 Carla M Borini Etichetti
Carla M Borini Etichetti Evelyn Arel Zalazar2†
Evelyn Arel Zalazar2† Javier Girardini
Javier Girardini- 1Instituto de Fisiología Experimental de Rosario, IFISE, CONICET-UNR, Rosario, Argentina
- 2Instituto de Inmunología Clínica y Experimental de Rosario, IDICER, CONICET-UNR, Rosario, Argentina
Missense mutations in the TP53 gene are among the most frequent alterations in human cancer. Consequently, many tumors show high expression of p53 point mutants, which may acquire novel activities that contribute to develop aggressive tumors. An unexpected aspect of mutant p53 function was uncovered by showing that some mutants can increase the malignant phenotype of tumor cells through alteration of the mevalonate pathway. Among metabolites generated through this pathway, isoprenoids are of particular interest, since they participate in a complex process of posttranslational modification known as prenylation. Recent evidence proposes that mutant p53 also enhances this process through transcriptional activation of ICMT, the gene encoding the methyl transferase responsible for the last step of protein prenylation. In this way, mutant p53 may act at different levels to promote prenylation of key proteins in tumorigenesis, including several members of the RAS and RHO families. Instead, wild type p53 acts in the opposite way, downregulating mevalonate pathway genes and ICMT. This oncogenic circuit also allows to establish potential connections with other metabolic pathways. The demand of acetyl-CoA for the mevalonate pathway may pose limitations in cell metabolism. Likewise, the dependence on S-adenosyl methionine for carboxymethylation, may expose cells to methionine stress. The involvement of protein prenylation in tumor progression offers a novel perspective to understand the antitumoral effects of mevalonate pathway inhibitors, such as statins, and to explore novel therapeutic strategies.
Introduction
TP53, the gene encoding the tumor suppressor p53, is one of the most frequently mutated genes in human cancer (1). More than 70% of TP53 alterations are missense mutations, leading to the conspicuous presence of p53 point mutants in tumors (2). Mounting evidence has supported the notion that these mutants cooperate with tumorigenesis though the acquisition of novel activities (3). Particularly, animal models provided compelling proof of the ability of p53 point mutants to promote the development of aggressive tumors. Intense research on the mechanisms underlying this effect has revealed a complex scenario (4). Mutant p53 can be considered as a pleiotropic factor that affects cell behavior by altering the function of different interactors. In this context, the presence of specific arrays of interactors combined with patterns of active signaling pathways may explain the manifold activities described for p53 mutants (5). Most mutations are found in the DNA Binding Domain, and a few codons concentrate the highest mutation frequencies. Although the development of aggressive tumors appears as a common biological outcome of most p53 mutants, some differences were also reported (6). The ability to cooperate with oncogenic mechanisms and the exclusive presence in tumor cells make mutant p53 an attractive therapeutic target. Therefore, much effort is concentrated in the study of its function. In this regard, the unexpected finding that mutant p53 alters the expression of mevalonate (MVA) pathway genes (7) opened new avenues to understand the importance of metabolism in tumor cell biology.
Taking Control of the Mevalonate Pathway, Mutant vs Wild Type p53
The pathological role of alterations on the MVA pathway was initially proposed based on the observation that inhibitors of the enzyme that catalyzes the rate-limiting step (3-hydroxy-3-methyl-glutaryl-CoA reductase, HMGCR), known as statins, reduced the proliferation of tumor cells (8, 9). This pathway allows the biosynthesis of cholesterol and isoprenoids from acetyl-CoA (Figure 1). The isoprenoid intermediates farnesyl and geranylgeranyl may be covalently attached to cysteine residues on the carboxyl terminus of proteins, in the first step of the protein prenylation pathway, a complex mechanism of posttranslational modification (10). The connection between mutant p53 and the MVA pathway was unveiled following the observation that several p53 point mutants promoted an aggressive phenotype in three-dimensional (3D) cultures of breast cancer cells (7). The finding that endogenous p53R273H enhanced the expression of at least 17 MVA pathway genes, along with evidence from elegant pharmacologic manipulation of the pathway, led to propose that enhanced flux through the MVA pathway was responsible for the phenotype associated to mutant p53. The expression of MVA pathway genes is under control of Sterol Responsive Element Binding Proteins (SREBPs), which induce transcription in response to low cholesterol levels (11). The recruitment of mutant p53 on the promoters of MVA pathway genes in the vicinity of Sterol Responsive Elements (SREs) as well as the ability of p53R273H to interact with SREBPs suggest that mutant p53 acts as a transcriptional co-activator. Supporting the idea that MVA pathway alteration cooperates with tumor progression, high expression of MVA pathway genes was correlated with poor clinical outcome in breast cancer patients (7).
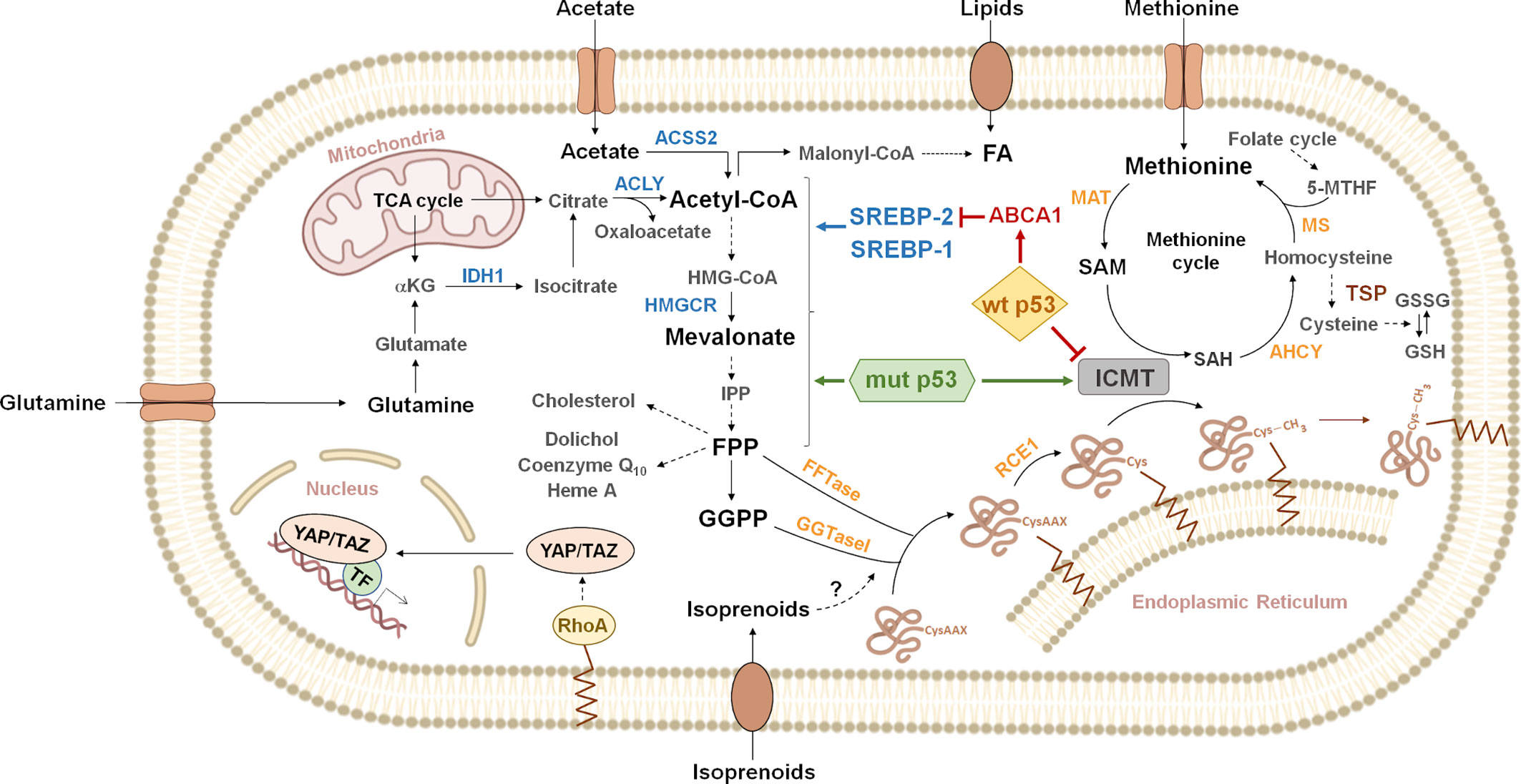
Figure 1 Overview of metabolic pathways connected to protein prenylation. Citrate generated from glutamine or the tricarboxylic acid cycle (TCA cycle) is cleaved by ATP-citrate lyase (ACLY) to acetyl-CoA and oxaloacetate. Acetyl-CoA can also be synthesized by cytoplasmic acetyl-CoA synthetase (ACSS2) from exogenous acetate. Acetyl-CoA, can be carboxylated to malonyl-CoA, to produce fatty acids (FA). Lipids can also be incorporated through exogenous uptake. Alternatively, acetyl-CoA enters the mevalonate pathway, where three molecules are condensed in a two-step reaction to produce 3-hydroxy-3-methylglutaryl CoA (HMG-CoA). HMG-CoA is then reduced by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) to produce mevalonate. Mevalonate is then converted into isopentenyl-diphosphate (IPP) through a series of enzymatic steps. IPP serves as a monomeric unit for the consequent synthesis of farnesyl diphosphate (FPP), geranylgeranyl diphosphate (GGPP) and other downstream metabolites (cholesterol, dolichol, coenzyme Q10, heme A, etc.). The isoprenoid moiety of FPP or GGPP may be covalently attached to cysteine residues on the carboxyl terminus of some proteins, in the first step of the protein prenylation pathway. For example, the activity of RHOA is regulated by geranylgeranylation, which localizes RHOA to the plasma membrane. RHOA promotes the nuclear localization and activity of the Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). Gain of function p53 mutants (mut p53) functionally interact with sterol regulatory element binding proteins (SREBPs) to drive increased expression of mevalonate pathway genes. In contrast, wild type p53 (wt p53) represses the mevalonate pathway genes through inhibition of SREBP-2 maturation, as a consequence of transcriptional induction of ATP binding cassette subfamily A member 1 (ABCA1). Additionally, p53 point mutants can induce isoprenylcysteine carboxymethyltransferase (ICMT) expression, while wt p53 exerts the opposite effect, through transcriptional repression. ICMT catalyzes protein carboxymethylation, the last step of the protein prenylation pathway. The methyl donor in this reaction is S-adenosyl methionine (SAM), which is produced from the essential aminoacid methionine in the rate-limiting reaction catalyzed by methionine adenosyl transferase (MAT). SAM is transformed into S-adenosyl homocysteine (SAH), which can be used to regenerate methionine through the methionine cycle. Homocysteine can be derived to the transsulfuration pathway (TSP) to synthesize glutathione (GSH). Dashed arrows represent multiple enzymatic steps. Indications on reversibility of enzymatic reactions and subcellular localization of some enzymes have been omitted for simplicity. Enzymes known to be regulated by SREBPs are shown in blue. IDH1, isocitrate dehydrogenase 1; α-KG, α-ketoglutarate; FFTase, farnesyl transferase; GGTaseI, geranylgeranyl transferase 1; RCE1, RAS converting enzyme 1; AHCY, adenosylhomocysteinase; MS, methionine synthase; 5-MTHF, 5-methyltetrahydrofolate; GSSG, oxidized glutathione; TF, transcription factors.
Protein geranylgeranylation appears to be crucial in the effect of mutant p53. Inhibition of geranylgeranyl transferase I (GGTaseI) attenuated the invasive morphology of MDA-MB-231 cells in 3D cultures, similar to endogenous mutant p53 downregulation. In contrast, inhibition of enzymes that derive the flux of the pathway to other molecules, such as squalene synthase (SQS) and farnesyl transferase (FTase), had no effect. Moreover, addition of geranylgeranyl diphosphate (GGPP) recovered the invasive morphology in cells where mutant p53 was downregulated (7). Furthermore, mutant p53 depletion or HMGCR inhibition by statins reduced the nuclear localization and activity of Yes-Associated Protein (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) (12), the transcriptional module of the Hippo pathway, through a mechanism that involves Ras homolog family member A (RHOA) prenylation. Hyperactivation of YAP and TAZ has been increasingly associated to proliferation and metastasis (13). Similarly, YAP/TAZ inactivation was not observed upon inhibition of SQS and FTase, but was phenocopied by GGTaseI inhibition. Moreover, adding GGPP reverted the effect of upstream inhibition of the MVA pathway (12).
The finding that wild type p53 (wt p53) repressed the expression of MVA pathway genes provides strong support to the idea that alteration of this pathway may be a critical event in tumor progression. In this case, an indirect mechanism was described, involving inhibition of SREBP-2 maturation (14). This effect was mediated by the transcriptional induction by wt p53 of ATP binding cassette subfamily A member 1 (ABCA1), which encodes a protein involved in the retrograde transport of cholesterol from the plasma membrane to the endoplasmic reticulum (ER). SREBPs are produced as inactive precursors anchored to the cytosolic side of the ER. Maturation can be stimulated by low cholesterol levels in the ER, which triggers a complex process that leads to proteolytic cleavage and nuclear import of SREBPs (15, 16). Analysis of cancer databases showed that ABCA1 expression was lower in colon, breast and liver carcinomas comparing with normal tissues. Likewise, Abca1 inactivation enhanced tumorigenesis in an experimental model of hepatocellular carcinoma (HCC). Additional evidence from animal models strongly supports the notion that ABCA1 is relevant for the tumor suppressive function of wt p53 (14). Noteworthy, wt p53 was also reported to repress the expression of SREBP1-c, suggesting that the interplay between the p53 pathway and SREBPs is even more complex (17, 18).
Post Prenylation Processing and the p53 Pathway
The posttranslational processing pathway known as prenylation involves three stages (Figure 1). First, the addition of farnesyl or geranylgeranyl, to a cysteine residue close to the carboxyl terminus of proteins (19), catalyzed by FTase or GGTaseI, respectively. The prenylated cysteine is typically part of a CAAX motif (C: cysteine; A: aliphatic amino acid; X: any amino acid), although other motifs such as CXC can also be targeted (20). Second, the terminal amino acids following the prenylated cysteine are removed by the specific peptidase RAS Converting Endoprotease 1 (RCE1) in the ER (21). Third, Isoprenylcysteine Carboxyl Methyltransferase (ICMT), also an integral membrane protein of the ER, catalyzes the methylation of the free carboxyl terminus on the cysteine. This modification provides an uncharged hydrophobic carboxyl terminus, which increases protein interaction with biological membranes and/or modifies its ability to interact with other proteins (22). Only one member of the ICMT methyltransferase class is encoded in mammalian genomes and lacks homology to other methyltransferases (23). Interestingly, methylation of prenylated proteins is absent in ICMT-/- cells, which indicates that ICMT is the only enzyme able to catalyze this reaction (24). A connection between the p53 pathway and post-prenylation processing was established by showing that wt and mutant p53 regulate ICMT expression (25). Several p53 point mutants induced ICMT expression in breast, colon, and lung cancer cell lines. This effect was associated to transcriptional activation, since mutant p53 was recruited on the ICMT promoter and was able to enhance its activity. Moreover, promoter activation and enhanced endogenous gene expression were observed in p53 null cells, showing that this activity is a novel function acquired by mutants. In contrast, wt p53 was also found on the ICMT promoter but repressed promoter activity and reduced mRNA and protein levels. Interestingly, the effects of wt and mutant p53 were shown to depend on different promoter regions, indicating that they act through different mechanisms. This evidence suggests that the acquisition of missense mutations on TP53 may exert a strong effect on ICMT expression by complementary mechanisms. The repressive function of wt p53 may be lost upon mutation of TP53, while the presence of point mutants may enhance gene expression by wt p53-independent mechanisms. Underlining the clinical relevance of the connection between the p53 pathway and post-prenylation processing, ICMT expression was found to be significantly reduced in patients classified as wt p53, but was increased in mutant p53 patients (25). The discussed evidence suggests that deregulated expression of ICMT may cooperate with tumor progression. In support to this idea, high ICMT levels were found in hepatocellular carcinoma patients and ICMT overexpression enhanced proliferation and migration in normal liver cells (26). Similarly, ICMT overexpression in H1299 non-small cell lung carcinoma (NSCLC) cells increased clonogenic potential in vitro and tumorigenesis in a xenograft model. Moreover, analysis of breast and lung cancer databases showed that high ICMT expression was correlated with reduced survival (25).
ICMT Targets in Oncogenesis
ICMT substrates are distributed among different families (Table 1), complicating the rationalization of its pathological effects. In addition to RAS and RHO families of GTPases, more than 200 CAAX proteins have been predicted based on structural analysis (70, 71). Polypeptides ending in CXC, as the doubly geranylgeranylated RAB family, are also modified by this pathway. The identification of RAS family members as ICMT substrates reinforced the notion that protein prenylation may play a role in cancer (72, 73). Deletion of Icmt reduced KRAS-induced transformation in vitro (74) and neoplastic lesions in a mouse model of myeloproliferative syndrome (75). Moreover, genetic ablation of ICMT in RAS-transformed human breast primary cells and human breast cancer cell lines harboring mutant RAS, reduced tumor formation in xenograft models (76). Intriguingly, Icmt inactivation in a KRAS-driven mouse model of pancreatic carcinoma increased the number of pancreatic neoplasias and promoted tumor progression (77). Impairment of Notch signaling through deregulation of RAB7 and RAB8 was suggested as responsible for this effect. Considering the impact of mutant p53 as a promoter of pancreatic cancer (78), It will be interesting to explore the interplay between the MVA pathway and protein prenylation in this pathology.
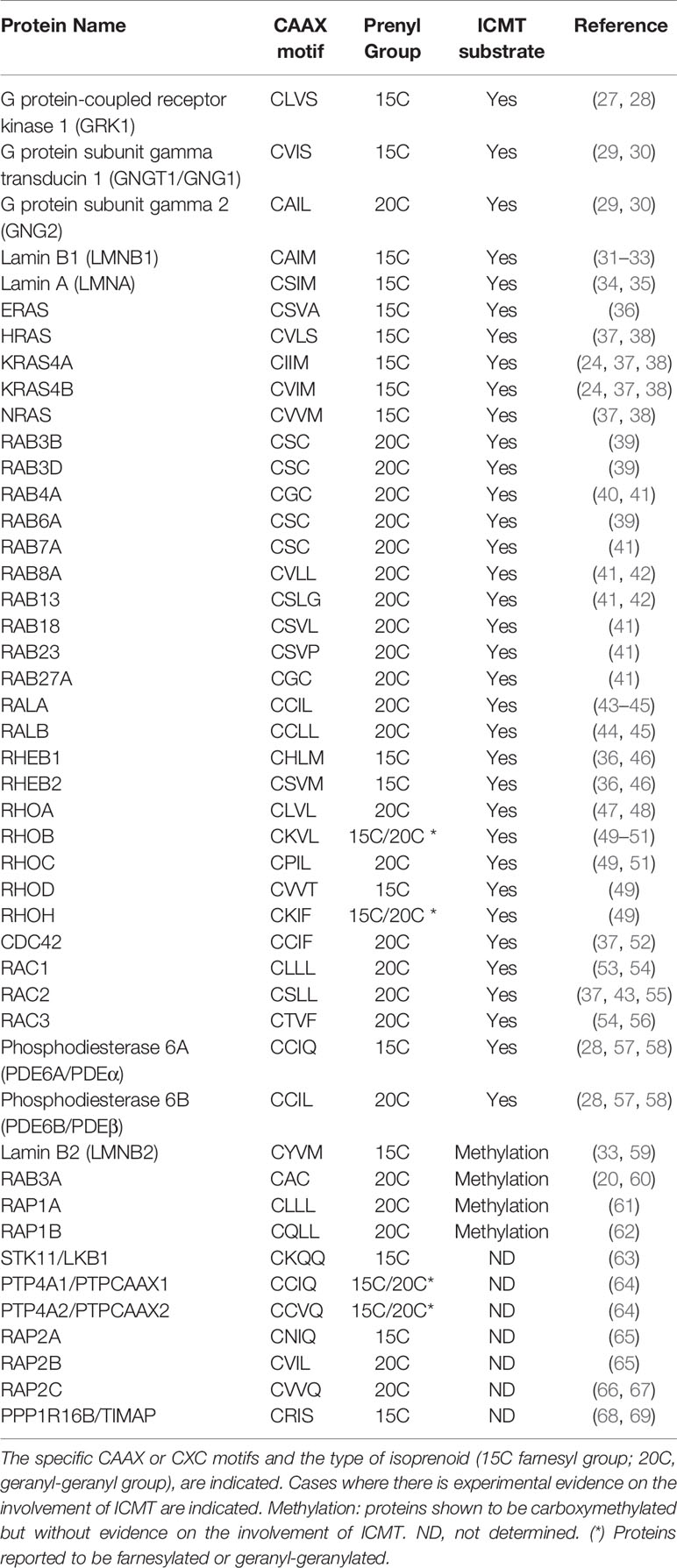
Table 1 List of prenylated proteins and ICMT substrates.
The deregulated action of ICMT on RHO GTPases may promote invasiveness and metastasis through alteration of cytoskeleton remodelling and cell motility. Accordingly, ICMT inhibition reduced migration and invasion in MDA-MB-231 cells (53), concomitant with decreased RHOA and RAC1 activity. The ability of miR-100 to attenuate lamellipodia formation, matrix metallopeptidase 2 (MMP2) activation and metastasis in hepatocellular carcinoma cells was associated to ICMT-RAC1 signaling inhibition (79). Likewise, reduced migration, invasion and metastasis were observed in HT-1080 fibrosarcoma cells upon ICMT inhibition (80), which was associated to RAB4A impaired function. ICMT overexpression in H1299 cells significantly affected actin cytoskeleton, suggesting an effect on RHO GTPases (25). Interestingly, some evidences reported differential effects of ICMT on subcellular localization and/or expression levels of protein substrates, arguing for a role in the concerted regulation of prenylated proteins. For example, ICMT inhibition reduced RHOA half-life, but enhanced RAS stability (74, 81). Lack of ICMT had different effects on the subcellular localization of RAS and RHO family members (49), and on the localization and stability of RALA and RALB. Dynamic regulation of protein carboxymethylation may have relevant consequences as suggested by the identification of carboxylesterase 1 (CES1), a carboxylesterase affecting the methylation status of RHOA. Interestingly, RHOA activity and cytoskeleton organization in breast cancer cells were similarly affected by CES1 silencing and ICMT overexpression (82).
Acetyl-CoA and Metabolic Stress in Tumor Cells
Availability of acetyl-CoA may be a critical aspect in tumor cells that sustain aggressive phenotypes by exploiting the MVA pathway. Acetyl-CoA is the starting point of the MVA pathway; however, it is also required for other important pathways, as fatty acids (FA) biosynthesis (Figure 1). An important source of acetyl-CoA is citrate produced in the mitochondria by the tricarboxylic acid (TCA) cycle, which can be converted in the cytosol into oxaloacetate and acetyl-CoA by ATP citrate-lyase (ACLY) (83). In addition, exogenous acetate may be directly converted into acetyl-CoA by cytoplasmic acetyl-CoA synthetase (ACSS2) (84). Glutamine uptake also allows the indirect production of acetyl-CoA through a series of reactions that take place in the cytosol (85, 86). A strong requirement of acetyl-CoA may expose tumor cells to the dependence on specific metabolic capabilities, forcing cells to shape their metabolism. Accordingly, there is evidence showing enhanced activity of ACLY (87) and ACSS2 (88) in cancer cells, as well as of isocitrate dehydrogenase 1 (IDH1) (89, 90), which catalyzes reductive carboxylation in the conversion of glutamine into acetyl-CoA. Expression of these genes is regulated by SREBPs suggesting the intriguing possibility that they may be induced by mutant p53 and repressed by wt p53 (91–95). Oxygen availability is frequently limited in the tumor microenvironment and nutrient uptake is highly conditioned by the degree of neovascularization (96). Entry of pyruvate into the mitochondria may be inhibited under hypoxic conditions (97), downregulating the TCA cycle and citrate production. Under these conditions, acetate and glutamine as alternative sources of acetyl-CoA may become critical. Moreover, if uptake of exogenous lipids is not able to satisfy the high demand in proliferating cells, active FA biosynthesis may be expected to compete with the MVA pathway for acetyl-CoA. In this scenario, strategies aimed at interfering with alternative acetyl-CoA sources may be effective to counteract cancer cell proliferation.
ICMT Links the Mevalonate Pathway With Methionine Metabolism
The methyl donor in protein carboxymethylation is S-adenosyl methionine (SAM), which is produced from the essential aminoacid methionine, in a reaction catalyzed by methionine adenosyl transferase (MAT). SAM is also the methyl donor in other reactions, including methylation of DNA, RNA, non-prenylated proteins and in polyamine biosynthesis. Upon methylation, SAM is transformed into S-adenosyl homocysteine (SAH), which can be used to regenerate methionine through the methionine cycle (98) (Figure 1). This cycle is closely interconnected with two other metabolic processes. Hydrolysis of SAH, catalyzed by adenosylhomocysteinase (AHCY), produces homocysteine, which can react with 5-methyl-tetra-hydrofolate (5-MTH) generated in the folate cycle, giving back methionine. Alternatively, homocysteine can be diverted to the transsulfuration pathway that ultimately leads to the synthesis of glutathione (GSH). Alteration of methionine cycle enzymes were related to cancer. For example, MAT2A and MAT2B, the genes coding for the subunits of the most abundant MAT isoenzyme, were found upregulated in tumors and cancer-initiating cells (99, 100). The close connection between SAM and the one-carbon metabolic network suggests that cell context and nutritional state may affect ICMT activity. Methionine availability may decrease SAM levels, thereby limiting ICMT catalyzed carboxymethylation. Therefore, limiting methionine uptake may have a selective inhibitory effect on cancer cells that benefit from ICMT hyperactivation. Accordingly, pioneering observations reported a marked requirement of methionine on transformed rat and human cells (101). Moreover, dietary methionine restriction reduced tumor growth and metastasis in animal models, and increased sensitivity to chemotherapeutic agents (98). Nevertheless, the molecular mechanisms underlying these effects are not yet clear.
Homocysteine is a key molecule in the one-carbon network, since it connects the methionine cycle with the folate cycle and GSH production. Under strong oxidative stress conditions, high availability of GSH may be required and, therefore, homocysteine may be preferentially driven to the transsulfuration pathway, precluding the possibility to regenerate methionine. Therefore, enhanced ICMT activity in cells under oxidative stress may further increase the dependency on methionine. Moreover, SAH acts as a negative feedback inhibitor of ICMT (102). Treatment with the AHCY inhibitor adenosine dihaldeyde (AdOx) produced accumulation of SAH (103) and reduced in vitro invasion and migration of cancer cell lines (104).
Discussion
Several metabolites produced by the MVA pathway may affect cell behavior, however, the positive effect of mutant p53 on the expression of MVA pathway genes and ICMT underline the relevance of isoprenoids in cancer. Conversely, the negative regulation exerted by wt p53 on SREBP-2 maturation and ICMT expression indicates that MVA pathway and carboxymethylation of prenylated proteins should be strictly regulated under physiological conditions. The concerted effects of mutant p53 on MVA and prenylation pathways allow tumor cells to connect both pathways, thereby fostering full modification of prenylated proteins playing key roles in oncogenesis. Still, selective alteration of each pathway may be enough to promote tumor progression. In this way, mutant p53 may activate alternative mechanisms useful to promote tumorigenesis in different contexts. Since exogenous isoprenoids may be incorporated into cancer cells and phosphorylated (105), the intriguing possibility that protein prenylation may be exploited by tumors independently from the MVA pathway may also be considered. Noteworthy, exogenous supplementation of geranylgeraniol counteracted the antitumoral effect of pitavastatin in a xenograft model of ovarian cancer cells (106). The correlation of ICMT expression with clinical outcome and the pro-oncogenic effects observed in experimental systems point at ICMT overexpression as a relevant event in tumor progression. Consequently, the potential of ICMT as a therapeutic target encouraged the identification of inhibitors. Isoprenylated cysteine analogs inhibited ICMT activity and showed antiproliferative effects, however, their mechanism of action is not clear since some of them act as modulators of RAS chaperones (107, 108). Indole-based molecules were also proposed, such as Cysmethynil (109), a competitive inhibitor with respect to isoprenylated cysteine and a non-competitive inhibitor with respect to SAM, which showed antitumor activity in vitro and in vivo (10, 110, 111). In summary, alteration of MVA pathway and protein prenylation by mutant p53 revealed interesting connections to explore. Understanding the role of less studied ICMT substrates in cancer and the study of mechanisms that regulate ICMT activity will be critical to dissect the molecular mechanisms underlying ICMT pathological effects.
Author Contributions
JG designed and wrote the manuscript. CBE contributed to manuscript writing and performed the figures. EAZ and NC contributed to the manuscript writing and figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to JG from Instituto Nacional del Cáncer (INC, Asistencia Financiera III) and from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT. PICT CABBIO 4758). CBE is a postdoctoral fellow from the National Council for Scientific and Technological Research of Argentina (CONICET). EAZ and NC are doctoral fellows from the National Council for Scientific and Technological Research of Argentina (CONICET).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.595034/full#supplementary-material
References
1. Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med (2016) 6(11):a026179. doi: 10.1101/cshperspect.a026179
2. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat (2016) 37:865–76. doi: 10.1002/humu.23035
3. Stein Y, Rotter V, Aloni-Grinstein R. Gain-of-function mutant p53: All the roads lead to tumorigenesis. Int J Mol Sci (2019) 20(24):6197. doi: 10.3390/ijms20246197
4. Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ (2019) 26(2):199–212. doi: 10.1038/s41418-018-0246-9
5. Girardini JE, Walerych D, Del Sal G. Cooperation of p53 mutations with other oncogenic alterations in cancer. SubcellBiochem (2014) 85:41–70. doi: 10.1007/978-94-017-9211-0_3
6. Muller PAJ, Vousden KH. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi: 10.1016/j.ccr.2014.01.021
7. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. doi: 10.1016/j.cell.2011.12.017
8. Quesney-Huneeus V, Wiley MH, Siperstein MD. Essential role for mevalonate synthesis in DNA replication. Proc Natl Acad Sci USA (1979) 76:5056–60. doi: 10.1073/pnas.76.10.5056
9. Habenicht AJR, Glomset JA, Ross R. Relation of cholesterol and mevalonic acid to the cell cycle in smooth muscle and Swiss 3T3 cells stimulated to divide by platelet-derived growth factor. J Biol Chem (1980) 255:5134–40.
10. Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol (2016) 17:110–22. doi: 10.1038/nrm.2015.11
11. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109(9):1125–31. doi: 10.1172/jci15593
12. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol (2014) 16:357–66. doi: 10.1038/ncb2936
13. Moroishi T, Hansen CG, Guan K-L. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer (2015) 15:73–9. doi: 10.1038/nrc3876
14. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JP, et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell (2019) 176(3):564–80.e19. doi: 10.1016/j.cell.2018.11.011
15. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metab (2008) 8(6):512–21. doi: 10.1016/j.cmet.2008.10.008
16. Brown MS, Goldstein JL. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell (1997) 89(3):331–40. doi: 10.1016/S0092-8674(00)80213-5
17. Vig S, Talwar P, Kaur K, Srivastava R, Srivastava AK, Datta M. Transcriptome profiling identifies p53 as a key player during calreticulin deficiency: Implications in lipid accumulation. Cell Cycle (2015) 14(14):2274–84. doi: 10.1080/15384101.2015.1046654
18. Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, et al. p53 activation in adipocytes of obese mice. J Biol Chem (2003) 278(28):25395–400. doi: 10.1074/jbc.M302364200
19. Liang PH, Ko TP, Wang AHJ. Structure, mechanism and function of prenyltransferases. Eur J Biochem (2002) 269(14):3339–54. doi: 10.1046/j.1432-1033.2002.03014.x
20. Smeland TE, Seabra MC, Goldstein JL, Brown MS. Geranylgeranylated Rab proteins terminating in Cys-Ala-Cys, but not Cys-Cys, are carboxyl-methylated by bovine brain membranes in vitro. Proc Natl Acad Sci USA (1994) 91(22):10712–6. doi: 10.1073/pnas.91.22.10712
21. Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, et al. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature (2013) 504(7479):301–5. doi: 10.1038/nature12754
22. Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer (2005) 5:405–12. doi: 10.1038/nrc1612
23. Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem (1998) 273(24):15030–4. doi: 10.1074/jbc.273.24.15030
24. Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Young SG. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. JBiolChem (2000) 275:17605–10. doi: 10.1074/jbc.C000079200
25. Borini Etichetti C, Di Benedetto C, Rossi C, Baglioni MV, Bicciato S, Del Sal G, et al. Isoprenylcysteine carboxy methyltransferase (ICMT) is associated with tumor aggressiveness and its expression is controlled by the p53 tumor suppressor. J Biol Chem (2019) 294(13):5060–73. doi: 10.1074/jbc.RA118.006037
26. Xu J, Zhu Y, Wang F, Zhou Y, Xia G, Xu W. ICMT contributes to hepatocellular carcinoma growth, survival, migration and chemoresistance via multiple oncogenic pathways. Biochem Biophys Res Commun (2019) 518(3):584–9. doi: 10.1016/j.bbrc.2019.08.094
27. Inglese J, Glickman JF, Lorenz W, Caron MG, Lefkowitz RJ. Isoprenylation of a protein kinase: Requirement of farnesylation/α-carboxyl methylation for full enzymatic activity of rhodopsin kinase. J Biol Chem (1992) 267(3):1422–5.
28. Christiansen JR, Pendse ND, Kolandaivelu S, Bergo MO, Young SG, Ramamurthy V. Deficiency of isoprenylcysteine carboxyl methyltransferase (ICMT) leads to progressive loss of photoreceptor function. J Neurosci (2016) 36(18):5107–14. doi: 10.1523/JNEUROSCI.0176-16.2016
29. Philips MR, Staud R, Pillinger M, Feoktistov A, Volker C, Stock JB, et al. Activation-dependent carboxyl methylation of neutrophil G-protein γ subunit. Proc Natl Acad Sci USA (1995) 92(6):2283–7. doi: 10.1073/pnas.92.6.2283
30. Michaelson D, Ahearn I, Bergo M, Young S, Philips M. Membrane trafficking of heterotrimeric G proteins via the endoplasmic reticulum and golgi. Mol Biol Cell (2002) 13(9):3294–302. doi: 10.1091/mbc.E02-02-0095
31. Maske CP, Hollinshead MS, Higbee NC, Bergo MO, Young SG, Vaux DJ. A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J Cell Biol (2003) 162(7):1223–32. doi: 10.1083/jcb.200303113
32. Malhas A, Lee CF, Sanders R, Saunders NJ, Vaux DJ. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J Cell Biol (2007) 176(5):593–603. doi: 10.1083/jcb.200607054
33. Adam SA, Butin-Israeli V, Cleland MM, Shimi T, Goldman RD. Disruption of lamin B1 and lamin B2 processing and localization by farnesyltransferase inhibitors. Nucl (U S) (2013) 4(2):142–50. doi: 10.4161/nucl.24089
34. Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci USA (2002) 99(20):13049–54. doi: 10.1073/pnas.192460799
35. Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell (2008) 19(12):5398–408. doi: 10.1091/mbc.E08-07-0704
36. Takahashi K, Nakagawa M, Young SG, Yamanaka S. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J Biol Chem (2005) 280(38):32768–74. doi: 10.1074/jbc.M506280200
37. Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, Wright L, et al. Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol Biol Cell (2005) 16:1606–16. doi: 10.1091/mbc.E04-11-0960
38. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol (2008) 9:517–31. doi: 10.1038/nrm2438
39. Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Gomes AQ. et al. Isoprenylcysteine Carboxyl Methyltransferase Deficiency in Mice. J Biol Chem (2001) 276:5841–5. doi: 10.1074/jbc.C000831200
40. Li GP, Stahl PD. Post-translational processing and membrane association of the two early endosome-associated rab GTP-binding proteins (rab4 and rab5). Arch Biochem Biophys (1993) 304(2):471–8. doi: 10.1006/abbi.1993.1377
41. Ka FL, Baron R, Ali BR, Magee AI, Seabra MC. Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J Biol Chem (2007)282(2):1487–97. doi: 10.1074/jbc.M605557200
42. Joberty G, Tavitian A, Zahraoui A. Isoprenylation of Rab proteins possessing a C-terminal CaaX motif. FEBS Lett (1993) 330(3):323–8. doi: 10.1016/0014-5793(93)80897-4
43. Kinsella BT, Erdman RA, Maltese WA. Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J Biol Chem (1991) 266(15):9786–94.
44. Falsetti SC, Wang D, Peng H, Carrico D, Cox AD, Der CJ, et al. Geranylgeranyltransferase I Inhibitors Target RalB To Inhibit Anchorage-Dependent Growth and Induce Apoptosis and RalA To Inhibit Anchorage-Independent Growth. Mol Cell Biol (2007) 27(22):8003–14. doi: 10.1128/mcb.00057-07
45. Gentry LR, Nishimura A, Cox AD, Martin TD, Tsygankov D, Nishida M. et al. Divergent roles of CAAX motif-signaled posttranslational modifications in the regulation and subcellular localization of Ral GTPases. J Biol Chem (2015) 290:22851–61. doi: 10.1074/jbc.M115.656710
46. Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem (1997) 272(16):10608–15. doi: 10.1074/jbc.272.16.10608
47. Yoshida Y, Kawata M, Katayama M, Horiuchi H, Kita Y, Takai Y. A geranylgeranyltransferase for rhoA p21 distinct from the farnesyltransferase for ras p21A. Biochem Biophys Res Commun (1991) 175(2):720–8. doi: 10.1016/0006-291X(91)91625-M
48. Katayama M, Kawata M, Yoshida Y, Horiuchi H, Yamamoto T, Matsuura Y, et al. The posttranslationally modified C-terminal structure of bovine aortic smooth muscle rhoA p21. J Biol Chem (1991) 266(19):12639–45.
49. Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, et al. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem (2008) 283:25150–63. doi: 10.1074/jbc.M800882200
50. Armstrong SA, Hannah VC, Goldstein JL, Brown MS. CAAX geranylgeranyl transferase transfers farnesyl as efficiently as geranylgeranyl to RhoB. J Biol Chem (1995) 270(14):7864–8. doi: 10.1074/jbc.270.14.7864
51. Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21(rho) proteins. J Biol Chem (1992) 267(28):20033–8.
52. Yamane HK, Farnsworth CC, Xie H, Evans T, Howald WN, Gelb MH, et al. Membrane-binding domain of the small G protein G25K contains an S-(all-trans-geranylgeranyl)cysteine methyl ester at its carboxyl terminus. Proc Natl Acad Sci USA (1991) 88(1):286–90. doi: 10.1073/pnas.88.1.286
53. Cushman I, Casey PJ. Role of isoprenylcysteine carboxylmethyltransferase-catalyzed methylation in Rho function and migration. JBiolChem (2009) 284:27964–73. doi: 10.1074/jbc.M109.025296
54. Joyce PL, Cox AD. Rac1 and Rac3 Are Targets for Geranylgeranyltransferase I Inhibitor-Mediated Inhibition of Signaling, Transformation, and Membrane Ruffling. Cancer Res (2003) 63(22):7959–67.
55. Philips MR, Pillinger MH, Staud R, Volker C, Rosenfeld MG, Weissmann G, et al. Carboxyl methylation of ras-related proteins during signal transduction in neutrophils. Sci (80- ) (1993) 259(5097):977–80. doi: 10.1126/science.8438158
56. Zhu WL, Hossain MS, Guo DY, Liu S, Tong H, Khakpoor A, et al. A role for Rac3 GTPase in the regulation of autophagy. J Biol Chem (2011) 286(40):35291–8. doi: 10.1074/jbc.M111.280990
57. Ong OC, Ota IM, Clarke S, Fung BKK. The membrane binding domain of rod cGMP phosphodiesterase is posttranslationally modified by methyl esterification at a C-terminal cysteine. Proc Natl Acad Sci USA (1989) 86(23):9238–42. doi: 10.1073/pnas.86.23.9238
58. Anant JS, Ong OC, Xie H, Clarke S, O’Brien PJ, Fung BKK. In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J Biol Chem (1992) 267(2):687–90.
59. Kitten GT, Nigg EA. The CaaX motif is required for isoprenylation, carboxyl methylation,and nuclear membrane association of lamin B2. J Cell Biol (1991) 113(1):13–23. doi: 10.1083/jcb.113.1.13
60. Farnsworth CC, Kawata M, Yoshida Y, Takai Y, Gelb MH, Glomset JA. C terminus of the small GTP-binding protein smg p25A contains two geranylgeranylated cysteine residues and a methyl ester. Proc Natl Acad Sci USA (1991) 88(14):6196–200. doi: 10.1073/pnas.88.14.6196
61. Buss JE, Quilliam LA, Kato K, Casey PJ, Solski PA, Wong G, et al. The COOH-terminal domain of the Rap1A (Krev-1) protein is isoprenylated and supports transformation by an H-Ras:Rap1A chimeric protein. Mol Cell Biol (1991) 11(3):1523–30. doi: 10.1128/mcb.11.3.1523
62. Kawata M, Farnsworth CC, Yoshida Y, Gelb MH, Glomset JA, Takai Y. Posttranslationally processed structure of the human platelet protein smg p21B: Evidence for geranylgeranylation and carboxyl methylation of the C-terminal cysteine. Proc Natl Acad Sci USA (1990) 87(22):8960–4. doi: 10.1073/pnas.87.22.8960
63. Houde VP, Ritorto MS, Gourlay R, Varghese J, Davies P, Shpiro N, et al. Investigation of LKB1 Ser431 phosphorylation and Cys 433 farnesylation using mouse knockin analysis reveals an unexpected role of prenylation in regulating AMPK activity. Biochem J (2014) 458(1):41–56. doi: 10.1042/BJ20131324
64. Cates CA, Michael RL, Stayrook KR, Harvey KA, Burke YD, Randall SK, et al. Prenylation of oncogenic human PTPcaax protein tyrosine phosphatases. Cancer Lett (1996) 110(1–2):49–55. doi: 10.1016/S0304-3835(96)04459-X
65. Farrell FX, Yamamoto K, Lapetina EG. Prenyl group identification of rap2 proteins: A ras superfamily member other than ras that is farnesylated. Biochem J (1993) 289(Pt 2)(Pt 2):349–55. doi: 10.1042/bj2890349
66. Paganini S, Guidetti GF, Catricalà S, Trionfini P, Panelli S, Balduini C, et al. Identification and biochemical characterization of Rap2C, a new member of the Rap family of small GTP-binding proteins. Biochimie (2006) 88(3–4):285–95. doi: 10.1016/j.biochi.2005.08.007
67. Chan LN, Hart C, Guo L, Nyberg T, Davies BSJ, Fong LG, et al. A novel approach to tag and identify geranylgeranylated proteins. Electrophoresis (2009) 30(20):3598–606. doi: 10.1002/elps.200900259
68. Cao W, Mattagajasingh SN, Xu H, Kim K, Fierlbeck W, Deng J, et al. TIMAP, a novel CAAX box protein regulated by TGF-β1 and expressed in endothelial cells. Am J Physiol - Cell Physiol (2002) 283(1):C327–37. doi: 10.1152/ajpcell.00442.2001
69. Boratkó A, Gergely P, Csortos C. RACK1 is involved in endothelial barrier regulation via its two novel interacting partners. Cell Commun Signal (2013) 11(1):2. doi: 10.1186/1478-811x-11-2
70. Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. Towards complete sets of farnesylated and geranylgeranylated proteins. PloS Comput Biol (2007) 3:634–48. doi: 10.1371/journal.pcbi.0030066
71. Nguyen UTT, Guo Z, Delon C, Wu Y, Deraeve C, Fränzel B, et al. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat Chem Biol (2009) 5(4):227–35. doi: 10.1038/nchembio.149
72. Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell (1989) 57:1167–77. doi: 10.1016/0092-8674(89)90054-8
73. Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci (1989) 86:8323–7. doi: 10.1073/pnas.86.21.8323
74. Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. JClinInvest (2004) 113:539–50. doi: 10.1172/JCI18829
75. Wahlstrom AM, Cutts BA, Liu M, Lindskog A, Karlsson C, Sjogren AK, et al. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood (2008) 112:1357–65. doi: 10.1182/blood-2007-06-094060
76. Lau H, Tang J, Casey PJ, Wang M. Isoprenylcysteine carboxylmethyltransferase is critical for malignant transformation and tumor maintenance by all RAS isoforms. Oncogene (2017) 36:3934–42. doi: 10.1038/onc.2016.508
77. Court H, Amoyel M, Hackman M, Lee KE, Xu R, Miller G, et al. Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. JClinInvest (2013) 123:4681–94. doi: 10.1172/JCI65764
78. Weissmueller S, Manchado E, Saborowski M, Morris JP, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell (2014) 157:382–94. doi: 10.1016/j.cell.2014.01.066
79. Zhou HC, Fang JH, Luo X, Zhang L, Yang J, Zhang C, et al. Downregulation of microRNA-100 enhances the ICMT-Rac1 signaling and promotes metastasis of hepatocellular carcinoma cells. Oncotarget (2014) 5:12177–88. doi: 10.18632/oncotarget.2601
80. Do MT, Chai TF, Casey PJ, Wang M. Isoprenylcysteine carboxylmethyltransferase function is essential for RAB4A-mediated integrin β3 recycling, cell migration and cancer metastasis. Oncogene (2017) 36:5757–67. doi: 10.1038/onc.2017.183
81. Backlund PS. Post-translational processing of RhoA. Carboxyl methylation of the carboxyl-terminal prenylcysteine increases the half-life of RhoA. J Biol Chem (1997) 272:33175–80. doi: 10.1074/jbc.272.52.33175
82. Cushman I, Cushman SM, Potter PM, Casey PJ. Control of RhoA methylation by carboxylesterase I. J Biol Chem (2013) 288:19177–83. doi: 10.1074/jbc.M113.467407
83. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res (2012) 72(15):3709–14. doi: 10.1158/0008-5472.CAN-11-4112
84. Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab (2014) 2:23. doi: 10.1186/2049-3002-2-23
85. Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenasedependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA (2011) 108(49):19611–6. doi: 10.1073/pnas.1117773108
86. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2012) 481(7381):380–4. doi: 10.1038/nature10602
87. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res (2008) 68(20):8547–54. doi: 10.1158/0008-5472.CAN-08-1235
88. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell (2014) 159(7):1591–602. doi: 10.1016/j.cell.2014.11.020
89. Ma QL, Wang JH, Wang YG, Hu C, Mu QT, Yu MX, et al. High IDH1 expression is associated with a poor prognosis in cytogenetically normal acute myeloid leukemia. Int J Cancer (2015) 137(5):1058–65. doi: 10.1002/ijc.29395
90. Dahl ES, Buj R, Leon KE, Newell JM, Imamura Y, Bitler BG, et al. Targeting IDH1 as a prosenescent therapy in high-grade serous ovarian cancer. Mol Cancer Res (2019) 17(8):1710–20. doi: 10.1158/1541-7786.MCR-18-1233
91. Sato R, Okamoto A, Inoue J, Miyamoto W, Sakai Y, Emoto N, et al. Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J Biol Chem (2000) 275(17):12497–502. doi: 10.1074/jbc.275.17.12497
92. Moon YA, Lee JJ, Park SW, Ahn YH, Kim KS. The roles of sterol regulatory element-binding proteins in the transactivation of the rat ATP citrate-lyase promoter. J Biol Chem (2000) 275(17):12497–502. doi: 10.1074/jbc.M001066200
93. Ricoult SJH, Dibble CC, Asara JM, Manning BD. Sterol Regulatory Element Binding Protein Regulates the Expression and Metabolic Functions of Wild-Type and Oncogenic IDH1. Mol Cell Biol (2016) 36(18):2384–95. doi: 10.1128/mcb.00163-16
94. Shechter I, Dai P, Huo L, Guan G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: Evidence that IDH1 may regulate lipogenesis in hepatic cells. J Lipid Res (2003) 44(11):2169–80. doi: 10.1194/jlr.M300285-JLR200
95. Xu H, Luo J, Ma G, Zhang X, Yao D, Li M, et al. Acyl-CoA synthetase short-chain family member 2 (ACSS2) is regulatedby SREBP-1 and plays a role in fatty acid synthesis in caprine mammary epithelial cells. J Cell Physiol (2018) 233(2):1005–16. doi: 10.1002/jcp.25954
96. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
97. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3(3):187–97. doi: 10.1016/j.cmet.2006.01.012
98. Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer (2019) 19(11):625–37. doi: 10.1038/s41568-019-0187-8
99. Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med (2019) 25(5):825–37. doi: 10.1038/s41591-019-0423-5
100. Murín R, Vidomanová E, Kowtharapu BS, Hatok J, Dobrota D. Role of S-adenosylmethionine cycle in carcinogenesis. Gen Physiol Biophys (2017)36(5):513–20. doi: 10.4149/gpb_2017031
101. Hoffman RM, Erbe RW. High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc Natl Acad Sci USA (1976) 73(5):1523–7. doi: 10.1073/pnas.73.5.1523
102. Wnuk SF, Yuan CS, Borchardt RT, Balzarini J, De Clercq E, Robins MJ. Anticancer and antiviral effects and inactivation of S-adenosyl-L- homocysteine hydrolase with 5’-carboxaldehydes and oximes synthesized from adenosine and sugar-modified analogues. J Med Chem (1997) 40(11):1608–18. doi: 10.1021/jm960828p
103. Patel-Thombre U, Borchardt RT. Adenine Nucleoside Dialdehydes: Potent Inhibitors of Bovine Liver S-Adenosylhomocysteine Hydrolase. Biochemistry (1985) 24(5):1130–6. doi: 10.1021/bi00326a010
104. Kim JH, Kim JH, Kim SC, Yi YS, Yang WS, Yang Y, et al. Adenosine dialdehyde suppresses MMP-9-mediated invasion of cancer cells by blocking the Ras/Raf-1/ERK/AP-1 signaling pathway. Biochem Pharmacol (2013) 86:1285–300. doi: 10.1016/j.bcp.2013.08.022
105. Onono F, Subramanian T, Sunkara M, Subramanian KL, Peter Spielmann H, Morris AJ. Efficient use of exogenous isoprenols for protein isoprenylation by MDA-MB-231 cells is regulated independently of the mevalonate pathway. J Biol Chem (2013) 288(38):27444–55. doi: 10.1074/jbc.M113.482307
106. De Wolf E, Abdullah MI, Jones SM, Menezes K, Moss DM, Drijfhout FP, et al. Dietary geranylgeraniol can limit the activity of pitavastatin as a potential treatment for drug-resistant ovarian cancer. Sci Rep (2017) 7(1):5410. doi: 10.1038/s41598-017-05595-4
107. Yang WS, Yeo SG, Yang S, Kim KH, Yoo BC, Cho JY. Isoprenyl carboxyl methyltransferase inhibitors: a brief review including recent patents. Amino Acids (2017) 49(9):1469–85. doi: 10.1007/s00726-017-2454-x
108. Kloog Y, Elad-Sfadia G, Haklai R, Mor A. Ras chaperones: New targets for cancer and immunotherapy. Enzymes (2013) 33 Pt A:267–89. doi: 10.1016/B978-0-12-416749-0.00012-9
109. Winter-Vann AM, Baron RA, Wong W, dela CJ, York JD, Gooden DM, et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci USA (2005) 102:4336–41. doi: 10.1073/pnas.0408107102
110. Zhu Y, Hu Q, Li H. Isoprenylcysteine carboxylmethyltransferase is associated withnasopharyngeal carcinoma chemoresistance and Ras activation. Biochem Biophys Res Commun (2019) 516(3):784–9. doi: 10.1016/j.bbrc.2019.06.074
Keywords: metastasis, carboxymethylation, actin cytoskeleton, CAAX proteins, cancer, methionine restriction, methionine stress
Citation: Borini Etichetti CM, Arel Zalazar E, Cocordano N and Girardini J (2020) Beyond the Mevalonate Pathway: Control of Post-Prenylation Processing by Mutant p53. Front. Oncol. 10:595034. doi: 10.3389/fonc.2020.595034
Received: 14 August 2020; Accepted: 08 October 2020;
Published: 05 November 2020.
Edited by:
Dawid Walerych, Polish Academy of Sciences, PolandReviewed by:
Apollonia Tullo, National Research Council, ItalyJorg Kobarg, Campinas State University, Brazil
Copyright © 2020 Borini Etichetti, Arel Zalazar, Cocordano and Girardini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Girardini, girardini@idicer-conicet.gob.ar
†These authors have contributed equally to this work