Using Intermittent Fasting as a Non-pharmacological Strategy to Alleviate Obesity-Induced Hypothalamic Molecular Pathway Disruption
 Luciana da Costa Oliveira1
Luciana da Costa Oliveira1  Gustavo Paroschi Morais1 Eduardo R. Ropelle2
Gustavo Paroschi Morais1 Eduardo R. Ropelle2  Leandro P. de Moura2 Dennys E. Cintra2 José R. Pauli2
Leandro P. de Moura2 Dennys E. Cintra2 José R. Pauli2  Ellen C. de Freitas3
Ellen C. de Freitas3  Rodrigo Rorato4*†
Rodrigo Rorato4*†  Adelino Sanchez R. da Silva1,3*†
Adelino Sanchez R. da Silva1,3*†
- 1Postgraduate Program in Rehabilitation and Functional Performance, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 2Laboratory of Molecular Biology of Exercise, School of Applied Sciences, University of Campinas, São Paulo, Brazil
- 3School of Physical Education and Sport of Ribeirão Preto, University of São Paulo, São Paulo, Brazil
- 4Postgraduate Program in Molecular Biology, Laboratory of Stress Neuroendocrinology, Department of Biophysics, Paulista Medical School, Federal University of São Paulo, São Paulo, Brazil
Intermittent fasting (IF) is a popular intervention used to fight overweight/obesity. This condition is accompanied by hypothalamic inflammation, limiting the proper signaling of molecular pathways, with consequent dysregulation of food intake and energy homeostasis. This mini-review explored the therapeutic modulation potential of IF regarding the disruption of these molecular pathways. IF seems to modulate inflammatory pathways in the brain, which may also be correlated with the brain-microbiota axis, improving hypothalamic signaling of leptin and insulin, and inducing the autophagic pathway in hypothalamic neurons, contributing to weight loss in obesity. Evidence also suggests that when an IF protocol is performed without respecting the circadian cycle, it can lead to dysregulation in the expression of circadian cycle regulatory genes, with potential health damage. In conclusion, IF may have the potential to be an adjuvant treatment to improve the reestablishment of hypothalamic responses in obesity.
Introduction
The neuronal circuits controlling food intake and the endocrine mechanisms involved in this complex modulation network have been widely investigated to clarify the factors associated with the regulation of energy homeostasis. The hypothalamus is considered the central point of this regulatory system. Therefore, impairment of the hypothalamic response generated by signaling disruption in crucial signaling molecules has been associated with the development of morbid obesity, highlighting the importance of controlling the hypothalamic function for health (1–3).
Different central nervous system regions mediate the regulation of food intake, body weight, and energy homeostasis. In this context, the mid-basal portion of the hypothalamus, where the arcuate nucleus is located, is composed of different subpopulations, including the orexigenic neurons, which are directly involved in the hunger stimulus, and also anorectic neurons, which are mainly involved in response to satiety signals (4). The agouti-related peptide (AgRP) orexigenic neuron and the pro-opium melanocortin anorectic neuron (POMC) are two essential components of energy expenditure, hunger, and satiety control neurocircuits, integrating central and peripheral energy status with metabolic signals (5). It is essential to highlight that the hypothalamus contains other neuronal groups involved in controlling food intake and energy expenditure (6), which are not the focus of this review.
Agouti-related peptide orexigenic neurons co-express the messenger ribonucleic acid (RNA) for the neuropeptide Y (NPY) and the neurotransmitter gamma-aminobutyric acid (GABA). Studies reveal that the intracerebroventricular administration of AgRP (7) or its overexpression is associated with increased food intake (8). In contrast, POMC anorectic neurons are co-located with those expressing the cocaine-and amphetamine-regulated transcript (CART) (2, 9). After its synthesis, POMC is cleaved by different enzymes, generating several peptides responsible for the POMC functions (10). Neurons expressing endogenous melanocortin ligands for POMC and AgRP neuropeptides (antagonists) and neurons containing melanocortin receptors compose the central melanocortin system (8, 11). This system is strictly involved in the control of food intake, glucose metabolism, and energy homeostasis (12, 13), in conjunction with anorectic hormones, primarily leptin and insulin, composing a complex neuroendocrine system to maintain the correct energy and body weight balance, as recently described by Yang et al. (14).
Both POMC and AgRP neurons have the leptin receptor (LepR). When leptin binds to POMC neuronal cell receptors, neuronal depolarization and activation initiate multiple signal translations related to satiety responses. The leptin-mediated signaling is transduced into the nucleus, producing the anorexic POMC and CART neurotransmitters (15, 16). In addition, a cross-inhibitory reaction between AGRP and POMC neurons induces a reduction in orexigenic neurotransmitters in the AGRP neurons.
Insulin is also a crucial hormone for maintaining energy homeostasis by inhibiting pathways associated with NPY/AgRP neurons and their ramifications (16, 17). Therefore, impairments in the central signaling pathways of insulin (18–20) and leptin (21, 22) are associated with energy imbalance and obesity development. In this context, intermittent fasting (IF) is a protocol popularly used as a strategy to promote weight loss (23) and has become a tremendous scientific topic of interest to elucidate the mechanisms that regulate the hypothalamic molecular responses that will reduce body weight and prevent obesity (24, 25).
Previous investigations in human and animal models analyzed the effects of IF on leptin and insulin sensitivity (26, 27), inflammatory pathways (28, 29), the brain-microbiota axis (30, 31), circadian cycle (32, 33), and autophagic pathway (34). All these factors seem to be related to adaptations in POMC and AgRP neuropeptides (35) that can improve energy homeostasis through pathways that are not yet fully understood. The present review explored the molecular and physiological adaptations of leptin, insulin, POMC, and AgRP neuropeptides to IF protocols, mostly performed in animal obesity models.
Leptin and Insulin in Energy Homeostasis: Molecular Pathways Linked to Pro-opium Melanocortin Anorectic Neuron and Agouti-Related Peptide Responses
Insulin and leptin are the main anorectic hormones that act on the arcuate nucleus, activating POMC neurons and inhibiting AgRP neurons (36). Several studies indicate that the loss of hypothalamic insulin signaling (18–20) and leptin (21, 22) can induce changes in energy homeostasis, excessive food intake (hyperphagia), and body weight gain, leading to obesity development. The arcuate nucleus is densely rich in leptin receptors (37). The intracellular signaling cascade begins after leptin binds to its receptors in neuronal cells. An internal conformational alteration in the LepR attracts the next downstream protein, JAK2 (Janus kinase 2) (38). JAK is a cytoplasmic cytokine receptor that can autophosphorylate and promote the phosphorylation of its intracellular tyrosine residue Y-938, associated with the recruitment of the phosphatase SHP2 and its extracellular regulator ERK2, and of the residue Y-1077, which recruits the STAT5 transcriptional and signal transduction activator pathway. The primary effects of leptin on energy homeostasis involve the phosphorylation of the Y-1138 tyrosine residue, which creates a STAT3 binding and recruitment site (39). After its binding and subsequent activation, the STAT3 is transferred to the nucleus of the neuronal cell and promotes the transcription of genes, such as the neuropeptide POMC (40, 41).
Regarding insulin, despite having been discovered in 1921 (42), the complete elucidation of its molecular signaling is still in progress. However, it is known that the insulin receptor (IR) is a tetrameric enzyme that comprises two extracellular alpha subunits and two transmembrane beta subunits. Once the hormone interacts with its receptor, there is activation and consequent phosphorylation of the generated substrates (IR family), leading to activation of its main pathway, the phosphoinositide 3-kinase (Pi3K) pathway, a heterodimeric lipid kinase that binds to tyrosine residues via its SH2 domain, generating PI membrane phosphates with PkB/Akt recruitment (43).
Both insulin and leptin can stimulate the Pi3K pathway in the arcuate nucleus with subsequent phosphorylation of their target proteins, leading to hyperpolarization and activation of POMC neurons (44) and inhibition of AgRP (17). Mice with genetic Pi3K deletion in POMC cells did not show activation of POMC neurons in response to insulin or intracerebroventricular leptin administration (45). However, Pi3K deletion in AgRP neurons seems to induce energy expenditure reduction, insulin and leptin resistance, and weight gain (17).
The mechanistic target of rapamycin (mTOR) is one of the Pi3K target molecules through activation of Akt in the hypothalamus (Pi3K/Akt/mTOR pathway) (46). Both leptin and insulin activate hypothalamic mTOR (47, 48), a serine-threonine kinase with an essential role in brain development (49), which is found in approximately 90% of NPY/AgRP neurons and 45% of POMC/CART in the arcuate nucleus (50). mTOR is known for acting as a metabolic energy sensor and can integrate the variations in the nutrient serum levels with the endocrine responses (51). Thus, in food deprivation (fasting) and with drastic drops in serum glucose and insulin levels, there is a decrease in the phosphorylation of the mTOR active form. On the other hand, increased serum levels of leptin (51) and insulin (52) leads to increased mTOR protein content and expression and reduced food intake in the fed state.
The study of Kocalis et al. (53) observed that the deletion of Rictor-mTOR complex (mTORC2) activation, specifically in POMC neurons, can induce hyperphagia and increase adiposity. Interestingly, the specific deletion in AgRP neurons did not affect energy balance, although it led to mild glucose intolerance. It is known that the p70S6k-mTOR kinase further leads to phosphorylation of AMP-dependent protein kinase α2 (AMPK α2) on serine 491, inhibiting its action and thus limiting the effects of leptin on food intake (52). Thus, in parallel with mTOR activation by food intake, the anorectic hormones leptin and insulin reduce the AMP-dependent protein kinase (AMPK) activity, specifically the AMPKα2 subunit (54, 55). Like mTOR, AMPK is also known as a metabolic energy sensor, being considered an essential protein in the complex system of intracellular energy regulation, which is based on the adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio (56).
In conditions of depletion of energy reserves such as hypoglycemia and fasting, AMPK is activated in the hypothalamus (57, 58), leading to increased gene expression of NPY/AgRP in neurons and stimulating food intake (54). AMPK is inhibited in the hypothalamic arcuate nucleus in the fed state in response to increased leptin, insulin, and high levels of serum glucose (54), which consequently inhibits the autophagic pathway in NPY/AgRP neurons, leading to a reduction in food intake by inducing the feeling of satiety and, thus contributing to the eutrophic phenotype (44, 50, 59). Other molecules and pathways are also stimulated by anorectic hormones and contribute to energy homeostasis. Further details about the molecules involved in the signaling pathway of insulin and leptin actions in POMC and AgRP neurons in eutrophic conditions were described in the review article by Varela and Horvath (16).
Regarding the PI3K-mTOR-AMPK pathway, anorexigenic hormones increase the activity of the Pi3K and mTOR pathways, leading to the activation of POMC neurons (44, 45). Additionally, leptin and insulin reduce AMPKα2 activity in the hypothalamic region (15, 54). The p70S6k-mTOR kinase can inhibit the AMPK pathway in the hypothalamus, acting as a counter-regulatory protein (52). The mechanisms by which these molecules modulate the expression of hypothalamic neuropeptides are not fully understood; however, evidence suggests that the autophagic pathway plays a crucial role in this regulation (59–61). Furthermore, Claret et al. (55) showed that mice with genetic deletion of AMPKα2 in AgRP neurons were grown with the eutrophic phenotype. Interestingly, the specific genetic deletion in POMC neurons led to increased body fat and reduced caloric expenditure despite remaining sensitive to leptin. These results suggest that AMPK also plays a regulatory role in POMC neurons by unknown mechanisms. Therefore, evidence suggests that the hypothalamic autophagic pathway is crucial for activating orexigenic and anorectic neurons (59, 61, 62). Figure 1 summarizes the data described so far.
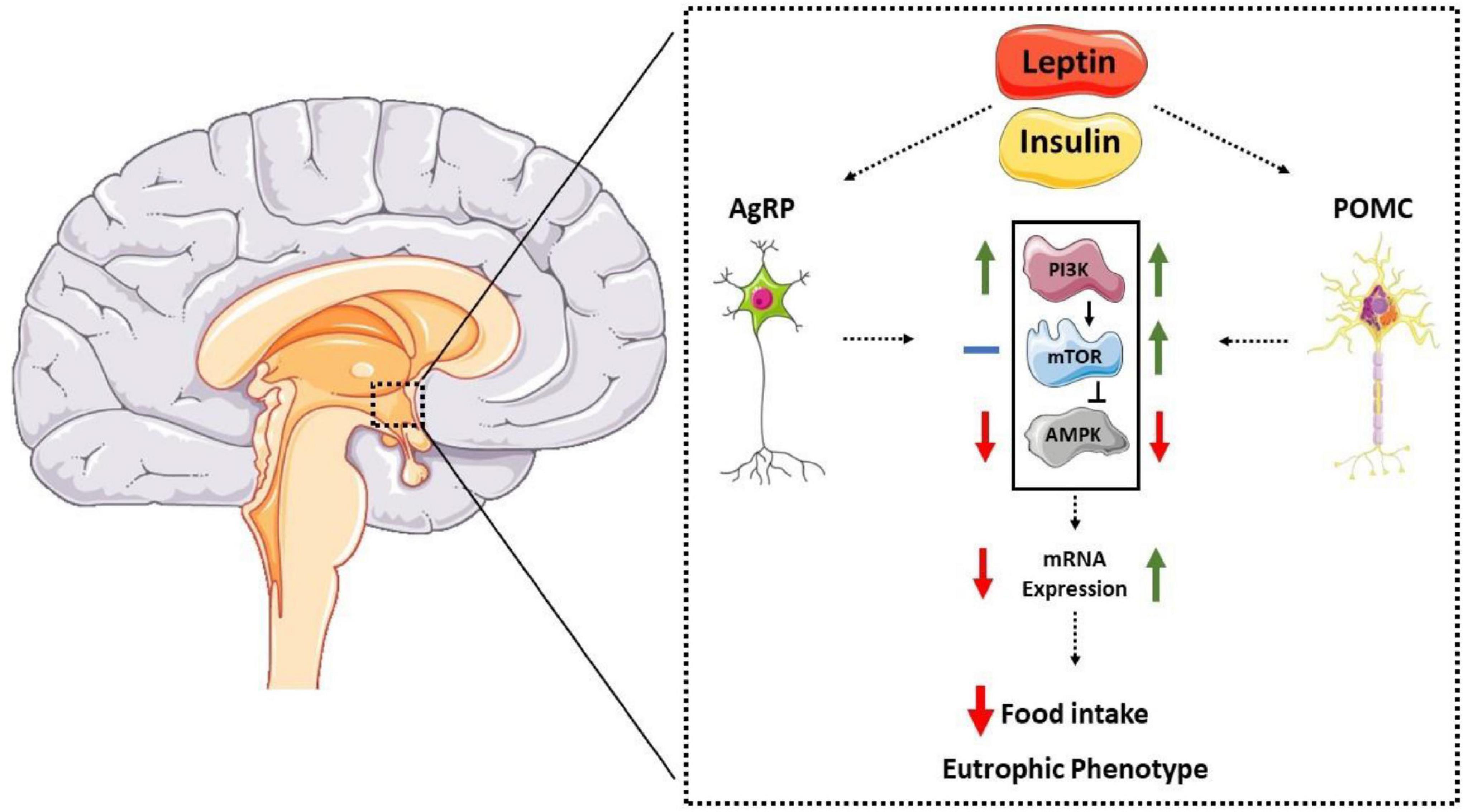
Figure 1. Representative diagram of the interaction between anorectic hormones, molecular pathways, and neuropeptides POMC and AgRP. Anorexigenic hormones (leptin and insulin) act on the hypothalamic arcuate nucleus, reducing AMPK activation in AgRP neurons, thus reducing its expression. In POMC neurons, hormones increase mTOR activity and reduce AMPK, increasing its expression and reducing food intake and eutrophic phenotype.
Autophagic Pathway in Pro-opium Melanocortin Anorectic Neuron and Agouti-Related Peptide Neurons: A Pivotal Point in Energy Homeostasis
It is well known that the neuronal autophagic pathway is crucial for maintaining cellular homeostasis both under basal conditions and in response to stress signals (63, 64). Several studies indicate that the imbalance between activation and inhibition of the autophagic pathway in the central nervous system (CNS) is associated with dysregulation of body energy homeostasis and obesity induction and a greater predisposition to the development of various neurodegenerative diseases (61, 65, 66).
Classically, autophagy can be divided into microautophagy, chaperone-mediated autophagy, and macroautophagy, the latter being the most prevalent and commonly referred to as autophagy (67, 68). The autophagic process begins with capturing cytoplasmic organelles or macromolecules surrounded by a vesicular membrane lining called the autophagosome, which fuses with the lysosome to form autophagolysosome (or autolysosome), and lysosomal enzymes then degrade the sequestered material (69). The autophagosome formation begins with a pre-phagophore structure, which elongates and expands to form the phagophore, which, in turn, will mature in the membrane vesicle, surrounding the substrate that will be degraded (69).
The regulation of this entire autophagic process occurs by activating autophagic molecular complexes, starting with activation of the ULK1 (Unc-51 like autophagy activating kinase 1) complex, followed by the activation of phosphatidylinositol 3-kinase (PI3K), which forms a complex with Beclin 1 after dissociating from lymphoma B cell 2 (BCL-2) (70). Thus, the formed complex activates several proteins of the autophagic family (ATGs) that participate in phagophore elongation and activate the LC3-I protein (light chain 3 of protein 1 associated with microtubules), forming LC3-II. LC3-II is responsible for closing the phagophore and interacting with the p62 protein, which targets the material that the autolysosome will degrade (71, 72). Figure 2 shows the schematic model of the autophagic pathway.
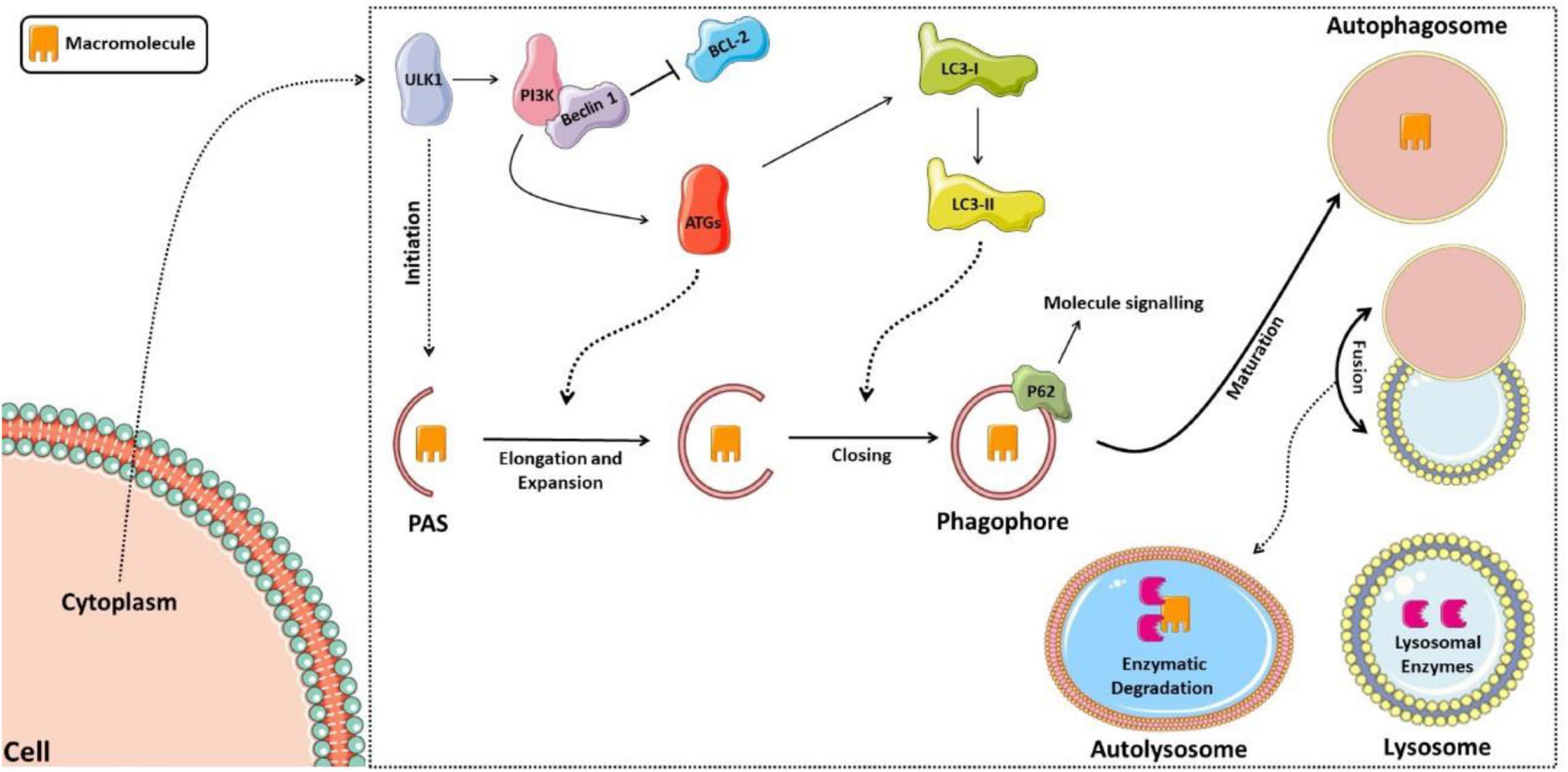
Figure 2. Schematic model of the autophagic pathway. The process starts with activation of the ULK1 complex, then activation of phosphatidylinositol 3-kinase (PI3K), which forms a complex with Beclin 1 after it dissociates from lymphoma B cell 2 (BCL-2). Thus, the complex formed activates several proteins of the autophagic family (ATGs), which participate in the elongation of the phagophore and activation of the LC3-I protein (light chain 3 of protein 1 associated with microtubules), forming LC3-II, responsible for closing the phagophore and interacting with the p62 protein, signaling the material to be degraded. The phagophore matures into the autophagosome, which fuses with the lysosome forming the autolysosome, in which lysosomal enzymes will then degrade the sequestered material.
Evidence suggests that the hypothalamic autophagic pathway is crucial in activating orexigenic and anorectic neurons (59, 61, 62). AMPK is an activating molecule of the autophagic pathway, while mTOR leads to inhibition (73). AMPK can activate the autophagic pathway in vivo and in vitro by phosphorylating raptor-mTOR (mTORC1) (74) and starting the autophagic pathway initiator complex ULK-1 in AgRP neurons (59). Therefore, AMPK and mTOR directly interact to regulate the autophagic pathway through its complex initiator, ULK-1 (73).
Specifically, in AgRP neurons, the deletion of Rictor (rapamycin-insensitive companion of TOR), a key molecule in the regulation of the MTORC2 complex, did not change the energy balance (53). However, the deletion of AMPK led to the eutrophic phenotype (55), suggesting that AMPK plays a more expressive role than mTOR in the energy regulation pathways of this neuronal subgroup. An elegant study published by Kaushik et al. (61) showed that inhibiting the autophagic pathway, specifically in AgRP neurons, in both cells and mice, through the Agt7 gene deletion, significantly reduces food intake and adiposity. In POMC neurons, studies with knockout mice showed that the activity of mTORC1 and mTORC2 complexes are vital factors for the anorectic effects induced by leptin on the neuron and maintenance of the eutrophic phenotype (53, 75).
While the reduction in AMPK activity in AgRP (59) neurons and the increase in mTOR activity in POMC neurons (53, 75) lead to the eutrophic phenotype and considering that AMPK inactivation and mTOR elevation lead to inhibition of the autophagic pathway (73, 76), hypothetically, inhibition of the autophagic pathway in neurons may be associated with the eutrophic phenotype. However, a study showed that the selective deletion of autophagy-related protein 7 (Atg7) in mouse POMC neurons, interestingly, leads to a reduction in melanocyte-stimulating hormone (MSH) and is also associated with increased adiposity and food intake through mechanisms involving resistance to lipolysis (60). In addition, Meng and Cai (77) observed that the suppression of Atg7 in the mediobasal hypothalamus using site-specific lentiviral delivery of shRNA, without distinction of neuronal subgroups, was accompanied by an increase in hypothalamic inflammation, with activation of IKKB and, consequently, increased food intake and reduced energy expenditure (77).
Supporting these data, the AMPK deletion specifically in POMC neurons (55) and the RICTOR/mTORC2 deletion in the arcuate nucleus (53) are also associated with reduced caloric expenditure and obesity. Together, these data reveal that AMPK and mTOR are correlated with the autophagy pathway, which orchestrates a series of coordinated molecular phosphorylations in neuronal subgroups to provide adequate control of hypothalamic inflammation and energy homeostasis, reinforcing that the hypothalamic molecular pathway of obese individuals needs to be further investigated. Table 1 presents the metabolic phenotypes found according to the deletion or inhibition of molecular pathways in the neuronal subgroups.
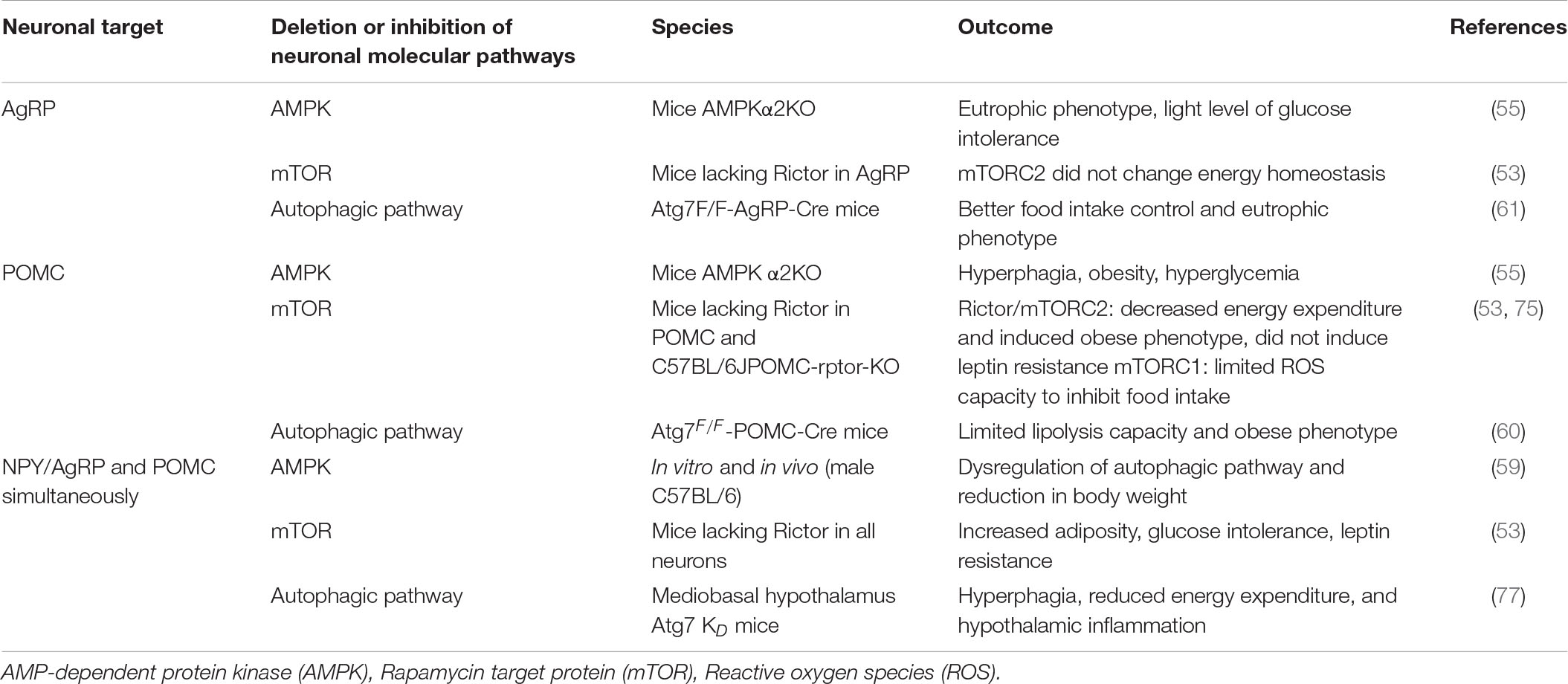
Table 1. Molecular pathway deletion or inhibition in the neuronal subgroups and the outcomes.
What Is Intermittent Fasting?
Intermittent fasting (IF) and caloric restriction are two distinct forms of dietary restriction associated with improving several metabolic parameters, including body weight control (78). Previous studies have shown that the obligation to maintain a daily calorie restriction reduces adherence to the caloric restriction protocols (79). Thus, the presence of ad libitum feeding windows in IF protocols emerged as an alternative protocol for dietary restriction interventions. The stress promoted by the low caloric intake is replaced by the metabolic stress induced by intermittent windows of prolonged fasting or alternate days of deficient caloric intake (78, 80). However, it is essential to highlight that both interventions must be carried out with professional supervision. Overfeeding episodes can occur after the fasting window, with the risk of developing eating disorders such as binge eating (81).
Furthermore, caloric restriction programs can also increase the predisposition to the development of psychological disorders (79). The review of Cerqueira et al. (82) pointed out that in animals fed with standardized diets balanced in macro and micronutrients, calorie restriction protocols with daily consumption of 40–60% of energy requirements are associated with micronutrient deficiencies. Deficiency of vitamin B12 and vitamin K, among others, depending on the diet consumed, is observed when the restriction protocols are chronically applied without supplementation with vitamins and minerals (79).
Despite its high popularity, there is no standardization of IF protocols (80). It is established that protocols do not impose water restrictions. All include periods of food restriction, which may refer to total deprivation from food consumption during some hours of the day (fasting window) or a full day containing no-energy food. Recently, some papers (80, 83) have been considering IF in three specific categories: (a) Complete Alternate Day Fasting (ADF) – consists of days with ad libitum feeding intercalated by whole days of food restriction, (b) Modified alternate-day fasting (MADF) or alternate-day modified fasting (ADMF) – with two non-consecutive days of total food restriction within the week, or two days of food intake of about 20% of the total caloric necessity with meals distributed throughout the day (c) Time-restricted feeding (TRF) – consisting of a protocol with a fasting window (usually 16 h) followed by a food intake window of approximately 8 h, with the meals distributed within this period, according to individual needs. The main point of the protocols is not to change the average weekly caloric intake but to change the frequency of food consumption (78, 80).
In this sense, the application of IF protocols generally does not change the average calorie intake due to post-fasting compensatory overfeeding. Thus, only a slight reduction in the average percentage of daily intake (84, 85) contributes to the protocol being considered an alternative strategy to improve weight loss and induce positive metabolic adaptations generated by energy stress (84, 85). However, it is essential to mention the warning that IF protocols are not recommended in cases of malnutrition, pregnancy, gastric ulcers, elite athletes, and patients at risk of hypoglycemia, among others (86–88).
Unlike the globally disseminated IF protocols for weight loss and improvement in health-related aspects (23, 24, 89), Ramadan fasting is a protocol with spiritual purpose practiced by Muslim followers of Islam (90). Once a year, according to the Islamic calendar, Muslims abstain from any food or drink, including water, during the period of daylight, having all their meals in the evening or just before sunrise (91). This practice extends for about 30 consecutive days once a year during the Islamic lunar month, which can occur in different seasons depending on the year (92) and the latitude of the geographic region. The fasting window can vary from 11:00 am to 6:00 pm (93). During this practice, most Muslims eat about two bulky meals within 24 h, one just after sunset and the other just before sunrise (93), resulting in a slight but significant reduction in the total calorie intake (94, 95) regarding loss or maintenance of body weight (96). Table 2 illustrates the main differences between intermittent fasting, caloric restriction, and Ramadan fasting.
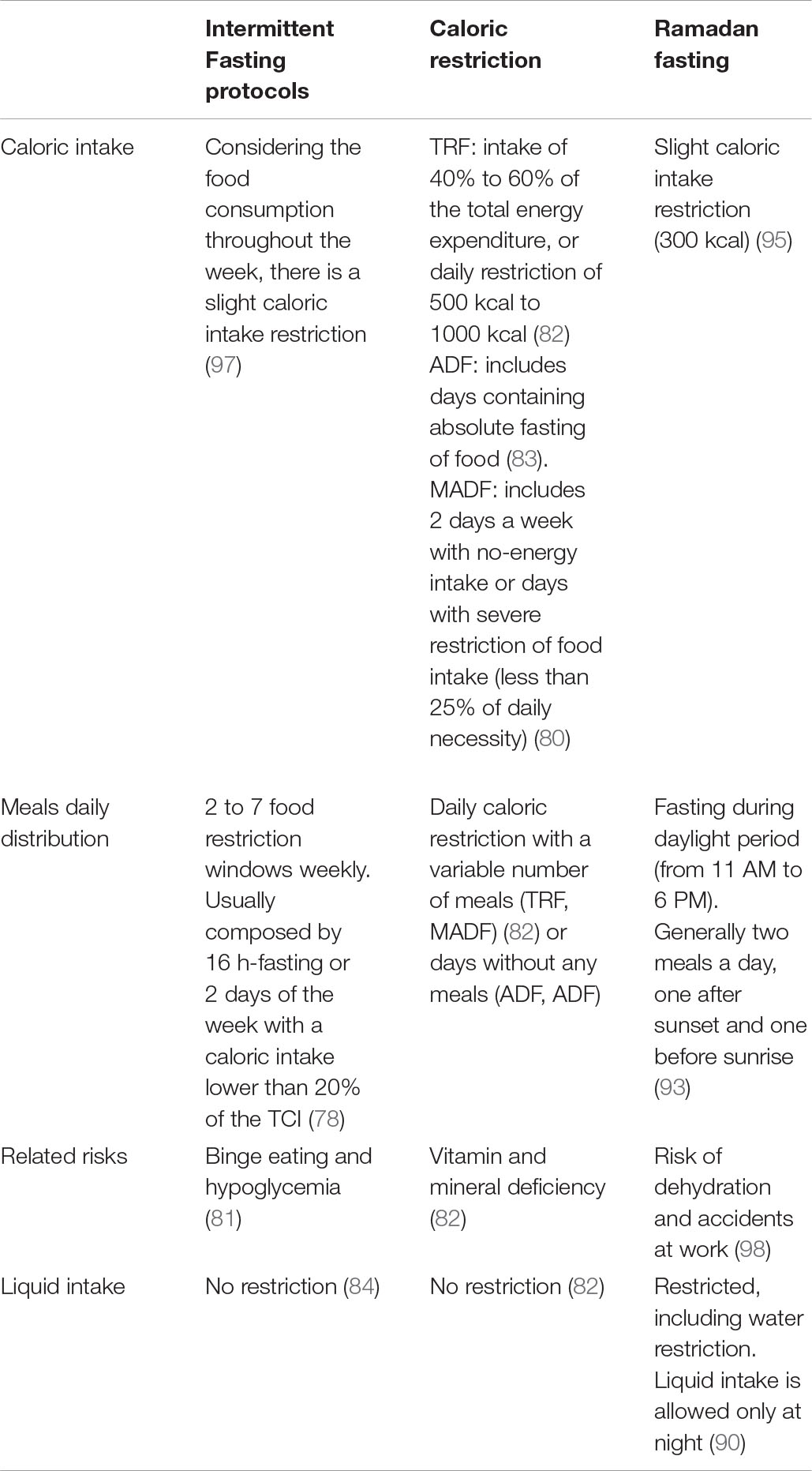
Table 2. Main differences between intermittent fasting, caloric restriction, and Ramadan fasting.
Intermittent Fasting as a Possible Adjuvant in the Treatment of Obesity: Modulations in Neuroinflammatory, and Leptin and Insulin Pathways
Obesity is a multifactorial disease usually associated with hyperphagia, hyperinsulinemia, and hyperleptinemia. The high levels of leptin and insulin in the cerebrospinal fluid of obese individuals indicate a chronic state of resistance to the actions of these hormones in the CNS (19, 99). It is essential to highlight the diet quality profile as a significant possible factor in the pathophysiology of obesity. Increased exposure to a high-fat diet (HFD) is associated with a reduction in hypothalamic mTORC1 and leptin resistance (100). There is evidence that an acute lipid infusion for 24 h or exposure to a HFD over 8 – 20 weeks induces markers of inflammation in the hypothalamic NPY/AgRP neurons, which may contribute to a significant alteration in NPY/AgRP expression or content (101) and also, 6 days of exposure to a high-fat diet can induce leptin resistance in mice with a predisposition to obesity (102).
Several studies indicate that the practice of IF for periods longer than 1 month can improve insulin resistance and reduce its serum levels, contributing to the regulation of glucose metabolism (26, 103–105). A recent meta-analysis evaluated 545 participants, most overweight or obese, and observed that IF protocols are associated with reducing the body mass index (BMI) and leptin serum levels, lowering fasting blood glucose, and improving insulin resistance. These results suggest that IF may contribute to prevention/improvement in the resistance of the anorectic hormone observed in obese individuals (106).
Although not fully elucidated, the mechanisms by which IF acts in the insulin signaling pathway are probably different from those observed in caloric restriction protocols since benefits associated with IF can be observed even when there is no reduction in calorie intake and weight loss (84, 103, 107, 108). In addition, there is some evidence that IF protocols may produce more significant beneficial effects on glucose regulation and fasting insulin (103, 108).
It is known that obesity is associated with the chronic low-grade inflammatory process, not only peripheral but also central, highlighted by increased expression of several inflammatory proteins related to impairments in the hypothalamic signaling of leptin and insulin, such as the suppressor of insulin signaling cytokine 3 (SOCS3) (103, 109, 110). Despite SOCS3 being part of a negative feedback system related to this signaling cascade, when it reaches a high concentration induced in an inflammatory scenario, SOCS3 significantly impairs the anorexic leptin cascade. This cytokine can bind to an intracellular region of LepR, attenuating the ability of JAK2 to autophosphorylate and recruit the STAT3 pathway (111). In addition, the C-terminal portion of SOCS can recruit the ubiquitin transferase system, promoting the degradation of JAK receptor complexes (112). Thus, SOCS3 impairs the reduction in the activity of the AMPK protein threonine 172 by leptin (113), stimulating autophagic activity in AgRP neurons and appetite (59). It is also known that SOCS3 can impair the insulin signaling pathway by binding directly to the insulin receptor (114) and/or degrading both substrates of insulin receptors 1 and 2 (IRS1/2) (115). The study of Mori et al. (109) observed that hypothalamic suppression of SOCS3 could prevent central insulin resistance generated by the chronic high-fat diet.
In this sense, although several studies show that IF protocols can reduce plasma levels of pro-inflammatory proteins in obese or overweight individuals (27, 116), few studies have assessed the adaptation of inflammatory proteins in the hypothalamic region. Spezani et al. (27) evaluated the effects of a 24-h fasting protocol interspersed with days of ad libitum high-fructose diet in mice with induced obesity (eight-week protocol with a high-fructose diet). After 4 weeks of intervention, a reduction in the expression of hypothalamic SOCS3 was observed. However, animals fed a standard diet and submitted to an IF protocol showed an increase in SOCS3 compared to control animals with a standard diet without applying the IF protocol (27). Controversially, the study of Zangh et al. (117) did not observe changes in the expression of hypothalamic SOCS3 or alteration in plasma insulin in female mice fed with a standard diet and submitted to chronic IF protocols for 24 h performed only one to two times a week, during a period of 13 or 42 days.
It is also known that the increase in tumor necrosis factor-alpha (TNFα) attenuates the anorectic effect of leptin and increases the expression of SOCS3 in the hypothalamus (118). Despite studies showing that caloric restriction (116) and Ramadan fasting (119) can reduce plasma TNFα levels, particularly in obese or overweight individuals, a recently published meta-analysis (28) evaluated serum levels of inflammatory markers in response to different IF or caloric restriction protocols. After applying the exclusion criteria, the meta-analysis included only one study with obese individuals and IF (alternating every 24-h between consuming 25% or 125% of energy needs), which did not reduce TNFα levels (120). However, it is crucial to consider the lack of papers published in this area.
Regarding animal studies, Spezani et al. (27) evaluated the effects of IF for 24 h. The authors observed that obese mice submitted to fasting curiously showed a greater expression of hypothalamic TNFα when compared to the control group. Therefore, the data are still contradictory, and further studies are needed to assess the content and expression of TNFα, specifically in the hypothalamus of obese animals submitted to different IF protocols.
Another relevant inflammatory pathway involved in the etiology of obesity is the IKKb/NF-kb pathway (110). Zhang et al. (110) showed that mice submitted to a high-fat diet developed obesity accompanied by increased concentrations of IKKB, which can activate the nuclear factor kb (Nf-kb), leading to endoplasmic reticulum stress in the hypothalamus and consequent resistance to leptin and insulin (110). IKKB phosphorylation in the mid-basal portion of the hypothalamus can impair the action of insulin by inducing tyrosine phosphorylation and the consequent inactivation of the insulin receptor (IR). In addition, it can limit the activity of its target proteins: phosphatidylinositol-3-kinase (Pi3K) and protein kinase B (Akt) (110, 121), which are involved in the control of the hypothalamic autophagic pathway (70). In a complementary way, the increase in IKKB contributes to the elevation of the expression of hypothalamic SOCS3 (110), impairing the central signaling of leptin and insulin (41, 122). However, although IKKB is a protein widely studied in obesity models, no investigations have evaluated IKKB in the hypothalamic region in response to IF protocols.
The IKKb/NF-kb inflammatory cascade can also be activated by lipopolysaccharides (LPS) when bound to their Toll 4 membrane receptor (TLR-4) (123, 124). Previous studies have shown that obese individuals present increased levels of LPS in the bloodstream, causing a condition called metabolic endotoxemia, which is associated with systemic inflammation and an increased risk of developing chronic diseases (125), favoring the development/worsening of obesity (125, 126). Additionally, prolonged treatment with LPS seems to increase JNK and limit the hypophagic effects in response to central insulin administration, regardless of the increase in body weight (127). Although not fully understood, the increase in LPS plasmatic levels is probably due to intestinal dysbiosis and changes in the permeability of the intestinal wall (128, 129).
Dietary factors seem to modulate endotoxemia, and the use of prebiotics could contribute to attenuating its progression (31), while chronic exposure to a high-fat diet intake could worsen progression (125). In this sense, a recent review article proposed that IF protocols can also be used as a nutritional strategy, affecting the brain-microbiota axis of obese individuals (31). An elegant study demonstrated that the removal of the intestinal microbiota with the use of antibiotics reduced the protective effects of IF on the cognitive function of the evaluated mice, with the subsequent administration of microbiota metabolites, such as short-chain fatty acids and 3-acid propionic indole, which were able to improve cognitive function and insulin sensitivity (30). Together, we hypothesized that IF might play a supporting role in attenuating inflammation in the CNS through actions on the microbiota-brain axis. However, this hypothesis needs to be evaluated.
Additionally, it is known that during acute fasting periods, there is an increase in β-hydroxybutyrate (βHB) production (130), leading to increased phosphorylation of IRS1 and Akt in their active forms, a reduction in serum insulin levels, and a better response to the intraperitoneal insulin tolerance test (131), as well as being able to modify hypothalamic leptin and insulin signaling pathways in type 2 diabetic rats (132). It is essential to highlight that βHB is also involved in inflammatory control (133). The oral administration in Crohn’s disease patients exerts an anti-inflammatory response through downregulation of NF-kb (134). Cerniuc et al. (135), evaluating an IF protocol (2 non-consecutive days of total fasting per week) in healthy women, also identified a significant increase in blood βHB levels. To date, despite not directly evaluating insulin response associated with βHB production and hypothalamic responses in IF protocols, data suggest that the increase in butyrate levels may also contribute to improving insulin sensitivity in response to IF.
Although the relationship of neuroinflammation with insulin and leptin signaling in response to IF protocols need to be further explored, data suggest that: (1) the mechanisms of action of IF seem to be different from those observed in calorie restriction protocols (103, 108); (2) IF appears to be able to improve insulin and leptin sensitivity (105, 106); (3) IF seems to be able to modulate inflammatory pathways in the brain (27, 29) and attenuate the levels of LPSs in the plasma (29), which we hypothesize could be associated with an improvement in hormonal and neuronal sensitivity; and (4) the increase in βHB production in IF (135) may also contribute to better insulin sensitivity considering the relationship of βHB and the insulin pathway (131, 132).
The Influence of the Circadian Cycle on the Modulation of Leptin and Insulin Pathways in Different Intermittent Fasting Protocols
Although several studies show that IF protocols are capable of improving insulin and leptin sensitivity, it is essential to emphasize that the time when the fasting window and the eating window are performed significantly interferes with metabolic responses and autophagic stimulation due to their influence on the hormonal rhythm guided by the circadian cycle (33, 136). However, the habit of skipping breakfast is associated with greater consumption of food at night (breakfast skipping and late-night eating pattern), increasing the risk of developing insulin resistance and cardiometabolic risk (33, 137, 138).
Jamshed and coworkers (33) submitted overweight or obese individuals to IF protocols. The authors evaluated the differences between the protocol carried out with the food window from 8 AM to 2 PM (Early Time-Restricted Food – eTRE) with a second protocol containing the last meal at 8 PM, both on a controlled diet. After four consecutive days of intervention, the authors observed increased BMAL1 expression in the morning, activation of Akt2, reduced fasting plasma insulin, and glucose concentrations in the eTRE group compared to the group that had the last meal at 8 PM (33). These results corroborate another study carried out with humans by the same research group that observed that the eTRE group improved insulin sensitivity, assessed by the glucose tolerance test, compared to the group fed at night (107).
The IF protocol performed without respecting the circadian cycle can induce a dysregulation in the expression of the circadian cycle leading to a significant increase in the natural peak of mRNA expression of genes involved in glucose regulation (i.e., Gck, Slc2a2, and Pdk4) and also lead to a higher plasma leptin levels when compared to an IF protocol applied to respect the circadian cycle (with a distributed feeding window in the active period of mice) (32). On the order hand, an IF protocol respecting the circadian cycle seems to reverse the obese and hyperphagic phenotype of heterozygous knockout mice of brain-derived neurotrophic factor (BDNF) and re-established insulin sensitivity and brain BDNF levels after 3 weeks of intervention (139). Thus, due to its influence on the circadian cycle, the effects of IF on endocrine responses and body weight may vary according to the time of day in which each food and fasting window is held. It seems better for healthy improvements not to skip breakfast and start the fasting window close to sunset to improve sensitivity to anorectic hormones and help prevent obesity (137, 140, 141).
Therefore, it is essential to point out that these data warn us regarding popular IF models disseminated in social media that encourage avoiding breakfast and starting the eating window at lunch. It is essential to reinforce the importance of a scientific basis to achieve better dietary prescriptions at the individual and population levels. This topic was deeply explored in the recent review published by Moon et al. (142).
Intermittent Fasting and the Autophagic Pathway
Autophagy is an essential mediator of physiological responses associated with the generation of ROS and cellular protein damage (143), being directly involved in maintaining energy homeostasis through the increased expression of neuropeptides (144) and in the control of the neuronal inflammatory response (77). Energy stress and IF-induced oxidative stress can activate the autophagic pathway (145) through the increase in sirtuins (SIRTs) (146, 147), associated with increased phosphorylation of AMPK in threonine (148, 149), correlated with phosphorylation of the ULK1 protein and autophagic pathway (150). Therefore, the application of IF protocols is a non-pharmacological alternative capable of activating the autophagic pathway (33, 34).
During prolonged fasting, lower glycemic values and changes in the adenosine monophosphate/adenosine triphosphate (AMP/ATP) ratio induce SIRTs activation in tissues such as the kidney, skeletal muscle, and blood samples from overweight individuals (145, 147, 151). Sirtuins are known to induce the autophagic pathway through phosphorylation of AMPK, FOXO1, or deacetylation of autophagic family proteins (145, 149). A study evaluating the IF and autophagic pathway in adults observed an increase in serum levels of SIRT1 and the autophagosomal membrane component LC3A, thus suggesting autophagic stimulation, accompanied by improved insulin sensitivity (33).
AMP-dependent protein kinase is an essential protein associated with the neuronal autophagic pathway and energy homeostasis (59). An interesting study by Kaushik et al. (61) observed that acute fasting could increase the content of free fatty acids in the hypothalamus, with consequent phosphorylation and activation of AMPK and ULK1, increasing autophagic flow in AgRP neurons with hunger induction. Furthermore, the authors observed that impairment of the autophagic pathway in cultures of hypothalamic cells through the deletion of the protein related to autophagy 7 led to a reduction in AgRP levels, food intake, and adiposity (61). The increased availability of fatty acids can induce the hypothalamic autophagic pathway and increase NPY expression (144).
Additionally, the acute fasting protocol can also lead to phosphorylation of mTOR and its target protein, ribosomal protein kinase S6 (S6K) at serines 240 and 244 in the hypothalamus, thus inactivating the mTOR/S6k pathway (50). During fasting, the increase in AMPK associated with the reduction in mTOR contributes to the regulation of food intake (50, 52) and activation of ULK1 and the autophagic pathway (150). These data corroborate the work of Chaix et al. (152), who evaluated IF protocols in obese mice, and observed an increase in the levels of the homolog of ATG8, Gabarap1, a key regulator of autophagic flow during fasting. The animals showed significant weight loss compared to the control group, although food intake did not show any significant difference.
Although several studies have shown the effects of caloric restriction programs or fasting periods on the autophagic pathway, it is essential to emphasize that, to our knowledge, few studies have assessed the impact of IF protocols on the autophagic pathway. To date, we understand that fasting periods in general lead to increased availability of free fatty acids in the hypothalamic region, reduced levels of glucose and serum amino acids, leading to activation of sirtuins (33) and the AMPK pathway (148), and inactivation of the mTOR pathway, thus stimulating the autophagic complex (50, 73). There are no studies evaluating the autophagic pathway in the hypothalamic nucleus in response to chronic IF protocols.
Intermittent Fasting, Pro-opium Melanocortin Anorectic Neuron, and Agouti-Related Peptide Neuropeptides
The mechanisms by which IF alters the expression of hypothalamic neuropeptides are not fully understood; however, it is known that IF can improve sensitivity to leptin and insulin (106) and stimulate the autophagic pathway (33, 145), which is related to the activation of hypothalamic neuropeptides and energy homeostasis (27, 55, 59, 60). Another relevant factor is the increase in reactive oxygen species induced by the IF protocol (146). ROS in the hypothalamus is also a factor that leads to the electrical activation of neuropeptides POMC and inactivation of Npy/AgRP (153).
The suppression of reactive oxygen species decreases the activation of POMC cells and increases the activity of NPY/AgRP neuropeptides (153). During fasting, the mechanisms of oxidative protection performed in the mitochondria protect the exacerbated increase in ROS in AgRP neurons. Uncoupling protein 2 (UCP2) is a protein abundantly expressed in arcuate nucleus neurons associated with energy homeostasis and involved in controlling oxidative stress in mitochondria (154, 155). A study shows that the function of UCP2 via AMPK seems to be a key point for the electrical activation of NPY/AgRP neurons during a fasting period (156). In contrast, ROS levels are low during fasting in POMC neurons, and the transient increase in ROS favors satiety and the action of leptin via mTOR (75, 157). However, these data were evaluated using acute fasting protocols. There is still no evidence about the content of ROS in neuronal groups and its impact on energy homeostasis in response to IF protocols.
Low serum leptin values during fasting contribute to an increase in the expression of AgRP and NPY (156, 158, 159), stimulating the autophagic pathway and consequently inhibiting activation of the hypothalamic-pituitary-thyroid axis to reduce caloric expenditure and save energy (160). However, contrary to the results observed after a single fasting period window, the study of Chausse et al. (161) submitted eutrophic rats to a 24-h IF protocol and observed that rats that underwent IF for 3 weeks, even with an excellent response to leptin, curiously showed increased expression of the orexigenic neuropeptide AgRP both during fasting periods and on feeding days. Increased energy expenditure, reduced energy efficiency factor, and lower weight gain were also observed when compared to the control group. These findings suggest that AgRP neuron responses to the IF protocol may differ from those observed after a single fasting window, and further studies are needed.
It seems that during the IF protocol, the response of neuropeptides can change. A study with eutrophic mice evaluating the response to IF protocols after 13 days and again after 42 days found that the first intervention increased food intake in the feeding windows accompanied by an increase in NPY mRNA expression. However, interestingly, after 42 days, there was a reduction in NPY mRNA expression that returned to baseline values. There was no difference compared to the control group (without IF intervention), thus suggesting a new late hypothalamic adaptation of NPY in response to the chronic application of the IF protocol (117), which may be related to the popularly described hunger adaptation. Regarding POMC neuropeptides, there were no significant changes.
Additionally, the time that the fasting window is performed can also influence the expression of neuropeptides. Animals exposed to a high-fat diet for 2 weeks and then submitted to the IF protocol of 16 h for 1 week, with the feeding period performed during the rest period, disregarding the circadian cycle, showed increased hypothalamic expression of the orexigenic genes NPY and AgRP. Higher food intake and higher serum levels of leptin were also observed, thus suggesting possible resistance to leptin compared to animals that followed the IF protocol with the feeding window performed in the active period (referring to the night period for mice). Therefore, it can be concluded that when the IF is carried out with the food period during the rest period, disregarding the physiological circadian cycle, there is a possibility that the protocol can trigger dysregulation of the neuroendocrine mechanisms of hunger control, which may harm health (32).
Few studies have assessed the effects of IF and neuropeptide expression in obese individuals. Gotthardt et al. (35) studied obese mice submitted to an IF protocol where the mice were food deprived every other 24-h period beginning at 9:00 AM (fasting day), 2 h into the light cycle, for 4 weeks. The results showed increased expression of mRNA of hypothalamic NPY and increased energy expenditure compared to the control group that consumed a high-fat diet ad libitum. Regarding the expression of POMC neurons, the group that performed the IF showed a significant reduction in the expression of the POMC neuropeptide when compared to the control group with an ad libitum high-fat diet, which was also accompanied by a decrease in serum levels of leptin, improvement in insulin sensitivity, and weight loss.
Interestingly, a study evaluating the effects of IF in animals fed with a standard diet observed that after 4 weeks of application of the IF protocol (24 h fed, 24 h fasting), the group with standard diet and fasting presented reduced expression of POMC when compared to its respective control (standard diet without fasting) (27). However, the authors also looked at the effects of IF on two other types of diet: obese animals fed a high-fat diet and obese animals fed a high fructose diet. After 4 weeks of applying the IF protocol (24 h fed, 24 h fasting), the fasted obese animals showed increased expression of POMC in both protocols (27).
Therefore, we suggest that the content of neuropeptides in IF seems to occur differently from that observed in caloric restriction protocols, and the adaptations of the CNS seem to differ according to (1) the duration of time that the IF protocol is being applied, with the orexigenic neuropeptide NPY being able to return to baseline values as a late adaptation (117); (2) it is essential that the distribution of feeding and fasting periods respects the circadian cycle to avoid possible health risks (32); (3) concerning POMC neuropeptides, the IF protocol interestingly seems to reduce the expression of POMC neurons in some models (27, 35), but the results are still contradictory. Therefore, further studies are necessary to elucidate the effects of IF on hypothalamic responses and energy homeostasis. In addition, it is necessary to investigate long-term changes. Figure 3 summarizes the possible effects of IF as an adjuvant treatment to partially rescue hypothalamic responses in obesity.
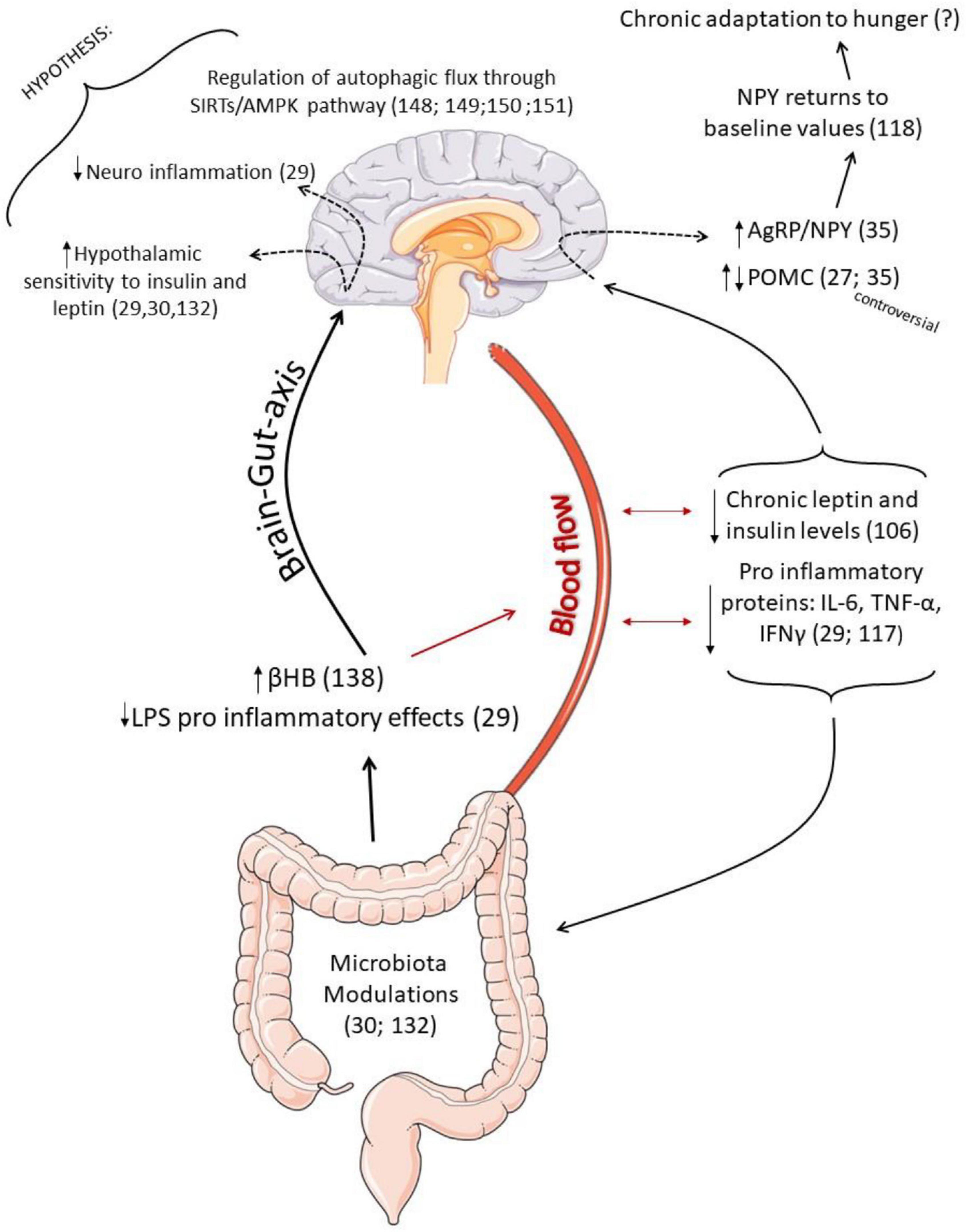
Figure 3. Schematic representation of the possible effects of intermittent fasting (IF) as an adjuvant treatment to partially rescue hypothalamic responses in obesity. Once IF prevents the pro-inflammatory effects in the hippocampus caused by LPS (29), and it is also able to increase the production of β-Hydroxybutyrate (βHB) (136), correlated to inflammatory control (134) and insulin sensitivity, it is possible to hypothesize that modulations in the microbiota may be helpful to reduce hypothalamic inflammation and increase hormonal sensitivity. Low serum leptin and insulin values during fasting contribute to an increase in the expression of AgRP and NPY. However, interestingly, the NPY mRNA expression returned to baseline values as a chronic response to IF, which may be is an adaptation to hunger. Furthermore, some studies found an increase in the expression of POMC neuropeptide, which is controversial. Such neuronal sensitivity to the IF protocol is perhaps also associated with better regulation of hypothalamic autophagic response since IF can activate the autophagic pathway in other tissues (146), and autophagic flux is an essential mediator of neuropeptide responses.
Conclusion and Future Perspectives
Evidence indicates that IF protocols can be used as a strategy to promote weight loss, as they induce an increase in energy expenditure (35, 161) and improve the peripheral response to anorectic hormones (33, 162), which can significantly interfere with the hypothalamic autophagic pathway (33) and also in the expression of neuropeptides (27, 35). Thus, the literature reviewed allows us to hypothesize that IF could help reestablish, at least in part, the control of hypothalamic molecular responses in obese individuals, alleviating neuroinflammation and improving hypothalamic sensitivity anorectic hormones, thus helping to enhance reestablishment of energy homeostasis. However, when the IF protocol is performed without considering the circadian cycle, it can impair energy metabolism regulation (32). These associations require more research, mainly when obese individuals submitted to long periods of IF are evaluated regarding the responses of the autophagic pathway and hypothalamic neuropeptides. In conclusion, considering the favorable results of IF in obesity, the protocol may be an adjuvant treatment to partially rescue hypothalamic responses in obesity.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
The present work received financial support from the São Paulo Research Foundation (FAPESP; process numbers 2017/25492-4 and 2020/08192-0), National Council for Scientific and Technological Development (CNPq; process number 301279/2019-5), and the Coordination for the Improvement of Higher Education Personnel (CAPES; finance code 001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sainsbury A, Zhang L. Role of the arcuate nucleus of the hypothalamus in regulation of body weight during energy deficit. Mol Cell Endocrinol. (2010) 316:109–19. doi: 10.1016/j.mce.2009.09.025
2. Schwartz MW, Woods SC, Porte D Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. (2000) 404:661.
3. Thaler JP, Schwartz MW. Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. (2010) 151:4109–15. doi: 10.1210/en.2010-0336
5. Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab. (2009) 297:E1247–59. doi: 10.1152/ajpendo.00274.2009
6. Williams G, Bing C, Cai XJ, Harrold JA, King PJ, Liu XH. The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol Behav. (2001) 74:683–701. doi: 10.1016/s0031-9384(01)00612-6
7. Rossi M, Kim M, Morgan D, Small CJ, Edwards CM, Sunter D, et al. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. (1998) 139:4428–31. doi: 10.1210/endo.139.10.6332
8. Ollmann MM, Wilson BD, Yang Y-K, Kerns JA, Chen Y, Gantz I, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. (1997) 278:135–8. doi: 10.1126/science.278.5335.135
9. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. (1999) 5:1066–70. doi: 10.1038/12506
10. Eberle AN. Proopiomelanocortin and the melanocortin peptides. In: RD Cone editor. The Melanocortin Receptors. Totowa, NJ: Humana Press Inc (2000). p. 3–67. doi: 10.1385/1-59259-031-4:3
11. Cai M, Hruby VJ. The melanocortin receptor system: a target for multiple degenerative diseases. Curr Protein Pept Sci. (2016) 17:488–96. doi: 10.2174/1389203717666160226145330
12. Fan W, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. (1997) 385:165–8. doi: 10.1038/385165a0
13. Butler AA. The melanocortin system and energy balance. Peptides. (2006) 27:281–90. doi: 10.1016/j.peptides.2005.02.029
14. Yang Y, Xu Y. The central melanocortin system and human obesity. J Mol Cell Biol. (2020) 12:785–97. doi: 10.1093/jmcb/mjaa048
16. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. (2012) 13:1079–86. doi: 10.1038/embor.2012.174
17. Huang Y, He Z, Gao Y, Lieu L, Yao T, Sun J, et al. Phosphoinositide 3-kinase is integral for the acute activity of leptin and insulin in male arcuate NPY/AgRP neurons. J Endocr Soc. (2018) 2:518–32. doi: 10.1210/js.2018-00061
18. Burks DJ, de Mora JF, Schubert M, Withers DJ. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. (2000) 407:377. doi: 10.1038/35030105
19. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. (2000) 289:2122–5. doi: 10.1126/science.289.5487.2122
20. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. (2002) 5:566. doi: 10.1038/nn0602-861
21. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. (2004) 42:983–91. doi: 10.1016/j.neuron.2004.06.004
22. Bates SH, Stearns WH, Dundon TA, Schubert M. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. (2003) 421:856. doi: 10.1038/nature01388
23. Harris L, Hamilton S, Azevedo LB, Olajide J, De Brún C, Waller G, et al. Intermittent fasting interventions for treatment of overweight and obesity in adults: a systematic review and meta-analysis. JBI Database Syst Rev Implement Rep. (2018) 16:507–47. doi: 10.11124/JBISRIR-2016-003248
24. Horne BD, Muhlestein JB, Anderson JL. Health effects of intermittent fasting: hormesis or harm? A systematic review. Am J Clin Nutr. (2015) 102:464–70. doi: 10.3945/ajcn.115.109553
25. Faris MA, Jahrami H, BaHammam A, Kalaji Z, Madkour M, Hassanein M. A systematic review, meta-analysis, and meta-regression of the impact of diurnal intermittent fasting during Ramadan on glucometabolic markers in healthy subjects. Diabetes Res Clin Pract. (2020) 165:108226. doi: 10.1016/j.diabres.2020.108226
26. Higashida K, Fujimoto E, Higuchi M, Terada S. Effects of alternate-day fasting on high-fat diet-induced insulin resistance in rat skeletal muscle. Life Sci. (2013) 93:208–13. doi: 10.1016/j.lfs.2013.06.007
27. Spezani R, da Silva RR, Martins FF, de Souza Marinho T, Aguila MB, Mandarim-de-Lacerda CA. Intermittent fasting, adipokines, insulin sensitivity, and hypothalamic neuropeptides in a dietary overload with high-fat or high-fructose diet in mice. J Nutr Biochem. (2020) 83:108419. doi: 10.1016/j.jnutbio.2020.108419
28. Wang X, Yang Q, Liao Q, Li M, Zhang P, Santos HO, et al. Effects of intermittent fasting diets on plasma concentrations of inflammatory biomarkers: a systematic review and meta-analysis of randomized controlled trials. Nutrition. (2020) 79–80:110974. doi: 10.1016/j.nut.2020.110974
29. Vasconcelos AR, Yshii LM, Viel TA, Buck HS, Mattson MP, Scavone C, et al. Intermittent fasting attenuates lipopolysaccharide-induced neuroinflammation and memory impairment. J Neuroinflammation. (2014) 11:85. doi: 10.1186/1742-2094-11-85
30. Liu Z, Dai X, Zhang H, Shi R, Hui Y, Jin X, et al. Gut microbiota mediates intermittent-fasting alleviation of diabetes-induced cognitive impairment. Nat Commun. (2020) 11:855. doi: 10.1038/s41467-020-14676-4
31. Frank J, Gupta A, Osadchiy V, Mayer EA. Brain–Gut–Microbiome Interactions and Intermittent Fasting in Obesity. Nutrients. (2021) 13:584. doi: 10.3390/nu13020584
32. Yasumoto Y, Hashimoto C, Nakao R, Yamazaki H, Hiroyama H, Nemoto T, et al. Short-term feeding at the wrong time is sufficient to desynchronize peripheral clocks and induce obesity with hyperphagia, physical inactivity and metabolic disorders in mice. Metabolism. (2016) 65:714–27. doi: 10.1016/j.metabol.2016.02.003
33. Jamshed H, Beyl RA, Della Manna DL, Yang ES, Ravussin E, Peterson CM. Early time-restricted feeding improves 24-hour glucose levels and affects markers of the circadian clock, aging, and autophagy in humans. Nutrients. (2019) 11:1234. doi: 10.3390/nu11061234
34. Mariño G, Pietrocola F, Madeo F, Kroemer G. Caloric restriction mimetics: natural/physiological pharmacological autophagy inducers. Autophagy. (2014) 10:1879–82. doi: 10.4161/auto.36413
35. Gotthardt JD, Verpeut JL, Yeomans BL, Yang JA, Yasrebi A, Roepke TA, et al. Intermittent fasting promotes Fat loss with lean mass retention, increased hypothalamic norepinephrine content, and increased neuropeptide Y gene expression in diet-induced obese male mice. Endocrinology. (2015) 157:679–91. doi: 10.1210/en.2015-1622
36. Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. (2005) 2:411–20. doi: 10.1016/j.cmet.2005.10.009
37. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Investig. (1996) 98:1101. doi: 10.1172/JCI118891
38. Münzberg H, Björnholm M, Bates S, Myers M. Leptin receptor action and mechanisms of leptin resistance. Cell Mol Life Sci. (2005) 62:642.
39. Banks AS, Davis SM, Bates SH, Myers MG. Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. (2000) 275:14563–72. doi: 10.1074/jbc.275.19.14563
41. Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. (2006) 17:365–71. doi: 10.1016/j.tem.2006.09.007
42. Brownsey RW, Boone AN, Allard MF. Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovasc Res. (1997) 34:3–24. doi: 10.1016/s0008-6363(97)00051-5
44. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. (2005) 115:951. doi: 10.1172/JCI24301
45. Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. (2008) 118:1796–805. doi: 10.1172/jci32964
46. Varela L, Martínez-Sánchez N, Gallego R, Vázquez MJ, Roa J, Gándara M, et al. Hypothalamic mTOR pathway mediates thyroid hormone-induced hyperphagia in hyperthyroidism. J Pathol. (2012) 227:209–22. doi: 10.1002/path.3984
47. Maya-Monteiro CM, Bozza PT. Leptin and mTOR: partners in metabolism and inflammation. Cell Cycle. (2008) 7:1713–7. doi: 10.4161/cc.7.12.6157
48. Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest. (2008) 118:2959–68. doi: 10.1172/JCI34277
49. Lee DY. Roles of mTOR signaling in brain development. Exp Neurobiol. (2015) 24:177. doi: 10.5607/en.2015.24.3.177
50. Cota D, Proulx K, Smith KAB, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science. (2006) 312:927–30. doi: 10.1126/science.1124147
51. Hu F, Xu Y, Liu F. Hypothalamic roles of mTOR complex I: integration of nutrient and hormone signals to regulate energy homeostasis. Am J Physiol Endocrinol Metab. (2016) 310:E994–1002. doi: 10.1152/ajpendo.00121.2016
52. Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. (2012) 16:104–12. doi: 10.1016/j.cmet.2012.05.010
53. Kocalis HE, Hagan SL, George L, Turney MK, Siuta MA, Laryea GN, et al. Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Mol Metab. (2014) 3:394–407. doi: 10.1016/j.molmet.2014.01.014
54. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. (2004) 428:569–74. doi: 10.1038/nature02440
55. Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. (2007) 117:2325–36. doi: 10.1172/JCI31516
56. Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. (2001) 23:1112–9. doi: 10.1002/bies.10009
57. Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, et al. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia. (2005) 48:2170–8. doi: 10.1007/s00125-005-1913-1
58. McCrimmon RJ, Shaw M, Fan X, Cheng H, Ding Y, Vella MC, et al. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes. (2008) 57:444–50. doi: 10.2337/db07-0837
59. Oh TS, Cho H, Cho JH, Yu S-W, Kim E-K. Hypothalamic AMPK-induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy. (2016) 12:2009–25. doi: 10.1080/15548627.2016.1215382
60. Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. (2012) 13:258–65. doi: 10.1038/embor.2011.260
61. Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. (2011) 14:173–83. doi: 10.1016/j.cmet.2011.06.008
62. Malik SA, Mariño G, BenYounès A, Shen S, Harper F, Maiuri MC, et al. Neuroendocrine regulation of autophagy by leptin. Cell Cycle. (2011) 10:2917–23. doi: 10.4161/cc.10.17.17067
63. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. (2013) 15:713–20. doi: 10.1038/ncb2788
64. Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. (2010) 21:683–90. doi: 10.1016/j.semcdb.2010.03.002
65. Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. (2008) 28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008
66. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. (2006) 441:885–9. doi: 10.1038/nature04724
67. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. (2015) 16:461–72. doi: 10.1038/nrm4024
68. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. (2010) 466:68. doi: 10.1038/nature09204
69. Sanchez AM, Bernardi H, Py G, Candau RB. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R956–69. doi: 10.1152/ajpregu.00187.2014
70. Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin–phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. (2001) 2:330–5. doi: 10.1093/embo-reports/kve061
71. Mejías-Peña Y, Rodriguez-Miguelez P, Fernandez-Gonzalo R, Martínez-Flórez S, Almar M, de Paz JA, et al. Effects of aerobic training on markers of autophagy in the elderly. Age. (2016) 38:33. doi: 10.1007/s11357-016-9897-y
72. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. (2000) 19:5720–8. doi: 10.1093/emboj/19.21.5720
73. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132. doi: 10.1038/ncb2152
74. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
75. Haissaguerre M, Ferrière A, Simon V, Saucisse N, Dupuy N, André C, et al. mTORC1-dependent increase in oxidative metabolism in POMC neurons regulates food intake and action of leptin. Mol Metab. (2018) 12:98–106. doi: 10.1016/j.molmet.2018.04.002
76. Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. (2008) 90:313–23. doi: 10.1016/j.biochi.2007.08.014
77. Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IκB kinase β (IKKβ)/NF-κB pathway. J Biol Chem. (2011) 286:32324–32. doi: 10.1074/jbc.m111.254417
78. Varady KA, Hellerstein MK. Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr. (2007) 86:7–13. doi: 10.1093/ajcn/86.1.7
79. Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. (2003) 78:361–9. doi: 10.1093/ajcn/78.3.361
80. Halpern B, Mendes TB. Intermittent fasting for obesity and related disorders: unveiling myths, facts, and presumptions. Arch Endocrinol Metab. (2021) 65:14–23. doi: 10.20945/2359-3997000000322
81. Dalle Grave R. Regular eating, not intermittent fasting, is the best strategy for a healthy eating control. IJEDO (2020) 2:5–7. doi: 10.32044/ijedo.2020.02
82. Cerqueira FM, Kowaltowski AJ. Commonly adopted caloric restriction protocols often involve malnutrition. Ageing Res Rev. (2010) 9:424–30. doi: 10.1016/j.arr.2010.05.002
83. Hoddy KK, Marlatt KL, Çetinkaya H, Ravussin E. Intermittent fasting and metabolic health: from religious fast to time-restricted feeding. Obesity. (2020) 28:S29–37. doi: 10.1002/oby.22829
84. Anson RM, Jones B, de Cabod R. The diet restriction paradigm: a brief review of the effects of every-other-day feeding. Age. (2005) 27:17–25. doi: 10.1007/s11357-005-3286-2
85. Varady KA, Bhutani S, Church EC, Klempel MC. Short-term modified alternate-day fasting: a novel dietary strategy for weight loss and cardioprotection in obese adults. Am J Clin Nutr. (2009) 90:1138–43. doi: 10.3945/ajcn.2009.28380
86. Mirghani HM, Weerasinghe SD, Smith JR, Ezimokhai M. The effect of intermittent maternal fasting on human fetal breathing movements. J Obstet Gynaecol. (2004) 24:635–7. doi: 10.1080/01443610400007844
87. Correia JM, Santos I, Pezarat-Correia P, Minderico C, Mendonca GV. Effects of intermittent fasting on specific exercise performance outcomes: a systematic review including meta-analysis. Nutrients. (2020) 12:1390. doi: 10.3390/nu12051390
88. Pakkir Maideen NM, Jumale A, Balasubramaniam R. Adverse health effects associated with Islamic fasting-A literature review. J Nutr Fasting Health. (2017) 5:113–8.
89. Saad R. Effects of intermittent fasting on health, aging, and disease. N Engl J Med. (2020) 382:1773.
90. Rouhani MH, Azadbakht L. Is Ramadan fasting related to health outcomes? A review on the related evidence. J Res Med Sci. (2014) 19:987–92.
91. Mohamed AI, Abdi AM, Abdilahi MM. Ramadan intermittent fasting and its beneficial effects of health: a review article. Cent Afr J Public Health. (2020) 6:288. doi: 10.11648/j.cajph.20200605.17
92. Roky R, Houti I, Moussamih S, Qotbi S, Aadil N. Physiological and chronobiological changes during Ramadan intermittent fasting. Ann Nutr Metab. (2004) 48:296–303. doi: 10.1159/000081076
94. Afrasiabi A, Hassanzadeh S, Sattarivand R, Nouri M, Mahbood S. Effects of low fat and low calorie diet on plasma lipid levels in the fasting month of Ramadan. Saudi Med J. (2003) 24:184–8.
95. Mafauzy M, Mohammed W, Anum M, Zulkifli A, Ruhani A. A study of the fasting diabetic patients during the month of Ramadan. Med J Malaysia. (1990) 45:14–7.
96. Fakhrzadeh H, Larijani B, Sanjari M, Baradar-Jalili R, Amini MR. Effect of Ramadan fasting on clinical and biochemical parameters in healthy adults. Ann Saudi Med. (2003) 23:223–6. doi: 10.5144/0256-4947.2003.223
97. Cerqueira FM, da Cunha FM, Caldeira da Silva CC, Chausse B, Romano RL, Garcia CC, et al. Long-term intermittent feeding, but not caloric restriction, leads to redox imbalance, insulin receptor nitration, and glucose intolerance. Free Radic Biol Med. (2011) 51:1454–60. doi: 10.1016/j.freeradbiomed.2011.07.006
98. Schmahl FW, Metzler B. The health risks of occupational stress in islamic industrial workers during the Ramadan fasting period. Pol J Occup Med Environ Health. (1991) 4:219–28.
99. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. (2001) 50:2786–91. doi: 10.2337/diabetes.50.12.2786
100. Cota D, Matter EK, Woods SC, Seeley RJ. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci. (2008) 28:7202–8. doi: 10.1523/JNEUROSCI.1389-08.2008
101. Dalvi PS, Chalmers JA, Luo V, Han DY, Wellhauser L, Liu Y, et al. High fat induces acute and chronic inflammation in the hypothalamus: effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int J Obes. (2017) 41:149–58. doi: 10.1038/ijo.2016.183
102. Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. (2004) 145:4880–9. doi: 10.1210/en.2004-0726
103. Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, Hagepanos A, et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci U S A. (2003) 100:6216–20. doi: 10.1073/pnas.1035720100
104. Wan R, Ahmet I, Brown M, Cheng A, Kamimura N, Talan M, et al. Cardioprotective effect of intermittent fasting is associated with an elevation of adiponectin levels in rats. J Nutr Biochem. (2010) 21:413–7. doi: 10.1016/j.jnutbio.2009.01.020
105. Pedersen CR, Hagemann I, Bock T, Buschard K. Intermittent feeding and fasting reduces diabetes incidence in BB rats. Autoimmunity. (1999) 30:243–50. doi: 10.3109/08916939908993805
106. Cho Y, Hong N, Kim KW, Cho SJ, Lee M, Lee YH, et al. The effectiveness of intermittent fasting to reduce body mass index and glucose metabolism: a systematic review and meta-analysis. J Clin Med. (2019) 8:1645. doi: 10.3390/jcm8101645
107. Sutton EF, Beyl R, Early KS, Cefalu WT, Ravussin E, Peterson CM. Early time-restricted feeding improves insulin sensitivity, blood pressure, and oxidative stress even without weight loss in men with prediabetes. Cell Metab. (2018) 27:1212–21.e3. doi: 10.1016/j.cmet.2018.04.010
108. Gabel K, Kroeger CM, Trepanowski JF, Hoddy KK, Cienfuegos S, Kalam F, et al. Differential effects of alternate-day fasting versus daily calorie restriction on insulin resistance. Obesity. (2019) 27:1443–50. doi: 10.1002/oby.22564
109. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. (2004) 10:739–43. doi: 10.1038/nm1071
110. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
111. Eyckerman S, Broekaert D, Verhee A, Vandekerckhove J, Tavernier J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett. (2000) 486:33–7. doi: 10.1016/s0014-5793(00)02205-5
112. Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. (2002) 2:410–6. doi: 10.1038/nri818
113. Steinberg GR, Watt MJ, Fam BC, Proietto J, Andrikopoulos S, Allen AM, et al. Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice. Endocrinology. (2006) 147:3906–14. doi: 10.1210/en.2005-1587
114. Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. (2000) 275:15985–91. doi: 10.1074/jbc.275.21.15985
115. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. (2002) 277:42394–8. doi: 10.1074/jbc.C200444200
116. Lee IS, Shin G, Choue R. A 12-week regimen of caloric restriction improves levels of adipokines and pro-inflammatory cytokines in Korean women with BMIs greater than 23 kg/m2. Inflamm Res. (2010) 59:399–405. doi: 10.1007/s00011-009-0113-8
117. Zhang LN, Mitchell SE, Hambly C, Morgan DG, Clapham JC, Speakman JR. Physiological and behavioral responses to intermittent starvation in C57BL/6J mice. Physiol Behav. (2012) 105:376–87. doi: 10.1016/j.physbeh.2011.08.035
118. Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, et al. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. (2011) 152:1314–26. doi: 10.1210/en.2010-0659
119. Mushtaq R, Akram A, Khwaja S, Ahmed S. The role of inflammatory markers following Ramadan Fasting. Pak J Med Sci. (2019) 35:77–81. doi: 10.12669/pjms.35.1.95
120. Trepanowski JF, Kroeger CM, Barnosky A, Klempel M, Bhutani S, Hoddy KK, et al. Effects of alternate-day fasting or daily calorie restriction on body composition, fat distribution, and circulating adipokines: secondary analysis of a randomized controlled trial. Clin Nutr. (2018) 37:1871–8. doi: 10.1016/j.clnu.2017.11.018
121. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. (2009) 296:E1003–12. doi: 10.1152/ajpendo.90377.2008
122. Bjørbæk C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. (1998) 1:619–25. doi: 10.1016/s1097-2765(00)80062-3
123. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
124. Cheong R, Hoffmann A, Levchenko A. Understanding NF−κB signaling via mathematical modeling. Mol Syst Biol. (2008) 4:192. doi: 10.1038/msb.2008.30
125. Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: is it real and is it relevant? Biochimie. (2016) 124:11–20. doi: 10.1016/j.biochi.2015.06.020
126. Bliss ES, Whiteside E. The gut-brain axis, the human gut microbiota and their integration in the development of obesity. Front Physiol. (2018) 9:900. doi: 10.3389/fphys.2018.00900
127. Rorato R, Borges BC, Uchoa ET, Antunes-Rodrigues J, Elias CF, Elias LLKLPS-. Induced low-grade inflammation increases hypothalamic jnk expression and causes central insulin resistance irrespective of body weight changes. Int J Mol Sci. (2017) 18:1431. doi: 10.3390/ijms18071431
128. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. doi: 10.2337/db07-1403
129. Xiao S, Fei N, Pang X, Shen J, Wang L, Zhang B, et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol. (2014) 87:357–67. doi: 10.1111/1574-6941.12228
130. Rojas-Morales P, Tapia E, Pedraza-Chaverri J. β-Hydroxybutyrate: a signaling metabolite in starvation response? Cell Signal. (2016) 28:917–23. doi: 10.1016/j.cellsig.2016.04.005
131. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58:1509–17. doi: 10.2337/db08-1637
132. Park S, Kim DS, Daily JW. Central infusion of ketone bodies modulates body weight and hepatic insulin sensitivity by modifying hypothalamic leptin and insulin signaling pathways in type 2 diabetic rats. Brain Res. (2011) 1401:95–103. doi: 10.1016/j.brainres.2011.05.040
133. Cavaleri F, Bashar E. Potential synergies of β-hydroxybutyrate and butyrate on the modulation of metabolism, inflammation, cognition, and general health. J Nutr Metab. (2018) 2018:7195760. doi: 10.1155/2018/7195760
134. Sabatino AD, Morera R, Ciccocioppo R, Cazzola P, Gotti S, Tinozzi FP, et al. Oral butyrate for mildly to moderately active Crohn’s disease. Aliment Pharmacol Ther. (2005) 22:789–94. doi: 10.1111/j.1365-2036.2005.02639.x
135. Cerniuc C, Fischer T, Baumeister A, Bordewick Dell U. Impact of intermittent fasting (5: 2) on ketone body production in healthy female subjects. Ernährungs Umschau. (2019) 66:2–9.
136. Froy O, Miskin R. Effect of feeding regimens on circadian rhythms: implications for aging and longevity. Aging. (2010) 2:7–27. doi: 10.18632/aging.100116
137. Kutsuma A, Nakajima K, Suwa K. Potential association between breakfast skipping and concomitant late-night-dinner eating with metabolic syndrome and proteinuria in the Japanese population. Scientifica. (2014) 2014:253581. doi: 10.1155/2014/253581
138. Schlundt DG, Hill JO, Sbrocco T, Pope-Cordle J, Sharp T. The role of breakfast in the treatment of obesity: a randomized clinical trial. Am J Clin Nutr. (1992) 55:645–51. doi: 10.1093/ajcn/55.3.645
139. Duan W, Guo Z, Jiang H, Ware M, Mattson MP. Reversal of behavioral and metabolic abnormalities, and insulin resistance syndrome, by dietary restriction in mice deficient in brain-derived neurotrophic factor. Endocrinology. (2003) 144:2446–53. doi: 10.1210/en.2002-0113
140. Froy O, Chapnik N, Miskin R. Effect of intermittent fasting on circadian rhythms in mice depends on feeding time. Mech Ageing Dev. (2009) 130:154–60. doi: 10.1016/j.mad.2008.10.006
141. Lee JS, Mishra G, Hayashi K, Watanabe E, Mori K, Kawakubo K. Combined eating behaviors and overweight: eating quickly, late evening meals, and skipping breakfast. Eat Behav. (2016) 21:84–8. doi: 10.1016/j.eatbeh.2016.01.009
142. Moon S, Kang J, Kim SH, Chung HS, Kim YJ, Yu JM, et al. Beneficial effects of time-restricted eating on metabolic diseases: a systemic review and meta-analysis. Nutrients. (2020) 12:1267. doi: 10.3390/nu12051267
143. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. (2012) 441:523–40. doi: 10.1042/BJ20111451
144. Reginato A, Siqueira BP, Miyamoto J, Portovedo M, Costa SO, de Fante T, et al. Acute effects of fatty acids on autophagy in NPY neurones. J Neuroendocrinol. (2020) 32:e12900. doi: 10.1111/jne.12900
145. Ning Y-C, Cai G-Y, Zhuo L, Gao JJ, Dong D, Cui S, et al. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech Ageing Dev. (2013) 134:570–9. doi: 10.1016/j.mad.2013.11.006
146. Wegman MP, Guo MH, Bennion DM, Shankar MN, Chrzanowski SM, Goldberg LA, et al. Practicality of intermittent fasting in humans and its effect on oxidative stress and genes related to aging and metabolism. Rejuvenation Res. (2015) 18:162–72. doi: 10.1089/rej.2014.1624
147. Madkour MI, T El-Serafi A, Jahrami HA, Sherif NM, Hassan RE, Awadallah S, et al. Ramadan diurnal intermittent fasting modulates SOD2, TFAM, Nrf2, and sirtuins (SIRT1, SIRT3) gene expressions in subjects with overweight and obesity. Diabetes Res Clin Pract. (2019) 155:107801. doi: 10.1016/j.diabres.2019.107801
148. Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. (2010) 11:213–9. doi: 10.1016/j.cmet.2010.02.006
149. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
150. Egan D, Kim J, Shaw RJ, Guan K-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. (2011) 7:643–4. doi: 10.4161/auto.7.6.15123
151. Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. (2013) 27:2072–85. doi: 10.1101/gad.227439.113
152. Chaix A, Lin T, Le HD, Chang MW, Panda S. Time-restricted feeding prevents obesity and metabolic syndrome in mice lacking a circadian clock. Cell Metab. (2019) 29:303–19.e4. doi: 10.1016/j.cmet.2018.08.004
153. Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med. (2011) 17:1121–7. doi: 10.1038/nm.2421
154. Horvath TL, Warden CH, Hajos M, Lombardi A, Goglia F, Diano S. Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci. (1999) 19:10417–27. doi: 10.1523/JNEUROSCI.19-23-10417.1999
155. Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. (1997) 15:269–72. doi: 10.1038/ng0397-269
156. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. (2008) 454:846–51. doi: 10.1038/nature07181
157. Sherman H, Genzer Y, Cohen R, Chapnik N, Madar Z, Froy O. Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J. (2012) 26:3493–502. doi: 10.1096/fj.12-208868
158. Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. (1999) 140:814–7. doi: 10.1210/endo.140.2.6491
159. Weigle DS, Duell PB, Connor WE, Steiner RA, Soules MR, Kuijper JL. Effect of fasting, refeeding, and dietary fat restriction on plasma leptin levels. J Clin Endocrinol Metab. (1997) 82:561–5. doi: 10.1210/jcem.82.2.3757
160. Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, et al. Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology. (2002) 143:3846–53. doi: 10.1210/en.2002-220338
161. Chausse B, Solon C, Caldeira da Silva CC, Masselli Dos Reis IG, Manchado-Gobatto FB, Gobatto CA, et al. Intermittent fasting induces hypothalamic modifications resulting in low feeding efficiency, low body mass and overeating. Endocrinology. (2014) 155:2456–66. doi: 10.1210/en.2013-2057
Keywords: intermittent fasting (IF), hypothalamus, obesity, hypothalamic inflammation, non-pharmaceutical intervention
Citation: Oliveira LC, Morais GP, Ropelle ER, de Moura LP, Cintra DE, Pauli JR, de Freitas EC, Rorato R and da Silva ASR (2022) Using Intermittent Fasting as a Non-pharmacological Strategy to Alleviate Obesity-Induced Hypothalamic Molecular Pathway Disruption. Front. Nutr. 9:858320. doi: 10.3389/fnut.2022.858320
Received: 19 January 2022; Accepted: 25 February 2022;
Published: 30 March 2022.
Edited by:
Hideaki Oike, National Agriculture and Food Research Organization (NARO), JapanReviewed by:
Colleen M. Novak, Kent State University, United StatesAmanda Brandon, The University of Sydney, Australia
Copyright © 2022 Oliveira, Morais, Ropelle, de Moura, Cintra, Pauli, de Freitas, Rorato and da Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adelino Sanchez R. da Silva, adelinosanchez@usp.br; Rodrigo Rorato, rorato@unifesp.br
†These authors have contributed equally to this work