 Runjiao Zhang
Runjiao Zhang Li Dong
Li Dong Jinpu Yu
Jinpu Yu- 1Cancer Molecular Diagnostics Core, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center of Caner, Key Laboratory of Cancer Prevention and Therapy, Key Laboratory of Cancer Immunology and Biotherapy, Tianjin, China
- 2Tianjin's Clinical Research Center for Cancer, Tianjin, China
Driver oncogene alterations have always been one of leading causes in the process of occurrence and development of tumors. And the effects of driver oncogene alterations on tumorigenesis and progression in different kinds of tumors have been studied heatedly. And the roles that the driver oncogenes alterations play have been elucidated clearly in previous studies. The phenomenon of concomitant driver oncogenes mutations and driver genes fusions has gained much concentration in the past two decades. And a growing number of studies reported this phenomenon, either coexistence or mutually exclusivity. Here we reviewed on the phenomenon of concomitant mutations in three common types of carcinomas—lung cancer, thyroid cancer, and leukemia, which have been studied relatively more detailed and more general compared with others.
Introduction
Genetic mutations are an important molecular background in tumors, and the most common types of alterations are point mutations and fusions. Currently, a large number of researches have been conducted to deeply study the influences of driver oncogenes (e.g. ALK, RAS, RAF, etc.) on the occurrence and development of different kinds of cancers. It has been correspondingly clear that driver oncogene mutations play diverse roles and are of vital importance in different types of tumors in the process of carcinogenesis, development, invasiveness, and metastasis. Also, effects of common driver gene fusions (e.g. RET fusion, etc.) on the oncogenesis and progression of different kinds of tumors have been demonstrated clearly by lots of studies. Nevertheless, studies and cognitions on the phenomenon of concomitant pathogenic mutations and fusions of driver oncogenes in tumors are relatively limited and not very completely investigated. Furthermore, in different types of carcinomas, dual driver mutations and driver gene fusions has different effect on the occurrence, development, invasiveness, and metastasis of tumors; what’s more, the occurrent frequency of this dual mutations and fusions in diverse neoplasms is also different. A portion of studies have been conducted to study simultaneous proto-oncogene mutations and driver gene fusions in different kinds of tumors in the past few decades. Herein we review on the coexistence of pathogenic mutations and fusions of driver oncogenes in lung cancer, thyroid cancer and leukemia, in which this phenomenon have been identified. Furthermore, we discuss the influences on tumors of this phenomenon, including clinical pathological features and prognosis of patients harboring dual mutations and fusions.
Concomitant Oncogene Mutations and Rearrangements in Lung Cancer
Lung cancer is the most commonly diagnosed carcinoma among different kinds of malignancies and is also the leading cause of cancer-related death both in China and worldwide (1–4). Lung cancer consists of two groups, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). And NSCLC is further divided into subtypes including large cell carcinoma (LCC), squamous cell lung cancer (SC), and adenocarcinoma (AC) (5). A large proportion of lung cancers are associated with tobacco smoking, while evidence shows that lung cancer in lifelong non-smokers appears to be a distinct disease caused by oncogene driver mutations which are different from the genetic pathways observed in lung cancer of smokers (6–13).
Epidermal growth factor receptor (EGFR) is a transmembrane protein with cytoplasmic kinase activity that transduces important growth factor signaling from the extracellular milieu to cells (14). And EGFR mutations are common in lung cancer. Driver genomic fusions [e.g. anaplastic lymphoma kinase (ALK), ROS1, and KIF5B-RET fusion, etc.] are thought not to be correlated with smoking-associated mutations and frequently served as driver events of smoking-signature-low lung adenocarcinomas. EGFR mutations are more common in Asian women, while ALK fusions showed no gender or racial difference which the age of onset was younger, and occurred more commonly in mucus-type lung cancer in pathology. The phenomenon of coexisting driver mutations and fusions has conventionally been considered to be mutually exclusive (15–25). However, cumulative studies have revealed that concomitant occurrence of driver gene mutations and gene fusions accounts for a small number of NSCLC cases (26–37). And concomitant EGFR mutations and ALK fusions is the most common form among all kinds of coexistence of driver mutations and driver oncogene fusions in NSCLC. In accordance with ALK fusions in lung cancer, the phenomenon of dual driver mutations and gene fusions has also been detected and elucidated in the never smoking patients with NSCLC (mainly lung adenocarcinomas) in several studies. Zhao et al. recruited 5,816 patients with lung cancer from Shanghai, China, and all of the patients are asked to undergo both EGFR mutation and ALK fusion analysis. They found that 2,392 (41.1%) patients had EGFR mutations, 503 (8.7%) had ALK fusions, and 26 (0.45%) had simultaneous EGFR mutations and ALK fusion (38). Won et al. analyzed the EGFR and ALK status in 1,458 cases of lung cancer including NSCLC (n = 1,445) and small-cell carcinoma (n = 13) using direct sequencing and FISH, respectively, and the cohort enrolled in the study are from Seoul, Korea. In this cohort, the EGFR mutation and ALK fusion rates in NSCLC patients were 42.4% (612/1,445) and 6.3% (91/1,445), respectively, and concomitant EGFR and ALK alteration was detected in 4 (0.3%) of the 1,445 NSCLC patients (35). Yang et al. carried out a study in Guangdong, China, which screened a total of 977 patients with NSCLC for the presence of EGFR mutations, ALK fusion and coexistence of EGFR mutations and ALK fusion. And 336 (34.4%) and 70 (7.2%) patients had EGFR mutations or ALK fusion, respectively. Meanwhile, thirteen (1.3%) patients harbored concomitant EGFR mutations and ALK fusion, all of which were adenocarcinomas and never or light smokers (36). Lee et al. detected EGFR mutations and ALK fusion among 444 Korean lung adenocarcinoma patients. They found 228 (51.4%) patients harbored EGFR mutations and 34 (7.7%) had ALK fusion, meanwhile four patients (0.9%) were found to have both EGFR mutations and ALK fusion (Table 1) (6).
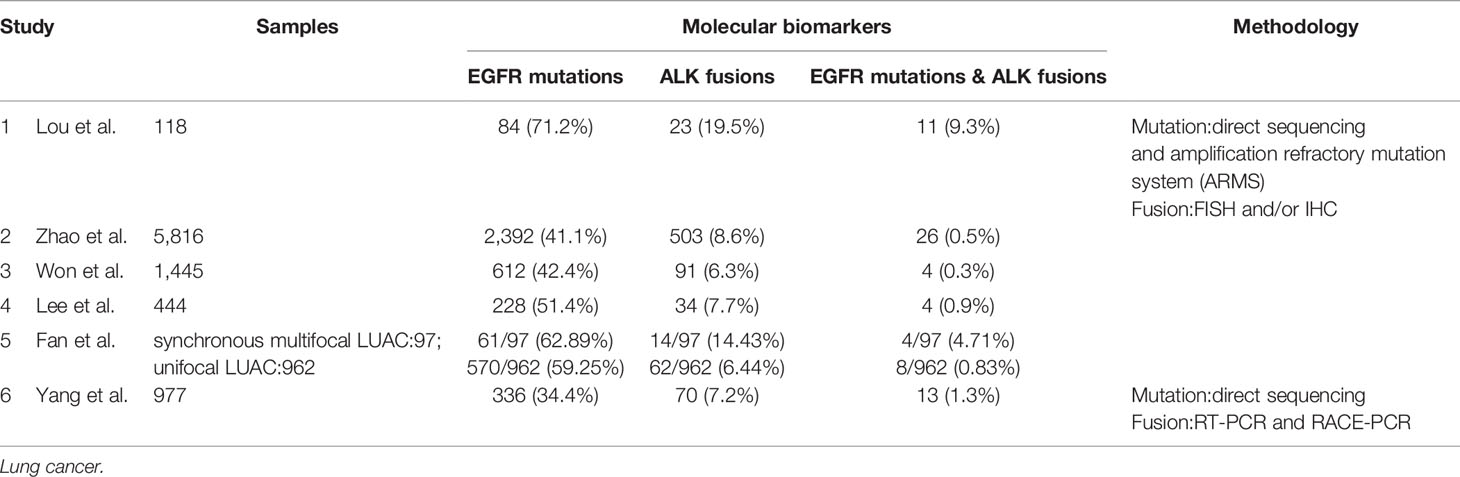
Table 1 Summary of studies on concomitant oncogene mutations and rearrangements in lung cancer.
With the widespread popularization and utilization of low-dose chest computed tomography (CT) as early-stage lung cancer screening, the incidence reported of lung cancer patients who present with multiple lesions ranges from 0.2 to 20%, especially those of multiple lung adenocarcinomas which is a rare molecular subtype of lung adenocarcinomas. A research launched by Fan et al. collected 1,059 patients with lung adenocarcinomas from Hubei, China to detect EGFR and ALK alterations. A total of 97 multiple synchronous lesions were observed among 1,059 LUAC patients, among which patients with concomitant EGFR mutations and ALK fusion were 4.12% (4/97). Comparatively, patients with unifocal LUAC harboring EGFR/ALK co-alterations were 0.83% (8/962). Apparently, the incidence of EGFR/ALK co-alterations in the multifocal LUAC was significantly higher than that in the unifocal LUAC (39). In accordance with the study by Fan et al., another study from Shanghai, China by Wu et al. reported that the rate of EGFR/ALK co-alterations in patients with synchronous multiple lung ground-glass opacity nodules was as high as 8.57% (40).
Although concomitant EGFR mutations and ALK fusion in lung cancer had once been considered to be mutually exclusive, the coexistence of EGFR mutations and ALK fusion in patients with lung cancer has been detected in a series of studies in recent years. Most of these studies are clinical researches, and their research contents mainly include the three aspects below: the correlation between concomitant mutations and clinical pathologic features of patients, influence of dual mutations on patients’ prognoses, potential mechanisms of TKI resistance and guidance of therapeutic regimens.
In regard to the relationship between the status of EGFR and ALK (EGFR mutations, ALK fusions and EGFR/ALK co-alterations) and the clinical pathological features of patients with lung cancer, there are not many studies conducted. Lou et al. reported 11 patients with dual mutations, 84 EGFR mutations and 23 ALK-positive patients with median survival time of 18.5, 21.3, and 23.7 months (p = 0.06), respectively. There was no statistical difference among the three groups, but the survival time of patients with dual mutations was the shortest, and there was a significant difference between ALK-positive group and EGFR/ALK co-alterations group (29). Elisa Brega and Guilherme Brandao also support the results that patients with more than two gene alterations living shorter than those with no or one gene alteration. Although the overall survival (OS) of patients with coexistent mutations is shorter than that of patients with single alteration of EGFR or ALK, the prognosis of patients with dual mutations is still better than that of the patients receiving chemotherapy alone after rational targeted therapy (41).
Main research content of the studies mentioned above is the responses to EGFR TKIs or/and ALK inhibitors among patients with concomitant EGFR/ALK alterations. First generation EGFR TKIs such as gefitinib, erlotinib, and icotinib provide survival benefits over conventional chemotherapy and have revolutionized the therapy of patients with NSCLC with EGFR-activating mutations, as has crizotinib, a TKI targeting ALK fusions, for ALK-positive patients. As for the efficacy of EGFR TKIs and crizotinib in EGFR/ALK double-positive patients, it has been controversial. Some reports have indicated that EGFR TKIs had a better response than ALK inhibitors in terms of objective response rate (ORR) and progression free survival (PFS) (27, 29, 30), but others have come to the opposite conclusion (31, 34, 35, 42). Wu et al. came to the conclusion that for NSCLC patients with coexistence of EGFR mutations and ALK fusion, first-line EGFR TKIs may be a reasonable care. Whether to administer application of crizotinib subsequently depends on ALK fusion status, and relative levels of EGFR and ALK phosphorylation (29). This may provide a new thinking that detecting the abundance of EGFR mutations and ALK fusion and the levels of phosphorylation of downstream proteins can determine whether or not to use sequential therapy or in combination with crizotinib, so that the most effective therapeutic regimens might be optimized for patients with concomitant EGFR and ALK alterations in the future. While their study is single-institution, small-sample, and retrospective, so their result can not accurately reflect the population with EGFR/ALK co-alterations at large. Zhao et al. found that first generation EGFR TKIs and ALK inhibitors in patients with concomitant EGFR mutations and ALK fusion were equally efficacious as in patients with single gene alterations. EGFR/ALK co-altered patients also appeared to have longer overall survival (OS) than patients with EGFR mutant disease after the sequential treatment with EGFR and ALK TKIs. So multi-drug combined therapy might be the best option for patients with simultaneous mutations. Nevertheless, the results have no statistical significance and this might be due to a small number of samples recruited were qualified to be included into the study (38).
The mechanisms of resistance to EGFR or ALK TKIs is another focus that researchers mainly concentrated on. Lou et al. demonstrated that relatively higher level of phospho-EGFR was the mechanism of resistance to crizotinib and the relatively higher level of phospho-ALK may be molecular mechanisms of resistance to EGFR TKIs (29). And Takaaki et al. held the same opinion that activation of EGFR signaling as a bypass signaling mechanism can contribute to ALK inhibitor resistance. In the presence of EGF, crizotinib was still able to inhibit ALK phosphorylation but not AKT, S6 and ERK1/2 phosphorylation (43). This can reasonably explain why single TKI (EGFR TKIs or ALK inhibitors) is not effective in the therapy of patients with dual mutations, and provide treatment advices for patients with concomitant mutations.
Concomitant Oncogene Mutations and Rearrangements in Thyroid Cancer
Thyroid carcinoma is the most common type of endocrine malignancies and can be classified into papillary thyroid carcinomas (PTCs), follicular thyroid carcinomas (FTCs), undifferentiated carcinomas, medullary thyroid carcinomas (MTCs), Hürthle cell carcinomas (HTCs), and poorly differentiated or anaplastic thyroid carcinomas (ATCs) (44). And in all of the multiple types of differentiated thyroid carcinoma, PTC is the most common one which accounts for more than 80% of thyroid malignancies (45). Identified oncogene mutations in thyroid neoplasms mainly consist of BRAF and RAS. BRAF mutations are the most common genetic alteration occurring in thyroid carcinomas especially in PTC, accounting for 28–83%, with an overall rate of 44% (46–48). The most common type of BRAF mutations found in PTC is a T to A substitution at nucleotide 1,799 in exon 15, which results in the conversion of a valine to glutamic acid at codon 600 (V600E) of the BRAF protein (49, 50). All of the relevant studies mainly demonstrate the influence of BRAF V600E on thyroid cancer. BRAF mutation has been illustrated to be associated with poor prognosis among PTC patients (51–53). BRAF V600E mutation occurs more frequently in the advanced stages (stage III or IV) of thyroid cancer (54). And the association between advanced stages and BRAF V600E becomes significant while compared with tumors with all other oncogene mutations. Taking only BRAF mutation positive tumors into account, the percentage of BRAF V600E alleles was directly correlated with disease stages (55). Among patients with BRAF V600E, the age of preliminary diagnosis is relatively younger (<45 years old) than others (56).
Gene fusions detected in patients with thyroid cancer are mainly RET and NTRK fusions. And RET/PTC is one of the most common kind of fusions, which mainly consists of RET/PTC-1 (CCDC6-RET), RET/PTC-2 (PRKAR1A-RET), and RET/PTC-3 (NCOA4-RET) (57). RET/PTC1 and RET/PTC3 are intrachromosomal fusions of the long arm of chromosome 10 and can be induced in vitro by irradiating normal thyroid cells (58, 59). The incidence of RET/PTC fusions in patients with PTC ranges from 2.5 to 67.0% depending on the cohorts studied. Most patients with RET/PTC fusions in their primary PTCs were younger than 45 years of age (60–64). And the distribution of stages varies in different cohorts that had been studied.
The phenomenon of concomitant BRAF mutations and gene fusions in thyroid malignancies has been controversial up to now. Some scholars hold the opinion that dual BRAF mutations and gene fusions can exist in the same thyroid tumor. In addition, they deem that this phenomenon is associated with clinical pathological features of the tumor and it may become a prognostic indicator for patients with thyroid cancer. A study from Texas M. D. Anderson Cancer Center by Henderson et al. reported concomitant BRAF and RET/PTC mutations in 5/54 (9.3%) recurrent PTC. And the prevalence of tumors with this concomitant driver mutations and fusions found in the recurrent population far exceeded the frequency historically reported for patients with primary PTC. They also found that patients with dual mutations and fusions were significantly older and had more advanced tumors than patients with a BRAF mutation or RET/PTC alone (56). Guerra et al. recruited 72 patients from Salerno, Italy and they demonstrated that patients with both BRAF mutations and RET/PTC accounted for 19.4% (14/72) among all of the patients of PTC. Different from Henderson’s discovery, they found that tumors with dual mutations were equally distributed among stages. And they found that the presence of dual mutations is associated with a higher rate of recurrence (55). Zou et al. found six patients with CPTC and one with tall-cell variant to have simultaneous BRAF V600E and RET/PTC-1 from an 85-participant cohort. What they found is consistent with the discovery of Henderson, all the seven patients with concomitant BRAF V600E and RET/PTC-1 were in the advanced stages (stage III or IV). And the association of concomitant mutations and fusions with advanced stages was statistically significant. They also elucidated that patients with concomitant mutations and fusions had much poorer clinical outcomes than those with a single or no mutation (Table 2) (54).
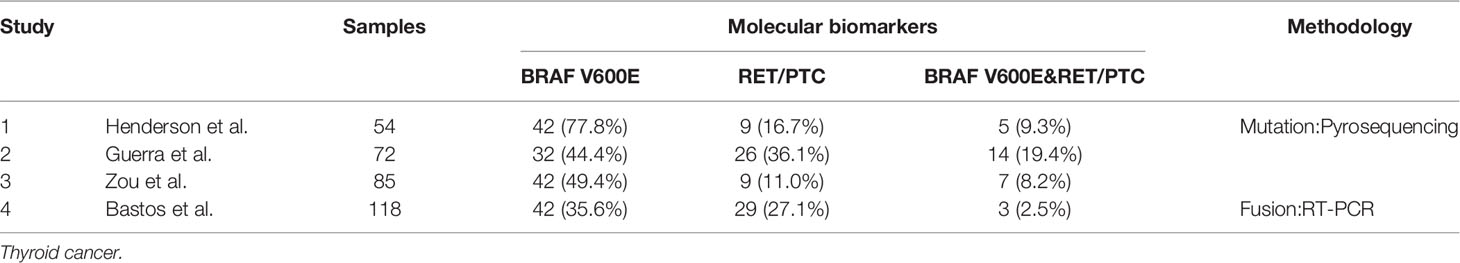
Table 2 Summary of studies on concomitant oncogene mutations and rearrangements in thyroid cancer.
Nevertheless, others think that gene fusions are mutually exclusive with BRAF mutations in patients harboring PTC, as well as with each other. And mutations at more than one of these genes are unlikely to provide an additional biological advantage. Paula et al. conducted a study including 50 patients with PTC from Porto, Portugal and detected 46% (23/50) of them harboring BRAF V600E. And meanwhile, seven of 39 PTC (18%) were validated to have RET/PTC fusions; none of these 7 PTC cases had the BRAF V600E mutation. So they thought that BRAF V600E mutation appeared to be an alternative event to RET/PTC in PTC (65). Liang et al. detected the mutation conditions of BRAF mutations and RET fusions among 355 Chinese patients with primary PTC. They found 72.4% (257/355) of the cases carried BRAF mutations and 8.5% (30/355) of cases were characterized with in-frame gene fusions. In order to further validate whether BRAF mutations is mutually exclusive with RET fusions, they screened patients with known BRAF point mutations for RET fusions transcripts by using RT-PCR, and none of RET fusions was identified in patients with BRAF point mutations (66). The Cancer Genome Atlas Research Network analyzed 496 patients with thyroid malignancies [324 (65.3%) classical-type, 99 (20.0%) follicular-variant, 35 (7.1%) tall-cell variant, 9 (1.8%) uncommon PTC variants and 29 without histological annotations] in The Cancer Genome Atlas (TCGA) project by comprehensive multi-platform analysis and drew the conclusion that fusions were mutually exclusive with each other and with proto-oncogenes mutations (67).
Concomitant Oncogene Mutations and Rearrangements in Leukemia
Leukemia is a genetically and clinically heterogeneous disease (68, 69). It consists of four main subtypes: acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL). Genomic aberrations are known to play an important role in the pathogenesis of leukemia, and cytogenetic aberrations have become well established diagnostic and prognostic markers. In the past few decades, mounting gene alterations have been detected and proved to play a role in the occurrence and development of leukemia. Meanwhile, some co-occurring mutations and fusions have also been found, especially in acute leukemia (AML and ALL). Hidemasa et al. recruited pediatric patients with MLL-rearranged (MLL-r) AML (n = 56) alongside data from the TARGET study’s pediatric cohorts with MLL-r AML (n = 104), non–MLL-r AML (n = 581), and adult MLL-r AML (n = 81) into their study, and they found that KRAS mutations were most frequent in pediatric patients with high-risk MLL fusions (MLL-MLLLT10, MLL-MLLT4, and MLL-MLLT1), accounting for 26.25% (42/160) in MLL-r AML. What’s more, an adverse prognostic impact of KRAS mutations was confirmed in adult MLL-r AML; KRAS mutations were associated with adverse prognoses in pediatric patients with both high-risk (MLLT10+MLLT4+MLLT1; n = 60) and intermediate-to-low-risk (MLLT3+ELL+others; n = 100) MLL fusions (70). The t (8;21) translocation is one of the most frequent cytogenetic abnormalities in acute myeloid leukemia (AML), resulting in the RUNX1/RUNX1T1 fusion. The transcription factor ZBTB7A is important for hematopoietic lineage fate decisions and for regulation of glycolysis. On a functional level, ZBTB7A mutations disrupt the transcriptional repressor potential and the anti-proliferative effect of ZBTB7A. Luise et al. revealed a kind of concomitant mutations and fusions - ZBTB7A mutations with t (8;21) translocation. ZBTB7A acts as a tumor suppressor in RUNX1-RUNX1T1 AML, so the specific association of ZBTB7A’s loss-of-function-mutations with t (8;21) rearranged AML can point towards leukemogenic cooperativity between mutant ZBTB7A and the RUNX1/RUNX1T1 fusion (71). And in their cohort, 13 (23.2%) samples were found to have ZBTB7A mutations among 56 AML patients with RUNX1-RUNX1T1. In addition, concomitant ZBTB7A mutations with t (8;21) translocation has also been reported in many relevant articles accounting for 4.8–23.2% in patients with RUNX1-RUNX1T1 fusion (Table 3) (72–76).
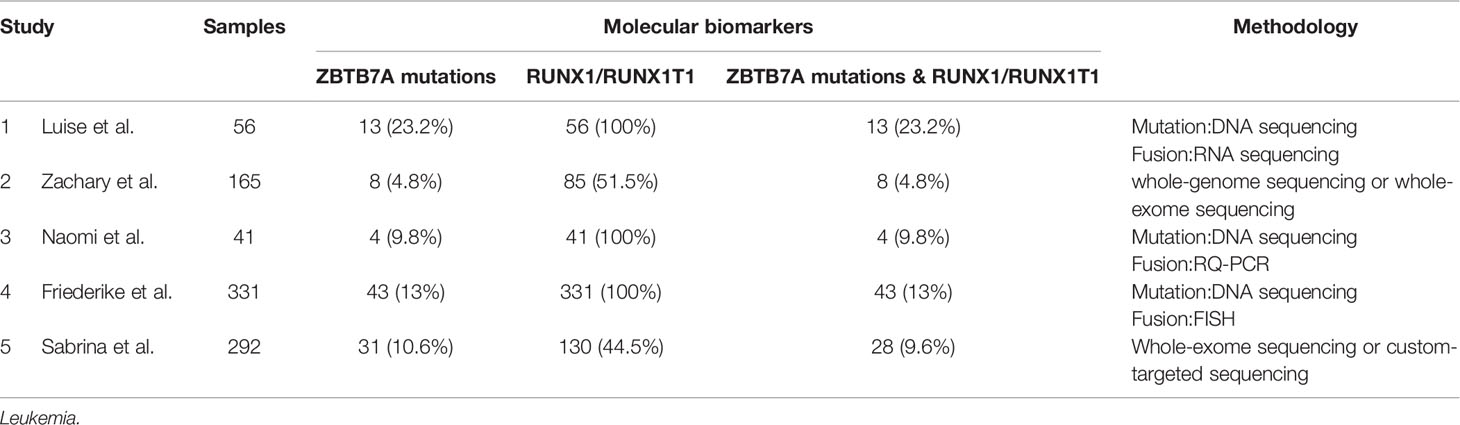
Table 3 Summary of studies on concomitant oncogene mutations and rearrangements in leukemia.
Concomitant Oncogene Mutations and Rearrangement in Other Solid Malignancies
The phenomenon of concomitant mutations and fusions of driver genes is relatively uncommon compared with single driver oncogene mutations and fusions. However, with the deepening of the sequencing depth in next generation sequencing technology and a wider application range of NGS, more and more dual mutations and fusions have been noticed by scholars. The most obvious is in lung cancer, thyroid cancer and leukemia, a fair amount of studies focused on this phenomenon and conducted corresponding clinical studies. Although there are relatively few articles reporting the co-occurrence of driver mutations and fusions in other solid tumors, like breast cancer, colorectal cancer, prostate cancer and melanoma, some cases of concomitant mutations and fusions of driver genes have attracted scholars’ attention.
Mutations in BRCA1 confer a high risk for breast cancer, and BRCA1 is involved in many cellular processes as well as in repairing double-stranded DNA breaks (DSBs) mediated by homologous recombination (77–79). Kevin et al. and Stephens et al. deemed that deficiencies in BRCA1 would cause increased chromosomal instability in a tumor cell due to impaired DNA repair pathways and NHEJ dysfunction. The resulting chromosomal lesions may potentially lead to the creation of gene fusions that can be detected in the transcriptome. And they found the concomitant BRCA1 mutations and gene fusions in breast cancer cells (80, 81). Regretfully, no corresponding large sample clinical study was conducted to identify this phenomenon. Similarly, Manuel et al. held the opinion that defects in DNA repair may lead to an increase of chromosomal rearrangements and thus to the occurrence of the somatic fusion of TMPRSS2 to ETS oncogenes in prostate cancer, and they detected that DNA repair genes (BRCA2, ESCO1, and POLI, etc.) mutations exist in TMPRSS2-ERG fusion-positive samples (82). In addition, no pathogenic mutations and fusions of driver genes has been found in breast cancer and prostate cancer up to now. Activation of RAS/MAPK pathways through mutations of RAS family members and BRAF are classical driver mutations of colorectal cancer (83). And NTRK1 and NTRK3 fusions have been described as oncogenic driver alterations in colorectal cancer (84–87). While the studies on the genomic landscape of colorectal cancers revealed that NTRK fusions are mutually exclusive with driver mutations like KRAS and BRAF mutations (85, 88–90). In melanoma, James et al., Iwei et al., and Thomas et al. thought that fusion proteins and most common oncogenic drivers such as BRAF, NRAS and HRAS are mutually exclusive, so that NTRK fusion proteins might be more common in BRAF or NRAS wild-type melanoma (91–93). However, there is still one study conducted by Lezcano et al. detected a case that harbored NTRK1 fusion as well as an additional activating NRAS Q61 mutation (94).
Disparity in the Incidence Rates of Concomitant Driver Mutations and Fusions
The phenomenon of concomitant mutations and fusions in cancer indeed exists. The incidence rates of concomitant driver mutations and fusion vary in cancers, while the frequency is relatively low, and associated with different type of cancers or gene varieties. The possible reasons affecting the incidence of concomitant mutations and fusions are as follows: (1) Ethnic factors: Concomitant driver mutations and fusions may have racial differences. And the racial differences mainly reflect in NSCLC. Among all the studies about NSCLC mentioned above, four of the five cohorts are from China and one from Korea, that is all the data are from Asians, which may suggest that concomitant driver mutations and fusions in NSCLC is more common in Asian populations. (2) Detecting technologies: The methods and platforms that are utilized in the detection of fusions varied in different studies, and these methods’ interval of sensitivity has a wide range. The incidence of dual driver mutations and fusions may be low by direct sequencing and FISH, while next generation sequencing can increase the detection rate. And the reaction conditions and primer selection in the detection of fusions are main influence factors, and inappropriate conditions and primer may lead to false positive or false negative results, thus bringing out incorrect judgements. It is believed that with the improvement of detection depth and sensitivity, the incidence of concomitant driver mutations and fusions will increase in the future. (3) Sample selection: DNA samples are usually applied to mutation detection while fusion detection requires RNA samples. Most of the samples extracted from tumors are single sample type—DNA or RNA, so that mutations and fusions cannot be detected appropriately at the same time. If DNA and RNA could be detected simultaneously, more samples with concomitant driver mutations and fusions will be found. (4) Population inclusion: Patients with different subtypes of tumors and different stages may present disparate genotypes. In NSCLC patients, most of the dual mutations have been reported in advanced and/or metastatic patients in the literatures, and this may be the result of the accumulation of genetic changes during the development of the disease. Therefore, the selection of different stages of samples may lead to totally opposite conclusions. So, these questions need to be taken into account in the following researches. (5) Source of tissue: We found that the phenomenon of concomitant mutations and fusions of driver genes are mainly detected in lung cancer, thyroid cancer, and leukemia. These three types of carcinomas are more susceptible to carcinogenic factors. Lung cancers are vulnerable to tobacco-smoking, thyroid cancers are closely related to radiation, and leukemia may be associated with chemicals. And in the process of exposure to different carcinogenic factors, there tends to be genomic instability in cancer cells, leading to an increased frequency of gene fusions. Conversely, for other carcinomas caused by intrinsic factors, gene fusions may be relatively uncommon.
Clinical Significance of Concomitant Driver Mutations and Fusions
In summary, the concomitant driver mutations and fusions may occur in specific molecular subtypes of tumor and advanced-stage tumors, and it is recommended to carry out a multiple centers-based large sample retrospective study by multi-point biopsy or liquid biopsy combined with next generation sequencing (NGS), so as to obtain the precise results of the frequency of concomitant mutations and fusions, and further to identify the correlation with multiple clinical and prognostic features. Besides, functional experiments and animal models are necessary to explore whether there are differences in biological behaviors and signaling pathway activated between concomitant driver mutations and fusions and single driver gene alterations. Therefore, we can lay the theoretical foundation for the occurrence of this phenomenon. In this way, the clinical significance of concomitant mutations and fusions should be elucidated and individualized target therapy should be proposed to benefit more patients from the development of precision medicine.
Author Contributions
RZ and JY conceptualized and designed the study. RZ wrote the article. LD critically revised the article. JY gave the final approval of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 81702280, 81472473, 81872143), National Science and Technology support Program of China (Grant No. 2015BAI12B15, 2018ZX09201015), National Key Research and Development program of China: The Net construction of human genetic resource Bio-bank in North China (2016YFC1201703), Projects of Science and Technology of Tianjin (Grant No. 13ZCZCSY20300, 18JCQNJC82700) and Key project of Tianjin Health and Family Planning Commission (Grant No. 16KG126).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
3. Chen W, Zheng R, Zeng H, Zhang S, He J. Annual report on status of cancer in China, 2011. Chin J Cancer Res (2015) 27(1):2–12. doi: 10.3978/j.issn.1000-9604.2015.01.06
4. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin (2011) 61(2):69–90. doi: 10.3322/caac.20107
5. Okazaki I, Ishikawa S, Sohara Y. Genes associated with succeptibility to lung adenocarcinoma among never smokers suggest the mechanism of disease. Anticancer Res (2014) 34(10):5229–40.
6. Lee YJ, Kim JH, Kim SK, Ha SJ, Mok TS, Mitsudomi T, et al. Lung cancer in never smokers: change of a mindset in the molecular era. Lung Cancer (2011) 72(1):9–15. doi: 10.1016/j.lungcan.2010.12.013
7. Burns TF, Rudin CM. Lung cancer in ‘never-smokers’: beyond EGFR mutations and EGFR-TK inhibitors. Oncol (Williston Park) (2010) 24(1):48–9.
8. Doran E, Jackman D. Fire without smoke: lung cancer in ‘never-smokers’. Oncol (Williston Park) (2010) 24(1):40, 43.
9. Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers–a review. Eur J Cancer (2012) 48(9):1299–311. doi: 10.1016/j.ejca.2012.03.007
10. Kim HR, Shim HS, Chung JH, Lee YJ, Hong YK, Rha SY, et al. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer (2012) 118(3):729–39. doi: 10.1002/cncr.26311
11. Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol (2008) 9(7):676–82. doi: 10.1016/S1470-2045(08)70174-8
12. Gazdar AF, Shigematsu H, Herz J, Minna JD. Mutations and addiction to EGFR: the Achilles ‘heal’ of lung cancers? Trends Mol Med (2004) 10(10):481–6. doi: 10.1016/j.molmed.2004.08.008
13. Han XH, Zhang NN, Ma L, Lin DM, Hao XZ, Liu YT, et al. Immunohistochemistry reliably detects ALK rearrangements in patients with advanced non-small-cell lung cancer. Virchows Arch (2013) 463(4):583–91. doi: 10.1007/s00428-013-1472-7
14. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol (2011) 6:49–69. doi: 10.1146/annurev-pathol-011110-130206
15. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol (2009) 27(26):4247–53. doi: 10.1200/JCO.2009.22.6993
16. Rodig SJ, Mino-Kenudson M, Dacic S, Yeap BY, Shaw A, Barletta JA, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res (2009) 15(16):5216–23. doi: 10.1158/1078-0432.CCR-09-0802
17. Inamura K, Takeuchi K, Togashi Y, Hatano S, Ninomiya H, Motoi N, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol (2009) 22(4):508–15. doi: 10.1038/modpathol.2009.2
18. Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res (2008) 14(13):4275–83. doi: 10.1158/1078-0432.CCR-08-0168
19. Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U.S.A. (2008) 105(50):19893–7. doi: 10.1073/pnas.0805381105
20. Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol (2008) 3(1):13–7. doi: 10.1097/JTO.0b013e31815e8b60
21. Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer (2009) 115(8):1723–33. doi: 10.1002/cncr.24181
22. Shinmura K, Kageyama S, Tao H, Bunai T, Suzuki M, Kamo T, et al. EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-, ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung Cancer (2008) 61(2):163–9. doi: 10.1016/j.lungcan.2007.12.013
23. Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res (2008) 14(20):6618–24. doi: 10.1158/1078-0432.CCR-08-1018
24. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448(7153):561–6. doi: 10.1038/nature05945
25. Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol (2009) 27(26):4232–5. doi: 10.1200/JCO.2009.23.6661
26. Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer (2014) 86(2):291–5. doi: 10.1016/j.lungcan.2014.09.011
27. Kuo YW, Wu SG, Ho CC, Shih JY. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol (2010) 5(12):2039–40. doi: 10.1097/JTO.0b013e3181f43274
28. Lo Russo G, Imbimbo M, Corrao G, Proto C, Signorelli D, Vitali M, et al. Concomitant EML4-ALK rearrangement and EGFR mutation in non-small cell lung cancer patients: a literature review of 100 cases. Oncotarget (2017) 8(35):59889–900. doi: 10.18632/oncotarget.17431
29. Lou NN, Zhang XC, Chen HJ, Zhou Q, Yan LX, Xie Z, et al. Clinical outcomes of advanced non-small-cell lung cancer patients with EGFR mutation, ALK rearrangement and EGFR/ALK co-alterations. Oncotarget (2016) 7(40):65185–95. doi: 10.18632/oncotarget.11218
30. Popat S, Vieira de Araujo A, Min T, Swansbury J, Dainton M, Wotherspoon A, et al. Lung adenocarcinoma with concurrent exon 19 EGFR mutation and ALK rearrangement responding to erlotinib. J Thorac Oncol (2011) 6(11):1962–3. doi: 10.1097/JTO.0b013e31822eec5e
31. Sahnane N, Frattini M, Bernasconi B, Zappa F, Schiavone G, Wannesson L, et al. EGFR and KRAS Mutations in ALK-Positive Lung Adenocarcinomas: Biological and Clinical Effect. Clin Lung Cancer (2016) 17(1):56–61. doi: 10.1016/j.cllc.2015.08.001
32. Sweis RF, Thomas S, Bank B, Fishkin P, Mooney C, Salgia R. Concurrent EGFR Mutation and ALK Translocation in Non-Small Cell Lung Cancer. Cureus (2016) 8(2):e513. doi: 10.7759/cureus.513
33. Tanaka H, Hayashi A, Morimoto T, Taima K, Tanaka Y, Shimada M, et al. A case of lung adenocarcinoma harboring EGFR mutation and EML4-ALK fusion gene. BMC Cancer (2012) 12:558. doi: 10.1186/1471-2407-12-558
34. Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, et al. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer (2011) 71(2):241–3. doi: 10.1016/j.lungcan.2010.11.014
35. Won JK, Keam B, Koh J, Cho HJ, Jeon YK, Kim TM, et al. Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol (2015) 26(2):348–54. doi: 10.1093/annonc/mdu530
36. Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res (2014) 20(5):1383–92. doi: 10.1158/1078-0432.CCR-13-0699
37. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res (2013) 19(15):4273–81. doi: 10.1158/1078-0432.CCR-13-0318
38. Zhao Y, Wang S, Zhang B, Qiao R, Xu J, Zhang L, et al. Clinical Management of Non-Small Cell Lung Cancer with Concomitant EGFR Mutations and ALK Rearrangements: Efficacy of EGFR Tyrosine Kinase Inhibitors and Crizotinib. Targ Oncol (2019) 14(2):169–78. doi: 10.1007/s11523-019-00628-6
39. Fan J, Dai X, Wang Z, Huang B, Shi H, Luo D, et al. Concomitant EGFR Mutation and EML4-ALK Rearrangement in Lung Adenocarcinoma Is More Frequent in Multifocal Lesions. Clin Lung Cancer (2019) 20(4):e517–30. doi: 10.1016/j.cllc.2019.04.008
40. Wu C, Zhao C, Yang Y, He Y, Hou L, Li X, et al. High Discrepancy of Driver Mutations in Patients with NSCLC and Synchronous Multiple Lung Ground-Glass Nodules. J Thorac Oncol (2015) 10(5):778–83. doi: 10.1097/JTO.0000000000000487
41. Brega E, Brandao G. Non-Small Cell Lung Carcinoma Biomarker Testing: The Pathologist’s Perspective. Front Oncol (2014) 4:182. doi: 10.3389/fonc.2014.00182
42. Lee JK, Kim TM, Koh Y, Lee SH, Kim DW, Jeon YK, et al. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer (2012) 77(2):460–3. doi: 10.1016/j.lungcan.2012.04.012
43. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res (2011) 71(18):6051–60. doi: 10.1158/0008-5472.CAN-11-1340
44. Fahiminiya S, de Kock L, Foulkes WD. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med (2016) 375(23):2306–7. doi: 10.1056/NEJMc1613118
45. McLeod DS, Sawka AM, Cooper DS. Controversies in primary treatment of low-risk papillary thyroid cancer. Lancet (2013) 381(9871):1046–57. doi: 10.1016/S0140-6736(12)62205-3
46. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res (2003) 63(7):1454–7. doi: 1016/j.jacc.2011.10.507
47. Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst (2003) 95(8):625–7. doi: 10.1093/jnci/95.8.625
48. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer (2005) 12(2):245–62. doi: 10.1677/erc.1.0978
49. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol (2004) 5(11):875–85. doi: 10.1038/nrm1498
50. Wojciechowska K, Lewinski A. BRAF mutations in papillary thyroid carcinoma. Endocr Regul (2006) 40(4):129–38.
51. Lupi C, Giannini R, Ugolini C, Proietti A, Berti P, Minuto M, et al. Association of BRAF V600E mutation with poor clinicopathological outcomes in 500 consecutive cases of papillary thyroid carcinoma. J Clin Endocrinol Metab (2007) 92(11):4085–90. doi: 10.1210/jc.2007-1179
52. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab (2005) 90(12):6373–9. doi: 10.1210/jc.2005-0987
53. Puxeddu E, Moretti S. Clinical prognosis in BRAF-mutated PTC. Arq Bras Endocrinol Metabol (2007) 51(5):736–47. doi: 10.1590/S0004-27302007000500011
54. Zou M, Baitei EY, Alzahrani AS, BinHumaid FS, Alkhafaji D, Al-Rijjal RA, et al. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid (2014) 24(8):1256–66. doi: 10.1089/thy.2013.0610
55. Guerra A, Zeppa P, Bifulco M, Vitale M. Concomitant BRAF(V600E) mutation and RET/PTC rearrangement is a frequent occurrence in papillary thyroid carcinoma. Thyroid (2014) 24(2):254–9. doi: 10.1089/thy.2013.0235
56. Henderson YC, Shellenberger TD, Williams MD, El-Naggar AK, Fredrick MJ, Cieply KM, et al. High rate of BRAF and RET/PTC dual mutations associated with recurrent papillary thyroid carcinoma. Clin Cancer Res (2009) 15(2):485–91. doi: 10.1158/1078-0432.CCR-08-0933
57. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol (2016) 12(4):192–202. doi: 10.1038/nrendo.2016.11
58. Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab (2005) 90(4):2364–9. doi: 10.1210/jc.2004-1811
59. Ameziane-El-Hassani R, Boufraqech M, Lagente-Chevallier O, Weyemi U, Talbot M, Metivier D, et al. Role of H2O2 in RET/PTC1 chromosomal rearrangement produced by ionizing radiation in human thyroid cells. Cancer Res (2010) 70(10):4123–32. doi: 10.1158/0008-5472.CAN-09-4336
60. Tallini G, Santoro M, Helie M, Carlomagno F, Salvatore G, Chiappetta G, et al. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin Cancer Res (1998) 4(2):287–94.
61. Nakazawa T, Kondo T, Kobayashi Y, Takamura N, Murata S, Kameyama K, et al. RET gene rearrangements (RET/PTC1 and RET/PTC3) in papillary thyroid carcinomas from an iodine-rich country (Japan). Cancer (2005) 104(5):943–51. doi: 10.1002/cncr.21270
62. Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin Endocrinol Metab (2000) 85(3):1170–5. doi: 10.1210/jc.85.3.1170
63. Elisei R, Romei C, Vorontsova T, Cosci B, Veremeychik V, Kuchinskaya E, et al. RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. J Clin Endocrinol Metab (2001) 86(7):3211–6. doi: 10.1210/jcem.86.7.7678
64. Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res (1997) 57(9):1690–4.
65. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene (2003) 22(29):4578–80. doi: 10.1038/sj.onc.1206706
66. Liang J, Cai W, Feng D, Teng H, Mao F, Jiang Y, et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol (2018) 244(2):215–26. doi: 10.1002/path.5005
67. Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell (2014) 159(3):676–90. doi: 10.1016/j.cell.2014.09.050
68. Cancer Genome Atlas Research N., Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
69. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
70. Matsuo H, Yoshida K, Nakatani K, Harata Y, Higashitani M, Ito Y, et al. Fusion partner-specific mutation profiles and KRAS mutations as adverse prognostic factors in MLL-rearranged AML. Blood Adv (2020) 4(19):4623–31. doi: 10.1182/bloodadvances.2020002457
71. Hartmann L, Dutta S, Opatz S, Vosberg S, Reiter K, Leubolt G, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun (2016) 7:11733. doi: 10.1038/ncomms11733
72. Lavallee VP, Lemieux S, Boucher G, Gendron P, Boivin I, Armstrong RN, et al. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood (2016) 127(20):2498–501. doi: 10.1182/blood-2016-03-703868
73. Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet (2016) 48(12):1551–6. doi: 10.1038/ng.3709
74. Kawashima N, Akashi A, Nagata Y, Kihara R, Ishikawa Y, Asou N, et al. Clinical significance of ASXL2 and ZBTB7A mutations and C-terminally truncated RUNX1-RUNX1T1 expression in AML patients with t(8;21) enrolled in the JALSG AML201 study. Ann Hematol (2019) 98(1):83–91. doi: 10.1007/s00277-018-3492-5
75. Christen F, Hoyer K, Yoshida K, Hou HA, Waldhueter N, Heuser M, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood (2019) 133(10):1140–51. doi: 10.1182/blood-2018-05-852822
76. Opatz S, Bamopoulos SA, Metzeler KH, Herold T, Ksienzyk B, Braundl K, et al. The clinical mutatome of core binding factor leukemia. Leukemia (2020) 34(6):1553–62. doi: 10.1038/s41375-019-0697-0
77. Begg CB, Haile RW, Borg A, Malone KE, Concannon P, Thomas DC, et al. Variation of breast cancer risk among BRCA1/2 carriers. JAMA (2008) 299(2):194–201. doi: 10.1001/jama.2007.55-a
78. Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst (2007) 99(23):1811–4. doi: 10.1093/jnci/djm203
79. Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell (1999) 4(4):511–8. doi: 10.1016/S1097-2765(00)80202-6
80. Ha KC, Lalonde E, Li L, Cavallone L, Natrajan R, Lambros MB, et al. Identification of gene fusion transcripts by transcriptome sequencing in BRCA1-mutated breast cancers and cell lines. BMC Med Genomics (2011) 4:75. doi: 10.1186/1755-8794-4-75
81. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature (2009) 462(7276):1005–10. doi: 10.1038/nature08645
82. Luedeke M, Linnert CM, Hofer MD, Surowy HM, Rinckleb AE, Hoegel J, et al. Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol Biomarkers Prev (2009) 18(11):3030–5. doi: 10.1158/1055-9965.EPI-09-0772
83. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol (2011) 6:479–507. doi: 10.1146/annurev-pathol-011110-130235
84. Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature (1986) 319(6056):743–8. doi: 10.1038/319743a0
85. Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, van Roosmalen MJ, Sieuwerts AM, Stangl C, et al. A Systematic Analysis of Oncogenic Gene Fusions in Primary Colon Cancer. Cancer Res (2017) 77(14):3814–22. doi: 10.1158/0008-5472.CAN-16-3563
86. Ardini E, Bosotti R, Borgia AL, De Ponti C, Somaschini A, Cammarota R, et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol Oncol (2014) 8(8):1495–507. doi: 10.1016/j.molonc.2014.06.001
87. Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, et al. Sensitivity to Entrectinib Associated With a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. J Natl Cancer Inst (2016) 108(1). doi: 10.1093/jnci/djv306
88. Lasota J, Chlopek M, Lamoureux J, Christiansen J, Kowalik A, Wasag B, et al. Colonic Adenocarcinomas Harboring NTRK Fusion Genes: A Clinicopathologic and Molecular Genetic Study of 16 Cases and Review of the Literature. Am J Surg Pathol (2020) 44(2):162–73. doi: 10.1097/PAS.0000000000001377
89. Chan AW, Pan Y, Tong JH, Lung RW, Kwan JS, Chow C, et al. Receptor tyrosine kinase fusions act as a significant alternative driver of the serrated pathway in colorectal cancer development. J Pathol (2020) 251(1):74–86. doi: 10.1002/path.5418
90. Lasota J, Chlopek M, Wasag B, Kowalik A, Christiansen J, Lamoureux J, et al. Colorectal Adenocarcinomas Harboring ALK Fusion Genes: A Clinicopathologic and Molecular Genetic Study of 12 Cases and Review of the Literature. Am J Surg Pathol (2020) 44(9):1224–34. doi: 10.1097/PAS.0000000000001512
91. Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun (2014) 5:3116. doi: 10.1038/ncomms4116
92. Yeh I, Botton T, Talevich E, Shain AH, Sparatta AJ, de la Fouchardiere A, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun (2015) 6:7174. doi: 10.1038/ncomms8174
93. Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol (2020) 33(1):38–46. doi: 10.1038/s41379-019-0324-7
Keywords: concomitant, mutation, fusion, lung cancer, thyroid cancer, leukemia
Citation: Zhang R, Dong L and Yu J (2021) Concomitant Pathogenic Mutations and Fusions of Driver Oncogenes in Tumors. Front. Oncol. 10:544579. doi: 10.3389/fonc.2020.544579
Received: 21 March 2020; Accepted: 27 November 2020;
Published: 15 January 2021.
Edited by:
Timothy F. Burns, University of Pittsburgh, United StatesReviewed by:
Ali M. Ardekani, Avicenna Research Institute (ARI), IranHugo de Jonge, University of Pavia, Italy
Copyright © 2021 Zhang, Dong and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinpu Yu, yujinpu@yahoo.com