 Anthony Quagliano
Anthony Quagliano Anilkumar Gopalakrishnapillai
Anilkumar Gopalakrishnapillai Sonali P. Barwe
Sonali P. Barwe- 1Nemours/Alfred I. duPont Hospital for Children, Wilmington, DE, United States
- 2Department of Biological Sciences, University of Delaware, Newark, DE, United States
The development of resistance to anti-cancer therapeutics remains one of the core issues preventing the improvement of survival rates in cancer. Therapy resistance can arise in a multitude of ways, including the accumulation of epigenetic alterations in cancer cells. By remodeling DNA methylation patterns or modifying histone proteins during oncogenesis, cancer cells reorient their epigenomic landscapes in order to aggressively resist anti-cancer therapy. To combat these chemoresistant effects, epigenetic modifiers such as DNA hypomethylating agents, histone deacetylase inhibitors, histone demethylase inhibitors, along with others have been used. While these modifiers have achieved moderate success when used either alone or in combination with one another, the most positive outcomes were achieved when they were used in conjunction with conventional anti-cancer therapies. Epigenome modifying drugs have succeeded in sensitizing cancer cells to anti-cancer therapy via a variety of mechanisms: disrupting pro-survival/anti-apoptotic signaling, restoring cell cycle control and preventing DNA damage repair, suppressing immune system evasion, regulating altered metabolism, disengaging pro-survival microenvironmental interactions and increasing protein expression for targeted therapies. In this review, we explore different mechanisms by which epigenetic modifiers induce sensitivity to anti-cancer therapies and encourage the further identification of the specific genes involved with sensitization to facilitate development of clinical trials.
Epigenetics and Cancer
The term “epigenetics” refers to the study of heritable phenotypic changes that do not involve mutations in DNA sequence (1). These changes are centered around alterations in gene activity and expression; through a variety of processes including DNA methylation and histone modifications (2). DNA methylation is the covalent addition of a methyl group to the C-5 position of DNA cytosine rings by DNA methyltransferases. Gene promoter hypermethylation often results in transcription depletion leading to decreased gene expression (3). Conversely, hypomethylation of ABCB1 promoter resulted in upregulation of ABCB1 protein and acquisition of taxane resistance via efficient drug efflux (4). In exceptional cases, promoter methylation of genes, like TERT gene encoding telomerase reverse transcriptase, leads to increased transcription and protein expression (5). Methylation in gene bodies also affects transcription; demethylation of gene bodies results in a decrease in gene transcription (6). These patterns of DNA methylation are retained during cell division and can persist across generations.
Histones are modified in multiple ways. These modifications alter chromatin structure and affect gene transcription by regulating the access of transcription machinery to DNA. For an excellent review on the different types of histone modifications, refer to Audia and Campbell (7). Enzymes that modify histone proteins also facilitate post-translational modifications in non-histone proteins, thereby affecting gene expression (8, 9). Acetylation of NFκB and methylation of tumor suppressor protein p53 promotes nuclear localization of these proteins and increases transcriptional activity of respective gene targets (10, 11). Due to the prominent role of these proteins in cancer progression and therapy resistance, targeting their post-translational modifications could have therapeutic benefit (12, 13).
Drastic alterations in the epigenetic landscapes occur in cancer cells (14). Aberrant epigenetic patterns function as key drivers in cancer initiation and progression; often a result of the silencing of tumor suppressor genes or induced overexpression of oncogenes (15). Several tumor suppressors, such as RASSF1A and CASP8, are frequently inactivated in multiple cancer subtypes via epigenetic downregulation rather than by genetic mutation. For an excellent review on this specific function in oncogenesis, see Kazanets et al. (16). On the other hand, certain oncogenes, such as c-Myc and insulin-like growth factor receptor 2 (IGF-2) are upregulated by epigenetic mechanisms (17). These epigenetic changes result in global dysregulation of gene expression; thereby solidifying the development of disease states (18). Anomalous epigenetic alterations can also lead to the acquisition of therapy resistance (19, 20). Figure 1 outlines how epigenetic-induced gene expression changes can give rise to multiple mechanisms of therapy resistance.
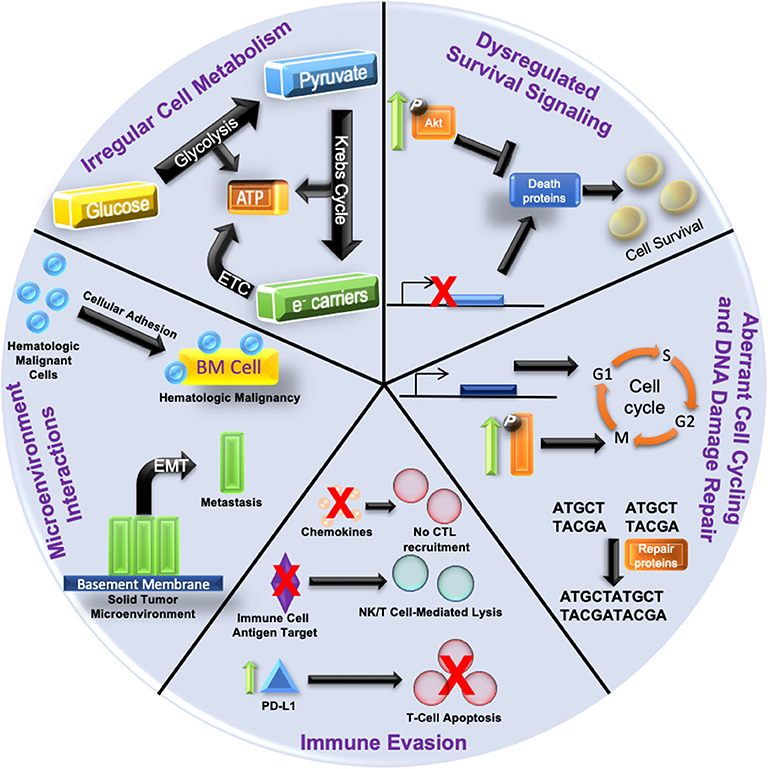
Figure 1. Hallmarks of Epigenetic Alteration-Induced Therapy Resistance Epigenetic dysregulation is a driving force in oncogenesis and the development of therapy resistance. (1) Increased pro-survival signaling (depicted by enhanced phosphorylation and activation of kinases such as Akt) can inhibit the expression of death proteins to promote cancer cell survival. The gene expression of death proteins (shown as transcriptional inhibition at the gene promoter) can also be disrupted, culminating in increased cell survival. (2) Aberrant cell cycling is caused by the over/under-expression of proliferative/checkpoint proteins (blue block with promoter), or increased activation of signaling pathways (shown by increased phosphorylation/activation) related to proliferation. DNA damage repair is augmented by an increased expression of repair proteins and disruption of checkpoint signaling. (3) Aberrant intracellular signaling can also alter cytokine expression and lead to reduced cytotoxic T lymphocyte (CTL) recruitment. Silencing of immune cell antigen targets can also suppress immune targeting of cancer cells (NK, natural killer). Increased expression of PD-L1 (green arrow and blue triangle) on cancer cells can augment the immune checkpoint response, resulting in T cell apoptosis. (4) Increased cellular adhesion within the bone marrow microenvironment (yellow block) in hematologic malignancies activates intracellular signaling pathways that protect malignant cells (blue spheres) from anti-cancer. Epithelial to mesenchymal transition (EMT) dislodges cancer cells (green blocks) from the solid tumor microenvironment and is the first step in metastasis. (5) Irregular cellular metabolism via overactive glucose metabolism leads to the Warburg effect favoring anabolic glycolysis over oxidative phosphorylation, and can render cells resistant to chemotherapeutics or antimetabolites. Resistance mechanisms are not restricted to just one of the categories; often with multiple categories being involved simultaneously.
Efforts to revert these epigenetic changes via the use of epigenome modifying drugs have achieved some success, specifically when used in conjunction with other therapies. While these modifiers are “non-specific” in that they affect gene expression on a global level, their action elicits “specific” effects in malignant cells. This is due to the altered epigenome that is acquired during oncogenesis, highlighted by expression changes in tumor suppressor genes (silenced) and oncogenes (augmented) that are responsible for cancer progression or therapy resistance. Thereby, treatment with epigenetic drugs elicits a “specific” effect on cancerous cells by reverting these unique expression changes. Additionally, sensitivity to epigenetic modifiers can be genomic loci specific, possibly due in part to the three-dimensional chromatin structure (21–23). Thus, epigenetic modifiers possess the unique ability to be effective in a broad category of patients; albeit via altering the expression of a set of genes in a patient- specific manner (24).
In order to better understand the uses and indications of epigenetic modifiers in these combinations, it is necessary to uncover mechanisms of epigenetic drug-induced sensitization to anti-cancer therapy. Below, we summarize the gene expression changes induced by specific epigenetic modifiers (listed in Table 1), and how they have a variety of intracellular/extracellular consequences to potentiate the effectiveness of subsequent anti-cancer therapies.
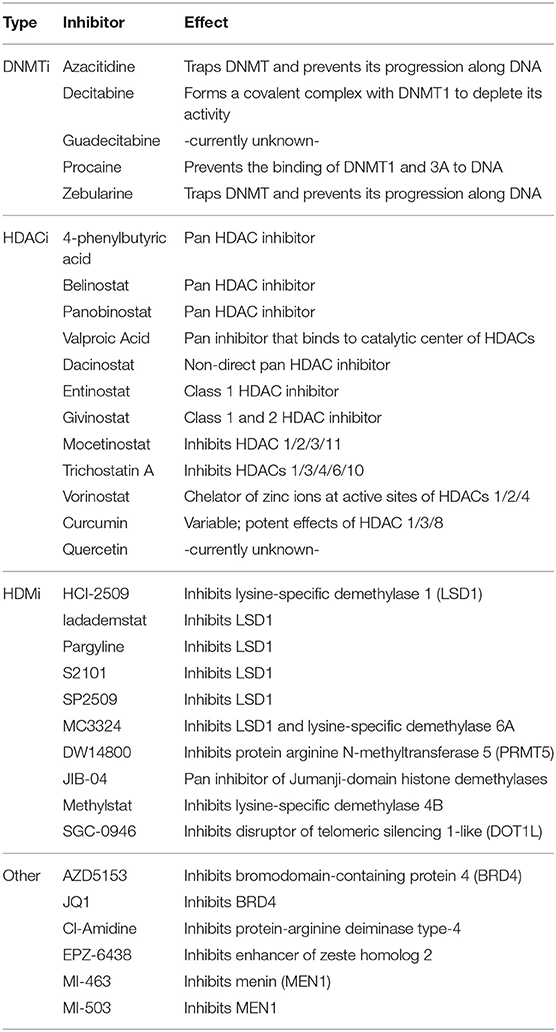
Table 1. Epigenetic modifiers discussed in this review.
Epigenetic Drug-Induced Sensitization Mechanisms
Disruption of Pro-survival Signaling
Epigenetic alterations during oncogenesis can dysregulate the expression of growth factor receptors (25). Increased expression of these receptors drives the development of therapy resistance due to the over-activation of their downstream pathways such as PI3K/Akt and subsequent inhibition of cell death (26). While targeted therapies against growth factor receptors have been used to mitigate their effects, the use of such therapies is limited by the rapid development of resistance. Using epigenetic modifiers to control the expression of growth factor receptors is a promising alternative. In breast cancer, dacinostat (HDACi) disrupted epidermal growth factor (EGF)-mediated signaling, which is associated with increased metastasis and cell survival. This was achieved by reducing HER2 (human EGF receptor-2) protein expression via two independent epigenetic mechanisms: first by decreasing HER2 mRNA level independent of alterations in promoter activity and secondly by increasing proteasomal degradation due to dissociation from its chaperone protein HSP90 via enhanced acetylation (27). In breast cancer as well, treatment with lapatinib (HER2/EGFR kinase inhibitor) and entinostat (HDACi) synergistically disrupted Akt signaling and promoted apoptosis (28). Though the mechanism of entinostat and lapatinib synergy is unknown, it is suggested that this effect is due to entinostat inhibiting lapatinib-induced expression of HER3; a HER2 heterodimerizing partner responsible for resistance to HER2 targeted therapies (29).
Hormone-dependent cancers such as breast and prostate respond to anti-hormone therapy by induction of apoptosis (30). Resistance to such therapy is acquired by downregulation of estrogen receptor (ER) or androgen receptor (AR) via epigenetic mechanisms. Thus, epigenetic drugs have been used to induce ER and AR expression to mediate sensitization to endocrine therapy in breast and prostate cancer, respectively (31–33).
Activation of pro-death pathways has been utilized as a therapeutic target to promote apoptosis in malignant cells. The binding of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to its receptors death domain containing receptor (DR) triggers pro-death signaling and induces apoptosis via the caspase cascade (34). Reduced receptor expression is frequently observed in cancer cells resistant to this pathway. Vorinostat (HDACi) sensitized breast cancer cells to TRAIL-induced apoptosis by increasing expression of DR5 (35, 36). Treatment with epigenetic drugs can also lead to hyperactivation of pro-death pathways such as the unfolded protein response (UPR) pathway. UPR is activated to protect cells from endoplasmic reticulum-stress mediated cell death (37). However, when the pathway becomes hyperactivated, this response actually leads to the activation of apoptotic pathways, making it a target in cancer cells. Treatment with methylstat (inhibitor of KDM4B, a lysine-specific histone demethylase) dissociated the UPR-activating initiation factor eIF2α and synergized with PI3K inhibition to hyperactivate UPR gene transcription, culminating in apoptosis (38).
Through modulation of the expression of growth factor receptors or augmenting apoptosis-inducing pathways, epigenetic modifiers can disrupt pro-survival signaling in cancer cells as an efficient mechanism of sensitization (Table 2).
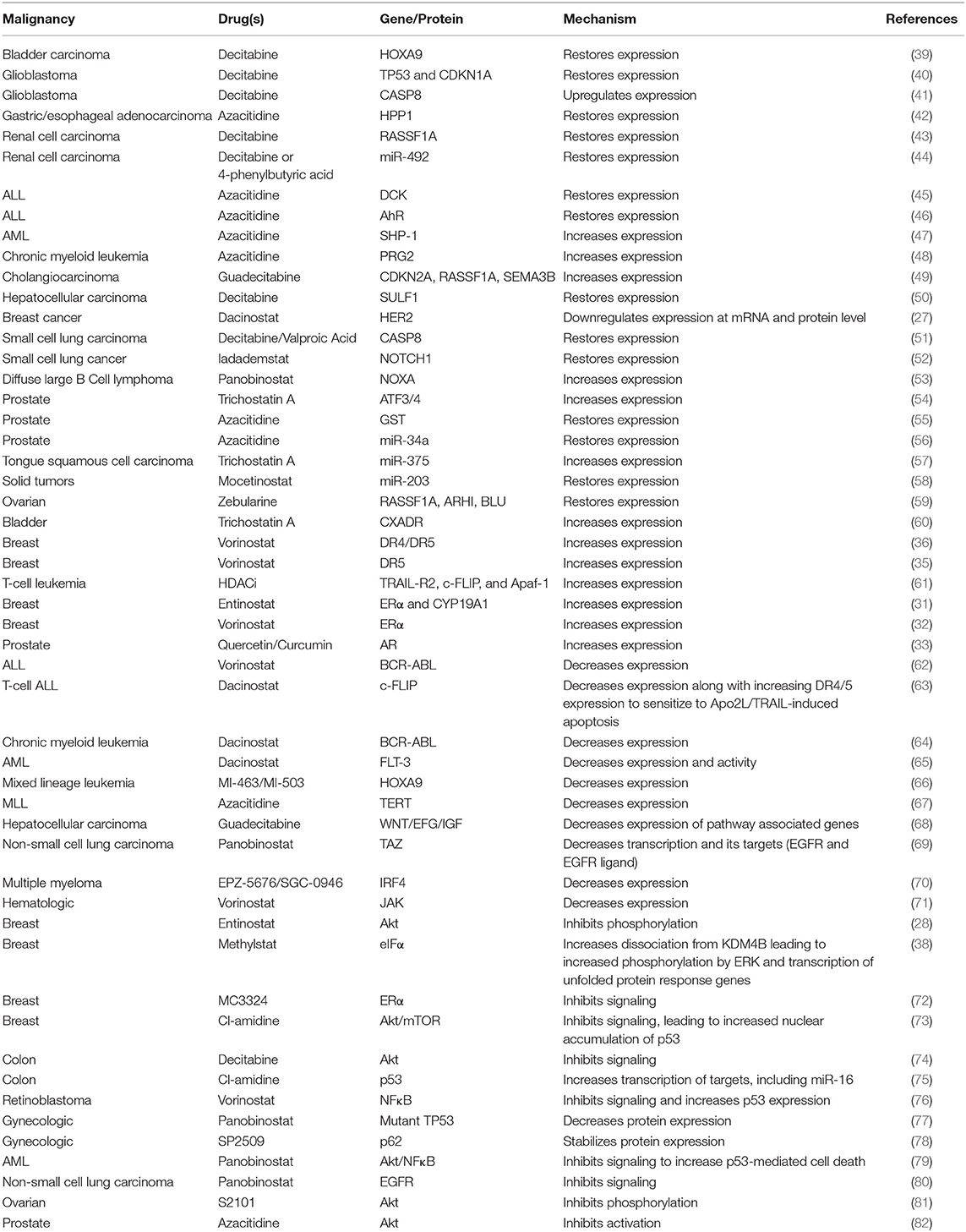
Table 2. Epigenetic modifier-induced disruption of pro-survival signaling.
Restoration of Cell Cycle Control and Disruption of DNA Damage Repair
Cancer cells often rely on a dysregulated cell cycle for their continued proliferation (83). Epigenetic modifiers can restore tight control of the cell cycle and proliferation by mediating a reversal of dysregulated gene expression as a mechanism to potentiate therapy. Entinostat (HDACi) downregulated the expression of MYC, E2F, and other G2M cell cycle genes to sensitize breast cancer cells to doxorubicin-induced growth arrest, however, how these genes are downregulated is unknown (84). Previously, Lee et al. showed entinostat treatment in breast cancer inhibited Akt signaling (28). Since Akt signaling controls cell cycle (85), it is likely that Akt is involved in entinostat-mediated doxorubicin sensitization. While entinostat alone inhibited the expression of cell cycle proteins, its combination with decitabine (DNMTi) in pancreatic cancer increased expression of p21 to reinstitute cell cycle control and inhibit tumor growth, likely due to increased acetylation of histone H3 and demethylation of the p21 promoter (86).
Epigenetic modifiers can potentially mitigate the effects of fusion oncoproteins. Gene fusions formed as a result of chromosomal translocations are often responsible for oncogenesis and therapy resistance (87, 88). In Ewing sarcoma, the EWS/Fli1 fusion gene is a key oncogenic driver. Treatment with JIB-04 (pan inhibitor of Jumanji-domain histone demethylases) simultaneously increased expression of cell-cycle inhibitor genes while suppressing expression of proteins that promote cell cycle, possibly through a disruption of EWS/Fli1 fusion gene program (89).
Hyperactive DNA damage repair pathways in cancer cells promote resistance to DNA damaging chemotherapeutics and radiation (90). In neuroblastoma, treatment with vorinostat (HDACi) diminished the expression of Ku-86, a key protein in non-homologous end joining DNA damage repair, to potentiate the anti-neoplastic effects of DNA damaging radiation (91). How vorinostat affects Ku-86 expression requires further study. Expression of DNA damage repair proteins like 53BP1 and RAD51 was also downregulated following treatment with pevonedistat (NEDD8-activating enzyme inhibitor) and belinostat (HDACi) in acute myeloid leukemia (AML). Downregulation of these proteins occurred in response to pevonedistat-mediated inhibition of belinostat-induced NFκB signaling and belinostat-mediated inhibition of pevonedistat-induced Chk1/Wee1 signaling, identifying a reliance of the two drugs on each other to disrupt DNA damage repair (92).
Restoring control of cell cycle progression and diminishing the activation of DNA damage repair pathways is a promising mechanism to improve responses to treatment. Epigenetic modifiers offer a unique route to achieving this objective (Table 3).
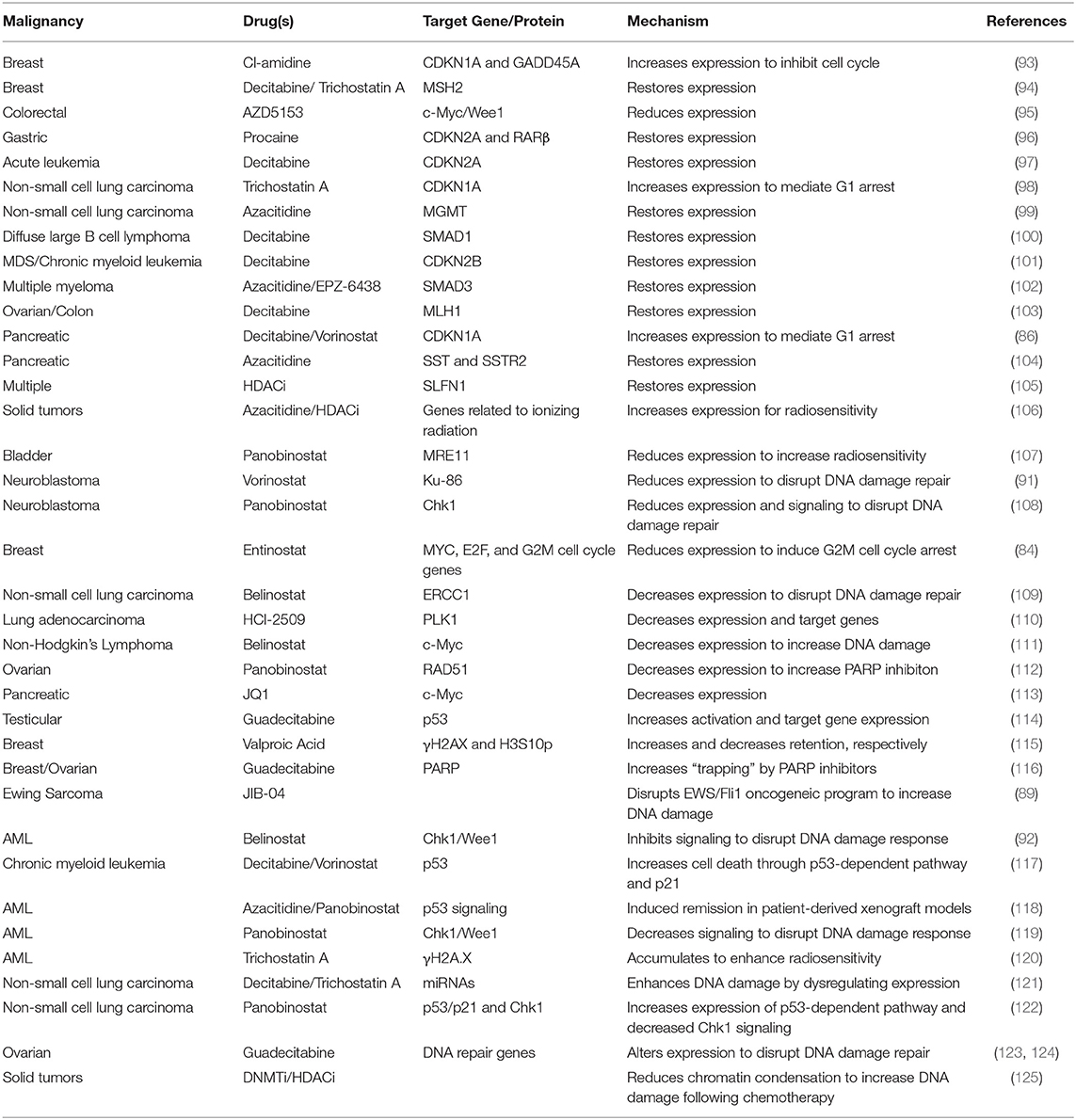
Table 3. Restoration of cell cycle control and disruption of DNA damage repair by epigenetic modifiers.
Suppress Immune Evasion/Augmenting Immune Responses
The immune system plays a pleiotropic role in cancer progression. Infiltration of immune cells into the tumor microenvironment releases a plethora of cytokines and growth factors that contribute to tumor proliferation, survival, and metastasis. Concurrently, activation of immune cells to target cancer is a promising strategy to utilize the host immune system in the fight against cancer (126). Like other anti-cancer treatments, malignant cells develop a resistance to immunotherapies by evading or suppressing the immune system and its activation via aberrant epigenetics (127, 128). Treatment with epigenetic modifiers has proved successful in augmenting immunotherapy. For a detailed review on this topic, please refer to Gomez et al. (129).
Epigenetic modifiers trigger increased expression of proteins for targeted therapies including immunotherapies. Trichostatin A (HDACi) up-regulated the mRNA and protein levels of both MIC-A and ULBP-2 in glioblastoma, which are recognized by natural killer (NK) cells to increase NK cell-mediated lysis (130). Entinostat (HDACi) blocked regulatory T cells (which negatively regulate the immune system and limit the efficacy of immunotherapy) in renal cell carcinoma via increased STAT3 acetylation, possibly due to increased CBP/p300 expression that acetylates STAT3 (131). In ovarian and colon cancer, azacitidine (DNMTi) increased the expression of multiple cancer cell-specific antigens. Since these antigens can be recognized by the host immune system, they represent prime targets for immunotherapies (132). The increased expression of cancer antigens also provides ample opportunity for the development of anti-cancer vaccines directing the host immune system to target these antigens. Such advances are currently in their infancy but have the potential for exceptional breakthrough in cancer treatment, especially when combined with epigenetic modifiers.
In non-small cell lung cancer, azacitidine (DNMTi) and givinostat (HDACi) induced Type I interferon signaling through transcriptional downregulation of MYC to increase the expression of the T cell chemoattractant CCL5, thereby reversing tumor immune evasion by promoting T cell infiltration. This combination also shifted host T cells from exhausted states (characterized by loss of effector function due to prolonged antigen stimulation) to memory and effector states [capable of durable responses to immune checkpoint blockades) via activation of associated genes (133)].
Cancer cells exploit the “immune checkpoint” function to evade the immune system (134) by expression of programmed cell death-1 (PD-1) or anti–cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) resulting in increased apoptosis of T cells. Immune checkpoint blockers such as nivolumab (monoclonal antibody blocking PD-1) and ipilimumab (monoclonal antibody blocking CTLA-4) have emerged as an attractive mechanism to decrease immune system evasion and tumor cell survival. Co-administration of azacitidine (DNMTi) and entinostat (HDACi) alongside immune checkpoint blockers improved treatment outcome in a preclinical metastatic cancer model via their inhibitory action on the myeloid derived suppressor cells within the tumor microenvironment (135).
Exploitation of the immune system to successfully diminish tumor burden is a promising avenue of improving anti-cancer therapy. The use of epigenetic modifiers offers a distinctive method to potentiate these therapies (Table 4).
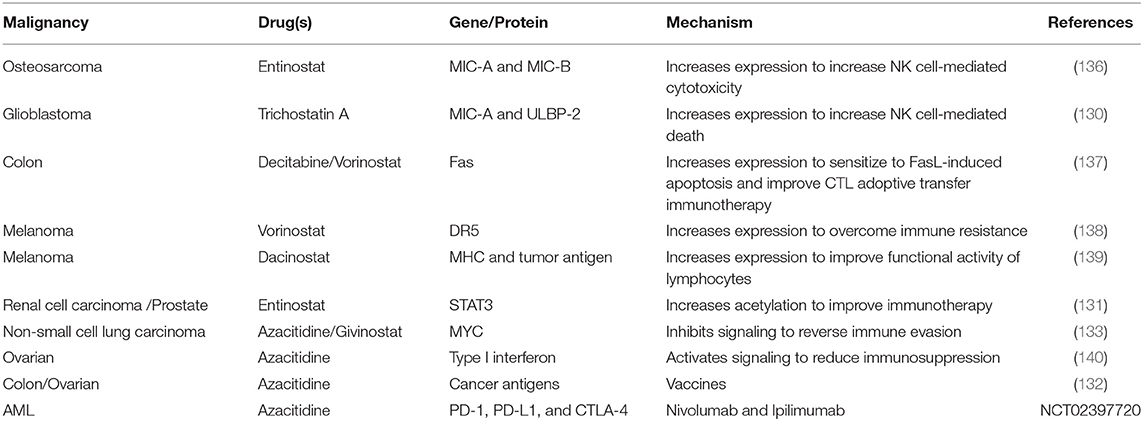
Table 4. Suppression of immune evasion/augmented immune responses following epigenetic modifier treatment.
Modulation of Microenvironmental Interactions
Cellular and extracellular matrix interactions within the tumor microenvironment are crucial for cancer development and progression. Epigenetic dysregulation in cancer is known to control adhesion through a variety of mechanisms (141–143). Thus, the use of epigenetic modifiers could provide a way to mollify these alterations. In solid tumors, disengagement from the microenvironment has severe consequences for the patient, as it is the first step in metastasis (144). Therefore, increasing cell adhesion proves beneficial to localize the tumor to the primary site.
In a majority of solid tumors, carcinomas arise from epithelial cells undergoing epithelial to mesenchymal transition (EMT), which causes loss of epithelial polarity/adhesion and increased migratory/invasiveness potential (145). Following EMT, cancer cells acquire stem-cell like properties and a higher rate of metastasis (146). EMT is controlled by multiple epigenetic mechanisms, including DNA methylation and histone modifications (147). The expression of the classical cell adhesion molecule and EMT suppressor E-cadherin is downregulated via promoter hypermethylation in cancer cells (148), or repressed by transcription factor Snail (149) in conjunction with histone modifiers such as lysine-specific histone demethylase 1 (LSD1) recruited by Snail (150). In breast cancer cells, EMT was suppressed by the LSD1 inhibitor pargyline (151). It is important to note, that the same study identified LSD1 to inhibit M1 macrophage infiltration into tumors, which is known to promote tumor progression and therapy resistance (152).
Targeting the SNAIL/LSD1 complex to prevent EMT via depletion of SNAIL expression was accomplished by the BRD4 inhibitor JQ1 in breast cancer. JQ1 repressed the expression of Gli1, an important mediator of SNAIL transcription. This prevented SNAIL-mediated repression of epithelial markers such as E-cadherin and prevented EMT (150, 153). Combined, these two studies provide a powerful indication of how the use of epigenetic modifiers can perturb EMT to prevent metastasis and improve treatment efficacy in solid tumors.
In hematologic malignancies, interactions within the bone marrow microenvironment transition malignant cells into chemoresistant states (154). Disruption of these interactions mobilizes cells from the bone marrow into the peripheral blood, thereby sensitizing them to therapy. In acute lymphoblastic leukemia (ALL), azacitidine (DNMTi) and panobinostat (HDACi) combined to disrupt cellular adhesion within the bone marrow microenvironment in ALL by decreasing the surface expression of the tetraspanin protein CD81, resulting in increased chemosensitivity (155, 156).
Hypoxia within the tumor microenvironment can often promote therapy resistance (157). This therapy resistance can be attributed to multiple factors including aberrant micro RNA (miRNA) expression and dysregulated epigenetic machinery (158, 159). Thus, gene expression alterations are accumulated and therapy resistance can occur in a variety of mechanisms such as those described in Figure 1. Due to the aberrant epigenetics involved, the use of epigenetic modifiers could sensitize cancer cells by reverting these hypoxic effects. However, further study is required to elucidate their effectiveness.
The role of microenvironmental interactions and their effect on cancer progression has been well-defined, however, the use of epigenetic modifiers to attenuate these effects has not been exploited. More studies across all cancer subtypes are necessary to achieve a greater understanding of how microenvironmental interactions can be modulated by epigenetic therapy.
Reprogramming of Cellular Metabolism
Through a variety of genetic and epigenetic mechanisms, metabolic reprogramming can render cancer cells resistant to chemotherapeutics (160–162). These changes can often result as a compensatory mechanism in response to the exposure of certain chemotherapeutics (162). Therefore, targeting aberrant cellular metabolism is a promising method of circumventing therapy resistance.
Due to the epigenetic regulation involved with aberrant metabolism, epigenetic modifiers could prove highly successful in mitigating the resultant chemoresistant effects. Treatment with entinostat (HDACi) combined with cisplatin upregulated the gene expression of thioredoxin-interacting protein (TXNIP), which inhibited the cellular uptake of glucose and increased DNA damage (163). This occurred via an increase in TXNIP promoter activity, however, this increase was only achievable with the two drugs in combination. In AML, treatment with the DNMTi azacitidine combined with the Bcl-2 inhibitor venetoclax disrupted cellular metabolism by decreasing glutathione levels, thereby diminishing electron transport chain complex II activity and oxidative phosphorylation (164).
Epigenetic modifiers can also augment the effectiveness of established antimetabolites like pemetrexed, which targets enzymes like thymidylate synthase (TI) catalyzing purine and pyrimidine synthesis. TI expression can be augmented post treatment with pemetrexed, thus leading to resistance (165). In non-small cell lung cancer, pemetrexed treatment followed by givinostat (HDACi) downregulated the mRNA and protein expression of TI, thereby overcoming therapy resistance and resulting in a synergistic increase in cell death (166).
While there has been strong evidence of the role played by epigenetic-induced metabolic changes in cancer cells in promoting therapy resistance, the study of how epigenetic modifiers can mitigate these effects has yet to be explored in depth. More examination into these effects is required in order to better overcome resistance to therapies.
Opportunities for Development of Rational Combinations With Epigenetic Therapy
The impact of epigenetic modifiers on global gene expression results in modulation of several genes, both promoting and inhibiting therapy resistance, thereby necessitating and offering opportunity to combine with targeted therapies. This is exemplified by a study in ovarian cancer that identified overexpression of CD146, a cell surface marker involved in tumor dissemination, following treatment with vorinostat (HDACi). This increased expression was exploited by combining vorinostat with anti-CD146 monoclonal antibody treatment to synergistically induce cell death via inhibition of CD146-mediated Akt signaling (167). Vorinostat (HDACi) along with decitabine (DNMTi) was also observed to significantly increase the expression of the tyrosine kinase AXL in AML. This led to the identification of a novel triple therapy with the AXL inhibitor BGB324 facilitating synergistic activation of cell death (168). Therefore, mechanistic understanding of epigenetic drug action is essential for developing rational combinations with targeted therapies.
Need for Clinical Trials
The use of epigenetic modifiers is a robust method for improving treatment efficacy in cancer. Through a variety of mechanisms, epigenetic therapy has the potential to augment the effectiveness of cancer treatments to improve overall survival in patients. In many of the examples presented above, a combination of epigenetic modifiers was used to induce specific changes that potentiate the effects of anti-cancer therapeutics in cancer cells. However, despite a plethora of clinical trials involving the use of epigenetic modifiers, very few have focused on the use of a combination of epigenetic modifiers along with anti-cancer therapy (Table 5). The combination therapies identified in this review underline the need and provide the basis for the development of future clinical trials to study their effectiveness.
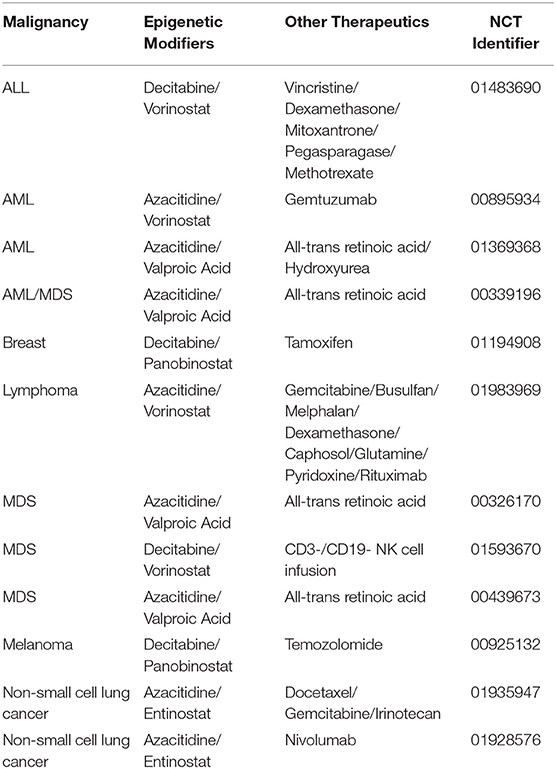
Table 5. List of clinical trials utilizing multiple epigenetic modifiers in combination with traditional therapy.
Additionally, it is worth mentioning that not only the use of epigenetic modifiers (either alone or in combination with one another) in conjunction with chemotherapeutics should be studied, but the protocols in which they are administered should be considered as well. Simultaneous exposures have been shown to have an inhibitory effect on cell viability compared to sequential treatment (169). Additionally, a study of the use of azacitidine and panobinostat in B-ALL identified that following treatment in mice; leukemic cells were mobilized from the bone marrow into the peripheral blood. This mobilization was responsible for the improved efficacy of subsequent chemotherapy treatment, thus suggesting that staggering the treatments had a significant effect (170). More study on this effect as well as its potential in other cancer subtypes must be performed to exploit the efficacy of epigenetic treatments.
Conclusions
Aberrant epigenetics is responsible for the development and progression of several cancers. These alterations can be the driving forces of therapy resistance and survival. Treatment with epigenetic modifiers offers a unique route to diminishing these effects and re-sensitizing cancer cells to traditional therapies. While there have been some clinical trials studying the efficacy of epigenetic modifiers in cancer, more studies focusing on identifying specific gene targets are required, particularly with a combination of epigenetic modifiers in conjunction with other therapies. By precisely identifying sensitization biomarkers, epigenetic/chemotherapeutic/immunotherapeutic combination therapies can achieve greater translational success (171). Follow-up studies using comprehensive analyses like RNAseq, global methylation, and chromatin immunoprecipitation-Seq are required to identify pathways of sensitization.
It is also imperative to include analyses of non-coding regions of the DNA, such as miRNA. While epigenetic alterations during oncogenesis directly affect the transcription of coding genes, these variations can have an effect on the expression of miRNAs (172, 173), which are non-coding RNAs that function in RNA silencing and post-transcriptional regulation of gene expression. miRNAs can mediate either tumor suppressive or oncogenic effects depending on their gene target (174). Examination of alterations in miRNA expression following treatment with epigenetic modifiers could identify additional sensitization mechanisms and therapeutic markers.
Studies investigating the development of inhibitors of atypical histone modifications, such as citrullination, phosphorylation, sumoylation, ubiquitylation, and ribosylation; are needed because these modifications are also known to regulate gene transcription and contribute to cancer progression (175–178). The mechanisms outlined in this review offer not only a rationale for successful combinations and mechanisms, but also identify indications for their use in specific patients based on the markers being modulated, in line with the advancements in personalized medicine. Further studies on the mechanisms of epigenetic modifier action in cancer are needed to identify markers that can detect and predict clinical response.
Author Contributions
AQ wrote the draft manuscript and generated figures and tables. AG and SB edited the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by Andrew McDonough B+ Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. (2009) 27:351–7. doi: 10.1055/s-0029-1237423
2. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–3. doi: 10.1101/gad.1787609
3. Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. (2011) 2:607–17. doi: 10.1177/1947601910393957
4. Sumarpo A, Ito K, Saiki Y, Ishizawa K, Wang R, Chen N, et al. Genetic and epigenetic aberrations of ABCB1 synergistically boost the acquisition of taxane resistance in esophageal squamous cancer cells. Biochem Biophys Res Commun. (2020) 526:586–91. doi: 10.1016/j.bbrc.2020.03.114
5. Lee DD, Leao R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. (2019) 129:1801. doi: 10.1172/JCI128527
6. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. (2014) 26:577–90. doi: 10.1016/j.ccr.2014.07.028
7. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. (2016) 8:a019521. doi: 10.1101/cshperspect.a019521
8. Das C, Kundu TK. Transcriptional regulation by the acetylation of nonhistone proteins in humans – a new target for therapeutics. IUBMB Life. (2005) 57:137–49. doi: 10.1080/15216540500090629
9. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
10. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, et al. p53 is regulated by the lysine demethylase LSD1. Nature. (2007) 449:105–8. doi: 10.1038/nature06092
11. Park J, Lee JH, La M, Jang MJ, Chae GW, Kim SB, et al. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J Mol Biol. (2007) 368:388–97. doi: 10.1016/j.jmb.2007.02.047
12. Scoumanne A, Chen X. Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol Histopathol. (2008) 23:1143–9. doi: 10.14670/HH-23.1143
13. Sung B, Pandey MK, Ahn KS, Yi T, Chaturvedi MM, Liu M, et al. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. (2008) 111:4880–91. doi: 10.1182/blood-2007-10-117994
14. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. (2012) 22:9–20. doi: 10.1016/j.ccr.2012.06.008
15. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. (2011) 11:726–34. doi: 10.1038/nrc3130
16. Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: paradigms, puzzles, and potential. Biochim Biophys Acta. (2016) 1865:275–88. doi: 10.1016/j.bbcan.2016.04.001
17. Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. (2012) 81:303–11. doi: 10.1111/j.1399-0004.2011.01809.x
18. Hassler MR, Egger G. Epigenomics of cancer - emerging new concepts. Biochimie. (2012) 94:2219–30. doi: 10.1016/j.biochi.2012.05.007
19. Crea F, Danesi R, Farrar WL. Cancer stem cell epigenetics and chemoresistance. Epigenomics. (2009) 1:63–79. doi: 10.2217/epi.09.4
20. Zeller C, Brown R. Therapeutic modulation of epigenetic drivers of drug resistance in ovarian cancer. Ther Adv Med Oncol. (2010) 2:319–29. doi: 10.1177/1758834010375759
21. Farhy C, Hariharan S, Ylanko J, Orozco L, Zeng FY, Pass I, et al. Improving drug discovery using image-based multiparametric analysis of the epigenetic landscape. Elife. (2019) 8:e49683. doi: 10.7554/eLife.49683.sa2
22. Achinger-Kawecka J, Valdes-Mora F, Luu PL, Giles KA, Caldon CE, Qu W, et al. Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat Commun. (2020) 11:320. doi: 10.1038/s41467-019-14098-x
23. Zhang T, Pilko A, Wollman R. Loci specific epigenetic drug sensitivity. Nucleic Acids Res. (2020) 48:4797–810. doi: 10.1093/nar/gkaa210
24. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. (2016) 17:630–41. doi: 10.1038/nrg.2016.93
25. Tiash S, Chowdhury E. Growth factor receptors: promising drug targets in cancer. J Cancer Metast Treat. (2015) 1:190. doi: 10.4103/2394-4722.163151
26. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. (2019) 18:26. doi: 10.1186/s12943-019-0954-x
27. Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther. (2003) 2:971–84.
28. Lee J, Bartholomeusz C, Mansour O, Humphries J, Hortobagyi GN, Ordentlich P, et al. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res Treat. (2014) 146:259–72. doi: 10.1007/s10549-014-3014-7
29. Novotny CJ, Pollari S, Park JH, Lemmon MA, Shen W, Shokat KM. Overcoming resistance to HER2 inhibitors through state-specific kinase binding. Nat Chem Biol. (2016) 12:923–30. doi: 10.1038/nchembio.2171
30. Maximov PY, Abderrahman B, Curpan RF, Hawsawi YM, Fan P, Jordan VC. A unifying biology of sex steroid-induced apoptosis in prostate and breast cancers. Endocr Relat Cancer. (2018) 25:R83–113. doi: 10.1530/ERC-17-0416
31. Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer Res. (2011) 71:1893–903. doi: 10.1158/0008-5472.CAN-10-2458
32. Stark K, Burger A, Wu J, Shelton P, Polin L, Li J. Reactivation of estrogen receptor alpha by vorinostat sensitizes mesenchymal-like triple-negative breast cancer to aminoflavone, a ligand of the aryl hydrocarbon receptor. PLoS ONE. (2013) 8:e74525. doi: 10.1371/journal.pone.0074525
33. Sharma V, Kumar L, Mohanty SK, Maikhuri JP, Rajender S, Gupta G. Sensitization of androgen refractory prostate cancer cells to anti-androgens through re-expression of epigenetically repressed androgen receptor - synergistic action of quercetin and curcumin. Mol Cell Endocrinol. (2016) 431:12–23. doi: 10.1016/j.mce.2016.04.024
34. von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. (2017) 17:352–66. doi: 10.1038/nrc.2017.28
35. Butler LM, Liapis V, Bouralexis S, Welldon K, Hay S, Thai le M, et al. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int JCancer. (2006) 119:944–54. doi: 10.1002/ijc.21939
36. Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci USA. (2008) 105:11317–22. doi: 10.1073/pnas.0801868105
37. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol Cell. (2019) 111:1–17. doi: 10.1111/boc.201800050
38. Wang W, Oguz G, Lee PL, Bao Y, Wang P, Terp MG, et al. KDM4B-regulated unfolded protein response as a therapeutic vulnerability in PTEN-deficient breast cancer. J Exp Med. (2018) 215:2833–49. doi: 10.1084/jem.20180439
39. Xylinas E, Hassler MR, Zhuang D, Krzywinski M, Erdem Z, Robinson BD, et al. An epigenomic approach to improving response to neoadjuvant cisplatin chemotherapy in bladder cancer. Biomolecules. (2016) 6:37. doi: 10.3390/biom6030037
40. Cui Y, Naz A, Thompson DH, Irudayaraj J. Decitabine nanoconjugate sensitizes human glioblastoma cells to temozolomide. Mol Pharm. (2015) 12:1279–88. doi: 10.1021/mp500815b
41. Eramo A, Pallini R, Lotti F, Sette G, Ricci-Vitiani L, Bartucci M, et al. Effective treatment of glioblastoma tumors by combined administration of decitabine and TRAIL. Cancer Res. (2005) 65:425–425. doi: 10.1158/0008-5472.CAN-05-1724
42. Schneider BJ, Shah MA, Klute K, Ocean A, Popa E, Altorki N, et al. Phase I study of epigenetic priming with azacitidine prior to standard neoadjuvant chemotherapy for patients with resectable gastric and esophageal adenocarcinoma: evidence of tumor hypomethylation as an indicator of major histopathologic response. Clin Cancer Res. (2017) 23:2673–80. doi: 10.1158/1078-0432.CCR-16-1896
43. Reu FJ, Leaman DW, Maitra RR, Bae SI, Cherkassky L, Fox MW, et al. Expression of RASSF1A, an epigenetically silenced tumor suppressor, overcomes resistance to apoptosis induction by interferons. Cancer Res. (2006) 66:2785–93. doi: 10.1158/0008-5472.CAN-05-2303
44. Wu A, Wu K, Li M, Bao L, Shen X, Li S, et al. Upregulation of microRNA-492 induced by epigenetic drug treatment inhibits the malignant phenotype of clear cell renal cell carcinoma in vitro. Mol Med Rep. (2015) 12:1413–20. doi: 10.3892/mmr.2015.3550
45. Avramis VI, Mecum RA, Nyce J, Steele DA, Holcenberg JS. Pharmacodynamic and DNA methylation studies of high-dose 1-beta-D-arabinofuranosyl cytosine before and after in vivo 5-azacytidine treatment in pediatric patients with refractory acute lymphocytic leukemia. Cancer Chemother Pharmacol. (1989) 24:203–10. doi: 10.1007/BF00257619
46. Andrade AF, Borges KS, Castro-Gamero AM, Silveira VS, Suazo VK, Oliveira JC, et al. Zebularine induces chemosensitization to methotrexate and efficiently decreases AhR gene methylation in childhood acute lymphoblastic leukemia cells. Anticancer Drugs. (2014) 25:72–81. doi: 10.1097/CAD.0000000000000028
47. Al-Jamal HA, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer. (2015) 15:869. doi: 10.1186/s12885-015-1695-x
48. Al-Jamal HAN, Johan MF, Mat Jusoh SA, Ismail I, Wan Taib WR. Re-expression of bone marrow proteoglycan-2 by 5-azacytidine is associated with STAT3 inactivation and sensitivity response to imatinib in resistant CML cells. Asian Pac J Cancer Prev. (2018) 19:1585–90. doi: 10.22034/APJCP.2018.19.6.1585
49. Surriga O, Schwartz GK. Abstract 2940: chemosensitization of cholangiocarcinoma by guadecitabine. Cancer Res. (2018) 78:2940. doi: 10.1158/1538-7445.AM2018-2940
50. Shire A, Lomberk G, Lai JP, Zou H, Tsuchiya N, Aderca I, et al. Restoration of epigenetically silenced SULF1 expression by 5-aza-2-deoxycytidine sensitizes hepatocellular carcinoma cells to chemotherapy-induced apoptosis. Med Epigenet. (2015) 3:1–18. doi: 10.1159/000375461
51. Kaminskyy VO, Surova OV, Vaculova A, Zhivotovsky B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis. (2011) 32:1450–8. doi: 10.1093/carcin/bgr135
52. Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal. (2019) 12:eaau2922. doi: 10.1126/scisignal.aau2922
53. Liu Y, Mondello P, Erazo T, Tannan NB, Asgari Z, de Stanchina E, et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc Natl Acad Sci USA. (2018) 115:12034–9. doi: 10.1073/pnas.1806928115
54. Xu Q, Liu X, Zhu S, Hu X, Niu H, Zhang X, et al. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostate cancer cells to histone deacetylase inhibitor Trichostatin A. J Cell Mol Med. (2018) 22:1909–22. doi: 10.1111/jcmm.13475
55. Sabatino MA, Geroni C, Ganzinelli M, Ceruti R, Broggini M. Zebularine partially reverses GST methylation in prostate cancer cells and restores sensitivity to the DNA minor groove binder brostallicin. Epigenetics. (2013) 8:656–65. doi: 10.4161/epi.24916
56. Liao H, Xiao Y, Hu Y, Xiao Y, Yin Z, Liu L, et al. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol Rep. (2016) 35:64–72. doi: 10.3892/or.2015.4331
57. Jia L, Zhang S, Huang Y, Zheng Y, Gan Y. Trichostatin A increases radiosensitization of tongue squamous cell carcinoma via miR375. Oncol Rep. (2017) 37:305–12. doi: 10.3892/or.2016.5261
58. Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. (2015) 7:831–47. doi: 10.15252/emmm.201404396
59. Balch C, Yan P, Craft T, Young S, Skalnik DG, Huang TH, et al. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol Cancer Ther. (2005) 4:1505–14. doi: 10.1158/1535-7163.MCT-05-0216
60. El-Zawahry A, Lu P, White SJ, Voelkel-Johnson C. In vitro efficacy of AdTRAIL gene therapy of bladder cancer is enhanced by trichostatin A-mediated restoration of CAR expression and downregulation of cFLIP and Bcl-XL. Cancer Gene Ther. (2006) 13:281–9. doi: 10.1038/sj.cgt.7700905
61. Morales JC, Ruiz-Magana MJ, Carranza D, Ortiz-Ferron G, Ruiz-Ruiz C. HDAC inhibitors with different gene regulation activities depend on the mitochondrial pathway for the sensitization of leukemic T cells to TRAIL-induced apoptosis. Cancer Lett. (2010) 297:91–100. doi: 10.1016/j.canlet.2010.04.029
62. Okabe S, Tauchi T, Kimura S, Maekawa T, Ohyashiki K. The analysis of HDAC inhibitor, vorinostat efficacy against wild type and BCR-ABL mutant positive leukemia cells in monotherapy and in combination with a pan-aurora kinase inhibitor, MK-0457. Blood. (2008) 112:5025. doi: 10.1182/blood.V112.11.5025.5025
63. Guo F, Sigua C, Tao J, Bali P, George P, Li Y, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. (2004) 64:2580–9. doi: 10.1158/0008-5472.CAN-03-2629
64. Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. (2003) 63:5126–35.
65. Bali P, George P, Cohen P, Tao J, Guo F, Sigua C, et al. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clin Cancer Res. (2004) 10:4991–7. doi: 10.1158/1078-0432.CCR-04-0210
66. Wu Y, Doepner M, Hojnacki T, Feng Z, Katona BW, He X, et al. Disruption of the menin-MLL interaction triggers menin protein degradation via ubiquitin-proteasome pathway. Am J Cancer Res. (2019) 9:1682–94.
67. Zhang X, Li B, de Jonge N, Bjorkholm M, Xu D. The DNA methylation inhibitor induces telomere dysfunction and apoptosis of leukemia cells that is attenuated by telomerase over-expression. Oncotarget. (2015) 6:4888–900. doi: 10.18632/oncotarget.2917
68. Kuang Y, El-Khoueiry A, Taverna P, Ljungman M, Neamati N. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol Oncol. (2015) 9:1799–814. doi: 10.1016/j.molonc.2015.06.002
69. Lee WY, Chen PC, Wu WS, Wu HC, Lan CH, Huang YH, et al. Panobinostat sensitizes KRAS-mutant non-small-cell lung cancer to gefitinib by targeting TAZ. Int J Cancer. (2017) 141:1921–31. doi: 10.1002/ijc.30888
70. Ishiguro K, Kitajima H, Niinuma T, Ishida T, Maruyama R, Ikeda H, et al. DOT1L inhibition blocks multiple myeloma cell proliferation by suppressing IRF4-MYC signaling. Haematologica. (2019) 104:155–65. doi: 10.3324/haematol.2018.191262
71. Civallero M, Cosenza M, Pozzi S, Sacchi S. Ruxolitinib combined with vorinostat suppresses tumor growth and alters metabolic phenotype in hematological diseases. Oncotarget. (2017) 8:103797–814. doi: 10.18632/oncotarget.21951
72. Benedetti R, Dell'Aversana C, De Marchi T, Rotili D, Liu NQ, Novakovic B, et al. Inhibition of histone demethylases LSD1 and UTX regulates ERalpha signaling in breast cancer. Cancers. (2019) 11:2027. doi: 10.3390/cancers11122027
73. Li F, Miao L, Xue T, Qin H, Mondal S, Thompson PR, et al. Inhibiting PAD2 enhances the anti-tumor effect of docetaxel in tamoxifen-resistant breast cancer cells. J Exp Clin Cancer Res. (2019) 38:414. doi: 10.1186/s13046-019-1404-8
74. Lou YF, Zou ZZ, Chen PJ, Huang GB, Li B, Zheng DQ, et al. Combination of gefitinib and DNA methylation inhibitor decitabine exerts synergistic anti-cancer activity in colon cancer cells. PLoS ONE. (2014) 9:e97719. doi: 10.1371/journal.pone.0097719
75. Cui X, Witalison EE, Chumanevich AP, Chumanevich AA, Poudyal D, Subramanian V, et al. The induction of microRNA-16 in colon cancer cells by protein arginine deiminase inhibition causes a p53-dependent cell cycle arrest. PLoS ONE. (2013) 8:e53791. doi: 10.1371/journal.pone.0053791
76. Poulaki V, Mitsiades CS, Kotoula V, Negri J, McMullan C, Miller JW, et al. Molecular sequelae of histone deacetylase inhibition in human retinoblastoma cell lines: clinical implications. Invest Ophthalmol Vis Sci. (2009) 50:4072–9. doi: 10.1167/iovs.09-3517
77. Meng X, Yang S, Li Y, Li Y, Devor EJ, Bi J, et al. Combination of proteasome and histone deacetylase inhibitors overcomes the impact of gain-of-function p53 mutations. Dis Mark. (2018) 2018:3810108. doi: 10.1155/2018/3810108
78. Chao A, Lin CY, Chao AN, Tsai CL, Chen MY, Lee LY, et al. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget. (2017) 8:74434–50. doi: 10.18632/oncotarget.20158
79. Jiang XJ, Huang KK, Yang M, Qiao L, Wang Q, Ye JY, et al. Synergistic effect of panobinostat and bortezomib on chemoresistant acute myelogenous leukemia cells via AKT and NF-kappaB pathways. Cancer Lett. (2012) 326:135–42. doi: 10.1016/j.canlet.2012.07.030
80. Wang L, Syn NL, Subhash VV, Any Y, Thuya WL, Cheow ESH, et al. Pan-HDAC inhibition by panobinostat mediates chemosensitization to carboplatin in non-small cell lung cancer via attenuation of EGFR signaling. Cancer Lett. (2018) 417:152–60. doi: 10.1016/j.canlet.2017.12.030
81. Feng S, Jin Y, Cui M, Zheng J. Lysine-Specific Demethylase 1 (LSD1) Inhibitor S2101 induces autophagy via the AKT/mTOR pathway in SKOV3 ovarian cancer cells. Med Sci Monit. (2016) 22:4742–8. doi: 10.12659/MSM.898825
82. Festuccia C, Gravina GL, D'Alessandro AM, Muzi P, Millimaggi D, Dolo V, et al. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr Relat Cancer. (2009) 16:401–13. doi: 10.1677/ERC-08-0130
83. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. (2003) 24:139–45. doi: 10.1016/S0165-6147(03)00026-9
84. Merino VF, Cho S, Nguyen N, Sadik H, Narayan A, Talbot C Jr, et al. Induction of cell cycle arrest and inflammatory genes by combined treatment with epigenetic, differentiating, and chemotherapeutic agents in triple-negative breast cancer. Breast Cancer Res. (2018) 20:145. doi: 10.1186/s13058-018-1068-x
85. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. (2003) 17:590–603. doi: 10.1038/sj.leu.2402824
86. Susanto JM, Colvin EK, Pinese M, Chang DK, Pajic M, Mawson A, et al. The epigenetic agents suberoylanilide hydroxamic acid and 5AZA2' deoxycytidine decrease cell proliferation, induce cell death and delay the growth of MiaPaCa2 pancreatic cancer cells in vivo. Int J Oncol. (2015) 46:2223–30. doi: 10.3892/ijo.2015.2894
87. Robin TP, Smith A, McKinsey E, Reaves L, Jedlicka P, Ford HL. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol Cancer Res. (2012) 10:1098–108. doi: 10.1158/1541-7786.MCR-12-0086
88. Knott MML, Holting TLB, Ohmura S, Kirchner T, Cidre-Aranaz F, Grunewald TGP. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer Metastasis Rev. (2019) 38:625–42. doi: 10.1007/s10555-019-09839-9
89. Parrish JK, McCann TS, Sechler M, Sobral LM, Ren W, Jones KL, et al. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget. (2018) 9:33110–23. doi: 10.18632/oncotarget.26011
90. Turgeon MO, Perry NJS, Poulogiannis G. DNA damage, repair, and cancer metabolism. Front Oncol. (2018) 8:15. doi: 10.3389/fonc.2018.00015
91. Mueller S, Yang X, Sottero TL, Gragg A, Prasad G, Polley MY, et al. Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: efficacy and underlying mechanisms. Cancer Lett. (2011) 306:223–9. doi: 10.1016/j.canlet.2011.03.010
92. Zhou L, Chen S, Zhang Y, Kmieciak M, Leng Y, Li L, et al. The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood. (2016) 127:2219–30. doi: 10.1182/blood-2015-06-653717
93. McElwee JL, Mohanan S, Griffith OL, Breuer HC, Anguish LJ, Cherrington BD, et al. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer. (2012) 12:500. doi: 10.1186/1471-2407-12-500
94. Ponnusamy L, Mahalingaiah PKS, Chang YW, Singh KP. Reversal of epigenetic aberrations associated with the acquisition of doxorubicin resistance restores drug sensitivity in breast cancer cells. Eur J Pharm Sci. (2018) 123:56–69. doi: 10.1016/j.ejps.2018.07.028
95. Zhang P, Li R, Xiao H, Liu W, Zeng X, Xie G, et al. BRD4 Inhibitor AZD5153 suppresses the proliferation of colorectal cancer cells and sensitizes the anticancer effect of PARP Inhibitor. Int J Biol Sci. (2019) 15:1942–54. doi: 10.7150/ijbs.34162
96. Li YC, Wang Y, Li DD, Zhang Y, Zhao TC, Li CF. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J Cell Biochem. (2018) 119:2440–9. doi: 10.1002/jcb.26407
97. Niitsu N, Hayashi Y, Sugita K, Honma Y. Sensitization by 5-aza-2'-deoxycytidine of leukaemia cells with MLL abnormalities to induction of differentiation by all-trans retinoic acid and 1alpha,25-dihydroxyvitamin D3. Br J Haematol. (2001) 112:315–26. doi: 10.1046/j.1365-2141.2001.02523.x
98. Mukhopadhyay NK, Weisberg E, Gilchrist D, Bueno R, Sugarbaker DJ, Jaklitsch MT. Effectiveness of trichostatin A as a potential candidate for anticancer therapy in non-small-cell lung cancer. Ann Thorac Surg. (2006) 81:1034–42. doi: 10.1016/j.athoracsur.2005.06.059
99. Fuller M, Klein M, Schmidt E, Rohde C, Gollner S, Schulze I, et al. 5-azacytidine enhances efficacy of multiple chemotherapy drugs in AML and lung cancer with modulation of CpG methylation. Int J Oncol. (2015) 46:1192–204. doi: 10.3892/ijo.2014.2792
100. Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. (2013) 3:1002–19. doi: 10.1158/2159-8290.CD-13-0117
101. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O'Brien S, Cortes J, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. (2007) 109:52–7. doi: 10.1182/blood-2006-05-021162
102. Dimopoulos K, Sogaard Helbo A, Fibiger Munch-Petersen H, Sjo L, Christensen J, Sommer Kristensen L, et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol. (2018) 12:180–95. doi: 10.1002/1878-0261.12157
103. Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2'-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. (2000) 60:6039–44.
104. Gailhouste L, Liew LC, Hatada I, Nakagama H, Ochiya T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. (2018) 9:468. doi: 10.1038/s41419-018-0487-z
105. Tang SW, Thomas A, Murai J, Trepel JB, Bates SE, Rajapakse VN, et al. Overcoming resistance to DNA-targeted agents by epigenetic activation of Schlafen 11 (SLFN11) expression with class i histone deacetylase inhibitors. Clin Cancer Res. (2018) 24:1944–53. doi: 10.1158/1078-0432.CCR-17-0443
106. Ren J, Chu Y, Ma H, Zhang Y, Zhang X, Zhao D, et al. Epigenetic interventions increase the radiation sensitivity of cancer cells. Curr Pharm Des. (2014) 20:1857–65. doi: 10.2174/13816128113199990529
107. Groselj B, Ruan JL, Scott H, Gorrill J, Nicholson J, Kelly J, et al. Radiosensitization In vivo by histone deacetylase inhibition with no increase in early normal tissue radiation toxicity. Mol Cancer Ther. (2018) 17:381–92. doi: 10.1158/1535-7163.MCT-17-0011
108. Wang G, Edwards H, Caldwell JT, Buck SA, Qing WY, Taub JW, et al. Panobinostat synergistically enhances the cytotoxic effects of cisplatin, doxorubicin or etoposide on high-risk neuroblastoma cells. PLoS ONE. (2013) 8:e76662. doi: 10.1371/journal.pone.0076662
109. To KK, Tong WS, Fu LW. Reversal of platinum drug resistance by the histone deacetylase inhibitor belinostat. Lung Cancer. (2017) 103:58–65. doi: 10.1016/j.lungcan.2016.11.019
110. Dalvi PS, Macheleidt IF, Lim SY, Meemboor S, Muller M, Eischeid-Scholz H, et al. LSD1 Inhibition attenuates tumor growth by disrupting PLK1 mitotic pathway. Mol Cancer Res. (2019) 17:1326–37. doi: 10.1158/1541-7786.MCR-18-0971
111. Nguyen T, Parker R, Hawkins E, Holkova B, Yazbeck V, Kolluri A, et al. Synergistic interactions between PLK1 and HDAC inhibitors in non-Hodgkin's lymphoma cells occur in vitro and in vivo and proceed through multiple mechanisms. Oncotarget. (2017) 8:31478–93. doi: 10.18632/oncotarget.15649
112. Wilson AJ, Sarfo-Kantanka K, Barrack T, Steck A, Saskowski J, Crispens MA, et al. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol Oncol. (2016) 143:143–51. doi: 10.1016/j.ygyno.2016.07.088
113. Sahai V, Kumar K, Knab LM, Chow CR, Raza SS, Bentrem DJ, et al. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol Cancer Ther. (2014) 13:1907–17. doi: 10.1158/1535-7163.MCT-13-0925
114. Albany C, Hever-Jardine MP, von Herrmann KM, Yim CY, Tam J, Warzecha JM, et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget. (2017) 8:2949–59. doi: 10.18632/oncotarget.13811
115. Sharda A, Rashid M, Shah SG, Sharma AK, Singh SR, Gera P, et al. Elevated HDAC activity and altered histone phospho-acetylation confer acquired radio-resistant phenotype to breast cancer cells. Clin Epigenet. (2020) 12:4. doi: 10.1186/s13148-019-0800-4
116. Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An effective epigenetic-PARP inhibitor combination therapy for breast and ovarian cancers independent of BRCA mutations. Clin Cancer Res. (2018) 24:3163–75. doi: 10.1158/1078-0432.CCR-18-0204
117. Brodska B, Holoubek A, Otevrelova P, Kuzelova K. Combined treatment with low concentrations of decitabine and SAHA causes cell death in leukemic cell lines but not in normal peripheral blood lymphocytes. Biomed Res Int. (2013) 2013:659254. doi: 10.1155/2013/659254
118. Gopalakrishnapillai A, Kolb EA, McCahan SM, Barwe SP. Epigenetic drug combination induces remission in mouse xenograft models of pediatric acute myeloid leukemia. Leuk Res. (2017) 58:91–7. doi: 10.1016/j.leukres.2017.05.004
119. Qi W, Zhang W, Edwards H, Chu R, Madlambayan GJ, Taub JW, et al. Synergistic anti-leukemic interactions between panobinostat and MK-1775 in acute myeloid leukemia ex vivo. Cancer Biol Ther. (2015) 16:1784–93. doi: 10.1080/15384047.2015.1095406
120. Karagiannis TC, Harikrishnan KN, El-Osta A. The histone deacetylase inhibitor, Trichostatin A, enhances radiation sensitivity and accumulation of gammaH2A.X. Cancer Biol Ther. (2005) 4:787–93. doi: 10.4161/cbt.4.7.1922
121. El-Awady RA, Hersi F, Al-Tunaiji H, Saleh EM, Abdel-Wahab AH, Al Homssi A, et al. Epigenetics and miRNA as predictive markers and targets for lung cancer chemotherapy. Cancer Biol Ther. (2015) 16:1056–70. doi: 10.1080/15384047.2015.1046023
122. Greve G, Schiffmann I, Pfeifer D, Pantic M, Schuler J, Lubbert M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer. (2015) 15:947. doi: 10.1186/s12885-015-1967-5
123. Fang F, Cardenas H, Huang H, Jiang G, Perkins SM, Zhang C, et al. Genomic and Epigenomic signatures in ovarian cancer associated with resensitization to platinum drugs. Cancer Res. (2018) 78:631–44. doi: 10.1158/0008-5472.CAN-17-1492
124. Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A Randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin Cancer Res. (2020) 26:1009–16. doi: 10.1158/1078-0432.CCR-19-1638
125. Li J, Hao D, Wang L, Wang H, Wang Y, Zhao Z, et al. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy. Sci Rep. (2017) 7:4035. doi: 10.1038/s41598-017-04406-0
126. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
127. Sylvestre M, Tarte K, Roulois D. Epigenetic mechanisms driving tumor supportive microenvironment differentiation and function: a role in cancer therapy? Epigenomics. (2020) 12:157–69. doi: 10.2217/epi-2019-0165
128. Xiao Q, Nobre A, Pineiro P, Berciano-Guerrero MA, Alba E, Cobo M, et al. Genetic and epigenetic biomarkers of immune checkpoint blockade response. J Clin Med. (2020) 9:286. doi: 10.3390/jcm9010286
129. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. (2019). doi: 10.1016/j.semcancer.2019.12.019. [Epub ahead of print].
130. Horing E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. (2013) 33:1351–60.
131. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS ONE. (2012) 7:e30815. doi: 10.1371/journal.pone.0030815
132. Siebenkas C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE. (2017) 12:e0179501. doi: 10.1371/journal.pone.0179501
133. Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. (2017) 171:1284–300 e1221. doi: 10.1016/j.cell.2017.10.022
134. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. (2019) 18:10. doi: 10.1186/s12943-018-0928-4
135. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. (2014) 111:11774–9. doi: 10.1073/pnas.1410626111
136. Kiany S, Huang G, Kleinerman ES. Effect of entinostat on NK cell-mediated cytotoxicity against osteosarcoma cells and osteosarcoma lung metastasis. Oncoimmunology. (2017) 6:e1333214. doi: 10.1080/2162402X.2017.1333214
137. Yang D, Torres CM, Bardhan K, Zimmerman M, McGaha TL, Liu K. Decitabine and vorinostat cooperate to sensitize colon carcinoma cells to Fas ligand-induced apoptosis in vitro and tumor suppression in vivo. J Immunol. (2012) 188:4441–9. doi: 10.4049/jimmunol.1103035
138. Jazirehi AR, Kurdistani SK, Economou JS. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J Immunol. (2014) 192:3981–9. doi: 10.4049/jimmunol.1302532
139. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. (2009) 69:8693–9. doi: 10.1158/0008-5472.CAN-09-1456
140. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci USA. (2017) 114:E10981–990. doi: 10.1073/pnas.1712514114
141. Katto J, Mahlknecht U. Epigenetic regulation of cellular adhesion in cancer. Carcinogenesis. (2011) 32:1414–8. doi: 10.1093/carcin/bgr120
142. Dreger H, Ludwig A, Weller A, Stangl V, Baumann G, Meiners S, et al. Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension. (2012) 60:1176–83. doi: 10.1161/HYPERTENSIONAHA.112.191098
143. Furukawa Y, Kikuchi J. Epigenetic regulation of cell adhesion-mediated drug resistance acquisition in multiple myeloma. Rinsho Ketsueki. (2016) 57:546–55.
144. Emon B, Bauer J, Jain Y, Jung B, Saif T. Biophysics of tumor microenvironment and cancer metastasis - a mini review. Comput Struct Biotechnol J. (2018) 16:279–87. doi: 10.1016/j.csbj.2018.07.003
145. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
146. Pal M, Chen H, Lee BH, Lee JYH, Yip YS, Tan NS, et al. Epithelial-mesenchymal transition of cancer cells using bioengineered hybrid scaffold composed of hydrogel/3D-fibrous framework. Sci Rep. (2019) 9:8997. doi: 10.1038/s41598-019-45384-9
147. O'Leary K, Shia A, Schmid P. Epigenetic regulation of EMT in non-small cell lung cancer. Curr Cancer Drug Targets. (2018) 18:89–96. doi: 10.2174/1568009617666170203162556
148. Shargh SA, Sakizli M, Khalaj V, Movafagh A, Yazdi H, Hagigatjou E, et al. Downregulation of E-cadherin expression in breast cancer by promoter hypermethylation and its relation with progression and prognosis of tumor. Med Oncol. (2014) 31:250. doi: 10.1007/s12032-014-0250-y
149. Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. (2000) 2:84–9. doi: 10.1038/35000034
150. Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. (2010) 29:4896–904. doi: 10.1038/onc.2010.234
151. Boulding T, McCuaig RD, Tan A, Hardy K, Wu F, Dunn J, et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci Rep. (2018) 8:73. doi: 10.1038/s41598-017-17913-x
152. Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim H, et al. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J Immunother Cancer. (2019) 7:147. doi: 10.1186/s40425-019-0610-4
153. Lu L, Chen Z, Lin X, Tian L, Su Q, An P, et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. (2020) 27:255–68. doi: 10.1038/s41418-019-0353-2
154. Barwe SP, Quagliano A, Gopalakrishnapillai A. Eviction from the sanctuary: development of targeted therapy against cell adhesion molecules in acute lymphoblastic leukemia. Semin Oncol. (2017) 44:101–12. doi: 10.1053/j.seminoncol.2017.06.005
155. Quagliano A, Gopalakrishnapillai A, Barwe SP. Epigenetic drug combination overcomes osteoblast-induced chemoprotection in pediatric acute lymphoid leukemia. Leuk Res. (2017) 56:36–43. doi: 10.1016/j.leukres.2017.01.030
156. Quagliano A, Gopalakrisnapillai A, Kolb EA, Barwe S. Epigenetic drug combination overcomes bone marrow microenvironment-induced chemoprotection in pediatric acute lymphoblastic leukemia via modulation of CD81. Blood. (2018) 132:3957. doi: 10.1182/blood-2018-99-120275
157. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. (2019) 18:157. doi: 10.1186/s12943-019-1089-9
158. Liao WLS, Sun HS, Tsai SJ. Hypoxia-induced tumor malignancy and drug resistance: role of microRNAs. Biomark Genom Med. (2014) 6:1–11. doi: 10.1016/j.bgm.2014.01.003
159. Macedo-Silva C, Miranda-Goncalves V, Henrique R, Jeronimo C, Bravo I. The critical role of hypoxic microenvironment and epigenetic deregulation in esophageal cancer radioresistance. Genes. (2019) 10:927. doi: 10.3390/genes10110927
160. Miranda-Goncalves V, Lameirinhas A, Henrique R, Jeronimo C. Metabolism and epigenetic interplay in cancer: regulation and putative therapeutic targets. Front Genet. (2018) 9:427. doi: 10.3389/fgene.2018.00427
161. Marengo B, Garbarino O, Speciale A, Monteleone L, Traverso N, Domenicotti C. MYC expression and metabolic redox changes in cancer cells: a synergy able to induce chemoresistance. Oxid Med Cell Longev. (2019) 2019:7346492. doi: 10.1155/2019/7346492
162. Desbats MA, Giacomini I, Prayer-Galetti T, Montopoli M. Metabolic plasticity in chemotherapy resistance. Front Oncol. (2020) 10:281. doi: 10.3389/fonc.2020.00281
163. Feingold PL, Surman DR, Brown K, Xu Y, McDuffie LA, Shukla V, et al. Induction of thioredoxin-interacting protein by a histone deacetylase inhibitor, entinostat, is associated with DNA damage and apoptosis in esophageal adenocarcinoma. Mol Cancer Ther. (2018) 17:2013–23. doi: 10.1158/1535-7163.MCT-17-1240
164. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. (2018) 24:1859–66. doi: 10.1038/s41591-018-0233-1
165. Takezawa K, Okamoto I, Okamoto W, Takeda M, Sakai K, Tsukioka S, et al. Thymidylate synthase as a determinant of pemetrexed sensitivity in non-small cell lung cancer. Br J Cancer. (2011) 104:1594–601. doi: 10.1038/bjc.2011.129
166. Del Bufalo D, Desideri M, De Luca T, Di Martile M, Gabellini C, Monica V, et al. Histone deacetylase inhibition synergistically enhances pemetrexed cytotoxicity through induction of apoptosis and autophagy in non-small cell lung cancer. Mol Cancer. (2014) 13:230. doi: 10.1186/1476-4598-13-230
167. Ma X, Wang J, Liu J, Mo Q, Yan X, Ma D, et al. Targeting CD146 in combination with vorinostat for the treatment of ovarian cancer cells. Oncol Lett. (2017) 13:1681–7. doi: 10.3892/ol.2017.5630
168. Young CS, Clarke KM, Kettyle LM, Thompson A, Mills KI. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget. (2017) 8:51429–46. doi: 10.18632/oncotarget.18009
169. Leonard SM, Perry T, Woodman CB, Kearns P. Sequential treatment with cytarabine and decitabine has an increased anti-leukemia effect compared to cytarabine alone in xenograft models of childhood acute myeloid leukemia. PLoS ONE. (2014) 9:e87475. doi: 10.1371/journal.pone.0087475
170. Quagliano A, Gopalakrishnapillai A, Kolb EA, Barwe S. Modulation of CD81 by epigenetic drug combination sensitizes acute lymphoblastic leukemia via decreased BTK signaling. Blood. (2019) 134:2628. doi: 10.1182/blood-2019-129198
171. Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. (2019) 20:109–27. doi: 10.1038/s41576-018-0074-2
172. Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. (2007) 61:24–9R. doi: 10.1203/pdr.0b013e3180457684
173. Tomasetti M, Gaetani S, Monaco F, Neuzil J, Santarelli L. Epigenetic regulation of miRNA expression in malignant mesothelioma: miRNAs as biomarkers of early diagnosis and therapy. Front Oncol. (2019) 9:1293. doi: 10.3389/fonc.2019.01293
174. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
175. Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA. (2003) 100:13225–30. doi: 10.1073/pnas.1735528100
176. Martinez-Zamudio R, Ha HC. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol. (2012) 32:2490–502. doi: 10.1128/MCB.06667-11
177. Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. (2012) 7:1098–108. doi: 10.4161/epi.21975
Keywords: epigenetic aberrations, chemoresistance, mechanism, cancer, epigenetic drugs, epigenetic combination therapies
Citation: Quagliano A, Gopalakrishnapillai A and Barwe SP (2020) Understanding the Mechanisms by Which Epigenetic Modifiers Avert Therapy Resistance in Cancer. Front. Oncol. 10:992. doi: 10.3389/fonc.2020.00992
Received: 14 March 2020; Accepted: 19 May 2020;
Published: 24 June 2020.
Edited by:
Atsushi Fujimura, Okayama University, JapanReviewed by:
Hamid Morjani, Université de Reims Champagne-Ardenne, FranceAntonis Kirmizis, University of Cyprus, Cyprus
Copyright © 2020 Quagliano, Gopalakrishnapillai and Barwe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonali P. Barwe, sbarwe@nemours.org