 Gerardo Ferrara
Gerardo Ferrara Giuseppe Argenziano
Giuseppe Argenziano- 1Anatomic Pathology Unit, Macerata General Hospital, Macerata, Italy
- 2Department of Dermatology, ‘Luigi Vanvitelli’ University School of Medicine, Naples, Italy
The “multidimensional” World Health Organization (WHO) classification 2018 of melanocytic tumors encompasses nine melanoma pathways (seven of which for cutaneous melanoma) according to a progression model in which morphologically intermediate melanocytic tumors are cosidered as simulators and/or precursors to melanoma. These “intermediates” can be subclassified into: i) a “classical” subgroup (superficial/thin compound: dysplastic nevus), which is placed within the morphologic and molecular progression spectrum of classical (Clark’s and McGovern’s) melanoma subtypes (superficial spreading and, possibly, nodular); and ii) a “non-classical” subgroup (thick compound/dermal: “melanocytomas”) whose genetic pathways diverge from classical melanoma subtypes. Such a progression model is aimed at giving a conceptual framework for a histopathological classification; however, routine clinicopathological practice strongly suggests that most melanomas arise de novo and that the vast majority of nevi are clinically stable or even involuting over time. Clinicopathological correlation can help identify some severely atypical but benign tumors (e.g.: sclerosing nevus with pseudomelanomatous features) as well as some deceptively bland melanomas (e.g.: lentiginous melanoma; nested melanoma), thereby addressing some ambiguous cases to a correct clinical management. The recently available adjuvant therapy regimens for melanoma raise the problem of a careful distinction between severely atypical (high grade) melanocytoma and “classical” melanoma: conventional morphology can guide an algorithmic approach based on an antibody panel (anti-mutated BRAF, BAP1, PRAME, ALK, TRKA, MET, HRAS-WT, ROS; beta catenin; R1alpha; p16; HMB45; Ki67), a first-line molecular study (identification of hot spot mutations of BRAF and NRAS) and an advanced molecular study (sequencing of NF1, KIT, BRAF, MAP2K1, GNAQ, GNA11, PLCB4, CYSLTR2, HRAS; fusions studies of BRAF, RET, MAP3K8, PRKCA); as a final step, next-generation sequencing can identify melanocytic tumors with rare genetic signatures and melanocytic tumors with a high tumor mutation burden which should be definitely ascribed to the category of classical melanoma with the respective therapeutic options.
Introduction
The histopathological diagnosis and classification of melanocytic skin tumors is probably the greatest conceptual and practical challenge in modern dermatopathology and is expected to rapidly evolve in the next future, with the WHO 2018 classification being the basis for the forthcoming studies (1). One major problem, however, is that the histopathological diagnosis itself is not based upon the search of a single (or a few), objective, and easily reproducible morphological diagnostic feature(s) but rather, it is born by a constellation of diagnostic criteria whose implementation, meaning, and relative weight considerably vary case by case and is responsible for a worrisome list of diagnostic pitfalls (Table 1). Thus, the histopathological diagnosis of melanocytic skin neoplasms, being based upon the simultaneous evaluation of several criteria, is no more than an assessment of probability and, as such, is often a matter of a sizable disagreement and inter-observer variability (2). In addition, and even more importantly, the time-honored “unifying concept of melanoma” (melanoma as a single entity evolving with a well-defined and repetitive “sequence of events”) (3) has been questioned, because both clinicopathological (4) and molecular studies (5) point toward the existence of melanocytic neoplasms of low malignant potential (putative low-grade melanocytic malignancies different from “classical” melanoma).

Table 1 Main settings of diagnostic difficulties in melanocytic skin neoplasms.
In order to face with these problems in routine histopathological practice, the WHO Working Group supports the use of descriptive and provisional terminology, i.e: i) “intraepidermal atypical melanocytic proliferation of uncertain significance (IAMPUS)”: a melanocytic neoplasms raising the differential diagnosis with melanoma in situ; ii) “superficial atypical melanocytic proliferation of uncertain significance (SAMPUS)”: a thin compound melanocytic neoplasm whose differential diagnosis is with early invasive, radial growth phase (thin non-mitogenic and non-tumorigenic) melanoma; iii) “melanocytic tumor of uncertain malignant potential (MELTUMP)”: a compound or dermal-based neoplasm whose differential diagnosis includes melanoma in vertical growth phase (typified by dermal mitotic figures and/or by dermal nests/sheets which are larger than the larger junctional nest) (6). Based on the these definitions, such a descriptive terminology applies to simulators (morphologically atypical nevi and deceptively bland melanomas) (2) as well as to biological “intermediates” (melanocytic neoplasms of low malignant potential) (4); and a strong suggestion is made that several neoplasms belonging to both categories may be in fact precursors to melanoma. The present review is aimed at giving some suggestions in the multidisciplinary approach based on the WHO 2018 classification.
The Pathways to Melanoma
The WHO 2018 classification of melanocytic tumors sets forth nine pathways to melanoma (6), seven of which being primary cutaneous (Table 2), by largely transposing a previously proposed “multidimensional” pathogenetic scheme based on: i) the role of ultraviolet (UV) radiation; ii) the cell (or tissue) of origin; iii) driving and/or recurrent genomic changes (7).
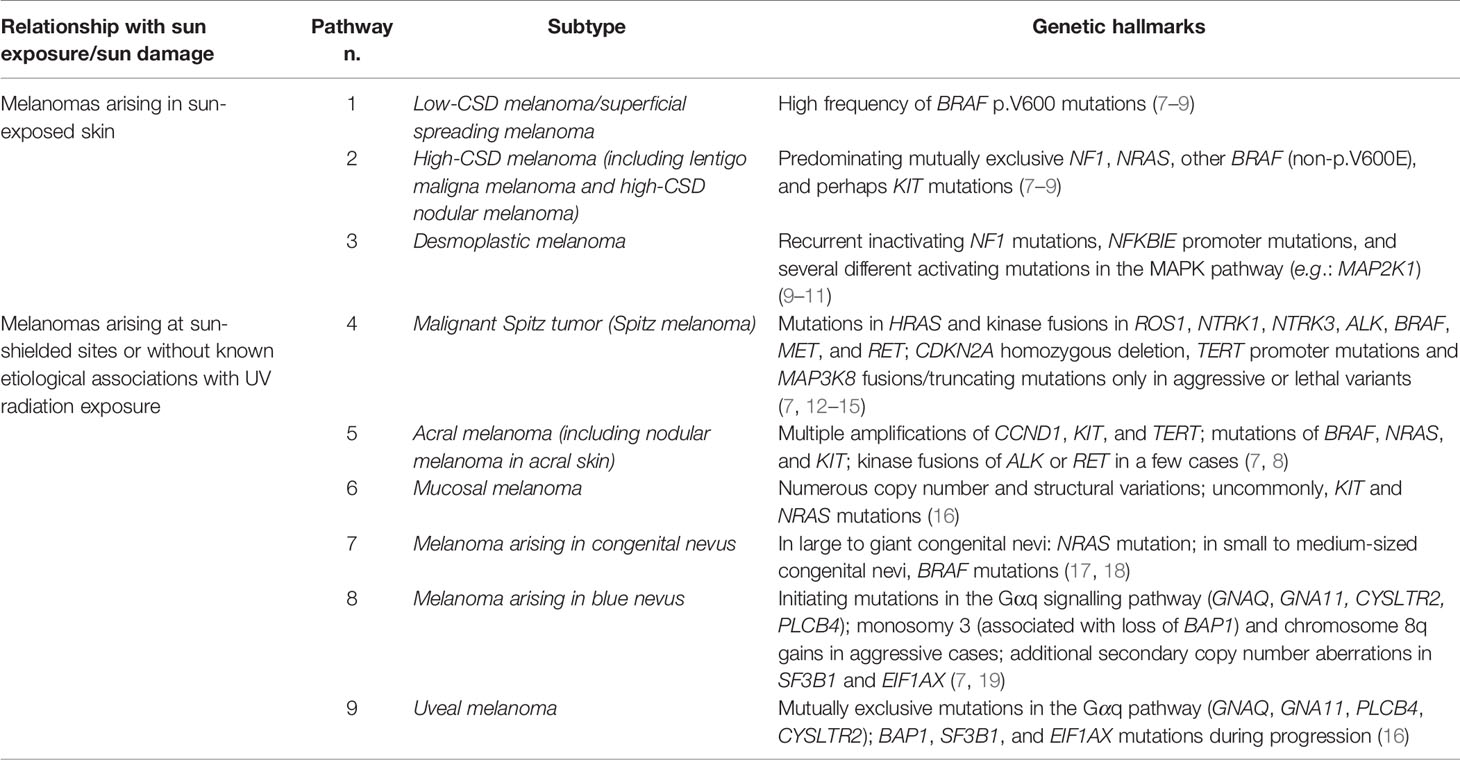
Table 2 The WHO 2018 classification of melanoma according to pathways.
The most common melanomas in Whites arise from epithelium-associated melanocytes in cutaneous sites with some degree of cumulative sun damage (CSD); these neoplasms are characterized by a high number of point mutations, mostly consisting in the so-called “UV signature” (cytosine to thymidine transitions at dipyrimidine sites); as a rule, the higher the degree of CSD the higher the tumor mutation burden (TMB) (on average: 30 mutations/megabase in high-CSD melanoma; 15 mutations/megabase in low-CSD melanoma) (10). Desmoplastic melanoma is a subtype of high-CSD characterized by a particularly high TMB (on average: 62 mutations/megabase) (11). The degree of CSD is related with the histopathological evidence of dermal solar elastosis, graded according to a three-tiered scale (grade 1: single elastic fibers; grade 2: bunches of fibers; grade 3 basophilic masses) (6).
The other subtypes of melanoma are UV-unrelated. The most common melanomas in non-White population arise from epithelium-asssociated melanocytes on acral skin (palms, soles, nail apparatus) or mucous membranes and are characterized by an early onset of major chromoscomal rearrangements, such as chromotripsis, with gene copy number changes, including multiple high-level amplifications (8). Spitz melanoma and melanomas arising from non-epithelium associated melanocytes (uveal melanoma, melanoma arising in blue nevus and in congenital nevus) also have a very low TMB, but lack the highly rearranged genomes of acral and mucosal melanomas (7, 20). The separation among melanomas with different TMBs is clinically relevant because the TMB may be predictive of response to immune checkpoint inhibitors (21, 22); parenthetically, the assessment of the TMB may be even proposed as a tool for the management of some cases of severely atypical MELTUMP (see below).
Next generation sequencing (NGS) studies have identified many recurrently mutated genes in melanoma, incuding well known genes (PTEN, MAP2K1-2, RB1) and recently identified genes (ARID2, PPP6C, RAC1, DDX3X, IDH1) (23, 24); however, most of these genes are involved in melanoma progression, rather than in melanoma initiation. Based on the presence of specific driver mutations, The Cancer Genome Atlas (TCGA) classified melanomas into four molecular subtypes: BRAF-mutated, RAS-mutated, NF1-mutated, and triple wild-type (lack of mutations in all three genes); among the latter were cases characterized by KIT mutations and by early onset of somatic copy number variations in terms of both gene amplifications in KIT, CCND1, CDK4, MITF, and TERT and gene deletion/loss-of-function of TP53 and CDKN2A (9).
TCGA molecular subtypes correspond to most cases of the classical (Clark’s and McGovern’s) (25, 26) types of melanoma and roughly identify melanoma pathways 1–3 of the WHO 2018 classification; melanoma arising in congenital nevus may be also genetically related to classical melanoma because they harbor multiple DNA copy number changes (17) superimposed to NRAS mutation. By contrast, the genetic profiles of Spitz melanoma (mutations in HRAS and kinase fusions in ROS1, NTRK1, NTRK3, ALK, BRAF, MET, and RET) (12, 13) as well as of melanoma arising in blue nevus (mutations in the Gαq signalling pathway) (19, 27) are not encompassed within the TCGA classification. Such cases will unlikely harbor numerous DNA copy number changes or a high TMB; thus they may be genetically considered as “non-classical” subtypes of melanoma.
Nevi as Potential Precursors to Melanoma
As a rule, all nevi may be virtually simulators of melanoma (and vice versa). In addition, the recent identification of the presence of shared genomic abnormalities between some melanomas and associated nevi has provided support for a potential role of some nevi (28) as both simulators and precursors. However, only some of the WHO 2018 pathways to melanoma may have their putative startpoint in nevi harboring the same mutation:
- Pathway 1: the vast majority of acquired nevi possess single driver mutations of either BRAF V600E or NRAS Q61R/L (29);
- Pathway 4: some Spitz nevi harbor HRAS mutation or translocations with kinase gene fusions involving ALK, ROS, RET, MET, and NTRK (12, 13).
- Pathway 7: NRAS mutation is most frequently observed in congenital melanocytic nevi (18);
- Pathway 8: some blue nevi harbor the GNAQ or GNA11 mutation (19, 27).
In contrast to melanomas, which acquire additional driver mutations, nevi usually enter a suppressive state of replicative senescence which is regulated by the tumor suppressor gene CDKN2A via its proteins, p14 and p16, and various transcriptional controls of the cell cycle (30, 31). Therefore, the above-listed mutations, as a single event, appear to be insufficient for melanomagenesis, but bear partially transformed melanocytes which may have an increased susceptibility to additional pathogenic mutation(s) (16). Such a progression model also encompasses neoplasms that have an intermediate number of pathogenetic mutations between nevi and melanomas: within this category, the WHO Working Group lists atypical junctional/thin compound neoplasms (dysplastic nevus and melanoma in situ) as well as papulonodular tumorigenic dermal proliferations (“melanocytomas”), and both categories are subclassified into low-grade and high-grade (16). Like Pathway 1 to melanoma, dysplastic nevi are associated with activating mutations of BRAF or NRAS (18, 29); additional mutation of the TERT promoter and, sometimes, hemizygous loss of CDKN2A are involved in the morphological progression to a “classical” (superficial spreading) melanoma in situ (32).
Many melanocytomas are instead dermal-based, thick, “combined” melanocytic tumors in which an activating mutation of BRAF (or, much less commonly, NRAS) is followed by a second genetic hit with expansion of a morphologically peculiar (“non-classical”) clone of melanocytes. Morphology of this secondary clone strictly depends on the type of second genetic hit: inactivation of the BAP1 (BRCA1-associated protein) gene is the hallmark of BAP1-inactivated nevus (BIN) (33, 34); gain-of-function mutations of CTNNB1 or loss of APC is found in deep penetrating nevus (DPN) (35, 36); loss-of-function of PRKAR1A is typical of pigmented epithelioid melanocytoma (PEM) (37, 38). However, several melanocytomas arise de novo (without a pre-exsisting common nevus): for example, cases of “pure” (non-combined) PEM are also genetically peculiar because often they harbor kinase (most commonly PRKA, but also NTRK1 and NTRK3) (38) fusions as the initiating event. Most of these dermal-based tumors are clinically stable; however, they can display various degrees of histopathological atypia (39–42). Increasing atypical histopathological features may correlate with increased risk of disease progression (43), but available data are too weak because of the relative rarity of these tumors and the need of long-term follow-up data. Since the initiating genetic change of such neoplasms is often an activating mutation of BRAF or NRAS, the three above-mentioned types of melanocytomas are placed within Pathway 1 of melanomagenesis, whose endpoint is superficial spreading melanoma; however, cases of superficial spreading melanoma dysplaying the genetic signature of the above-listed melanocytomas are exceedingly rare. Therefore, in real life such melanocytomas are probably unrelated to the vast majority of classical (Clark’s and McGovern’s) (25, 26) types of melanoma. Figure 1 shows a case of early superficial spreading melanoma over a combined BIN, with the malignant component being BAP1-positive, and being thus unrelated with the dermal melanocytoma.
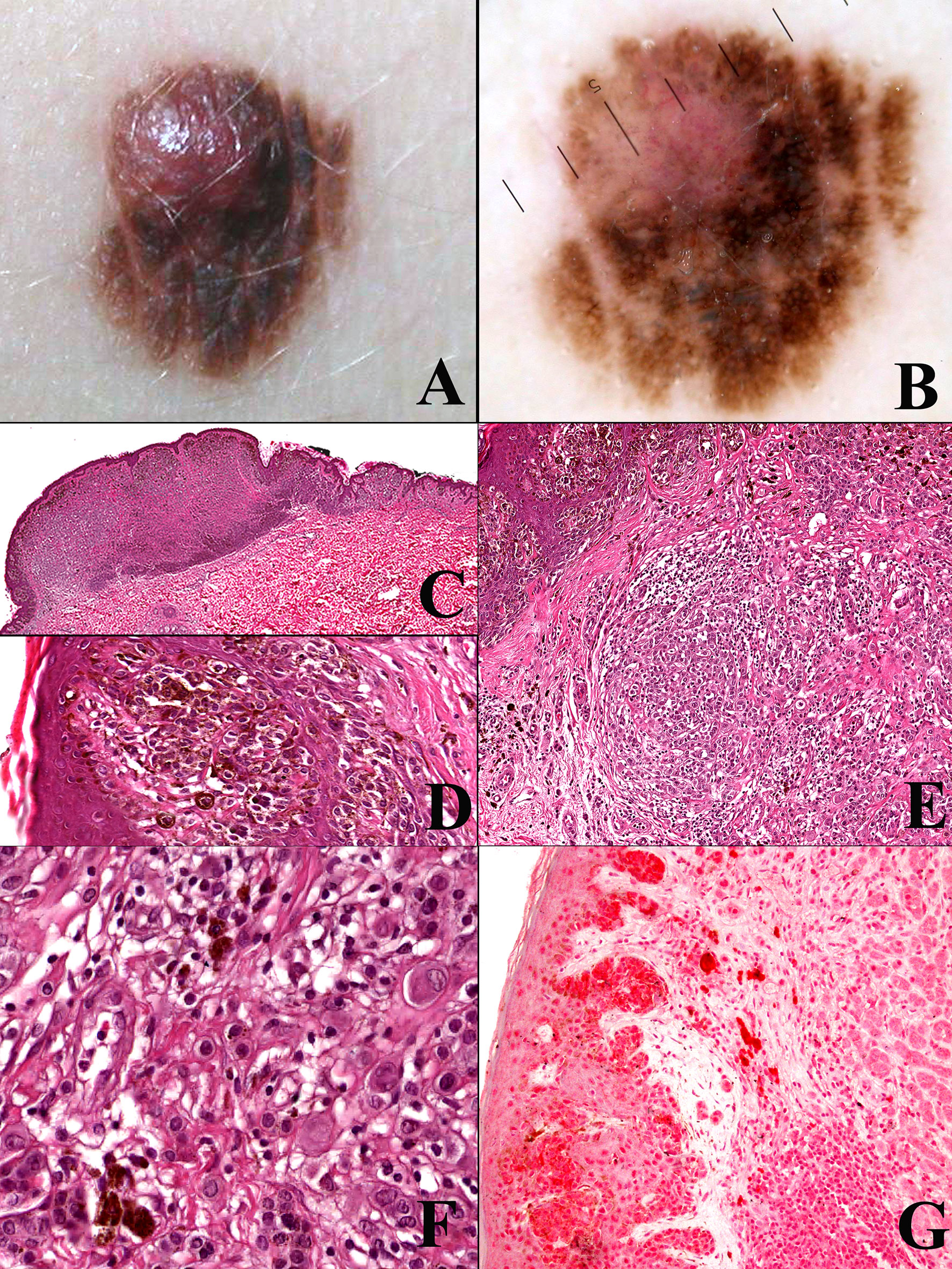
Figure 1 Man, 54 years; a severely atypical melanocytic tumor of the abdomen characterized by a flat pigmented area with an eccentric nodule (A). On dermoscopy, the flat area is typified by a prominent and focally irregular pigment network, whereas the nodular area is characterized by an atypical vascular pattern (B). Histopathologically, the tumor is strikingly asymmetric (C; hematoxylin–eosin, ×25), with a broad highly cellular “shoulder” composed by junctional melanocytes arranged in irregular nests and in single unit (D; hematoxylin–eosin, ×400); the severely atypical junctional component spans above the dermal nodule, the latter being characterized by a lymphoid cell infiltrate (E; hematoxylin–eosin, ×250) and nests of nevocytes intermingled with moderately pleomorphic epithelioid melanocytes with “inclusion-like” cytoplasms (F; hematoxylin–eosin, ×400); all the melanocytic components of this tumor were BRAFv600e mutated protein positive (not shown) and only the dermal epithelioid cell component disclosed loss of the nuclear expression of BAP1 (G; ×250). The tumor was interpreted as an early melanoma developing as a neoplastic progression of a common nevus and not as a progression of a BIN.
According to Table 2.06 of the WHO classification (16), even the other pathways to melanoma starting from the respective nevi have their own “melanocytomas”, namely: atypical Spitz tumor (Pathway 4), (atypical proliferative) nodule in congenital nevus (Pathway 7), and (atypical) cellular blue nevus (Pathway 8). It has been suggested that these entities share with BIN, DPN, and PEM the existence of a “spectrum within the spectrum” (43), namely: a set of atypical histopathological features which can be variously combined with each other, thereby bearing a “spectrum” of lesions with increasing risk of disease progression up to overtly malignant neoplasms. However, the WHO Working Group underlines that regarding Pathway 7, there is no convincing evidence that bona fide proliferative nodules in congenital nevi evolve into melanoma (44); and that regarding Pathway 8, a histopathological diagnosis of malignancy is straightforward for melanoma arising in blue nevus (45). Instead, regarding atypical Spitz tumor, it is acknowledged that there is the need of a “risk stratification” (46), evidently because neoplasms belonging to the Spitz lineage distribute along a spectrum of increasing histopathological atypia, with their malignant end being Spitz melanoma (14, 15).
Interestingly, atypical Spitz tumor shares at least with PEM a peculiar biological behavior, featuring a high incidence of nodal metastases with a very low incidence of distant metastases (41, 47): such as unique biological property that strongly favors ultrasonograpy monitoring over sentinel node biopsy in the clinical management of such cases (47, 48). Based on these data, PEM and atypical Spitz tumor might represent melanocytic tumors of low-grade (mostly lymphotropic) malignancy different from “classical” melanoma: it seems thus reasonable to include atypical Spitz tumor into the “melanocytoma” rubric, as suggested since the beginning (49). Interestingly enough, the list of putative low-grade melanocytic malignancies with a peculiar genetic and morphologic profile has been growing for the last years and has thus been increasingly supporting the concept itself (50–53). An example of CRTC1-TRIM11 (50) fused melanocytoma is provided in Figure 2; like several other melanocytomas, such a putatively low-grade malignant melanocytic tumor does not likely progress from a common nevus.
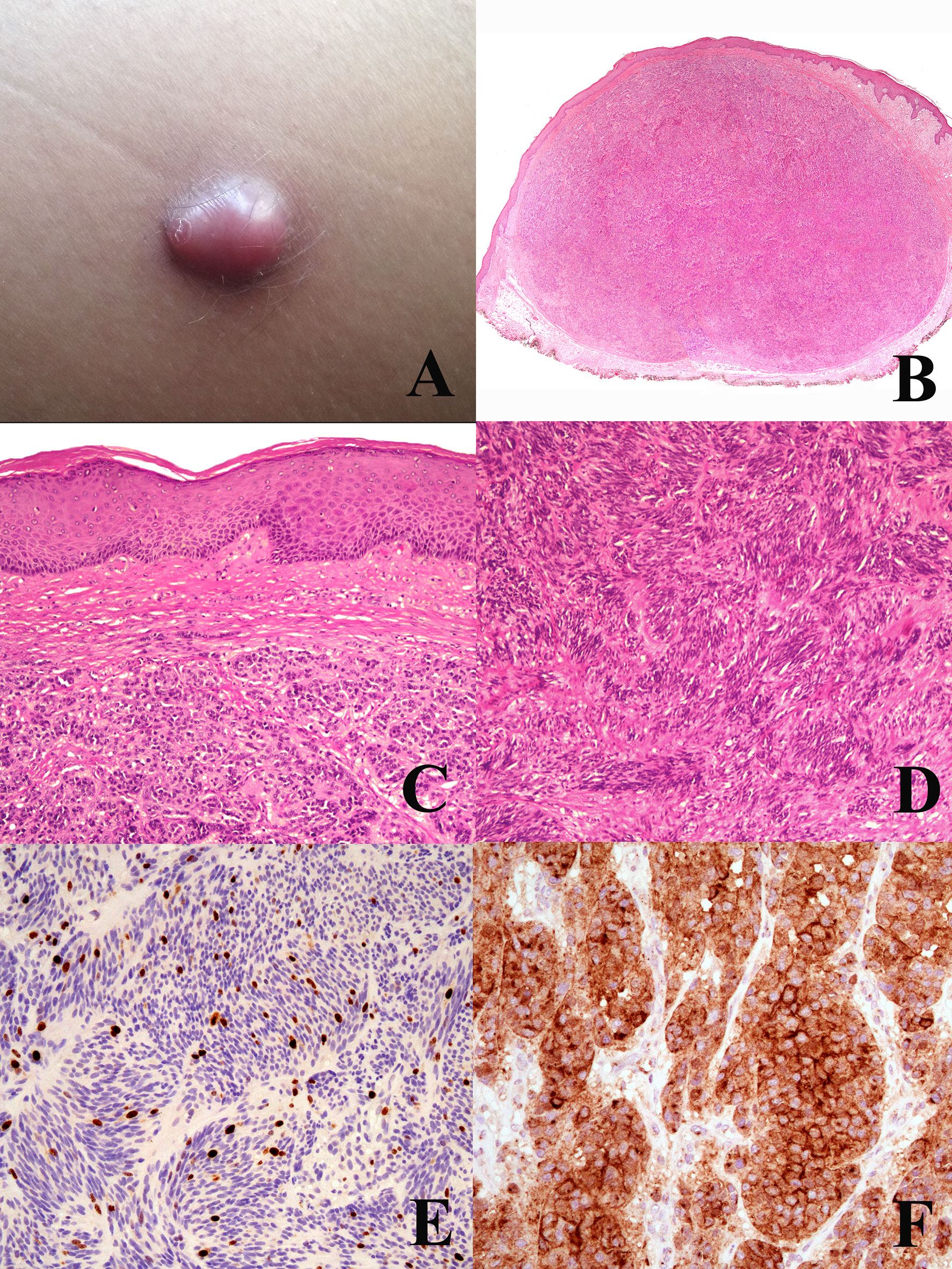
Figure 2 Woman, 44 years; a reddish nodule of the thigh (A). Histopathology shows an expansile dermal nodule (B hematoxylin–eosin, ×25) composed by nests of epithelioid cells (C hematoxylin–eosin, ×250) and fascicles of spingle cells separated by thin fibrotic bands (D hematoxylin–eosin, ×250); the proliferation rate (Ki67-positive cells) is 5%, with no clusters of proliferating cells (E; ×250); the tumor cells are diffusely positive for TRKA (F; ×400). Molecular studies allowed to exclude the possibility of a dermal clear cell sarcoma and to establish a diagnosis of CRTC1-TRIM1 fused melanocytoma. Courtesy of Dr. Arnaud de la Fouchardière, Lyon, F.
For the above, intermediate melanocytic tumors may be subclassified into: i) a “classical” subgroup (dysplastic nevus and melanoma in situ), which is placed within the morphologic and molecular progression spectrum of “classical” melanoma subtypes (superficial spreading and, possibly, nodular; WHO 2018 Pathway 1); and ii) a “non-classical” subgroup (“melanocytomas”) whose genetic pathways diverge from “classical” melanoma subtypes. Among the latter are probably low-grade melanocytic malignancies whose list has been increasing for the last years and whose risk stratification needs a careful and systematic approach (48).
Not surprisingly, neoplasms belonging to the WHO 2018 intermediate category are prone to a lower interobserver agreement and are classified as ambiguous by multiple pathologists. Thus, the intermediate rubric also encompasses the provisional categories IAMPUS, SAMPUS, and MELTUMP (6), whose definitions (see above) imply a “subjective” diagnostic uncertainty, rather than a morphologic subset of melanocytic neoplasms. Immunohistochemical and genetic investigations may help classify the WHO 2018 provisional entities into the proper subgroup of melanoytic tumors: this goal is of paramount importance because the “provisional” terminology should be adopted as less as possible (48).
The WHO 2018 Progression Model: What Matters in Routine Practice
The WHO 2018 progression model is aimed at giving a framework for a histopathological classification; it is therefore a relatively simplifed linear scheme which must be accepted with the awareness that not only are there multiple pathways to melanomagenesis but also that some of the intermediate steps may be bypassed and that other non-linear pathways exist. The most frequent and most important non-linear pattern is by far melanoma de novo of the “classical” type. In a meta-analysis carried out by Pampena et al. on 38 observational cohort and case–control studies, only 29.1% of melanomas likely arose from a preexisting nevus and 70.9% arose de novo (54). Studies on nevus-associated melanoma based on histopathology alone may have several biases: a benign component may be absent in the tissue levels examined or, else, it may be completely destroyed by the malignant growth; on the contrary, peripheral or deep areas of melanoma may have a deceptive “nevus-like” appearance (“pseudomaturation”). Dermoscopy and dermoscopic digital monitoring can help differentiate between melanoma characterized by a homogeneous remodeling of the tumor (likely melanoma de novo; Figures 3A–D) and melanoma characterized by focal changes (“dermoscopic island”; likely nevus-associated melanoma) (55) (Figures 3E–H). An early melanoma may be missed if grossing of the specimen is carried out blind to the clinicodermoscopic features of a given melanocytic lesion (56). Dermoscopic digital monitoring also shows that the overwhelming majority of nevi are stable and are more likely to involute according to one of the following: i) a fading pattern (progressive replacement of the nevus by normal skin); ii) a haloed pattern (progressive replacement of the nevus by centripetal extension of a peripheral white vitiligo-like ring); iii) a regression-like pattern (replacement of the nevus by dermoscopic regression structures (peppering, white scarlike ares) (57). The regression-like pattern is seldom documented with dermoscopic monitoring, but is peculiar enough to allow a clinicopathological differential diagnosis between melanoma with regression and its main benign simulator, the so-called “sclerosing nevus with pseudomelanomatous features” or “compound nevus with regression-like fibrosis” (58, 59). The latter is a kind of “chronically recurrent nevus” following chronic unnoticed trauma, and has been described mainly, albeit not exclusively, in the convex area of the back of young to middle aged patients. Histopathologically, this neoplasm is usually large and asymmetric with a typical “trizonal” pattern featuring: i) an irregular junctional component with irregular epidermal hyperplasia and areas of prevailing single cell proliferation; ii) a significant area of dermal sclerosis with architecturally atypical melanocytic nests; iii) a residual, bland-appearing nevus tissue (very often with congenital nevus-like features) around and deep into the cicatricial tissue (Figure 4). The presence of a clear-cut benign dermal component is the main clue to the diagnosis, because regressing melanoma is usually not associated with a nevus. Such a severely atypical melanocytic tumor, in our experience often cautiously diagnosed as MELTUMP, can be indeed diagnosed with confidence when considering the proper clinicopathological setting; together with the many nevi in special sites (nevi with site-related atypia), it is an example of histopathological atypia probably unrelated with a signficantly higher risk of progression toward melanoma. This entity also underlines the role of clinically identifiable “environmental modifiers” (trauma, epilation, acute sun exposure) which may increase the histopathological features of atypia in nevi (2, 34) presumably without any impact in melanomagenesis.
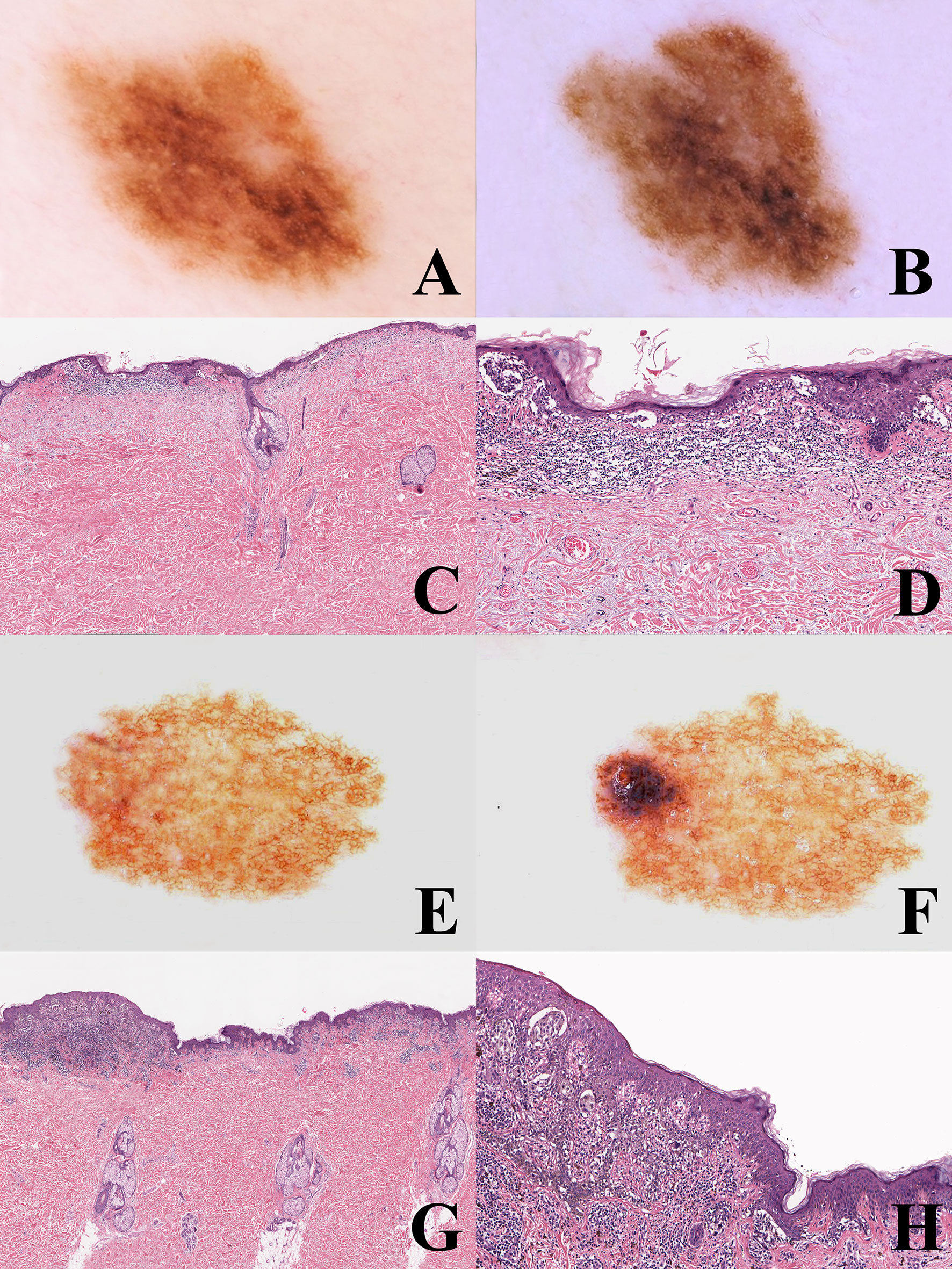
Figure 3 (A–D) man, 53 years; a pigmented lesion of the back with a slightly irregular pigment network (A); after six months, the tumor appears as uniformly enlarged, with increasingly irregular pigment network (B). Histopathologically, the tumor is strikingly asymmetric (C; hematoxylin–eosin, ×25), with a lichenoid infiltrate at the base of its more severely atypical half (D; hematoxylin–eosin, ×100). Even if the histopathological picture might be interpreted as a melanoma in situ developing in the background of a dysplastic nevus, the homogeneous remodeling of the tumor documented with dermoscopic digital monitoring favored the diagnosis of melanoma de novo. E-H: Woman, 35 years; a pigmented lesion of the back with a thin and regular pigment network at the baseline (E); after eight months, a raised bluish areas is evident at the periphery (“dermoscopic island”) (F). Histopathologically the tumor shares with the previous case the striking asymmetry (G) hematoxylin-eosin, ×25) and the presence of a lichenoid infiltrate at the base of its more severely atypical half (H) hematoxylin-eosin, ×100). However, dermoscopic digital follow up data clarify that this case likely represents an early melanoma in situ over a junctional dysplastic nevus.
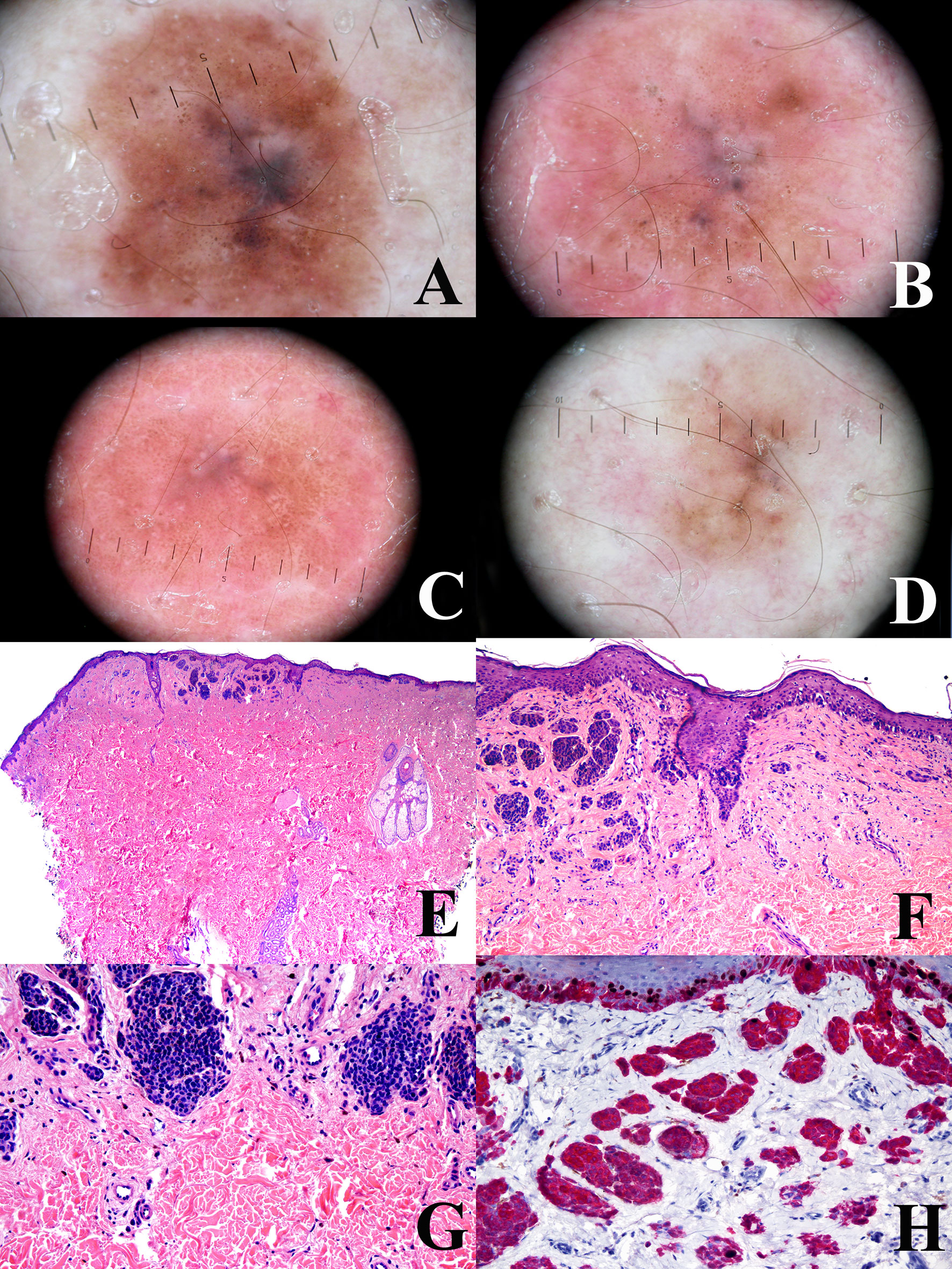
Figure 4 Man, 38 years at the time of the surgical excision of a pigmented lesion of the scapular area; at the baseline, the tumor shows a a relatively regular peripheral pigment network associated with slightly eccentric globules and a central bluish area (A) the tumor shows a progressive and relatively symmetric fading after 1 year (B), four years (C), and 6 years (D). The tumor discloses a “trizonal” histopathological pattern (E; hematoxylin–eosin, ×25), with an atypical junctional component, a scar-like dermal thickening (F; hematoxylin–eosin, ×100) and a very bland-appearing deep dermal component (G; hematoxylin–eosin, ×100); the proliferation rate (Ki67-positive dermal melanocytes, evaluated with a KI67/MART1 double stain) is very low (H; ×250). These histopathological features are consistent with the so-called “sclerosing nevus with pseudomelanomatus features”. Such a histopathological diagnosis is in keeping with the slowly progressive and relatively symmetrical involution of the tumor, as documented with dermoscopic digital monitoring. Clinical images provided by Dr. Luigi Ligrone, Salerno, I.
As also underlined by the WHO Working Group in a paper published shortly after the 2018 Classification, the risk of an individual nevus progressing to melanoma has been estimated to be in the order of one in 33,000 or less per year (60). Therefore, from a practical point of view, we can conclude that:
1. the vast majority of nevi are, at worse, clinicopathological simulators and not precursors to melanoma;
2. besides esthetic reasons, indication to their excision is solely related to the impossibility to rule out melanoma on clinical grounds alone;
3. with the possible (but not universally accepted) exception of medium (1.5–20 cm) and large/giant (>20 cm) congenital nevi, which carry a definite size-related melanoma risk [up to 15% (61)], by no means the excision of a nevus must be viewed as a tool of primary prevention (“prophylactic excision”).
These statements also apply to dysplastic nevus and dysplastic nevus syndrome. The WHO Working Group defines dysplastic nevus as a clinically atypical, histopathologically benign junctional or compound melanocytic tumor, >4 mm in breadth on fixed sections (>5 mm clinically), with architectural disorder plus cytological atypia (62). The former is typified by irregular (horizontally oriented, bridging adjacent rete, and/or varying in shape and size) and/or dyscohesive nests of intraepidermal melanocytes plus increased density of non-nested junctional melanocytes (e.g. more melanocytes than keratinocytes in an area ≥1 mm2); the latter is evaluated on the basis of the highest degree of cytological atypia present in more than a few melanocytes as low grade (nuclei ≤1.5× larger than basilar keratinocytes, with small or absent nucleoli and uniformly hyperchromatic or dispersed chromatin, and with “random” variation in size and shape) or high grade (nuclei ≥ larger than basilar keratinocytes, with prominent nucleoli and coarse or peripherally condensed chromatin, and with slightly confluent variation in size and shape) (62). It is stated that nevi with high-grade dysplasia and/or with additional genetic alterations such as TERT promoter mutation should be considered for complete excision (62); this implies that a nevus with high-grade displasia needs no re-excision if already excised with clear margins.
Some studies are reported in which the degree of dysplasia is related with an increased melanoma risk (63–66); however, with the sole exception of a retrosective review considering the personal history of melanoma (66), these studies were histopathologically based, i.e.: they did not take into account the clinical features of risk of the individual patients (familial history of melanoma, skin type, personal history of sunburns, number of nevi, number of clinically atypical nevi). Thus, from a practical point of view, a histopathological diagnosis of dysplastic nevus must be evaluated in the clinical context in order to assess the risk of the individual patient to develop a melanoma; and, since genetic findings are relatively inconsistent to date (62), the diagnosis of dysplastic nevus syndrome (aka: Familial Atypical Multiple Mole and Melanoma, FAMMM; OMIN #155600) is largely based on clinical criteria, i.e.: number of nevi, number of clinically atypical and/or large nevi, personal/famlial history of melanoma (64, 66).
Excluded from the rubric of dysplastic nevus is lentiginous nevus, because being very common, unassociated with a relevant risk of progression to melanoma, and prone to poor diagnostic riproducibility (67). Lentiginous nevus is defined as a benign, junctional, or compound melanocytic tumor, <4 mm in width (on fixed sections), usually symmetrical but with poorly defined borders, with increased density of regularly spaced, non-nested junctional melanocytes around the tips and sides of the rete ridges, with no to mild cytological atypia and minor/variable features also seen in dysplastic nevi (67). These definitional features must be kept in mind because not uncommon in clinical practice are broad and irregular lentiginous melanocytic proliferations of the trunk and the proximal limbs, mostly found in elderly patients, which are probably the clinicopathological counterpart of lentigo maligna on non-chronically sun-exposed skin and are called lentiginous melanoma (68, 69). Dermoscopic digital monitoring of some of these lesions has demonstrated a homogeneous remodelling over many years, thereby suggesting that these are very slow-growing melanomas de novo and not the evolution to melanoma from lentiginous nevi (Figures 5A–E). In our experience on lentiginous melanoma, histopathological criteria alone are often weak and may result in a provisional diagnosis of IAMPUS or SAMPUS; the clinical picture of these cases is, however, very often unequivocal for melanoma and must be therefore incorporated into the decision-making process regarding their management.
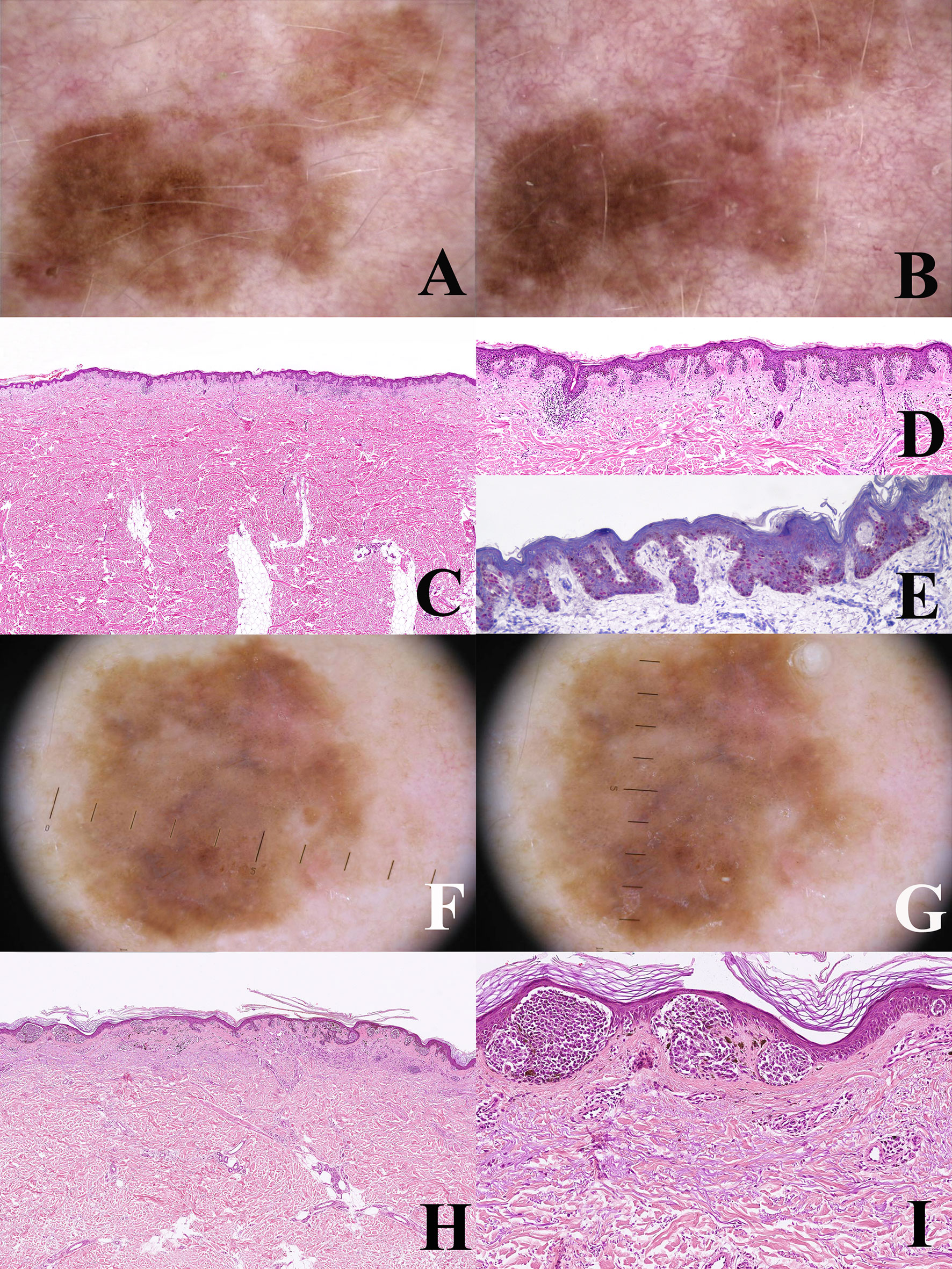
Figure 5 (A–E) Man 52 years. Dermoscopy of a large pigmented lesion of the back with an irregular pigment network at the baseline (A) after one year, the lesion shows an increase in size with a homogeneous remodeling and a more prominent pigment network (B) such a slow clinical evolution is akin to a lentigo maligna of chronically sun-exposed skin and virtually excludes a diagnosis of nevus. Histopathologically, the tumor has a dysplastic nevus-like silhouette (C; hematoxylin–eosin, ×25) but is severely atypical because of the striking predominance of tightly packed single melanocytes at the junction (D; hematoxylin–eosin, ×100). PRAME immunostain shows a strong and diffuse nuclear positivity in intraepidermal melanocytes (E) ×250), as expected in melanoma. Clinicopathological features of the lesion are diagnostic for lentiginous melanoma in situ. (F–I) Man, 59 years. A large pigmented lesion of the abdomen, dermoscopically characterized by tiny eccentically grouped globules and structureless peripheral areas (F) after seven months the peripheral strucureless areas show a clear-cut increase in size (G). Histopathologically there are some areas with a dysplastic nevus-like silhouette, but the epidermis is largely atrophic (H; hematoxylin–eosin, ×25) and junctional nests are very large and irregular (I; hematoxylin–eosin, ×250). These features suggest a diagnosis of melanoma in situ with a focally “nested” architecture.
Nested melanoma (of the elderly) is another example of deceptively bland melanoma (70) whose recognition often depends on a thorough clinicopathological correlation. Like lentiginous melanoma, it is often removed from the trunk and limbs in elderly patients as being large, growing and dermoscopically atypical flat pigmented tumor (71); histopathology features a junctional nesting which is not invariably irregular enough to allow a confident histopathological diagnosis; thus, the result is often a provisionla diagnosis of high-grade dysplasia, IAMPUS, or SAMPUS which, however, is not consistent with the clinical picture. Dermoscopic features of nested melanoma (70) suggest that it conceivably a slow growing melanoma de novo, rather than a melanoma evolving from a nevus (Figures 5F–I).
A Management-Based Approach: The MPATH-Dx System and Beyond
A histopathological diagnosis is aimed at giving a Mutidisciplinary Team the main (albeit not the sole) information for the clinical management. However, such an approach centered on histopathology having some major limitations, more or less explicitly underlined by the WHO Working Group, namely:
1. the diagnostic terminology varies depending on the individual cultural background and on local giudelines (72);
2. the diagnostic interobserver reproducibility is poor even among experts (73);
3. all the available evidence-based clinical guidelines are set upon a dichotomic diagnostic approach (all melanocytic tumors are either nevi or melanomas) and upon a unifying concept of melanoma (all melanocytic malignancies have the same biological behavior which can be predicted on the basis of a universally applicable set of histopathological parameters) (3).
In 2014, the Melanocytic Pathology Assessment Tool and Hierarchy for Diagnosis (MPATH-Dx) schema was proposed in an effort to reduce uncertainty and offer guidelines, mostly for melanocytic tumors different from melanoma (the “classical” melanocytic malignancy with its own evidence-based guidelines) (74): notably, the original schema excluded some melanocytic tumors (pigmented spindle cell; Spitz; epithelioid blue; cellular blue; deep penetrating/plexiform spindle cell) from Class 1 (no apparent risk), thereby anticipating the WHO 2018 concept of intermediate melanocytic tumors. The MPAT-Dx system stratified melanocytomas into four classes (Classes 2 to 5) of melanocytic tumors, with the first two being discriminated on the basis of the degree of histopathological atypia, and the last two discriminated on the basis of Breslow’s thickness. The latter criterion, however, should not be applied to melanocytomas, because they are morphologically, genetically, and biologically different from “classical” melanoma with its “classical” prognostic parameters.
In order to specifically address the clinical management of dermal-based tumorigenic “intermediate” melanocytic tumors, practical recommendations have been delivered by the ESP, the EORTC, and the EURACAN (48). Morphological evaluation of these tumors is based on the evaluation of a list of general criteria, both architectural (diameter >6 mm; asymmetry; epidermal effacement; ulceration; high dermal cellularity; tumor clones; loss of grenz zone; absence of vertical “maturation”; expansile nodule formation; destriucive growth pattern; deep subcutaneous extension; pagetoid spread) and cytological (cellular pleomorphism; macro-eosinophilic nucleoli; variable density of nuclear chromatin; irregular nuclear membrane; >1 mitosis/mm2; overlapping nuclei; tumor necrosis). Melanocytomas are then stratified into “low-grade” (few criteria present) and “high grade” (roughly up to half of them present), with excision margins estimated as adequate at 2 mm for the former and at 5–10 mm for the latter. Since a 2-mm excision margin is recommended for every melanocytic tumor, no further excision is required for low-grade melanocytomas. Pigmented epithelioid melanocytoma is by definition an intermediate-high-grade tumor; sentinel node staging is recommended only for “unclassified atypical dermal tumors” and for cases in which a Spitz melanoma cannot be ruled out; cases labeled as MELTUMP should be managed as per melanoma of the same thickness.
The ESP-EORTC-EURACAN recommendations concerning Spitz melanoma should be applied also on the basis of the recent observation that a “spitzoid” morphology is not invariably associated with a “Spitz” genetic signature (14, 15); in other words, malignant Spitz tumor (Spitz melanoma) is different from “spitzoid” melanoma, which can be regarded as a melanocytic malignancy with “Spitz-like” morphology but genetically ascribed to a “classical” melanoma subtype because of the presence of a specific driver mutation, or numerous DNA copy number changes, or a high TMB. Figure 6 illustrates the clinicopathological features of an ulcerated melanocytic malignancy histopathologically composed of large epithelioid cells with Spitz-like features, but immunohstichemically typified as a “classical” melanoma because of its immunohistochemical positivity to the anti-BRAF mutated protein VE1 antibody. Parenthetically, PEM-like (75, 76) and DPN-like melanomas (77, 78) might be differentiated from their “melanocytoma counterpart” based on immunohistochemical and/or genetic findings akin to “classical” melanoma.

Figure 6 Woman, 22 years. An ulcerated nodule of the right flank (A) dermoscopically characterized by keratoacanthoma-like features with vessels surrounded by a white halo (B). Histopathologically, the tumor has an irregularly nodular, exophytic silhouette with an epidermal “collarette”, a superficial crust, and a “brisk” inflammatory infiltrate in the dermis (C; hematoxylin–eosin, ×25); the superficial nests are very irregularly confluent with no sharp circumscription from the overlying epidermis (D; hematoxylin–eosin, ×250); dermal melanocytes show a “spitzoid” morphology, with spindle (E; hematoxylin–eosin, ×400) and epthelioid (F; hematoxylin–eosin, ×400) cells, both with reatively abundant and eosinophilic cytoplasms. In spite of the severe architectural atypia, the proliferation rate of the tumor (Ki67-positive dermal melanocytes) is low (G) ×250); however, the tumor is not an atypical Spitz tumor, but a classical nodular melanoma because it is positive to the antibody anti-BRAFv600e-mutated protein (H) ×250).
Based on the above, a new problem is thus rising in dermatopathology, i.e.: the differential diagnosis between severely atypical melanocytoma and melanocytoma-like “classical” melanoma. This is not merely a speculative problem, because both a severely atypical melanocytoma and a melanocytoma-like “classical” melanoma will likely spread to the regional nodes, but only the latter will be candidates to sentinel node biopsy and, possibly, to an adjuvant therapy with BRAF-inhibitors or with immune checkpoint inhibitors (79, 80). This means that underdiagnosing a “classical” melanoma as a severely atypical melanocytoma may address the patient to an improper wait-and-watch strategy. Many melanocytomas (comprising Spitz tumors) currently lack an identifiable genetic “signature”; by definition, however, they lack BRAF-mutation and a high TMB which are predictive parameters for neoadjuvant therapy (79, 80). Thus, the differential diagnosis between a severely atypical melanocytoma with no known genetic signature and a classical “melanocytoma-like” melanoma may be approached by looking for predictive (rather than diagnostic) paramenters; the same might apply for cases provisionally labeled as MELTUMP or as unclassified atypical dermal lesion (48).
A Therapy-Oriented Diagnostic Approach
When dealing with an atypical melanocytic tumor of the skin, the first step can be the differential diagnosis between a “classical” type of melanocytic tumor and a “melanocytoma” (comprising Spitz tumor). Immunohistochemistry can assist such a differential diagnosis as follows:
- The anti BRAF-mutated protein VE1 antibody identifies the subset of melanocytic tumors of the “classical” type harboring the BRAFv600e mutation (or a “combined” melanocytoma) (48, 81);
- The immunostain for BAP1 can document loss of the consitutive nuclear immunoreactivity in BAP1-inactivated melanocytic tumors (33, 34);
- The anti PRAME immunostain can assist the differential diagnosis between benign and malignant “traditional” melanocytic tumors (82); in our experience, particularly for lentiginous neoplasms and for the differential diagnosis between congenital nevus and nevoid melanoma;
- The anti-ALK, anti-TRKA, anti-MET, anti-HRAS-WT, and anti-ROS1 antibodies identify the subset of melanocytic tumors of the Spitz lineage with the respective kinase gene changes (48, 83, 84);
- The anti-beta catenin immunostain identifies the aberrant nuclear positivity definitional for DPN and related tumors (36);
- Tha anti-R1alpha can document loss of constitutive nuclear immunoreactivity in PEM with inactivating mutation or epigenetic inactivation of PRKAR1A (85).
An immunohistochemical panel aimed at a risk stratification can encompass:
- p16, which may disclose uneven immunoreactivity or “clonal” loss as an atypical feature (2, 48);
- HMB45, which may be unevenly distributed, with loss of the “gradient” pattern seen in benign tumors (2);
- Cell cycle-related protein Ki67, which may show a high rate of expression and/or “proliferative clusters” in atypical lesions (2).
The traditional four-probe (targeting MYB, RREB, Cep6, and CCND11) plus the anti-CDKN2A/Cep9 dual probe FISH examination may help refine the risk stratification of melanocytic tumors as recently proposed (86).
If morphology and immunohistochemistry are not contributory in assigning the melanocytic tumor to a given lineage, molecular analysis guided by morphology may be implemented as follows:
- Identification of hotspot mutations of BRAF (codon 600) and NRAS [exon 2 (odons 12, 13), exon 3 (codons 59, 61), and of exon 4 (codons 117, 146)];
- Sequencing techniques for the following: NF1, KIT (exons 11, 13, 17, and 18), BRAF (rare mutations), NRAS (rare mutations), and MAP2K1 (exons 2 and 3; in-frame deletion) for “classical” melanocytic tumors; GNAQ (exons 4 and 5), GNA11 (exons 4 and 5), PLCB4, and CYSLTR2 for dendritic melanocytic tumors (WHO 2018 Pathways 8 and 9); HRAS (exons 2 and 3) for a subset of Sptz tumors; TERT promoter for a subset of aggressive malignencies (some characterized by a 'Spitz-like' morphology);
- Fluorescence in situ hybridization (FISH) or reverse transcriptase polymerase chain reaction (RT-PCR) examination for fusions involving: BRAF and RET for Spitz tumors; MAP3K8 for morphologically malignant epithelioid cell Spitz neoplasms (87, 88); PRKCA for PEM.
As per ESP-EORTC-EURACAN guidelines, if the immunohistochemical screening implies additional procedures, immuno-positive cases (of Spitz neoplasms) should be confirmed for the respective genomic aberration by molecular examinations (48); this is, however, a theroretically uncommon scenario.
As a final step for an approach akin to tumor-agnostic therapy, NGS analysis can help identify melanocytic tumors with “rare” genetic signatures, and—even more important—melanocytic tumors with a high TMB which should be definitely ascribed to the category of classical melanoma with the relative therapeutic options. Specialized referral centers must be involved for sequencing, fusion studies, and NGS examination (48).
A visual summary of the above-proposed algorithmic diagnostic approach is given in Figure 7.
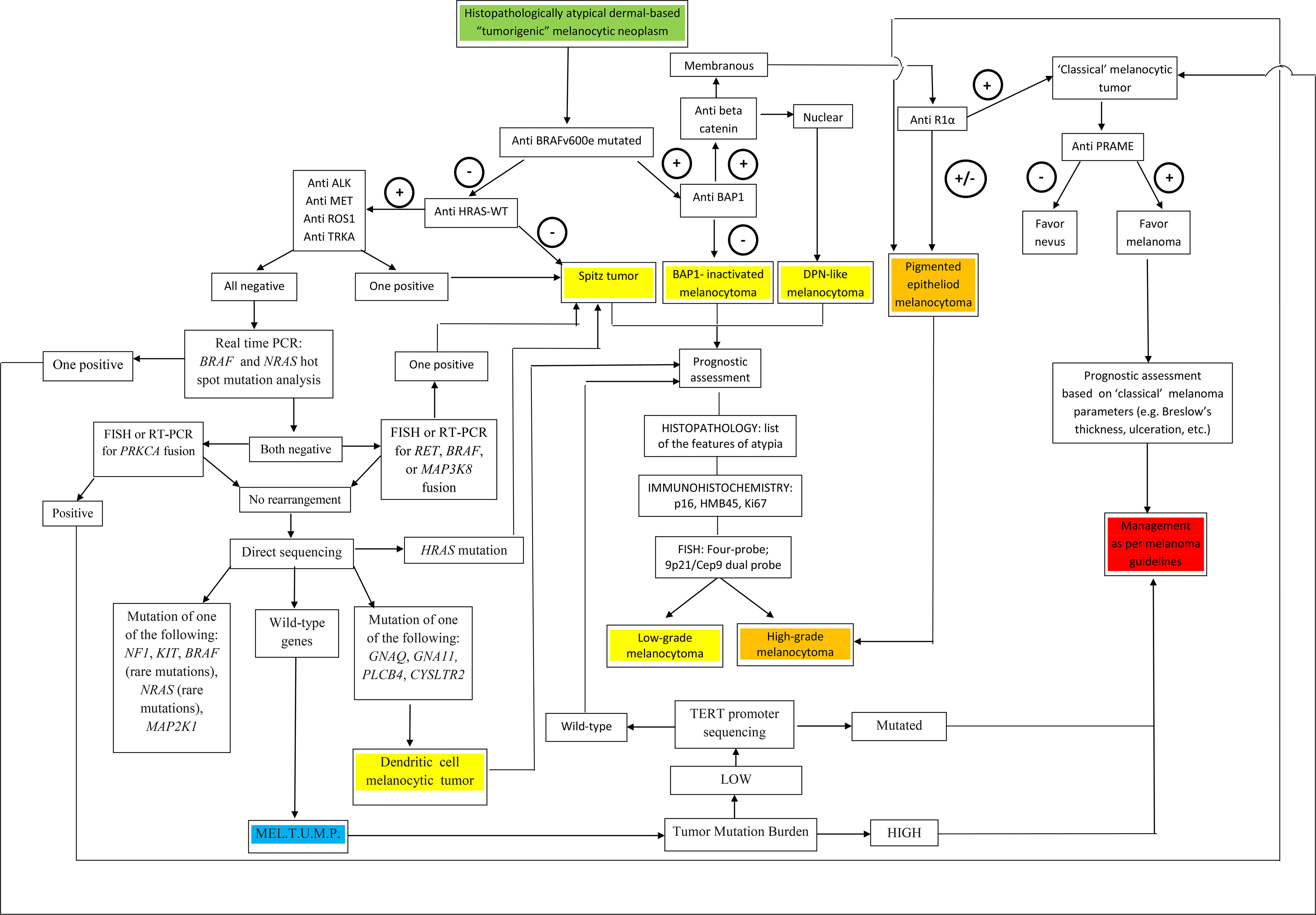
Figure 7 A flow chart illustrating a therapy-oriented morphomolecular approach to atypical dermal-based tumorigenic melanocytic neoplasms. Of paramount importance are: i) the distinction between melanocytomas (recognized as such by specific genetic signatures) and melanocytic tumors of uncertain malignant potential (MEL.T.U.M.P.; provisionally defined as tumors with unknown driver mutations); ii) among melanocytomas, the distinction between low-grade and high-grade tumors; iii) among MELTUMP, the distinction between tumors with a low tumor mutation burden and tumors with a a high tumor mutation burden, the latter being best managed as per “classical” melanoma.
Take-Home Message
The traditional “dichotomic” (benign vs malignant) view of melanocytic tumors and the concept of melanoma as a “unique” clinicopathological entity no longer fit with the routine diagnostic approach. Along with “classical” (Clark’s and McGovern’s) subtypes of melanoma, other melanocytic malignancies, each charcaterized by peculiar biological behavior probably exist, must be distinguished from “classical” melanoma subypes and require specific clinical guidelines. Clinicopathological correlation can allow both reducing the histopathological diagnostic uncertainty and addressing patients to a proper management.
Author Contributions
Both authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Elder DE, Massi D, Scolyer RA, Willemze R. Who Classification of Skin Tumours, 4th Edition. Lyon: IARC (2018).
2. Ferrara G. “The Histopathological Gray Zone”. In: Argenziano G, Lallas A, Longo C, Moscarella E, Kyrgidis A, Ferrara G, editors. Cutaneous Melanoma: A Pocket Guide for Diagnosis and Management. London, UK: Academic Press (2017). p. 155–89.
3. Ackerman AB. Malignant Melanoma: A Unifying Concept. Hum Pathol (1980) 11:591–5. doi: 10.1016/s0046-8177(80)80069-4
4. Cerroni L, Barnhill R, Elder D, Gottlieb G, Heenan P, Kutzner H, et al. Melanocytic Tumors of Uncertain Malignant Potential: Results of a Tutorial Held at the XXIX Symposium of the International Society of Dermatopathology in Graz, October 2008. Am J Surg Pathol (2010) 34:314–26. doi: 10.1097/PAS.0b013e3181cf7fa0
5. Harbst K, Staaf J, Lauss M, Karlsson A, Måsbäck A, Johansson I, et al. Molecular Profiling Reveals Low- and High-Grade Forms of Primary Melanoma. Clin Cancer Res (2012) 18:4026–36. doi: 10.1158/1078-0432.CCR-12-0343
6. Elder DE, Barnhil R, Bastian BC, Cook MG, de la Fouchardière A, Gerami P, et al. “Melanocytic Tumour Classification and the Pathway Concept”. In: Elder DE, Massi D, Scolyer RA, Willemze R, editors. Who Classification of Skin Tumours, 4th Edition. Lyon, F: IARC (2018). p. 66–71.
7. Bastian BC. The Molecular Pathology of Melanoma: An Integrated Taxonomy of Melanocytic Neoplasia. Annu Rev Pathol (2014) 9:239–71. doi: 10.1146/annurev-pathol-012513-104658
8. Curtin JA, Fridyland J, Kageshita T, Patel HN, Busam K, Kutzner H, et al. Distinct Sets of Genetic Alterations in Melanoma. N Engl J Med (2005) 353:2135–47. doi: 10.1056/NEJMoa050092
9. Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell (2015) 161:1681–96. doi: 10.1016/j.cell.2015.05.044
10. Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pomputtapong N, Wu C, et al. Exome Sequencing Identifies Recurrent Mutations in NF1 and RASopathy Genes in Sun-Exposed Melanomas. Nat Genet (2015) 47:996–1002. doi: 10.1038/ng.3361
11. Shain AH, Garrido M, Botton T, Talevich E, Yeh I, Sanborn IZ, et al. Exome Sequencing of Desmoplastic Melanoma Identifies Recurrent NFKBIE Promoter Mutations and Diverse Activating Mutations in the MAPK Pathway. Nat Genet (2015) 47:1194–9. doi: 10.1038/ng.3382
12. van Dijk MC, Bernsen MR, Ruiter DJ. Analysis of Mutations in B-RAF, N-RAS, and H-RAS Genes in the Differential Diagnosis of Spitz Nevus and Spitzoid Melanoma. Am J Surg Pathol (2005) 29:1145–51. doi: 10.1097/01.pas.0000157749.18591.9e
13. Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase Fusions are Frequent in Spitz Tumours and Spitzoid Melanomas. Nat Commun (2014) 5:3116. doi: 10.1038/ncomms4116
14. Lazova R, Pornputtapong N, Halaban R, Bosenberg M, Bai Y, Chai H, et al. Spitz Nevi and Spitzoid Melanomas: Exome Sequencing and Comparison With Conventional Melanocytic Nevi and Melanomas. Mod Pathol (2015) 30:640–9. doi: 10.1038/modpathol.2016.237
15. Raghavan SS, Peternel S, Mully TW, North JP, Pincus LB, LeBoit PE, et al. Spitz Melanoma is a Distinct Subset of Spitzoid Melanoma. Mod Pathol (2020) 33:1122–34. doi: 10.1038/s41379-019-0445-z
16. Bastian BC, de la Fouchardière A, Elder DE, Gerami P, Lazar AJ, Massi D, et al. “Genomic Landscape of Melanoma”. In: Elder DE, Massi D, Scolyer RA, Willemze R, editors. Who Classification of Skin Tumours, 4th Edition. Lyon, F: IARC. (2018) p. 72–5.
17. Bastian BC, Xiong J, Frieden IJ, Williams ML, Chou P, Busam K, et al. Genetic Changes in Neoplasms Arising in Congenital Melanocytic Nevi: Differences Between Nodular Proliferations and Melanoma. Am J Pathol (2002) 161:1163–9. doi: 10.1016/S0002-9440(10)64393-3
18. Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital Melanocytic Nevi Frequently Harbor NRAS Mutations But No BRAF Mutations. J Invest Dermatol (2007) 127:179–82. doi: 10.1038/sj.jid.5700490
19. Pérez-Alea M, Vivancos A, Caratú G, Matito J, Ferrer B, Hernandez-Losa J, et al. Genetic Profile of GNAQ-mutated Blue Melanocytic Neoplasms Reveals Mutations in Genes Linked to Genomic Instability and the PI3K Pathway. Oncotarget (2016) 7:28086–95. doi: 10.18632/oncotarget.8578
20. Newell F, Wilmott JS, Johanson PA, Nones K, Addala V, Mukhopadhyay P, et al. Whole-Genome Sequencing of Acral Melanoma Reveals Genomic Complexity and Diversity. Nat Commun (2020) 11:5259. doi: 10.1038/s41467-020-18988-3
21. Boussemart L, Johnson A, Schrock AB, Pal SK, Frampton GM, Fabrizio D, et al. Tumor Mutational Burden and Response to PD-1 Inhibitors in a Case Series of Patients With Metastatic Desmoplastic Melanoma. J Am Acad Dermatol (2019) 80:1780–2. doi: 10.1016/j.jaad.2018.12.020
22. Chan TA, Yarchoan M, Jafee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann Oncol (2019) 30:44–56. doi: 10.1093/annonc/mdy495
23. Palmieri G, Ombra M, Colombino M, Casula M, Sini M, Manca A, et al. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front Oncol (2015) 5:183. doi: 10.3389/fonc.2015.00183
24. Palmieri G, Colombino M, Casula M, Manca A, Mandalà M, Cossu A. Molecular Pathways in Melanomagenesis: What We Learned From Next-Generation Sequencing Approaches. Curr Oncol Rep (2018) 20:86. doi: 10.1007/s11912-018-0733-7
25. Clark WH Jr., From L, Bernardino EA, Mihm MC. The Histogenesis and Biologic Behaviour of Primary Human Malignant Melanoma of the Skin. Cancer Res (1969) 29:705–27.
26. McGovern VJ. The Classification of Melanoma and Its Relationship With Prognosis. Pathology (1970) 2:85–98. doi: 10.3109/00313027009077330
27. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature (2009) 457:599–602. doi: 10.1038/nature07586
28. Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The Genetic Evolution of Melanoma From Precursor Lesions. N Engl J Med (2015) 373:1926–36. doi: 10.1056/NEJMoa1502583
29. Colebatch AJ, Ferguson P, Newell F, Kazakoff S. Molecular Genomic Profiling of Melanocytic Nevi. J Invest Dermatol (2019) 139:1762–8. doi: 10.1016/j.jid.2018.12.033
30. LaPak KM, Burd CE. The Molecular Balancing Act of 16INK4a in Cancer and Aging. Mol Cancer Res (2014) 12:167–83. doi: 10.1158/1541-7786.MCR-13-0350
31. Ross AL, Sanchez MI, Grichnik JM. Nevus Senescence. ISRN Dermatol (2011) 2011:642157. doi: 10.5402/2011/642157
32. Shain AH, Yeh I, Kovalyshin I, Sriharan A, Talevich E, Gagnon A, et al. The Genetic Evolution of Melanoma From Precursor Lesions. N Engl J Med (2015) 373:1926–36. doi: 10.1056/NEJMoa1502583
33. Zhang AJ, Rush PS, Tsao H. Duncan LM BRCA1-Associated Protein (BAP1)-Inactivated Melanocytic Tumors. J Cutan Pathol (2019) 46:965–72. doi: 10.1111/cup.13530
34. Ferrara G, Mariani MP, Auriemma M. BAP1-Inactivated Melanocytic Tumour With Borderline Histopathological Features (BAP1-Inactivated Melanocytoma): A Case Report and a Reappraisal. Australas J Dermatol (2021) 62:e88–91. doi: 10.1111/ajd.13408.
35. Yeh I, Lang UE, Durieux E, Tee MK, Jorapur A, Shain H, et al. Combined Activation of MAP Kinase Pathway and β-Catenin Signaling Cause Deep Penetrating Nevi. Nat Commun (2017) 8:644. doi: 10.1038/s41467-017-00758-3
36. de la Fouchardière A, Caillot C, Jacquemus J, Durieux E, Houlier A, Haddad V, et al. β-Catenin Nuclear Expression Discriminates Deep Penetrating Nevi From Other Cutaneous Melanocytic Tumors. Virchows Arch (2019) 474:539–50. doi: 10.1007/s00428-019-02533-9
37. Cohen JN, Joseph NM, North JP, Onodera C, Zembowicz A, LeBoit PE. Genomic Analysis of Pigmented Epithelioid Melanocytomas Reveals Recurrent Alterations in PRKAR1A, and PRKCA Genes. Am J Surg Pathol (2017) 41:1333–46. doi: 10.1097/PAS.0000000000000902
38. Isales MC, Mohan LS, Quan VL, Garfield EM, Zhang B, Shi K, et al. Distinct Genomic Patterns in Pigmented Epithelioid Melanocytoma: A Molecular and Histologic Analysis of 16 Cases. Am J Surg Pathol (2019) 43:480–8. doi: 10.1097/PAS.0000000000001195
39. Yélamos O, Navarrete-Dechent C, Marchetti MA, Rogers T, Apalla Z, Bahadoran P, et al. Clinical and Dermoscopic Features of Cutaneous BAP1 Inactivated Melanocytic Tumors: Results of a Multicenter Case-Control Study by the International Dermoscopy Society (Ids). J Am Acad Dermatol (2019) 80:1585–93. doi: 10.1016/j.jaad.2018.09.014
40. Cosgarea I, Griewank KG, Ungureanu L, Tamayo A, Siepman T. Deep Penetrating Nevus and Borderline Deep Penetrating Nevus: A Literature Review. Front Oncol (2020) 10:837. doi: 10.3389/fonc.2020.00837
41. Zembowicz A, Carney JA, Mihm MC. Pigmented Epithelioid Melanocytoma: A Low-Grade Melanocytic Tumor With Metastatic Potential Indistinguishable From Animal-Type Melanoma and Epithelioid Blue Nevus. Am J Surg Pathol (2004) 28:31–40. doi: 10.1097/00000478-200401000-00002
42. Cohen JN, Yeh I, Mully TW, LeBoit PE, McCalmont TH. Genomic and Clinicopathologic Characteristics of PRKAR1A-Inactivated Melanomas: Toward Genetic Distinctions of Animal-type Melanoma/Pigment Synthesizing Melanoma am. J Surg Pathol (2020) 44:805–16. doi: 10.1097/PAS.0000000000001458
43. Ferrara G, Bradamante M. Melanocytic Skin Tumors: Does the Molecular Progression Model Fit With the Routine Clinicopathological Practice? Dermatol Pract Concept (2019) 10:e2020001. doi: 10.5826/dpc.1001a01
44. Massi G, Bastian BC, PE L, VG P, Xu X. “Proliferative Nodules in Congenital Melanocytic Naevus”. In: (WHO Classification of Skin Tumours, 4th Edition, eds. Elder D. E., Massi D., Scolyer R. A., Willemze R (Lyon F. F: IARC), (2018). p. 136.
45. de la Fouchardière A, Scolyer R, Calonje E, Fullen DR, Gerami P, Requena L, et al. “Melanoma Arsing in Blue Naevus”. In: (WHO Classification of Skin Tumours, 4th Edition, eds. Elder D. E., Massi D., Scolyer R. A., Willemze R (Lyon F. F: IARC), (2018). p. 124–5.
46. Barnhill R, Bahrami A, Bastian BC, Busam KJ, Cerroni L, de la Fouchardière A, et al. “Malignant Spitz Tumor (Spitz Melanoma)”. In: (WHO Classification of Skin Tumours, 4th Edition, eds. Elder D. E., Massi D., Scolyer R. A., Willemze R (Lyon F. F: IARC), (2018). p. 108–10.
47. Lallas A, Kyrgidis A, Ferrara G, Kittler H, Apalla Z, Castagnetti F, et al. Atypical Spitz Tumours and Sentinel Lymph Node Biopsy: A Systematic Review. Lancet Oncol (2014) 15:e176–83. doi: 10.1016/S1470-2045(13)70608-9
48. de la Fouchardiere A, Blokx W, van Kempen LC, Luzar B, Piperno-Neumann S, Puig S, et al. Esp, EORTC, and EURACAN Expert Opinion: Practical Recommendations for the Pathological Diagnosis and Clinical Management of Intermediate Melanocytic Tumors and Rare Melanoma Variants. Virch Arch (2021). doi: 10.1007/s00428-020-03005-1. Online ahead of print.
49. Zembowicz A, Scolyer RA. Nevus/Melanocytoma/Melanoma: An Emerging Paradigm for Classification of Melanocytic Neoplasms? Arch Pathol Lab Med (2011) 135:300–6. doi: 10.1043/2010-0146-RA.1
50. Cellier L, Perron E, Pissaloux D, Karanian M, Haddad V, Alberti L, et al. Cutaneous Melanocytoma With CRTC1-TRIM11 Fusion: Report of 5 Cases Resembling Clear Cell Sarcoma. Am J Surg Pathol (2018) 42:382–91. doi: 10.1097/PAS.0000000000000996
51. Macagno N, Pissaloux D, Etchevers H, Haddad V, Vergier B, Sierra-Fortuny S, et al. Cutaneous Melanocytic Tumors With Concomitant NRASQ61R and IDH1R132C Mutations. A Report of Six Cases. Am J Surg Pathol (2020) 44:1398–405. doi: 10.1097/PAS.0000000000001500
52. de la Fouchardiere A, Pissaloux D, Tirode F, Karanian M, Fletcher CDM, Hanna J. Clear Cell Tumor With Melanocytic Differentiation and ACTIN-MITF Translocation: Report of 7 Cases of a Novel Entity. Am J Surg Pathol (2020) 45:962–8. doi: 10.1097/PAS.0000000000001630
53. de la Fouchardiere A, Pissaloux D, Tirode F, Hanna J. Clear Cell Tumor With Melanocytic Differentiation and MITF-CREM Translocation: A Novel Entity Similar to Clear Cell Sarcoma. Virchows Arch (2021). doi: 10.1007/s00428-021-03027-3. Online ahead of print.
54. Pampena R, Kyrgidis A, Lallas A, Moscarella E, Argenziano G, Longo C. A Meta-Analysis of Nevus-Associated Melanoma: Prevalence and Practical Implications. J Am Acad Dermatol (2017) 77:938–45. doi: 10.1016/j.jaad.2017.06.149
55. Borsari S, Longo C, Ferrari C, Benati E, Bassoli S, Schianchi S, et al. Dermoscopi Island: A New Descriptor for Thin Melanoma. Arch Dermatol (2010) 146:1257–62. doi: 10.1001/archdermatol.2010.311
56. Ferrara G, Argenziano G, Giorgio CM, Zalaudek I, Kittler H. Dermoscopic-Pathologic Correlation: Apropos of Six Equivocal Cases. Semin Cutan Med Surg (2009) 28:157–64. doi: 10.1016/j.sder.2009.06.003
57. Terushkin V, Scope A, Halpern AC, Marghoob AA. Pathways to Involution of Nevi: Insights From Dermoscopic Follow-Up. Arch Dermatol (2010) 146:459–60. doi: 10.1001/archdermatol.2010.20
58. Fabrizi G, Pennacchia I, Pagliarello C, Massi G. Sclerosing Nevus With Pseudomelanomatous Features. J Cutan Pathol (2008) 35:995–1002. doi: 10.1111/j.1600-0560.2008.01176.x
59. Ferrara G, Amantea A, Argenziano G, Broganelli P, Cesinaro AM, Donati P, et al. Sclerosing Nevus With Pseudomelanomatous Features and Regressing Melanoma With Nevoid Features. J Cutan Pathol (2009) 36:913–5. doi: 10.1111/j.1600-0560.2008.01176.x
60. Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma. Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Patter. Arch Pathol Lab Med (2020) 144:500–22. doi: 10.5858/arpa.2019-0561-RA
61. Marghoob AA. Congenital Melanocytic Nevi. Evaluation and Management. Dermatol Clin (2002) 20:697–16. doi: 10.1016/s0733-8635(02)00030-x. viii.
62. Elder DE, Barnhill R, Bastian BC, Duncan LM, Massi D, Mihm MC Jr, et al. “Dysplastic Naevus”. In: (WHO Classification of Skin Tumours, 4th Edition, eds. Elder D. E., Massi D., Scolyer R. A., Willemze R (Lyon F. F: IARC), (2018). p. 82–6.
63. Shors AR, Kim S, White A, Arenyi Z, Barnhil RL, Duray P, et al. Dysplastic Naevi With Moderate to Severe Histological Dysplasia: A Risk Factor for Melanoma. Br J Dermatol (2006) 155:988–93. doi: 10.1111/j.1365-2133.2006.07466.x
64. Xiong MY, Rabkin MS, Piepkorn MW, Barnhill RL, Argenyi Z, Erickson L, et al. Diameter of Dysplastic Nevi is a More Robust Biomarker of Increased Melanoma Risk Than Degree of Histologic Dysplasia: A Case-Control Study. J Am Acad Dermatol (2014) 71:1257–68.e4. doi: 10.1016/j.jaad.2014.07.030
65. Arumi-Uria M, McNutt NS, Finnerty B. Grading of Atypia in Nevi: Correlation With Melanoma Risk. Mod Pathol (2003) 16:764–71. doi: 10.1097/01.MP.0000082394.91761.E5
66. Slade J, Marghoob AA, Salopek TG, Rigel DS, Kopf AW, Bart RS. Atypical Mole Syndrome: Risk Factor for Cutaneous Malignant Melanoma and Implications for Management. J Am Acad Dermatol (1995) 32:479–94. doi: 10.1016/0190-9622(95)90073-x
67. Wick MR, Elenitsas R, Kim J, Kossard R. Simple Lentigo and Lentiginous Melanocytic Naevus. In: (WHO Classification of Skin Tumours, 4th Edition, eds. Elder D. E., Massi D., Scolyer R. A., Willemze R (Lyon F. F: IARC), (2018). p. 78–9.
68. King R, Page RN, Googe PB, Mihm MC Jr. Lentiginous Melanoma: A Histologic Pattern of Melanoma To Be Distinguished From Lentiginous Nevus. Mod Pathol (2005) 18:1397–401. doi: 10.1038/modpathol.3800454
69. Ferrara G, Zalaudek I, Argenziano G. Lentiginous Melanoma: A Distincive Clinicopathological Entity. Histopathology (2008) 52:523–5. doi: 10.1111/j.1365-2559.2008.02943.x
70. Kutzner H, Metzler G, Argenyi Z, Requena L, Palmedo G, Mentzel T, et al. Histological and Genetic Evidence for a Variant of Superficial Spreading Melanoma Composed Predominantly of Large Nests. Mod Pathol (2012) 25:838–45. doi: 10.1038/modpathol.2012.35
71. Dri A, Conforti C, Zelin E, Toffoli L, Signoretto D, Zacchi A, et al. Nested Melanoma: When Dermoscopy Turns Histopathology Into Question. Int J Dermatol (2020) 60:e70–2. doi: 10.1111/ijd.15218
72. Piepkorn MW, Longton GM, Reish LM, Elder DE, Pepe MS, Kerr KF, et al. Assessment of Second-Opinion Strategies for Diagnoses of Cutaneous Melanocytic Lesions. JAMA Netw Open (2019) 10:e1912597. doi: 10.1001/jamanetworkopen.2019.12597
73. Ferrara G, Argenyi Z, Argenziano G, Cerio R, Cerroni L, Di Blasi A, et al. The Influence of Clinical Information in the Histopathologic Diagnosis of Melanocytic Skin Neoplasms. PlosONE (2009) 4:e5375. doi: 10.1371/journal.pone.0005375
74. Piepkorn MW, Barnhill RL, Elder DE, Knezevich SR, Carney PA, Reish LM, et al. The MPATH-Dx Reporting Schema for Melanocytic Proliferations and Melanoma. J Am Acad Dermatol (2014) 70:131–41. doi: 10.1016/j.jaad.2013.07.027
75. Cohen J, Spies J, Ross FNP-C, Bolke A, McCalmont T. Heavily Pigmented Epithelioid Melanoma With Loss of Protein Kinase A Regulatory Subunit-α Expression. Am J Dermatopathol (2020) 40:912–6. doi: 10.1097/DAD.0000000000001185
76. Cohen JN, Yeh I, Mully TW, LeBoit PE, McCalmont TH. Genomic and Clinicopathologic Characteristics of PRKAR1A-inactivated Melanomas: Toward Genetic Distinctions of Animal-Type Melanoma/Pigment Synthesizing Melanoma. Am J Surg Pathol (2020) 44:805–16. doi: 10.1097/PAS.0000000000001458
77. Magro CM, Abraham RM, Guo R, Li S, Wang X, Proper S, et al. Deep Penetrating Nevus-Like Borderline Tumors: A Unique Subset of Ambiguous Melanocytic Tumors With Malignant Potential and Normal Cytogenetics. Eur J Dermatol (2014) 24:594–602. doi: 10.1684/ejd.2014.2393
78. Isales MC, Khan AU, Zhang B, Compres EV, Kim D, Tan TL, et al. Molecular Analysis of Atypical Deep Penetrating Nevus Progressing to Melanoma. J Cutan Pathol (2020) 47:1150–4. doi: 10.1111/cup.13775
79. Baetz TD, Fletcher GG, Knight G, McWhirter E, Rajagopal S, Song X, et al. Systemic Adjuvant Therapy for Adult Patients At High Risk for Recurrent Melanoma: A Systematic Review. Cancer Treat Rev (2020) 87:102032. doi: 10.1016/j.ctrv.2020.102032
80. Pham TV, Boichard A, Goodman A, Riviere P, Yeerna H, Tamayo P, et al. Role of Ultraviolet Mutational Signature Versus Tumor Mutation Burden in Predicting Response to Immunotherapy. Mol Oncol (2020) 14:1680–94. doi: 10.1002/1878-0261.12748
81. Long GV, Wilmott JS, Capper D, Preusser M, Zhang YE, Thompson JF, et al. Immunohistochemistry is Highly Sensitive and Specific for the Detection of V600E BRAF Mutation in Melanoma. Am J Surg Pathol (2013) 37:61–5. doi: 10.1097/PAS.0b013e31826485c0
82. Lezcano C, Jungbluth AA, Nehal KS, Hollman TJ. PRAME Expression in Melanocytic Tumors. Busam KJ Am J Surg Pathol (2018) 42:1456–65. doi: 10.1097/PAS.0000000000001134
83. Kiuru M, Jungbluth A, Kutzner H, Wiesner T, Busam KJ. Spitz Tumors: Comparison of Histological Features in Relationship to Immunohistochemical Staining for ALK and NTRK1. Int J Surg Pathol (2016) 24:200–6. doi: 10.1177/1066896916630375
84. Quan VL, Panah E, Zhang B, Shi K, Mohan LS, Gerami P. The Role of Gene Fusions in Melanocytic Neplasms. J Cutan Pathol (2019) 46:878–87. doi: 10.1111/cup.13521
85. Zembowicz A, Knoepp SM, Bei T, Stergiopoulos S, Eng C, Mihm MC, et al. Loss of Expression of Protein Kinase a Regulatory Subunit 1alpha in Pigmented Epithelioid Melanocytoma But Not in Melanoma or Other Melanocytic Lesions. Am J Surg Pathol (2007) 31:1764–75. doi: 10.1097/PAS.0b013e318057faa7
86. Ferrara G, De Vanna AC. Fluorescence in Situ Hybridization for Melanoma Diagnosis: A Review and a Reappraisal. Am J Dermatopathol (2016) 38:253–69. doi: 10.1097/DAD.0000000000000380
87. Houlier A, Pissaloux D, Masse I, Tirose F, Karanian M, Pincus LB, et al. Melanocytic Tumors With MAP3K8 Fusions: Report of 33 Cases With Morphological-Genetic Correlations. Mod Pathol (2019) 33:846–57. doi: 10.1038/s41379-019-0384-8
Keywords: melanoma, melanocytoma, dysplastic nevus, clinicopathological correlation, histopathology, immunohistochemistry, molecular biology
Citation: Ferrara G and Argenziano G (2021) The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions From Routine Practice. Front. Oncol. 11:675296. doi: 10.3389/fonc.2021.675296
Received: 02 March 2021; Accepted: 31 May 2021;
Published: 02 July 2021.
Edited by:
Nihal Ahmad, University of Wisconsin-Madison, United StatesReviewed by:
Chandra K. Singh, University of Wisconsin-Madison, United StatesGagan Chhabra, University of Wisconsin-Madison, United States
Copyright © 2021 Ferrara and Argenziano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerardo Ferrara, gerardo.ferrara@libero.it