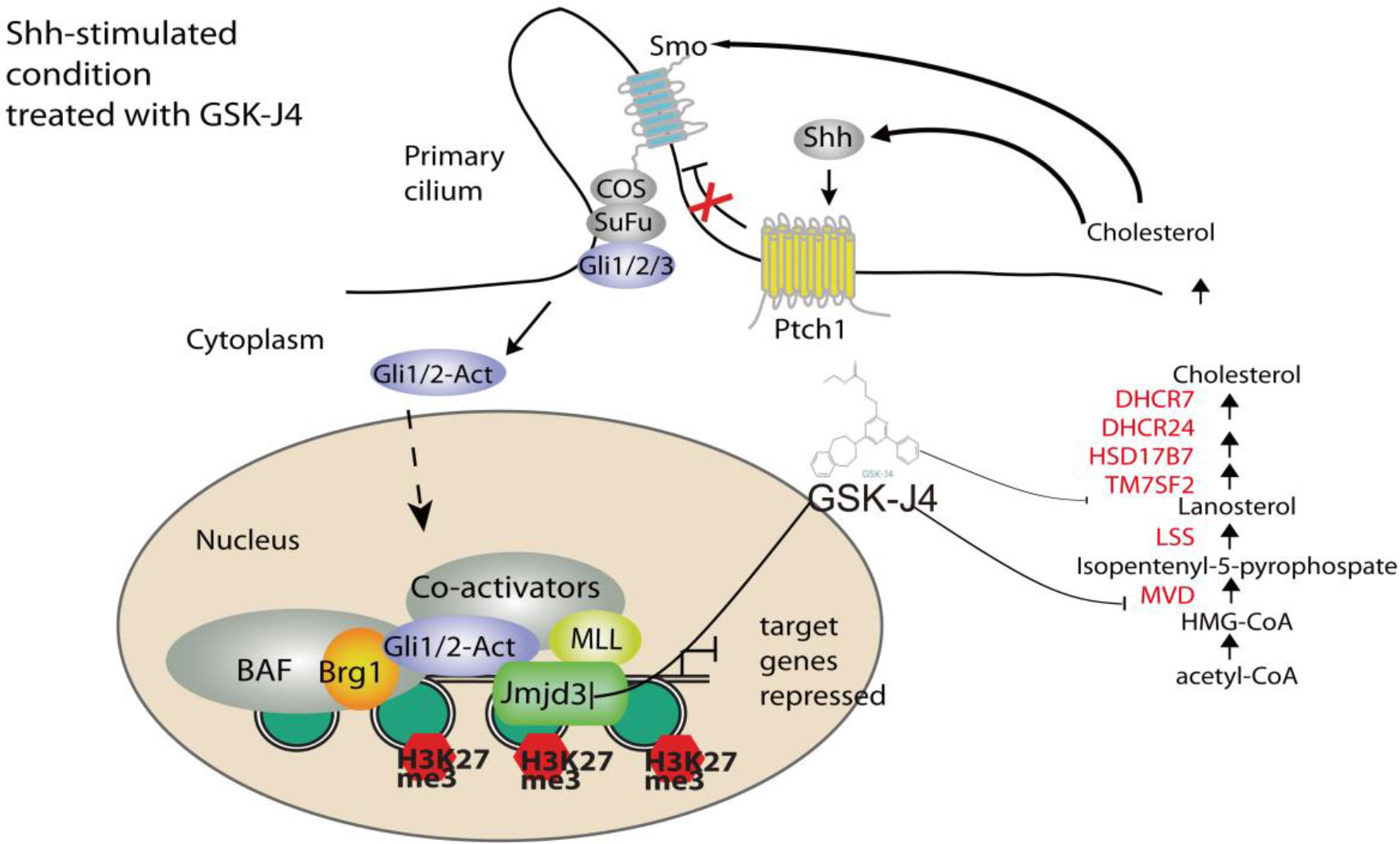
Figure 8 A model of Shh signaling regulated by small molecule compound GSK-J4 targeting the epigenetic switch. At basal condition, the processed transcriptional factor Gli3R represses Shh target gene and maintains high level of H3K27me3 on the regulatory region of target genes. In the presence of Shh, Gli3R is replaced with Gli1/2-Act, which recruits the coactivators Brg1 and Jmjd3. Decreases of H3K27me3 turn on the target gene expression. While acetyl-CoA is present, serial enzymes including MVD, DHCR7, and DHCR24, LSS convert acetyl-CoA to form cholesterol. With the treatment of GSK-J4, activity of Jmjd3 is inhibited and H3K27me3 increases, which leads to impaired Shh signaling and cholesterol biosynthesis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The animal study was reviewed and approved by University of Texas Southwestern Medical Center.
Author contributions
JW and XS designed the experiments. HD, XG, XS, NF, YL and BL performed the experiments, collected the data and analysed the results. XS, SL, and JW wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Grants for Scientific Research of BSKY (XJ2020039) from Anhui Medical University, the Natural Science Foundation of China (82073124), and Scientific Research Platform and Base Upgrading Plan of Anhui Medical University (2021xkjT048).
Acknowledgments
We appreciate the Jmjd3-/- MEF cells from Dr. Akira from Japan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1057147/full#supplementary-material
References
1. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell (2008) 15(6):801–12. doi: 10.1016/j.devcel.2008.11.010
2. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial shh signaling and ectopic activation of the shh pathway. Development (2002) 129(20):4753–61. doi: 10.1242/dev.129.20.4753
3. Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development (2001) 128(24):5161–72. doi: 10.1242/dev.128.24.5161
4. Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development (1999) 126(14):3089–100. doi: 10.1242/dev.126.14.3089
5. Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron (1999) 22(1):103–14. doi: 10.1016/S0896-6273(00)80682-0
6. Wallace VA. Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol (1999) 9(8):445–8. doi: 10.1016/S0960-9822(99)80195-X
7. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature (2012) 488(7409):43–8. doi: 10.1038/nature11213
8. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science (2011) 331(6016):435–9. doi: 10.1126/science.1198056
9. Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the hedgehog pathway. Cancer Res (2006) 66(20):10171–8. doi: 10.1158/0008-5472.CAN-06-0657
10. Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, et al. An epigenetic switch induced by shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun (2014) 5:5425. doi: 10.1038/ncomms6425
11. Shi X, Wang Q, Gu J, Xuan Z, Wu JI. SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying shh-type medulloblastoma development. Oncogene (2016) 35(44):5746–58. doi: 10.1038/onc.2016.108
12. de Bont JM, Packer RJ, Michiels EM, den Boer ML, Pieters R. Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro Oncol (2008) 10(6):1040–60. doi: 10.1215/15228517-2008-059
13. Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol (2011) 8(9):540–9. doi: 10.1038/nrclinonc.2011.95
14. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol (2012) 123(4):465–72. doi: 10.1007/s00401-011-0922-z
15. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature (2012) 488(7409):49–56. doi: 10.1038/nature11327
16. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature (2012) 488(7409):106–10. doi: 10.1038/nature11329
17. Roussel MF, Robinson G. Medulloblastoma: advances and challenges. F1000 Biol Rep (2011) 3:5. doi: 10.3410/B3-5
18. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature (2010) 468(7327):1095–9. doi: 10.1038/nature09587
19. Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form shh-induced medulloblastoma. Cancer Cell (2008) 14(2):123–34. doi: 10.1016/j.ccr.2008.07.005
20. Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell (2008) 14(2):135–45. doi: 10.1016/j.ccr.2008.07.003
21. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell (2012) 21(2):168–80. doi: 10.1016/j.ccr.2011.12.023
22. Zagozewski J, Shahriary GM, Morrison LC, Saulnier O, Stromecki M, Fresnoza A, et al. An OTX2-PAX3 signaling axis regulates group 3 medulloblastoma cell fate. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-17357-4
23. Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature (2019) 572(7767):67–+. doi: 10.1038/s41586-019-1158-7
24. Hendrikse LD, Haldipur P, Saulnier O, Millman J, Sjoboen AH, Erickson AW, et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature (2022) 609(7929):1021. doi: 10.1038/s41586-022-05215-w
25. Smith KS, Bihannic L, Gudenas BL, Haldipur P, Tao R, Gao QS, et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature (2022) 609(7929):1012–+. doi: 10.1038/s41586-022-05208-9
26. Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol (2007) 6(12):1073–85. doi: 10.1016/S1474-4422(07)70289-2
27. Huang SY, Yang JY. Targeting the hedgehog pathway in pediatric medulloblastoma. Cancers (Basel) (2015) 7(4):2110–23. doi: 10.3390/cancers7040880
28. Yi JQ, Kim B, Shi XM, Zhan XM, Lu QR, Xuan ZY, et al. PRC2 heterogeneity drives tumor growth in medulloblastoma. Cancer Res (2022) 82(16):2874–86. doi: 10.1158/0008-5472.CAN-21-4313
29. Yi JQ, Shi XM, Xuan ZY, Wu J. Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett (2021) 499:188–200. doi: 10.1016/j.canlet.2020.11.031
30. Yi JQ, Wu J. Epigenetic regulation in medulloblastoma. Mol Cell Neurosci (2018) 87:65–76. doi: 10.1016/j.mcn.2017.09.003
31. Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. Dual role of brg chromatin remodeling factor in sonic hedgehog signaling during neural development. Proc Natl Acad Sci U.S.A. (2011) 108(31):12758–63. doi: 10.1073/pnas.1018510108
32. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature (2012) 488(7411):404–8. doi: 10.1038/nature11262
33. Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med (2014) 20(12):1394–6. doi: 10.1038/nm.3716
34. Hong BJ, Park WY, Kim HR, Moon JW, Lee HY, Park JH, et al. Oncogenic KRAS sensitizes lung adenocarcinoma to GSK-J4-Induced metabolic and oxidative stress. Cancer Res (2019) 79(22):5849–59. doi: 10.1158/0008-5472.CAN-18-3511
35. Lhuissier E, Aury-Landas J, Allas L, Boittin M, Boumediene K, Bauge C. Antiproliferative effect of the histone demethylase inhibitor GSK-J4 in chondrosarcomas. IUBMB Life (2019) 71(11):1711–9. doi: 10.1002/iub.2110
36. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 11(10):936–44. doi: 10.1038/ni.1920
37. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of smoothened activity. Proc Natl Acad Sci U.S.A. (2002) 99(22):14071–6. doi: 10.1073/pnas.182542899
38. Shi X, Zhan X, Wu J. A positive feedback loop between Gli1 and tyrosine kinase hck amplifies shh signaling activities in medulloblastoma. Oncogenesis (2015) 4:e176. doi: 10.1038/oncsis.2015.38
39. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat Protoc (2016) 11(9):1650–67. doi: 10.1038/nprot.2016.095
40. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (2014) 30(7):923–30. doi: 10.1093/bioinformatics/btt656
41. Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol (1963) 17:299–313. doi: 10.1083/jcb.17.2.299
42. Singh P, Saxena R, Srinivas G, Pande G, Chattopadhyay A. Cholesterol biosynthesis and homeostasis in regulation of the cell cycle. PloS One (2013) 8(3):e58833. doi: 10.1371/journal.pone.0058833
43. Xiao X, Tang JJ, Peng C, Wang Y, Fu L, Qiu ZP, et al. Cholesterol modification of smoothened is required for hedgehog signaling. Mol Cell (2017) 66(1):154–162 e110. doi: 10.1016/j.molcel.2017.02.015
Citation: Deng H, Guo X, Feng N, Luo Y, Liu B, Liu S, Wu JI and Shi X (2022) Targeting H3K27me3 demethylase to inhibit Shh signaling and cholesterol metabolism in medulloblastoma growth. Front. Oncol. 12:1057147. doi: 10.3389/fonc.2022.1057147
Received: 29 September 2022; Accepted: 21 November 2022;
Published: 01 December 2022.
Edited by:
Hatim E. Sabaawy, University of Colorado, United StatesReviewed by:
Luz Jubierre Zapater, Memorial Sloan Kettering Cancer Center, United StatesYirizhati Aili, First Affiliated Hospital of Xinjiang Medical University, China
Copyright © 2022 Deng, Guo, Feng, Luo, Liu, Liu, Wu and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuzhen Liu, shuzhen.liu@utsouthwestern.edu; Jiang I. Wu, jiang9.wu@utsouthwestern.edu; Xuanming Shi, xuanming.shi@ahmu.edu.cn
†These authors have contributed equally to this work