 Zeyaul Islam
Zeyaul Islam Ameena Mohamed Ali
Ameena Mohamed Ali Adviti Naik
Adviti Naik Mohamed Eldaw
Mohamed Eldaw Julie Decock
Julie Decock Prasanna R. Kolatkar
Prasanna R. Kolatkar- 1Diabetes Center, Qatar Biomedical Research Institute (QBRI), Hamad Bin Khalifa University (HBKU), Qatar Foundation, Doha, Qatar
- 2Translational Cancer and Immunity Center, Qatar Biomedical Research Institute (QBRI), Hamad Bin Khalifa University (HBKU), Qatar Foundation, Doha, Qatar
Higher eukaryotic development is a complex and tightly regulated process, whereby transcription factors (TFs) play a key role in controlling the gene regulatory networks. Dysregulation of these regulatory networks has also been associated with carcinogenesis. Transcription factors are key enablers of cancer stemness, which support the maintenance and function of cancer stem cells that are believed to act as seeds for cancer initiation, progression and metastasis, and treatment resistance. One key area of research is to understand how these factors interact and collaborate to define cellular fate during embryogenesis as well as during tumor development. This review focuses on understanding the role of TFs in cell development and cancer. The molecular mechanisms of cell fate decision are of key importance in efforts towards developing better protocols for directed differentiation of cells in research and medicine. We also discuss the dysregulation of TFs and their role in cancer progression and metastasis, exploring TF networks as direct or indirect targets for therapeutic intervention, as well as specific TFs’ potential as biomarkers for predicting and monitoring treatment responses.
Introduction
To establish and maintain specific cell lineage during development, a complex and tightly regulated gene expression network is active under the control of both intrinsic and extrinsic signaling pathways that culminate in the activation of transcription factors (1). Transcription factors (TFs) play a major role in regulating gene expression by recognizing and directly binding to specific DNA sequences. This binding then results in direct and/or indirect transcription activation of downstream genes, bringing RNA polymerase and other transcriptional machinery to the promoter sequence (2–4). TFs can also regulate expression through the recruitment of corepressors or by interfering with the binding of other TFs (5, 6).
During the early stages of embryo development, asymmetrical cell divisions along a basolateral cleavage plane create inner cell mass (ICM) and outer cell mass of trophectoderm (TE) lineage (7, 8). This hallmark event in early mammalian development is mainly dictated by two transcription factors; the POU-family transcription factor octamer-binding transcription factor 3/4 (Oct3/4) and the caudal-type homeobox protein 2 (Cdx2). Oct3/4 and Cdx2 establish a mutually exclusive expression pattern and form a complex for reciprocal repression of their target genes, suggesting reciprocal inhibition between lineage specific TFs during early stages of mammalian differentiation and development where Cdx2 drives lineage towards trophectoderm (9, 10). In addition, the Tea-domain family member 4 (Tead4) transcription factor regulates TE lineage specification (11, 12) through differential subcellular regulation (11). Furthermore, the early ICM contains a cell subpopulation that leads to the formation of two lineages, the epiblast (EPI) and primitive endoderm (PE), which are differentially regulated by Nanog (a homeobox TF in EPI) or Gata6 (Gata binding factor 6 in PE). Nanog and Gata6 are expressed in the ICM in an apparently random and mutually exclusive manner (13, 14). Nanog, together with Sall4, is vital to maintain the self-renewal capability of the ICM and of embryonic stem (ES) cells, derived from the inner cell mass (ICM) (15–18). Sall4 regulates Oct4 by binding to its conserved region, thus availing it for maintenance of ES cell pluripotency (18, 19) while Sall1 is implicated in the development of kidney, heart, and other systems (20). ES cells can maintain pluripotency and generate somatic and germ cell lineages of the developing embryo (17), and ESC pluripotency is governed by the core transcription factors Nanog, Oct4, Klf4, and Sox2 among others (17). Other key developmental transcription factors include the FoxA, Pax, and Pparγ families. FoxA family proteins are well known early developmental transcription factors (21) and are hence also known as “pioneer factors”. The Pax family TFs are involved in maintenance of progenitor cells across a wide variety of tissues including the thymus, pancreas and eye (20). Pparγ proteins are involved in many functions including cell proliferation and development of several tissues, and gliomas (20).
Given the vital role of TFs in determining cell fate, the extrapolation to tumor development and progression is easily made. Epithelial to mesenchymal transition (EMT) is not only an essential embryonic process during which apical-basal polarized epithelial cells convert into motile front-back polarized mesenchymal cells, but it is also crucial for ‘tissue invasion and metastasis’, a hallmark of cancer. The plasticity of cancer cells to switch between an epithelial and mesenchymal phenotype bestows them with the ability to survive the hostile tumor microenvironment and to colonize distant organs. Multiple theories exist on the identity of cancer cells with such abilities, supporting either the presence of a specific subpopulation of cancer stem cells (CSCs) within the bulk tumor or a subset of cancer cells with high plasticity, or a combination of both theories. Irrespective of the theories on CSC origin, cancer cells with stemness features are associated with the ability to self-renew and propagate unlimited. Less-differentiated tumors contain higher amount of CSCs as compared to well-differentiated tumors (22). Moreover, CSCs have been involved in tumor initiation, metastasis, and resistance to chemotherapy and radiotherapy (22, 23). The expression profiles of TFs involved in CSCs maintenance are similar to what is found in ESCs as compared to what is observed in adult stem cells (24, 25). The aim of this review is to summarize the current knowledge and highlight differences in the role of transcription factors that are involved in cell fate control during normal tissue as well as tumor development. Transcription factors involved in early as well as key developmental stages and those with strong cancer links were specifically chosen for this review. This review also tries to give a wider breadth of different types of TFs to better capture the diversity of involved TFs rather than focus on any single family of TFs or type of cancer.
Forkhead Box A
The Fox family encompasses more than 170 transcription factors with a conserved winged-helix DNA-binding domain (DBD) (26–28). These proteins participate in cellular processes ranging from development to immunity and metabolism (26, 27, 29–31). The Fox family can be stratified into 19 subfamilies, FoxA to FoxS, based on protein sequence homology (32). Fox proteins share a signature 80–100 amino acid DNA-binding domain known as forkhead box but significantly differ in other regions, allowing for differential expression, regulation, and functional diversification (29, 33).
The FoxA subfamily, known as hepatocyte nuclear factor 3 (Hnf3), comprises three members, FoxA1, FoxA2, and FoxA3, that can remodel nucleosomes and facilitate DNA binding of other TFs (21, 34). FOXA members have been depicted as pioneer factors because of their ability to bind transcription factor binding sites (Tfbs) located in condensed, inactive chromatin in order to initiate chromatin remodeling and support other TFs in accessing chromatin to prompt their tissue-specific functions such as estrogen and androgen modulation (21, 34–36).
Increased expression of Fox proteins has been observed in a wide range of cancers and is commonly associated with advanced cancer stages and poor survival via increased proliferation (37). Several studies have demonstrated a role for FoxA1 and FoxA2 in the regulation of cell cycle progression, proliferation and differentiation, genomic instability and DNA damage repair, metabolism, angiogenesis, invasion, and senescence (Figure 1). In comparison, sparse data is available on the role of FoxA3 in cancer. One recent study demonstrated an increase in FoxA3 expression in esophageal cancer, which was associated with increased invasion, distant metastasis, disease stage, and a shorter overall survival (38). However, these TFs are correlated with oncogenic but also conversely tumor-suppressive functions (inhibiting metastasis) depending on how they interact with the transcriptional networks of tissue-specific cancers (29, 39, 40). Table 1 summarizes the expression patterns of FoxA protein in various cancers.
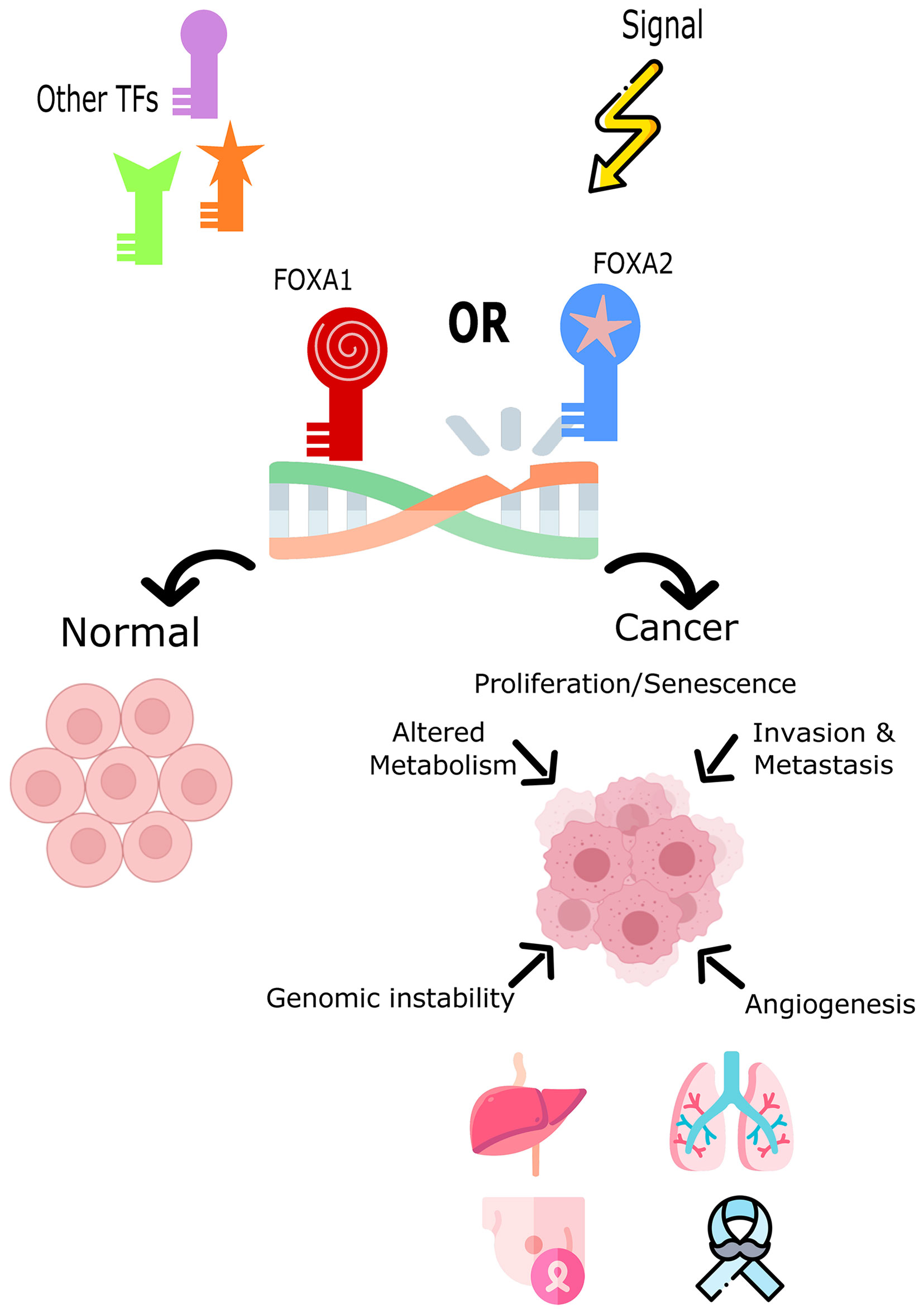
Figure 1 The role of FoxA1 and FoxA2 in cell fate decision or tumor induction. Both TFs impact the cell’s development toward normal cycle and differentiation or toward cancer and tumorigenesis. FOXA1 and FOXA2 overexpression, mutation, or down-regulation is associated with different cancers such as, lung, liver, breast, and prostate cancers.
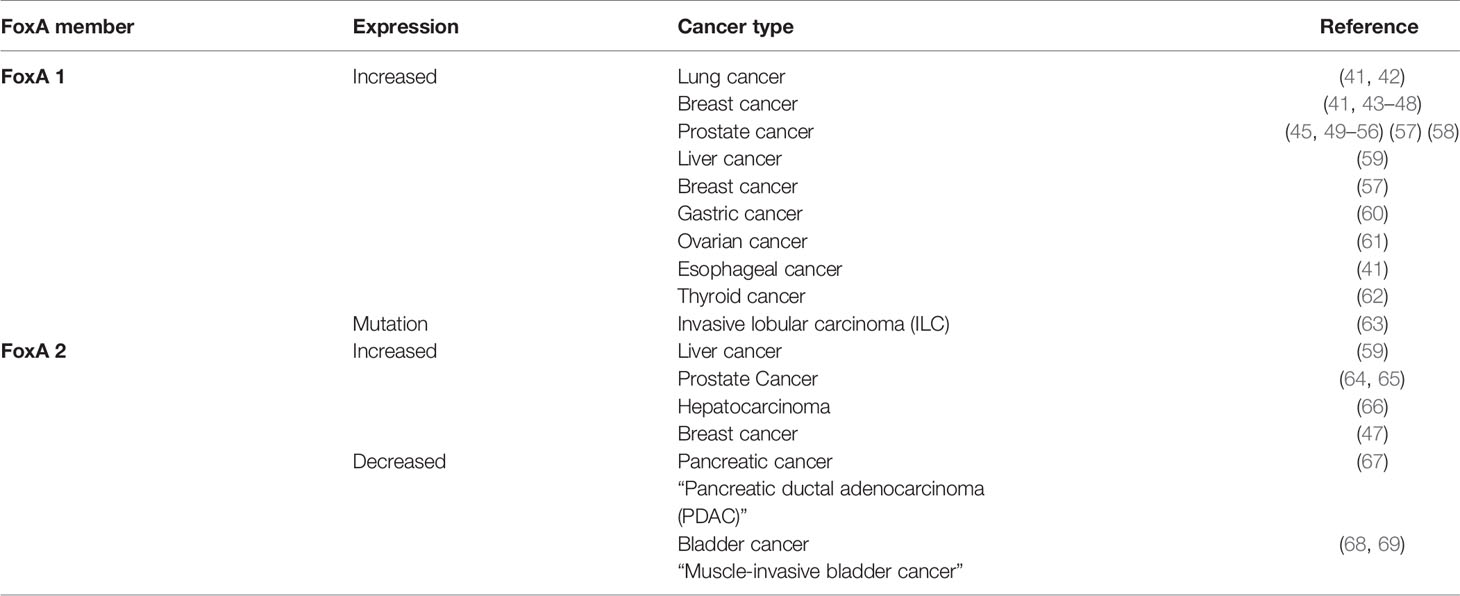
Table 1 The association between each member of FoxA family and different cancers.
OCT4
Oct4, also known as Pou5f1, is one of the core transcription factors that regulates ESC pluripotency (80). It contains three domains; the DNA binding POU domain, C-terminal transactivation domain, and variable N-terminal domain and binds an octamer sequence motif (ATGCAAAT) to regulate the expression of its target genes (Figure 2A) (81, 82). The expression level of Oct4 is critical for ESCs’ fate determination whereby an intermediary level maintains the self-renewal capacity of ESCs, a decrease in the expression level supports differentiation into trophectoderm lineage, and a subsequent increase induces differentiation into primitive endoderm or mesoderm (83).
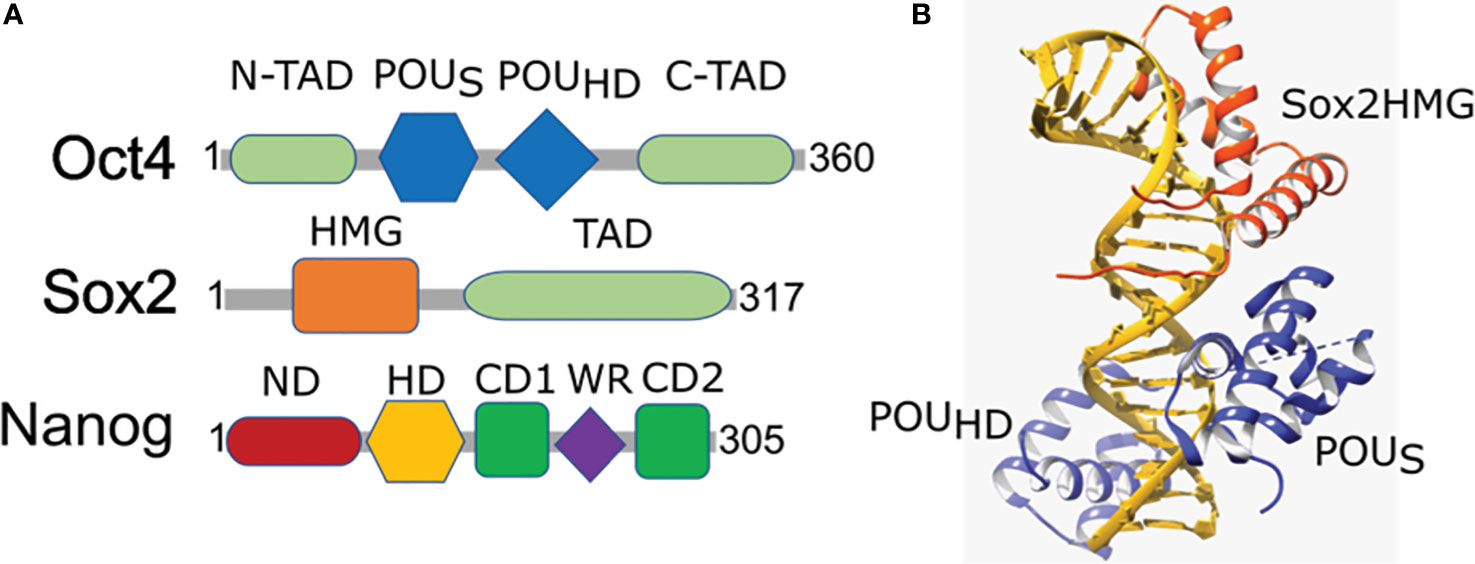
Figure 2 Domain organization and structural arrangement of Oct4, Sox2, and Nanog. (A) Oct4 has DNA-binding domains (a POU-specific DNA-binding domain (POUS) and a POU-homeodomain (POUHD)) interacting independently with DNA as well as transactivation domains located N-terminal (N-TAD) and C-terminal (C-TAD). Sox2 is a High mobility group (HMG) family member and has a single HMG DNA-binding domain and a transactivation domain (TAD). Nanog has N-terminal containing a DNA-binding homeodomain (HD) and an N-terminal domain (ND), C-terminal containing a dimerization domain (blue) referred to as the tryptophan repeat (WR), that separates C-terminal domain 1 (CD1) from C-terminal domain 2 (CD2). (B) Ternary structure of Oct–Sox–DNA (PDBID: 1O4X). Sox2 binding to DNA and Oct4 is enabled by the HMG domain (orange) that cooperates in binding of Oct4 POU domain (blue) onto the DNA (golden).
Oct4 expression has been observed in numerous cancers, with increased expression in more aggressive tumors and decreased expression being associated with regression of tumor potential (84). For instance, high expression of Oct4 in combination with other core pluripotency factors has been linked with pancreatic carcinogenesis, whereas silencing of Oct4 results in decreased proliferation, migration, invasion, and chemoresistance (85). Accordingly, multiple studies have demonstrated a correlation between Oct4 expression and treatment resistance and poor survival. For example, Oct4 expression is increased in docetaxel and mitoxantrone-resistant prostate cancer, cisplatin-resistant lung and ovarian cancer, radiation-resistant cervical cancer and chemo-resistant oral squamous carcinoma cancers (82, 86, 87). Conversely, Oct4-knockdown has been shown to increase the sensitivity to cisplatin treatment and radiotherapy in lung and ovarian cancers and to temozolomide in glioma-initiating cells (88–91). In contrast, testicular germ cell tumors display an inverse relation between Oct4 expression and resistance to cisplatin, although the exact mechanism is not yet clear (92, 93). Although increased Oct4 is generally associated with better outcome, in these tumors Oct4 expression was correlated with worse survival, similar to what has been observed in hypopharyngeal squamous cell carcinoma (94). This discrepancy in prognostic connotation underlines the biological complexity of this TF in cancer biology. In analogy with most stem cell factors, the level of Oct4 expression appears to be critical in cancer whereby either increased or decreased expression can perturb distinct cancer-related pathways.
SOX2
Sox2 is well established as a key transcription factor for self-renewal and pluripotency of neural stem cells and undifferentiated ESCs and is an integral part of embryogenesis, organogenesis, and overall animal development (95–98). It is a member of the Sox family of proteins that contain a DNA-binding high mobility group (HMG) domain that forms a concave surface enabling binding to DNA in a sequence-specific manner (99–101). The subsequent conformational change then unwinds the DNA, which helps to recruit other TFs, coactivators, or repressors (Figure 2B). The differential partnership of a functional Sox2 DNA binding site in combination with a second binding site for a partner protein determines the overall transcriptional activation or repression (102). Furthermore, Sox2 and Oct4 co-binding is required for gene activation at several loci providing more support that Sox2 and its partner factors are recruited to unique target sequences in specific conformations for transcriptional regulation (102). As such, partner switching plays an important role in differential gene expression. For example, during endodermal differentiation, the Sox2–Oct4 complex switches into a Sox17–Oct4 complex (103) as a result of Sox2 and Sox17 competing for binding to Oct4 and forming stable complexes on specific regions to determine the cell fate (104).
Of note, Sox2 is considered a proto-oncogene whereby Sox2 gene amplification, mutation, and overexpression can lead to multiple malignant conditions with metastasis (105–109). Sox2 amplification is positively correlated with increased proliferation, tumor burden metastasis, and poor prognosis (110–114). In tongue squamous cell carcinoma, Sox2 overexpression was associated with EMT progression, suggesting its involvement in regulation of cancer cell motility (115). Furthermore, Sox2 has been associated with tumorigenicity, illustrating its role in cancer stemness. For example, overexpression of Sox2 speeds up tumorigenicity in I-type neuroblastoma cells (116). Conversely, Sox2 silencing reduced tumorigenicity of glioblastoma and lung cancer tumor initiating cells (117, 118) and of epithelial ovarian cancer (EOC) cells (115). Downregulation of Sox2 reduces the tumorigenicity of cancer stem cells and regulates the expression of various cancer genes in lung cancer, including c-MYC, WNT1, WNT2, and NOTCH1 (119). In addition, Sox2 downregulation in breast cancer and glioma cells results in decreased proliferation by cell cycle arrest (120).
NANOG
Nanog is a homeodomain protein that is critical for mammalian development and specification of the ICM in the early embryo (16, 121). It forms dimers through its tryptophan-rich (WR) domain that is essential for ESCs’ self-renewal and pluripotency (122, 123). In addition, the dimer interacts with Kruppel-like zinc finger transcription factor Zfp281 (122) that functions as a transcriptional repressor for Nanog (124) while Patz1 (also a Kruppel-like zinc finger transcription factor) has the opposite effect as a transcriptional activator of Nanog (125). Nanog contains several phosphorylation sites at Ser/Thr-Pro motifs, which enable Nanog to be recognized and bound by the prolyl isomerase Pin1, leading to Nanog protein stabilization by preventing proteasome-mediated degradation (126). Phosphorylation and stabilization of Nanog by focal adhesion kinase (Fak) and protein kinase Cϵ (PKCϵ) has also been associated with tumor development (127). More specifically, PKCϵ-mediated phosphorylation translocates Nanog to the nucleus and activates miR-21 to promote breast tumor development and progression (128). Nanog is specifically expressed in ESCs, germ fibroblasts, and several tumor cell lines (129, 130). Knockdown of Nanog in gastric cancer cells reduced their proliferative and metastatic capacity, possibly as a result of increased apoptosis and cell cycle arrest (131). Similarly, Nanog was shown to exhibit anti-tumorigenic effects in glioblastoma (132), breast (133), and prostate (134) carcinoma cells. Furthermore, Nanog was found to promote chemoresistance and to increase cell migration and EMT (135, 136).
Krüppel-Like Factor 4
Klf4 is a three-zinc finger TF with two nuclear localization signals (NLSs) discovered in 1996 and also known as gut-enriched krüppel-like factor (GKLF). KLF4 is highly expressed in skin and intestinal epithelial cells and is involved in the regulation of cellular proliferation and terminal differentiation of several different tissues such as intestinal, eye, and skin tissues. Moreover, Klf4 is a well-known key factor required to produce induced pluripotent stem cells (iPSCs) (137, 138), first discovered by Takahashi and Yamanaka (139). Dhaliwal et al. highlighted Klf4’s role to maintain pluripotency and prevent embryonic stem cell differentiation. It is maintained post-transcriptionally by Nanog and Sox2 where Sox2 co-expression enables KLF4 stability.
Klf4 is an important regulator of adipogenesis and together with Krox20 (early growth response protein 2) induces expression of C/EBPβ through binding to C/EBPβ promoter regions in conjunction with histone acetyltransferase p300. Prior induction of Klf4 via cAMP regulates C/EBPβ expression, indicating a synergistic interaction. Conversely, knockdown of C/EBPβ results in overexpression of Klf4 and Krox20 identifying C/EBPβ as a downstream target (140). Klf4 knockdown is directly correlated with dysregulation of adipogenesis characterized by differentiation fat markers including peroxisome proliferator-activated receptor (PPARγ) as it is mediated through C/EBPβ (140). Pparγ, in turn, regulates Klf4 expression via binding of the PPAR response element (PPRE) in its promoter making it a key transcription regulator of lipid metabolism (70). Pparγ binding to Klf4 promoter induces the tumor suppression activity by affecting the complex pathways involving Klf4 in tumorigenesis as well as adipogenesis.
In cancer, particularly non-small cell lung cancer (NSCLC), Klf4 expression is downregulated in comparison to the surrounding normal tissues, indicative of a tumor suppressive function. Likewise, Klf4 has been found to act as a tumor suppressor in gastrointestinal cancer where it is associated with growth arrest through inhibition of G1/S cell cycle progression (71, 141). Klf4 has been reported to be a downstream target of methyltransferase like 3 (METTL3) using METTL3-depleted T24 bladder cancer cells. The cooperation of METTL3 with the reader protein YTH N6-Methyladenosine RNA Binding Protein 2 (YTHDF2) leads to the degradation of Klf4, which diminishes the tumor suppression activity of Klf4 and consequently induces cancer progression (72). Further, Klf4 negatively regulates serine/threonine kinase 33 (Stk33) by direct binding to its promoter, resulting in the inhibition of Stk33-induced EMT, a pivotal step in metastasis (73). In line with this, Klf4 expression has been correlated with inhibition of c-Jun N-terminal kinase (Jnk) which reportedly triggers EMT during cancer metastasis. In hepatocellular carcinoma (HCC), Klf4 was shown to regulate the expression of CD9/CD81, exosomal tetraspanin surface proteins that mediate cellular interaction and have been found to be involved in cancer (142). More specifically, CD9/CD81 were identified as transcriptional targets for Klf4 with a Klf4 binding site in their promoter regions. The expression of Klf4 was positively associated with the expression of CD9/CD81, and negatively affected downstream MAPK/JNK signaling, suggesting targeting Klf4-CD9/CD81-Jnk for future therapy. Table 2 shows the expression levels of KLF4 involved in cancer processes.
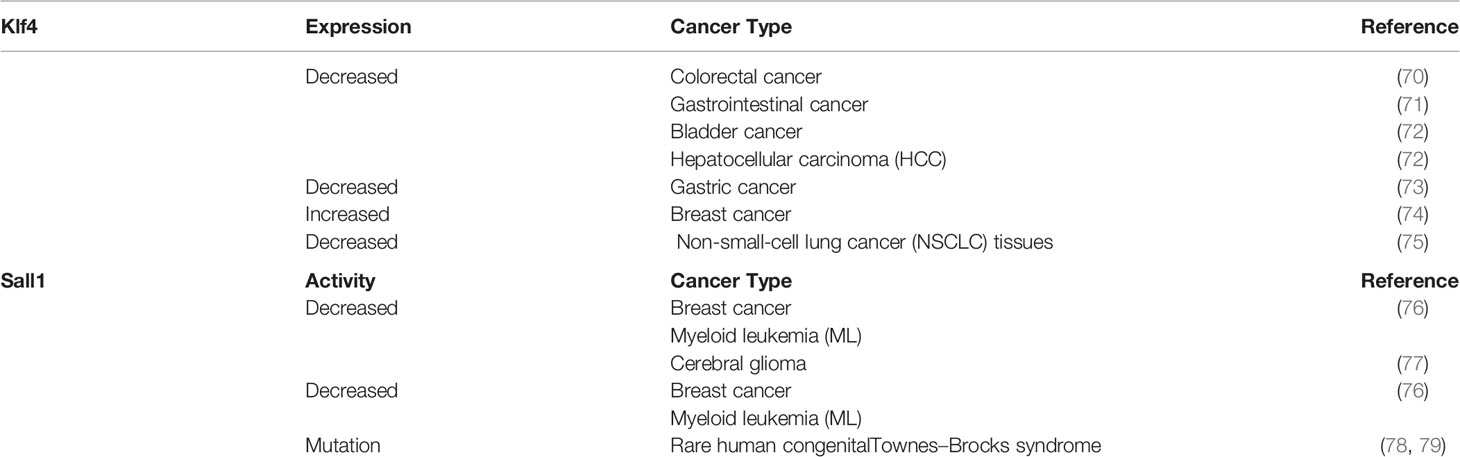
Table 2 Expression of KLF4 and SALL1 in cancer suppression and carcinogenesis.
It is important to note that few studies have reported Klf4 to be a tumor promoting factor. For instance, Klf4 has been shown to mediate estrogen-induced mitogenic effects as it accumulates upon estrogen-induced downregulation of the ubiquitin protein ligase Von Hippel-Lindau (VHL) (74). Overexpression of KLF4 was shown to promote osteosarcoma cancer stem cells (143) and act as a tumor promoting gene in nasopharyngeal carcinoma (144). Finally, Klf4 promotes breast tumor development and is upregulated in 70% of breast tumors (71).
Spalt-Like Transcription Factor 1
Sall1, together with Sall2, Sall3, and Sall4, forms the Sall family of zinc finger proteins containing cysteine–histidine residues (C2H2) (CX2–4CX12HX2–6H). Sall proteins are involved in organ development. Sall1 and Sall4 are specifically found to have an association with the rare human congenital Townes–Brocks syndrome that affects multiple organs (78, 79). Sall1 likely manifests this syndrome due to its role in kidney, heart, limbs, and central nervous system development (78). In this review, we focus on Sall1 as it is has been more frequently studied in the context of cancer as compared to the other Sall proteins.
The role of Sall1 in cell reprogramming was demonstrated through a Genome-Scale CRISPRa Screen (145), in which the expression of Sall1 was monitored individually and synergistically with Nanog. This study confirmed the capacity of Sall1 to reprogram primed epiblast stem cells (EpiSCs) and embryonic fibroblasts (MEFs) to iPSC, resulting in reprogramming the cell to ground state. In addition, Sall1 combined with Nanog maintained ESC state and regulated ESC reprogramming and differentiation. Concurrent overexpression of Nanog and Sall1 bestowed cells with the ability to form ESC colonies, whereas Sall1 alone was incapable of maintaining the ground state relative to Nanog’s ability. This work also showed that Sall1 and Nanog can delay differentiation of ESCs into EpiSCs via delayed upregulation of the differentiation markers Fgf5 (fibroblast growth factor 5) and Otx2 (orthodenticle homeobox 2).
In cancer, Sall1 has been found to be downregulated in breast cancer, glioblastoma (77), and myeloid leukemia, supporting its role as a tumor suppressor (76). In support of such a tumor suppressor role, Sall1 has been found to be a target of oncogenic miRNAs. For instance, Sall1 was found to be a potential target of the oncogenic miR-4286 in prostate cancer whereby miR-4286 knockdown abrogated Sall1’s ability to induce apoptosis and inhibit proliferation. Another study reported an inverse correlation, although not significant, between Sall1 and the oncogenic miR-181a-2 that is involved in microsatellite instability (146). Table 2 highlights SAL1 expression in cancer modulation.
In addition to the zinc finger domains which are important for DNA binding, Sall1 is characterized by a rich glutamine domain responsible for dimerization. This domain comprises an N-terminal region with tumor-suppression and transcription repression activity, enabled by interaction with nucleosome-remodeling deacetylase complex (NuRD) (78), resulting in decreased tumor growth and proliferation, cell cycle arrest, and metastasis regression. Furthermore, overexpression of Sall1 negatively impacts cell cycle progression and proliferation through the suppression of β-catenin, antagonizing the Wnt/β-catenin signaling pathway accordingly by targeting Wnt downstream targets Cyclin D1 (Ccnd1) and c-Myc oncogene. In addition, Sall1 is affecting the progression of cancer through the upregulation of the epithelial marker E-cadherin and downregulation of the mesenchymal markers vimentin and N-cadherin, driving mesenchymal-to-epithelial transition (77).
Gata TFs
The Gata family of TFs comprises zinc-finger DNA-binding proteins that control the development of diverse tissues, especially during hematopoiesis. They share conserved C2H2-type zinc-finger motifs (Cys-X2-C-X17-Cys-X2-Cys) that are involved in DNA-binding by recognizing the Gata element (A/TGATAA/G) (147). Based on expression pattern, they can be subdivided into two groups: Gata1, Gata2, and Gata3 forming the group of hematopoietic Gata factors, and Gata4, Gata5, and Gata6 grouped as endodermal Gata factors (148, 149). X-linked congenital anemia and thrombocytopenia have been linked to a point mutation within the N-terminal zinc finger of Gata1 that abolishes the interaction of Gata1 with the hematopoietic expressed transcription co-factor Fog1 (150). Gata3 plays an essential role in development and mammary gland function by maintaining the luminal cell lineage, and is expressed in differentiated luminal epithelial cells lining the breast ductal structures (151, 152). Gata3 gene deletion affects the mammary gland morphogenesis and in adults results in loss of luminal lineage (151, 153).
Since Gata proteins are heavily involved in regulating cell proliferation and survival of non-cancerous cells, it is evident how they can play a role in cancer. Altered expression or mutations of Gata factors are associated with a broad range of tumors including leukemia, colorectal, lung, breast, and brain tumors [Zhang et al., Rodriguez et al., Gao et al., Usary et al., Akiyama et al., Gong et al.]. Two mutations in the coding region (zinc finger domain) of Gata2 have been identified in a subset of human chronic myelogenous leukemia (CML). These mutations altered transactivation activity of Gata2 and its inhibitory effects on the activity of PU.1, a major regulator of myelopoiesis (154). In breast cancer, Gata3 expression is associated with invasive growth and poor prognosis (155). Its expression is maintained between primary and metastatic breast carcinoma and could potentially be used as a marker for metastatic breast carcinoma (156). Gata3 has also been suggested as a specific marker for urothelial carcinoma (157). Association of Gata3 with favorable clinicopathological parameters may indicate prognostic significance for Gata3 through its ability to promote luminal progenitor cells differentiation (158). Genomic analysis of breast cancer reveals high-frequency mutation in Gata3; however, most mutations were limited to a single allele, and expression of both mutated and wild-type alleles is approximately equivalent (159–161).
Pax TFs
Pax TFs are involved in multiple lineages to regulate cell fate during development and differentiation (162). They are sequence-specific DNA-binding proteins that are essential during early development and organogenesis (163). In general, Pax proteins are characterized by the presence of three conservative elements: two DNA-binding domains, the paired domain (PD) and homeodomain (HD), and the short octopeptide sequence (OP) located between PD and HD (Figure 3A). The paired domain, named after its first identification in the Drosophila gene paired (164), is the defining feature of this class of genes, while the OP and HD domains may be dispensable (Figure 3B).
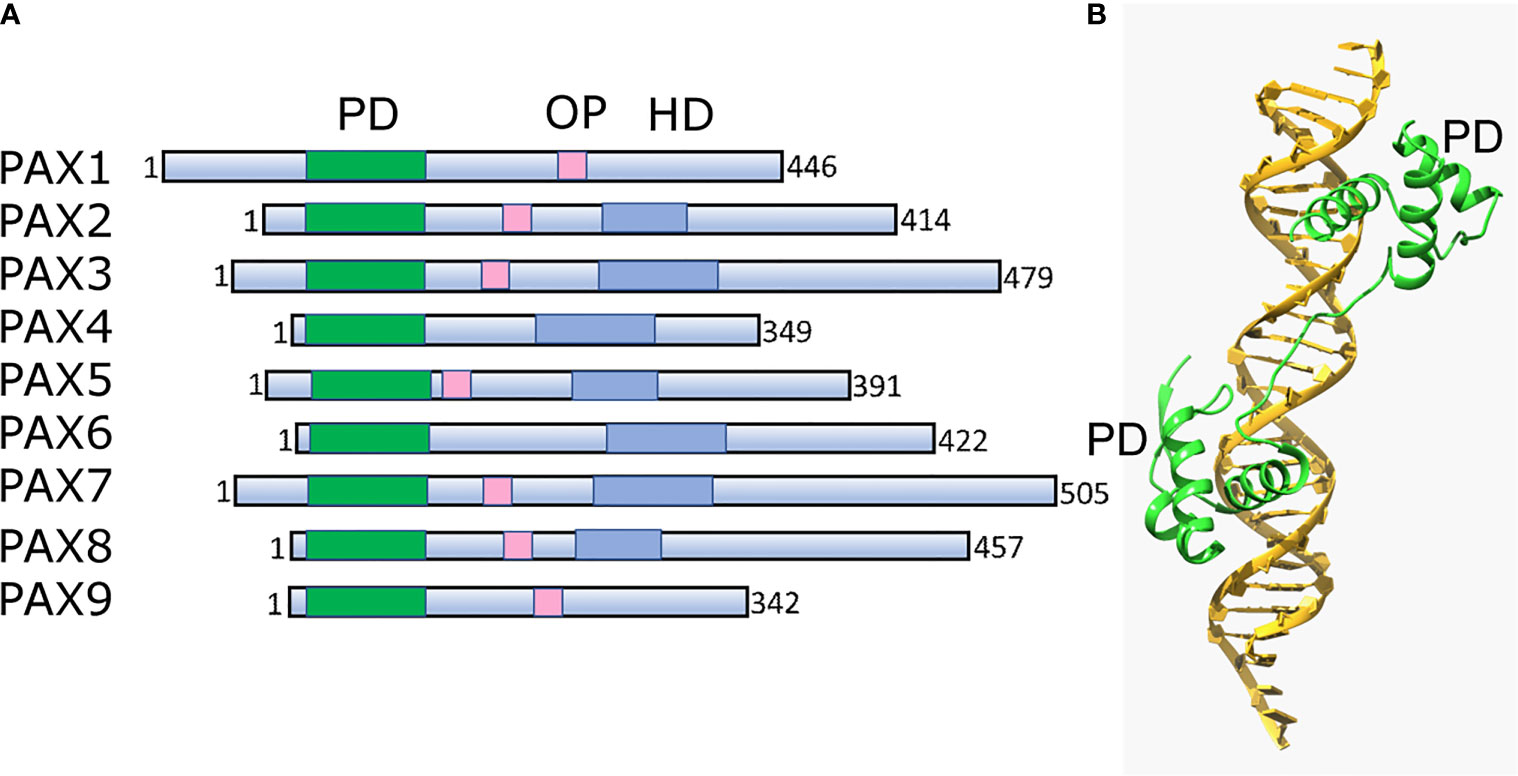
Figure 3 Structural architecture of Pax family and its binding to DNA. (A) Schematic drawing of conserved structural domains of Pax family members. The domains include the conserved paired domain (PD, green), the defining domain for this family. The Pax family members selectively contain other domains such as the homeodomain (HD, blue) and the octapeptide (OP, pink). (B) Ribbon diagram of Pax6 paired domain (PD)–DNA complex (PDBID: 6PAX). Crystal structure of the human Pax6 PD (primarily helix-turn-helix motif)-DNA complex reveals the region/subdomain involved in DNA binding.
The Pax family comprises nine members (Pax1–Pax9) in humans, subdivided into subgroups I–IV based on the presence, absence, or truncation of domains. Pax1 is expressed in cortical cells of the embryonic and adult thymus, where it participates in the maturation of thymocytes (165). It is often hypermethylated in cervical cancer and is a potential novel diagnostic biomarker (166). Pax2 binds to the promoter of a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), a metalloprotease that plays a crucial role in cancer progression and metastasis (167). It has been shown to regulate ADAM10 protein expression in renal cancer where it is expressed in 73% of cancer cells (168). Pax2 downregulation has been shown to lead to growth inhibition of cancer cells, and reactivation of Pax2 is also observed in clear cell renal cell carcinoma, a tumor type characterized by loss of VHL tumor suppressor function (169). Pax2 is also involved in cell proliferation and carcinogenesis in the endometrium, where it is activated by estrogen and tamoxifen, possibly due to cancer-linked hypomethylation of the Pax2 promoter (170). To date, very little information is available on Pax3 expression and function in cancer. In alveolar rhabdomyosarcoma, a pediatric soft tissue cancer related to the striated muscle lineage and characterized by the chromosomal translocations, chromosomal translocation events result in rearrangement of Pax3 and Pax7, juxtaposing these TFs with members of the fork head transcription factor family, and resulting in altered function from the chimeric gene product (171). Pax4 is involved in the differentiation and development of pancreatic islets. The high expression of Pax4 and the alternative splice variant Pax4v are critical in development of insulinoma through the upregulation of the anti-apoptotic gene bcl-xl (172, 173). Pax5 plays a vital role in all stages of B lymphocyte development (174). Reprogramming of mature B cells into pluripotent stem cells requires either ectopic expression of the myeloid transcription factor CCAAT/enhancer-binding-protein-alpha (C/EBPalpha) or Pax5 (175), in addition to core pluripotency TFs. Pax5 also mediates enhancer–promoter interactions and is able to alter genome topology, even in untranscribed regions (176). Pax6 regulates the neuroectoderm formation from ESCs, neural stem cell proliferation, neural stem cell self-renewal, neurogenesis and is critical for the development of the central nervous system (177, 178). Pax6 forms a complex with Sox2 on the lens-specific enhancer elements known as delta-crystallin minimal enhancer (DC5). Pax6 alone shows a poor binding on DC5; however, it cooperatively forms a stable ternary complex with Sox2 to the DC5 cis element, correlating with the enhancer activation required for eye development (95, 179). Pax6 is overexpressed in pancreatic carcinoma cell lines and promotes cancer progression by directly binding and activating the MET tyrosine kinase receptor (180). In contrast, Pax6 suppresses glioblastoma cell growth by downregulating the expression of the gene encoding vascular endothelial growth factor A (VEGFA) (181). Pax7 plays an important role in skeletal muscle formation (182). PAX8 is abundantly expressed in renal tissues and is a nephric-lineage TF required for the formation of the kidney (183). Pax8 expression is also frequently observed in renal, bladder, ovarian, and thyroid cancer cells. Silencing of Pax8 leads to a reduction in the expression of E2F1 and proteasome-dependent destabilization of the tumor suppressor retinoblastoma protein (RB) (184). Pax8 is also involved in telomerase regulation, telomerase reverse transcriptase and telomerase RNA component, in glioma (185). In thyroid carcinoma, Pax8 exists as a gene fusion with peroxisome proliferator activated receptor gamma (Pax8/PPARG gene fusion), resulting in an oncogenic Pax8–PPARγ fusion protein (186). Similar to Pax1, Pax9 is expressed in embryonic and adult thymus (165). In lung cancer, amplification of Pax9 promotes cell proliferation of lung cancer cells (187). Conversely, inhibition of Pax9 in oral squamous cell carcinoma triggers the induction of apoptosis corroborating its critical role in cell growth and survival, and thus disrupting the function could be a potential avenue for cancer treatment (188).
PPARγ TFs
Pparγ TFs, together with C/EBPs and the basic helix–loop–helix family (ADD1/SREBP1c), play a crucial role in adipogenesis, a process that involves cellular differentiation and morphological changes in cell size and lipid content (189–191). Pparγ is a member of the nuclear hormone receptor superfamily and requires heterodimerization with retinoid X receptor or Rxr to bind DNA and be transcriptionally active (192, 193). It can be present as two protein isoforms through alternate promoters and splicing whereby Pparγ 2 the dominant isoform is in fat cells with an extra 30 amino acids at the N-terminus compared to Pparγ1 (192).
Given its prominent role in adipogenesis, it is not surprising that increased expression of Pparγ/Rxr has been found in liposarcomas that were triggered to undergo terminal differentiation in vitro by thiazolidinediones or TZDs (class of antidiabetic drugs) and Rxr-specific retinoids (194). These results suggest that these compounds may be useful drugs to differentiate liposarcomas through maximal activation of the Pparγ pathway (194). Additionally, thiazolidinedione could be used as a non-toxic alternative to conventional chemotherapy for the treatment of locally advanced liposarcoma (194). Nevertheless, TZDs have shown only modest therapeutic benefit in clinical trials over the past 15 years (195). Factors affecting drug efficacy could include compound-specific effects, the necessity of Pparγ activation or other targets, the tumor stage at the time of drug exposure, the age of the patient, and finally the influence of TZDs on cancer cell paracrine activity (195–197). In addition, Pparγ can inhibit β-catenin that activates Pdk1 and Cyclin D1 (198) and upregulates Myc (199).
On the other hand, several studies revealed a pro-tumorigenic role for Pparγ in urinary bladder cancer, promoting tumorigenesis, metastasis, and angiogenesis (200–202) through several pathways including adipose differentiation and cell cycle arrest. The pro-tumorigenic function of Pparγ can be induced by increased inflammation through the upregulation of IL-6/STAT3 (203), Cox2, and PGE2 (204). Other pathways also give rise to cancers due to Pparγ mutations with partial loss of function or chimeric mutations such as in colon cancer (205), prostate cancer (206), and thyroid tumors (207, 208) where Pparγ levels were associated with tumor grade and invasive ability. Figure 4 illustrates the dual role of Pparγ within the cell.
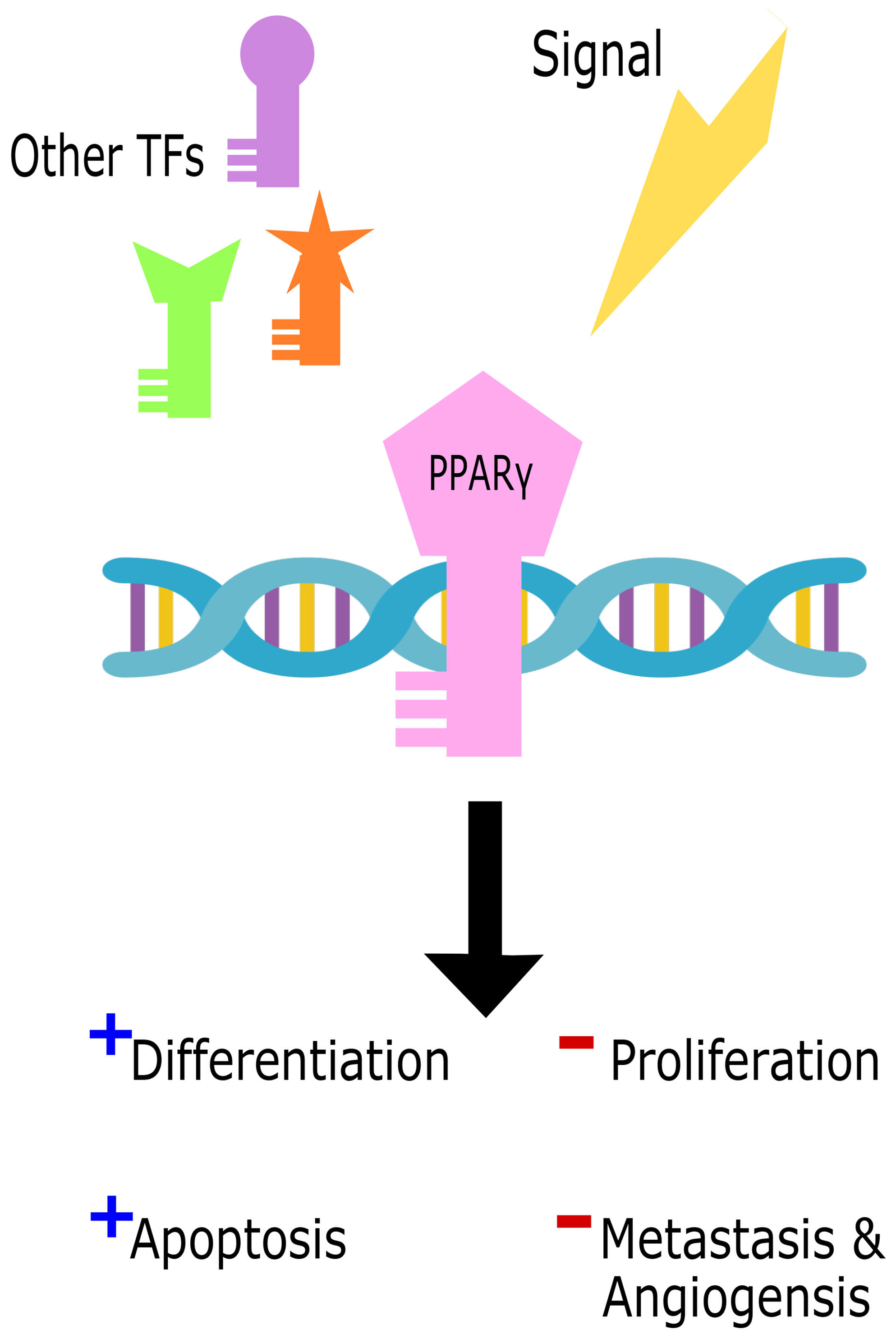
Figure 4 The dual role of Pparγ in the cell. Cell signals in parallel with other transcription factors (TFs) trigger Pparγ binding to DNA to initiate either tumor suppressive promoting functions.
Thus, Pparγ exhibits a context-dependent pro- or anti-tumorigenic behavior, which needs to be carefully considered prior to therapeutic intervention.
Pok Family
The POK transcription repressors (also named POZ-ZF transcription factors) are a major family of transcription factors which have a dual role in development and cancer. Apart from their involvement in several fundamental biological processes, they also participate in hematopoiesis, adipogenesis, chondrogenesis, DNA repair, development of oligodendrocytes, osteoclast, and unfolded protein response (209). The POK family (present in approximately 43 human genes) is composed of one or more C-terminal C2H2 Krüppel-type zinc finger domains, which are DNA binding domains, coupled with an N-terminal POZ/BTB (broad-complex, tramtrack, and Bric a brac) domain used for protein–protein interactions, allowing recruitment of corepressor complexes. The hinge region between the POZ/BTB and ZF domains and the C-terminal end of the ZF domain are often targeted for post-translational modification and regulation (210). The members of this family enable their regulation by binding of the zing finger domain in their target genes followed by recruitment of various cofactors (NCoR, SMRT, Sin3a) through the N-terminal domain for chromatin remodeling and transcriptional silencing or activation (211).
This family includes Bcl-6, PLZF, PATZ1 (also named MAZR), Kaiso, and many others (212, 213). Members include PLZF (promyelocytic leukemia zinc finger), which is involved in limb and skeleton development (214), regulates spermatogenesis (215) as well as natural killer T-cell (NKT) development (216). PLZF is linked to tumor suppression via its transcriptional repression of the c-myc oncogene (217). PATZ1/MAZR (AT-hook containing zinger finger protein 1) is similar and has been implicated in spermatogenesis (218), pluripotency maintenance (125), and in different developmental processes, including neural development (219) and T cell differentiation (220). However PATZ1/MAZR has also been described to act as an oncogene or tumor suppressor in experimental tumors and human cancer (221). Bcl-6 (B cell lymphoma 6) is critical in B cell development and is also dysregulated in B cell lymphoma (222, 223). Kaiso is involved in intestinal cell fate by regulating Notch signaling (224) and promotes EMT in prostate cancer by regulating miR-200c (225).
Thus this family of proteins carries out key steps in developmental pathways, and dysfunction can lead to carcinogenesis through several pathways involved in cell fate decisions, cell cycle control and apoptosis.
Targeting Transcription Factors in Cancer: Potential and Challenges
A plethora of evidence has identified CSC transcription factors that can drive tumorigenesis. CSCs additionally display resistance to chemotherapy (226) and radiotherapy (227, 228), thus rendering them capable of repopulating tumors in pre-treated relapsing patients. As such, strategies to target CSCs are lucrative to improve treatment response and disease-free survival. However, until recently, this class of proteins were considered “undruggable” (229). Firstly, transcription factors’ function broadly as master regulators in an immense repertoire of signaling pathways regulating normal tissue homeostasis, thus highlighting a need for targeted inhibition in cancer cells. Secondly, the lack of enzymatic activity and hence binding sites has rendered designing small molecule inhibitors challenging. In addition, the redundancy and functional compensation of transcription factors may limit the efficacy of single agent therapy. Consequently, the majority of currently available CSC TF modulators are non-selective or target upstream molecules (Table 3). For example, fursultiamine (thiamine tetrahydrofurfuryl disulfide, TTFD), a derivative of vitamin B, has been reported to suppress the expression of several CSC TFs including Oct4, Sox2, and Nanog resulting in reduced stem cell properties in esophageal carcinoma spheroids and mice xenografts (237). In addition, TTFD treatment also improved the response to concurrent chemoradiotherapy in the same mouse model. This combination modality has been investigated in a phase II clinical trial (NCT02423811) of esophageal squamous cell carcinoma, and the results are still pending. Similarly, a synthetic compound PT‐262 (7‐chloro‐6‐piperidin‐1‐yl‐quinoline‐5, 8‐dione) has been shown to inhibit the expression of Oct4 and Nanog, concurrent to suppressing the growth of lung tumor xenografts in mice (238). Furthermore, few drugs have been identified that affect upstream regulators of CSC TFs (Table 3). MLN4924, also known as pevonedistat, is a neddylation inhibitor that induces the accumulation of MSX2, a known transcription repressor of Sox2. MLN4924-mediated Sox2 downregulation has been shown to suppress stem cell properties and to exert broad anti-cancer effects both in in vitro and in vivo models (239, 240). Several phase I/II clinical trials are investigating single agent pevonedistat and its combination with standard chemotherapy in mesothelioma (NCT03319537), acute myeloid leukemia (AML) (NCT03009240, NCT00911066, NCT03330821, NCT03009240, NCT03459859, NCT03772925), acute lymphoblastic leukemia (NCT03349281), chronic lymphocytic leukemia (NCT03479268), relapsed or refractory lymphoma or multiple myeloma (NCT00722488, NCT03323034, NCT03770260), melanoma (NCT01011530) and non-hematologic malignancies (NCT00677170, NCT01862328) such as advanced non-small cell lung cancer (NSCLC, NCT03965689, NCT03228186) and intrahepatic cholangiocarcinoma (NCT04175912). In line with promising observations from pre-clinical studies (241), numerous phase I/II clinical trials are assessing the combination of pevonedistat with 5-azacytadine in newly diagnosed or relapsed/refractory AML or myelodysplastic syndrome (NCT03813147, NCT02782468, NCT04172844, NCT03238248, NCT02610777). This combination has now progressed into phase III trials in newly diagnosed AML not eligible for intensive chemotherapy (NCT04090736) and higher-risk myelodysplastic syndromes, chronic myelomonocytic leukemia, or low-blast AML (NCT03268954). Although single agent pevonedistat indicated modest clinical benefit (242–246), a combination of pevonedistat treatment with carboplatin and paclitaxel in advanced solid tumors (NCT01862328, 35% objective response rate) (247) or with 5-azacytadine in treatment-naïve AML patients (NCT01814826, >50% ORR) (248) showed promising anti-tumor activity. Both these studies did not indicate any additional toxicity to those elicited by chemotherapy or 5-azacytadine treatment alone. However, transient elevations in liver function tests were dose limiting for pevonedistat treatment. Likewise, modulators of calcium signaling such as thapsigargin, a Sarco/Endoplasmic Reticulum Ca(2+)-ATPases (SERCA) inhibitor induce a rise in cytosolic Ca2+ levels, which activates Akt-mediated phosphorylation and subsequently inhibits the oncogenic fusion transcription factor Pax3-FoxO1 (249). Accordingly, thapsigargin treatment suppresses the growth of Pax3-FoxO1 expressing alveolar rhabdomyosarcoma cell lines and xenografts.
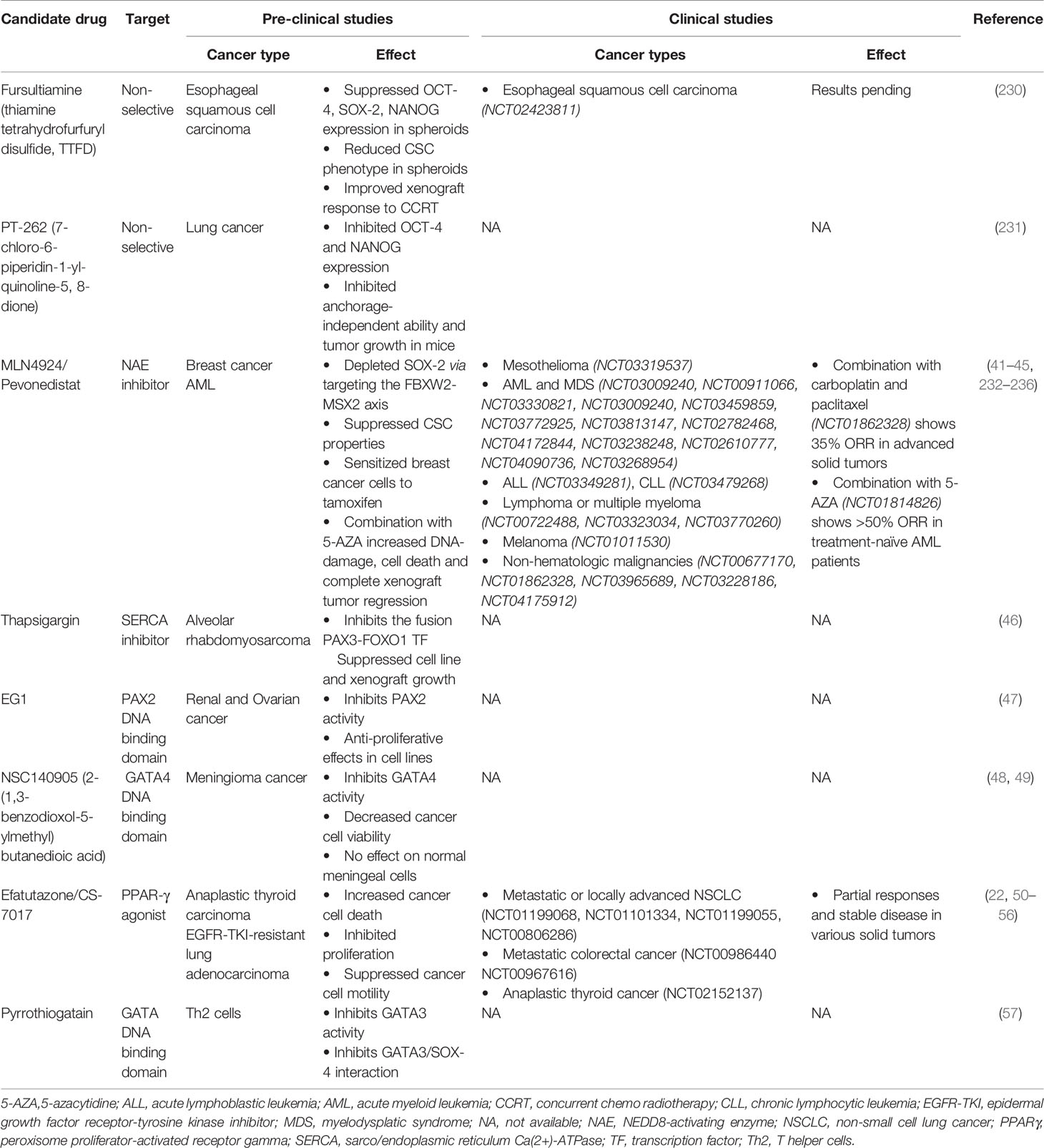
Table 3 Inhibitors of cancer stem cell transcription factors.
Due to challenges in designing selective inhibitors for transcription factors, potential strategies have focused on disrupting their binding to DNA (Table 3). In this regard, a compound termed EG1 was reported to target the DNA binding domain of Pax2, thereby blocking its transcriptional activity. EG1 treatment has demonstrated anti-proliferative effects in Pax2 expressing renal and ovarian cancer cell lines; however, its efficacy in vivo has not yet been reported (250). Similarly, pyrrothiogatain has been identified as a DNA-binding inhibitor of the Gata family, particularly of Gata2–Gata5, in addition to inhibiting the interaction of Sox4 and Gata3 (251). However, its effect on cancer cells remains to be investigated. Furthermore, the synthetic derivative of succinic acid NSC140905 [2-(1,3-benzodioxol-5-ylmethyl)butanedioic acid] was reported to bind to the DNA-binding domain of Gata4, thus blocking its transcriptional activity (252). Of note, treatment of meningioma cancer cells with NSC140905 decreased cancer cell viability but did not affect normal human meningeal cells in vitro (253). The potential of Sox decoy molecules, which target their DNA binding activity, has also been demonstrated to inhibit Sox18-induced genes in the COS-7 cell line (254). These decoys have been designed to resist nuclease digestion, degradation, and thermal denaturation in vitro but remain to be investigated in pre-clinical cancer models.
As transcriptions factors typically interact with numerous proteins downstream of signaling cascades, targeting such partner proteins may potentially affect their transcriptional activity. For instance, FoxA1 interacts with the cyclin-dependent kinase 1 (Cdk1) cell cycle regulator in certain types of breast cancer cells (255). Additionally, in silico analyses has indicated that Cdk-mediated phosphorylation of FoxA1 may potentially regulate FoxA1 binding to DNA. Consequently, treating these cell lines with Cdk inhibitors suppresses FoxA1 binding to DNA (255). Theoretically, this may also negatively affect cancer cell proliferation and hence, requires further investigation. In contrast, strategies targeting the CSC TF Pparγ focus on activating this tumor suppressor to mitigate oncogenesis (256). Pparγ agonists, particularly the thiazolidinedione class of ligands (troglitazone, rosiglitazone, and pioglitazone), have been commonly used as anti-diabetic drugs. Although these drugs have shown pre-clinical anti-proliferative effects in numerous cancer types (257–259), their administration in clinical trials has indicated limited efficacy (260, 261). A novel, third generation thiazolidinedione, efatutazone or CS-7017, is significantly more potent than its predecessors in inducing Ppar response element activation and anti-tumor activity, and thus might exhibit a higher efficacy in clinical setting (262, 263). Pre-clinical studies have shown that efatutazone in combination with chemotherapy can increase cancer cell death, inhibit proliferation, and suppress cancer cell motility of particularly epidermal growth factor receptor-tyrosine kinase inhibitor (Egfr-Tki)-resistant lung adenocarcinoma cells (230–232, 264). Clinically, single agent efatutazone therapy and efatutazone therapy in combination with chemotherapy have induced partial responses and stable disease in various solid tumors (20, 233–235). Although efatutazone treatment demonstrated acceptable tolerability, peripheral edema was commonly observed as an adverse effect, with patients often requiring diuretics. Furthermore, numerous ongoing phase I and II clinical trials are assessing the synergistic efficacy of efatutazone with the Egfr-Tki Erlotinib (NCT01199068, NCT01101334) or carboplatin/paclitaxel (NCT01199055, NCT00806286) in metastatic or locally advanced NSCLC, with irinotecan, leucovorin, and 5-fluorouracil chemotherapy in metastatic colorectal cancer (NCT00986440 NCT00967616) and with paclitaxel in anaplastic thyroid cancer (NCT02152137).
Emerging technologies in high-throughput screening are shifting the “undruggable” paradigm towards identifying selective modulators of cancer-associated transcription factor activity (236). Moreover, efforts towards designing targeted delivery of small molecules, including synthetic compounds, short-interfering RNA or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) genome editing tools, could transform cancer treatment to specifically target transcription factors and their mutant alleles in tumor cells with minimal off-target effects. Finally, the functional redundancy of CSC TFs could be overcome by combining TF modulators with or without standard cancer treatment, which has already been indicated by the improved efficacy of clinical trials combining CSC TF modulators with chemotherapy.
Conclusion
This review attempts to summarize the choices of regulated cell fate decisions versus dysfunction leading to cancer meted out by several transcription factors. Key TFs were chosen which are known to have important cell fate roles as well as dysfunction during carcinogenesis. This review covers early players in stem cell development such as Oct4 and Sox2 as well as other TFs in early differentiation events such as Gata, Pax, and Pparγ. Different criteria including expression levels and mutations in critical functional domains are described and how they exert their effects for several different cancers. Finally, this review describes the potential for drugging different cancers using various compounds which specifically could mitigate the “stemness” of cancers. Understanding how the TFs conspire for normal cellular development versus malignant outcomes will be critical in developing better selective ligands that can target cancer with fewer side effects in the future.
Author Contributions
PK had the principal idea to combine applications of transcription factors with stem cell development and tumorigenesis for the review. ZI, AA, and ME also added many specifics about stem cells and pluripotency details as well as tumorigenesis and metastasis due to the relevant transcription factors. AN and JD were the principal contributors for the clinical relevance to cancer as well as other cancer specifics. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by two intramural grants (IGP) from the Qatar Biomedical Research Institute, awarded to PK and JD, respectively. In addition work was also supported by a QNRF NPRP grant NPRP11S-0120-180346 to PK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arnold SJ, Robertson EJ. Making a Commitment: Cell Lineage Allocation and Axis Patterning in the Early Mouse Embryo. Nat Rev Mol Cell Biol (2009) 10:91–103. doi: 10.1038/nrm2618
2. Allen BL, Taatjes DJ. The Mediator Complex: A Central Integrator of Transcription. Nat Rev Mol Cell Biol (2015) 16:155–66. doi: 10.1038/nrm3951
3. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell (2018) 172:650–65. doi: 10.1016/j.cell.2018.01.029
4. Soutourina J. Transcription Regulation by the Mediator Complex. Nat Rev Mol Cell Biol (2018) 19:262–74. doi: 10.1038/nrm.2017.115
5. Moody SE, Perez D, Pan T, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The Transcriptional Repressor Snail Promotes Mammary Tumor Recurrence. Cancer Cell (2005) 8:197–209. doi: 10.1016/j.ccr.2005.07.009
6. Reinke LM, Xu Y, Cheng C. Snail Represses the Splicing Regulator Epithelial Splicing Regulatory Protein 1 to Promote Epithelial-Mesenchymal Transition. J Biol Chem (2012) 287:36435–42. doi: 10.1074/jbc.M112.397125
7. Rossant J, Chazaud C, Yamanaka Y, Jones M, Robertson EJ, Smith A, et al. Lineage Allocation and Asymmetries in the Early Mouse Embryo. Philos Trans R Soc B Biol Sci (2003) 358:1341–48. doi: 10.1098/rstb.2003.1329
8. Mihajlović AI, Bruce AW. The First Cell-Fate Decision of Mouse Preimplantation Embryo Development: Integrating Cell Position and Polarity. Open Biol (2017) 7:170210. doi: 10.1098/rsob.170210
9. Niwa H, Sekita Y, Tsend-Ayush E, Grützner F. Platypus Pou5f1 Reveals the First Steps in the Evolution of Trophectoderm Differentiation and Pluripotency in Mammals. Evol Dev (2008) 10:671–82. doi: 10.1111/j.1525-142X.2008.00280.x
10. Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, et al. Interaction Between Oct3/4 and Cdx2 Determines Trophectoderm Differentiation. Cell (2005) 123:917–29. doi: 10.1016/j.cell.2005.08.040
11. Home P, Saha B, Ray S, Dutta D, Gunewardena S, Yoo B, et al. Altered Subcellular Localization of Transcription Factor TEAD4 Regulates First Mammalian Cell Lineage Commitment. Proc Natl Acad Sci (2012) 109:7362–7. doi: 10.1073/pnas.1201595109
12. Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, et al. Transcription Factor TEAD4 Specifies the Trophectoderm Lineage at the Beginning of Mammalian Development. Development (2007) 134:3827–36. doi: 10.1242/dev.010223
13. Bessonnard S, Mot L, Gonze D, Barriol M, Dennis C, Goldbeter A, et al. Gata6, Nanog and Erk Signaling Control Cell Fate in the Inner Cell Mass Through a Tristable Regulatory Network. Dev (2014) 141:3637–48. doi: 10.1242/dev.109678
14. Chazaud C, Yamanaka Y, Pawson T, Rossant J. Early Lineage Segregation Between Epiblast and Primitive Endoderm in Mouse Blastocysts Through the Grb2-MAPK Pathway. Dev Cell (2006) 10:615–24. doi: 10.1016/j.devcel.2006.02.020
15. Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, et al. Functional Expression Cloning of Nanog, a Pluripotency Sustaining Factor in Embryonic Stem Cells. Cell (2003) 113:643–55. doi: 10.1016/S0092-8674(03)00392-1
16. Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The Homeoprotein Nanog Is Required for Maintenance of Pluripotency in Mouse Epiblast and ES Cells. Cell (2003) 113:631–42. doi: 10.1016/S0092-8674(03)00393-3
17. Saunders A, Faiola F, Wang J. Concise Review: Pursuing Self-Renewal and Pluripotency With the Stem Cell Factor Nanog. Stem Cells (2013) 31:1227–36. doi: 10.1002/stem.1384
18. Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, et al. Sall4 Modulates Embryonic Stem Cell Pluripotency and Early Embryonic Development by the Transcriptional Regulation of Pou5f1. Nat Cell Biol (2006) 8:1114–23. doi: 10.1038/ncb1481
19. Lim CY, Tam WL, Zhang J, Ang HS, Jia H, Lipovich L, et al. Sall4 Regulates Distinct Transcription Circuitries in Different Blastocyst-Derived Stem Cell Lineages. Cell Stem Cell (2008) 3:543–54. doi: 10.1016/j.stem.2008.08.004
20. Komatsu Y, Yoshino T, Yamazaki K, Yuki S, Machida N, Sasaki T, et al. Phase 1 Study of Efatutazone, a Novel Oral Peroxisome Proliferator-Activated Receptor Gamma Agonist, in Combination With FOLFIRI as Second-Line Therapy in Patients With Metastatic Colorectal Cancer. Invest New Drugs (2014) 32:473–80. doi: 10.1007/s10637-013-0056-3
21. Friedman JR, Kaestner KH. The Foxa Family of Transcription Factors in Development and Metabolism. Cell Mol Life Sci (2006) 63:2317–28. doi: 10.1007/s00018-006-6095-6
22. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and Molecular Heterogeneity of Breast Cancers Correlates With Their Cancer Stem Cell Content. Cell (2010) 140:62–73. doi: 10.1016/j.cell.2009.12.007
23. Nagata S, Hirano K, Kanemori M, Sun LT, Tada T. Self-Renewal and Pluripotency Acquired Through Somatic Reprogramming to Human Cancer Stem Cells. PloS One (2012) 7:e48699. doi: 10.1371/journal.pone.0048699
24. Zhao W, Li Y, Zhang X. Stemness-Related Markers in Cancer. Cancer Transl Med (2017) 3:87–95. doi: 10.4103/ctm.ctm_69_16
25. Pádua D, Figueira P, Ribeiro I, Almeida R, Mesquita P. The Relevance of Transcription Factors in Gastric and Colorectal Cancer Stem Cells Identification and Eradication. Front Cell Dev Biol (2020) 8:442. doi: 10.3389/fcell.2020.00442
26. Hannenhalli S, Kaestner KH. The Evolution of Fox Genes and Their Role in Development and Disease. Nat Rev Genet (2009) 10:233–40. doi: 10.1038/nrg2523
27. Lam EWF, Brosens JJ, Gomes AR, Koo C-Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat Rev Cancer (2013) 13:482–95. doi: 10.1038/nrc3539
28. Carlsson P, Mahlapuu M. Forkhead Transcription Factors: Key Players in Development and Metabolism. Dev Biol (2002) 250:1–23. doi: 10.1006/dbio.2002.0780
29. Zhang W, Duan N, Song T, Li Z, Zhang C, Chen X. The Emerging Roles of Forkhead Box (Fox) Proteins in Osteosarcoma. J Cancer (2017) 8:1619–28. doi: 10.7150/jca.18778
30. Zhu H, Xi Q, Liu L, Wang J, Gu M. Quantitative Assessment of Common Genetic Variants on FOXE1 and Differentiated Thyroid Cancer Risk. PloS One (2014) 9:e87332. doi: 10.1371/journal.pone.0087332
31. Tuteja G, Kaestner KH. Snapshot:Forkhead Transcription Factors I. Cell (2007) 130:1160. doi: 10.1016/j.cell.2007.09.005
32. Tuteja G, Kaestner KH. Snapshot: Forkhead Transcription Factors Ii. Cell (2007) 131:192–2.e1. doi: 10.1016/j.cell.2007.09.016
33. Benayoun BA, Caburet S, Veitia RA. Forkhead Transcription Factors: Key Players in Health and Disease. Trends Genet (2011) 27:224–32. doi: 10.1016/j.tig.2011.03.003
34. Jägle S, Busch H, Freihen V, Beyes S, Schrempp M, Boerries M, et al. SNAIL1-Mediated Downregulation of FOXA Proteins Facilitates the Inactivation of Transcriptional Enhancer Elements at Key Epithelial Genes in Colorectal Cancer Cells. PloS Genet (2017) 13:e1007109. doi: 10.1371/journal.pgen.1007109
35. Iwafuchi-Doi M, Zaret KS. Pioneer Transcription Factors in Cell Reprogramming. Genes Dev (2014) 28:2679–92. doi: 10.1101/gad.253443.114
36. Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of Compacted Chromatin by Early Developmental Transcription Factors HNF3 (FoxA) and GATA-4. Mol Cell (2002) 9:279–89. doi: 10.1016/S1097-2765(02)00459-8
37. Bach D-H, Long NP, Luu T-T-T, Anh NH, Kwon SW, Lee SK. The Dominant Role of Forkhead Box Proteins in Cancer. Int J Mol Sci (2018) 19:3279. doi: 10.3390/ijms19103279
38. Chen B, Yu J, Lu L, Dong F, Zhou F, Tao X, et al. Upregulated Forkhead-Box A3 Elevates the Expression of Forkhead-Box A1 and Forkhead-Box A2 to Promote Metastasis in Esophageal Cancer. Oncol Lett (2019) 17:4351–60. doi: 10.3892/ol.2019.10078
39. Choi EJ, Seo EJ, Kim DK, Lee SI, Kwon YW, Jang IH, et al. FOXP1 Functions as an Oncogene in Promoting Cancer Stem Cell-Like Characteristics in Ovarian Cancer Cells. Oncotarget (2016) 7:3506–19. doi: 10.18632/oncotarget.6510
40. Qiu M, Bao W, Wang J, Yang T, He X, Liao Y, et al. FOXA1 Promotes Tumor Cell Proliferation Through AR Involving the Notch Pathway in Endometrial Cancer. BMC Cancer (2014) 14:78. doi: 10.1186/1471-2407-14-78
41. Lin L, Miller CT, Contreras JI, Prescott MS, Dagenais SL, Wu R, et al. The Hepatocyte Nuclear Factor 3 Alpha Gene, HNF3alpha (FOXA1), on Chromosome Band 14q13 is Amplified and Overexpressed in Esophageal and Lung Adenocarcinomas. Cancer Res (2002) 62:5273–9.
42. Huang C, Liu J, Xiong B, Yonemura Y, Yang X. Expression and Prognosis Analyses of Forkhead Box A (FOXA) Family in Human Lung Cancer. Gene (2019) 685:202–10. doi: 10.1016/j.gene.2018.11.022
43. Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. Location Analysis of Estrogen Receptor Target Promoters Reveals That FOXA1 Defines a Domain of the Estrogen Response. Proc Natl Acad Sci (2005) 102:11651–6. doi: 10.1073/pnas.0505575102
44. Thorat MA, Marchio C, Morimiya A, Savage K, Nakshatri H, Reis-Filho JS, et al. Forkhead Box A1 Expression in Breast Cancer is Associated With Luminal Subtype and Good Prognosis. J Clin Pathol (2007) 61:327–32. doi: 10.1136/jcp.2007.052431
45. Augello MA, Hickey TE, Knudsen KE. FOXA1: Master of Steroid Receptor Function in Cancer. EMBO J (2011) 30:3885–94. doi: 10.1038/emboj.2011.340
46. Perez-Balaguer A, Ortiz-Martínez F, García-Martínez A, Pomares-Navarro C, Lerma E, Peiró G. Foxa2 mRNA Expression is Associated With Relapse in Patients With Triple-Negative/Basal-like Breast Carcinoma. Breast Cancer Res Treat (2015) 153:465–74. doi: 10.1007/s10549-015-3553-6
47. Slebe F, Rojo F, Vinaixa M, García-Rocha M, Testoni G, Guiu M, et al. Foxa and LIPG Endothelial Lipase Control the Uptake of Extracellular Lipids for Breast Cancer Growth. Nat Commun (2016) 7:11199. doi: 10.1038/ncomms11199
48. Schaner ME, Ross DT, Ciaravino G, Sørlie T, Troyanskaya O, Diehn M, et al. Gene Expression Patterns in Ovarian Carcinomas. Mol Biol Cell (2003) 14:4376–86. doi: 10.1091/mbc.e03-05-0279
49. Mirosevich J, Gao N, Matusik RJ. Expression of Foxa Transcription Factors in the Developing and Adult Murine Prostate. Prostate (2005) 62:339–52. doi: 10.1002/pros.20131
50. Jain RK, Mehta RJ, Nakshatri H, Idrees MT, Badve SS. High-Level Expression of Forkhead-Box Protein A1 in Metastatic Prostate Cancer. Histopathology (2011) 58:766–72. doi: 10.1111/j.1365-2559.2011.03796.x
51. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual Role of FoxA1 in Androgen Receptor Binding to Chromatin, Androgen Signalling and Prostate Cancer. EMBO J (2011) 30:3962–76. doi: 10.1038/emboj.2011.328
52. Jozwik KM, Carroll JS. Pioneer Factors in Hormone-Dependent Cancers. Nat Rev Cancer (2012) 12:381–5. doi: 10.1038/nrc3263
53. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J-P, et al. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat Genet (2012) 44:685–9. doi: 10.1038/ng.2279
54. Jin H-J, Zhao JC, Wu L, Kim J, Yu J. Cooperativity and Equilibrium With FOXA1 Define the Androgen Receptor Transcriptional Program. Nat Commun (2014) 5:3972. doi: 10.1038/ncomms4972
55. Liu S, Zhao Z, Wang D, Hu D, Li Y. Decreased FOXF1 Promotes Hepatocellular Carcinoma Tumorigenesis, Invasion, and Stemness and is Associated With Poor Clinical Outcome. Onco Targets Ther (2016) 9:1743–52. doi: 10.2147/OTT.S95002
56. Parolia A, Cieslik M, Chu S-C, Xiao L, Ouchi T, Zhang Y, et al. Distinct Structural Classes of Activating FOXA1 Alterations in Advanced Prostate Cancer. Nature (2019) 571:413–8. doi: 10.1038/s41586-019-1347-4
57. Katoh M, Igarashi M, Fukuda H, Nakagama H, Katoh M. Cancer Genetics and Genomics of Human FOX Family Genes. Cancer Lett (2013) 328:198–206. doi: 10.1016/j.canlet.2012.09.017
58. Kim J, Jin H, Zhao JC, Yang YA, Li Y, Yang X, et al. FOXA1 Inhibits Prostate Cancer Neuroendocrine Differentiation. Oncogene (2017) 36:4072–80. doi: 10.1038/onc.2017.50
59. Zhao Y, Li Z. Interplay of Estrogen Receptors and FOXA Factors in the Liver Cancer. Mol Cell Endocrinol (2015) 418:334–9. doi: 10.1016/j.mce.2015.01.043
60. Zhang W, Ren H, Zhang P, Tang Y, Wu M. Forkhead Box Protein A1 is a Prognostic Predictor and Promotes Tumor Growth of Gastric Cancer. Onco Targets Ther (2015) 8:3029–39. doi: 10.2147/OTT.S91035
61. Wang L-L, Xiu Y-L, Chen X, Sun K-X, Chen S, Wu D-D, et al. The Transcription Factor FOXA1 Induces Epithelial Ovarian Cancer Tumorigenesis and Progression. Tumor Biol (2017) 39:101042831770621. doi: 10.1177/1010428317706210
62. Nucera C, Eeckhoute J, Finn S, Carroll JS, Ligon AH, Priolo C, et al. Foxa1 Is a Potential Oncogene in Anaplastic Thyroid Carcinoma. Clin Cancer Res (2009) 15:3680–9. doi: 10.1158/1078-0432.CCR-08-3155
63. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell (2015) 163:506–19. doi: 10.1016/j.cell.2015.09.033
64. Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-Dependent Concerted Activity of HIF and FoxA2 Regulates Formation of Neuroendocrine Phenotype and Neuroendocrine Prostate Tumors. Cancer Cell (2010) 18:23–38. doi: 10.1016/j.ccr.2010.05.024
65. Eisinger-Mathason TSK, Simon MC. Hif-1α Partners With FoxA2, a Neuroendocrine-Specific Transcription Factor, to Promote Tumorigenesis. Cancer Cell (2010) 18:3–4. doi: 10.1016/j.ccr.2010.06.007
66. Yamamura N, Fugo K, Kishimoto T. Forkhead Box Protein A2, a Pioneer Factor for Hepatogenesis, is Involved in the Expression of Hepatic Phenotype of Alpha-Fetoprotein-Producing Adenocarcinoma. Pathol - Res Pract (2017) 213:1082–8. doi: 10.1016/j.prp.2017.07.024
67. Vorvis C, Hatziapostolou M, Mahurkar-Joshi S, Koutsioumpa M, Williams J, Donahue TR, et al. Transcriptomic and CRISPR/Cas9 Technologies Reveal FOXA2 as a Tumor Suppressor Gene in Pancreatic Cancer. Am J Physiol Liver Physiol (2016) 310:G1124–37. doi: 10.1152/ajpgi.00035.2016
68. DeGraff DJ, Clark PE, Cates JM, Yamashita H, Robinson VL, Yu X, et al. Loss of the Urothelial Differentiation Marker Foxa1 Is Associated With High Grade, Late Stage Bladder Cancer and Increased Tumor Proliferation. PloS One (2012) 7:e36669. doi: 10.1371/journal.pone.0036669
69. Yamashita H, Amponsa VO, Warrick JI, Zheng Z, Clark PE, Raman JD, et al. On a FOX Hunt: Functions of FOX Transcriptional Regulators in Bladder Cancer. Nat Rev Urol (2017) 14:98–106. doi: 10.1038/nrurol.2016.239
70. Li S, Zhou Q, He H, Zhao Y, Liu Z. Peroxisome Proliferator-activated Receptor γ Agonists Induce Cell Cycle Arrest Through Transcriptional Regulation of Krüppel-like Factor 4 (Klf4). J Biol Chem (2013) 288:4076–84. doi: 10.1074/jbc.M111.317487
71. Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-Like Factor 4 Is Acetylated by p300 and Regulates Gene Transcription Via Modulation of Histone Acetylation. J Biol Chem (2007) 282:33994–4002. doi: 10.1074/jbc.M701847200
72. Xie H, Li J, Ying Y, Yan H, Jin K, Ma X, et al. METTL3/YTHDF2 M 6 A Axis Promotes Tumorigenesis by Degrading SETD7 and KLF4 mRNAs in Bladder Cancer. J Cell Mol Med (2020) 24:4092–104. doi: 10.1111/jcmm.15063
73. Kong F, Sun T, Kong X, Xie D, Li Z, Xie K. Krüppel-Like Factor 4 Suppresses Serine/Threonine Kinase 33 Activation and Metastasis of Gastric Cancer Through Reversing Epithelial–Mesenchymal Transition. Clin Cancer Res (2018) 24:2440–51. doi: 10.1158/1078-0432.CCR-17-3346
74. Hu D, Zhou Z, Davidson NE, Huang Y, Wan Y. Novel Insight Into KLF4 Proteolytic Regulation in Estrogen Receptor Signaling and Breast Carcinogenesis. J Biol Chem (2012) 287:13584–97. doi: 10.1074/jbc.M112.343566
75. Wu Y, Lin L, Wang X, Li Y, Liu Z, Ye W, et al. Overexpression of Krüppel-Like Factor 4 Suppresses Migration and Invasion of Non-Small Cell Lung Cancer Through C-Jun-NH2-Terminal Kinase/Epithelial-Mesenchymal Transition Signaling Pathway. Front Pharmacol (2019) 10:1512. doi: 10.3389/fphar.2019.01512
76. Li Z, Zhao S, Wang H, Zhang B, Zhang P. miR-4286 Promotes Prostate Cancer Progression Via Targeting the Expression of SALL1. J Gene Med (2019) n/a:e3127. doi: 10.1002/jgm.3127
77. Chi D, Zhang W, Jia Y, Cong D, Hu S. Spalt-Like Transcription Factor 1 (Sall1) Gene Expression Inhibits Cell Proliferation and Cell Migration of Human Glioma Cells Through the Wnt/β-Catenin Signaling Pathway. Med Sci Monit Basic Res (2019) 25:128–38. doi: 10.12659/MSMBR.915067
78. Baba Y, Watabe Y, Sagara H, Watanabe S. Sall1 Plays Pivotal Roles for Lens Fiber Cell Differentiation in Mouse. Biochem Biophys Res Commun (2019) 512:927–33. doi: 10.1016/j.bbrc.2019.03.098
79. Valikodath NG, Jain S, Miller M, Kaufman LM. Ocular Features of Townes-Brocks Syndrome. J Am Assoc Pediatr Ophthalmol Strabismus (2020) 24:115–8. doi: 10.1016/j.jaapos.2019.12.004
80. Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, et al. Activation of the Pluripotency Factor OCT4 in Smooth Muscle Cells is Atheroprotective. Nat Med (2016) 22:657–65. doi: 10.1038/nm.4109
81. Zhao S, Yuan Q, Hao H, Guo Y, Liu S, Zhang Y, et al. Expression of OCT4 Pseudogenes in Human Tumours: Lessons From Glioma and Breast Carcinoma. J Pathol (2011) 223:672–82. doi: 10.1002/path.2827
82. Linn DE, Yang X, Sun F, Xie Y, Chen H, Jiang R, et al. A Role for OCT4 in Tumor Initiation of Drug-Resistant Prostate Cancer Cells. Genes Cancer (2010) 1:908–16. doi: 10.1177/1947601910388271
83. Niwa H, Miyazaki J, Smith AG. Quantitative Expression of Oct-3/4 Defines Differentiation, Dedifferentiation or Self-Renewal of ES Cells. Nat Genet (2000) 24:372–6. doi: 10.1038/74199
84. Monferrer E, Burgos-Panadero R, Blanquer-Maceiras M, Cañete A, Navarro S, Noguera R. High Oct4 Expression: Implications in the Pathogenesis of Neuroblastic Tumours. BMC Cancer (2019) 19:1. doi: 10.1186/s12885-018-5219-3
85. Wang Y-J, Herlyn M. The Emerging Roles of Oct4 in Tumor-Initiating Cells. Am J Physiol Cell Physiol (2015) 309:C709–18. doi: 10.1152/ajpcell.00212.2015
86. Shen L, Huang X, Xie X, Su J, Yuan J, Chen X. High Expression of SOX2 and OCT4 Indicates Radiation Resistance and an Independent Negative Prognosis in Cervical Squamous Cell Carcinoma. J Histochem Cytochem (2014) 62:499–509. doi: 10.1369/0022155414532654
87. Tsai L-L, Yu C-C, Chang Y-C, Yu C-H, Chou M-Y. Markedly Increased Oct4 and Nanog Expression Correlates With Cisplatin Resistance in Oral Squamous Cell Carcinoma. J Oral Pathol Med (2011) 40:621–8. doi: 10.1111/j.1600-0714.2011.01015.x
88. Liu X, Ma M, Duan X, Zhang H, Yang M. Knockdown of OCT4 may Sensitize NSCLC Cells to Cisplatin. Clin Transl Oncol (2017) 19:587–92. doi: 10.1007/s12094-016-1569-y
89. Samardzija C, Luwor RB, Volchek M, Quinn MA, Findlay JK, Ahmed N. A Critical Role of Oct4A in Mediating Metastasis and Disease-Free Survival in a Mouse Model of Ovarian Cancer. Mol Cancer (2015) 14:152. doi: 10.1186/s12943-015-0417-y
90. Ikushima H, Todo T, Ino Y, Takahashi M, Saito N, Miyazawa K, et al. Glioma-Initiating Cells Retain Their Tumorigenicity Through Integration of the Sox Axis and Oct4 Protein. J Biol Chem (2011) 286:41434–41. doi: 10.1074/jbc.M111.300863
91. Cortes-Dericks L, Farashahi Yazd E, Mowla SJ, Schmid RA, Karoubi G. Suppression of OCT4B Enhances Sensitivity of Lung Adenocarcinoma A549 Cells to Cisplatin Via Increased Apoptosis. Anticancer Res (2013) 33:5365–73.
92. Le Q, Lin J, Xie X, Yu X, Cai Y, Shentu Y, et al. Role of OCT4 in Cisplatin Treatment of Testicular Embryonal Carcinoma. Trop J Pharm Res (2018) 17:1353. doi: 10.4314/tjpr.v17i7.18
93. Abada PB, Howell SB. Cisplatin Induces Resistance by Triggering Differentiation of Testicular Embryonal Carcinoma Cells. PloS One (2014) 9:e87444. doi: 10.1371/journal.pone.0087444
94. Villodre ES, Kipper FC, Pereira MB, Lenz G. Roles of OCT4 in Tumorigenesis, Cancer Therapy Resistance and Prognosis. Cancer Treat Rev (2016) 51:1–9. doi: 10.1016/j.ctrv.2016.10.003
95. Chaudhary S, Islam Z, Mishra V, Rawat S, Ashraf GM, Kolatkar PR. Sox2: A Regulatory Factor in Tumorigenesis and Metastasis. Curr Protein Pept Sci (2019) 20:495–504. doi: 10.2174/1389203720666190325102255
96. Hawkins K. Cell Signalling Pathways Underlying Induced Pluripotent Stem Cell Reprogramming. World J Stem Cells (2014) 6:620. doi: 10.4252/wjsc.v6.i5.620
97. Zhang S. Sox2, a Key Factor in the Regulation of Pluripotency and Neural Differentiation. World J Stem Cells (2014) 6:305. doi: 10.4252/wjsc.v6.i3.305
98. Basu-Roy U, Ambrosetti D, Favaro R, Nicolis SK, Mansukhani A, Basilico C. The Transcription Factor Sox2 is Required for Osteoblast Self-Renewal. Cell Death Differ (2010) 17:1345–53. doi: 10.1038/cdd.2010.57
99. Remenyi A. Crystal Structure of a POU/HMG/DNA Ternary Complex Suggests Differential Assembly of Oct4 and Sox2 on Two Enhancers. Genes Dev (2003) 17:2048–59. doi: 10.1101/gad.269303
100. Williams DC, Cai M, Clore GM. Molecular Basis for Synergistic Transcriptional Activation by Oct1 and Sox2 Revealed From the Solution Structure of the 42-Kda Oct1·Sox2· Hoxb1 -Dna Ternary Transcription Factor Complex. J Biol Chem (2004) 279:1449–57. doi: 10.1074/jbc.M309790200
101. Werner MH, Huth JR, Gronenborn AM, Marius Clore G. Molecular Basis of Human 46X, Y Sex Reversal Revealed From the Three-Dimensional Solution Structure of the Human SRY-DNA Complex. Cell (1995) 81:705–14. doi: 10.1016/0092-8674(95)90532-4
102. Yuan H, Corbi N, Basilico C, Dailey L. Developmental-Specific Activity of the FGF-4 Enhancer Requires the Synergistic Action of Sox2 and Oct-3. Genes Dev (1995) 9:2635–45. doi: 10.1101/gad.9.21.2635
103. Aksoy I, Jauch R, Chen J, Dyla M, Divakar U, Bogu GK, et al. Oct4 Switches Partnering From Sox2 to Sox17 to Reinterpret the Enhancer Code and Specify Endoderm. EMBO J (2013) 32:938–53. doi: 10.1038/emboj.2013.31
104. Jauch R, Aksoy I, Hutchins AP, Ng CKL, Tian XF, Chen J, et al. Conversion of Sox17 Into a Pluripotency Reprogramming Factor by Reengineering its Association With Oct4 on DNA. Stem Cells (2011) 29:940–51. doi: 10.1002/stem.639
105. Lundberg IV, Edin S, Eklöf V, Öberg Å, Palmqvist R, Wikberg ML. SOX2 Expression is Associated With a Cancer Stem Cell State and Down-Regulation of CDX2 in Colorectal Cancer. BMC Cancer (2016) 16:471. doi: 10.1186/s12885-016-2509-5
106. Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 Oncogenes Are Coamplified and Cooperate to Activate Hedgehog Signaling in Lung Squamous Cell Carcinoma. Cancer Cell (2014) 25:139–51. doi: 10.1016/j.ccr.2014.01.008
107. Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembelé D, et al. Sox2 Is an Oncogene Activated by Recurrent 3q26.3 Amplifications in Human Lung Squamous Cell Carcinomas. PloS One (2010) 5:e8960. doi: 10.1371/journal.pone.0008960
108. McCaughan F, Pole JCM, Bankier AT, Konfortov BA, Carroll B, Falzon M, et al. Progressive 3q Amplification Consistently Targets SOX2 in Preinvasive Squamous Lung Cancer. Am J Respir Crit Care Med (2010) 182:83–91. doi: 10.1164/rccm.201001-0005OC
109. Wilbertz T, Wagner P, Petersen K, Stiedl A-C, Scheble VJ, Maier S, et al. SOX2 Gene Amplification and Protein Overexpression are Associated With Better Outcome in Squamous Cell Lung Cancer. Mod Pathol (2011) 24:944–53. doi: 10.1038/modpathol.2011.49
110. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an Amplified Lineage-Survival Oncogene in Lung and Esophageal Squamous Cell Carcinomas. Nat Genet (2009) 41:1238–42. doi: 10.1038/ng.465
111. Xiang R, Liao D, Cheng T, Zhou H, Shi Q, Chuang TS, et al. Downregulation of Transcription Factor SOX2 in Cancer Stem Cells Suppresses Growth and Metastasis of Lung Cancer. Br J Cancer (2011) 104:1410–7. doi: 10.1038/bjc.2011.94
112. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive Genomic Analysis Identifies SOX2 as a Frequently Amplified Gene in Small-Cell Lung Cancer. Nat Genet (2012) 44:1111–6. doi: 10.1038/ng.2405
113. Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, et al. Correlation of CD133, OCT4, and SOX2 in Rectal Cancer and Their Association With Distant Recurrence After Chemoradiotherapy. Ann Surg Oncol (2009) 16:3488–98. doi: 10.1245/s10434-009-0617-z
114. Lu Y, Futtner C, Rock JR, Xu X, Whitworth W, Hogan BLM, et al. Evidence That Sox2 Overexpression is Oncogenic in the Lung. PloS One (2010) 5:e11022. doi: 10.1371/journal.pone.0011022
115. Liu X, Qiao B, Zhao T, Hu F, Lam AK, Tao Q. Sox2 Promotes Tumor Aggressiveness and Epithelial−Mesenchymal Transition in Tongue Squamous Cell Carcinoma. Int J Mol Med (2018) 42:1418–26. doi: 10.3892/ijmm.2018.3742
116. Yang S, Zheng J, Xiao X, Xu T, Tang W, Zhu H, et al. SOX2 Promotes Tumorigenicity and Inhibits the Differentiation of I-type Neuroblastoma Cells. Int J Oncol (2015) 46:317–23. doi: 10.3892/ijo.2014.2713
117. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, et al. Cancer Stem Cells in Squamous Cell Carcinoma Switch Between Two Distinct Phenotypes That Are Preferentially Migratory or Proliferative. Cancer Res (2011) 71:5317–26. doi: 10.1158/0008-5472.CAN-11-1059
118. Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, et al. SOX2 Promotes Tumor Metastasis by Stimulating Epithelial-to-Mesenchymal Transition Via Regulation of WNT/β-Catenin Signal Network. Cancer Lett (2013) 336:379–89. doi: 10.1016/j.canlet.2013.03.027
119. Chen S, Xu Y, Chen Y, Li X, Mou W, Wang L, et al. Sox2 Gene Regulates the Transcriptional Network of Oncogenes and Affects Tumorigenesis of Human Lung Cancer Cells. PloS One (2012) 7:e36326. doi: 10.1371/journal.pone.0036326
120. Oppel F, Müller N, Schackert G, Hendruschk S, Martin D, Geiger KD, et al. Sox2-RNAi Attenuates S-phase Entry and Induces RhoA-dependent Switch to Protease-Independent Amoeboid Migration in Human Glioma Cells. Mol Cancer (2011) 10:137. doi: 10.1186/1476-4598-10-137
121. Rossant J, Tam PPL. Blastocyst Lineage Formation, Early Embryonic Asymmetries and Axis Patterning in the Mouse. Development (2009) 136:701–13. doi: 10.1242/dev.017178
122. Wang J, Levasseur DN, Orkin SH. Requirement of Nanog Dimerization for Stem Cell Self-Renewal and Pluripotency. Proc Natl Acad Sci (2008) 105:6326–31. doi: 10.1073/pnas.0802288105
123. Mullin NP, Yates A, Rowe AJ, Nijmeijer B, Colby D, Barlow PN, et al. The Pluripotency Rheostat Nanog Functions as a Dimer. Biochem J (2008) 411:227–31. doi: 10.1042/BJ20080134
124. Fidalgo M, Shekar PC, Ang Y-S, Fujiwara Y, Orkin SH, Wang J. Zfp281 Functions as a Transcriptional Repressor for Pluripotency of Mouse Embryonic Stem Cells. Stem Cells (2011) 29:1705–16. doi: 10.1002/stem.736
125. Ow JR, Ma H, Jean A, Goh Z, Lee YH, Chong YM, et al. Patz1 Regulates Embryonic Stem Cell Identity. Stem Cells Dev (2014) 23:1062–73. doi: 10.1089/scd.2013.0430
126. Moretto-Zita M, Jin H, Shen Z, Zhao T, Briggs SP, Xu Y. Phosphorylation Stabilizes Nanog by Promoting its Interaction With Pin1. Proc Natl Acad Sci (2010) 107:13312–7. doi: 10.1073/pnas.1005847107
127. Ho B, Olson G, Figel S, Gelman I, Cance WG, Golubovskaya VM. Nanog Increases Focal Adhesion Kinase (Fak) Promoter Activity and Expression and Directly Binds to FAK Protein to Be Phosphorylated. J Biol Chem (2012) 287:18656–73. doi: 10.1074/jbc.M111.322883
128. Bourguignon LYW, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 Interaction With Protein Kinase Cϵ Promotes Oncogenic Signaling by the Stem Cell Marker Nanog and the Production of MicroRNA-21, Leading to Down-regulation of the Tumor Suppressor Protein Pdcd4, Anti-apoptosis, and Chemotherapy Resistance. J Biol Chem (2009) 284:26533–46. doi: 10.1074/jbc.M109.027466
129. Ambady S, Malcuit C, Kashpur O, Kole D, Holmes WF, Hedblom E, et al. Expression of NANOG and NANOGP8 in a Variety of Undifferentiated and Differentiated Human Cells. Int J Dev Biol (2010) 54:1743–54. doi: 10.1387/ijdb.103192sa
130. Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, et al. Functional Evidence That the Self-Renewal Gene NANOG Regulates Human Tumor Development. Stem Cells (2009) 27:993–1005. doi: 10.1002/stem.29
131. Ji W, Jiang Z. Effect of shRNA-mediated Inhibition of Nanog Gene Expression on the Behavior of Human Gastric Cancer Cells. Oncol Lett (2013) 6:367–74. doi: 10.3892/ol.2013.1394
132. Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Suzuki K, et al. Resveratrol Promotes Proteasome-Dependent Degradation of Nanog Via p53 Activation and Induces Differentiation of Glioma Stem Cells. Stem Cell Res (2013) 11:601–10. doi: 10.1016/j.scr.2013.04.004
133. Han J, Zhang F, Yu M, Zhao P, Ji W, Zhang H, et al. RNA Interference-Mediated Silencing of NANOG Reduces Cell Proliferation and Induces G0/G1 Cell Cycle Arrest in Breast Cancer Cells. Cancer Lett (2012) 321:80–8. doi: 10.1016/j.canlet.2012.02.021
134. Jeter CR, Liu B, Lu Y, Chao H-P, Zhang D, Liu X, et al. NANOG Reprograms Prostate Cancer Cells to Castration Resistance Via Dynamically Repressing and Engaging the AR/FOXA1 Signaling Axis. Cell Discov (2016) 2:16041. doi: 10.1038/celldisc.2016.41
135. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG Promotes Cancer Stem Cell Characteristics and Prostate Cancer Resistance to Androgen Deprivation. Oncogene (2011) 30:3833–45. doi: 10.1038/onc.2011.114
136. Sun C, Sun L, Jiang K, Gao D-M, Kang X-N, Wang C, et al. NANOG Promotes Liver Cancer Cell Invasion by Inducing Epithelial-Mesenchymal Transition Through NODAL/SMAD3 Signaling Pathway. Int J Biochem Cell Biol (2013) 45:1099–108. doi: 10.1016/j.biocel.2013.02.017
137. Ghaleb AM, Yang VW. Krüppel-Like Factor 4 (KLF4): What We Currently Know. Gene (2017) 611:27–37. doi: 10.1016/j.gene.2017.02.025
138. Jia Y, Ying X, Zhou J, Chen Y, Luo X, Xie S, et al. The Novel KLF4/PLAC8 Signaling Pathway Regulates Lung Cancer Growth. Cell Death Dis (2018) 9:603. doi: 10.1038/s41419-018-0580-3
139. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
140. Birsoy K, Chen Z, Friedman J. Transcriptional Regulation of Adipogenesis by KLF4. Cell Metab (2008) 7:339–47. doi: 10.1016/j.cmet.2008.02.001
141. Ghaleb AM, Yang VW. The Pathobiology of Krüppel-like Factors in Colorectal Cancer. Curr Colorectal Cancer Rep (2008) 4:59–64. doi: 10.1007/s11888-008-0011-4
142. Li Y, Yu S, Li L, Chen J, Quan M, Li Q, et al. KLF4-Mediated Upregulation of CD9 and CD81 Suppresses Hepatocellular Carcinoma Development Via JNK Signaling. Cell Death Dis (2020) 11:299. doi: 10.1038/s41419-020-2479-z
143. Qi X-T, Li Y-L, Zhang Y-Q, Xu T, Lu B, Fang L, et al. KLF4 Functions as an Oncogene in Promoting Cancer Stem Cell-Like Characteristics in Osteosarcoma Cells. Acta Pharmacol Sin (2019) 40:546–55. doi: 10.1038/s41401-018-0050-6
144. Mai J, Zhong ZY, Guo GF, Chen XX, Xiang YQ, Li X, et al. Polo-Like Kinase 1 Phosphorylates and Stabilizes KLF4 to Promote Tumorigenesis in Nasopharyngeal Carcinoma. Theranostics (2019) 9:3541–54. doi: 10.7150/thno.32908
145. Yang J, Rajan SS, Friedrich MJ, Lan G, Zou X, Ponstingl H, et al. Genome-Scale CRISPRa Screen Identifies Novel Factors for Cellular Reprogramming. Stem Cell Rep (2019) 12:757–71. doi: 10.1016/j.stemcr.2019.02.010
146. Kondelova A, Alburquerque-González B, Vychytilova-Faltejskova P, García-Solano J, Prochazka V, Kala Z, et al. miR-181a-2* Expression is Different Amongst Carcinomas From the Colorectal Serrated Route. Mutagenesis (2020) 35:233–41. doi: 10.1093/mutage/gez039
148. Zheng R, Blobel GA. Gata Transcription Factors and Cancer. Genes Cancer (2010) 1:1178–88. doi: 10.1177/1947601911404223
149. Lentjes MHFM, Niessen HEC, Akiyama Y, de Bruïne AP, Melotte V, van Engeland M. The Emerging Role of GATA Transcription Factors in Development and Disease. Expert Rev Mol Med (2016) 18:e3. doi: 10.1017/erm.2016.2
150. Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial Dyserythropoietic Anaemia and Thrombocytopenia Due to an Inherited Mutation in GATA1. Nat Genet (2000) 24:266–70. doi: 10.1038/73480
151. Kouros-Mehr H, Werb Z. Candidate Regulators of Mammary Branching Morphogenesis Identified by Genome-Wide Transcript Analysis. Dev Dyn (2006) 235:3404–12. doi: 10.1002/dvdy.20978
152. Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. Gata-3 Maintains the Differentiation of the Luminal Cell Fate in the Mammary Gland. Cell (2006) 127:1041–55. doi: 10.1016/j.cell.2006.09.048
153. Asselin-Labat M-L, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, et al. Gata-3 is an Essential Regulator of Mammary-Gland Morphogenesis and Luminal-Cell Differentiation. Nat Cell Biol (2007) 9:201–9. doi: 10.1038/ncb1530
154. Zhang S-J, Ma L-Y, Huang Q-H, Li G, Gu B-W, Gao X-D, et al. Gain-of-Function Mutation of GATA-2 in Acute Myeloid Transformation of Chronic Myeloid Leukemia. Proc Natl Acad Sci (2008) 105:2076–81. doi: 10.1073/pnas.0711824105
155. Mehra R, Varambally S, Ding L, Shen R, Sabel MS, Ghosh D, et al. Identification of GATA3 as a Breast Cancer Prognostic Marker by Global Gene Expression Meta-Analysis. Cancer Res (2005) 65:11259–64. doi: 10.1158/0008-5472.CAN-05-2495
156. Cimino-Mathews A, Ye X, Meeker A, Argani P, Emens LA. Metastatic Triple-Negative Breast Cancers at First Relapse Have Fewer Tumor-Infiltrating Lymphocytes Than Their Matched Primary Breast Tumors: A Pilot Study. Hum Pathol (2013) 44:2055–63. doi: 10.1016/j.humpath.2013.03.010
157. Shaoxian T, Baohua Y, Xiaoli X, Yufan C, Xiaoyu T, Hongfen L, et al. Characterisation of GATA3 Expression in Invasive Breast Cancer: Differences in Histological Subtypes and Immunohistochemically Defined Molecular Subtypes. J Clin Pathol (2017) 70:926–34. doi: 10.1136/jclinpath-2016-204137
158. Cakir A, Isik Gonul I, Ekinci O, Cetin B, Benekli M, Uluoglu O. GATA3 Expression and its Relationship With Clinicopathological Parameters in Invasive Breast Carcinomas. Pathol Res Pract (2017) 213:227–34. doi: 10.1016/j.prp.2016.12.010
159. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The Landscape of Cancer Genes and Mutational Processes in Breast Cancer. Nature (2012) 486:400–4. doi: 10.1038/nature11017
160. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence Analysis of Mutations and Translocations Across Breast Cancer Subtypes. Nature (2012) 486:405–9. doi: 10.1038/nature11154
161. The Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490:61–70. doi: 10.1038/nature11412
162. Robson EJD, He S-J, Eccles MR. A PANorama of PAX Genes in Cancer and Development. Nat Rev Cancer (2006) 6:52–62. doi: 10.1038/nrc1778
163. Monsoro-Burq AH. PAX Transcription Factors in Neural Crest Development. Semin Cell Dev Biol (2015) 44:87–96. doi: 10.1016/j.semcdb.2015.09.015
164. Frigerio G, Burri M, Bopp D, Baumgartner S, Noll M. Structure of the Segmentation Gene Paired and the Drosophila PRD Gene Set as Part of a Gene Network. Cell (1986) 47:735–46. doi: 10.1016/0092-8674(86)90516-7
165. Peters H, Doll U, Niessing J. Differential Expression of the Chicken Pax-1 and Pax-9 Gene: In Situ Hybridization and Immunohistochemical Analysis. Dev Dyn (1995) 203:1–16. doi: 10.1002/aja.1002030102
166. Lai H-C, Lin Y-W, Huang THM, Yan P, Huang R-L, Wang H-C, et al. Identification of Novel DNA Methylation Markers in Cervical Cancer. Int J Cancer (2008) 123:161–7. doi: 10.1002/ijc.23519
167. Doberstein K, Pfeilschifter J, Gutwein P. The Transcription Factor PAX2 Regulates ADAM10 Expression in Renal Cell Carcinoma. Carcinogenesis (2011) 32:1713–23. doi: 10.1093/carcin/bgr195
168. Gnarra JR, Dressler GR. Expression of Pax-2 in Human Renal Cell Carcinoma and Growth Inhibition by Antisense Oligonucleotides. Cancer Res (1995) 55:4092–8.
169. Luu V-D, Boysen G, Struckmann K, Casagrande S, von Teichman A, Wild PJ, et al. Loss of VHL and Hypoxia Provokes PAX2 Up-Regulation in Clear Cell Renal Cell Carcinoma. Clin Cancer Res (2009) 15:3297–304. doi: 10.1158/1078-0432.CCR-08-2779
170. Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y, et al. Hypomethylation-Linked Activation of PAX2 Mediates Tamoxifen-Stimulated Endometrial Carcinogenesis. Nature (2005) 438:981–7. doi: 10.1038/nature04225
171. Barr FG. Gene Fusions Involving PAX and FOX Family Members in Alveolar Rhabdomyosarcoma. Oncogene (2001) 20:5736–46. doi: 10.1038/sj.onc.1204599
172. Miyamoto T, Kakizawa T, Ichikawa K, Nishio S, Kajikawa S, Hashizume K. Expression of Dominant Negative Form of PAX4 in Human Insulinoma. Biochem Biophys Res Commun (2001) 282:34–40. doi: 10.1006/bbrc.2001.4552
173. Brun T, Duhamel DL, Hu He KH, Wollheim CB, Gauthier BR. The Transcription Factor PAX4 Acts as a Survival Gene in INS-1E Insulinoma Cells. Oncogene (2007) 26:4261–71. doi: 10.1038/sj.onc.1210205
174. Sanz E, Alvarez-Mon M, Martínez-A C, de la Hera A. Human Cord Blood CD34+Pax-5+ B-Cell Progenitors: Single-Cell Analyses of Their Gene Expression Profiles. Blood (2003) 101:3424–30. doi: 10.1182/blood-2002-07-2244
175. Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, et al. Direct Reprogramming of Terminally Differentiated Mature B Lymphocytes to Pluripotency. Cell (2008) 133:250–64. doi: 10.1016/j.cell.2008.03.028
176. Johanson TM, Lun ATL, Coughlan HD, Tan T, Smyth GK, Nutt SL, et al. Transcription-Factor-Mediated Supervision of Global Genome Architecture Maintains B Cell Identity. Nat Immunol (2018) 19:1257–64. doi: 10.1038/s41590-018-0234-8
177. Sansom SN, Griffiths DS, Faedo A, Kleinjan D-J, Ruan Y, Smith J, et al. The Level of the Transcription Factor Pax6 is Essential for Controlling the Balance Between Neural Stem Cell Self-Renewal and Neurogenesis. PloS Genet (2009) 5:e1000511. doi: 10.1371/journal.pgen.1000511
178. Zhang X, Huang CT, Chen J, Pankratz MT, Xi J, Li J, et al. Pax6 is a Human Neuroectoderm Cell Fate Determinant. Cell Stem Cell (2010) 7:90–100. doi: 10.1016/j.stem.2010.04.017
179. Kamachi Y, Uchikawa M, Tanouchi A, Sekido R, Kondoh H. Pax6 and SOX2 Form a co-DNA-binding Partner Complex That Regulates Initiation of Lens Development. Genes Dev (2001) 15:1272–86. doi: 10.1101/gad.887101
180. Mascarenhas JB, Young KP, Littlejohn EL, Yoo BK, Salgia R, Lang D. PAX6 is Expressed in Pancreatic Cancer and Actively Participates in Cancer Progression Through Activation of the MET Tyrosine Kinase Receptor Gene. J Biol Chem (2009) 284:27524–32. doi: 10.1074/jbc.M109.047209
181. Zhou Y-H, Hu Y, Mayes D, Siegel E, Kim JG, Mathews MS, et al. PAX6 Suppression of Glioma Angiogenesis and the Expression of Vascular Endothelial Growth Factor a. J Neurooncol (2010) 96:191–200. doi: 10.1007/s11060-009-9963-8
182. Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 Is Required for the Specification of Myogenic Satellite Cells. Cell (2000) 102:777–86. doi: 10.1016/S0092-8674(00)00066-0
183. Tong G-X, Yu WM, Beaubier NT, Weeden EM, Hamele-Bena D, Mansukhani MM, et al. Expression of PAX8 in Normal and Neoplastic Renal Tissues: An Immunohistochemical Study. Mod Pathol (2009) 22:1218–27. doi: 10.1038/modpathol.2009.88
184. Li CG, Nyman JE, Braithwaite AW, Eccles MR. PAX8 Promotes Tumor Cell Growth by Transcriptionally Regulating E2F1 and Stabilizing RB Protein. Oncogene (2011) 30:4824–34. doi: 10.1038/onc.2011.190
185. Chen Y-J, Campbell HG, Wiles AK, Eccles MR, Reddel RR, Braithwaite AW, et al. Pax8 Regulates Telomerase Reverse Transcriptase and Telomerase Rna Component in Glioma. Cancer Res (2008) 68:5724–32. doi: 10.1158/0008-5472.CAN-08-0058
186. Raman P, Koenig RJ. Pax-8-PPAR-γ Fusion Protein in Thyroid Carcinoma. Nat Rev Endocrinol (2014) 10:616–23. doi: 10.1038/nrendo.2014.115
187. Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KCQ, Lakshmi B, et al. Oncogenic Cooperation and Coamplification of Developmental Transcription Factor Genes in Lung Cancer. Proc Natl Acad Sci U.S.A. (2007) 104:16663–8. doi: 10.1073/pnas.0708286104
188. Lee J-C, Sharma M, Lee Y-H, Lee N-H, Kim S-Y, Yun J-S, et al. Pax9 Mediated Cell Survival in Oral Squamous Carcinoma Cell Enhanced by C-Myb. Cell Biochem Funct (2008) 26:892–9. doi: 10.1002/cbf.1522
189. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional Regulation of Adipogenesis. Genes Dev (2000) 14:1293–307. doi: 10.1101/gad.14.11.1293
190. Sears IB, MacGinnitie MA, Kovacs LG, Graves RA. Differentiation-Dependent Expression of the Brown Adipocyte Uncoupling Protein Gene: Regulation by Peroxisome Proliferator-Activated Receptor Gamma. Mol Cell Biol (1996) 16:3410–9. doi: 10.1128/MCB.16.7.3410
191. Kim JB, Spiegelman BM. ADD1/SREBP1 Promotes Adipocyte Differentiation and Gene Expression Linked to Fatty Acid Metabolism. Genes Dev (1996) 10:1096–107. doi: 10.1101/gad.10.9.1096
192. Tontonoz P, Hu E, Spiegelman BM. Regulation of Adipocyte Gene Expression and Differentiation by Peroxisome Proliferator Activated Receptor γ. Curr Opin Genet Dev (1995) 5:571–6. doi: 10.1016/0959-437X(95)80025-5
193. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR Gamma 2: Tissue-Specific Regulator of an Adipocyte Enhancer. Genes Dev (1994) 8:1224–34. doi: 10.1101/gad.8.10.1224
194. Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CDM, et al. Terminal Differentiation of Human Liposarcoma Cells Induced by Ligands for Peroxisome Proliferator-Activated Receptor and the Retinoid X Receptor. Proc Natl Acad Sci (1997) 94:237–41. doi: 10.1073/pnas.94.1.237
195. Fröhlich E, Wahl R. Chemotherapy and Chemoprevention by Thiazolidinediones. BioMed Res Int (2015) 2015:1–14. doi: 10.1155/2015/845340
196. Behera R, Kaur A, Webster MR, Kim S, Ndoye A, Kugel CH, et al. Inhibition of Age-Related Therapy Resistance in Melanoma by Rosiglitazone-Mediated Induction of Klotho. Clin Cancer Res (2017) 23:3181–90. doi: 10.1158/1078-0432.CCR-17-0201
197. Pich C, Meylan P, Mastelic-Gavillet B, Nguyen NT, Loyon R, Trang BK, et al. Induction of Paracrine Signaling in Metastatic Melanoma Cells by Pparγ Agonist Rosiglitazone Activates Stromal Cells and Enhances Tumor Growth. Cancer Res (2018) 78:6447–61. doi: 10.1158/0008-5472.CAN-18-0912 canres.0912.2018.
198. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, et al. Wnt Signaling Directs a Metabolic Program of Glycolysis and Angiogenesis in Colon Cancer. EMBO J (2014) 33:1454–73. doi: 10.15252/embj.201488598
199. Niehrs C, Acebron SP. Mitotic and Mitogenic Wnt Signalling. EMBO J (2012) 31:2705–13. doi: 10.1038/emboj.2012.124
200. Zhang Z, Xu H, Ji J, Shi X, Lyu J, Zhu Y, et al. Heterogeneity of PTEN and PPAR−γ in Cancer and Their Prognostic Application to Bladder Cancer. Exp Ther Med (2019) 18:3177–83. doi: 10.3892/etm.2019.7879
201. Lee C, Ramirez JA, Guitart J, Diaz LK. Expression of Cyclooxygenase-2 and Peroxisome Proliferator-Activated Receptor Gamma During Malignant Melanoma Progression. J Cutan Pathol (2008) 35:989–94. doi: 10.1111/j.1600-0560.2007.00939.x
202. Yoshimura R, Matsuyama M, Segawa Y, Hase T, Mitsuhashi M, Tsuchida K, et al. Expression of Peroxisome Proliferator-Activated Receptors (Ppars) in Human Urinary Bladder Carcinoma and Growth Inhibition by its Agonists. Int J Cancer (2003) 104:597–602. doi: 10.1002/ijc.10980
203. Mao F, Xu M, Zuo X, Yu J, Xu W, Moussalli MJ, et al. 15-Lipoxygenase-1 Suppression of Colitis-Associated Colon Cancer Through Inhibition of the IL-6/STAT3 Signaling Pathway. FASEB J (2015) 29:2359–70. doi: 10.1096/fj.14-264515
204. Wang D, Fu L, Ning W, Guo L, Sun X, Dey SK, et al. Peroxisome Proliferator-Activated Receptor δ Promotes Colonic Inflammation and Tumor Growth. Proc Natl Acad Sci U S A (2014) 111:7084–9. doi: 10.1073/pnas.1324233111
205. Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, et al. Loss-of-Function Mutations in Pparγ Associated With Human Colon Cancer. Mol Cell (1999) 3:799–804. doi: 10.1016/S1097-2765(01)80012-5
206. Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, et al. Effects of Ligand Activation of Peroxisome Proliferator-Activated Receptor Gamma in Human Prostate Cancer. Proc Natl Acad Sci (2000) 97:10990–5. doi: 10.1073/pnas.180329197
207. Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, Tallini G, et al. Ras Point Mutations and PAX8-Pparγ Rearrangement in Thyroid Tumors: Evidence for Distinct Molecular Pathways in Thyroid Follicular Carcinoma. J Clin Endocrinol Metab (2003) 88:2318–26. doi: 10.1210/jc.2002-021907
208. Marques AR, Espadinha C, Catarino AL, Moniz S, Pereira T, Sobrinho LG, et al. Expression of PAX8-Pparγ1 Rearrangements in Both Follicular Thyroid Carcinomas and Adenomas. J Clin Endocrinol Metab (2002) 87:3947–52. doi: 10.1210/jcem.87.8.8756
209. Gupta S, Singh AK, Prajapati KS, Kushwaha PP, Shuaib M, Kumar S. Emerging Role of ZBTB7A as an Oncogenic Driver and Transcriptional Repressor. Cancer Lett (2020) 483:22–34. doi: 10.1016/j.canlet.2020.04.015
210. Lunardi A, Guarnerio J, Wang G, Maeda T, Pandolfi PP. Role of LRF/Pokemon in Lineage Fate Decisions. Blood (2013) 121:2845–53. doi: 10.1182/blood-2012-11-292037
211. Beaulieu AM, Sant’Angelo DB. The BTB-ZF Family of Transcription Factors: Key Regulators of Lineage Commitment and Effector Function Development in the Immune System. J Immunol (2011) 187:2841–7. doi: 10.4049/jimmunol.1004006
212. Kelly KF, Daniel JM. POZ for Effect–POZ-ZF Transcription Factors in Cancer and Development. Trends Cell Biol (2006) 16:578–87. doi: 10.1016/j.tcb.2006.09.003
213. Costoya JA. Functional Analysis of the Role of POK Transcriptional Repressors. Brief Funct Genomic Proteomic (2007) 6:8–18. doi: 10.1093/bfgp/elm002
214. Barna M, Hawe N, Niswander L, Pandolfi PP. Plzf Regulates Limb and Axial Skeletal Patterning. Nat Genet (2000) 25:166–72. doi: 10.1038/76014
215. Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, Sukhwani M, et al. Essential Role of Plzf in Maintenance of Spermatogonial Stem Cells. Nat Genet (2004) 36:653–9. doi: 10.1038/ng1367
216. Mao A-P, Constantinides MG, Mathew R, Zuo Z, Chen X, Weirauch MT, et al. Multiple Layers of Transcriptional Regulation by PLZF in NKT-cell Development. Proc Natl Acad Sci U S A (2016) 113:7602–7. doi: 10.1073/pnas.1601504113
217. McConnell MJ, Chevallier N, Berkofsky-Fessler W, Giltnane JM, Malani RB, Staudt LM, et al. Growth Suppression by Acute Promyelocytic Leukemia-Associated Protein PLZF is Mediated by Repression of C-Myc Expression. Mol Cell Biol (2003) 23:9375–88. doi: 10.1128/mcb.23.24.9375-9388.2003
218. Fedele M, Franco R, Salvatore G, Paronetto MP, Barbagallo F, Pero R, et al. PATZ1 Gene has a Critical Role in the Spermatogenesis and Testicular Tumours. J Pathol (2008) 215:39–47. doi: 10.1002/path.2323
219. Mancinelli S, Vitiello M, Donnini M, Mantile F, Palma G, Luciano A, et al. The Transcription Regulator Patz1 is Essential for Neural Stem Cell Maintenance and Proliferation. Front Cell Dev Biol (2021) 9:657149. doi: 10.3389/fcell.2021.657149
220. Andersen L, Gülich AF, Alteneder M, Preglej T, Orola MJ, Dhele N, et al. The Transcription Factor MAZR/PATZ1 Regulates the Development of FOXP3+ Regulatory T Cells. Cell Rep (2019) 29:4447–59. doi: 10.1016/j.celrep.2019.11.089
221. Fedele M, Crescenzi E, Cerchia L. The POZ/BTB and AT-Hook Containing Zinc Finger 1 (Patz1) Transcription Regulator: Physiological Functions and Disease Involvement. Int J Mol Sci (2017) 18:2524. doi: 10.3390/ijms18122524
222. Yang H, Green MR. Epigenetic Programing of B-Cell Lymphoma by BCL6 and Its Genetic Deregulation. Front Cell Dev Biol (2019) 7:272. doi: 10.3389/fcell.2019.00272
223. Lu X, Fernando TM, Lossos C, Yusufova N, Liu F, Fontán L, et al. PRMT5 Interacts With the BCL6 Oncoprotein and is Required for Germinal Center Formation and Lymphoma Cell Survival. Blood (2018) 132:2026–39. doi: 10.1182/blood-2018-02-831438
224. Robinson SC, Klobucar K, Pierre CC, Ansari A, Zhenilo S, Prokhortchouk E, et al. Kaiso Differentially Regulates Components of the Notch Signaling Pathway in Intestinal Cells. Cell Commun Signal (2017) 15:24. doi: 10.1186/s12964-017-0178-x
225. Abisoye-Ogunniyan A, Lin H, Ghebremedhin A, Salam AB, Karanam B, Theodore S, et al. Transcriptional Repressor Kaiso Promotes Epithelial to Mesenchymal Transition and Metastasis in Prostate Cancer Through Direct Regulation of Mir-200c. Cancer Lett (2018) 431:1–10. doi: 10.1016/j.canlet.2018.04.044
226. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci (2017) 18:2574. doi: 10.3390/ijms18122574
227. Arnold CR, Mangesius J, Skvortsova II, Ganswindt U. The Role of Cancer Stem Cells in Radiation Resistance. Front Oncol (2020) 10:164. doi: 10.3389/fonc.2020.00164
228. Chen X, Liao R, Li D, Sun J. Induced Cancer Stem Cells Generated by Radiochemotherapy and Their Therapeutic Implications. Oncotarget (2017) 8:17301–12. doi: 10.18632/oncotarget.14230
229. Bushweller JH. Targeting Transcription Factors in Cancer - From Undruggable to Reality. Nat Rev Cancer (2019) 19:611–24. doi: 10.1038/s41568-019-0196-7
230. Serizawa M, Murakami H, Watanabe M, Takahashi T, Yamamoto N, Koh Y. Peroxisome Proliferator-Activated Receptor Gamma Agonist Efatutazone Impairs Transforming Growth Factor beta2-induced Motility of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistant Lung Cancer Cells. Cancer Sci (2014) 105:683–9. doi: 10.1111/cas.12411
231. Ni J, Zhou LL, Ding L, Zhao X, Cao H, Fan F, et al. Ppargamma Agonist Efatutazone and Gefitinib Synergistically Inhibit the Proliferation of EGFR-TKI-resistant Lung Adenocarcinoma Cells Via the PPARgamma/PTEN/Akt Pathway. Exp Cell Res (2017) 361:246–56. doi: 10.1016/j.yexcr.2017.10.024
232. Ni J, Zhou LL, Ding L, Zhang XQ, Zhao X, Li H, et al. Efatutazone and T0901317 Exert Synergistically Therapeutic Effects in Acquired Gefitinib-Resistant Lung Adenocarcinoma Cells. Cancer Med (2018) 7:1955–66. doi: 10.1002/cam4.1440
233. Pishvaian MJ, Marshall JL, Wagner AJ, Hwang JJ, Malik S, Cotarla I, et al. A Phase 1 Study of Efatutazone, an Oral Peroxisome Proliferator-Activated Receptor Gamma Agonist, Administered to Patients With Advanced Malignancies. Cancer (2012) 118:5403–13. doi: 10.1002/cncr.27526
234. Murakami H, Ono A, Takahashi T, Onozawa Y, Tsushima T, Yamazaki K, et al. Phase I Study of Efatutazone, an Oral PPARgamma Agonist, in Patients With Metastatic Solid Tumors. Anticancer Res (2014) 34:5133–41.
235. Smallridge RC, Copland JA, Brose MS, Wadsworth JT, Houvras Y, Menefee ME, et al. Efatutazone, an Oral PPAR-gamma Agonist, in Combination With Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. J Clin Endocrinol Metab (2013) 98:2392–400. doi: 10.1210/jc.2013-1106
236. Shiroma Y, Takahashi RU, Yamamoto Y, Tahara H. Targeting DNA Binding Proteins for Cancer Therapy. Cancer Sci (2020) 111:1058–64. doi: 10.1111/cas.14355
237. Lin CH, Li HY, Liu YP, Kuo PF, Wang WC, Lin FC, et al. High-CLDN4 ESCC Cells Harbor Stem-Like Properties and Indicate for Poor Concurrent Chemoradiation Therapy Response in Esophageal Squamous Cell Carcinoma. Ther Adv Med Oncol (2019) 11:1758835919875324. doi: 10.1177/1758835919875324
238. Chen Y-T, Li J-Y, Pan P-Y, Chen C-P, Chao J-I. A Novel Compound PT-262 Inhibits Oct4 and Nanog on the Survivin Blockage and Apoptosis Induction in Human Lung Cancer. FASEB J (2016) 30:652.7–7. doi: 10.1096/fasebj.30.1_supplement.652.7
239. Yin Y, Xie CM, Li H, Tan M, Chen G, Schiff R, et al. The FBXW2-MSX2-SOX2 Axis Regulates Stem Cell Property and Drug Resistance of Cancer Cells. Proc Natl Acad Sci U S A (2019) 116:20528–38. doi: 10.1073/pnas.1905973116
240. Zhou L, Jia L. Targeting Protein Neddylation for Cancer Therapy. Adv Exp Med Biol (2020) 1217:297–315. doi: 10.1007/978-981-15-1025-0_18
241. Smith PG, Traore T, Grossman S, Narayanan U, Carew JS, Lublinksky A, et al. Azacitidine/Decitabine Synergism With the NEDD8-Activating Enzyme Inhibitor MLN4924 in Pre-Clinical Aml Models. Blood (2011) 118:578. doi: 10.1182/blood.V118.21.578.578
242. Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-Activating Enzyme Inhibitor, in Patients With Acute Myeloid Leukaemia and Myelodysplastic Syndromes: A Phase 1 Study. Br J Haematol (2015) 169:534–43. doi: 10.1111/bjh.13323
243. Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, et al. Phase I Study of the Investigational Nedd8-Activating Enzyme Inhibitor Pevonedistat (Tak-924/MLN4924) in Patients With Advanced Solid Tumors. Clin Cancer Res (2016) 22:847–57. doi: 10.1158/1078-0432.CCR-15-1338
244. Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD, et al. A Phase I Study of the Investigational NEDD8-activating Enzyme Inhibitor Pevonedistat (TAK-924/MLN4924) in Patients With Metastatic Melanoma. Invest New Drugs (2016) 34:439–49. doi: 10.1007/s10637-016-0348-5
245. Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients With Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin Cancer Res (2016) 22:34–43. doi: 10.1158/1078-0432.CCR-15-1237
246. Swords RT, Watts J, Erba HP, Altman JK, Maris M, Anwer F, et al. Expanded Safety Analysis of Pevonedistat, a First-in-Class NEDD8-activating Enzyme Inhibitor, in Patients With Acute Myeloid Leukemia and Myelodysplastic Syndromes. Blood Cancer J (2017) 7:e520. doi: 10.1038/bcj.2017.1
247. Lockhart AC, Bauer TM, Aggarwal C, Lee CB, Harvey RD, Cohen RB, et al. Phase Ib Study of Pevonedistat, a NEDD8-activating Enzyme Inhibitor, in Combination With Docetaxel, Carboplatin and Paclitaxel, or Gemcitabine, in Patients With Advanced Solid Tumors. Invest New Drugs (2019) 37:87–97. doi: 10.1007/s10637-018-0610-0
248. Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J, et al. Pevonedistat, a First-in-Class NEDD8-activating Enzyme Inhibitor, Combined With Azacitidine in Patients With AML. Blood (2018) 131:1415–24. doi: 10.1182/blood-2017-09-805895
249. Jothi M, Mal M, Keller C, Mal AK. Small Molecule Inhibition of PAX3-FOXO1 Through AKT Activation Suppresses Malignant Phenotypes of Alveolar Rhabdomyosarcoma. Mol Cancer Ther (2013) 12:2663–74. doi: 10.1158/1535-7163.MCT-13-0277
250. Grimley E, Liao C, Ranghini EJ, Nikolovska-Coleska Z, Dressler GR. Inhibition of Pax2 Transcription Activation With a Small Molecule That Targets the DNA Binding Domain. ACS Chem Biol (2017) 12:724–34. doi: 10.1021/acschembio.6b00782
251. Nomura S, Takahashi H, Suzuki J, Kuwahara M, Yamashita M, Sawasaki T. Pyrrothiogatain Acts as an Inhibitor of GATA Family Proteins and Inhibits Th2 Cell Differentiation In Vitro. Sci Rep (2019) 9:17335. doi: 10.1038/s41598-019-53856-1
252. El-Hachem N, Nemer G. Identification of New GATA4-small Molecule Inhibitors by Structure-Based Virtual Screening. Bioorg Med Chem (2011) 19:1734–42. doi: 10.1016/j.bmc.2011.01.022
253. Negroni C, Hilton DA, Ercolano E, Adams CL, Kurian KM, Baiz D, et al. Gata-4, a Potential Novel Therapeutic Target for High-Grade Meningioma, Regulates miR-497, a Potential Novel Circulating Biomarker for High-Grade Meningioma. EBioMedicine (2020) 59:102941. doi: 10.1016/j.ebiom.2020.102941
254. Klaus M, Prokoph N, Girbig M, Wang X, Huang YH, Srivastava Y, et al. Structure and Decoy-Mediated Inhibition of the SOX18/Prox1-DNA Interaction. Nucleic Acids Res (2016) 44:3922–35. doi: 10.1093/nar/gkw130
255. Wang S, Singh SK, Katika MR, Lopez-Aviles S, Hurtado A. High Throughput Chemical Screening Reveals Multiple Regulatory Proteins on FOXA1 in Breast Cancer Cell Lines. Int J Mol Sci (2018) 19:4123. doi: 10.3390/ijms19124123
256. Vella V, Nicolosi ML, Giuliano S, Bellomo M, Belfiore A, Malaguarnera R. PPAR-Gamma Agonists as Antineoplastic Agents in Cancers With Dysregulated Igf Axis. Front Endocrinol (2017) 8:31. doi: 10.3389/fendo.2017.00031
257. Yu J, Qiao L, Zimmermann L, Ebert MP, Zhang H, Lin W, et al. Troglitazone Inhibits Tumor Growth in Hepatocellular Carcinoma In Vitro and In Vivo. Hepatology (2006) 43:134–43. doi: 10.1002/hep.20994
258. Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and Reversal of Malignant Changes in Colon Cancer Through Ppargamma. Nat Med (1998) 4:1046–52. doi: 10.1038/2030
259. Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, et al. A New Ligand for the Peroxisome Proliferator-Activated Receptor-Gamma (PPAR-Gamma), GW7845, Inhibits Rat Mammary Carcinogenesis. Cancer Res (1999) 59:5671–3.
260. Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, et al. A Phase II Study of Troglitazone, an Activator of the PPARgamma Receptor, in Patients With Chemotherapy-Resistant Metastatic Colorectal Cancer. Cancer J (2002) 8:395–9. doi: 10.1097/00130404-200209000-00010
261. Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, et al. Rosiglitazone Versus Placebo for Men With Prostate Carcinoma and a Rising Serum Prostate-Specific Antigen Level After Radical Prostatectomy and/or Radiation Therapy. Cancer (2004) 101:1569–74. doi: 10.1002/cncr.20493
262. Shimazaki N, Togashi N, Hanai M, Isoyama T, Wada K, Fujita T, et al. Anti-Tumour Activity of CS-7017, a Selective Peroxisome Proliferator-Activated Receptor Gamma Agonist of Thiazolidinedione Class, in Human Tumour Xenografts and a Syngeneic Tumour Implant Model. Eur J Cancer (2008) 44:1734–43. doi: 10.1016/j.ejca.2008.04.016
263. Sawayama H, Ishimoto T, Watanabe M, Yoshida N, Sugihara H, Kurashige J, et al. Small Molecule Agonists of PPAR-gamma Exert Therapeutic Effects in Esophageal Cancer. Cancer Res (2014) 74:575–85. doi: 10.1158/0008-5472.CAN-13-1836
Keywords: transcription factors, cell fate, pluripotency, tumorigenesis, cancer mechanisms, clinical relevance
Citation: Islam Z, Ali AM, Naik A, Eldaw M, Decock J and Kolatkar PR (2021) Transcription Factors: The Fulcrum Between Cell Development and Carcinogenesis. Front. Oncol. 11:681377. doi: 10.3389/fonc.2021.681377
Received: 16 March 2021; Accepted: 26 May 2021;
Published: 14 June 2021.
Edited by:
Dario Palmieri, The Ohio State University, United StatesReviewed by:
Mariastella Zannini, Istituto per l’Endocrinologia e l’oncologia “Gaetano Salvatore (CNR), ItalyMonica Fedele, Istituto per l’Endocrinologia e l’oncologia “Gaetano Salvatore (CNR), Italy
Copyright © 2021 Islam, Ali, Naik, Eldaw, Decock and Kolatkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prasanna R. Kolatkar, pkolatkar@hbku.edu.qa