 Andrea Gaudio1*†
Andrea Gaudio1*† Fabio Gotta1†
Fabio Gotta1† Clarissa Ponti1,2Francesca Sanguineri1,2
Clarissa Ponti1,2Francesca Sanguineri1,2 Lucia Trevisan2,3Alessandro Geroldi2Serena Patrone2
Lucia Trevisan2,3Alessandro Geroldi2Serena Patrone2 Chiara Gemelli4Corrado Cabona5
Chiara Gemelli4Corrado Cabona5 Guja Astrea6
Guja Astrea6 Chiara Fiorillo2,7
Chiara Fiorillo2,7 Stefano Gustincich8
Stefano Gustincich8 Marina Grandis2,4
Marina Grandis2,4 Paola Mandich1,2
Paola Mandich1,2- 1IRCCS Ospedale Policlinico San Martino—UOC Genetica Medica, Genova, Italy
- 2Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal and Child Health, University of Genova, Genova, Italy
- 3IRCCS Ospedale Policlinico San Martino—SS Centro Tumori Ereditari, Genova, Italy
- 4IRCCS-Ospedale Policlinico San Martino—UOC Clinica Neurologica, Genova, Italy
- 5IRCCS-Ospedale Policlinico San Martino—UOC Neurofisiopatologia, Genova, Italy
- 6IRCCS Fondazione Stella Maris, Pisa, Italy
- 7IRCCS Istituto Giannina Gaslini—UOC Neuropsichiatria Infantile, Genova, Italy
- 8Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, Genova, Italy
Hereditary myopathies represent a clinically and genetically heterogeneous group of neuromuscular disorders, characterized by highly variable clinical presentations and frequently overlapping phenotypes with other neuromuscular disorders, likely influenced by genetic and environmental modifiers. Genetic testing is often challenging due to ambiguous clinical diagnosis. Here, we present the case of a family with clinical and Electromyography (EMG) features resembling a myotonia-like disorder in which Whole Exome Sequencing (WES) analysis revealed the co-segregation of two rare missense variants in UBR4 and HSPG2, genes previously associated with episodic ataxia 8 (EA8). A review of the literature highlighted a striking overlap between the clinical and the molecular features of our family and the previously described episodic ataxias (EAs), which raises concerns about the genotype–phenotype correlation, clinical variability, and the confounding overlap in these groups of disorders. This emphasizes the importance of thoroughly framing the patient's phenotype. The more clear-cut the diagnosis, the easier the identification of a genetic determinant, and the better the prognosis and the treatment of patients.
1. Introduction
Episodic ataxias (1) and hereditary myopathies (2) are two groups of clinically and genetically heterogeneous neuromuscular diseases. The main molecular determinants are damaging variants in genes encoding for ion channels subunits. Both are characterized by wide phenotypic variability, even intra-familiar, suggesting a possible influence of genetic and environmental modifiers on clinical manifestations.
Hereditary myopathies are a group of muscle disorders characterized by muscle stiffness, cramps, and weakness due to abnormal muscle structure and function. The clinical onset is usually in infancy and can vary among individuals, even within families. Diagnosis involves a combination of clinical evaluation, electromyography (EMG), muscle biopsy, and genetic testing.
EAs are a group of autosomal dominant inherited neuromuscular disorders characterized by variable spells of imbalance and ataxia often accompanied by additional ictal and interictal symptoms (1, 3, 4). The incidence is likely to be less than 1/100,000, but it may be underestimated due to clinical variability and to the incomplete knowledge of genetic causes (4). To date, nine subtypes have been described (EA 1-9) according to shared clinical and genetic features. These characteristics are quite heterogeneous and, in some cases, not fully elucidated (5). The two most common and best characterized forms are EA1 and EA2. The others are rarer and less molecularly depicted, and sometimes described only in single large families and never replicated (EA5) (5–7) (Table 1).

Table 1. Overview of the clinical and genetic features of EAs.
Here, we report the case of a family who was referred to our neuromuscular clinic for frequent cramps, especially at night, easy fatigability, muscle weakness, muscle hypertrophy, and twitching predominantly in ocular muscles.
Molecular analysis revealed the presence of two heterozygous missense variants in UBR4 and HSPG2, genes previously reported to co-segregate in a large EA8-diagnosed family (5). The co-segregation of these two variants in our family with a prevalent clinical myopathic phenotype raised concerns about the genotype–phenotype correlation of EA8. This prompted a careful literature review, which highlighted its wide and understudied phenotypic spectrum (Table 2), and the uncertainty in the definition of a precise genetic cause, which suggested a possible, or at least a confounding, overlap between EA8 and other neuromuscular disorders (8).
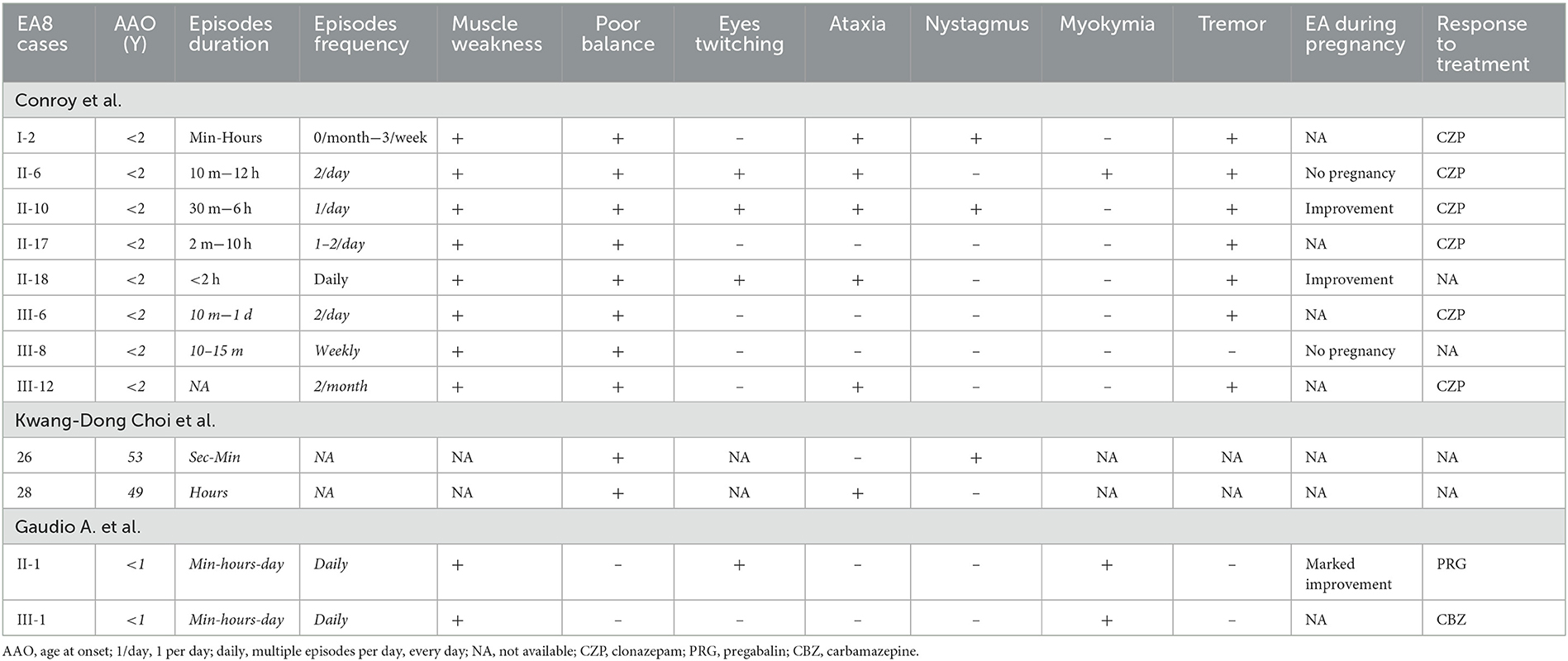
Table 2. Major differences between episodic ataxia 8 described cases and the index cases of the current paper.
2. Case report
2.1. Patients
The proband was referred to our neuromuscular clinic for a hereditary myopathy. She was born from unrelated parents. Her mother is healthy, and her father reported with cramps, myalgia, and elevated serum CK levels since childhood. The father died when he was 60 years old due pneumonia complications. The proband reported frequent episodes of cramps in the lower limbs, especially at night, easy fatigability, and muscle weakness associated with increased serum CK levels (2–3 times the upper normal limit) since the age of three. The symptoms are triggered by mild physical exercise, such as a short walk, tiredness, and stress. A muscular biopsy was performed at the age of 12, which did not show any pathological finding. At the age of 22, none of the muscular CT scan, anti-Ach antibodies, or echocardiography showed pathological results. The EMG showed decreased motor unit action potential (MUAP) amplitude during maximal voluntary contraction, and the ischemia-hyperpnea test revealed double discharges (doublets) of motor units with spontaneous resolution. She was then diagnosed with spasmophilia and treated with pregabalin, with improvement of the neuromuscular symptoms. At the age of 25, during pregnancy, she had a strong improvement of symptoms, despite interrupting the therapy. She gave birth to a healthy male, later evaluated at a pediatric hospital at the age of one for myalgia and elevated serum CK levels (915 U/L; NV <150 U/L). The neurological examination at the age of 27 was unremarkable except for muscular hypertrophy, especially in the lower limbs. At the last examination (28y), the proband reported palpebral and hand muscle myokymia and frequent falls due to sudden muscular failure. The neurological examination was normal, except for diffuse muscle hypertrophy, especially in the lower limbs and mild difficulties in standing up from a squatting position. The muscular strength and reflexes were normal, and no rippling or myotonic phenomenon was observed. The gait was unremarkable, while run and jump were slightly impaired. Clonazepam was included in therapy, instead of pregabalin, with improvement of symptoms.
Since the age of 3, her son has daily episodes of inconsolable crying, mostly at night, due to legs cramps, easy fatigability compared to healthy peers, and sporadic falls. The EMG and Electrocardiogram (ECG) were normal, and the muscle biopsy showed no pathological findings.
2.2. Molecular analysis
Informed consent was obtained from proband and relatives for all genetic investigations, according to Regione Liguria ethical committee guidelines. Myotonic dystrophy type 2 (ZNF9), as well as duplications/deletions of the Duchenne Muscular Dystrophy (DMD) gene in both the proband and her father were screened according to an established protocol for hyperCkemia (9) and the results were negative. Myotonic dystrophy type 1 genetic testing was also negative. A two-step custom Next Generation Sequencing (NGS) gene panel analysis was performed on the proband and her son. The first panel included 52 genes associated with myotonia and periodic paralysis; the second included 67 genes associated with hyperCkemia and limb girdle muscular dystrophies. Both results were negative.
Array comparative genomic hybridization (aCGH) analysis was performed in the child and his parents using Agilent Human Genome G3 SurePrint 4 × 180 K Microarray (Agilent Technologies, California, USA) and analyzed by Genomic Workbench (Agilent), which revealed a paternal duplication of 0.4–0.5 Mbp on 15q25.2(chr15:83913016-84363679 × 3—GRCh37). The Copy Number Variant (CNV) classified as Variant of Unknown Significance (VUS) included BNC1, SH3GL3, and ADAMTSL3, genes not associated with neuromuscular disorders.
Since family history was indicative of an inherited disorder, a WES analysis was performed. Genomic DNA of the proband and her child was isolated from peripheral blood according to standard protocols. DNA was enriched and the exonic sequences were captured with SOPHiA Whole Exome Solution kit (SOPHiA GeneticsTM) and sequenced on Illumina Novaseq6000 (IlluminaTM). Sequencing data were cleaned, processed, and aligned against hg38 genome assembly. Quality Control (QC), alignment, variant calling, and annotation were performed using Sophia DDM v.4 software (SOPHiA GeneticsTM). Variants were filtered and retained according to mean read depth > 10x, allele frequency in The Genome Aggregation Database (gnomAD) population <1%, allele frequency in an internal cohort <0.1%, segregation, and association with neuromuscular diseases. Eventually, only two variants were retained and prioritized according to American College of Medical Genetics and Genomics (ACMG) classification, co-segregation, and phenotype match. Both were validated via Sanger sequencing on an Abi Prism 3130XL sequencer (ThermofisherTM).
3. Results
WES analysis revealed the co-segregation of two missense heterozygous variants in UBR4 and HSPG2, namely, chr1:19140856:C>G—NM_020765.3: c.8525G>C (p.Gly2842Ala) and chr1:21831086:C>T—NM_005529.7: c.11567G>A (p.Gly3856Asp). Both variants have very rare frequencies in general population databases. Notably, computational in-silico tools unanimously predicted the pathogenicity of HSPG2 variant. Based on this evidence, both variants were classified as VUS, according to ACMG criteria (UBR4: PM2m, PP2s. HSPG2: PM2m, PP3s [Revel]). To evaluate family segregation, Sanger sequencing was performed on the proband's healthy mother, who resulted wild-type in both loci (Figure 1).
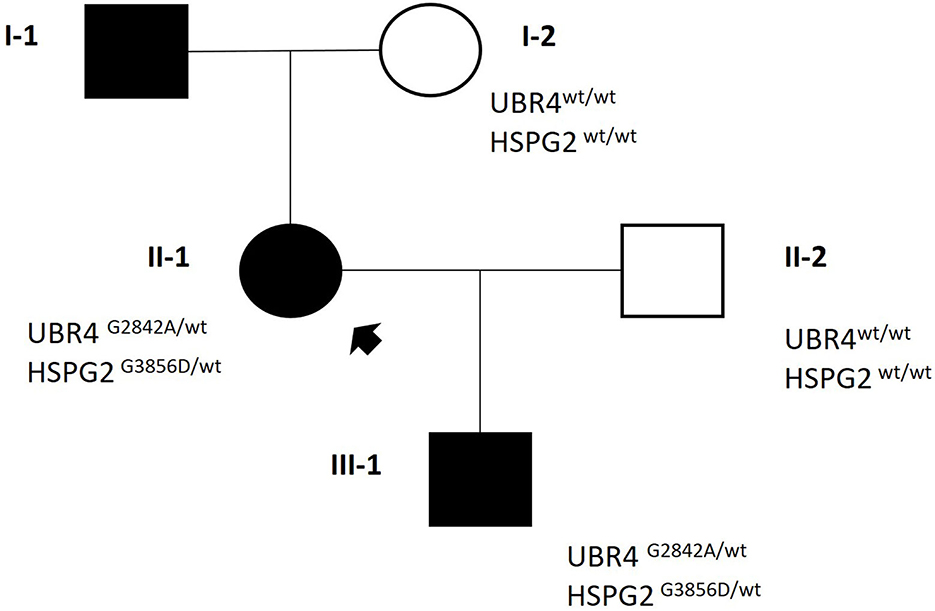
Figure 1. Family pedigree.
4. Discussion
Here, we present the case of a family who was referred to our clinic due to attacks of muscle cramps, pain, slight increased serum CK levels, hypertrophy, and transient muscle weakness. Although the cardinal features of myotonic syndromes (e.g., warm-up phenomenon and myotonic contraction) were missing, the set of symptoms and signs suggested a possible diagnosis of a genetically determined non-dystrophic myotonia-like disorder. However, the apparent myopathic origin of these clinical features were not confirmed by instrumental and morphological assessments (EMG, muscle CT scan, and muscle biopsy). Given the family history was suggestive of an inherited disorder, genetic testing was extensively performed, and WES analysis revealed the co-segregation of two variants in UBR4 and HSPG2, genes previously identified in a novel subtype of EA (5). Although the family phenotype was not clearly suggestive of EA, the molecular findings, and some peculiar clinical overlap with EA8 (e.g., cramps, muscle weakness, improvement during pregnancy, and responsiveness to clonazepam), led to a careful review of the available literature of this group of disorders.
EAs are a group of clinical and genetic multifaceted disorders, occasionally lacking its core feature, i.e., ataxia (10). Sometimes, the wide phenotypic spectrum is related to the variant's functional effect (gain- or loss-of-function), e.g., CACNA1A or SCNA2; sometimes, phenotypes are highly variable, even within the same functional effect, e.g., EA1 patients carrying p.T226R substitution differ from p.T226A or p.T226M carriers (11). Moreover, intra-familiar phenotypic variability is also reported (12), even in monozygotic twins (13). The broad extension of the clinical spectrum is often documented in single case reports with an atypical phenotype (4), raising doubts over the actual association of the clinical features with the reported variant, especially in the absence of extensive genetic studies. Furthermore, genetic modifiers as well as environmental factors are likely to influence the clinical outcome, but both have been poorly investigated so far (11).
According to Online Mendelian Inheritance in Man (OMIM), heterozygous missense variants in UBR4 are associated with EA8.
EA8 was first described in 2014 in a large Irish family with a rather different phenotype compared with the previously described EAs (4, 5, 11). The affected members manifested EA within the second year of life. The episodes were characterized by ataxia, dysarthria, nystagmus, myokymia, ocular twitching, and tremor and usually lasted minutes to hours, triggered by stress or tiredness. Peculiar features were the improvement of symptoms during pregnancy and the unusual responsiveness to clonazepam while being unresponsive to acetazolamide (4, 11). Linkage analysis identified a novel locus in 1p36.13-p34.3. Subsequent WES analysis and prioritization identified heterozygous missense variants in UBR4 and HSPG2. UBR4 was prioritized as the potential candidate since: (a) UBR4 had been reported in association with Ca2+-Calmodulin complex, as well as interacting with ITPR1, an Inositol 1,4,5-triphosphate receptor, both important in calcium signaling and thus potentially involved in the regulation of neuronal excitability (14); and (b) biallelic HSPG2 variants are associated with two recessively inherited disorders (dyssegmental dysplasia, Silverman–Handmaker type #MIM 224410; Schwartz–Jampel syndrome, type 1 #MIM 255800) while heterozygous carriers are asymptomatic.
Recent studies on drosophila highlighted the importance of UBR4 in the protein synthesis/degradation pathway (15–18). Being an E3-Ubiquitin ligase, its dysregulation affects the degradation of misfolded or corrupted proteins via proteosome. Moreover, the same model demonstrated UBR4 activity in promoting protein synthesis via direct impact on mRNA translation. The authors demonstrated that damaging UBR4 variants impair the balance between protein synthesis and degradation, promoting muscle hypertrophy via either increased protein synthesis or decreased ubiquitination and recycle. This supports the importance of UBR4 in signaling and in axonal and muscle development and function. However, to date, no functional study has been performed to establish a clear and exclusive role of UBR4 in EA8. Thus, the co-segregation of UBR4 and HSPG2 variants in the affected individuals cannot be ruled out and it may suggest that they play a synergistic role in EA8 pathogenesis.
Additionally, although rarer, milder phenotypes have been observed in Schwartz–Jampel syndrome in which the major signs are mostly myopathic (19). This strengthens the idea of a potential role of HSPG2 in the variable clinical manifestation of EA8.
HSPG2 encodes for perlecan, an extracellular matrix (ECM) proteoglycan (20, 21) fundamental for acetylcholinesterase (AchE) anchoring onto the neuromuscular junction (NMJ) (21–23). The complex perlecan's quaternary structure facilitates the binding of different extracellular molecules, such as integrins and α-destroglycan. When bound, perlecan becomes able to bind the AchE, anchoring it to the correct position at the NMJ. Studies revealed that an aberrant dosage of perlecan in the ECM affects its ability to correctly anchor the AchE to the NMJ (24), dysregulating AchE activity and preventing Ach firing and signaling (24). This has been suggested as the candidate pathomechanism underlying the two HSPG2-associated conditions with consequences on muscles such as cramps, easy fatigability, weakness, and progressive wasting (21, 24).
Since the initial report, another group identified disease-causing variants in UBR4 in two unrelated Korean cases, but the co-segregation with other variants, such as HSPG2, has not been investigated (25). These two cases differed from the Irish patients in age at onset and, in one patient, ataxia was not the major sign. Furthermore, Kwang-Dong and colleagues identified two additional unrelated patients with a missense heterozygous variant in UBR4 and a pathogenic mutation in CACNA1A. The clinical presentation was different from CACNA1A-associated EA2 patients and from the two with just a heterozygous missense variant in UBR4, especially in the earlier age at onset. The authors hypothesized a possible role for UBR4 as a genetic modifier for CACNA1A (25).
However, no functional study has been performed either to clearly establish the pathogenic involvement of UBR4 and HSPG2 in this disorder, nor to rule it out. It is still unclear whether or not they are linked to such a phenotype, and whether they act alone or synergistically in the pathogenesis. Given the above-mentioned wide variability, we hypothesized that in these condition modifiers do exist and that, in the reported family, HSPG2 could act as a possible genetic UBR4 modifier. The two encoded proteins share common pathways at the NMJ level, namely, signaling transduction. It is possible that not-completely functional proteins exert synergistic damage at different cellular and tissue levels depending on the position and consequence of the mutation, thereby varying the clinical manifestation. Additionally, we cannot rule out environmental modifiers, which are, unfortunately, more difficult to identify due to the limited patient cohort availability.
To date, the description of the EA8 clinical spectrum is highly variable and, unfortunately, is limited to just two families. It typically manifests in infancy, but adulthood age at onset has been described as well. The major symptom is episodic ataxia, which typically manifests as recurrent episodes of unsteadiness, clumsiness, and coordination difficulties. Moreover, frequent cramps and extensive muscle weakness have been described. The duration and frequency of these attacks vary from seconds to minutes, sometimes they can last for days to months. There is no clear definition of how an attack primarily manifests and distinguishing symptoms during attacks can be challenging, especially when episodes occur in close temporal proximity. Furthermore, it is unclear what triggers such attacks. Some triggers have been identified as physical exercise, stress, and changes in temperature, but, as well as in the other EA subtypes, there could be many more, including food, immunological status, and others that are yet to be identified. Some peculiar features highlighted by Conroy et al. are the marked improvement of symptoms during pregnancy and the lack of responsiveness to acetazolamide, which represents the most effective drug in the other EAs subtypes.
The family described here present no episodes of ataxia. The main clinical characteristics are frequent muscle cramps, often after mild physical activity, muscle hypertrophy, and sudden muscle weakness that culminates in falls. It is unclear if this manifestation is continuous or is the results of multiple temporally close attacks, which unfortunately are indistinguishable. Triggers can be identified in physical exercise and stress, but the influence of other secondary factors cannot be ruled out. Notably, the proband referred a dramatic resolution of symptoms during pregnancy, even without any medical treatment in the meantime. Instrumental ascertainments excluded primary muscular damage. Muscle biopsy revealed no specific alteration. EMG and the ischemia-hyperpnea test highlighted a decreased amplitude of MUAP and doublets, resembling a neuromuscular failure of probable muscular genesis.
There are often no clear-cut edges between the clinical manifestations in neuromuscular disorders, and sometimes this complicates the definition of the correct phenotype, misleading both clinicians and geneticists on what to give attention to and investigate. The current paper aims to argue the precise and comprehensive clinical description, genetic determinants, and pathophysiology underlying this complex group, especially EA8, while also raising doubts over a possible clinical overlap between different conditions.
On the one hand, the genetic studies cannot exclude the presence of complex rearrangements and additional genetic defects in uninvestigated genomic regions; on the other, we cannot rule out the involvement of both UBR4 and HSPG2 in the definition of the clinical features. However, much more effort is needed in order to disentangle the precise underlying molecular mechanism.
The uncertainty in establishing a clinical diagnosis, coupled with incomplete knowledge of the molecular and genetic backgrounds in such phenotypes reflects in challenging genetic testing and counseling. Altogether, this complicates the exact clinical picture definition and the correct genotype–phenotype inference in these overlapping disorders. Episodic ataxias may be misdiagnosed for a variety of reasons, including phenotype–genotype variability, clinical overlap with primary and secondary causes, and common mimicking disorders. Therefore, it is crucial to extend the molecular study of these genes, even in undiagnosed patients with family recurrence of myopathic clinical features or with presenting phenotypes overlapping any features of this complex and heterogeneous group of disease. The more clear-cut the clinical diagnosis, the easier the identification of a possible genetic determinant, and the better the treatment and prognosis of the subject.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Regione Liguria Ethical Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
AGa, FG, and PM designed and supervised the study. CP, FS, LT, CG, CC, GA, CF, and MG obtained consent from patients and performed the clinical evaluation and description. AGa, AGe, SP, and SG set up molecular and bioinformatics analysis. AGa, FG, CP, and PM wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the final manuscript.
Acknowledgments
The authors thank the patient and her family.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; CNV, Copy Number Variant; DMD, Duchenne Muscular Dystrophy; ECG, Electrocardiogram; EMG, Electromyography; gnomAD, The Genome Aggregation Database; NGS, Next Generation Sequencing; OMIM, Online Mendelian Inheritance in Man; QC, Quality Control; WES, Whole Exome Sequencing; VUS, Variant of Unknown Significance.
References
1. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, et al. CINCH investigators. Primary episodic ataxias: diagnosis, pathogenesis, and treatment. Brain. (2007) 130(Pt 10):2484–93. doi: 10.1093/brain/awm126
2. González J, Arlek M, Jorge Alfredo B, Ana María C. ‘Hereditary Myopathies'. Muscle Cell and Tissue—Current Status of Research Field. InTech (2018).
3. Tomlinson SE, Hanna MG, Kullmann DM. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol. (2009) 120:1768–76. doi: 10.1016/j.clinph.2009.07.003
4. Choi KD, Choi JH. Episodic ataxias: clinical and genetic features. J Mov Disord. (2016) 9:129–35. doi: 10.14802/jmd.16028
5. Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia: UBR4 the likely candidate. Eur J Hum Genet. (2014) 22:505–10. doi: 10.1038/ejhg.2013.173
6. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. (2000) 66:1531–9. doi: 10.1086/302909
7. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. (2005) 65:529–34. doi: 10.1212/01.WNL.0000172638.58172.5a
8. Hassan A. Episodic ataxias: primary and secondary etiologies, treatment, and classification approaches. Tremor Other Hyperkinet Mov (N Y). (2023) 13:9. doi: 10.5334/tohm.747
9. Gemelli C, Traverso M, Trevisan L, Fabbri S, Scarsi E, Carlini B, et al. An integrated approach to the evaluation of patients with asymptomatic or minimally symptomatic hyperCKemia. Muscle Nerve. (2022) 65:96–104. doi: 10.1002/mus.27448
10. Imbrici P, Accogli A, Blunck R, Altamura C, Iacomino M, D'adamo MC, et al. Musculoskeletal features without ataxia associated with a novel de novo mutation in KCNA1 impairing the voltage sensitivity of Kv1.1 channel. Biomedicines. (2021) 9:75. doi: 10.3390/biomedicines9010075
11. Giunti P, Mantuano E, Frontali M. Episodic ataxias: faux or real? Int J Mol Sci. (2020) 21:6472. doi: 10.3390/ijms21186472
12. Romaniello R, Zucca C, Tonelli A, Bonato S, Baschirotto C, Zanotta N, et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J Neurol Neurosurg Psychiatry. (2010) 81:840–3. doi: 10.1136/jnnp.2008.163402
13. Graves TD, Rajakulendran S, Zuberi SM, Morris HR, Schorge S, Hanna MG, et al. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology. (2010) 75:367–72. doi: 10.1212/WNL.0b013e3181ea9ee3
14. Schorge S, van de Leemput J, Singleton A, Houlden H, Hardy J. Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. (2010) 33:211–9. doi: 10.1016/j.tins.2010.02.005
15. Kim JG, Shin HC, Seo T, Nawale L, Han G, Kim BY, et al. Signaling pathways regulated by UBR Box-containing E3 Ligases. Int J Mol Sci. (2021) 22:8323. doi: 10.3390/ijms22158323
16. Parsons K, Nakatani Y, Nguyen MD. p600/UBR4 in the central nervous system. Cell Mol Life Sci. (2015) 72:1149–60. doi: 10.1007/s00018-014-1788-8
17. Kim ST, Lee YJ, Tasaki T, Hwang J, Kang MJ, Yi EC, et al. The N-recognin UBR4 of the N-end rule pathway is required for neurogenesis and homeostasis of cell surface proteins. PLoS One. (2018) 13:e0202260. doi: 10.1371/journal.pone.0202260
18. Hunt LC, Stover J, Haugen B, Shaw TI, Li Y, Pagala VR, et al. A key role for the ubiquitin ligase ubr4 in myofiber hypertrophy in drosophila and mice. Cell Rep. (2019) 28:1268–1281. doi: 10.1016/j.celrep.2019.06.094
19. Lin PY, Hung JH, Hsu CK, Chang YT, Sun YT. A novel pathogenic HSPG2 mutation in schwartz-jampel syndrome. Front Neurol. (2021) 12:632336. doi: 10.3389/fneur.2021.632336
20. Farach-Carson MC, Carson DD. Perlecan–a multifunctional extracellular proteoglycan scaffold. Glycobiology. (2007) 17:897–905. doi: 10.1093/glycob/cwm043
21. Arikawa-Hirasawa E, Rossi SG, Rotundo RL, Yamada Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat Neurosci. (2002) 5:119–23. doi: 10.1038/nn801
22. Steen MS, Froehner SC. PerleCan fix your muscle AChEs. Trends Neurosci. (2003) 26:241–2. doi: 10.1016/S0166-2236(03)00077-8
23. Stum M, Girard E, Bangratz M, Bernard V, Herbin M, Vignaud A, et al. Evidence of a dosage effect and a physiological endplate acetylcholinesterase deficiency in the first mouse models mimicking Schwartz-Jampel syndrome neuromyotonia. Hum Mol Genet. (2008) 17:3166–79. doi: 10.1093/hmg/ddn213
24. Martinez JR, Dhawan A, Farach-Carson MC. Modular proteoglycan perlecan/HSPG2: mutations, phenotypes, and functions. Genes (Basel). (2018) 9:556. doi: 10.3390/genes9110556
Keywords: hereditary myopathies, UBR4, HSPG2, episodic ataxia, genetic modifiers, genetic testing
Citation: Gaudio A, Gotta F, Ponti C, Sanguineri F, Trevisan L, Geroldi A, Patrone S, Gemelli C, Cabona C, Astrea G, Fiorillo C, Gustincich S, Grandis M and Mandich P (2023) Case report: Episodic ataxia without ataxia? Front. Neurol. 14:1224241. doi: 10.3389/fneur.2023.1224241
Received: 17 May 2023; Accepted: 03 October 2023;
Published: 26 October 2023.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Shahriar Nafissi, Tehran University of Medical Sciences, IranDario Ronchi, University of Milan, Italy
Copyright © 2023 Gaudio, Gotta, Ponti, Sanguineri, Trevisan, Geroldi, Patrone, Gemelli, Cabona, Astrea, Fiorillo, Gustincich, Grandis and Mandich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Gaudio, andrea.gaudio@hsanmartino.it
†These authors have contributed equally to this work and share first authorship