 Susanna Manenti1*
Susanna Manenti1* Mario Orrico1
Mario Orrico1 Stefano Masciocchi2
Stefano Masciocchi2 Alessandra Mandelli1
Alessandra Mandelli1 Annamaria Finardi1
Annamaria Finardi1 Roberto Furlan1
Roberto Furlan1- 1Clinical Neuroimmunology Unit, Division of Neuroscience, Institute of Experimental Neurology, IRCCS Ospedale San Raffaele, Milan, Italy
- 2Neuroimmunology Laboratory and Research Unit, IRCCS Mondino Foundation, Pavia, Italy
The approval of immune checkpoint inhibitors (ICIs) by the Food and Drug Administration (FDA) led to an improvement in the treatment of several types of cancer. The main targets of these drugs are cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death protein-1/programmed death-ligand 1 pathway (PD-1/PD-L1), which are important inhibitory molecules for the immune system. Besides being generally safer than common chemotherapy, the use of ICIs has been associated with several immune-related adverse effects (irAEs). Although rare, neurological adverse effects are reported within the irAEs in clinical trials, particularly in patients treated with anti-PD-1 antibodies or a combination of both anti-CTLA-4 and PD-1 drugs. The observations obtained from clinical trials suggest that the PD-1 axis may play a remarkable role in the regulation of neuroinflammation. Moreover, numerous studies in preclinical models have demonstrated the involvement of PD-1 in several neurological disorders. However, a comprehensive understanding of these cellular mechanisms remains elusive. Our review aims to summarize the most recent evidence concerning the regulation of neuroinflammation through PD-1/PD-L signaling, focusing on cell populations that are involved in this pathway.
Introduction to Neuroinflammation
Immune checkpoints, such as programmed cell death protein-1 (PD-1) and its ligands, are regulatory molecules that are fundamental to suppress the immune response and promote self-tolerance. PD-1 has two known ligands: PD-L1 (also called B7 homolog 1, B7-H1) and PD-L2 (or B7-DC). Both ligands have been characterized as powerful inhibitors in the context of tumor evasion from the immune system. Since 2014, six different inhibitors of PD-1 and PD-L1 were approved for cancer immunotherapy by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) (1), revolutionizing the treatment of certain cancers. However, in addition to their desired effects, immune-checkpoints inhibitors (ICIs) modify the balance of immune responses and induce specific off-target toxicities called immune-related adverse events (irAEs) (2). Several neurological immune-related adverse events were described during post-marketing surveillance with an estimated incidence of about 3–4% (3, 4). The reported neurological irAEs include encephalitis, aseptic meningitis, peripheral neuropathy, myasthenia gravis, and myositis. These clinical observations, combined with growing evidence about the role of PD-1 in neuroinflammatory disorders, suggest that the PD-1 axis may play a critical role not only in peripheral immune imbalance but also in the regulation of neuroinflammation, as highlighted in a previous work by Zhao et al. (5, 6). A comprehensive view of the cell-to-cell interactions and the molecular mechanisms underlying PD-l functions in neuroinflammation is, however, still missing. This review aims to summarize the most recent evidence concerning the regulation of human and murine neuroinflammatory disorders by PD-1 signaling, focusing on cell populations that are involved in this pathway.
PD-1/PD-L AXIS
Molecular Overview
Programmed cell death protein-1 is a 288 amino acid protein that belongs to the immunoglobulin superfamily and is a homolog to CD28. PD-1 is expressed in physiological conditions on a subset of thymocytes but can be induced upon activation in many types of immune cells, including T cells, B cells, natural killer (NK) cells, monocytes, and dendritic cells (DCs) (5). In its cytoplasmic tail, PD-1 has two tyrosine motifs, an immunoreceptor tyrosine-based switch-motif (ITSM) and an immunoreceptor tyrosine-based inhibitions motif (ITIM) (7). PD ligands are members of the B7 family of type 1 transmembrane proteins, which also include CD86 and CD80 (8). They have similar exon organization of the 5′UTR region, a signal sequence, IgV-like, IgC-like, and transmembrane domains, cytoplasmic exon 1, and cytoplasmic exon 2 with the 3′ untranslated region (8). However, the affinity of PD-L2 to PD-1 is three times stronger when compared to that of PD-L1, and this is probably due to tryptophan that is unique to PD-L2 (9). PD-1 ligands differ in their expression patterns: PD-L2 expression is restricted to professional antigen-presenting cells (APCs) (10), while PD-L1 is ubiquitously expressed in the inflamed tissues (11). To date, in physiological conditions, PD-L1 mRNA is largely present in various tissues, while PD-L1 protein is barely expressed on the cell surface, suggesting that PD-L1 mRNA is under tight post-transcriptional regulation. An exception is made in the context of human cancers, where PD-L1 protein is highly expressed by the tumor cells in an attempt to hide neoantigens from immune surveillance (12). The engagement of PD-1 by its ligands leads to the formation of PD-1 microclusters together with the T-cell or B-cell receptor (TCR or BCR). This leads to the recruitment of the Src homology phosphatase (SH)-2 domain-containing tyrosine phosphatase 2 (SHP2). Which then causes a decrease in the phosphorylation of the entire spectrum of TCR downstream signaling molecules. PD-1 engagement decreases the downstream signaling of both T- and B-cell receptors, respectively, by decreasing the phosphorylation of CD3z and protein kinase C q (PCK-q) and that of Igb, Syk, and phospholipase Cg2 (PLCg2). Furthermore, PD-1 engagement leads to the blockage of both the phosphatidylinositol-3 kinase and the serine-threonine kinase Akt through the recruitment of SHP2 (13). The downstream effects of PD-1 and PD-L1/L2 interaction comprehensively result in reduced proliferation of autoreactive T cells, suppression of effector T and B cells in parenchymal tissues, reduced cytokine production, induced T-cell anergy and exhaustion, reduced motility, and increased IL-10 production (14). The absence of PD-1 leads to an alteration of the signaling threshold during the development of T cells in the thymus, resulting in an increased presence of CD4/CD8 double-negative T cells. Furthermore, in several preclinical models, the blockade of the PD-1 pathway results in the development or exacerbation of autoimmune diseases depending on the genetic background they have (15–17). For example, C57BL/6 PD-1−/− mice develop lupus-like glomerulonephritis and arthritis starting at 6 months of age, while BALB/c knockout mice develop a dilated cardiomyopathy starting at 5 weeks of age (18–20). Among others, these findings suggest that the PD-1 axis plays an important role in central and peripheral tolerance, and a preventive role for several types of autoimmune disorders (Figure 1).
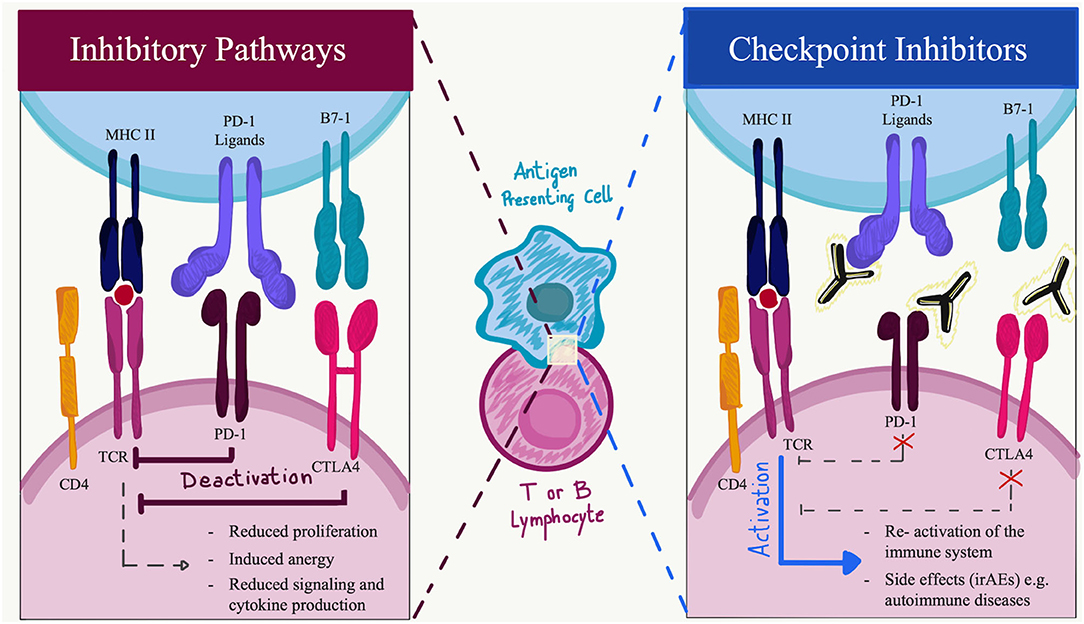
Figure 1. Immune checkpoint function during activation or inhibition pathways. Made with Adobe Draw.
PD-L1 and PD-L2 Expression in the CNS During Neuroinflammation
Preliminary evidence of the expression of PD-1 ligands in the central nervous system (CNS) comes from experimental autoimmune encephalomyelitis (EAE). In EAE mice, PD-L1 is overexpressed on microglial cells, astrocytes, and infiltrating mononuclear cells near the meninges, particularly in the areas with the highest inflammatory response (21). PD-L1 expression is also increased on the endothelium surrounding the cell infiltrates. Microglial cells, which represent 5–20% of all glial cells in the murine CNS, constitutively express a low level of PD-L1, and its expression can be upregulated in vitro when exposed to inflammatory conditions, for example, in the presence of IFN-γ or Th1 supernatants. PD-L1 expression by microglia can regulate immune responses by interacting with PD-1. Thus, one of the current hypotheses is that PD-L1 is expressed by microglia and infiltrating cells, which might act a strong immune inhibitory molecule that is able to curb T-cell activation and thus useful to maintain immune homeostasis in the CNS (11, 22).
The functions and distribution of PD-L2 are similar but not overlapping to PD-L1, and they still need to be fully elucidated in the CNS. Similar to PD-L1, PD-L2 inhibits T-cell proliferation by blocking cell cycle progression without increasing cell death. However, PD-L2 seems to be slightly less potent than PD-L1. Moreover, PD-L2 seems to be upregulated on small round cells in the brain, indicative of infiltrating macrophages or B cells (11).
To date, some authors reported that PD-L2 might bind to a second receptor different from PD-1, and is known as repulsive guidance molecule b (RGMb). RGMb, also called DRAGON, is a part of the RGM family, a group of glycosylphosphatidylinositol-anchored membrane proteins that bind bone morphogenetic proteins (BMPs) and neogenin. RGMb does not directly act as a signaling molecule, although it can function as a co-receptor modulating BMP signaling. RGMb is expressed mainly in the CNS and in particular on the surface of macrophages and other immune system cells. There is some evidence that the interaction of PD-L2 with RGMb through the BMP pathway might be co-stimulatory rather than inhibitory on T cells (23). This interaction seems to promote the development of respiratory tolerance, facilitating the proliferation of T cells. However, the potential role of RGMb has just begun to emerge, and further studies are needed to clarify its functions.
PD-1 Role During Neuroinflammation
The multifactorial nature of neuroinflammation unfolds through a complex, highly multicellular pathophysiological process that evolves according to the type and the duration of the disease. Many studies have tried to explain the role of the PD-1/PD-L1 pathway in CNS infections, often with contradictory results. In this section, we will discuss recent findings from various studies in murine models, focusing on the different cellular subsets that are involved (Table 1).
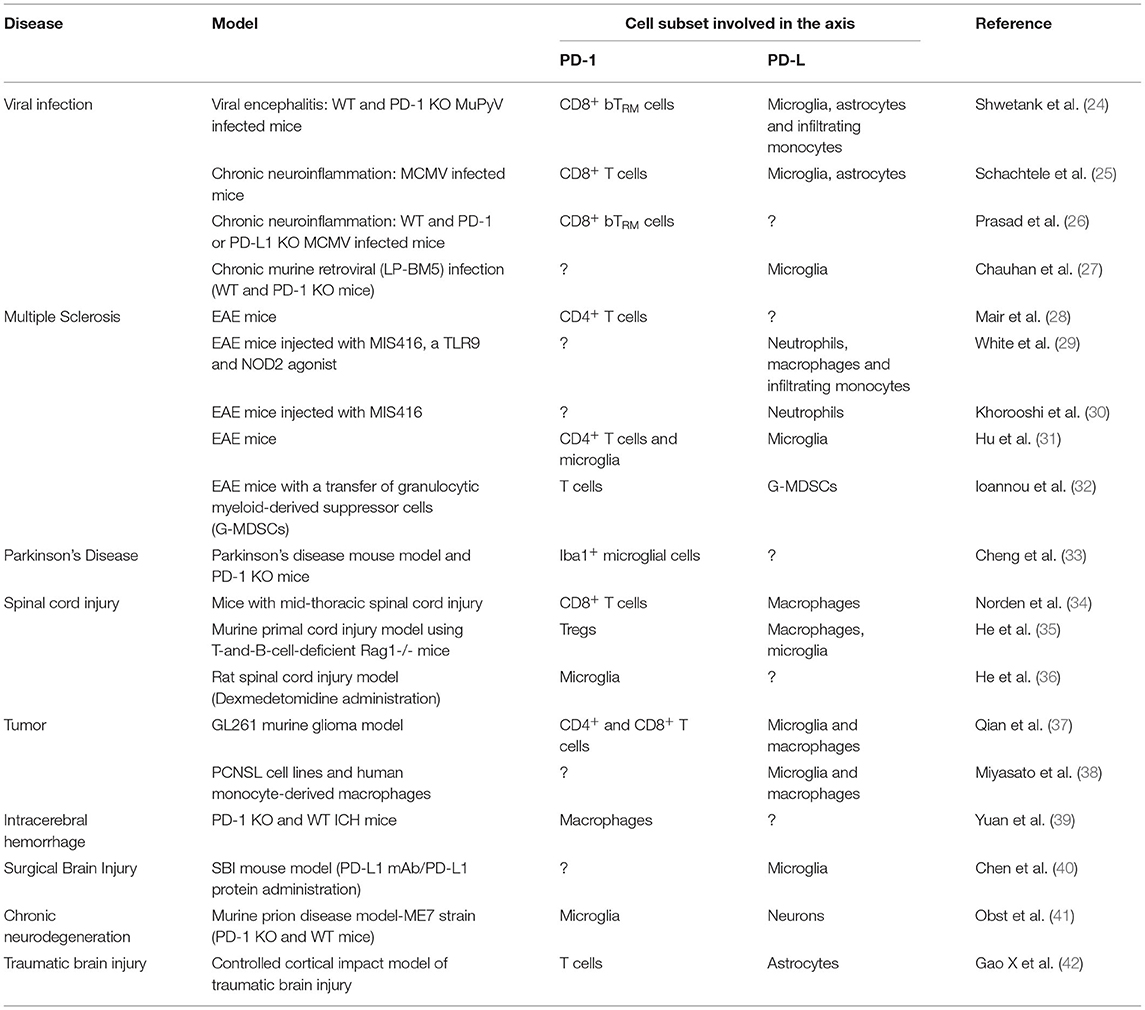
Table 1. PD-1 and its ligands in animal models of neurological disorders.
Lymphoid Cells
As highlighted in the Introduction section, PD-1 acts as an important immunosuppressive molecule that can exert several regulatory roles on T-cell effector functions. It has extensively been demonstrated that signaling through the PD-1 receptor results in the inhibition of TCR-mediated cell activation (e.g., proliferation and cytokine production), (8, 43, 44) but increasing evidence suggests that the PD-1 axis also has a crucial role in modulating T-cell activation in different models of neuroinflammatory diseases.
CD4 and CD8 T Lymphocytes
Mair et al. studied the role of PD-1 in the development and maintenance of T-cell adaptive tolerance [i.e., a process by which T cells acquire a hyporesponsive state when antigen stimulation persists in vivo (28)] after exposure to high levels of autoantigens in the model of EAE. This group reported that PD-1 expression is upregulated on adapted CD4+ T cells within the CNS, but PD-1 loss did not preclude T cells to acquire an adapted phenotype both in vivo and in vitro, indicating that the maintenance of their unresponsiveness in EAE is independent of PD-1 (28). This finding suggests that PD-l is not fundamental in this process, and it is in contrast to the majority of studies about PD-1 involvement in T-cell adapted state (45). Using the same disease mouse model (i.e., EAE), it was shown that CD4+ T cells, infiltrating the CNS at the peak of disease, upregulate PD-1, and this overexpression is associated with a reduction of Th1 cells due to nitric oxide release by PD-L1+ microglia. This evidence supports the role of PD-1 in EAE regulation through the suppression of Th1 cell differentiation (31).
The impairment of CD8+ T-cell function found in mice with spinal cord injury (SCI) was associated with the induction of regulatory pathways, including the upregulation of PD-1/PD-L1. Injured mice display an increased percentage of PD-1+ CD8 T cells which also have a higher expression of this receptor on their surface. In this way, PD-1 upregulation prevents the immune-inflammatory cascade and limits the spreading of inflammation at the site of damage (34).
Using the same mouse model, the study of He et al. showed that the anti-inflammatory function carried out by regulatory T cells (Tregs) that infiltrate the spinal cord in the SCI subacute phase is maintained, thanks to the overexpression of PD-1. Knockdown of PD-1 in Tregs caused decreased production of IL-10, TGF-β, and Foxp3, inducing a lower inhibitory activity of Tregs on pro-inflammatory macrophages/microglia (35).
In the glioma 261 model (GL261), through the analysis of cells that infiltrate the glioma, Qian et al. demonstrated that PD-1 expression is higher both on CD4+ and CD8+ T cells and increases during tumor progression. Besides, PD-1 expression, in cells that infiltrate the tumor, correlates with an increased apoptotic rate of T cells, suggesting a role for the PD-1 axis in the inhibition of T-cell function in glioma (37). Some evidence regarding the importance of the PD-1/PD-L1 pathway in controlling neuroinflammation also comes from different models of chronic neuroinflammation in mice recovering from a viral brain infection.
In mice with encephalitis induced by murine cytomegalovirus (MCMV) infection, expression of PD-1 is found on CD8+ T cells within the brain. Such expression contributes to their functional suppression and cytokine production inhibition in the post-encephalitic brain (25).
Brain-Resident Memory T Cells (BTRMs)
In 2017, Qian et al. assessed the importance of the PD-1 axis in the generation of brain-resident memory T cells (bTRMs) after viral infection. bTRMs are a population of tissue-resident CD8+ lymphocytes that persist for a long time in the brain and play a fundamental role in controlling pathogen clearance in the case of virus re-infection (37, 46).
Authors phenotypically characterize bTRMs by analyzing CD8+ lymphocytes residing in the brain of mice chronically infected with MCMV and describe that loss of PD-1 results in fewer bTRM cells within the brains of PD-1 KO mice (i.e., the reduced number of CD8+ cells expressing the integrin CD103, a marker of brain TRM). Furthermore, the characterization of PD-1 expression on CD103+CD8+ T cells isolated from the brain at 30 days post-infection reveals a higher frequency of PD-1+CD103+ cells in the brains of wild-type animals compared to PD-L1 KO mice. These results suggest that the upregulation of PD-1 receptors on bTRM cells may help to preserve their longevity (26). Moreover, Shwetank et al. confirmed that PD-1 signaling plays a pivotal role in the regulation of immune response in CNS persistent infection, revealing a multifaceted function of PD-1 on neuroinflammation regulation in mouse polyomavirus (MuPyV) encephalitis. These authors demonstrate that the intracerebral inoculation of MuPyV leads to the generation of a permanent population of virus-specific PD-1+bTRM cells in infected mice brains. During the acute phase of infection, PD-1 inhibits the effector functions of virus-specific CD8+ bTRM cells, limiting the severity of neuroinflammation but maintaining the control of re-encountered virus during persistent infection (24).
All these findings suggest that PD-1 signaling represents not only a mechanism to inhibit T-cell stimulation and proliferation but also to promote the long life of bTRM cells.
Evidence supporting the role of the PD-1 regulator in bTRM cells also comes from studies conducted in the human brain. In line with results obtained in mouse models, Smolders et al. demonstrated that in human white matter CD103+ CD8+ TRM cells, PD-1 is highly expressed (47) and that this cell population is enriched in the active lesions in multiple sclerosis (48).
Myeloid Cells
Both peripheral and CNS-resident myeloid cells are remarkably involved in the onset and development of neurological disorders. The pathogenesis of many neuroinflammatory diseases has been associated with impaired neuronal function due to the production of reactive oxygen species (ROS) or reactive nitrogen species (RNS). This consequently leads to an increase in oxidative stress (49). In mice, GM-CSF, IFNγ, and IL1ß are fundamental stimulations for the recruitment of myeloid cells in the CNS (50–53). Accordingly, receptors for IFNγ and GM-CSF are abundant on a variety of myeloid cells, such as neutrophils, DCs, macrophages, and monocytes (54). It has been demonstrated that GM-CSF plays a fundamental and non-redundant role in EAE pathology (55). IL1ß is involved both in the development of EAE and in the transmigration of myeloid cells into the CNS (56). Moreover, endothelial cells activated with IL1ß release GM-CSF which then converts monocytes into antigen-presenting cells (APCs). Mice depleted for both IL1ß and GM-CSF are resistant to EAE (57), highlighting the importance of myeloid cell recruitment in the CNS. On the same note, IFNγ seems to be fundamental for the gradual acquisition of a mature inflammatory phagocyte phenotype in Ly6Chi monocytes (50). Only in recent years, an increasing number of studies have highlighted the involvement of myeloid cells expressing PD-1/PD-L in the immune regulation of CNS disorders in different contexts. Ioannou et al. showed that granulocytic myeloid-derived suppressor cells (MDSC) expressing PD-L1 could be found in the peripheral lymphoid tissue of EAE mice. PD-L1 upregulation was stimulated by IFNγ, and the active transfer of these cells was able to ameliorate the disease, significantly decrease demyelination, and delay the onset of symptoms (32). As an example, studies conducted with MIS416, a small molecule that is a TLR9 and NOD2 agonist, show interesting results in the EAE animal model. MIS416 treatment produced an expansion of myeloid cells and an increased expression of PD-L1 on the peripheral myeloid subset (CD11b+, CD45high) that was recruited to the CNS (29). Moreover, MIS416 has shown success in reducing disease burden in mice with EAE when administered prophylactically or therapeutically. This protection depends on the rapid production of IFNγ, a cytokine that was also significantly increased in MIS416-treated secondary progressive MS patients (58). MIS416 leads to an upregulation of PD-L1 and MHCII on neutrophils, macrophages, and infiltrating monocytes and enhances the homeostatic recruitment of PD-L1-expressing myeloid cells to the CNS (30).
Monocytes and Macrophages
Infiltrating monocytes and monocyte-derived macrophages are fundamental for the pathogenesis and the development of inflammation inside the CNS. Differentiated inflammatory monocytes and their progeny are increasingly present in the inflamed tissue and represent the most important executors of GM-CSF-dependent pathogenesis (54). In 2019, Schwartz's group showed that immune checkpoint blockade targeting the PD-1/PD-L1 pathway might have beneficial results in a tau-driven disease model resembling Alzheimer's disease (AD). The use of PD-L1 blocking antibodies resulted in increased immunomodulatory monocyte-derived macrophages within the brain parenchyma, which ultimately led to the modification of brain pathology and the restoration of cognitive performance (59, 60).
Yuan et al. conducted a study exploiting an animal model of intracerebral hemorrhage (ICH), which accounts for 10–15% of all acute strokes, and showed that PD-1 expression is increased in perihematomal tissue and mainly on CD11b+ macrophages. The expression of PD-1 attenuates macrophage-mediated inflammation and brain injuries (39).
On the other hand, in an animal model of spinal cord injury, Norden's group showed that macrophages were highly activated in the spleen of mice, with an increased expression of PD-L1 and MHCII after the induction of the disease (34).
Neutrophils
The importance of neutrophils in neuroinflammatory disorders is gaining growing interest, and their involvement in the pathogenesis and progression of a stroke, MS, and Alzheimer's disease is now undeniable (61–64). With their capability to release ROS, enzymes, neutrophil extracellular traps (NETs), and cytokines in different pathophysiological conditions, neutrophils are involved not only in acute inflammation but also in causing chronic collateral damage in tissues (65). However, several studies reported a duality in the function of neutrophils, as they carry an active contribution both in a pro-inflammatory and an anti-inflammatory way. On the one end, neutrophils perform several crucial functions during all the stages of autoimmune progression, including antigen presentation, modulation of several cell types, and direct tissue damage (66). In human patients, the neutrophil-to-lymphocyte ratio has been proposed as a clinical marker both for MS and ischemic stroke (67). Accordingly, the percentage of neutrophils in the infiltrating cells in EAE, MS preclinical model, increased remarkably during the disease onset, remaining high until the peak stage and then drastically decreasing thereafter (68). Moreover, blocking a fundamental cytokine for the recruitment of neutrophils, namely CXCR2 (CXC motif chemokine receptor 2), resulted in reduced disease severity in EAE (69). On the other hand, several recent papers reported that neutrophils might exert a protective function in neuroinflammatory disorders, by slowing down the disease progression (70). On the same note, Khorooshi et al. reported that MIS416 injected intrathecally in EAE mice suppressed the disease and recruited neutrophils into the CNS. In the first phase of the EAE, these neutrophils were described as “protective,” as they were able to produce IL-10 and express PD-L1. When tested for in vitro proliferation assays, PD-L1+ neutrophils showed immunosuppressive features, slowing down lymphocyte proliferation, while PD-L1+ monocytes cannot (30). Moreover, Melero-Jerez et al. identified myeloid-derived suppressor cells expressing Arg1+PD-L1+ and Gr1+. These cells were induced after IFN-ß treatment and once again showed immunosuppressive features toward T lymphocytes (71).
Microglia
Microglial cells are CNS-resident myeloid cells that are extremely sensitive to alterations in the homeostasis of the brain. In autoimmune inflammation, microglia act as a direct link between the immune system and the CNS (21). Microglial cells express basal levels of PD-L1 (approximately 20%) in uninfected mice, but this expression can be increased to over 90% of the cells within 1 week following a viral brain infection (25). In 2018, Chauhan et al. exploited a model of chronic murine retroviral (LP-BM5) infection. They described that the infiltration rate of T lymphocytes and macrophages, as well as microglial activation, was remarkably increased in PD-1 KO mice compared to wild-type animals (27). A basal level of PD-L1 protein expression was observed on approximately 20% of microglial cells in uninfected mice, but it reached over 90% of the cells within 7 days following viral brain infection. In MCMV-induced brain infection, there is chronic neuroinflammation and the production of IFNγ by infiltrating T lymphocytes, which induces the upregulation of PD-L1 in activated brain-resident glial cells. The antiviral responses are controlled through functional inhibition of effector CD8+ T cells via the PD-L1 pathway, limiting the consequences of neuroinflammation (25). During neuroinflammation, both microglial cells and astrocytes upregulate PD-L1 along with MHC I and MHC II, suggesting a role for resident glial cells in limiting CNS pathology (72). PD-L1 expression on microglia and astrocytes, but not on oligodendrocytes, has also been reported to inhibit T-cell activation and limit immune-mediated tissue damage in a mouse model of acute viral encephalitis (MuPyV encephalitis). PD-1 acts to inhibit the effector functions of virus-specific CD8+ bTRM during MuPyV encephalitis (24). This study highlights the role of PD-L1 in mediating protection from viral-induced immunopathology associated with encephalomyelitis.
Several other studies with murine models illustrate the immunoregulatory role of microglial cells during chronic, persistent neuroinflammation. At the peak of the disease in the EAE model, Hu et al. found that microglial cells were increased in number and displayed an upregulation of PD-L1 along with MHC II and CD86. Using an ex vivo co-culture, microglia from EAE mice inhibited antigen-specific CD4+ T-cell proliferation, as well as Th1 differentiation via nitric oxide (whose production was dependent on PD-L1) (31).
Moreover, the adoptive transfer of M2-polarized microglia expressing PD-L1 attenuated the severity of an established EAE, demonstrating again the importance of the regulatory role of PD-L1 in neuroinflammation (73).
In a murine model of chronic neurodegeneration, a prion disease in the ME7 strain, PD-1 expression was increased on microglia cells compared to wild-type mice. Moreover, PD-1−/− mice did not show an increase in myeloid cell infiltration or a major change in the inflammatory profile. Furthermore, no changes were observed in the neurodegeneration of the pyramidal neurons in the hippocampus that indifferently expresses PD-L1 and PD-L2 both in healthy controls and in ME7 mice. The absence of PD-1 led only to a slight exacerbation in the behavioral phenotypes (41).
The involvement of the PD-1 axis has also been studied in a model of surgical brain injury (SBI). PD-L1 was significantly upregulated on microglial cells both in vitro and in vivo through the ERK signaling pathway. Consistently, the blockade of the PD-1 axis using a PD-L1 antibody significantly enhanced brain edema, exacerbated apoptosis, and increased neurodeficits post-SBI. On the other hand, the activation of PD-1/PD-L1 signaling with a PD-L1 recombinant protein significantly attenuated the inflammatory responses and brain edema post-SBI. Thus, the PD-1/PD-L1 pathway might be involved in a “self-protection mechanism” in SBI (40).
Dexmedetomidine, a drug used in a rat SCI model, inhibits neuroinflammation through the upregulation of PD-1 on microglia cells mediated by AMPK signaling. PD-1/PD-L1 interactions downregulate pro-inflammatory cytokine expression by activating microglia and inducing the M2 polarization of microglial cells (36). Besides suppressing T-cell activity, the PD-1/PD-L1 axis might also prevent the immune system from eliminating cancer cells. The role of PD-L1 on brain tumors might serve as an immune evasion strategy. Miyasato et al. reported that PD-L1 expression was upregulated on tumor-associated macrophages/microglia in the case of primary central nervous system lymphoma (38). In 2017, Saha et al. demonstrated that the triple-combination therapy of anti-cytotoxic T-lymphocyte-associated protein (CTLA)-4, anti-PD-1, and G47Δ-mIL12 (oncolytic HSV expressing murine IL-12) healed most mice in two glioma models. This curative effect was associated with macrophage infiltration and the subsequent M1-like polarization, along with an increase in the ratio of T-effector to T-regulatory cells (74). Immune checkpoint inhibitors function as tumor-regressing factors via the modulation of the interactions between immune and tumor cells. They lead to the reactivation of cytotoxic T cells which fight against cancer cells. Moreover, in 2018, Qian et al. identified the distribution of tumor-infiltrating T cells and PD-L1 expression in a model of murine glioma. The group showed that the IFNγ level was positively correlated with PD-L1 expression in the glioma microenvironment (37). Finally, in a preclinical model of Parkinson's disease, Cheng et al. hypothesized the involvement of the PD-1/PD-L axis in the pathogenesis of the disease. The knockout of PD-1 led to an exacerbation in the motor dysfunction of animals. This was explained by an increase in microglial activation and release of pro-inflammatory cytokines, which ultimately led to neuroinflammation in midbrains (33).
Astrocytes
Astrocytes are a very heterogeneous population in the CNS. Similar to microglia, they can switch to a reactive state and can modulate the progression of multiple neurologic disorders both positively and negatively (75). Similar to microglia, during neuroinflammation, astrocytes can upregulate PD-L1 along with MHC I and MHC II, suggesting a role for resident glial cells in limiting CNS pathology (72). Accordingly, Lipp et al. showed an upregulation of PD-L1 on hippocampal astrocytes in a model of axonal degeneration (76), suggesting a potential role in the inhibition of T cells. Gao et al. described the formation of dense areas of activated astrocytes expressing PD-L1 near the lesions in a traumatic brain injury model (42). In this model, astrocytes acted as gatekeepers, by blocking the infiltration of inflammatory Ly6chi monocytes/macrophages, but not the entrance of tissue repairing Ly6clowF4/80 (42) cells.
PD-1 Pathways in Human Neuroinflammatory Disorders
Immune-Related Adverse Effects of Checkpoint Inhibitors
Due to the high level of expression of PD-L1, monoclonal antibodies targeting the PD-1 axis have been developed to treat a wide variety of human tumors. Nivolumab was the first PD-1 checkpoint inhibitor approved for the treatment of metastatic melanoma (77). The approval of these drugs improved the treatment of several types of cancer. To date, the immune checkpoint blockade approach is generally safer than common chemotherapy. On the other hand, the possible side effects on the immune system are not completely understood, and they have been associated with irAEs. These side effects mostly involve compartments like the gastrointestinal tract, endocrine glands, skin, liver, and the CNS. Neurological autoimmune diseases are reported within the irAEs in the clinical trials, but at a low frequency (78). During post-marketing surveillance, neurological complications have been described with an estimated incidence of about 3–4% (3, 4). Some authors reported neuromuscular complications as the most common nervous system condition, (3) but cases of multiple sclerosis and meningoencephalitis have been increasingly reported in the treated patients (79, 80). The clinical management of neurological adverse events is not trivial, which can be attributed to the partial knowledge of the underlying immunological mechanisms (81). Systemic corticosteroid therapy is the most common first-line therapy for irAEs (82). Clear guidelines for the treatment of neurological adverse events are still missing. Typically, drugs interfering with the PD-1 axis have been associated with irAEs at a lower frequency compared to those binding other immune checkpoint inhibitors (i.e., CTLA-4). The risk rises when these therapies are combined (83). Neurological irAEs seem to show a different trend with an overall incidence of 6.1% in patients treated with anti-PD-1 antibodies, 3.8% with anti-CTLA-4 antibodies, and 12% in combination (84). Diamanti et al. recently reported common neurological manifestations of irAEs in a small cohort of 27 oncologic patients, including myositis, inflammatory polyradiculoneuropathies, and myasthenia gravis, alone or in combination. The authors suggest that in corticoresistant or severely affected patients, second-line treatments with IVIg or plasmapheresis may provide additional benefit (85).
PD-1 and Multiple Sclerosis
The immune-regulatory role of PD-1 in MS, the most common neuroinflammatory disease, is still to be fully elucidated. However, studies have demonstrated the importance of the PD-1 pathway in the development and progression of EAE, suggesting that this pathway might play a role in human disease as well. In people with MS, the PD-1 gene polymorphism (PD-1.3), which is related to reduced PD-1 activity, was associated with a progressive course of the disease, possibly due to a partial defect in PD-1–mediated inhibition of T-cell activation (86). Pawlak-Adamska et al. in a population-based case-control study with 203 MS patients investigated and selected four PD-1 single-nucleotide polymorphisms: rs36084323 (PD-1.1), rs11568821 (PD-1.3), rs2227981 (PD-1.5), and rs2227982 (PD-1.9). The study revealed that the polymorphic variations could be rather disease-modifying than MS risk factors (87).
The relative expression of PD-1 and PD-L1 in the PBMCs of MS patients seems to be significantly decreased compared to healthy donors (88). Javan et al. showed a general reduction in the expression of inhibitory receptors like PD-1, CTLA-4, and TIM-3 in the PBMCs of MS patients, particularly for PD-1 (89). Moreover, after treatment with autologous hematopoietic stem cell transplant in MS patients, Arruda et al. observed a temporary increase in the number of regulatory T cells and PD-1- expressing CD8+ T cells (90). The expansion of CD8+PD-1+ T and CD19+PD-1+ B cells was associated with better clinical outcomes. Interferon-beta, a primary immunomodulatory treatment for MS, enhances PD-L1 expression in vitro as well as in vivo on APCs (91). Koto et al. highlighted differences in the presence of circulating CD8+ PD-1+ T cells according to the stage of the disease. In fact, in the disease remission state, CD8+ PD-1+ T cells were decreased in the peripheral blood of patients with MS and resolved in patients treated with IFN-β treatment who showed immune-regulatory cytokine interleukin (IL)-10 expression. On the other hand, CD8+ PD-1+ T cells were enriched in the CSF of MS patients, which predicted a good response to subsequent IV steroid therapy (92).
Regarding the neuropathological analysis of post-mortem brain tissues, Pittet et al. showed that PD-L1 is largely expressed in MS lesions compared to controls and that it is colocalized with astrocytes or microglia/macrophage markers. On the contrary, PD-L2 expression was notably reduced on brain endothelial cells of MS brains, while being easily detectable in controls. In this case, only a small number of infiltrating CD8+ T lymphocytes in the lesions expressed PD-1 (93). One possible explanation is that during MS pathogenesis, the inflamed CNS attempts to protect itself against active T lymphocytes through the expression of PD-L1. However, this process is not effective since the majority of CD8+ T infiltrating lymphocytes lack PD-1 and are insensitive to PD-L1/L2 (93). On a different note, van Nierop et al. reported that post-mortem brains of patients with an advanced disease contained a high frequency of CD8+ T cells that expressed both co-inhibitory (TIM-3 and PD-1) and co-stimulatory (ICOS) T-cell receptors (94).
The usage of checkpoint inhibitors in clinics (such as ipilimumab, an anti-CTLA-4 antibody) was associated with MS development and an increase in MS activity (95). Other checkpoint inhibitors like nivolumab, ipilimumab, pembrolizumab, and atezolizumab were associated with MS relapse (80, 96). A recent meta-analysis described a rapid progression of MS in 14 patients with MS and concomitant immunotherapy (97). Gerdes et al., with quantitative NGS, showed that distinct clonal expansions of CD4+ and CD8+ T cells in the melanoma and CSF were found during ipilimumab treatment, and concomitant MS activity permitted conversion of RIS to MS (97). These data suggest that the protective antitumor response could be associated with inadvertent anti-CNS autoimmune response toward different antigens and MS reactivation (91).
PD-1 and Alzheimer's Disease
Alzheimer's disease is the most common cause of dementia in humans and is characterized by a decline in cognitive decline and neuronal loss. AD pathologies are characterized by two conditions: β-amyloid (Aβ)-containing extracellular plaques and tau-containing intracellular neurofibrillary tangles (98). The mechanism that underlines the deposition of β-amyloid and TAU protein remains indecipherable, and the immune system may play an essential role in the pathological process (99). Recent evidence suggests that systemic immunity should be boosted, rather than suppressed, to promote brain repair through an immune-dependent cascade. In this context, the PD-1 pathway has been proposed as a possible target therapy for Alzheimer's disease (100). In mouse models of AD, trafficking of blood-borne myeloid cells (monocyte-derived macrophages) to the CNS was shown to be neuroprotective. The expression of both PD-1 on T cells and PD-L1 on monocytes and macrophages decreases significantly in AD patients and patients with mild cognitive impairment compared to age and sex-matched healthy controls (101). Baruch et al. demonstrated that PD-1 blockade evokes a systemic IFN-γ-dependent immune response that enables the mobilization of monocyte-derived macrophages into the brain (100). This process is reminiscent of tissue-specific immune surveillance induced by ICI blockade in cancer therapy. Moreover, PD-1 signaling impairments inhibit IL-10 production, suggesting that positive PD-1 signaling increases IL-10 production. IL-10, in turn, can reduce inflammatory responses and ameliorate AD pathology as demonstrated in animal models (102).
Besides modulating peripheral blood cells, this route may also influence the resident cells of CNS like microglia and astrocytes. Recent work also showed that PD-L1 was increased in the CSF of AD patients (6). PD-L1 expression in astrocytes and PD-1 expression in microglia are close to amyloid plaques in AD patients and AD animal models. Kummer et al. demonstrated that PD-1/PD-L1 signaling is an important factor in Ab phagocytosis, with PD-1 knockout resulting in increased Ab levels, amyloid plaques, and cognitive deficits in APP/PS1 mice together with an inflammatory response in PD-1-deficient microglia (103). Instead, Latta-Mahieu et al. showed that inhibition of PD-1 checkpoint signaling in some different animal models by itself is not sufficient to reduce amyloid pathology (104). In conclusion, the PD-1/PD-L1 axis is a promising target for AD treatment but will need to be further examined to translate the data from animal models to clinical trials.
Discussion
In basic research, the PD-1/PD-L axis has been identified as being of primary importance to immunosurveillance. Studies in preclinical models suggest that this axis plays an important role in different pathological conditions involving the CNS, such as viral encephalitis, brain tumors, autoimmune disorders, and dementia. In most cases, the authors describe an upregulation of PD-1 or PD-Ls during the pathological condition. PD-L1 and PD-L2 seem to be expressed and upregulated mainly on myeloid-derived cells, namely, resident microglial cells and infiltrating macrophages and neutrophils. Conversely, the receptor PD-1 is predominantly found on T and B lymphoid cells, although there is some evidence that it can be upregulated also on activated microglia and macrophages. Under the current understanding of the PD-1/PD-L1 axis in the CNS, its role cannot be described uniquely as protective or pathogenic. In most cases, the upregulation of PD-L1 or PD-L2 helps in slowing down and limiting the inflammatory process, suggesting a protective mechanism (40, 73). However, the use of PD-L1 blocking antibodies might have beneficial results in other contexts, such as in a tau-driven disease model resembling Alzheimer's disease, as described by Schwartz's group (59). Moreover, in the first attempt at a therapeutic approach, Hirata et al. (105) transferred genetically modified dendritic cells presenting MOG peptide in the context of MHC class II and overexpressing PD-L1 before the EAE induction. This treatment reduced T-cell response to MOG, cell infiltration into the spinal cord, and ultimately the severity of the disease. Moreover, the administration of αPD-1 antibody that possesses selective toxicity both in vitro and in vivo for PD-1+ cells restored mobility in mice that were paralyzed by EAE (37, 106). Most of the current knowledge about neurological pathologies is based on studies of mice. Since murine models cannot reliably reproduce the complexity of pathologies observed in the central nervous system, it is widely recognized that this is a limitation in the understanding of neurological disorders. At the moment, only a few studies focused on neurological patients and with controversial results (89, 107).
From the presented results, the PD-1/PD-L axis appears to be regulating the immune response but is not involved in determining the disease or in causing exacerbation. Indeed, inhibition of this axis increases the severity of neuroinflammation, which occurs as a side effect of PD-1 axis inhibition in cancer (Figure 2A). Most results, however, rely on mouse models of acute inflammation and indicate the upregulation of the PD-1/PD-L axis as a counteracting mechanism trying to re-establish homeostasis. Indeed, anti-migratory therapies, diminishing the number of blood-derived CSN-infiltrating cells, are very efficacious in MS. This suggests that inhibitory checkpoints, including the PD-1 axis, can take care of the few residual inflammatory cells still infiltrating the CNS. On the other hand, if the PD-1/PD-L axis fails in the long term, its contribution to chronic CNS inflammation leading to neurodegeneration is not currently known (Figure 2B). The latter hypothesis, if confirmed, would highlight a potential therapeutic strategy in fostering, supporting, and reinforcing this axis to treat chronic neuroinflammation.
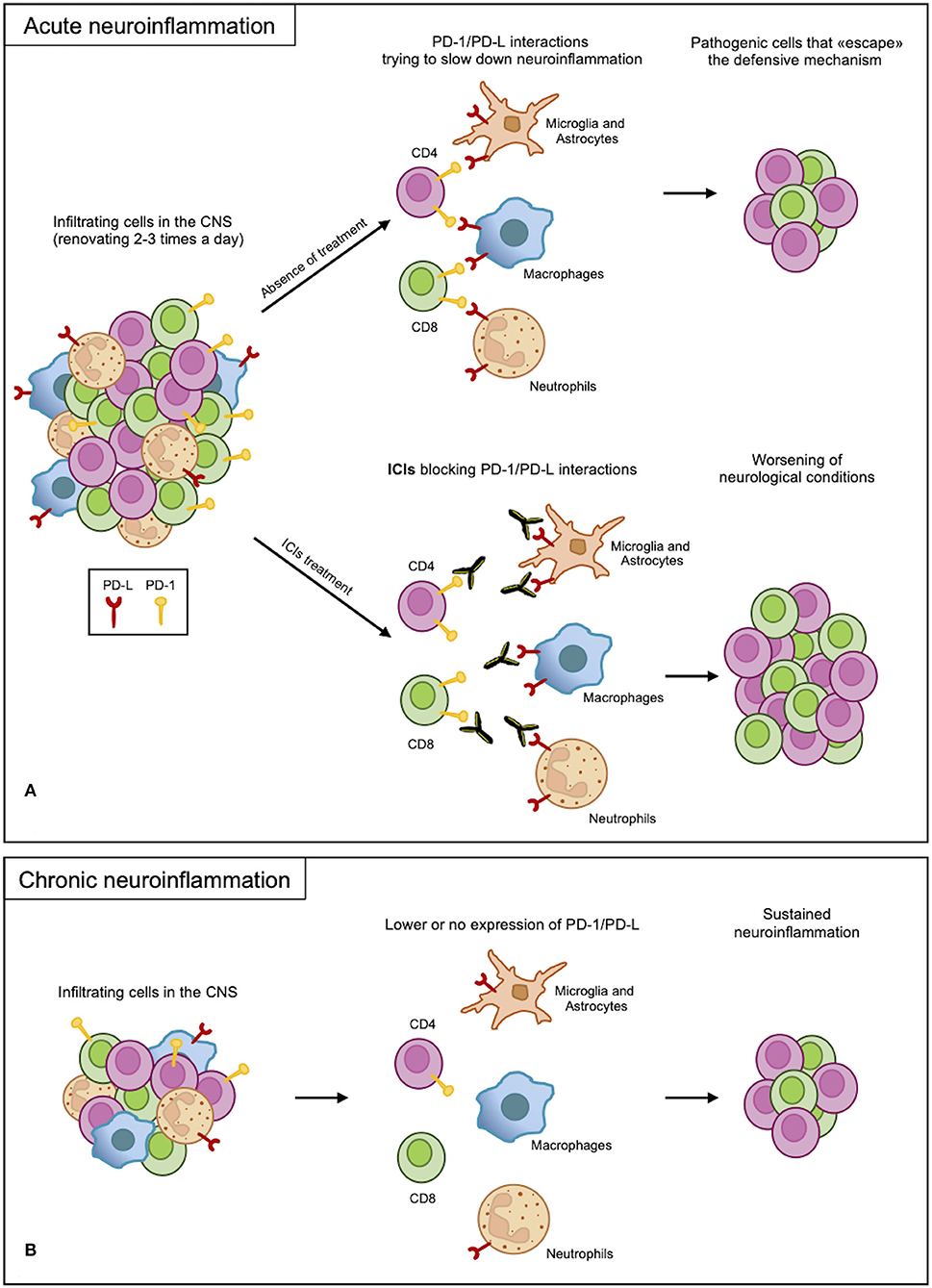
Figure 2. PD-1/PD-L axis in neuroinflammation. (A) In acute neuroinflammation, CNS infiltrates are renewed 2-3 times a day. Infiltrating myeloid and resident cells expressing PD-Ls attempt to slow down the inflammatory process through interactions with PD-1 expressed on the lymphoid cells. Due to an overabundance of cells, some pathogenic cells escape this protective mechanism, causing damage. However, blocking PD-1/PD-L interactions with ICI treatments remove this defense, leading to the worsening of the inflammation. (B) In chronic inflammation, the PD-1/PD-L axis may not function properly as a result of low or no expression of the receptor and ligand; therefore, the sustained inflammation continues.
This underlines the need for further investigations to better understand the role of the PD-1 axis during neuroinflammatory disorders.
Author Contributions
SMan, MO, SMas, AM, and AF wrote the review and correct the text. RF supervised the writing of the review and correct the final form of the article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. (2021) 23:1–11. doi: 10.1208/s12248-021-00574-0
2. Darnell EP, Mooradian MJ, Baruch EN, Yilmaz M, Reynolds KL. Immune-related adverse events (irAEs): diagnosis, management, and clinical pearls. Curr Oncol Rep. (2020) 22:1–11. doi: 10.1007/s11912-020-0897-9
3. Kao JC, Liao B, Markovic SN, Klein CJ, Naddaf E, Staff NP, et al. Neurological complications associated with anti–programmed death 1 (PD-1) antibodies. JAMA Neurol. (2017) 74:1216–22. doi: 10.1001/jamaneurol.2017.1912
4. Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer. (2016) 60:210–25. doi: 10.1016/j.ejca.2016.02.024
5. Zhao S, Li F, Leak RK, Chen J, Hu X. Regulation of neuroinflammation through programed death-1/programed death ligand signaling in neurological disorders. Front Cell Neurosci. (2014) 8:271. doi: 10.3389/fncel.2014.00271
6. Zhao J, Roberts A, Wang Z, Savage J, Ji R-R. Emerging role of PD-1 in the central nervous system and brain diseases. Neurosci Bull. (2021) 37:1188–202. doi: 10.1007/s12264-021-00683-y
7. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
8. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
9. Philips EA, Garcia-España A, Tocheva AS, Ahearn IM, Adam KR, Pan R, et al. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J Biol Chem. (2020) 295:4372–80. doi: 10.1074/jbc.AC119.011747
10. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
11. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. (2003) 33:2706–16. doi: 10.1002/eji.200324228
12. Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin cancer Res. (2005) 11:2947–53. doi: 10.1158/1078-0432.CCR-04-1469
13. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
14. Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1–induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. (2010) 16:452. doi: 10.1038/nm.2106
15. Salama AD, Chitnis T, Imitola J, Ansari MJI, Akiba H, Tushima F, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. (2003) 198:71–8. doi: 10.1084/jem.20022119
16. Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, et al. Intestinal tolerance is converted to autoimmune enteritis upon PD-1 ligand blockade. J Immunol. (2009) 182:2102–12. doi: 10.4049/jimmunol.0802769
17. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR–induced stop signal. Nat Immunol. (2009) 10:1185. doi: 10.1038/ni.1790
18. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
19. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. (2005) 102:11823–8. doi: 10.1073/pnas.0505497102
20. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
21. Chauhan P, Lokensgard JR. Glial cell expression of PD-L1. Int J Mol Sci. (2019) 20:1677. doi: 10.3390/ijms20071677
22. Magnus T, Schreiner B, Korn T, Jack C, Guo H, Antel J, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci. (2005) 25:2537–46. doi: 10.1523/JNEUROSCI.4794-04.2005
23. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. (2014) 211:943–59. doi: 10.1084/jem.20130790
24. Shwetank, Frost EL, Mockus TE, Ren HM, Toprak M, Lauver MD, et al. PD-1 dynamically regulates inflammation and development of brain-resident memory CD8 T cells during persistent viral encephalitis. Front Immunol. (2019) 10:1–14. doi: 10.3389/fimmu.2019.00783
25. Schachtele SJ, Hu S, Sheng WS, Mutnal MB, Lokensgard JR. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia. (2014) 62:1582–94. doi: 10.1002/glia.22701
26. Prasad S, Hu S, Sheng WS, Chauhan P, Singh A, Lokensgard JR. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J Neuroinflammation. (2017) 14:1–13. doi: 10.1186/s12974-017-0860-3
27. Chauhan P, Sheng WS, Hu S, Prasad S, Lokensgard JR. Nitrosative damage during retrovirus infection-induced neuropathic pain. J Neuroinflammation. (2018) 15:66. doi: 10.1186/s12974-018-1107-7
28. Mair I, Besusso D, Saul L, Patel SD, Ravindran R, McPherson RC, et al. PD-1 expression is upregulated on adapted T cells in experimental autoimmune encephalomyelitis but is not required to maintain a hyporesponsive state. Eur J Immunol. (2019) 49:112–20. doi: 10.1002/eji.201847868
29. White MPJ, Webster G, Leonard F, La Flamme AC. Innate IFN-γ ameliorates experimental autoimmune encephalomyelitis and promotes myeloid expansion and PDL-1 expression. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-017-18543-z
30. Khorooshi R, Marczynska J, Dieu RS, Wais V, Hansen CR, Kavan S, et al. Innate signaling within the central nervous system recruits protective neutrophils. Acta Neuropathol Commun. (2020) 8:1–13. doi: 10.1186/s40478-019-0876-2
31. Hu J, He H, Yang Z, Zhu G, Kang L, Jing X, et al. Programmed death ligand-1 on microglia regulates Th1 differentiation via nitric oxide in experimental autoimmune encephalomyelitis. Neurosci Bull. (2016) 32:70–82. doi: 10.1007/s12264-015-0010-9
32. Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol. (2012) 188:1136–46. doi: 10.4049/jimmunol.1101816
33. Cheng Y-Y, Chen B-Y, Bian G-L, Ding Y-X, Chen L-W. Programmed death-1 deficiency aggravates motor dysfunction in MPTP model of parkinson's disease by inducing microglial activation and neuroinflammation in mice. Mol Neurobiol. (2022) 59:2642–55. doi: 10.1007/s12035-022-02758-x
34. Norden DM, Bethea JR, Jiang J. Impaired CD8 T cell antiviral immunity following acute spinal cord injury. J Neuroinflammation. (2018) 15:149. doi: 10.1186/s12974-018-1191-8
35. He X, Lin S, Yang L, Tan P, Ma P, Qiu P, et al. Programmed death protein 1 is essential for maintaining the anti-inflammatory function of infiltrating regulatory T cells in a murine spinal cord injury model. J Neuroimmunol. (2021) 354:577546. doi: 10.1016/j.jneuroim.2021.577546
36. He H, Zhou Y, Zhou Y, Zhuang J, He X, Wang S, et al. Dexmedetomidine mitigates microglia-mediated neuroinflammation through upregulation of programmed cell death protein 1 in a rat spinal cord injury model. J Neurotrauma. (2018) 35:2591–603. doi: 10.1089/neu.2017.5625
37. Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation. (2018) 15:1–13. doi: 10.1186/s12974-018-1330-2
38. Miyasato Y, Takashima Y, Takeya H, Yano H, Hayano A, Nakagawa T, et al. The expression of PD-1 ligands and IDO1 by macrophage/microglia in primary central nervous system lymphoma. J Clin Exp Hematop. (2018) 58:95–101. doi: 10.3960/jslrt.18001
39. Yuan B, Huang S, Gong S, Wang F, Lin L, Su T, et al. Programmed death (PD)-1 attenuates macrophage activation and brain inflammation via regulation of fibrinogen-like protein 2 (Fgl-2) after intracerebral hemorrhage in mice. Immunol Lett. (2016) 179:114–21. doi: 10.1016/j.imlet.2016.10.001
40. Chen Q, Xu L, Du T, Hou Y, Fan W, Wu Q, et al. Enhanced expression of PD-L1 on microglia after surgical brain injury exerts self-protection from inflammation and promotes neurological repair. Neurochem Res. (2019) 44:2470–81. doi: 10.1007/s11064-019-02864-8
41. Obst J, Mancuso R, Simon E, Gomez-Nicola D. PD-1 deficiency is not sufficient to induce myeloid mobilization to the brain or alter the inflammatory profile during chronic neurodegeneration. Brain Behav Immun. (2018) 73:708–16. doi: 10.1016/j.bbi.2018.08.006
42. Gao X, Li W, Syed F, Yuan F, Li P, Yu Q. PD-L1 signaling in reactive astrocytes counteracts neuroinflammation and ameliorates neuronal damage after traumatic brain injury. J Neuroinflammation. (2022) 19:1–18. doi: 10.1186/s12974-022-02398-x
43. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
44. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
45. Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz J, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. (2013) 210:757–74. doi: 10.1084/jem.20121416
46. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
47. Smolders J, Heutinck KM, Fransen NL, Remmerswaal E, Hombrink P, Ten Berge IJM, et al. Tissue-resident memory T cells populate the human brain. Nat Commun. (2018) 9:1–14. doi: 10.1038/s41467-018-07053-9
48. Hsiao C, Fransen NL, van den Bosch AMR, Brandwijk KIM, Huitinga I, Hamann J, et al. White matter lesions in multiple sclerosis are enriched for CD20dim CD8+ tissue-resident memory T cells. Eur J Immunol. (2021) 51:483–6. doi: 10.1002/eji.202048665
49. Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. (2004) 251:261–8. doi: 10.1007/s00415-004-0348-9
50. Amorim A, De Feo D, Friebel E, Ingelfinger F, Anderfuhren CD, Krishnarajah S, et al. IFNγ and GM-CSF control complementary differentiation programs in the monocyte-to-phagocyte transition during neuroinflammation. Nat Immunol. (2022) 23:217–28. doi: 10.1038/s41590-021-01117-7
51. Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front Immunol. (2015) 6:539. doi: 10.3389/fimmu.2015.00539
52. Willenborg DO, Staykova M, Fordham S, O'Brien N, Linares D. The contribution of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2007) 191:16–25. doi: 10.1016/j.jneuroim.2007.09.007
53. Kulkarni A, Ganesan P, O'Donnell LA. Interferon gamma: influence on neural stem cell function in neurodegenerative and neuroinflammatory disease. Clin Med Insights Pathol. (2016) 9:CPath-S40497. doi: 10.4137/CPath.S40497
54. Croxford AL, Spath S, Becher B. GM-CSF in neuroinflammation: licensing myeloid cells for tissue damage. Trends Immunol. (2015) 36:651–62. doi: 10.1016/j.it.2015.08.004
55. Monaghan KL, Wan ECK. The role of granulocyte-macrophage colony-stimulating factor in murine models of multiple sclerosis. Cells. (2020) 9:611. doi: 10.3390/cells9030611
56. Paré A, Mailhot B, Lévesque SA, Juzwik C, Doss PMIA, Lécuyer M-A, et al. IL-1β enables CNS access to CCR2hi monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc Natl Acad Sci. (2018) 115:E1194–203. doi: 10.1073/pnas.1714948115
57. Lévesque SA, Paré A, Mailhot B, Bellver-Landete V, Kébir H, Lécuyer M-A, et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med. (2016) 213:929–49. doi: 10.1084/jem.20151437
58. White M, Webster G, O'Sullivan D, Stone S, La Flamme AC. Targeting innate receptors with MIS416 reshapes Th responses and suppresses CNS disease in a mouse model of multiple sclerosis. PLoS ONE. (2014) 9:e87712. doi: 10.1371/journal.pone.0087712
59. Rosenzweig N, Dvir-Szternfeld R, Tsitsou-Kampeli A, Keren-Shaul H, Ben-Yehuda H, Weill-Raynal P, et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun. (2019) 10:1–15. doi: 10.1038/s41467-019-08352-5
60. Dvir-Szternfeld R, Castellani G, Arad M, Cahalon L, Colaiuta SP, Keren-Shaul H, et al. Alzheimer's disease modification mediated by bone marrow-derived macrophages via a TREM2-independent pathway in mouse model of amyloidosis. Nat Aging. (2022) 2:60–73. doi: 10.1038/s43587-021-00149-w
61. Woodberry T, Bouffler SE, Wilson AS, Buckland RL, Brüstle A. The emerging role of neutrophil granulocytes in multiple sclerosis. J Clin Med. (2018) 7:511. doi: 10.3390/jcm7120511
62. Stock AJ, Kasus-Jacobi A, Pereira HA. The role of neutrophil granule proteins in neuroinflammation and Alzheimer's disease. J Neuroinflammation. (2018) 15:1–15. doi: 10.1186/s12974-018-1284-4
63. Wanrooy BJ, Wen SW, Wong CHY. Dynamic roles of neutrophils in post-stroke neuroinflammation. Immunol Cell Biol. (2021) 99:924–35. doi: 10.1111/imcb.12463
64. Ruhnau J, Schulze J, Dressel A, Vogelgesang A. Thrombosis, neuroinflammation, and poststroke infection: the multifaceted role of neutrophils in stroke. J Immunol Res. (2017) 2017:5140679. doi: 10.1155/2017/5140679
65. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD. Neutrophils: new insights and open questions. Sci Immunol. (2018) 3:eaat4579. doi: 10.1126/sciimmunol.aat4579
66. De Bondt M, Hellings N, Opdenakker G, Struyf S. Neutrophils: underestimated players in the pathogenesis of Multiple Sclerosis (MS). Int J Mol Sci. (2020) 21:4558. doi: 10.3390/ijms21124558
67. Bisgaard AK, Pihl-Jensen G, Frederiksen JL. The neutrophil-to-lymphocyte ratio as disease actvity marker in multiple sclerosis and optic neuritis. Mult Scler Relat Disord. (2017) 18:213–7. doi: 10.1016/j.msard.2017.10.009
68. Wu F, Cao W, Yang Y, Liu A. Extensive infiltration of neutrophils in the acute phase of experimental autoimmune encephalomyelitis in C57BL/6 mice. Histochem Cell Biol. (2010) 133:313–22. doi: 10.1007/s00418-009-0673-2
69. Kroenke MA, Chensue SW, Segal BM. EAE mediated by a non-IFN-γ/non-IL-17 pathway. Eur J Immunol. (2010) 40:2340–8. doi: 10.1002/eji.201040489
70. Owens T, Benmamar-Badel A, Wlodarczyk A, Marczynska J, Mørch MT, Dubik M, et al. Protective roles for myeloid cells in neuroinflammation. Scand J Immunol. (2020) 92:e12963. doi: 10.1111/sji.12963
71. Melero-Jerez C, Suardíaz M, Lebrón-Galán R, Marín-Bañasco C, Oliver-Martos B, Machín-Díaz I, et al. The presence and suppressive activity of myeloid-derived suppressor cells are potentiated after interferon-β treatment in a murine model of multiple sclerosis. Neurobiol Dis. (2019) 127:13–31. doi: 10.1016/j.nbd.2019.02.014
72. Lokensgard JR, Schachtele SJ, Mutnal MB, Sheng WS, Prasad S, Hu S. Chronic reactive gliosis following regulatory T cell depletion during acute MCMV encephalitis. Glia. (2015) 63:1982–96. doi: 10.1002/glia.22868
73. Zhang X, Lund H, Mia S, Parsa R, Harris RA. Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia. (2014) 62:804–17. doi: 10.1002/glia.22643
74. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. (2017) 32:253–67. doi: 10.1016/j.ccell.2017.07.006
75. Valiente M, Priego N. The potential of astrocytes as immune modulators in brain tumors. Front Immunol. (2019) 10:1314. doi: 10.3389/fimmu.2019.01314
76. Lipp M, Brandt C, Dehghani F, Kwidzinski E, Bechmann I. PD-L1 (B7-H1) regulation in zones of axonal degeneration. Neurosci Lett. (2007) 425:156–61. doi: 10.1016/j.neulet.2007.07.053
77. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
78. De Velasco G, Je Y, Bossé D, Awad MM, Ott PA, Moreira RB, et al. Comprehensive meta-analysis of key immune-related adverse events from CTLA-4 and PD-1/PD-L1 inhibitors in cancer patients. Cancer Immunol Res. (2017) 5:312–8. doi: 10.1158/2326-6066.CIR-16-0237
79. Fellner A, Makranz C, Lotem M, Bokstein F, Taliansky A, Rosenberg S, et al. Neurologic complications of immune checkpoint inhibitors. J Neurooncol. (2018) 137:601–9. doi: 10.1007/s11060-018-2752-5
80. Garcia CR, Jayswal R, Adams V, Anthony LB, Villano JL. Multiple sclerosis outcomes after cancer immunotherapy. Clin Transl Oncol. (2019) 21:1336–42. doi: 10.1007/s12094-019-02060-8
81. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
82. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J Clin Oncol Off J Am Soc Clin Oncol. (2018) 36:1714. doi: 10.1200/JCO.2017.77.6385
83. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. (2016) 2:1346–53. doi: 10.1001/jamaoncol.2016.1051
84. Cuzzubbo S, Javeri F, Tissier M, Roumi A, Barlog C, Doridam J, et al. Neurological adverse events associated with immune checkpoint inhibitors: review of the literature. Eur J Cancer. (2017) 73:1–8. doi: 10.1016/j.ejca.2016.12.001
85. Diamanti L, Picca A, Bini P, Gastaldi M, Alfonsi E, Pichiecchio A, et al. Characterization and management of neurological adverse events during immune-checkpoint inhibitors treatment: an Italian multicentric experience. Neurol Sci. (2022) 43:2031–41. doi: 10.1007/s10072-021-05561-z
86. Kroner A, Schwab N, Ip CW, Ortler S, Göbel K, Nave K-A, et al. Accelerated course of experimental autoimmune encephalomyelitis in PD-1-deficient central nervous system myelin mutants. Am J Pathol. (2009) 174:2290–9. doi: 10.2353/ajpath.2009.081012
87. Pawlak-Adamska E, Nowak O, Karabon L, Pokryszko-Dragan A, Partyka A, Tomkiewicz A, et al. PD-1 gene polymorphic variation is linked with first symptom of disease and severity of relapsing-remitting form of MS. J Neuroimmunol. (2017) 305:115–27. doi: 10.1016/j.jneuroim.2017.02.006
88. Javan MR, Aslani S, Zamani MR, Rostamnejad J, Asadi M, Farhoodi M, et al. Downregulation of immunosuppressive molecules, PD-1 and PD-L1 but not PD-L2, in the patients with multiple sclerosis. Iran J Allergy Asthma Immunol. (2016) 15:296–302.
89. Mohammadzadeh A, Rad IA, Ahmadi-Salmasi B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J Neuroimmunol. (2018) 323:105–8. doi: 10.1016/j.jneuroim.2018.08.004
90. Arruda LCM, de Azevedo JTC, de Oliveira GL V, Scortegagna GT, Rodrigues ES, Palma PVB, et al. Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin Immunol. (2016) 169:47–57. doi: 10.1016/j.clim.2016.06.005
91. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung H-P, Weller M, et al. Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol. (2004) 155:172–82. doi: 10.1016/j.jneuroim.2004.06.013
92. Koto S, Chihara N, Akatani R, Nakano H, Hara A, Sekiguchi K, et al. Transcription factor c-maf promotes immunoregulation of programmed cell death 1-expressed CD8+ T cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflammation. (2022) 9: e1166. doi: 10.1212/NXI.0000000000001166
93. Pittet CL, Newcombe J, Antel JP, Arbour N. The majority of infiltrating CD8 T lymphocytes in multiple sclerosis lesions is insensitive to enhanced PD-L1 levels on CNS cells. Glia. (2011) 59:841–56. doi: 10.1002/glia.21158
94. van Nierop GP, van Luijn MM, Michels SS, Melief M-J, Janssen M, Langerak AW, et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. (2017) 134:383–401. doi: 10.1007/s00401-017-1744-4
95. Gettings EJ, Hackett CT, Scott TF. Severe relapse in a multiple sclerosis patient associated with ipilimumab treatment of melanoma. Mult Scler J. (2015) 21:670. doi: 10.1177/1352458514549403
96. Lu BY, Isitan C, Mahajan A, Chiang V, Huttner A, Mitzner JR, et al. Intracranial complications from immune checkpoint therapy in a patient with NSCLC and multiple sclerosis: case report. JTO Clin Res Reports. (2021) 2:100183. doi: 10.1016/j.jtocrr.2021.100183
97. Gerdes LA, Held K, Beltrán E, Berking C, Prinz JC, Junker A, et al. CTLA4 as immunological checkpoint in the development of multiple sclerosis. Ann Neurol. (2016) 80:294–300. doi: 10.1002/ana.24715
98. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer's disease. Nat Rev Dis Prim. (2021) 7:1–21. doi: 10.1038/s41572-021-00269-y
99. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimer's Dement Transl Res Clin Interv. (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
100. Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. (2016) 22:135–7. doi: 10.1038/nm.4022
101. Rossini PM, Rossi S, Babiloni C, Polich J. Clinical neurophysiology of aging brain: from normal aging to neurodegeneration. Prog Neurobiol. (2007) 83:375–400. doi: 10.1016/j.pneurobio.2007.07.010
102. Koronyo-Hamaoui M, Ko MK, Koronyo Y, Azoulay D, Seksenyan A, Kunis G, et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J Neurochem. (2009) 111:1409–24. doi: 10.1111/j.1471-4159.2009.06402.x
103. Kummer MP, Ising C, Kummer C, Sarlus H, Griep A, Vieira-Saecker A, et al. Microglial PD-1 stimulation by astrocytic PD-L1 suppresses neuroinflammation and Alzheimer's disease pathology. EMBO J. (2021) 40:e108662. doi: 10.15252/embj.2021108662
104. Latta-Mahieu M, Elmer B, Bretteville A, Wang Y, Lopez-Grancha M, Goniot P, et al. Systemic immune-checkpoint blockade with anti-PD1 antibodies does not alter cerebral amyloid-β burden in several amyloid transgenic mouse models. Glia. (2018) 66:492–504. doi: 10.1002/glia.23260
105. Hirata S, Senju S, Matsuyoshi H, Fukuma D, Uemura Y, Nishimura Y. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programmed death-1 ligand. J Immunol. (2005) 174:1888–97. doi: 10.4049/jimmunol.174.4.1888
106. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of PD-1-positive cells ameliorates autoimmune disease. Nat Biomed Eng. (2019) 3:292–305. doi: 10.1038/s41551-019-0360-0
Keywords: PD-1, PD-L1, PD-L2, neuroinflammation, multiple sclerosis, Alzheimer's disease
Citation: Manenti S, Orrico M, Masciocchi S, Mandelli A, Finardi A and Furlan R (2022) PD-1/PD-L Axis in Neuroinflammation: New Insights. Front. Neurol. 13:877936. doi: 10.3389/fneur.2022.877936
Received: 17 February 2022; Accepted: 29 April 2022;
Published: 09 June 2022.
Edited by:
Daniela Carnevale, Sapienza University of Rome, ItalyReviewed by:
Norio Chihara, Kobe University, JapanZorica Stojic-Vukanic, University of Belgrade, Serbia
Copyright © 2022 Manenti, Orrico, Masciocchi, Mandelli, Finardi and Furlan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanna Manenti, manenti.susanna@hsr.it