 Seyed Mehdi Havaei
Seyed Mehdi Havaei Marc G. Aucoin
Marc G. Aucoin Ali Jahanian-Najafabadi
Ali Jahanian-Najafabadi- 1Department of Pharmaceutical Biotechnology, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran
- 2Department of Chemical Engineering, Faculty of Engineering, University of Waterloo, Waterloo, ON, Canada
Cancer is one of the prominent causes of death worldwide. Despite the existence of various modalities for cancer treatment, many types of cancer remain uncured or develop resistance to therapeutic strategies. Furthermore, almost all chemotherapeutics cause a range of side effects because they affect normal cells in addition to malignant cells. Therefore, the development of novel therapeutic agents that are targeted specifically toward cancer cells is indispensable. Immunotoxins (ITs) are a class of tumor cell-targeted fusion proteins consisting of both a targeting moiety and a toxic moiety. The targeting moiety is usually an antibody/antibody fragment or a ligand of the immune system that can bind an antigen or receptor that is only expressed or overexpressed by cancer cells but not normal cells. The toxic moiety is usually a protein toxin (or derivative) of animal, plant, insect, or bacterial origin. To date, three ITs have gained Food and Drug Administration (FDA) approval for human use, including denileukin diftitox (FDA approval: 1999), tagraxofusp (FDA approval: 2018), and moxetumomab pasudotox (FDA approval: 2018). All of these ITs take advantage of bacterial protein toxins. The toxic moiety of the first two ITs is a truncated form of diphtheria toxin, and the third is a derivative of Pseudomonas exotoxin (PE). There is a growing list of ITs using PE, or its derivatives, being evaluated preclinically or clinically. Here, we will review these ITs to highlight the advances in PE-based anticancer strategies, as well as review the targeting moieties that are used to reduce the non-specific destruction of non-cancerous cells. Although we tried to be as comprehensive as possible, we have limited our review to those ITs that have proceeded to clinical trials and are still under active clinical evaluation.
Introduction
Cancer is the leading cause of death in developed countries and the second cause of death in developing countries (1, 2). The prevalence and mortality of cancer are rapidly growing because of aging, population growth, and predisposing behaviors such as smoking (2, 3). Over 7 million cancer-related deaths have been recorded worldwide, which is 13% of all deaths (4). Chemotherapy, radiotherapy, and surgery are the major treatment strategies for cancer (5, 6). Chemo- and radio-therapeutics target rapidly growing cancer cells; however, they are also cytotoxic to normal cells (7). Therefore, besides their success in eradicating tumor cells, conventional chemo- and radio-therapeutics result in a wide range of side effects such as alopecia, gastrointestinal symptoms, myelosuppression, and even secondary cancers (7). To reduce the side effects, various modalities have emerged that target cancer cells based on tumor-specific antigens or the use of cell-specific gene promoters (8). The aim of targeted therapy is to inhibit the proliferation of cancer cells either by delivery of growth inhibitory molecules or cytolethal agents to cancer cells or by controlled expression of cytolethal proteins via the use of a cancer-specific promoter (9).
In the former strategy, tumor-specific receptors, which are not, or are much less, expressed on normal cells are targeted by a monoclonal antibody, or an antibody fragment, which consequently blocks ligand–receptor interaction and intracellular signaling (10). Furthermore, the antibody/antibody fragment or the ligand can also be fused to a protein toxin to specifically kill the targeted cancer cell. Such chimeric molecules are called immunotoxins (ITs) (11). In the latter strategy, the coding sequence of a toxic protein is cloned under the control of a tumor-specific promoter and delivered to cancer cells (12). Tumor-specific promoters are derived from genes that are specifically and ectopically overexpressed in cancer cells (13). Although normal cells might uptake the expression cassette (depending on the gene delivery vehicle), transcription can be controlled by the cell type, resulting in expression in only cancer cells (14).
Targeted cancer therapy is an extremely broad area, and novel strategies are continually emerging. This review aims to highlight advances in applications of Pseudomonas exotoxin A (PE) protein and its derivatives for the production of ITs and their use in targeted cancer therapy. It is worth mentioning that the coding sequence of this bacterial toxin has been also used in many studies for cancer cell-specific gene therapy as summarized in Table 1. However, here, we only focus on various PE-derived ITs, one of which has been recently approved for clinical application.
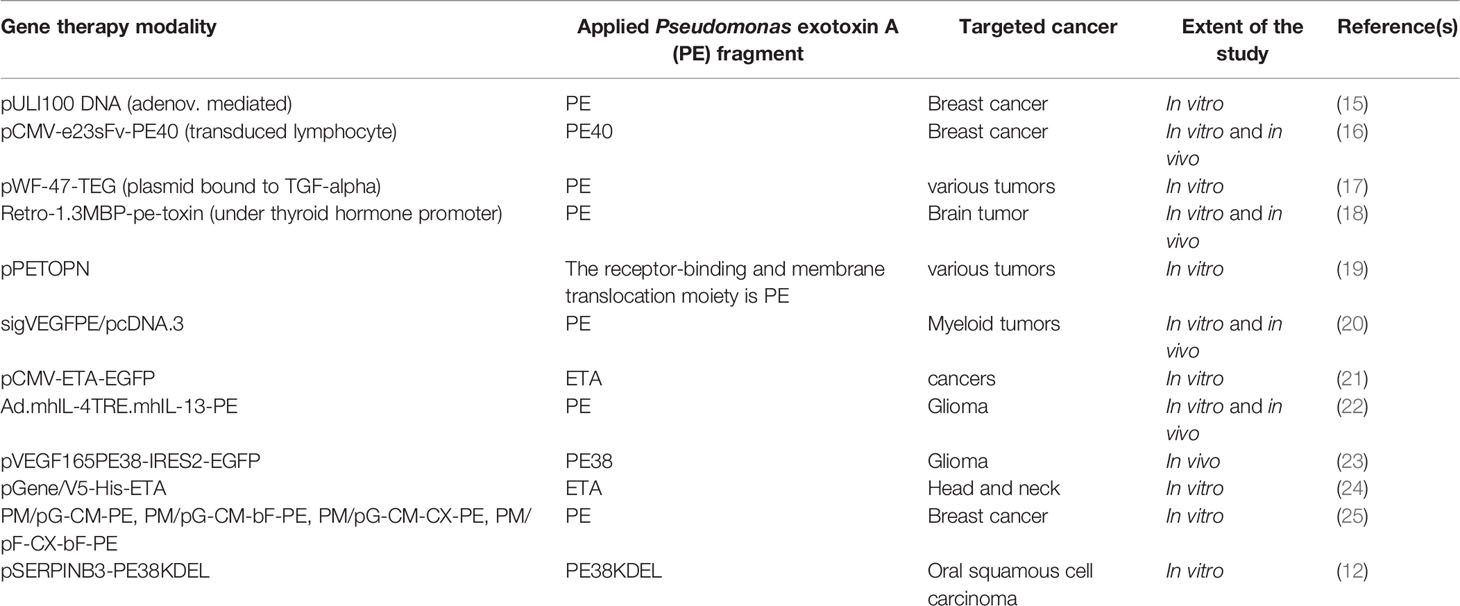
Table 1 A summary of studies on gene therapy applications of Pseudomonas exotoxin A or its derivatives usually encoded under the control of a tumor-specific promoter for targeted killing of corresponding cancer cells.
Immunotoxins
ITs were first defined as engineered proteins consisting of an antibody or antibody fragment, or a ligand of the immune system, such as a growth factor or cytokine, as the targeting moiety, fused to a cytolethal protein. However, recent studies have used other cancer-specific targeting molecules including natural or synthetic cell-penetrating peptides (26, 27) as well as natural and mutated antimicrobial peptides (28, 29). The toxic moiety has not deviated as much and has consisted of cytolethal proteins from plants, animals, fungi, or bacteria (30). Among the bacterial toxins, diphtheria toxin (9, 31, 32), Shiga toxin (33), Vibrio cholerae toxin (34), and PE (35, 36) have been used for construction of ITs. PE is one of the most potent bacterial toxins and is the most toxic virulence factor of the pathogenic bacterium Pseudomonas aeruginosa.
Pseudomonas Exotoxin A
PE is an NAD+-diphthamide-ADP-ribosyl transferase (EC 2.4.2.36), which falls within the family of mono-ADP-ribosyl transferases (37). PE ADP-ribosylates eukaryotic elongation factor-2 (eEF-2) on the ribosomes when it reaches the cytosol of eukaryotic cells (38). eEF-2 is a crucial factor involved in protein biosynthesis and promotes GTP-dependent translocation of mRNA from the ribosomal A-site to the P-site (39). ADP-ribosylation results in the inactivation of eEF-2 and subsequent termination of protein biosynthesis within the affected cell. Consequently, extrinsic and intrinsic apoptosis pathways are activated, which results in cell death (40–42).
PE is a 638-amino-acid protein that belongs to the AB toxins family (Figure 1A), where the A domain harbors enzymatic activity and the B domain acts as a cell-binding moiety (43, 44). The first 25 amino acids form a highly hydrophobic signal sequence that is removed during secretion (5). The remaining 613 amino acids make up three domains. Domain Ia (aa 1–252) is a receptor-binding domain and helps PE to recognize and attach to target cells. Domain II (aa 253–364) facilitates translocation of PE across the cell membrane and contains a furin cleavable motif (aa 274–280, RHRQPR^G). The last four residues (amino acids 400–404) of domain Ib (aa 365–404) along with domain III (405–613 aa) make up the catalytic part of the toxin (45). Upon secretion, the terminal amino acid residue of PE (lysine 613) is thought to be cleaved by a host plasma carboxypeptidase, which converts the REDLK (609–613) motif into REDL (609–612) (as reviewed by 37). PE interacts with the low-density lipoprotein receptor-related protein 1 (LRP-1) (aka CD91 or the α2-macroglobulin receptor) via its Ia domain and is subsequently internalized through receptor-mediated endocytosis. In the early endosome, PE dissociates from the receptor and undergoes a conformational change due to the acidic environment. This makes the furin-cleavable motif accessible, which allows furin to cleave PE into two fragments of about 28 kDa (aa1–279) and 37 kDa (aa280–613) (45). The smaller fragment consists of domain Ia and parts of domain II. The larger fragment contains parts of domain II, domains Ib, and domain III and has enzymatic activity. The 37-kDa fragment exploits a pathway from the late endosome to the trans-Golgi network (TGN) and from there to the ER via retrograde pathway following interaction of its C-terminal REDL (aa 609–612) motif with the KDEL receptors on the TGN (46, 47).
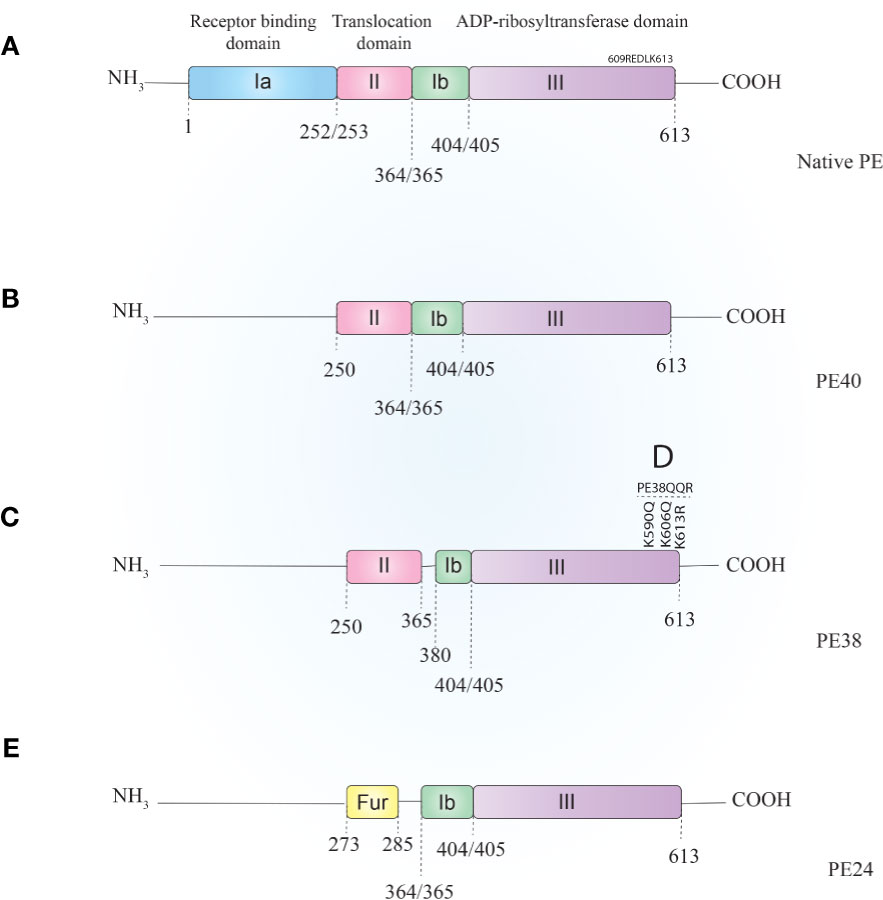
Figure 1 Schematic representation of Pseudomonas exotoxin A (A) and its most applicable derivatives. In order to reduce PE–ITs, non-specific toxicities, immunogenicity, and size, various PE derivatives have been evaluated, most of which are PE40 (B), PE38 (C), PE38QQR (D), and PE24 (E). PE, Pseudomonas exotoxin A; IT, immunotoxin.
Pseudomonas Exotoxin A-Derived Immunotoxins
To date, various ITs have been constructed by chemical conjugation or recombinant fusion of full or truncated PEs to target moieties (Figure 2). Chemically conjugated ITs lacked stability (due to vulnerable disulfide or thioether bonds used for chemical conjugation) and specificity (due to having the native binding domain of PE in addition to having the conjugated targeting moiety) (48, 49). Attempts have been made to produce more stable and specific PE–ITs via recombinant protein production.
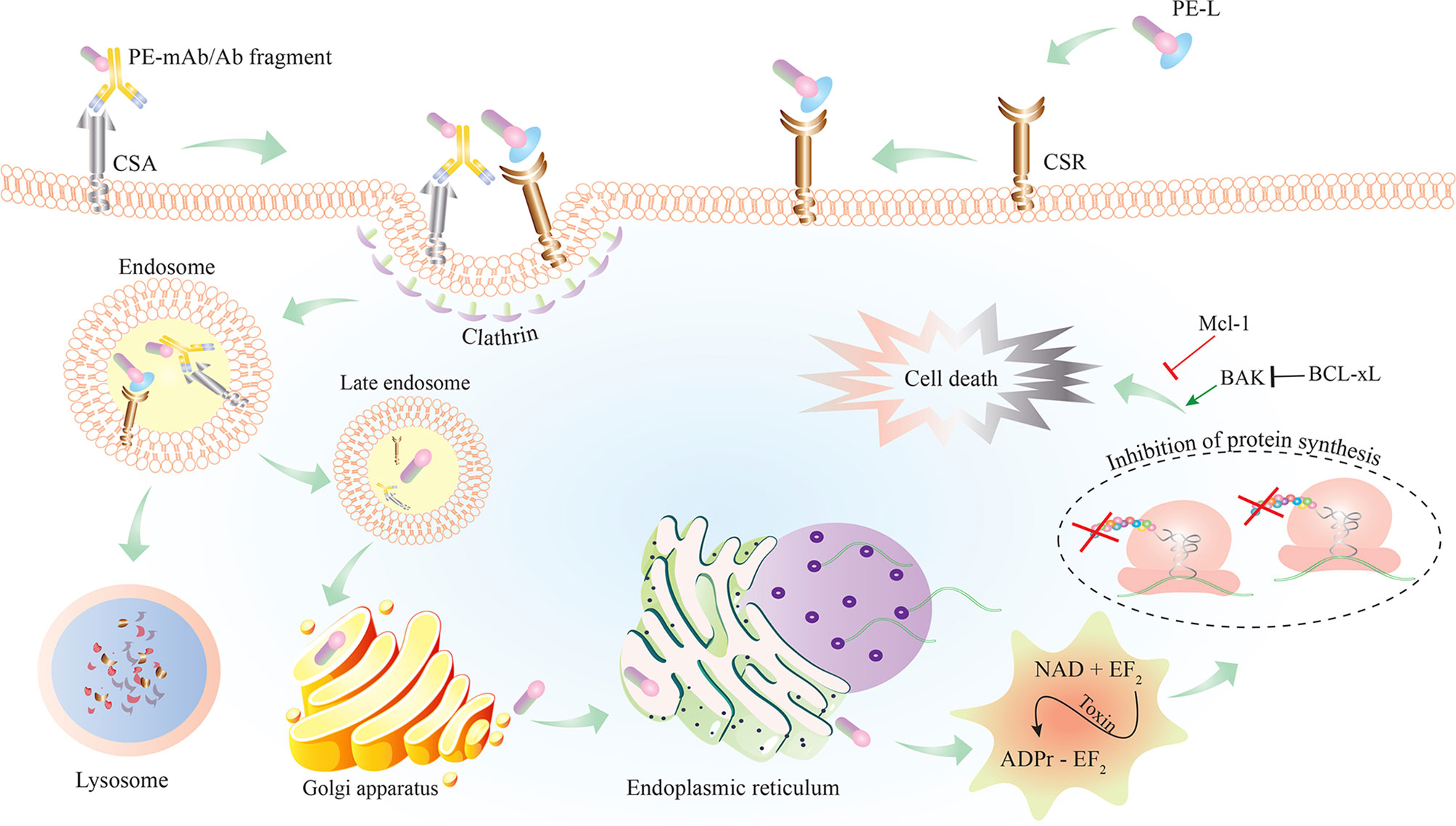
Figure 2 Schematic representation of PE–IT interaction with the cancer-specific antigen (CSA) or cancer-specific receptor (CSR) targeted on cancer cells and the subsequent intracellular events resulting in cell death. PE-L, Pseudomonas exotoxin A (PE) fused to a cancer-specific ligand; Ab, antibody; IT, immunotoxin.
PE40 and PE38 (Figures 1B, C, respectively) are the two most-used truncated forms of PE, constructed by the removal of the Ia domain, or the Ia and part of the Ib domains (aa 365–380), respectively. Moreover, to enhance ER localization of the PE-derived ITs, the C-terminus REDL motif of PE has been mutated to match the KDEL receptors on TGNs (PE-KDEL) (46, 50). Studies involving “PE-derived immunotoxins” retrieved by searching scientific databases have been collated in Supplementary Table 1. PE-derived ITs have been evaluated in vitro, in vivo, and/or in clinical studies. Although the list is comprehensive, this review has been limited to those therapeutics that have advanced to clinical evaluations and that are currently under active clinical trial or clinical application. In Table 2, PE-derived ITs that proceeded to phase I clinical trial are described. However, only a few of them have been safe enough to advance to subsequent phases.
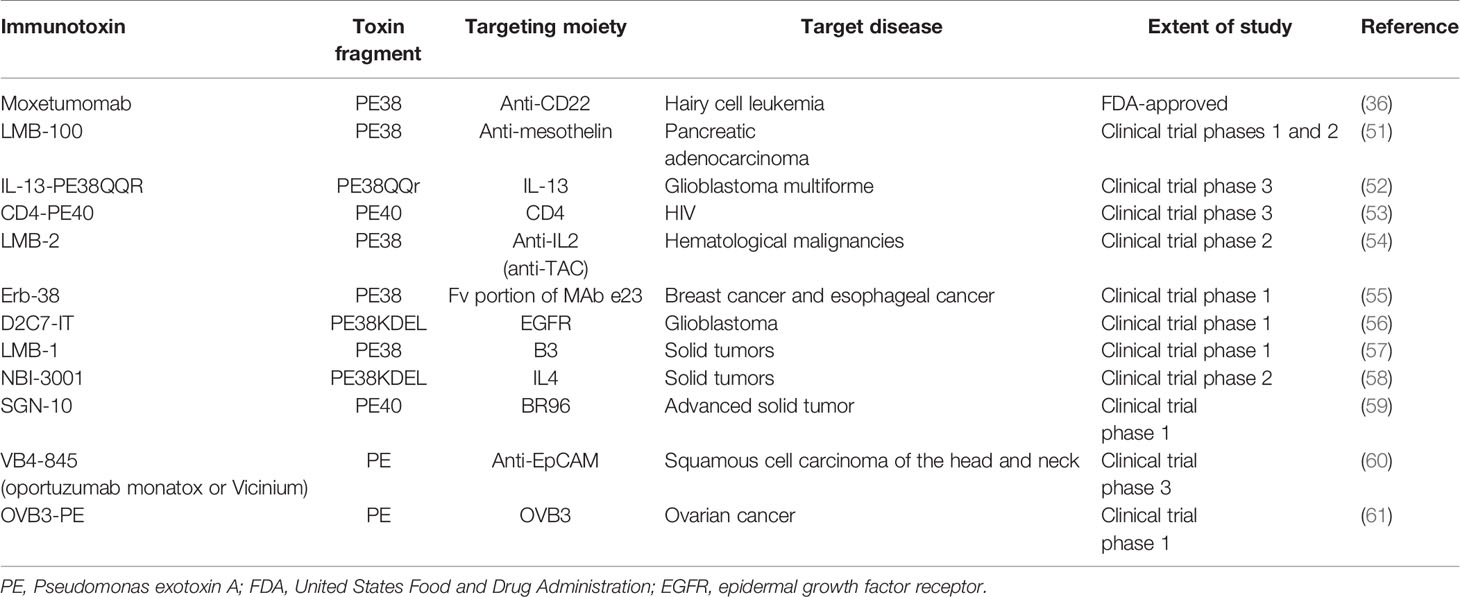
Table 2 PE-derived immunotoxins advanced to various phases of clinical evaluations or clinical application.
In the next sections, we review those PE–ITs that are currently under active clinical trials or approved for clinical application. In each case, first, the targeted receptor and its significance in the corresponding malignancy or disease will be addressed, and then, the IT(s) will be discussed.
Targeting CD22
CD22 is an inhibitory co-receptor of the B-cell receptor that is expressed on the surface of normal B lymphocytes and also many malignant B cells including chronic B-lymphocytic cells (B-CLL), B-lymphoma cells such as Burkitt’s lymphoma, and hairy cell leukemia (HCL). However, because CD22 is not expressed on stem cells, it has been considered for targeting the malignancies (62).
RFB4 is a monoclonal antibody against CD22, which was primarily used for the production of chemically conjugated PE–ITs (63). However, in order to enhance tumor penetration as well reduce non-specific cytotoxicity of the ITs, the chemically conjugated ITs were replaced by a recombinant IT (RFB4(dsFv)PE38) composed of disulfide stabilized variable fragments (dsFv) of the RFB4 antibody fused to PE38. The recombinant IT showed IC50 values of approximately 2 ng/ml on four Burkitt’s lymphoma cell lines, while being not toxic to CD22-negative cell lines (IC50 > 1,000 ng/ml) (64). Next, in vivo studies in nude mouse xenograft of human Burkitt’s lymphoma resulted in 80% and 100% tumor regression following administration of 200 or 275 µg/kg of the IT every other day for three doses. The cumulative MTD of the IT was 3.7 mg/kg for continuous infusion and 1.2 mg/kg for intermittent bolus dosing. In addition, i.v. infusion of three doses of 275 or 350 μg/kg every other day of RFB4(dsFv)PE38 resulted in complete remission (CR) in all animals. In the case of continuous infusion, 100 or 200 μg/kg/day resulted in CR (65).
Safety and specificity of the IT were also studied in cynomolgus monkeys that also express CD22, similar to humans. Three doses of 100 or 500 μg/kg injected every other day were well tolerated and showed mild laboratory abnormalities. The tolerated dose of 500 μg/kg was much higher than the dose needed to obtain CR in the mice xenografts (i.e., 275 μg/kg) (65).
The level of cytotoxicity of the IT was also confirmed on cell samples obtained from 28 patients with CD22-positive leukemias. Moreover, the sensitivity of CD22-positive tumor cells was correlated with CD22 expression on the cells’ surface (66).
RFB4(dsFv)-PE38 proceeded to clinical evaluations under the name of BL22 (aka CAT-3888). In a dose-escalation trial (Clinicaltrials.gov Identifier (CTI): NCT00021983) on 16 patients with chemotherapy-resistant HCL, 69% (11 patients) underwent CR and 12% (two patients) had partial remission (PR) following administration of 3–50 µg/kg of the IT, every other day for three times in a cycle, and within a range of 1 to 15 cycles in different patients. The three non-responsive patients had neutralizing antibodies before therapy or received the least amount of the therapeutic (up to 6 µg/kg/dose) (67). Moreover, only one case of dose-limiting cytokine release syndrome was reported. Therefore, the IT was further evaluated in a phase I trial (CTI: NCT00126646) on 46 CD22+ patients (four non-Hodgkin’s lymphoma (NHL), 11 chronic lymphocytic leukemia (CLL), and 31 HCL). The effective and tolerable dose of BL22 consisted of one cycle (three doses) of 40 μg/kg of the IT every other day. However, among the patients, only HCL patients responded with 19 (61%) CRs and six (19%) PRs (81% of overall response), likely because of the higher number of CD22 on the surface of HCL cells (5,000 to 70,000) compared with CLL (350–1,000) and NHL cells (unknown) (68).
BL22 proceeded to a phase II clinical trial (CTI: NCT00074048) consisting of 36 chemoresistant HCL patients. The patients received a single round of treatment consisting of 40 μg/kg of the IT every other day over 5 days (total of three doses). This resulted in nine cases (25%) with CR, one of which relapsed. After the re-treatment of 20 patients (56%), the total CRs increased to 17 CR (47%). Therefore, the high response rate seen in HCL patients during phase I testing was also confirmed by the phase II trial. In addition, for both phase I and phase II trials, the average dose per cycle was similar, ranging from 29 to 33 μg/kg three times in a cycle. In phase II, hemolytic uremic syndrome (HUS) occurred less than in phase I, and major toxicities were hypoalbuminemia, aspartate aminotransferase/alanine aminotransferase (AST/ALT) elevation, edema, myalgia, proteinuria, fatigue, nausea, and fever (69).
BL22 was also evaluated in patients with B-cell acute lymphoblastic leukemia (ALL) that relapsed after chemotherapy. A phase I dose-escalation trial (CTI: NCT00077493) on 23 pediatric patients who received 10–40 µg/kg of BL22 every other day for three or six doses repeated every 21 or 28 days revealed a significant reduction in peripheral blast counts, recovery of normal blood counts, or decreased blast infiltration of bone marrow and extramedullary sites in 16 subjects (70%). However, although the treatment was associated with an acceptable safety profile, and adverse events were rapidly reversible, higher doses might have been required to achieve maximal benefit (70).
As mentioned before, the very low efficiency of BL22 in CLL patients is likely due to the small quantity of CD22 on the surface of CLL cells. In an attempt to increase the cytotoxicity of BL22, a greater affinity toward CD22 was sought. Phage display was used to screen various mutations in the heavy chain (VH) complementarity-determining regions 3 (CDR3) of RFB4(dsFv). Among five selected mutants, a variant where the CDR3 Ser-Ser-Tyr (SSY) residues were replaced by Thr-His-Trp (THW) showed increased affinity (~14-fold) toward the target molecule compared with the parent protein. When the THW mutant was fused to PE38, the IC50 values on fresh leukemic cells from CLL and HCL patients decreased to 22–29 ng/ml compared with an IC50 > 1,000 ng/ml for BL22 on the same cells. This 50-fold difference in IC50 (71) prompted additional in vitro and in vivo evaluations of RFB4(dsFv) THW mutant-PE38, now also known as HA22 or CAT-8015 (72). HA22 cytotoxicity was evaluated on Burkitt’s lymphoma and CLL cell lines. An overall IC50 of 0.3–8.6 ng/ml, with only 10% cell survival following treatment with ≥50 ng/ml of HA22 was determined. Safety studies were performed in cynomolgus monkeys receiving similar doses of either HA22 or BL22, and no differences have been reported between the two ITs. This suggested that the increased affinity of CAT-8015 for CD22 was not associated with additional or exacerbated side effects. In vivo antitumor activity was evaluated in female athymic NCr nude mice model of human Burkitt’s lymphoma. Administration of three doses of 12.5 μg/kg of HA22 injected every other day resulted in a long-lasting cytostatic response. Moreover, when the dose increased to 150 μg/kg with the same dosing paradigm, a long-term inhibition in the growth of Burkitt’s lymphoma was achieved. Therefore, the non-clinical data suggested that a therapeutic window for HA22 in humans could be achieved (73).
HA22, under the name of moxetumomab pasudotox (aka Moxe), proceeded to a phase I dose-escalation trial (CTI: NCT00462189) consisting of 28 patients with chemotherapy-resistant HCL. The patients received cycles of three doses of 5 to 50 µg/kg every other day, for one to 16 cycles (median: four cycles). It must be noted that 50 µg/kg (administered in 12 of the patients) is one dose level higher than the MTD of BL22. No dose-limiting toxicity (DLT) was reported, and only two cases of reversible HUS were observed. Hypoalbuminemia (in 54% of total treatment cycles) and elevated aminotransferases (38% for both ALT and AST) were the most commonly observed drug-related adverse effects. Hence, HA22 showed an acceptable safety profile in patients at the given doses. Of the patients, 86% responded to the treatment, with 46% having CR, even with doses as low as 10 µg/kg for a single cycle, and 39% having PR. The median duration of response was 29 months, and only one patient relapsed within 1 year of treatment. Moreover, seven patients in CR were negative for minimal residual disease (MRD) (74). The phase I trial was further expanded to a long-term follow-up (CTI: NCT00586924) by the inclusion of 21 patients receiving 50 µg/kg of the IT every other day for three doses in 4-week cycles. Of the 21-patient extension, 91% responded to treatment, with 71% having CR, improving the overall study to an 88% overall response rate and 64% having CR. Among the 21 patients with CR, 11 (55%) cases were negative for MRD. The median CR duration was 82.7 months in MRD-negative CRs (75). Moxe was further evaluated in a phase III pivotal, multicenter, single-arm, open-label study at 32 centers in 14 countries (CTI: NCT01829711). A dose of 40 μg/kg was administered by intravenous (i.v.) infusion over 30 min on days 1, 3, and 5 of 28-day cycles for a maximum of six cycles, or documentation of MRD-negative CR, disease progression, initiation of alternate therapy, or unacceptable toxicity (75). The reason for the dose reduction (50 to 40 μg/kg) was due to the improvement of the Moxe production process, which resulted in higher IT activity (76). A primary report from this pivotal study at a median follow-up of 16.7 months (range: 2.1–48.8 months) showed enrolment of eighty patients, among which 33 (41%) achieved CR. Moreover, 24 (30%) of the CRs were durable (CR with hematologic remission >180 days), and 27 (33%) of the CRs were negative for MRD by immunohistochemistry. The median duration of MRD-positive CR was 5.9 months. The most common treatment-related adverse events leading to permanent discontinuation (n = 8) were HUS, capillary leak syndrome (CLS), and increased blood creatinine. All HUS and CLS events were reversible (77). In Sep 2018, Moxe (Lumoxiti™) was approved by the US FDA for the treatment of adult patients with relapsed or refractory HCL who received at least two prior systemic therapies, including treatment with a purine nucleoside analog (36). Lumoxiti™ obtained European Medicines Agency (EMA) approval for the treatment of HCL patients with the same conditions in Dec 2020 (78). The final report on the phase III study was recently released, which covered a median follow-up period of 24.6 months (range: 1.2–71.7 months). According to the report, the safety concerns were similar to the previous report, but the efficacy data were better, with the rate of durable CR increasing to 36% (vs. 30% in the primary report). In addition, a durable CR rate with HR ≥ 360 days was reported in 33% of the patients (79).
Like BL22, Moxe was also evaluated in pediatric B-lineage ALL malignancies, where blasts from the patients were shown to be sensitive to Moxe (70), and the data provided enough rationale for the IT to be clinically evaluated in children with resistant ALL (80). Therefore, the safety and efficacy of Moxe were evaluated in a phase I multicenter dose-escalation trial (CTI: NCT00659425) followed by a phase II study (CTI: NCT02227108) in children, adolescents, and young adults (ages 1–25 years) with ALL or NHL. However, due to the drug-related adverse effects and unacceptable clinical activity of the IT, the study was terminated prior to a planned interim analysis (81). Therefore, although Moxe was promising for the treatment of relapsed HCL patients and even got approval for the treatment of the disease, it was not deemed to be safe and effective enough for use in pediatric B-lineage ALL. Further investigations are warranted for the latter.
Targeting Interleukin 13 Receptor
IL-13 is predominantly a TH2-derived immunoregulatory cytokine. Since IL-13 receptors are significantly overexpressed on glioma cells (more than 30,000 receptors/cell), a PE-derived IT called IL-13-PE38QQR (Figure 1D) was constructed (82). The PE38 fragment in this IT harbored three mutations: C-terminal lysines at positions 590, 606, and 613 were mutated to glutamine, glutamine, and arginine, respectively. This modification of PE38 enhanced the toxicity of the protein and increased its expression in Escherichia coli during production (83). The cytotoxicity of the IT was assessed in nine glioma cell lines with IC50 values ranging between <0.1 and >300 ng/ml. Furthermore, the cytotoxicity was blocked in the presence of IL-13, which confirmed the specificity of the IT (84).
One of the limitations for targeting brain tumors is the delivery of therapeutic agents through the blood–brain barrier (BBB) (85). IL-13-PE38QQR has suffered this problem due to the size of the molecule. To overcome this limitation, convection-enhanced delivery (CED) has been used. CED facilitates the distribution of macromolecules into brain tissue by positive-pressure micro-infusion over a period of hours to days (86). In a human glioma xenograft model that received 100 µg/kg of IL-13-PE38QQR via CED, all six animals showed CR without acute histopathologic cytotoxicity (87). The safety of IL-13-PE38QQR was assessed in rats receiving 24-h infusion (200 µl totally) of a 10 μg/ml solution of the IT. All mice (n = 6) tolerated the infusion without any neurological changes (88).
The safety and efficacy of IL-13-PE38QQR were further evaluated clinically under the name cintredekin besudotox (CB). In a phase I trial (CTI: NCT00024570) consisting of 51 patients with recurrent malignant gliomas who received 0.25, 0.5, or 1 µg/ml of the IT via CED over 5–6 days of infusion, the 1 µg/ml dose caused DLT. The doses of 0.25 and 0.5 µg/ml were very well tolerated, and no patients displayed any toxicity. The adverse events were neurologic or psychiatric including headache, sensory disturbance, aphasia, asthenia, and convulsion (89). In a phase III clinical trial (CTI: NCT00076986), the efficacy of administration of CB via CED was compared with an approved treatment (Gliadel wafers containing 7.7 mg of carmustine) in adults with recurrent glioblastoma multiforme (GBM). Although the median survival duration in patients who received CB was higher than in patients who received standard treatment (9.1 vs. 8.8 months), the incidence of severe adverse effects, especially thromboembolic complications, resulting in death or termination of the treatment was higher in cases treated with CB. Poor results of CB could also be attributed to the need for exact placement of catheters, which were incorrect in up to 49% of the cases (52, 90). A later animal study comparing CED with bolus stereotactic administration resulted in higher volume distribution in the tumor tissue that lasted for a longer period of time (91); however, improper positioning of the catheter, the catheter configuration, the infusion rate, and the infusion volume could have all contributed to inadequate concentration of CB in tumors and consequently lower than expected efficiencies. Moreover, the GBM patients were not assessed for the density of IL-13 receptor on their tumor cells (52). Variation in the expression level of the receptor among the patients and on the tumors (as shown before (92, 93) could be another reason for the lackluster results.
In addition to GBM, a dose-escalation phase I trial (CTI: NCT00088061) to evaluate the safety and tissue distribution of the IL13-PE38QQR administered via CED in five children with diffuse intrinsic pontine glioma (DIPG) has been reported recently. However, despite interim prevention of DIPG progression in two of the patients, all patients showed disease progression 3 months after initial infusion and died (94). There has been no further report on any aspects of IL13-PE38QQR in DIPG patients.
Targeting Mesothelin
Mesothelin is a cell surface antigen that is highly expressed on pancreatic, colon, lung, and ovarian solid tumors as well as in mesothelioma and cholangiocarcinoma (95). The physiological role of mesothelin is not known, but it might be involved in tumorigenesis and metastasis (96–98). The antigen was first detected in a search for monoclonal antibodies interacting with ovarian cancer cells but not normal human tissues, via immunization of mice with a human ovarian cancer cell line, OVCAR3 (99). Later, it was shown that mesothelial cells are the only normal cells that express mesothelin, albeit at a much lower level than on malignant cells (100).
To target and destroy these tumors, K1, a monoclonal antibody against mesothelin, (99, 101) was conjugated to PE38QQR (K1-LysPE38QQR). In vitro, the IC50 values of 3–6 ng/ml were obtained for A431-K5 cells (a cell line engineered to express mesothelin). In vivo, complete tumor regression was observed in 50% of A431-K5 xenograft nude mice that received three doses of 0.75 mg/kg of the IT (102). Further studies aimed at the production of K1-based ITs by recombinant DNA technology. In this regard, the single-chain variable fragment (scFv) of the K1 antibody was fused to PE38. The scFv-PE38 showed reasonable cytotoxicity on A431-K5 cells with an IC50 of 0.6 ng/ml, whereas on various mesothelin-negative cells, IC50 ranged between 450 and 1,000 ng/ml. Unfortunately, the IT was very unstable with a half-life of 8 h at 37°C (103). Therefore, a series of attempts were made to produce an anti-mesothelin scFv with enhanced stability, binding characteristics (kD), and cytotoxicity (when fused to PE38) (103–105). Finally, an improved scFv with high stability (up to 40 h at 37°C) and high affinity (kd of 11 nM) was obtained and designated as SS scFv. In addition, when fused to PE38, the SS scFV-PE38 IT had an IC50 of 0.5 ng/ml on A431-K5 cells and 6–16 ng/ml on mesothelin-positive cancer cells. The IC50 ranged from 450 to over 1,000 ng/ml on mesothelin-negative cancer cells. An in vivo study in A431-K5 xenograft nude mice, i.v. injected with three doses of 2.6 or 4.3 µg of the IT every other day, showed complete tumor regression (103). Next, random mutations were introduced to hotspots in the complementarity-determining region 3 (CDR3) of the light chain of SS scFv to enhance its affinity. Finally, a variant named SS1 showed much higher affinity (15 times) and when fused with PE38 (SS1 scFv-PE38) showed an increase in in vitro cytotoxicity (13 times) (106). In another study, a disulfide bond stabilized (ds) bivalent SS Fv (SS (dsFv)2) was developed in which the VL and VH of each Fv were joined via a disulfide bond. The two Fvs were fused to PE by a (Gly4-Ser)3 linker. The SS (dsFv)2-PE38 IT had an enhanced half-life (47 vs. 27 min), affinity (40 times more), and in vitro cytotoxicity (10 times higher) when compared with SS dsFv-PE38. However, no significant difference in activity was reported between the two ITs in vivo (107). Combining advances, SS1(dsFv)-PE38 was constructed and evaluated on the primary culture of tumor cells obtained from patients with ovarian and cervical cancers. The IC50 of SS1(dsFv)-PE38 was 1–10 ng/ml for mesothelin-positive cells, while for negative cells, the IC50 was greater than 1,000 ng/ml (108). Moreover, tumor cells obtained from ascites of patients with peritoneal mesothelioma were killed by the IT with IC50 values ranging between 0.08 and 3.9 ng/ml (109).
In vivo evaluation of SS1(dsFv)-PE38 in nude mice xenografted with non-small cell lung cancer (NSCLC) cells resulted in almost complete tumor regression and significantly prevented metastasis (110). Safety and toxicology studies of the IT were performed in cynomolgus monkeys receiving three doses of 250 or 1,000 µg/kg every other day. Decreased appetite and physical activity were the only side effects observed with the 1,000 µg/kg dose. Microscopic evaluation of serosal membranes revealed micro-inflammatory lesions, predicting pleuritic, and/or pericarditis as possible DLTs (111).
SS1(dsFv)-PE38 was termed SS1P and evaluated in a phase I dose-escalation clinical trial (CTI: NCT00006981) on patients with advanced mesothelioma, ovarian, and pancreatic cancers. The patients in different cohorts received three doses of 8–60 µg/kg every other day for either six cycles (8–25 µg/kg doses) or three cycles (25–60 µg/kg doses). However, despite the reasonable safety of the IT, it showed limited efficacy due to anti-IT antibody formation in almost 88% of the patients (111). Considering the high concentration of anti-SS1P antibodies (defined as 75% or more neutralization of SS1P activity in vitro) developed in most of the patients, and the limited efficiency of the IT, later studies used different strategies including 1) administration of the SS1P by continuous infusion instead of bolus injection; 2) administration of SS1P in combination with other chemotherapeutics to enhance tumor penetration and lower the required IT dose; and 3) re-engineering SS1P to remove its B- and T-cell epitopes.
The first strategy was used in a phase I clinical trial (CTI: NCT00024674) consisting of 24 patients with various mesothelioma and ovarian and pancreatic carcinoma. Patients received 4 to 25 μg/kg/day of SS1P by continuous infusion over 10 days. This strategy did lead to slightly lower neutralizing antibodies following the infusion, and the total amount of SS1P administered to the patients was higher than that of the bolus injection. However, the response in patients did not differ significantly as compared with the previous trial (112).
As the second strategy, the combination of SS1P with other chemotherapeutics was targeted. The use of chemotherapeutics has been shown to increase the uptake of IT, presumably through endothelial damage (113), making this approach a potential strategy for SS1P. In an athymic nude mice study, where the mice were xenografted with A431-K5 cells, the use of a single dose of Taxol (50, 20, or 10 mg/kg), cisplatin (5 mg/kg), or cyclophosphamide (15 mg/kg) 24 h before administration of the first dose of SS1P was investigated. As expected, results showed an increased potency and enhanced tumor regression when SS1P administration was combined with a single dose of any of the chemotherapeutics. Interestingly, complete tumor regression for at least 40 days was reported when SS1P was administered after a single dose of 20 or 50 mg/kg of Taxol. Further analysis of the results indicated a synergistic effect for the combinations. However, in vitro assessment of the combinations on A431-K5 cell line revealed no enhanced cytotoxic effects when compared with the agent alone, indicating that the observed synergistic effects must not be related to direct effects of the agents on the cells (114). A subsequent mechanistic study showed that the promoting effect of Taxol on SS1P efficiency was not related to endothelial damage. In Taxol-sensitive tumors, mesothelin shedding into the tumor environment and in blood was significantly reduced (~10 times) over 5 days after a single injection of 20 mg/kg of Taxol. Hence, SS1P was not antagonized by shed mesothelin and could readily bind membrane mesothelin and kill tumor cells (115). A phase I dose-escalation clinical trial (CTI: NCT01445392) has also been reported for SS1P combination therapy, with standard doses of pemetrexed and cisplatin in patients with confirmed malignant pleural mesothelioma. However, although pretreatment of the patients with the chemotherapeutics followed by administration of different doses (25–55 µg/kg) of SS1P caused some partial responses, the production of neutralizing antibodies by the patients’ immune system hampered further administration of SS1P (116). Therefore, the third strategy, i.e., de-immunizing SS1P, was intensely pursued.
De-immunization of SS1P was expected to be promising since previous studies on de-immunization of PE (117, 118) or using lymphocyte depleting regimen (119) showed reduced production of anti-SS1P antibodies and consequently increased SS1P serum concentration. These findings were confirmed by a subsequent pilot clinical trial in patients with refractory malignant mesothelioma, in which the patients received the immune-depleting regimen before each cycle of SS1P administration to reduce their B and T lymphocytes. Lower serum anti-SS1P antibody, higher serum SS1P concentrations, and prolonged PRs were found in the pilot study, supporting the notion that reducing immune responses toward the SS1P could allow repeated administration of the IT and enhance overall responses (120). Among various mutants of SS1P (99, 105, 107, 111, 112, 114–119, 121–123), RG7787 is one of the most widely studied forms, which also proceeded to clinical evaluation.
RG7787 (also known as LMB-100) is a mutant of SS1P developed by 1) introducing seven point mutations to the catalytic domain of PE (domain III), which removed its human B-cell epitopes; 2) removing the majority of domain II of PE, which removed protease sites as well as additional B-cell epitopes; and 3) replacing the murine dsFv with a humanized anti-mesothelin Fab fragment (124). The first two modifications resulted in a new generation of PE–IT based on PE24 (Figure 1E), which, in addition to much lower immunogenicity, was resistant to lysosomal degradation and resulted in a substantial reduction in off-target toxicity (125).
The cytotoxicity and antitumor effects of LMB-100 were verified on different cancer types including breast (126), gastric (126), lung (127), ovarian (128), and colorectal (129) cancers, as well as pancreatic ductal adenocarcinoma (130–133). Almost all cell lines corresponding to these malignancies were affected by LMB-100 at concentrations in the picomolar range (as low as 6.8 pmol/lit) (130–133). Because in vivo evaluation in rodents and cynomolgus monkeys revealed a 5- to 10-fold higher MTD for LMB-100 when compared with the SS1P (124, 126), LMB-100 could be administered at higher doses (2.5 vs. 0.4 mg/kg/dose for SS1P). However, although the treatment of xenograft models with LMB-100 alone resulted in some PRs and tumor regression, no cases of CR were reported (124, 126). Therefore, LMB-100 was evaluated in combination with other chemotherapeutics, including taxanes (paclitaxel and nano-albumin bound (Nab)-paclitaxel) (126–128, 130, 134), platinum-based agents (cisplatin and oxaliplatin) (128, 129), actinomycin D (129, 135), and gemcitabine (134). Co-administration of LMB-100 with the taxanes enhanced the antitumor activity in a synergistic manner and even resulted in four CRs in gastric cancer (received paclitaxel) (126), as well as pancreatic cancer (received nab-paclitaxel) (134) xenografts. Likewise, actinomycin D co-administration synergistically potentiated the antitumor activity of LMB-100 in pancreatic (135) and colorectal (129) carcinoma xenografts and resulted in some CRs. Cisplatin co-administration in an ovarian xenograft model also enhanced antitumor activity of LMB-100, though in an additive manner and without any CRs (128). The combination of gemcitabine only slightly improved LMB-100 activity in pancreatic cancer xenografts (134), and oxaliplatin showed no enhancement in antitumor activity in colorectal xenografts (129). Taken together, enhancement of LMB-100 efficacy is dependent not only on the selected chemotherapeutics but also on the type of cancer itself. None of the above studies could clearly define the mechanisms behind the observed synergistic effects, though further mechanistic and pharmacokinetic studies are underway (136, 137).
LMB-100 monotherapy was also shown to be inefficient in a phase I dose-escalation clinical trial (CTI: NCT02798536) due to the development of anti-LMB-100 antibodies, as well as the lack of durable responses or CRs in patients with different mesothelin-positive cancers (138). Further clinical evaluation of LMB-100 alone or in combination with nab-paclitaxel in a phase I/II clinical study (CTI: NCT02810418) in pancreatic cancer patients was also underwhelming (139). The study included two arms based on the infusion rate of LMB-100. In arm A (A1 and A2) of the study, patients were infused over 30 min (short infusion), while in arm B (B1 and B2), patients were infused over 24 or 48 h (long infusion). Patients in A1 (dose-escalation phase I trial), A2 (phase II trial), and B2 received nab-paclitaxel (125 mg/m2) in addition to the IT. Despite the beneficial antitumor activity of the combination therapy, the amplification of toxic side effects by nab-paclitaxel, especially the CLS-related side effects, such as severe myalgia and cardiac toxicity, prevented its further use (51). A recent abstract presented at the annual meeting of the American Society of Clinical Oncology (2020) reported results corresponding to the B1 and B2 arms of the study. Despite being well tolerated by the patients, higher titers of anti-IT antibodies were reported compared with the short infusion (the A arm of the study). Clinically, the combination of LMB-100 and nab-paclitaxel was not beneficial for the treatment of pancreatic cancer.
Yet another strategy to overcome anti-LMB-100 antibody formation in patients has been the use of tofacitinib (a JAK inhibitor), which was previously used in an SS1P study and significantly lowered the production of anti-SS1P antibodies (140). Concurrent administration of LMB-100 and tofacitinib in patients with mesothelin-positive solid tumors has started recently (CTI: NCT04034238). However, according to a recent brief report on the study presented at the annual meeting of the American Society of Clinical Oncology (2021), the combination has yielded no objective responses and even raised certain safety concerns (141). Another strategy has been to combine LMB-100 with monoclonal antibodies against immune checkpoints, CTLA-4 and PD-1. In vivo and clinical observations have shown that administration of anti-CTLA-4 as well as anti-PD-1 after treatment with either SS1P or LMB-100 resulted in significant antitumor activity and tumor mass eradication (142–144). In this regard, three clinical trials are ongoing including administration of LMB-100 in combination with ipilimumab (anti-CTLA-4, CTI: NCT04840615) or pembrolizumab (anti-PD-1, CTI: NCT03644550) in malignant mesothelioma, as well as pembrolizumab in NSCLC (CTI: NCT04027946). There are no reports from these studies as of the writing of this review.
Targeting Epidermal Growth Factor Receptor
EGFRvIII is a variant of epidermal growth factor receptor (EGFR) missing 267 amino acids of the extracellular domain because of an in-frame deletion of exons 2–7 (145, 146). EGFRvIII is not capable of binding to EGF; however, due to its constitutive kinase activity, EGFRvIII-bearing cells grow rapidly and are invasively metastatic (147). Both wild-type EGFR (wtEGFR) and the EGFRvIII are highly expressed on the surface of GBM cells, while normal brain cells express none of these receptors (148). D2C7 is a monoclonal antibody that binds to both wtEGFR and EGFRvIII on the surface of GBM cells and is subsequently internalized. The antibody localizes to tumors expressing the receptors and has thus been investigated as part of a PE-derived IT (149). The targeting moiety was created by fusing the VH and VL domains of D2C7 via a (Gly4Ser)3 linker and further stabilized by the introduction of Cys mutations in both domains to form a disulfide bridge (D2C7-scdsFv). To create the IT, D2C7-scdsFv was fused to the PE38KDEL (D2C7-(scdsFv)-PE38KDEL). Cytotoxicity evaluation of the IT showed IC50 values ranging from 0.18 to 2.5 ng/ml when assessed on various human epidermoid carcinoma or glioblastoma cell lines. Moreover, the application of the IT in an intracranial tumor murine xenograft overexpressing human wtEGFR increased the survival of the animals by up to 310%. The survival rate was approximately 160% when the IT was administered via CED to intracranial tumor xenografts expressing both receptors. CED was also shown to be a reasonable route to provide sufficient concentrations of the IT in tumors (150, 151). Next, the D2C-(scdsFv)-PE38KDEL IT, or D2C7-IT in brief, was subjected to a toxicity study in rats to determine the MTD and the dose level in which no adverse effect (NOAEL) was observed. The rats received different concentrations of the IT up to 0.4 µg/rat via CED infusion over 72 h. The MTD was between 0.1 and 0.35 µg/rat, and the NOAEL was 0.05 µg (152, 153). D2C7-IT production was scaled (154) for phase I/II clinical trials involving patients with recurrent GB (CTI: NCT02303678). Phase II is still ongoing, but a brief report at the annual meeting of the American Society of Clinical Oncology (2020) presented preliminary data of the dose-escalation phase I, which involved doses ranging from 440 to 23,354 ng/ml. D7C2-IT was administered in patients (two patients in each dose level) via CED, and the patients were examined for DLTs. A dose of 6,920 ng/ml was deemed the most suitable therapeutic dose. Phase II was justified because three patients remained in PR for 28–54 months (56). Another clinical trial combining D7C2-IT with atezolizumab, an anti-PD-1 antibody, has been recently initiated (CTI: NCT04160494) (155) This trial was based on a previous animal study of mice xenografts that received D7C2-IT and either anti-CTLA-4 or anti-PD-1 antibodies. In the animal study, the combination with either antibody resulted in prolonged CRs. Moreover, the surviving animals were resistant to tumor development upon subcutaneous re-challenge, indicating a protective antitumor immunity (156). As of the date of this review, there has been no report on the results of this latest clinical trial.
Targeting Epithelial Cell Adhesion Molecule
Epithelial cell adhesion molecule (EpCAM, aka CD326) is a transmembrane glycoprotein mediating epithelial-specific intracellular cell adhesion (157). The glycoprotein has been shown to be stably overexpressed on the surface of some malignancies including bladder (158), prostate (159), ovary (160), colorectal, breast, and lung cancers (161–163), while its expression is limited on normal tissues corresponding to the malignancies (162). EpCAM is involved in cell migration, proliferation, and differentiation, as well as cell adhesion. The contribution of EpCAM to proliferation and promotion of tumor growth, and metastasis as well, has made it an attractive antigen for cancer cell-targeted antibodies and ITs (164–166).
Primary attempts to produce an IT targeting EpCAM-positive malignancies were made by chemical conjugation of a full anti-EpCAM antibody, MOC-31, to PE via chemical conjugation (167). The IT was effective on human small cell lung cancer (SCLC) in vitro and in vivo in rat xenograft models (168). A subsequent version of the IT was made by chemical conjugation of the MOC-31 antibody to PE40 (PE252-613) and termed MOC31-ETA252–613. The IT was cytotoxic to an array of lung cancer cell lines with IC50 values ranging from 10 to 3,500 pM. Furthermore, i.p. administration of four doses of the IT measuring 10 or 20 µg every other day in SCLC xenografts showed promising antitumor effects as well as an LD50 of 210 µg (169).
To enhance tumor penetration of the IT, a MOC-31 antibody-derived scFv (termed 4D5MOCB) was developed (170) and recombinantly fused to PE252-608KDEL, yielding 4D5MOCB-PE252-608KDEL (also referred to as 4D5MOCB-PE40KDEL). In vitro assessment of 4D5MOCB-PE40KDEL revealed specific cytotoxicity on EpCAM-positive cell lines with IC50 values of 0.005–0.2 pM. In vivo safety and toxicity evaluation of the IT in mice revealed no organ toxicity. Antitumor activity of the 4D5MOCB-PE40KDEL was verified in nude mice xenografted with SW2 (lung), HT29 (colon), or CAL27 (squamous) carcinoma cell lines. The mice received a total amount of either 45 or 30 µg of 4D5MOCB-PE40KDEL over 3 or 1 weeks, respectively. All animals tolerated the administered doses. Significant tumor regression was observed in all animals that received 45 µg of the IT, with five cases of durable CR (171). Toxicokinetics of the 4D5MOCB-PE40KDEL (now termed VB4-845) was assessed following intratumoral and i.v. administration in rats and cynomolgus monkeys. The MTD was determined to be 1 mg/kg in cynomolgus monkey, and local administration of the IT was safe even after repeated injections. However, in rats administered systemic doses, significant toxicities including vascular leak syndrome (VLS) and multiple organ ischemic necrosis resulting in animal death occurred (50). Despite the effect in rats, and considering the promising antitumor effects of VB4-845, as well as its safety via locoregional administration, it was concluded that the IT might be suitable for the treatment of locally accessible malignancies.
A dose-escalation phase I clinical trial involving 20 patients with squamous cell carcinoma of the head and neck (SCCHN) has been conducted. Patients received IT doses ranging at 100–930 μg once weekly for four consecutive weeks. The DLT was grade 3 elevated liver enzyme, and the MTD was determined to be 930 μg/dose. Moreover, a notable reduction in injected tumors size was observed in 10 patients, and four patients showed complete clinical resolution of their tumors (172).
Concurrently, another phase I clinical trial was also performed in patients with EpCAM-positive non-muscle-invasive bladder cancer either refractory to or intolerant of Bacillus Calmette–Guérin (BCG) therapy. The patients received intravesical injections of 0.1–30.16 mg/dose of VB4-845 once weekly for six consecutive doses. Only 20 patients experienced mild VB4-845 administration-related adverse effects; hence, no DLT and, consequently, no MTD was determined. In addition, an overall CR of 39% was observed, most of which occurred at doses higher than 1 mg (173). Thus, since the modality was found to be safe and efficient for the disease, it proceeded to a phase II clinical trial (CTI: NCT00462488) under the name oportuzumab monatox or Vicinium. In that study, 46 patients with non-invasive urothelial carcinoma in situ were split into two cohorts (cohorts 1 and 2) and received 30 mg/dose of intravesical Vicinium according to treatment schedules specific for each cohort for up to 1 year. Forty-two patients completed the treatment regimens. All patients tolerated the IT well. No one reported serious adverse effects related to the medication, and no one died during the study. Of the evaluable patients, 44% achieved a CR that was reduced to 40%, 27%, 18%, and 16% when evaluated at 3, 6, 9, and 12 months, respectively. However, seven patients of both cohorts remained with CR for 18–25 months. This study confirmed the safety and efficacy of Vicinium (60). Vicinium is being evaluated in a phase III clinical trial (CTI: NCT04859751) that started on March 23, 2021, and is expected to be completed around December 2023.
Another phase I clinical trial combining Vicinium with durvalumab [a monoclonal anti-PD-L1 antibody (174) to treat patients with high-grade non-muscle-invasive bladder cancer previously treated with BCG (CTI: NCT03258593)] is ongoing. Durvalumab is expected to potentiate Vicinium through an enhancement of immune responses resulting from a PD1–PD-L1 interaction blockade. Yet there is no report on the clinical effects and outcomes of the combination.
Conclusion
There have been 12 PE–ITs (Table 3) with promising effects in vitro and in animal studies; however, most could not be translated to the clinic. The inability to transfer the results from the laboratory to the clinic was based on safety or efficacy issues, or both. Safety concerns mostly arise due to non-specific uptake of an IT by non-target cells, which is partly due to the physiologic expression of targeted antigen/receptor by normal cells, though at low levels. Furthermore, immune responses against ITs, which are considered foreign proteins by the immune system, could result in both safety and efficacy issues. Recent publications fully review why the immunogenicity of PE-derived ITs and disease resistance renders this treatment inapplicable in a clinical setting (176, 177); however, given the approval of Moxe by the FDA, and promising clinical effects observed following combination therapies, especially with immune checkpoint inhibitors (178) as well as efforts that are underway to address the safety and efficacy issues (179), PE–ITs remain a promising research area for cancer-specific targeted therapeutic modalities.
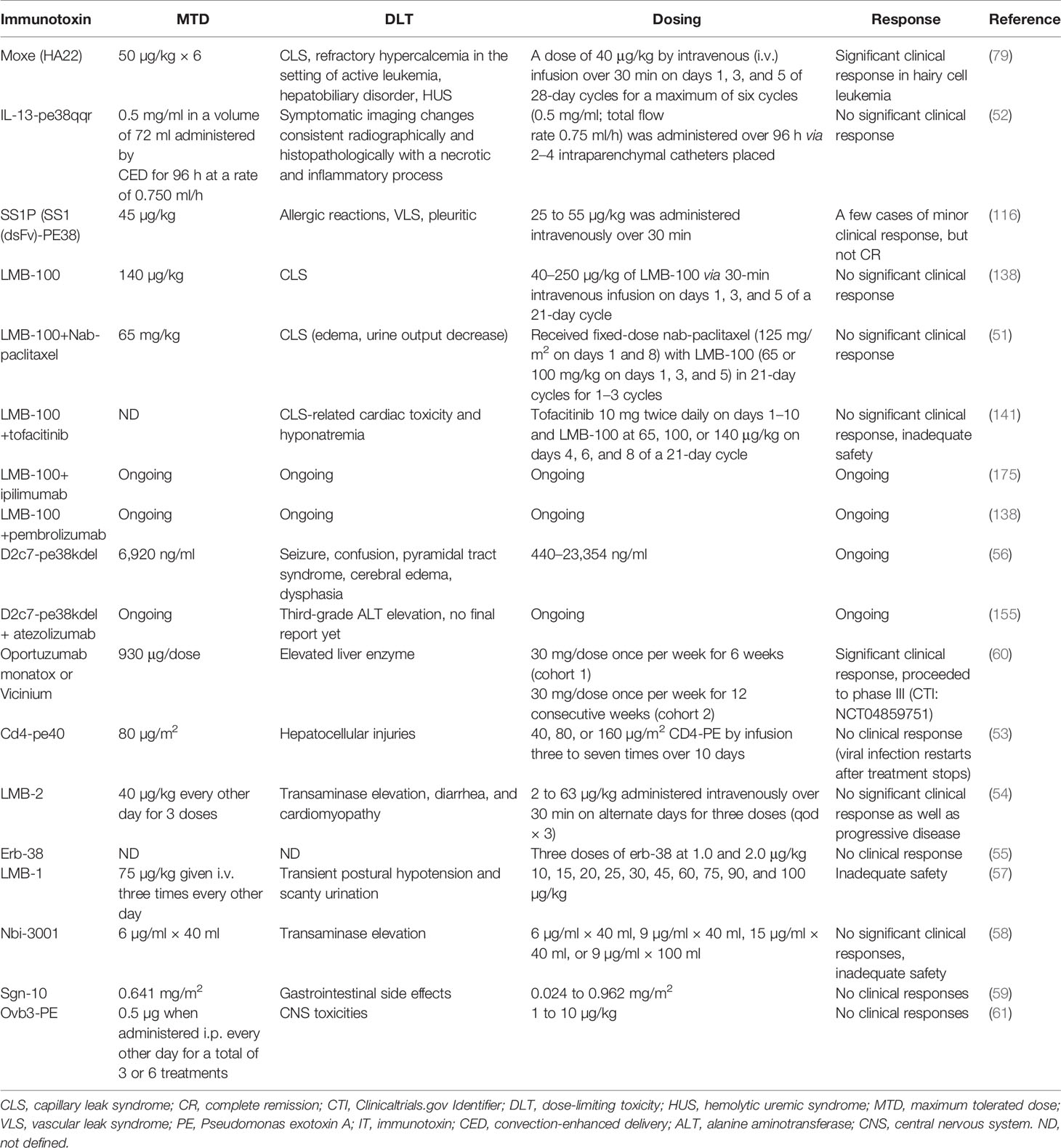
Table 3 Clinical characteristics of PE–ITs advanced to clinical trials, alone or in combination with other therapeutic agents.
Author Contributions
SH, MA, and AJ took part in drafting, revising, and preparing the manuscript. The manuscript was finalized and prepared for submission by AJ. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.781800/full#supplementary-material
Abbreviations
BBB, blood–brain barrier; BCG, Bacillus Calmette–Guérin; CED, convection-enhanced delivery; CLL, chronic lymphocytic leukemia; CR, complete remission; DIPG, diffuse intrinsic pontine glioma; DLT, dose-limiting toxicity; FDA, United States Food and Drug Administration; GB, glioblastoma; GBM, glioblastoma multiforme; HCL, hairy cell leukemia; HUS, hemolytic uremic syndrome; IT, immunotoxin; i.p., intraperitoneal; i.v., intravenous; MRD, minimal residual disease; MTD, maximum tolerated dose; NHL, non-Hodgkin’s lymphoma; SCCHN, squamous cell carcinoma of the head and neck; SCLC, small cell lung cancer; PE, Pseudomonas exotoxin A; PR, partial remission.
References
1. Ghavimi R, Mohammadi E, Akbari V, Shafiee F, Jahanian-Najafabadi A. In Silico Design of Two Novel Fusion Proteins, P28-IL-24 and P28-M4, Targeted to Breast Cancer Cells. Res Pharm Sci (2020) 15:200–8. doi: 10.4103/1735-5362.283820
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
4. Padma VV. An Overview of Targeted Cancer Therapy. BioMedicine (2015) 5:19. doi: 10.7603/s40681-015-0019-4
5. Wolf P, Elsässer-Beile U. Pseudomonas Exotoxin A: From Virulence Factor to Anti-Cancer Agent. Int J Med Microbiol (2009) 299:161–76. doi: 10.1016/j.ijmm.2008.08.003
6. Jahanian-Najafabadi A, Mirian M, Rohani F, Karami K, Hosseini Kharat M, Sadeghi-Aliabadi H. Novel Palladium Complex: Cytotoxicity Against Cisplatin-Resistant K562 Cells. Iran J Pharm Res (2019) 18:1323–31. doi: 10.22037/ijpr.2019.1100714
7. Gerber DE. Targeted Therapies: A New Generation of Cancer Treatments. Am Fam Physician (2008) 77(3):311–9.
8. Xu G, Mcleod HL. Strategies for Enzyme/Prodrug Cancer Therapy. Clin Cancer Res (2001) 1: (7):3314–24.
9. Shafiee F, Aucoin MG, Jahanian-Najafabadi A. Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front Microbiol (2019) 10:2340. doi: 10.3389/fmicb.2019.02340
10. Jahanian-Najafabadi A, Bouzari S, Oloomi M, Habibi Roudkenar M, Mayr LM. Attempts to Express the A1-GMCSF Immunotoxin in the Baculovirus Expression Vector System. Biosci Biotechnol Biochem (2012) 76:749–54. doi: 10.1271/bbb.110862
11. Allahyari H, Heidari S, Ghamgosha M, Saffarian P, Amani J. Immunotoxin: A New Tool for Cancer Therapy. Tumour Biol (2017) 39:1010428317692226. doi: 10.1177/1010428317692226
12. Wu J, Guo Q, Zhang G, Zhao L, Lv Y, Wang J, et al. Study on the Targeted Therapy of Oral Squamous Cell Carcinoma With a Plasmid Expressing PE38KDEL Toxin Under Control of the SERPINB3 Promoter. Cancer Med (2020) 9:2213–22. doi: 10.1002/cam4.2880
13. Javan B, Shahbazi M. Hypoxia-Inducible Tumour-Specific Promoters as a Dual-Targeting Transcriptional Regulation System for Cancer Gene Therapy. Ecancermedicalscience (2017) 11:751. doi: 10.3332/ecancer.2017.751
14. Lavie O, Edelman D, Levy T, Fishman A, Hubert A, Segev Y, et al. A Phase 1/2a, Dose-Escalation, Safety, Pharmacokinetic, and Preliminary Efficacy Study of Intraperitoneal Administration of BC-819 (H19-DTA) in Subjects With Recurrent Ovarian/Peritoneal Cancer. Arch Gynecol Obstet (2017) 295:751–61. doi: 10.1007/s00404-017-4293-0
15. Seth P, Brinkmann U, Schwartz GN, Katayose D, Gress R, Ira P, et al. Adenovirus-Mediated Gene Transfer to Human Breast Tumor Cells: An Approach for Cancer Gene Therapy and Bone Marrow Purging. Cancer Res (1996) 56:1346–51.
16. Zhang L, Zhao J, Wang Z, Wen WH, Zhang YH, Wang CJ, et al. Construction and Expression of Recombinant Antibody/Granzyme B Containing Truncated Translocating Peptide. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2003) 19(5):434–6.
17. Fominaya J, Uherek C, Wels W. A Chimeric Fusion Protein Containing Transforming Growth Factor-Alpha Mediates Gene Transfer via Binding to the EGF Receptor. Gene Ther (1998) 5:521–30. doi: 10.1038/sj.gt.3300614
18. Martín V, Cortés ML, Felipe P, de Farsetti A, Calcaterra NB, Izquierdo M. Cancer Gene Therapy by Thyroid Hormone-Mediated Expression of Toxin Genes. Cancer Res (2000) 60:321824.
19. Chen TY, Hsu CT, Chang KH, Ting CY, Whang-Peng J, Hui CF, et al. Development of DNA delivery system using Pseudomonas exotoxin A and a DNA binding region of human DNA topoisomerase I. Appl Microbiol Biotechnol (2000) 53:558–67. doi: 10.1007/s002530051657
20. Jin N, Chen W, Blazar BR, Ramakrishnan S, Vallera DA. Gene Therapy of Murine Solid Tumors With T Cells Transduced With a Retroviral Vascular Endothelial Growth Factor-Immunotoxin Target Gene. Hum Gene Ther (2002) 13:497–508. doi: 10.1089/10430340252809793
21. Glinka EM, Andryushchenko AS, Sapozhnikov AM, Zatsepina OV. Construction of the Plasmid for Expression of ETA-EGFP Fusion Protein Under Control of the Cytomegalovirus Promoter and Its Effects in HeLa Cells. Plasmid (2009) 62:119–27. doi: 10.1016/j.plasmid.2009.06.004
22. Candolfi M, Xiong W, Yagiz K, Liu C, Muhammad AKMG, Puntel M, et al. Gene Therapy-Mediated Delivery of Targeted Cytotoxins for Glioma Therapeutics. Proc Natl Acad Sci USA (2010) 107:20021–6. doi: 10.1073/pnas.1008261107
23. Hu C, Ji H, Chen S, Zhang H, Wang B, Zhou L, et al. Investigation of a Plasmid Containing a Novel Immunotoxin VEGF165-PE38 Gene for Antiangiogenic Therapy in a Malignant Glioma Model. Int J Cancer (2010) 127:2222–9. doi: 10.1002/ijc.25217
24. Schmidt M, Gruensfelder P, Roller J, Hagen R. Suicide Gene Therapy in Head and Neck Carcinoma Cells: An In Vitro Study. Int J Mol Med (2011) 27:591–7. doi: 10.3892/ijmm.2011.610
25. Moradian C, Rahbarizadeh F. Targeted Toxin Gene Therapy Of Breast Cancer Stem Cells Using CXCR1 Promoter And bFGF 5’UTR. Onco Targets Ther (2019) 12:8809–20. doi: 10.2147/OTT.S221223
26. Soleimani M, Mahnam K, Mirmohammad-Sadeghi H, Sadeghi-Aliabadi H, Jahanian-Najafabadi A. Theoretical Design of a New Chimeric Protein for the Treatment of Breast Cancer. Res Pharm Sci (2016) 11:187–99.
27. Ghavimi R, Akbari V, Jahanian-Najafabadi A. Production and Evaluation of In-Vitro and In-Vivo Effects of P28-IL24, a Promising Anti-Breast Cancer Fusion Protein. Int J Pept Res Ther (2021) 27:2583–94. doi: 10.1007/s10989-021-10275-z
28. Shafiee F, Rabbani M, Jahanian-Najafabadi A. Production and Evaluation of Cytotoxic Effects of DT386-BR2 Fusion Protein as a Novel Anti-Cancer Agent. J Microbiol Methods (2016) 130:100–5. doi: 10.1016/j.mimet.2016.09.004
29. Soleimani M, Sadeghi HM, Jahanian-Najafabadi A. A Bi-Functional Targeted P28-NRC Chimeric Protein With Enhanced Cytotoxic Effects on Breast Cancer Cell Lines. Iran J Pharm Res (2019) 18:735–44. doi: 10.22037/ijpr.2019.2392
30. Mei X, Chen J, Wang J, Zhu J. Immunotoxins: Targeted Toxin Delivery for Cancer Therapy. Pharm Front (2019) 01:e33–45. doi: 10.1055/s-0039-1700507
31. Shapira A, Benhar I. Toxin-Based Therapeutic Approaches. Toxins (Basel) (2010) 2:2519–83. doi: 10.3390/toxins2112519
32. Shafiee F, Rabbani M, Jahanian-Najafabadi A. Optimization of the Expression of DT386-BR2 Fusion Protein in Escherichia Coli Using Response Surface Methodology. Adv Biomed Res (2017) 6:22. doi: 10.4103/2277-9175.201334
33. Jahanian-Najafabadi A, Bouzari S, Oloomi M, Habibi Roudkenar M, Shokrgozar MAA. Assessment of Selective Toxicity of Insect Cell Expressed Recombinant A1-GMCSF Protein Toward GMCSF Receptor Bearing Tumor Cells. Res Pharm Sci (2012) 7:133–40.
34. Sarnovsky R, Tendler T, Makowski M, Kiley M, Antignani A, Traini R, et al. Initial Characterization of an Immunotoxin Constructed From Domains II and III of Cholera Exotoxin. Cancer Immunol Immunother (2010) 59:737–46. doi: 10.1007/s00262-009-0794-4
35. Yu Y, Li J, Zhu X, Tang X, Bao Y, Sun X, et al. Humanized CD7 Nanobody-Based Immunotoxins Exhibit Promising Anti-T-Cell Acute Lymphoblastic Leukemia Potential. Int J Nanomedicine (2017) 12:1969–83. doi: 10.2147/IJN.S127575
36. Dhillon S. Moxetumomab Pasudotox: First Global Approval. Drugs (2018) 78:1763–7. doi: 10.1007/s40265-018-1000-9
37. Michalska M, Wolf P. Pseudomonas Exotoxin A: Optimized by Evolution for Effective Killing. Front Microbiol (2015) 6:963. doi: 10.3389/fmicb.2015.00963
38. Iglewski BH, Liu PV, Kabat D. Mechanism of Action of pseudomonas Aeruginosa Exotoxin A: Adenosine Diphosphate-Ribosylation of Mammalian Elongation Factor 2 In Vitro and In Vivo. Infect Immun (1977) 15:138–44. doi: 10.1128/iai.15.1.138-144.1977
39. Proud CG. Peptide-Chain Elongation in Eukaryotes. Mol Biol Rep (1994) 19:161–70. doi: 10.1007/BF00986958
40. Jenkins CE, Swiatoniowski A, Issekutz AC, Lin TJ. Pseudomonas Aeruginosa Exotoxin A Induces Human Mast Cell Apoptosis by a Caspase-8 and -3-Dependent Mechanism. J Biol Chem (2004) 279:37201–7. doi: 10.1074/jbc.M405594200
41. Chang JH, Kwon HY. Expression of 14-3-3δ, Cdc2 and Cyclin B Proteins Related to Exotoxin A-Induced Apoptosis in HeLa S3 Cells. Int Immunopharmacol (2007) 7:1185–91. doi: 10.1016/j.intimp.2007.05.001
42. Du X, Youle RJ, FitzGerald DJ, Pastan I. Pseudomonas Exotoxin A-Mediated Apoptosis Is Bak Dependent and Preceded by the Degradation of Mcl-1. Mol Cell Biol (2010) 30:3444–52. doi: 10.1128/mcb.00813-09
43. Wedekind JE, Trame CB, Dorywalska M, Koehl P, Raschke TM, McKee M, et al. Refined Crystallographic Structure of Pseudomonas Aeruginosa Exotoxin A and Its Implications for the Molecular Mechanism of Toxicity. J Mol Biol (2001) 314:823–37. doi: 10.1006/jmbi.2001.5195
44. Odumosu O, Nicholas D, Yano H, Langridge W. AB Toxins: A Paradigm Switch From Deadly to Desirable. Toxins (Basel) (2010) 2:1612–45. doi: 10.3390/toxins2071612
45. Weldon JE, Pastan I. A Guide to Taming a Toxin - Recombinant Immunotoxins Constructed From Pseudomonas Exotoxin A for the Treatment of Cancer. FEBS J (2011) 278:4683–700. doi: 10.1111/j.1742-4658.2011.08182.x
46. Kreitman RJ, Pastan I. Importance of the Glutamate Residue of KDEL in Increasing the Cytotoxicity of Pseudomonas Exotoxin Derivatives and for Increased Binding to the KDEL Receptor. Biochem J (1995) 307:29–37. doi: 10.1042/bj3070029
47. Jackson ME, Simpson JC, Girod A, Pepperkok R, Roberts LM, Lord JM. The KDEL Retrieval System Is Exploited by Pseudomonas Exotoxin A, But Not by Shiga-Like Toxin-1, During Retrograde Transport From the Golgi Complex to the Endoplasmic Reticulum. J Cell Sci (1999) 112(Pt 4):467–75. doi: 10.1242/jcs.112.4.467
48. Dosio FD, Brusa P, Cattel L. Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage Between Components. Toxins (Basel) (2011) 3:848–83. doi: 10.3390/toxins3070848
49. Antignani A, Fitzgerald D. Immunotoxins: The Role of the Toxin. Toxins (Basel) (2013) 5:1486–502. doi: 10.3390/toxins5081486
50. Brown J, Rasamoelisolo M, Spearman M, Bosc D, Cizeau J, Entwistle J, et al. Preclinical Assessment of an Anti-EpCAM Immunotoxin: Locoregional Delivery Provides a Safer Alternative to Systemic Administration. Cancer Biother Radiopharm (2009) 24:477–87. doi: 10.1089/cbr.2008.0579
51. Alewine C, Ahmad M, Peer CJ, Hu ZI, Lee M-J, Yuno A, et al. Phase I/II Study of the Mesothelin-Targeted Immunotoxin LMB-100 With Nab-Paclitaxel for Patients With Advanced Pancreatic Adenocarcinoma. Clin Cancer Res (2020) 26:828–36. doi: 10.1158/1078-0432.CCR-19-2586
52. Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, et al. Phase III Randomized Trial of CED of IL13-PE38QQR vs Gliadel Wafers for Recurrent Glioblastoma. Neuro Oncol (2010) 12:871–81. doi: 10.1093/neuonc/nop054
53. Ramachandran RV, Katzenstein DA, Wood R, Batts DH, Merigan TC. Failure of Short-Term CD4-PE40 Infusions to Reduce Virus Load in Human Immunodeficiency Virus-Infected Persons. J Infect Dis (1994) 170:1009–13. doi: 10.1093/infdis/170.4.1009
54. Kreitman RJ, Stetler-Stevenson M, Jaffe ES, Conlon KC, Steinberg SM, Wilson W, et al. Complete Remissions of Adult T-cell Leukemia with Anti-CD25 Recombinant Immunotoxin LMB-2 and Chemotherapy to Block Immunogenicity. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:310–8. doi: 10.1158/1078-0432.CCR-15-1412
55. Pai-Scherf LH, Villa J, Pearson D, Watson T, Liu E, Willingham MC, et al. Hepatotoxicity in Cancer Patients Receiving erb-38, A Recombinant Immunotoxin That Targets the erbB2 Receptor. Clin Cancer Res (1999) 5:2311–5.
56. Desjardins A, Randazzo D, Chandramohan V, Peters KB, Johnson MO, Threatt S, et al. Phase I Trial of D2C7 Immunotoxin (D2C7-IT) Administered Intratumorally via Convection-Enhanced Delivery (CED) for Recurrent Malignant Glioma (MG). J Clin Oncol (2020) 38:2566. doi: 10.1200/JCO.2020.38.15_suppl.2566
57. Pai LH, Wittes R, Setser A, Willingham MC, Pastan I. Treatment of Advanced Solid Tumors With Immunotoxin LMB-1: An Antibody Linked to Pseudomonas exotoxin. Nat Med (1996) 2:350–3. doi: 10.1038/nm0396-350
58. Weber F, Asher A, Bucholz R, Berger M, Prados M, Chang S, et al. Safety, Tolerability, and Tumor Response of IL4-Pseudomonas exotoxin (NBI-3001) in Patients With Recurrent Malignant Glioma. J Neurooncol (2003) 64:125–37. doi: 10.1007/BF02700027
59. Posey JA, Khazaeli MB, Bookman MA, Nowrouzi A, Grizzle WE, Thornton J, et al. A Phase I Trial of the Single-Chain Immunotoxin SGN-10 (BR96 sFv-PE40) in Patients With Advanced Solid Tumors. Clin Cancer Res (2002) 8:3092–9.
60. Kowalski M, Guindon J, Brazas L, Moore C, Entwistle J, Cizeau J, et al. A Phase II Study of Oportuzumab Monatox: An Immunotoxin Therapy for Patients With Noninvasive Urothelial Carcinoma in Situ Previously Treated With Bacillus Calmette-Guérin. J Urol (2012) 188:1712–8. doi: 10.1016/j.juro.2012.07.020
61. Pai LH, Bookman MA, Ozols RF, Young RC, Smith JW 2nd, Longo DL, et al. Clinical Evaluation of Intraperitoneal Pseudomonas exotoxin Immunoconjugate OVB3-PE in Patients With Ovarian Cancer. J Clin Oncol (1991) 9:2095–103. doi: 10.1200/JCO.1991.9.12.2095
62. Walker JA, Smith KGC. Dependence of Surface Monoclonal Antibody Binding on Dynamic Changes in Fcγriib Expression. Immunology (2008) 124:412–8. doi: 10.1111/j.1365-2567.2007.02791.x
63. Mansfield E, Pastan I, Fitzgerald DJ. Characterization of RFB4-PseudomonasExotoxin A Immunotoxins Targeted to CD22 on B-Cell Malignancies. Bioconjugate Chem (1996) 7:1802. doi: 10.1021/bc960043y
64. Mansfield E, Amlot P, Pastan I, FitzGerald DJ. Recombinant RFB4 Immunotoxins Exhibit Potent Cytotoxic Activity for CD22-Bearing Cells and Tumors. Blood (1997) 90:2020–6. doi: 10.1182/blood.V90.5.2020
65. Kreitman RJ, Wang QC, FitzGerald DJ, Pastan I. Complete Regression of Human B-Cell Lymphoma Xenografts in Mice Treated With Recombinant Anti-CD22 Immunotoxin RFB4(dsFv)-PE38 at Doses Tolerated by Cynomolgus Monkeys. Int J Cancer (1999) 81:148–55. doi: 10.1002/(sici)1097-0215(19990331)81:1<148::aid-ijc24>3.0.co;2-l
66. Kreitman RJ, Margulies I, Stetler-Stevenson M, Wang QC, FitzGerald DJ, Pastan I. Cytotoxic Activity of Disulfide-Stabilized Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) Toward Fresh Malignant Cells From Patients With B-Cell Leukemias. Clin Cancer Res (2000) 6:1476–87.
67. Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, et al. Efficacy of the Anti-CD22 Recombinant Immunotoxin BL22 in Chemotherapy-Resistant Hairy-Cell Leukemia. N Engl J Med (2001) 345:241–7. doi: 10.1056/NEJM200107263450402
68. Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJP, Wilson WH, et al. Phase I Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With B-Cell Malignancies. J Clin Oncol (2005) 23:6719–29. doi: 10.1200/JCO.2005.11.437
69. Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, FitzGerald DJP, Wilson WH, et al. Phase II Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With Hairy Cell Leukemia. J Clin Oncol (2009) 27:2983–90. doi: 10.1200/JCO.2008.20.2630
70. Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, et al. Anti-CD22 Immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-Positive Hematologic Malignancies of Childhood: Preclinical Studies and Phase I Clinical Trial. Clin Cancer Res (2010) 16:1894–903. doi: 10.1158/1078-0432.CCR-09-2980
71. Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved Cytotoxic Activity Toward Cell Lines and Fresh Leukemia Cells of a Mutant Anti-CD22 Immunotoxin Obtained by Antibody Phage Display. Clin Cancer Res (2002) 8:995–1002.
72. Alderson RF, Escandon E, Chen T, Yeung P, Hodges D, Geng W, et al. Characterization of CAT-8015: A Pseudomonas Exotoxin Based Immunotoxin for the Treatment of CD22-Related Hematological Malignancies. Cancer Res (2006) 66:877 LP – 877.
73. Alderson RF, Kreitman RJ, Chen T, Yeung P, Herbst R, Fox J, et al. CAT-8015: A Second-Generation Pseudomonas Exotoxin a-Based Immunotherapy Targeting CD22-Expressing Hematologic Malignancies. Clin Cancer Res (2009) 15:832–9. doi: 10.1158/1078-0432.CCR-08-1456
74. Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Phase I Trial of Anti-CD22 Recombinant Immunotoxin Moxetumomab Pasudotox (CAT-8015 or HA22) in Patients With Hairy Cell Leukemia. J Clin Oncol (2012) 30:1822–8. doi: 10.1200/JCO.2011.38.1756
75. Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Minimal Residual Hairy Cell Leukemia Eradication With Moxetumomab Pasudotox: Phase 1 Results and Long-Term Follow-Up. Blood (2018) 131:2331–4. doi: 10.1182/blood-2017-09-803072
76. Kreitman RJ, Pastan I. Contextualizing the Use of Moxetumomab Pasudotox in the Treatment of Relapsed or Refractory Hairy Cell Leukemia. Oncologist (2020) 25:170–7. doi: 10.1634/theoncologist.2019-0370
77. Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Karlin L, Robak T, et al. Moxetumomab Pasudotox in Relapsed/Refractory Hairy Cell Leukemia. Leukemia (2018) 32:1768–77. doi: 10.1038/s41375-018-0210-1
78. Committee for Medicinal Products for Human Use. Lumoxiti Moxetumomab Pasudotox. Eur Med Agency (2020).
79. Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Robak T, le Coutre PD, et al. Moxetumomab Pasudotox in Heavily Pre-Treated Patients With Relapsed/Refractory Hairy Cell Leukemia (HCL): Long-Term Follow-Up From the Pivotal Trial. J Hematol Oncol (2021) 14:1–11. doi: 10.1186/s13045-020-01004-y
80. Mussai F, Campana D, Bhojwani D, Stetler-Stevenson M, Steinberg SM, Wayne AS, et al. Cytotoxicity of the Anti-CD22 Immunotoxin HA22 (CAT-8015) Against Paediatric Acute Lymphoblastic Leukaemia. Br J Haematol (2010) 150:352–8. doi: 10.1111/j.1365-2141.2010.08251.x
81. Shah NN, Bhojwani D, August K, Baruchel A, Bertrand Y, Boklan J, et al. Results From an International Phase 2 Study of the Anti-CD22 Immunotoxin Moxetumomab Pasudotox in Relapsed or Refractory Childhood B-Lineage Acute Lymphoblastic Leukemia. Pediatr Blood Cancer (2020) 67:1–9. doi: 10.1002/pbc.28112
82. Debinski W, Obiri NI, Pastan I, Puri RK. A Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin Is Highly Cytotoxic to Human Carcinoma Cells Expressing Receptors for Interleukin 13 and Interleukin 4. J Biol Chem (1995) 270:16775–80. doi: 10.1074/jbc.270.28.16775
83. Debinski W, Pastan I. An Immunotoxin With Increased Activity and Homogeneity Produced by Reducing the Number of Lysine Residues in Recombinant. Bioconjugate Chem (1994) 5:40–6. doi: 10.1021/bc00025a006
84. Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Advances in Brief: Human Glioma Cells Overexpress Receptors for Interleukin 13 and Are Extremely Sensitive to a Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin. Clin Cancer Res (1995) 1:1253–8.
85. Prabhu SS, Broaddus WC, Gillies GT, Loudon WG, Chen ZJ, Smith B. Distribution of Macromolecular Dyes in Brain Using Positive Pressure Infusion: A Model for Direct Controlled Delivery of Therapeutic Agents. Surg Neurol (1998) 50:367–75. doi: 10.1016/S0090-3019(97)00361-3
86. Lieberman DM, Laske DW, Morrison PF, Bankiewicz KS, Oldfield EH. Convection-Enhanced Distribution of Large Molecules in Gray Matter During Interstitial Drug Infusion. J Neurosurg (1995) 82:1021–9. doi: 10.3171/jns.1995.82.6.1021
87. Kunwar S. Convection Enhanced Delivery of IL13-PE38QQR for Treatment of Recurrent Malignant Glioma: Presentation of Interim Findings From Ongoing Phase 1 Studies. Acta Neurochir Suppl (2003) 88:105–11. doi: 10.1007/978-3-7091-6090-9_16
88. Souweidane MM, Occhiogrosso G, Mark EB, Edgar MA. Interstitial Infusion of IL13-PE38QQR in the Rat Brain Stem. J Neurooncol (2004) 67:287–93. doi: 10.1023/b:neon.0000024219.47447.91
89. Kunwar S, Prados MD, Chang SM, Berger MS, Lang FF, Piepmeier JM, et al. Direct Intracerebral Delivery of Cintredekin Besudotox (IL13-PE38QQR) in Recurrent Malignant Glioma: A Report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol (2007) 25:837–44. doi: 10.1200/JCO.2006.08.1117
90. Mueller S, Polley MY, Lee B, Kunwar S, Pedain C, Wembacher-Schröder E, et al. Effect of Imaging and Catheter Characteristics on Clinical Outcome for Patients in the PRECISE Study. J Neurooncol (2011) 101:267–77. doi: 10.1007/s11060-010-0255-0
91. Suzuki A, Leland P, Kobayashi H, Choyke PL, Jagoda EM, Inoue T, et al. Analysis of Biodistribution of Intracranially Infused Radiolabeled Interleukin-13 Receptor-Targeted Immunotoxin IL-13PE by SPECT/CT in an Orthotopic Mouse Model of Human Glioma. J Nucl Med (2014) 55:1323–9. doi: 10.2967/jnumed.114.138404
92. Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of Interleukin-13 Receptor α2 in Glioblastoma Multiforme: Implications for Targeted Therapies. Cancer Res (2007) 67:7983–6. doi: 10.1158/0008-5472.CAN-07-1493
93. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of Nine Tumour Antigens in a Series of Human Glioblastoma Multiforme: Interest of EGFRvIII, IL-13rα2, Gp100 and TRP-2 for Immunotherapy. J Neurooncol (2007) 81:139–48. doi: 10.1007/s11060-006-9220-3
94. Heiss JD, Jamshidi A, Shah S, Martin S, Wolters PL, Argersinger DP, et al. Phase I Trial of Convection-Enhanced Delivery of IL13-Pseudomonas Toxin in Children With Diffuse Intrinsic Pontine Glioma. J Neurosurg Pediatr (2019) 23:333–42. doi: 10.3171/2018.9.PEDS17225
95. Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov (2016) 6:133–46. doi: 10.1158/2159-8290.CD-15-0583
96. Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin Binding to CA125/MUC16 Promotes Pancreatic Cancer Cell Motility and Invasion via MMP-7 Activation. Sci Rep (2013) 3:1–10. doi: 10.1038/srep01870
97. Avula LR, Michael R, Salma E-B, Arons D, Albalawy R, Chen X, et al. Mesothelin Enhances Tumor Vascularity in Newly Forming Pancreatic Peritoneal Metastases Leela. Physiol Behav (2017) 176:139–48. doi: 10.1016/j.physbeh.2017.03.040
98. Shin S, Park S, Kim MH, Nam CM, Kim H, Choi YY, et al. Mesothelin Expression Is a Predictive Factor for Peritoneal Recurrence in Curatively Resected Stage III Gastric Cancer. Oncologist (2019) 24:1108–14. doi: 10.1634/theoncologist.2018-0896
99. Chang K, Pai LH, Batra JK, Pastan I, Willingham MC. Characterization of the Antigen (CAK1) Recognized by Monoclonal Antibody K1 Present on Ovarian Cancers and Normal Mesothelium. Cancer Res (1992) 52:181–6.
100. Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J Clin Oncol (2016) 34:4171–9. doi: 10.1200/JCO.2016.68.3672
101. Chang K, Pastan I. Molecular Cloning of Mesothelin, a Differentiation Antigen Present on Mesothelium, Mesotheliomas, and Ovarian Cancers. Proc Natl Acad Sci USA (1996) 93:136–40. doi: 10.1073/pnas.93.1.136
102. Hassan R, Viner JL, Wang QC, Margulies I, Kreitman RJ, Pastan I. Anti-Tumor Activity of K1-LysPE38QQR, an Immunotoxin Targeting Mesothelin, a Cell-Surface Antigen Overexpressed in Ovarian Cancer and Malignant Mesothelioma. J Immunother (2000) 23:473–9. doi: 10.1097/00002371-200007000-00011
103. Chowdhury PS, Vasmatzis G, Beers R, Lee B, Pastan I. Improved Stability and Yield of a Fv-Toxin Fusion Protein by Computer Design and Protein Engineering of the Fv. J Mol Biol (1998) 281:917–28. doi: 10.1006/jmbi.1998.1980
104. Chowdhury PS, Chang K, Pastan I. Isolation of Anti-Mesothelin Antibodies From a Phage Display Library. Mol Immunol (1997) 34:9–20. doi: 10.1016/s0161-5890(97)00011-4
105. Chowdury PS, Viner JL, Beers R, Pastan I. Isolation of a High-Affinity Stable Single-Chain Fv Specific for Mesothelin From DNA-Immunized Mice by Phage Display and Construction of a Recombinant Immunotoxin With Anti-Tumor Activity. Proc Natl Acad Sci USA (1998) 95:669–74. doi: 10.1073/pnas.95.2.669
106. Chowdhury PS, Pastan I. Improving Antibody Affinity by Mimicking Somatic Hypermutation In Vitro. Nat Biotechnol (1999) 17:568–72. doi: 10.1038/9872
107. Bera TK, Williams-Gould J, Beers R, Chowdhury P, Pastan I. Bivalent Disulfide-Stabilized Fragment Variable Immunotoxin Directed Against Mesotheliomas and Ovarian Cancer. Mol Cancer Ther (2001) 1:79–84.
108. Hassan R, Lerner MR, Benbrook D, Lightfoot SA, Brackett DJ, Wang Q. Antitumor Activity of SS ( dsFv ) PE38 and SS1 ( dsFv ) PE38 , Recombinant Antimesothelin Immunotoxins Against Human Gynecologic Cancers Grown in Organotypic Culture in Vitro. Clin Cancer Res (2002) 8:3520–6.
109. Li Q, Verschraegen CF, Mendoza J, Hassan R. Cytotoxic Activity of the Recombinant Anti-Mesothelin Immunotoxin, SS1(dsFv)PE38, Towards Tumor Cell Lines Established From Ascites of Patients With Peritoneal Mesotheliomas. Anticancer Res (2004) 24:1327–35.
110. Fan D, Yano S, Shinohara H, Solorzano C, Arsdall M, Bucana CD, et al. Targeted Therapy Against Human Lung Cancer in Nude Mice by High-Affinity Recombinant Antimesothelin Single-Chain Fv Immunotoxin. Mol Cancer Therap (2002) 1:595–600.
111. Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, et al. Phase I Study of SS1P, A Recombinant Anti-Mesothelin Immunotoxin Given as A Bolus I.V. Infusion to Patients With Mesothelin-Expressing Mesothelioma, Ovarian, and Pancreatic Cancers. Clin Cancer Res (2007) 13:5144–9. doi: 10.1158/1078-0432.CCR-07-0869
112. Kreitman RJ, Hassan R, FitzGerald DJ, Pastan I. Phase I Trial of Continuous Infusion Anti-Mesothelin Recombinant Immunotoxin SS1P. Clin Cancer Res (2009) 15:5274–9. doi: 10.1158/1078-0432.CCR-09-0062
113. Holden SA, Lan Y, Pardo AM, Wesolowski JS, Gillies SD. Augmentation of Antitumor Activity of an Antibody-Interleukin 2 Immunocytokine With Chemotherapeutic Agents. Clin Cancer Res (2001) 7:2862–9.
114. Zhang YJ, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, et al. Synergistic Antitumor Activity of Taxol and Immunotoxin SS1P in Tumor-Bearing Mice. Clin Cancer Res (2006) 12:4695–701. doi: 10.1158/1078-0432.CCR-06-0346
115. Zhang Y, Xiang L, Hassan R, Pastan I. Immunotoxin and Taxol Synergy Results From a Decrease in Shed Mesothelin Levels in the Extracellular Space of Tumors. Proc Natl Acad Sci USA (2007) 104:17099–104. doi: 10.1073/pnas.0708101104
116. Hassan R, Sharon E, Thomas A, Zhang J, Ling A, Miettinen M, et al. Phase 1 Study of the Antimesothelin Immunotoxin SS1P in Combination With Pemetrexed and Cisplatin for Front-Line Therapy of Pleural Mesothelioma and Correlation of Tumor Response With Serum Mesothelin, Megakaryocyte Potentiating Factor, and Cancer Antigen. Cancer (2014) 120:3311–9. doi: 10.1002/cncr.28875
117. Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, et al. Recombinant Immunotoxin Against B-Cell Malignancies With No Immunogenicity in Mice by Removal of B-Cell Epitopes. Proc Natl Acad Sci USA (2011) 108:5742–7. doi: 10.1073/pnas.1102746108
118. Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L, et al. Recombinant Immunotoxin Engineered for Low Immunogenicity and Antigenicity by Identifying and Silencing Human B-Cell Epitopes. Proc Natl Acad Sci USA (2012) 109:11782–7. doi: 10.1073/pnas.1209292109
119. Mossoba ME, Onda M, Taylor J, Massey PR, Treadwell S, Sharon E, et al. Pentostatin Plus Cyclophosphamide Safely and Effectively Prevents Immunotoxin Immunogenicity in Murine Hosts. Clin Cancer Res (2011) 17:3697–705. doi: 10.1158/1078-0432.CCR-11-0493
120. Hassan R, Miller AC, Sharon E, Thomas A, Reynolds JC, Ling A, et al. Major Cancer Regressions in Mesothelioma After Treatment With an Anti-Mesothelin Immunotoxin and Immune Suppression. Sci Transl Med (2013) 5:208ra147. doi: 10.1126/scitranslmed.3006941
121. Weldon JE, Skarzynski M, Therres JA, Ostovitz JR, Zhou H, Kreitman RJ, et al. Designing the Furin-Cleavable Linker in Recombinant Immunotoxins Based on Pseudomonas Exotoxin A. Bioconjug Chem (2015) 26:1120–8. doi: 10.1021/acs.bioconjchem.5b00190
122. Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, et al. Recombinant Immunotoxin With T Cell Epitope Mutations That Greatly Reduce Immunogenicity for Treatment of Mesothelin Expressing Tumors. Molecular Cancer Therapeutics (2015) 14:2789–96. doi: 10.1158/1535-7163.MCT-15-0532
123. Kaplan G, Lee F, Onda M, Kolyvas E, Bhardwaj G, Baker D, et al. Protection of the Furin Cleavage Site in Low-Toxicity Immunotoxins Based on Pseudomonas Exotoxin A. Toxins (Basel) (2016) 8:217. doi: 10.3390/toxins8080217
124. Niederfellner G, Bauss F, Imhof-Jung S, Hesse F, Kronenberg S, Staak R, et al. Abstract 4510: RG7787 - A Novel De-Immunized PE Based Fusion Protein for Therapy of Mesothelin-Positive Solid Tumors. Cancer Res (2014) 74:4510–0. doi: 10.1158/1538-7445.am2014-4510
125. Weldon JE, Xiang L, Zhang J, Beers R, Walker DA, Onda M, et al. A Recombinant Immunotoxin Against the Tumor-Associated Antigen Mesothelin Reengineered for High Activity, Low Off-Target Toxicity, and Reduced Antigenicity. Mol Cancer Ther (2013) 12:48–57. doi: 10.1158/1535-7163.MCT-12-0336
126. Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K, Pastan I. Efficacy of RG7787, a Next-Generation Mesothelin-Targeted Immunotoxin, Against Triple-Negative Breast and Gastric Cancers. Mol Cancer Ther (2014) 13:2653–61. doi: 10.1158/1535-7163.MCT-14-0132
127. Bauss F, Lechmann M, Krippendorff B, Staack R, Herting F, Festag M, et al. Characterization of a Re-Engineered, Mesothelin-Targeted Pseudomonas Exotoxin Fusion Protein for Lung Cancer Therapy. Mol Oncol (2016) 1–13. doi: 10.1016/j.molonc.2016.07.003
128. Kollmorgen G, Palme K, Seidl A, Scheiblich S, Birzele F, Wilson S, et al. A Re-Engineered Immunotoxin Shows Promising Preclinical Activity in Ovarian Cancer. Sci Rep (2017) 7:1–13. doi: 10.1038/s41598-017-17329-7
129. Cerise A, Bera TK, Liu X, Wei J, Pastan I. Anti-Mesothelin Recombinant Immunotoxin Therapy for Colorectal Cancer. Clin Colorectal Cancer (2019) 18:1–9. doi: 10.1016/j.clcc.2019.06.006
130. Hollevoet K, Mason-Osann E, Liu XF, Imhof-Jung S, Niederfellner G, Pastan I. In Vitro and In Vivo Activity of the Low-Immunogenic Antimesothelin Immunotoxin RG7787 in Pancreatic Cancer. Mol Cancer Ther (2014) 13:2040–9. doi: 10.1158/1535-7163.MCT-14-0089-T
131. Hollevoet K, Antignani A, Fitzgerald DJ, Pastan I. Combining the Antimesothelin Immunotoxin SS1P With the BH3- Mimetic ABT-737 Induces Cell Death in SS1P-Resistant Pancreatic Cancer Cells. J Immunother (2014) 37:8–15. doi: 10.1097/CJI.0000000000000010
132. Hollevoet K, Mason-Osann E, Muller F, Pastan I. Methylation-Associated Partial Down-Regulation of Mesothelin Causes Resistance to Anti-Mesothelin Immunotoxins in a Pancreatic Cancer Cell Line. PloS One (2015) 10:e0122462. doi: 10.1371/journal.pone.0122462
133. Liu X, Zhou Q, Hassan R, Pastan I. Panbinostat Decreases cFLIP and Enhances Killing of Cancer Cells by Immunotoxin LMB-100 by Stimulating the Extrinsic Apoptotic Pathway. Oncotarget (2017) 8:87307–16. doi: 10.18632/oncotarget.20263
134. Kolyvas E, Rudloff M, Poruchynsky M, Landsman R, Hollevoet K, Venzon D, et al. Mesothelin-Targeted Immunotoxin RG7787 has Synergistic Anti-Tumor Activity When Combined With Taxanes. Oncotarget (2017) 8:9189–99. doi: 10.18632/oncotarget.13984
135. Liu XF, Xiang L, Zhou Q, Carralot J, Prunotto M, Niederfellner G. Actinomycin D Enhances Killing of Cancer Cells by Immunotoxin RG7787 Through Activation of the Extrinsic Pathway of Apoptosis. Proc Natl Acad Sci USA (2016) 113:10666–71. doi: 10.1073/pnas.1611481113
136. El-Behaedi S, Landsman R, Rudloff M, Kolyvas E, Albalawy R, Zhang X, et al. Protein Synthesis Inhibition Activity of Mesothelin Targeting Immunotoxin LMB-100 Decreases and Secreted Growth Factors. Toxin (2018) 10(447). doi: 10.3390/toxins10110447
137. Zheng AZ, Okada R, Kobayashi H, Nagaya T. Site-Specific PEGylation of Anti-Mesothelin Recombinant Immunotoxins Increases Half-Life and Anti-Tumor Activity. Mol Cancer Therap (2019) 19:812–21. doi: 10.1158/1535-7163.MCT-19-0890
138. Hassan R, Alewine C, Mian I, Spreafico A, Siu LL, Gomez-Roca C, et al. Phase 1 Study of the Immunotoxin LMB-100 in Patients With Mesothelioma and Other Solid Tumors Expressing Mesothelin. Cancer (2020) 126:1–12. doi: 10.1002/cncr.33145
139. Pegna GJ, Ahmad MI, Yu Y, Yuno A, Lee M-J, Peer CJ, et al. Phase I Study of Mesothelin-Targeted Immunotoxin LMB-100 Given as a Long Infusion With or Without Nab-Paclitaxel. J Clin Oncol (2020) 38:3553. doi: 10.1200/JCO.2020.38.15_suppl.3553
140. Onda M, Ghoreschi K, Steward-tharp S, Thomas C, Shea JJO, Pastan IH, et al. Tofacitinib Suppresses Antibody Responses to Protein Therapeutics in Murine Hosts. J Immunol (2014) 191:48–55. doi: 10.4049/jimmunol.1400063
141. Skorupan N, Ahmad MI, Pegna GJ, Peer CJ, Trepel JB, Lee M-J, et al. Phase I Study of Mesothelin-Targeted Immunotoxin LMB-100 in Combination With Tofacitinib in Patients With Advanced Pancreatobiliary Cancer. J Clin Oncol (2021) 39:3051. doi: 10.1200/JCO.2021.39.15_suppl.3051
142. Leshem Y, O’Brien J, Liu X, Bera TK, Terabe M, Berzofsky JA, et al. Combining Local Immunotoxins Targeting Mesothelin With CTLA-4 Blockade Synergistically Eradicates Murine Cancer by Promoting Anticancer Immunity. Cancer Immunol Res (2017) 5:685–94. doi: 10.1158/2326-6066.CIR-16-0330
143. Leshem Y, King EM, Mazor R, Reiter Y, Pastan I. SS1P Immunotoxin Induces Markers of Immunogenic Cell Death and Enhances the Effect of the CTLA-4. Toxins (2018) 4:1–11. doi: 10.3390/toxins10110470
144. Jiang Q, Ghafoor A, Mian I, Rathkey D, Thomas A, Alewine C, et al. Enhanced Efficacy of Mesothelin-Targeted Immunotoxin LMB-100 and Anti – PD-1 Antibody in Patients With Mesothelioma and Mouse Tumor Models. Science Transl Med (2020) 12:7252. doi: 10.1126/scitranslmed.aaz7252
145. Luwor RB, Zhu HJ, Walker F, Vitali AA, Perera RM, Burgess AW, et al. The Tumor-Specific De2-7 Epidermal Growth Factor Receptor (EGFR) Promotes Cells Survival and Heterodimerizes With the Wild-Type EGFR. Oncogene (2004) 23:6095–104. doi: 10.1038/sj.onc.1207870
146. Johns TG, McKay MJ, Cvrljevic AN, Gan HK, Taylor C, Xu H, et al. MAb 806 Enhances the Efficacy of Ionizing Radiation in Glioma Xenografts Expressing The De2-7 Epidermal Growth Factor Receptor. Int J Radiat Oncol Biol Phys (2010) 78:572–8. doi: 10.1016/j.ijrobp.2010.03.027
147. Gomez J, Areeb Z, Stuart SF, Nguyen HPT, Paradiso L, Zulkifli A, et al. Egfrviii Promotes Cell Survival During Endoplasmic Reticulum Stress Through a Reticulocalbin 1-Dependent Mechanism. Cancers (Basel) (2021) 13:1–21. doi: 10.3390/cancers13061198
148. Panousis C, Rayzman VM, Johns TG, Renner C, Liu Z, Cartwright G, et al. Engineering and Characterisation of Chimeric Monoclonal Antibody 806 (Ch806) for Targeted Immunotherapy of Tumours Expressing De2-7 EGFR or Amplified EGFR. Br J Cancer (2005) 92:1069–77. doi: 10.1038/sj.bjc.6602470
149. Zalutsky MR, Boskovitz A, Kuan C-T, Pegram CN, Ayriss J, Wikstrand CJ, et al. Radioimmunotargeting of Malignant Glioma by Monoclonal Antibody D2C7 Reactive Against Both Wild-Type and Variant III Mutant Epidermal Growth Factor Receptors. Nucl Med Biol (2012) 39:23–34. doi: 10.1016/j.nucmedbio.2011.06.005
150. Chandramohan V, Bigner DD. A Novel Recombinant Immunotoxin-Based Therapy Targeting Wild-Type and Mutant EGFR Improves Survival in Murine Models of Glioblastoma. Oncoimmunology (2013) 2:1–2. doi: 10.4161/onci.26852
151. Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, et al. Construction of an Immunotoxin, D2C7-(scdsFv)-PE38KDEL, Targeting EGFRwt and EGFRvIII for Brain Tumor Therapy. Clin Cancer Res (2013) 19:4717–27. doi: 10.1158/1078-0432.CCR-12-3891
152. Bao X, Chandramohan V, Reynolds RP, Norton JN, Wetsel WC, Rodriguiz RM, et al. Preclinical Toxicity Evaluation of a Novel Immunotoxin, D2C7-(scdsFv)-PE38KDEL, Administered via Intracerebral Convection-Enhanced Delivery in Rats. Invest New Drugs (2016) 34:149–58. doi: 10.1007/s10637-015-0318-3
153. Bao X, Pastan I, Bigner DD, Chandramohan V. EGFR/EGFRvIII-Targeted Immunotoxin Therapy for the Treatment of Glioblastomas via Convection-Enhanced Delivery. Recept Clin Investig (2016) 3:e1430. doi: 10.14800/rci.1430
154. Chandramohan V, Pegram CN, Piao H, Szafranski SE, Kuan CT, Pastan IH, et al. Production and Quality Control Assessment of a GLP-Grade Immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a Phase I/II Clinical Trial. Appl Microbiol Biotechnol (2017) 101:2747–66. doi: 10.1007/s00253-016-8063-x
155. Desjardins A, Randazzo D, Chandramohan V, Peters K, Johnson M, Landi D, et al. Ctim-23. A Phase 1 Trial of D2C7-It in Combination With Atezolizumab in Recurrent Who Grade Iv Malignant Glioma (Mg). Neuro Oncol (2020) 22:ii38–8. doi: 10.1093/neuonc/noaa215.157
156. Chandramohan V, Bao X, Yu X, Parker S, McDowall C, Yu YR, et al. Improved Efficacy Against Malignant Brain Tumors With EGFRwt/EGFRvIII Targeting Immunotoxin and Checkpoint Inhibitor Combinations. J Immunother Cancer (2019) 7:1–14. doi: 10.1186/s40425-019-0614-0
157. Baeuerle PA, Gires O. EpCAM (CD326) Finding Its Role in Cancer. Br J Cancer (2007) 96:417–23. doi: 10.1038/sj.bjc.6603494
158. Patriarca C, Colombo P, Pio Taronna A, Wesseling J, Franchi G, Guddo F, et al. Cell Discohesion and Multifocality of Carcinoma in Situ of the Bladder: New Insight From the Adhesion Molecule Profile (E-Cadherin, Ep-CAM, and MUC1). Int J Surg Pathol (2009) 17:99–106. doi: 10.1177/1066896908326918
159. Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, et al. Frequent High-Level Expression of the Immunotherapeutic Target Ep-CAM in Colon, Stomach, Prostate and Lung Cancers. Br J Cancer (2006) 94:128–35. doi: 10.1038/sj.bjc.6602924
160. Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, et al. Frequent EpCam Protein Expression in Human Carcinomas. Hum Pathol (2004) 35:122–8. doi: 10.1016/j.humpath.2003.08.026
161. Provencher DM, Finstad CL, Saigo PE, Rubin SC, Hoskins WJ, Federici MG, et al. Comparison of Antigen Expression on Fresh and Cultured Ascites Cells and on Solid Tumors of Patients With Epithelial Ovarian Cancer. Gynecol Oncol (1993) 50:78–83. doi: 10.1006/gyno.1993.1167
162. Winter MJ, Nagtegaal ID, van Krieken JHJM, Litvinov SV. The Epithelial Cell Adhesion Molecule (Ep-CAM) as a Morphoregulatory Molecule Is a Tool in Surgical Pathology. Am J Pathol (2003) 163:2139–48. doi: 10.1016/S0002-9440(10)63570-5
163. Bellone S, Siegel ER, Cocco E, Cargnelutti M, Silasi D-A, Azodi M, et al. Overexpression of Epithelial Cell Adhesion Molecule in Primary, Metastatic, and Recurrent/Chemotherapy-Resistant Epithelial Ovarian Cancer: Implications for Epithelial Cell Adhesion Molecule-Specific Immunotherapy. Int J Gynecol Cancer (2009) 19:860–6. doi: 10.1111/IGC.0b013e3181a8331f
164. Osta WA, Chen Y, Mikhitarian K, Mitas M, Salem M, Hannun YA, et al. EpCAM Is Overexpressed in Breast Cancer and Is a Potential Target for Breast Cancer Gene Therapy. Cancer Res (2004) 64:5818–24. doi: 10.1158/0008-5472.CAN-04-0754
165. Trzpis M, McLaughlin PMJ, de Leij LMFH, Harmsen MC. Epithelial Cell Adhesion Molecule: More Than a Carcinoma Marker and Adhesion Molecule. Am J Pathol (2007) 171:386–95. doi: 10.2353/ajpath.2007.070152
166. Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, et al. Nuclear Signalling by Tumour-Associated Antigen EpCAM. Nat Cell Biol (2009) 11:162–71. doi: 10.1038/ncb1824
167. Myklebust AT, Godal A, Pharo A, Juell S, Fodstad O. Eradication of Small Cell Lung Cancer Cells From Human Bone Marrow With Immunotoxins. Cancer Res (1993) 53:3784–8.
168. Myklebust AT, Godal A, Fodstad O. Targeted Therapy With Immunotoxins in a Nude Rat Model for Leptomeningeal Growth of Human Small Cell Lung Cancer. Cancer Res (1994) 1:2146–50.
169. Zimmermann S, Wels W, Froesch BA, Gerstmayer B, Stahel RA, Zangemeister-Wittke U. A Novel Immunotoxin Recognising the Epithelial Glycoprotein-2 has Potent Antitumoural Activity on Chemotherapy-Resistant Lung Cancer. Cancer Immunol Immunother (1997) 44:1–9. doi: 10.1007/s002620050348
170. Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, et al. High Thermal Stability Is Essential for Tumor Targeting of Antibody Fragments: Engineering of a Humanized Anti-Epithelial Glycoprotein-2 (Epithelial Cell Adhesion Molecule) Single-Chain Fv Fragment. Cancer Res (1999) 59:5758–67.
171. Di Paolo C, Willuda J, Kubetzko S, Lauffer I, Tschudi D, Waibel R, et al. A Recombinant Immunotoxin Derived From a Humanized Epithelial Cell Adhesion Molecule-Specific Single-Chain Antibody Fragment has Potent and Selective Antitumor Activity. Clin Cancer Res (2003) 9:2837–48.
172. Macdonald GC, Rasamoelisolo M, Entwistle J, Cizeau J, Bosc D, Cuthbert W, et al. A Phase I Clinical Study of VB4-845: Weekly Intratumoral Administration of an Anti-EpCAM Recombinant Fusion Protein in Patients With Squamous Cell Carcinoma of the Head and Neck. Drug Des Dev Ther (2008) 2:105–14. doi: 10.2147/dddt.s3442
173. Kowalski M, Entwistle J, Cizeau J, Niforos D, Loewen S, Chapman W, et al. A Phase I Study of an Intravesically Administered Immunotoxin Targeting EpCAM for the Treatment of Nonmuscle-Invasive Bladder Cancer in BCGrefractory and BCG-Intolerant Patients. Drug Des Devel Ther (2010) 4:313–20. doi: 10.2147/DDDT.S14071
174. El Gharib K, Lilly E, Chebel R. Checkpoint Inhibitors in BCG-Unresponsive Nonmuscle Invasive Bladder Cancer: Can They Help Spare the Bladder? Immunotherapy (2021) 13:1105–11. doi: 10.2217/imt-2021-0030
175. Mazor R, King E, Pastan I. Anti-Drug Antibodies to LMB-100 Are Enhanced by mAbs Targeting OX40 and CTLA4 but Not by mAbs Targeting PD1 or PDL-1. Cell Immunol (2018) 334:38–41. doi: 10.1016/j.cellimm.2018.08.016
176. Dieffenbach M, Pastan I. Mechanisms of Resistance to Immunotoxins Containing Pseudomonas Exotoxin a in Cancer Therapy. Biomolecules (2020) 10:1–13. doi: 10.3390/biom10070979
177. Mazor R, Pastan I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front Immunol (2020) 11:1261. doi: 10.3389/fimmu.2020.01261
178. Leshem Y, Pastan I. Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models. Toxins (Basel) (2019) 11(20). doi: 10.3390/toxins11010020
Keywords: immunotoxin, Pseudomonas exotoxin A, cancer, targeted therapy, bacterial toxin
Citation: Havaei SM, Aucoin MG and Jahanian-Najafabadi A (2021) Pseudomonas Exotoxin-Based Immunotoxins: Over Three Decades of Efforts on Targeting Cancer Cells With the Toxin. Front. Oncol. 11:781800. doi: 10.3389/fonc.2021.781800
Received: 24 September 2021; Accepted: 16 November 2021;
Published: 16 December 2021.
Edited by:
Fabrizio Martelli, National Center for Drug Research and Evaluation, National Institute of Health (ISS), ItalyReviewed by:
Abid R. Mattoo, Ultragenyx Pharmaceutical, United StatesJianwei Zhu, Shanghai Jiao Tong University, China
Daniel A. Vallera, University of Minnesota Twin Cities, United States
Copyright © 2021 Havaei, Aucoin and Jahanian-Najafabadi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc G. Aucoin, marc.aucoin@uwaterloo.ca; Ali Jahanian-Najafabadi, jahanian@pharm.mui.ac.ir