 Luciano Cascione
Luciano Cascione Luca Aresu
Luca Aresu Michael Baudis2,4
Michael Baudis2,4 Francesco Bertoni
Francesco Bertoni- 1Institute of Oncology Research, Faculty of Biomedical Sciences, USI, Bellinzona, Switzerland
- 2SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland
- 3Department of Veterinary Science, University of Turin, Grugliasco, Italy
- 4Department of Molecular Life Science, University of Zurich, Zurich, Switzerland
- 5Oncology Institute of Southern Switzerland, Bellinzona, Switzerland
Copy number aberrations (CNV/CNA) represent a major contribution to the somatic mutation landscapes in cancers, and their identification can lead to the discovery of oncogenetic targets as well as improved disease (sub-) classification. Diffuse large B cell lymphoma (DLBCL) is the most common lymphoma in Western Countries and up to 40% of the affected individuals still succumb to the disease. DLBCL is an heterogenous group of disorders, and we call DLBCL today is not necessarily the same disease of a few years ago. This review focuses on types and frequencies of regional DNA CNVs in DLBCL, not otherwise specified, and in two particular conditions, the transformation from indolent lymphomas and the DLBCL in individuals with immunodeficiency.
Introduction
Copy number aberrations (CNV/CNA) represent a major contribution to the somatic mutation landscapes in cancers, and their identification can lead to the discovery of oncogenetic targets as well as improved disease (sub-) classification (1, 2). In malignant lymphomas, the contribution of partial and complete chromosomal CNV had been recognized early on through cytogenetic analyses (3, 4) and interphase fluorescence in-situ hybridization (FISH) studies (5, 6). The more systematic, genome-wide mapping of CNVs has been facilitated through the development of chromosomal comparative genomic hybridization (CGH) (7, 8) followed by array-based CGH technologies (aCGH) (9, 10) with increasingly higher spatial resolution, as well as through the widespread adoption of SNP-arrays (11) for copy number profiling. More recently the application of high throughput sequencing approaches (12, 13) has led to increasingly precise identification of regional gains or losses of genomic material (14–21), although the frequently used whole-exome sequencing strategies (WES) have limited precision for CNV mapping (22) compared to high-resolution genomic array technologies or whole-genome sequencing WGS). Diffuse large B cell lymphoma (DLBCL) is the most common lymphoma in Western Countries and up to 40% of the affected individuals still succumb to the disease (23–26). DLBCL is an heterogenous group of disorders as it has been demonstrated by studies that have explored transcriptome profiles and/or at DNA alterations in large series of cases (2, 3, 5, 7, 12, 17, 19, 23, 27–40). It is important to mention that the disease we call DLBCL is not necessarily the same of what we called DLBCL just a few years ago. Indeed, the so called “double” or “triple hit lymphomas”, a subgroup of cases with particularly poor prognosis and previously largely included within DLBCL, are now regarded a distinct entity (“High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements”) separate from the “DLBCL, not otherwise specified (NOS)” as expressed in the 2017 WHO classification (24, 31, 39, 41–43). A similar path was previously followed for primary mediastinal B-cell lymphoma (PMBCL), which, based on its very peculiar features (44, 45), was separated from DLBCL and it is considered a distinct clinicopathologic entity (24). Here, we will review the DLBCL genomics with a particular focus on types and frequencies of regional DNA CNVs in DLBCL, not otherwise specified and in two particular conditions, the transformation from indolent lymphomas and the DLBCL in individuals with immunodeficiency.
CNVs and DLBCL
Within DLBCL, at least two main subtypes have been recognized, in which the gene expression profiles show similarities with two types of normal B-cells: the germinal center B-cell like (GCB) subtype and an activated B cell-like (ABC) subtype (15, 46–51). Clinically, those subtypes are characterized by prognostic differences; patients with an ABC DLBCL have a worse outcome than those with GCB DLBCL when treated with the standard chemo-immunotherapy chemotherapy regimen R-CHOP (24, 48, 49). Genetically, GCB and ABC DLBCL present a series of subtype-specific lesions that explain can explain the different biology of the disease, but they also share others that, with a couple of exceptions (BCL6 and MEF2B alterations), are not DLBCL specific and can be observed in other lymphoma types or even in other cancers. Both GCB and ABC DLBCL present genetic alterations on genes encoding chromatin modifiers [KMT2D/MLL2 or KMT2C/MLL3 (mutations); CREEBBP (mutations or 16p13 deletions) or EP300 (mutations or 22q13 deletions)], the germinal center master regulator BCL6 (BCL6 chromosomal translocations, MEF2B mutations), proteins involved in DNA damage response ([TP53 (mutations or 17p13 deletions)], or proteins contributing to immune surveillance [B2M (mutations or 15q21 deletions); CD58 (mutations or 15q21 deletions)]. ABC DLBCL is characterized by lesions in genes involved in NF-κB pathway and B-cell receptor (BCR) signaling [TNFAIP3 (mutations or 6q23 deletions); MYD88, CD79A, CD79B, CARD11 (mutations)], cell cycle [CDKN2A/B (9p21 deletions)], terminal B cell differentiation [PRDM1 (mutations or 6q21 deletions); SPIB (19q13 gains and amplifications)], and apoptosis [BCL2 (18q21 gains or amplifications)]. In addition, ABC DLBCL have common gains affecting chromosome 3, which could might contribute to immune escape (FOXP1, 3p14), NF-κB pathway activation (NFKBIZ, 3q12) and B cell differentiation arrest (BCL6, 3q27) (4, 7, 13, 15, 17, 27, 30–32, 34, 36, 38, 40, 48, 49, 52–54). GCB DLBCL presents lesions leading to deregulated cell motility [GNA13 (mutations)], apoptosis [BCL2 (chromosomal translocations)], cell cycle [MYC (chromosomal translocations)], chromatin regulation [EZH2 (mutations)], immune escape TNFRSF14 (mutations or 1p36 deletions), PI3K/AKT signaling [PTEN (10q23 deletions); MIR17HG (13q31 gains or amplifications)], and DNA damage response [ING1 (deletions)]. As for ABC DLBCL, also GCB DLBCL present some recurrent gains affecting specific (gains of 2p16 with REL) or large and still not fully characterized regions (chromosomes 7 and 12) (15, 16, 49–51, 55, 56). Figure 1 shows examples of genomic profiles obtained in DLBCL.
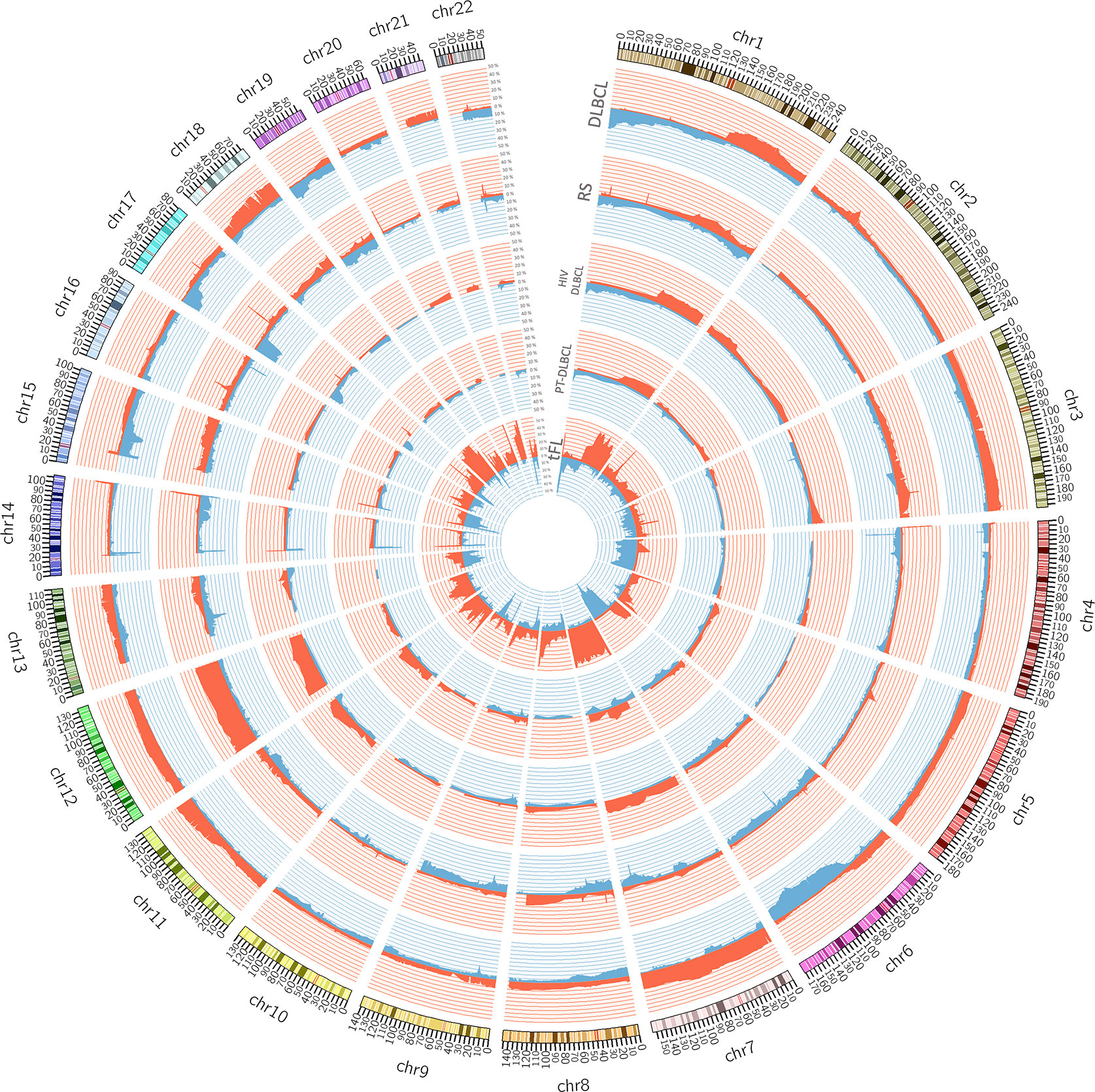
Figure 1 Circos plot summarizing all the copy number changes observed in de novo DLBCL (n. = 22), Richter syndrome (RS, n. = 59), HIV-DLBCL (n. = 50), PT-DLBCL (n. = 44), and transformed FL (tFL, n. = 79). For each histology, the layers represent the frequency of copy number loss (blue) and gain (red). Data are obtained from published papers (28, 30, 33, 57, 58). The plot has been generated using Circos tool (v. 0.69) (53).
The inferior outcome given by the ABC COO alongside the discovery of pathways specifically deregulated in this subtype led to clinical studies designed to target the activation of NF-κB pathway activation. Unfortunately, no advantages for the experimental arms were observed in any of the phase III trials that were looking for improvements in patients classified as ABC DLBCL using gene-expression profiling (34, 59, 60). A possible explanation of these negative results could be not only that treatments that have been explored are not optimal but also that the GCB and ABC subtypes defined at RNA level still comprise too heterogenous patients populations. The latter possibility is strongly sustained by recent studies that have looked at the genetic heterogeneity of DLBCL patients and have led to three novel subclassifications (19–21, 54).
A first classification identifies five clusters (C1-C5) (19) (Table 1). C1 (18% of DLBCL) has cases with BCL6 chromosomal translocations, active NOTCH signaling (NOTCH2 mutations, SPEN inactivation), active NF-κB pathway (TNFAIP3 mutations or deletions, BCL10 mutations), and immune escape mechanisms (inactivation of CD70, CD58, FAS, and structural variations of PD-L1 and PD-L2). C2 (21% of DLBCL) is a mixture of GCB and ABC DLBCL, which share lesions in genes involved in the DNA damage response (TP53 inactivation), cell cycle (inactivation of CDKN2A and RB1), PI3K/AKT signaling (MIR17HG amplifications), and apoptosis (MCL1 gain or amplifications). C3 (13% of all DLBCL) includes GCB-DLBCL with lesions affecting chromatin regulation (EZH2 mutations, KMT2D mutations, CREBBP or EP300 mutations or deletions), PI3K/AKT signaling (PTEN deletions or mutations, mTOR mutations, MIR17HG amplifications), apoptosis (BCL2 chromosomal translocations), cell motility (GNA13 mutations), and germinal center program (MEF2B or IRF8 mutations). The GCB DLBCL C4 (17% of all DLBCL) contains cases with genetic lesions affecting chromatin structure (mutations in linker and core histone genes), immune escape (CD83, CD58, and CD70), NF-κB pathway (mutations of CARD11, NFKBIE, and NFKBIA), BCR and PI3K signaling (mutations of RHOA and SGK1), cell motility (GNA13 mutations), and RAS/JAK/STAT signaling (BRAF and STAT3 mutations). The last one, C5 (21% of all DLBCL) comprises ABC DLBCL cases with BCL2 gains, concordant MYD88 L265P/CD79B plus additional lesions such as gains of 3q, 19q13.42 and inactivation of PRDM1.
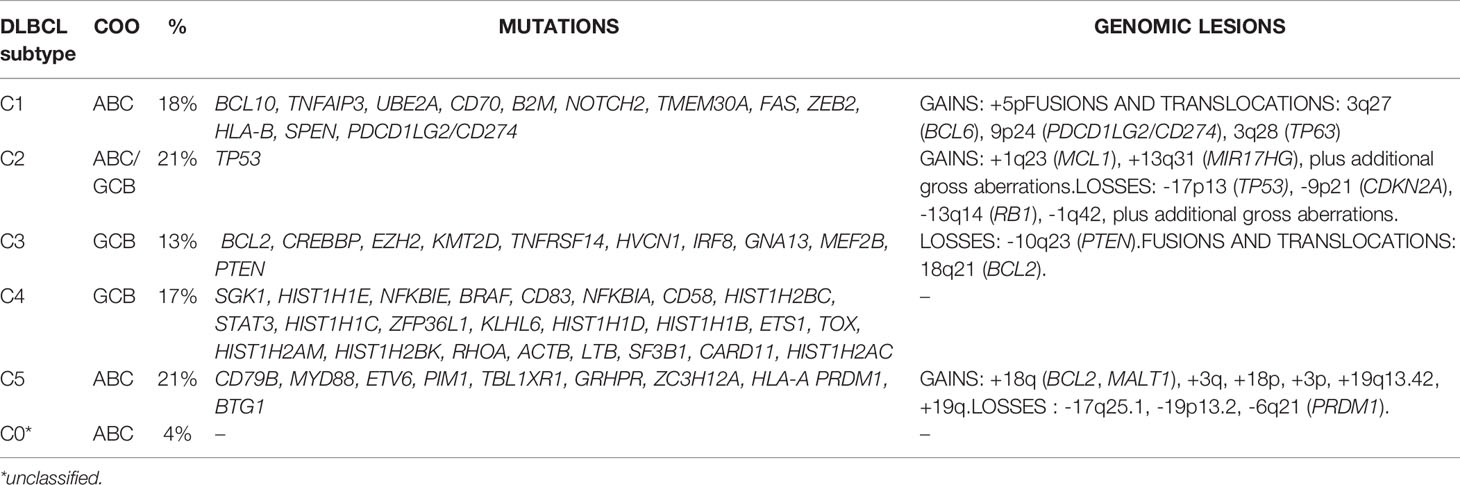
Table 1 DLBCL subtypes according to Chapuy et al. (19).
The second classification originally identified four subtypes (EZB, MCD, N1, and BN2) (20), which more recently have been extended to six (21) (Table 2). Cluster EZB (22% of DLBCL) resembles C3 and the genomic lesions of GCB DLBCL with lesions in genes coding for proteins involved in chromatin regulation (EZH2 mutations, KMT2D mutations, CREEBBP or EP300 mutations or deletions), apoptosis (BCL2 translocations), immune escape (TNFRSF14 mutations or deletions), cell motility (GNA13 mutations), JAK/STAT signaling (STAT6 mutations or amplifications, SOCS1 mutations or deletions), PI3K/AKT signaling (PTEN deletions, mTOR mutations, and MIR17HG amplifications), immune escape (inactivation of TNFRSF14, CIITA, HLA-DMA), and REL amplifications. The MCD cluster (8% of DLBCL), similar to the C5, contains almost exclusively ABC-DLBCL with aberrant activation of the chronic BCR and NF-κB signaling (mutations of MYD88, CD79A, CD79B, and CARD11), impaired terminal B cell differentiation (PRDM1 mutations or deletions, SPIB gains or amplifications), deregulated cell cycle (CDKN2A/B deletions), and immune escape (mutations or deletions of HLA-A, HLA-B, HLA-C, and CD58). The N1 subtype (2% of DLBCL) mostly contains ABC DLBCL with Notch activation (NOTCH1 mutations), NF-κB pathway (TNFAIP3 mutations or deletions), and impaired terminal B cell (lesions of IRF4, ID3, and BCOR). The BN2 (15% of DLBCL), similar to C1, contains both GCB and ABC DLBCL and it is enriched of cases with Notch activation (NOTCH2 mutations or amplifications, mutations of DTX1 or SPEN), BCL6 translocations, NF-κB signaling (inactivation of TNFAIP3 or TN1P1 and gains or amplification of PRKCB and BCL10), immune escape (CD70 inactivation), cell cycle (CCND3 mutations), and cell migration (CXCR5). Since with this classification almost half of DLBCL cases did not fit in any defined subgroup (20), two additional subtypes have been proposed (ST2 and A53) (21). The ST2 subtype (6% of DLBCL) is consists mostly of GCB DLBCL and is characterized by mutations in TET2, SGK1 and JAK/STAT (SOCS1 and STAT3 mutations), and homing effectors (GNA13 and P2RY8). The A53 subtype is enriched of ABC DLBCL and is characterized by TP53 mutations and deletions, with extensive aneuploidy, plus deletions of the B2M locus, amplifications of CNPY3 (6p21), 6q losses (TNFAIP3 and PRDM1), gain/amplification of 3q (NFKBIZ) and BCL2 amplifications. Moreover, following the development of a double-hit gene expression signature identifying GCB-DLBCL patients with no evidence of a dual hit at FISH analysis but an outcome similar to the double-hit patients (36), the EZB group has been divided based on the presence (EZB-MYC+) or absence (EZB-MYC-) of a double hit (DHIT) signature (21).

Table 2 DLBCL subtypes according to Wright et al. (21).
Starting from a series of 928 cases that included also not de novo DLBCL and that were analyzed with a targeted panel of 293 genes, the last classification identifies five subgroups, with names based on their most common lesion (MYD88, BCL2, SOCS1/SGK1, TET2/SGK1, and NOTCH2), leaving 27% of cases unclassified (54) (Table 3). The MYD88 cluster (16%) contains mostly ABC, and genes commonly mutated are MYD88 (L265P), PIM1, CD79B, and ETV6 with also CDKN2A losses. The cluster overlaps with C5 and MCD from the other classifications (19, 21) and contains primary extranodal DLBCL (CNS; testis, breast). The BCL2 cluster (19%) includes mostly GCB DLBCL and the majority of the cases that bear a BCL2 translocation. It has high frequency of mutations of EZH2, BCL2, CREBBP, TNFRSF14, KMT2D, and MEF2B. The cluster overlaps with previously described C3 and EZB (19, 21) and contains most of the transformed FL included in the series. The SOCS1/SGK1 group (12%) presents mutations of SOCS1, CD83, SGK1, NFKBIA, HIST1H1E, and STAT3. The TET2/SGK1 cluster (11%) includes cases with mutations of TET2, SGK1, KLHL6, ZFP36L1, BRAF, MAP2K1, and KRAS. Both the SOCS1/SGK1 and the TET2/SGK1 clusters contain mostly GCB and overlap with the ST2 and C4 of the other classifications. Importantly, the SOCS1/SGK1 cluster also includes the PMBCL cases included in the study (STAT3 and SOCS1 mutations). The last cluster (NOTCH2, 15%) presents mutations of NOTCH2, BCL10, TNFAIP3, CCND3, SPEN, TMEM30A, FAS, and CD70, and cases with BCL6 translocations. It has both GCB and ABC and it overlaps with the previously reported BN2 and C1 clusters (19, 21).

Table 3 DLBCL subtypes according to Lacy et al. (54).
Although similar genetic features are picked up by the three classifications (19–21, 54), the final overlaps are only partial (Table 4), largely due to the approaches used by the Investigators to tackle the issue of DLBCL heterogeneity. However, the two large ABC and GCB subtypes have now been split in subgroups of cases bearing more similar genomic landscapes, and, thus, perhaps sharing more similar responses to targeted therapies (Table 4). New generation of clinical trials can now be designed to assess targeted agents, for example in addition to R-CHOP, in much better genetically defined subgroups of patients.
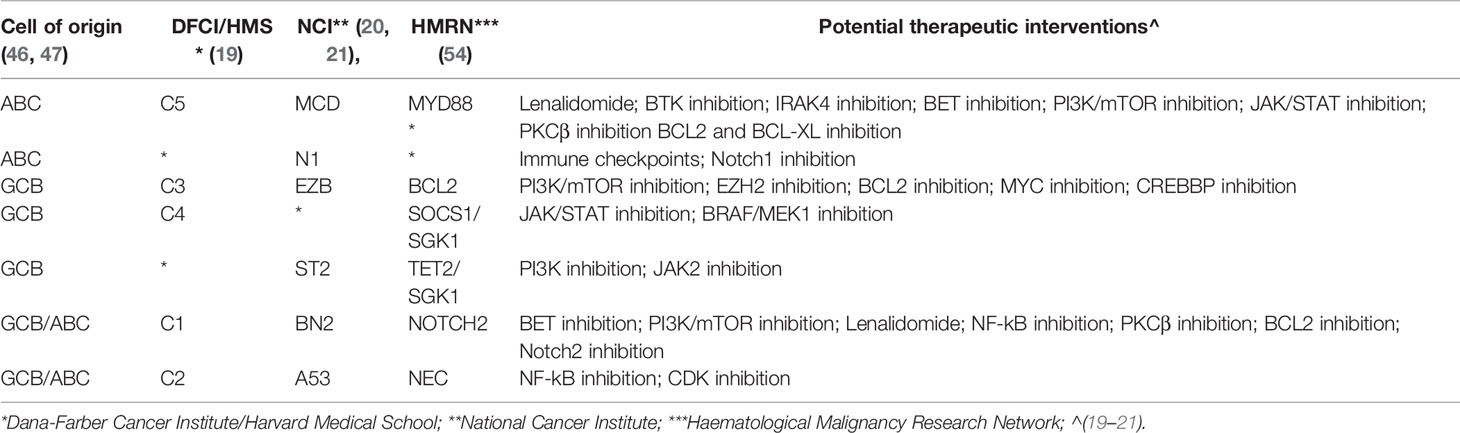
Table 4 Overlaps among DLBCL classifications and potential therapeutic interventions.
Interestingly, the genetics of the individual subtypes suggest that some DLBCL derive from the transformation of indolent lymphomas and/or that they follow specific pathogenetic mechanisms at least partially shared by other lymphoid neoplasms. These connections are evident for C5, MCD, MYD88 (primary extranodal DLBCL of the central nervous system or of the testis; transformed Waldenström macroglobulinemia), C3, EZB, BCL2 [follicular lymphoma (FL); transformed FL; Burkitt lymphoma), N1 [(NOTCH1 mutated chronic lymphocytic leukemia (CLL)], C2, BN2, NOTCH2 (transformed MZL), and ST2 (nodular lymphocyte-predominant Hodgkin lymphoma; T cell/histiocyte-rich large B cell lymphoma) (19–21, 54).
Copy Number Changes and Transformation From Indolent Lymphomas to DLBCL
Copy number changes play important role in the transformation from indolent lymphomas to DLBCL and their presence can also be associated with a higher risk of transformation. Deregulation of MYC via DNA gains, amplifications or chromosomal translocation is the most frequent event occurring at the transformation from FL to DLBCL, followed by inactivation, mainly by DNA loss, of CDKN2A/B, of B2M (losses or mutations) and activating mutations of P1M1 (28, 61). Transformed FL also have higher frequency of 3q and 11q gains than FL (28). Transformed FL and GCB DLBCL are phenotypically similar but their genomic profiles are not the same (28). Here, they present similar frequencies of 1p losses and 2p gains, but overall fewer occurrences of 13q gains (MIR17HG) or losses (ING1), as well of PTEN losses at 10q. Deletions of TNFAIP3 and of CDKN2A are more common in transformed FCL than in GCB DLBCL (28).
A quite similar pattern is observed in the transformation from CLL to DLBCL (Richter syndrome) with the deletion at the CDKN2A/B locus as the most common acquired event (33, 37, 62). Despite the morphological appearance, as a whole, Richter syndrome has a CNV pattern that differs from de novo DLBCL, largely due to the under-representation of DNA gains and losses that are common in the latter disorder. Richter syndrome samples have a higher frequency of deletions at 7q31-q36 (still undefined role) and of the CLL related losses at 13q14.3 and 11q22.3 as well as trisomy 12 (Figure 2). Interestingly, copy number changes define two main subtypes of Richter syndrome (33). A first group (50% of Richter syndrome) bears TP53 inactivation (by loss or by somatic mutations) and/or CDKN2A loss, alongside MYC gain/amplifications, 13q14.3 loss and additional lesions (33). A second group has almost exclusively trisomy 12 (33).
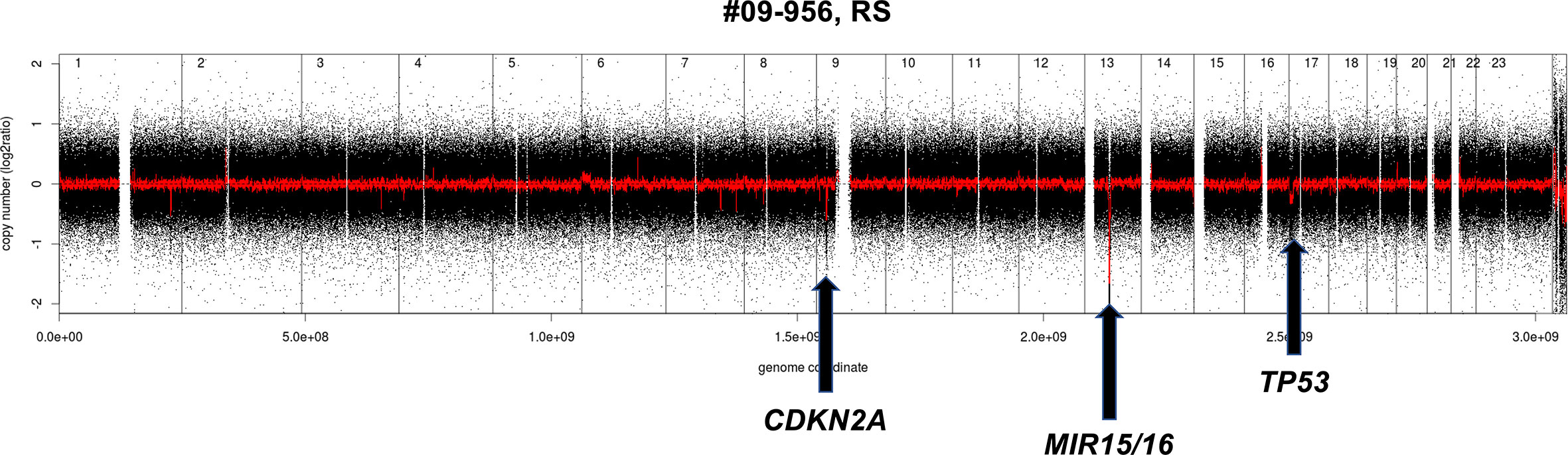
Figure 2 Example of genomic profile of a RS case bearing the typical CDKN2A and MIR15/16 deletion. Profiles obtained using the Affymetrix Genome-Wide Human SNP Array Version 6.0 [modified from (33)]. Black, raw copy number values; red, smoothed copy number values. X-axis, genomic mapping; Y-axis, log2 copy-number values.
Regarding the risk of transformation to DLBCL, deletions at 1p35, 6q and copy neutral LOH at 16p have been associated with higher risk of transformation to DLBCL in FL patients (32, 63). The presence of losses at 17p (TP53), 15q (MGA), and gains at 2p (MYCN, REL) and the lack of 13q14.3 deletions targeting MIR15/MIR16 appeared linked with a higher risk of transformation to Richter syndrome from CLL (33).
Copy Number Changes and Immunodeficiency-Related DLBCL
As there are differences in recurring CNV patterns between GCB and ABC DLBCL as well as between Richter syndrome and de novo DLBCL, a similar observation can be made when comparing the genomic profiles of DLBCL in immunocompetent individuals versus immunodeficiency related DLBCL. This became evident from studies comparing DLBCL obtained in persons with human immunodeficiency virus (HIV) infection in the pre-HAART (highly active antiretroviral therapy) (HIV-DLBCL) era, and in recipients of solid organ transplants (PT-DLBCL) with DLBCL from immunocompetent individuals, all analyzed with the same platform and data mining workflow (30, 57). First, a higher frequency of DNA breakages within fragile sites is seen in immunodeficiency related DLBCL than in immunocompetent cases, with perhaps a higher contribution of these changes to the etiology of the disease. Since viral DNA can insert in fragile sites, the immunodeficiency can expose the individuals to a multitude of viruses, which could infect B cells and integrate in the genome, preferentially at fragile sites (35, 38, 40, 52, 64–67).
Despite their phenotypic reminiscence of post-GC B-cells (29, 68), PT-DLBCL have a pattern of DNA gains and losses that is different from ABC DLBCL, lacking gains of 3q and 18q (BCL2, NFATC1) and losses of 6q (PRDM1 and TNFAIP3) (57). Pre-HAART HIV-DLBCL show genomic profiles that are intermediate between ABC and GCB DLBCL, with more similarities towards the latter. Indeed, HIV-DLBCL has GCB DLBCL lesions such as gains of 2p, 7q, and 12q, as well as losses of 1p, but it also carries 3q and 18q gains, commonly associated with ABC DLBC, and lacks the 10q deletions involving PTEN (30).
While gains of 1q, 11q and of chromosome 7 as well as 17p losses are present in both immunodeficiency related and immunocompetent DLBCL, deletions at 13q14 are usually absent (30, 57) suggesting a possible role in immune escape for the inactivation of MIR15/MIR16 or of RB1, whose loci on 13q are frequently co-deleted in DLBCL (69). Interestingly, the loss of RB1 has been associated with T-cells exclusion in prostate cancer (70). Similarly, PT-DLBCL do not show copy neutral LOH (CN-LOH) affecting 6p, a common feature in different lymphomas including DLBCL and HIV-DLBCL. CN-LOH on 6p is believed to contribute to the silencing of the major histocompatibility complex (71) and DLBCL can indeed show absence or reduced expression of MHC class II proteins (27, 71–73). Importantly, the low MHC-II expression is associated with a decreased number of infiltrating T cells and reduced cytotoxic CD8+ T cells activation (27). Thus, it seems that the immune escape induced by the 6p copy neutral LOH is not required by PT-DLBCL but still needed by HIV-DLBCL. This could be due to the iatrogenic immunosuppression lowering both CD4+ and CD8+ T cells in the first lymphoma type while the viral infection causes a more pronounced loss of CD4+ than of cytotoxic CD8+ T-cells. Similarly, PT-DLBCL also have fewer B2M mutations—another immune escape mechanism—than immunocompetent DLBCL (74). It is also worth mentioning that among immunodeficiency related DLBCL the presence of Epstein Barr virus (EBV) is associated with a lower number of genomic lesions, both in terms of copy number changes and of somatic mutations (30, 57, 74, 75).
A global view of the different genomic profiles of DLBCL, Richter syndrome, immunodeficiency related DLBCL and transformed FL can be seen in Figure 3.
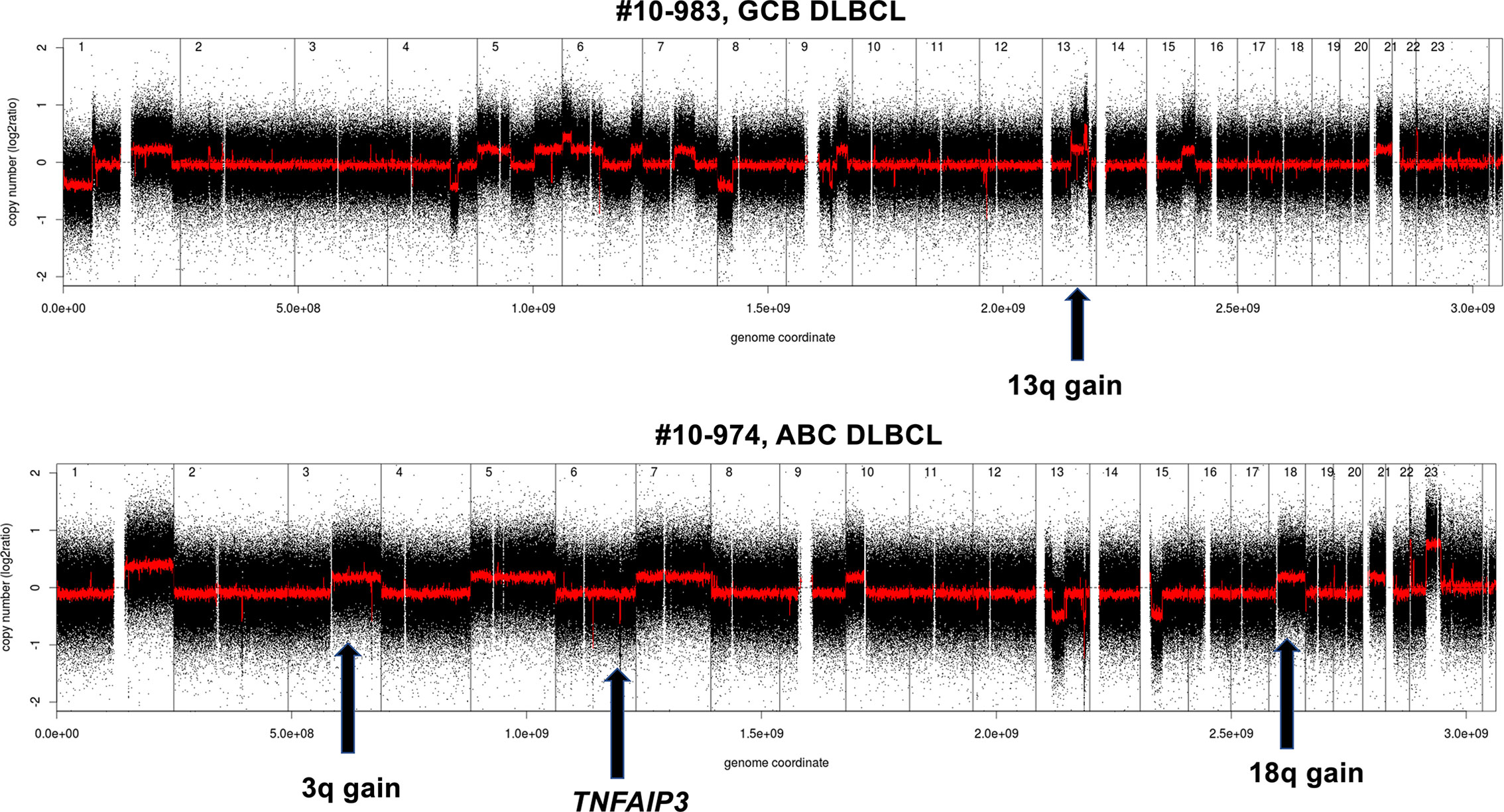
Figure 3 Examples of genomic profile of two DLBCL cases bearing GCB (above) or ABC (below) lesions among others. Profiles obtained using the Affymetrix Genome-Wide Human SNP Array Version 6.0 [modified from (33)]. Black, raw copy number values; red, smoothed copy number values. X-axis, genomic mapping; Y-axis, log2 copy-number values.
Conclusions
Data obtained in all these last years using genome wide technologies that allow for the molecular study of transcriptome profiles and of DNA changes (CNVs or somatic mutations) have led to building a much more precise framework to explain the heterogenous biology and clinical course of DLBCL cases. Although novel approaches such as the use of liquid biopsies are becoming increasingly feasible at least in the context of clinical trials, reproducible and commonly agreed genetic classification systems have to be defined. This is necessary to compare results from future individual clinical trials and to then transfer the findings to the right patients in the clinical practice. Indeed, the identification of group of patients with homogenous patterns of genetic lesions leading to the deregulation of specific pathways represents an opportunity to study novel agents in a more targeted approach than done so far, hopefully overcoming the disappointing results obtained trying to target the ABC DLBCL subtype defined based on gene expression profiling.
Author Contributions
All authors participated to the design of the review, literature revision, manuscript writing, and final revision. All authors contributed to the article and approved the submitted version.
Funding
Partially supported Oncosuisse (02296-08-2008, 1939-8-2006), and Swiss National Science Foundation (Sinergia 147620), Bern, Switzerland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baudis M. Genomic imbalances in 5918 malignant epithelial tumors: an explorative meta-analysis of chromosomal CGH data. BMC Cancer (2007) 7:226. doi: 10.1186/1471-2407-7-226
2. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463(7283):899–905. doi: 10.1038/nature08822
3. Bloomfield CD, Arthur DC, Frizzera G, Levine EG, Peterson BA, Gajl-Peczalska KJ. Nonrandom chromosome abnormalities in lymphoma. Cancer Res (1983) 43(6):2975–84.
4. Levine EG, Bloomfield CD. Cytogenetics of non-Hodgkin’s lymphoma. J Natl Cancer Inst Monogr (1990) 10):7–12.
5. Döhner H, Pohl S, Bulgay-Mörschel M, Stilgenbauer S, Bentz M, Lichter P. Trisomy 12 in chronic lymphoid leukemias–a metaphase and interphase cytogenetic analysis. Leukemia (1993) 7(4):516–20.
6. Younes A, Pugh W, Goodacre A, Katz R, Rodriguez MA, Hill D, et al. Polysomy of chromosome 12 in 60 patients with non-Hodgkin’s lymphoma assessed by fluorescence in situ hybridization: differences between follicular and diffuse large cell lymphoma. Genes Chromosomes Cancer (1994) 9(3):161–7. doi: 10.1002/gcc.2870090303
7. Bentz M, Huck K, du Manoir S, Joos S, Werner CA, Fischer K, et al. Comparative genomic hybridization in chronic B-cell leukemias shows a high incidence of chromosomal gains and losses. Blood (1995) 85(12):3610–8. doi: 10.1182/blood.V85.12.3610.bloodjournal85123610
8. Joos S, Otaño-Joos MI, Ziegler S, Brüderlein S, du Manoir S, Bentz M, et al. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood (1996) 87(4):1571–8. doi: 10.1182/blood.V87.4.1571.bloodjournal8741571
9. Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Döhner H, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer (1997) 20(4):399–407. doi: 10.1002/(SICI)1098-2264(199712)20:4<399::AID-GCC12>3.0.CO;2-I
10. Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet (1998) 20(2):207–11. doi: 10.1038/2524
11. Zhao X, Li C, Paez JG, Chin K, Jänne PA, Chen TH, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res (2004) 64(9):3060–71. doi: 10.1158/0008-5472.CAN-03-3308
12. Campbell PJ, Stephens PJ, Pleasance ED, O’Meara S, Li H, Santarius T, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet (2008) 40(6):722–9. doi: 10.1038/ng.128
13. Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature (2008) 456(7218):66–72. doi: 10.1038/nature07485
14. Monni O, Joensuu H, Franssila K, Knuutila S. DNA copy number changes in diffuse large B-cell lymphoma–comparative genomic hybridization study. Blood (1996) 87(12):5269–78. doi: 10.1182/blood.V87.12.5269.bloodjournal87125269
15. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A (2008) 105(36):13520–5. doi: 10.1073/pnas.0804295105
16. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet (2011) 43(9):830–7. doi: 10.1038/ng.892
17. Guo Y, Takeuchi I, Karnan S, Miyata T, Ohshima K, Seto M. Array-comparative genomic hybridization profiling of immunohistochemical subgroups of diffuse large B-cell lymphoma shows distinct genomic alterations. Cancer Sci (2014) 105(4):481–9. doi: 10.1111/cas.12378
18. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171(2):481–494 e415. doi: 10.1016/j.cell.2017.09.027
19. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90 doi: 10.1038/s41591-018-0016-8
20. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
21. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell (2020) 37(4):551–568 e514. doi: 10.1016/j.ccell.2020.03.015
22. Nam JY, Kim NK, Kim SC, Joung JG, Xi R, Lee S, et al. Evaluation of somatic copy number estimation tools for whole-exome sequencing data. Brief Bioinform (2016) 17(2):185–92. doi: 10.1093/bib/bbv055
23. Gascoyne RD, Campo E, Jaffe ES, Chan WC, Chan JKC, Rosenwald A, et al. Diffuse large B-cell lymphoma, NOS. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri A, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Siebert R, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th Edition. Lyon, France: IARC Press (2017). p. 291–7.
24. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri A, Stein H, et al eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th Edition. Lyon, France: IARC Press (2017).
25. Li S, Young KH, Medeiros LJ. Diffuse large B-cell lymphoma. Pathology (2018) 50(1):74–87. doi: 10.1016/j.pathol.2017.09.006
26. Mondello P, Mian M. Frontline treatment of diffuse large B-cell lymphoma: Beyond R-CHOP. Hematol Oncol (2019) 37(4):333–44. doi: 10.1002/hon.2613
27. Booman M, Szuhai K, Rosenwald A, Hartmann E, Kluin-Nelemans H, de Jong D, et al. Genomic alterations and gene expression in primary diffuse large B-cell lymphomas of immune-privileged sites: the importance of apoptosis and immunomodulatory pathways. J Pathol (2008) 216(2):209–17. doi: 10.1002/path.2399
28. Bouska A, McKeithan TW, Deffenbacher KE, Lachel C, Wright GW, Iqbal J, et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood (2014) 123(11):1681–90. doi: 10.1182/blood-2013-05-500595
29. Capello D, Rossi D, Gaidano G. Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol (2005) 23(2):61–7. doi: 10.1002/hon.751
30. Capello D, Scandurra M, Poretti G, Rancoita PM, Mian M, Gloghini A, et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Br J Haematol (2010) 148(2):245–55. doi: 10.1111/j.1365-2141.2009.07943.x
31. Cheah CY, Oki Y, Westin JR. and Turturro, F. A clinician’s guide to double hit lymphomas. Br J Haematol (2015) 168(6):784–95. doi: 10.1111/bjh.13276
32. Cheung KJ, Shah SP, Steidl C, Johnson N, Relander T, Telenius A, et al. Genome-wide profiling of follicular lymphoma by array comparative genomic hybridization reveals prognostically significant DNA copy number imbalances. Blood (2009) 113(1):137–48. doi: 10.1182/blood-2008-02-140616
33. Chigrinova E, Rinaldi A, Kwee I, Rossi D, Rancoita PM, Strefford JC, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood (2013) 122(15):2673–82. doi: 10.1182/blood-2013-03-489518
34. Davies A, Cummin TE, Barrans S, Maishman T, Mamot C, Novak U, et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): an open-label, randomised, phase 3 trial. Lancet Oncol (2019) 20(5):649–62. doi: 10.1016/S1470-2045(18)30935-5
35. Doolittle-Hall JM, Cunningham Glasspoole DL, Seaman WT, Webster-Cyriaque J. Meta-Analysis of DNA Tumor-Viral Integration Site Selection Indicates a Role for Repeats, Gene Expression and Epigenetics. Cancers (Basel) (2015) 7(4):2217–35. doi: 10.3390/cancers7040887
36. Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol (2019) 37(3):190–201. doi: 10.1200/JCO.18.01583
37. Fabbri G, Khiabanian H, Holmes AB, Wang J, Messina M, Mullighan CG, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med (2013) 210(11):2273–88. doi: 10.1084/jem.20131448
38. Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett (2007) 252(2):157–70. doi: 10.1016/j.canlet.2006.11.010
39. Friedberg JW. How I treat double-hit lymphoma. Blood (2017) 130(5):590–6. doi: 10.1182/blood-2017-04-737320
40. Gao G, Johnson SH, Vasmatzis G, Pauley CE, Tombers NM, Kasperbauer JL, et al. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer (2017) 56(1):59–74. doi: 10.1002/gcc.22415
41. Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, et al. Double-hit B-cell lymphomas. Blood (2011) 117(8):2319–31. doi: 10.1182/blood-2010-09-297879
42. Sesques P, Johnson NA. Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood (2017) 129(3):280–8. doi: 10.1182/blood-2016-02-636316
43. Riedell PA, Smith SM. Double hit and double expressors in lymphoma: Definition and treatment. Cancer (2018) 124(24):4622–32. doi: 10.1002/cncr.31646
44. Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med (2003) 198(6):851–62. doi: 10.1084/jem.20031074
45. Savage KJ, Monti S, Kutok JL, Cattoretti G, Neuberg D, De Leval L, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood (2003) 102(12):3871–9. doi: 10.1182/blood-2003-06-1841
46. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
47. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med (2002) 346(25):1937–47. doi: 10.1056/NEJMoa012914
48. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med (2008) 359(22):2313–23. doi: 10.1056/NEJMoa0802885
49. Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med (2010) 362(15):1417–29. doi: 10.1056/NEJMra0807082
50. Rui L, Schmitz R, Ceribelli M, Staudt LM. Malignant pirates of the immune system. Nat Immunol (2011) 12(10):933–40. doi: 10.1038/ni.2094
51. Shaffer AL,3, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol (2012) 30:565–610. doi: 10.1146/annurev-immunol-020711-075027
52. Katano H, Sato Y, Hoshino S, Tachikawa N, Oka S, Morishita Y, et al. Integration of HIV-1 caused STAT3-associated B cell lymphoma in an AIDS patient. Microbes Infect (2007) 9(14-15):1581–9. doi: 10.1016/j.micinf.2007.09.008
53. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res (2009) 19(9):1639–45. doi: 10.1101/gr.092759.109
54. Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood (2020) 135(20):1759–71. doi: 10.1182/blood.2019003535
55. Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood (2002) 99(7):2285–90. doi: 10.1182/blood.V99.7.2285
56. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature (2011) 471(7337):189–95. doi: 10.1038/nature09730
57. Rinaldi A, Capello D, Scandurra M, Greiner TC, Chan WC, Bhagat G, et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. Br J Haematol (2010) 149(4):569–77. doi: 10.1111/j.1365-2141.2010.08125.x
58. Scandurra M, Mian M, Greiner TC, Rancoita PM, De Campos CP, Chan WC, et al. Genomic lesions associated with a different clinical outcome in diffuse large B-Cell lymphoma treated with R-CHOP-21. Br J Haematol (2010) 151(3):221–31. doi: 10.1111/j.1365-2141.2010.08326.x
59. Iacoboni G, Zucca E, Ghielmini M, Stathis A. Methodology of clinical trials evaluating the incorporation of new drugs in the first-line treatment of patients with diffuse large B-cell lymphoma (DLBCL): a critical review. Ann Oncol (2018) 29(5):1120–9. doi: 10.1093/annonc/mdy113
60. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol (2019) 37(15):1285–95. doi: 10.1200/JCO.18.02403
61. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep (2014) 6(1):130–40. doi: 10.1016/j.celrep.2013.12.027
62. Beà S, López-Guillermo A, Ribas M, Puig X, Pinyol M, Carrió A, et al. Genetic imbalances in progressed B-cell chronic lymphocytic leukemia and transformed large-cell lymphoma (Richter’s syndrome). Am J Pathol (2002) 161(3):957–68. doi: 10.1016/S0002-9440(10)64256-3
63. O’Shea D, O’Riain C, Gupta M, Waters R, Yang Y, Wrench D, et al. Regions of acquired uniparental disomy at diagnosis of follicular lymphoma are associated with both overall survival and risk of transformation. Blood (2009) 113(10):2298–301. doi: 10.1182/blood-2008-08-174953
64. Rassool FV, Le Beau MM, Neilly ME, van Melle E, Espinosa R,3, McKeithan TW. Increased genetic instability of the common fragile site at 3p14 after integration of exogenous DNA. Am J Hum Genet (1992) 50(6):1243–51.
65. Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene (2003) 22(8):1225–37. doi: 10.1038/sj.onc.1206170
66. Luo WJ, Takakuwa T, Ham MF, Wada N, Liu A, Fujita S, et al. Epstein-Barr virus is integrated between REL and BCL-11A in American Burkitt lymphoma cell line (NAB-2). Lab Invest (2004) 84(9):1193–9. doi: 10.1038/labinvest.3700152
67. Tang D, Li B, Xu T, Hu R, Tan D, Song X, et al. VISDB: a manually curated database of viral integration sites in the human genome. Nucleic Acids Res (2020) 48(D1):D633–d641. doi: 10.1093/nar/gkz867
68. Vakiani E, Basso K, Klein U, Mansukhani MM, Narayan G, Smith PM, et al. Genetic and phenotypic analysis of B-cell post-transplant lymphoproliferative disorders provides insights into disease biology. Hematol Oncol (2008) 26(4):199–211. doi: 10.1002/hon.859
69. Mian M, Scandurra M, Chigrinova E, Shen Y, Inghirami G, Greiner TC, et al. Clinical and molecular characterization of diffuse large B-cell lymphomas with 13q14.3 deletion. Ann Oncol (2012) 23(3):729–35. doi: 10.1093/annonc/mdr289
70. Olson B, Bao R, Fessler J, Luke J, Gajewski TF, Patnaik A. Abstract 5737: Genomic drivers of cancer are enriched and mutually exclusive within non-T cell-inflamed tumors. Cancer Res (2018) 78(13 Supplement):5737–7. doi: 10.1158/1538-7445.AM2018-5737
71. Riemersma SA, Jordanova ES, Schop RF, Philippo K, Looijenga LH, Schuuring E, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood (2000) 96(10):3569–77. doi: 10.1182/blood.V96.10.3569
72. Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood (2004) 103(11):4251–8. doi: 10.1182/blood-2003-07-2365
73. Roberts RA, Rimsza LM, Staudt LM, Rosenwald A, Chan WC, Dave S, et al. Gene expression differences between low and high stage diffuse large B cell lymphoma. Blood (2006) 108(111):243a. doi: 10.1182/blood.V108.11.809.809
74. Menter T, Juskevicius D, Alikian M, Steiger J, Dirnhofer S, Tzankov A, et al. Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br J Haematol (2017) 178(1):48–56. doi: 10.1111/bjh.14633
Keywords: copy number aberrations, genetic alteration, lymphoma, diffuse large B cell lymphoma, hematological malignancies, MYC, TP53, CDKN2A
Citation: Cascione L, Aresu L, Baudis M and Bertoni F (2020) DNA Copy Number Changes in Diffuse Large B Cell Lymphomas. Front. Oncol. 10:584095. doi: 10.3389/fonc.2020.584095
Received: 16 July 2020; Accepted: 29 October 2020;
Published: 02 December 2020.
Edited by:
Luca Agnelli, University of Milan, ItalyReviewed by:
Antonello Domenico Cabras, Istituto Nazionale dei Tumori (IRCCS), ItalyAnnalisa Chiappella, Istituto Nazionale dei Tumori (IRCCS), Italy
Copyright © 2020 Cascione, Aresu, Baudis and Bertoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Bertoni, frbertoni@mac.com