 Giuliana Capece
Giuliana Capece Mauro Ceroni
Mauro Ceroni Enrico Alfonsi3
Enrico Alfonsi3 Ilaria Palmieri
Ilaria Palmieri Cristina Cereda
Cristina Cereda Luca Diamanti
Luca Diamanti- 1Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy
- 2General Neurology Unit, IRCCS Mondino Foundation, Pavia, Italy
- 3Clinical Neurophysiology Unit, IRCCS Mondino Foundation, Pavia, Italy
- 4Genomic and Post-genomic Centre, IRCCS Mondino Foundation, Pavia, Italy
- 5Department of Molecular Medicine, University of Pavia, Pavia, Italy
- 6Neuro-Oncology Unit, IRCCS Mondino Foundation, Pavia, Italy
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease affecting motor neurons. Although its etiology is still unknown, many genes have been found to be implicated in ALS pathogenesis. The Cu/Zn superoxide dismutase (SOD1) gene was the first to be identified. Currently, more than 230 mutations in the SOD1 gene have been reported. p.D90A (p. Asp90Ala) is the most common SOD1 mutation worldwide. It shows both autosomal and recessive inheritance in different populations. To date, five Italian patients with the heterozygous p.D90A mutation have been reported. None of them complained of laryngological symptoms as the initial manifestation of ALS, although they had atypical clinical features. We describe a long-survival patient carrying heterozygous p.D90A mutation who presented with severe laryngospasm due to bilateral vocal cord paralysis. We suggest that genetic analysis may help to diagnose ALS with insidious onset like hoarseness, laryngospasm, and other type of voice disturbances.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disorder that affects the upper and lower motor neurons in the spinal cord, brainstem, and motor cortex (1). Its etiology is still unknown, but it is now accepted to be based on a complex interplay between environmental factors and genetic background (1). Most cases of ALS are sporadic (SALS), while about 10% of cases are familial (FALS), even though SALS and FALS are clinically identical and have both a genetic basis (1). In 1993, Rosen et al. discovered the first ALS-associated gene, SOD1, that is located on chromosome 21q22.11 and encodes a Cu/Zn-binding superoxide dismutase (2–4). To date, more than 230 mutations have been reported to be ALS-associated, but it is still controversial whatever all of them are disease-causative (3, 4).
Currently, p.D90A (p.Asp90Ala) is the most common SOD1 mutation (3, 4) and is inherited as both a recessive and dominant trait in different populations (5). In northern Scandinavia, p.D90A heterozygous carriers are unaffected, while they developed ALS in Belgium (5).
In Italy, five patients carrying the heterozygous p.D90A mutation have been reported (6–9).
We herein report the heterozygous p.D90A mutation in a long-survival patient with ALS and laryngospasm as the initial manifestation.
Case Presentation
A 58-year-old male was admitted to our department in May 2016 because of a 12-year history of progressive muscle weakness in the upper limbs. Family history for neurodegenerative diseases was negative. He was diagnosed with Gilbert syndrome and mild tricuspid valve regurgitation and he suffered from tachyarrhythmia and dyspepsia due to esophagitis. He presented with hypophonia due to a bilateral vocal fold paralysis occurred in 1999. Electromyography (EMG) showed a bilateral axonal neuropathy involving the superior and recurrent laryngeal nerves, especially on the left. As laryngeal spasms caused episodes of respiratory failure, he underwent a left laser posterior cordotomy. He began to suffer from low back pain after an accidental fall due to a disc herniation at L5-S1. Furthermore, mild postural tremor affecting the hands appeared and neurological examination showed muscle weakness in the proximal segments of the upper limbs. He was initially diagnosed with spinobulbar atrophy. Genetic tests for Kennedy's disease were negative. Furthermore, Brown-Vialetto-Van Laere syndrome was ruled out ex juvantibus by administering high-dose riboflavin and then by molecular analysis.
At first neurological examination, he had bilateral muscle weakness and hypotrophy of the proximal segments of the upper limbs, especially of the deltoids (Medical Research Council, MRC 3+ on the right side, 4− on the left side), of the both biceps brachii (MRC 3+) and triceps brachii (3−). Muscle strength was preserved in the distal segments of the arms. Triceps reflex was absent, while the other tendon reflexes of the upper limbs were normal. Lower limbs show no pathological signs except bilaterally decreased ankle jerk reflexes. Flexor plantar responses were assessed bilaterally. No fasciculations were detected. Cranial nerves were normal and there are no signs of sensory impairment. Magnetic Resonance Imaging (MRI) of the chest and cervical spine showed bilateral changes in the muscles of the rotator cuff, especially on the right, depending on a chronic neurogenic damage, and degeneration of the inferior roots of the brachial plexus. EMG and electroneuronography (ENoG) revealed diffuse motor axonopathy in spinal and bulbar regions with fasciculation potentials and denervation activity at rest, especially in the right deltoid, in the left thyroarytenoid muscle and in the thoracic paraspinal muscles on both sides. Sensory evoked potentials (SEPs) were normal. Routine laboratory investigations showed high levels of serum creatine kinase (359 UI/l; n.v.: 39–308). A diagnosis of motor neuron disease with predominant lower motor neuron phenotype was made.
The patient's disease progression was monitored at 3-month intervals by testing muscle strength and performing ALSFRS-R (ALS-Functional Rating Scale Revised). Disease course was slowly progressive and he was clinically stable (Figure 1).
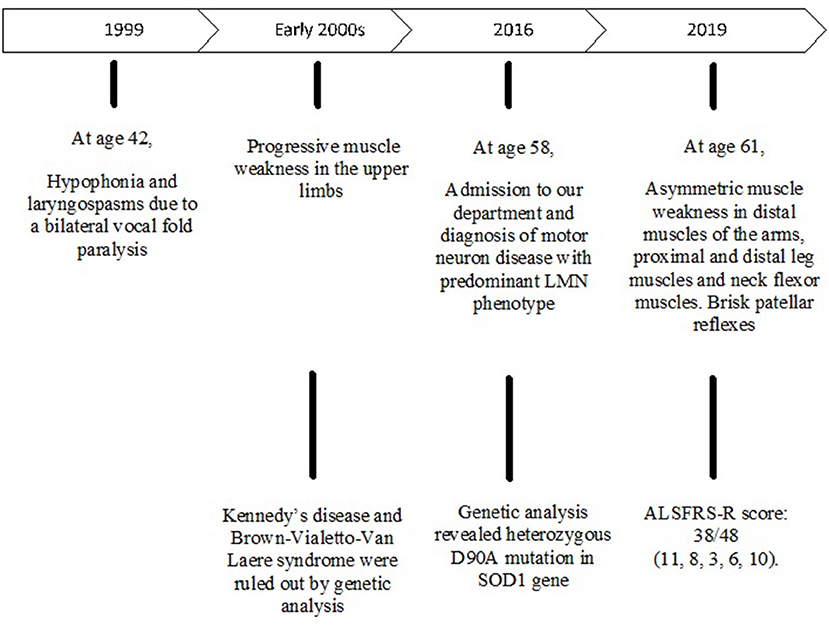
Figure 1. Sanger sequencing electropherogram; “*” shows the variant c.272A>C in the exon 4 of SOD1 in the patient.
After a 3-year follow up, clinical examination showed weakness of neck flexor muscles (MRC 4), of proximal segments of the upper limbs (MRC 3–4), of the opponens pollicis muscle bilaterally (MRC 4), of the left iliopsoas, of the left peroneal muscles and of the left plantar flexors muscles (MRC 4, 5). Right plantar response was indifferent, while patellar reflexes spread to adductor muscles of the thigh. He complained of dysphagia and weight loss. According to the revised El Escorial criteria, a diagnosis of probable ALS-laboratory supported was made. The ALSFRS-R score was 38/48 (4, 7, 9–11).
Genetic Analysis
After obtaining a written informed consent, Next Generation Sequencing (NGS) analysis was performed, using a customized panel of 174 genes related to neurodegenerative diseases as described in the Supplementary Material. We identified the heterozygous variant g.12669A>C, c.272A>C in the exon 4 of the SOD1 gene resulting in the amino acid change p.Asp90Ala (Figure 2). The c.272A>C mutation was then confirmed by Sanger sequencing.
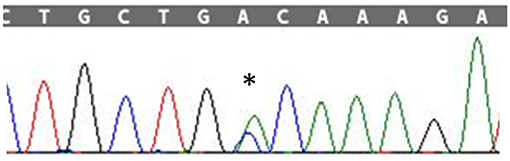
Figure 2. Timeline with relevant data from the episode of care.
Discussion
In this study we described an ALS patient presenting with laryngospasm as disease onset and a predominant lower motor neuron phenotype involving the proximal segments of the upper limbs. The clinical course was slowly progressive, according to previous evidence of longer survival in patients with a lower motor neuron phenotype (12).
Genetic analysis showed a heterozygous p.D90A mutation of the SOD1 gene.
To date, five Italian patients with the heterozygous p.D90A have been reported (6–9) (Table 1). They had a spinal onset involving lower and/or upper limbs typically with muscle weakness at an average age of 46, 6 years. Neurological examination showed both upper and lower motor neuron signs. Two patients complained of sensory disturbances. The disease onset did not show vocal cord paralysis in any patient except our case (6–9). Although this variant is not associated with a distinct phenotype, Origone et al. concluded that patients carrying the heterozygous p.D90A mutation have atypical clinical features. So, they suggested to perform molecular analysis looking for SOD1 mutations in both SALS and FALS patients with atypical disease onset (8).

Table 1. Clinical features of ALS patients carrying the heterozygous p.D90A-SOD1 mutation in the Italian population.
Although voice disturbances are rare, they are an insidious type of ALS onset (11).
Laryngological symptoms included: hoarseness, dysphonia, hypophonia/aphonia, non-productive cough, and life-threatening conditions as inspiratory stridor and laryngospasm.
Chen and Garrett demonstrated that a significant number of ALS patients with bulbar onset are initially referred to otolaryngologists. One thousand seven hundred fifty-nine patients were evaluated at a voice center from 1998 to 2003: <1% later received a diagnosis of ALS. In contrast, 20% of 220 ALS patients seen at the neurological clinic had bulbar onset and about half of them complained of dysphonia. When dysphonia was found, patients were initially referred to an otolaryngologist rather than a neurologist. Unfortunately, they were often misdiagnosed due to previous misleading diagnoses, dysarthria mistaken for dysphonia, subtle symptoms, and signs of neuromuscular disease overlooked by physicians. Therefore, the authors concluded that ALS diagnosis is a significative challenge in the otolaryngology practice (11).
Vocal cord problems may present with acute dyspnoea due to glottic narrowing or even glottic occlusion (10). In ALS, glottic narrowing has been supposed to depend on two types of vocal cord dysfunction: the supranuclear non-paralytic type causing an overactivity of vocal cord adductors and/or the infranuclear paralytic type consisting in neurogenic atrophy and weakness of the posterior cricoarytenoid muscle (10). Glottic narrowing is clinically symptomatic, resulting in inspiratory stridor, especially at an early disease stage in patients with a good vital capacity (10).
Laryngospasm is defined as a rapid and involuntary closure of the larynx (10). It is well-known that it may occur during intubation/extubation procedures and during disease progression probably due to a combination of gastro-esophageal reflux disease (GORD), aspiration of gastric contents, and dysphagia (10, 19). Diet modifications and pharmacological therapy with prokinetic agents and proton pump inhibitors (PPI) are recommended (19).
Instrumental assessment consists in laryngoscopy and laryngeal electromyography (10).
Changing to an upright position of the trunk, stabilizing the body through the fixation of the arms and slowly breathing are sufficient maneuvers to shorten the spasm episode (19).
Treatment of life-threatening vocal cord disturbances ranges from intubation and cricothyroidotomy to tracheotomy according to patient's advance directives (10).
Among neuromuscular diseases, laryngospasm is common at an early stage of Kennedy disease, while it is essentially a rare type of ALS onset (20).
In addition to our patient, literature reported other cases of ALS patients experiencing laryngospasm as initial manifestation of ALS (10, 11, 20).
It should be emphasized that vocal cord dysfunction occurs also in ALS patients without major bulbar involvement, as in the case of our patient (10, 21).
SALS and FALS patients presenting with laryngological onset has been reported to carry a missense mutation in the SOD1 gene, as shown in Table 2 (13–18).
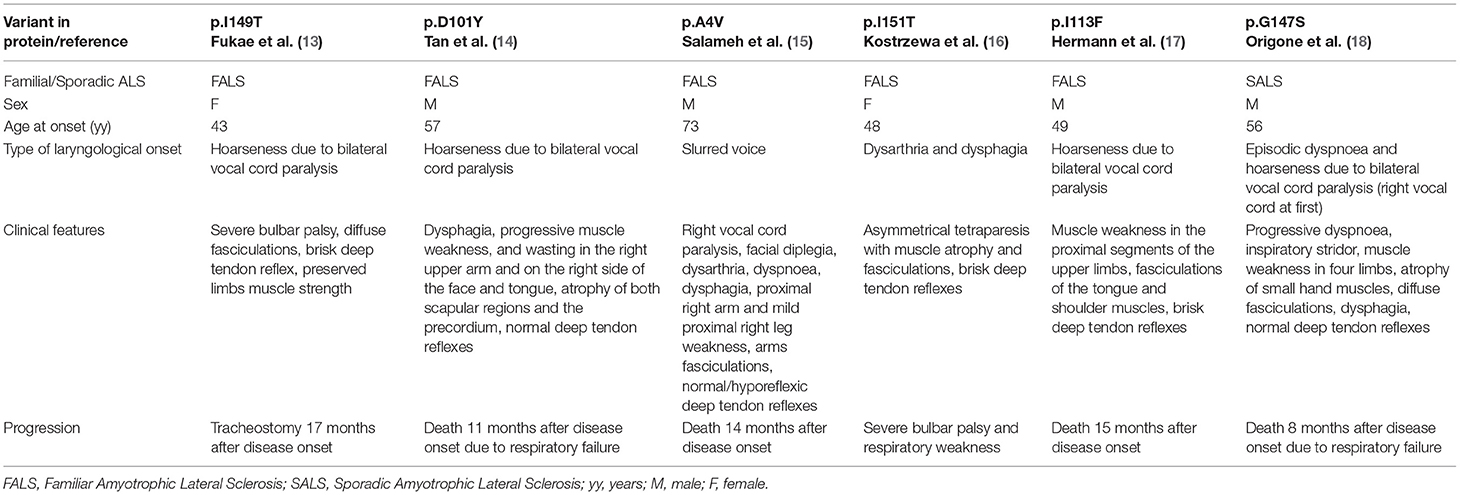
Table 2. Previously reported ALS patients carrying missense SOD1 mutations and presenting with voice disturbance.
Hermann et al. described a patient with hoarseness and muscle weakness in the proximal segments of the upper limbs at onset. As our patient, he had a bilateral compromission of vocal folds and a predominant lower motor neuron phenotype although upper motor neuron signs were present too. However, the disease progression was different from our case and the patient died 15 months after disease onset. Genetic analysis revealed a heterozygous missense mutation c.337 A>T in exon 4 of the SOD1 gene. Authors suggested a pathogenic role for this mutation (17).
Table 2 summarizes other patients affected by motor neuron disease with voice disturbances as disease onset. Most of them presented with hoarseness and had a rapid progressive disease course and none of them carried the same mutation as our case. Some of them showed a predominant lower motor neuron phenotype (14, 15, 18).
Conclusion
ALS onset may be insidious and subtle including laryngological symptoms like hoarseness, dysphonia, inspiratory stridor, and laryngospasm. Therefore, many ALS patients are initially referred to an otolaryngologist rather than a neurologist. Although the role of genetic testing is still controversial in clinical practice, our findings suggest that molecular analysis of the SOD1 gene may be indicated in ALS patients with laryngological onset.
Data Availability Statement
Some of the original contributions presented in the study are publicly available. This data can be found here: ZENODO: https://zenodo.org/record/5361094. The rest of the data can be provided by request to the corresponding author.
Ethics Statement
Written informed consent was obtained from the individual for the publication of any potentially identifiable images or data included in this article.
Author Contributions
GC, LD, EA, and MC contributed to writing of the manuscript and to critical revision of the manuscript. CC and IP contributed to genetic analysis. All authors gave important contributions to the final form of the manuscript.
Funding
We want to thank the Italian Ministry of Health (Ricerca Corrente 2020-2021).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.708885/full#supplementary-material
References
1. Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. (2006) 7:710–23. doi: 10.1038/nrn1971
2. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 362:59–62. doi: 10.1038/362059a0.
3. Mathis S, Goizet C, Soulages A, Vallat J-M, Masson GL. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. (2019) 399:217–26. doi: 10.1016/j.jns.2019.02.030
4. Diamanti L, Gagliardi S, Cereda C, Ceroni M. Genetics of ALS and correlations between genotype and phenotype in ALS — a focus on italian population. In: Current Advances in Amyotrophic Lateral Sclerosis. InTech (2013). Available online at: https://www.intechopen.com/books/current-advances-in-amyotrophic-lateral-sclerosis/genetics-of-als-and-correlations-between-genotype-and-phenotype-in-als-a-focus-on-italian-population (accessed August 25, 2019). doi: 10.5772/56547
5. Robberecht W, Aguirre T, Van Den Bosch L, Tilkin P, Cassiman JJ, Matthijs G. D90A heterozygosity in the SOD1 gene is associated with familial and apparently sporadic amyotrophic lateral sclerosis. Neurology. (1996) 47:1336–9. doi: 10.1212/WNL.47.5.1336
6. Battistini S, Giannini F, Greco G, Bibbò G, Ferrera L, Marini V, et al. SOD1 mutations in amyotrophic lateral sclerosis. J Neurol. (2005) 252:782–8. doi: 10.1007/s00415-005-0742-y
7. Giannini F, Battistini S, Mancuso M, Greco G, Ricci C, Volpi N, et al. D90A-SOD1 mutation in ALS: the first report of heterozygous Italian patients and unusual findings. Amyotroph Lateral Scler. (2010) 11:216–9. doi: 10.3109/17482960902721642
8. Origone P, Caponnetto C, Mascolo M, Mandich P. Heterozygous D90A-SOD1 mutation in an Italian ALS patient with atypical presentation. Amyotroph Lateral Scler. (2009) 10:492–492. doi: 10.3109/17482960903055966
9. Luigetti M, Conte A, Madia F, Marangi G, Zollino M, Mancuso I, et al. Heterozygous SOD1 D90A mutation presenting as slowly progressive predominant upper motor neuron amyotrophic lateral sclerosis. Neurol Sci. (2009) 30:517–20. doi: 10.1007/s10072-009-0125-8
10. van der Graaff MM, Grolman W, Westermann EJ, Boogaardt HC, Koelman H, van der Kooi AJ, et al. Vocal cord dysfunction in amyotrophic lateral sclerosis. Arch Neurol. (2009) 66:1329–33. doi: 10.1001/archneurol.2009.250
11. Chen A, Garrett CG. Otolaryngologic presentations of amyotrophic lateral sclerosis. Otolaryngol Neck Surg. (2005) 132:500–4. doi: 10.1016/j.otohns.2004.09.092
12. Schito P, Ceccardi G, Calvo A, Falzone YM, Moglia C, Lunetta C, et al. Clinical features and outcomes of the flail arm and flail leg and pure lower motor neuron MND variants: a multicentre Italian study. J Neurol Neurosurg Psychiatry. (2020) 91:1001–3. doi: 10.1136/jnnp-2020-323542
13. Fukae J, Kubo S, Hattori N, Komatsu K, Kato M, Aoki M, et al. Hoarseness due to bilateral vocal cord paralysis as an initial manifestation of familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2005) 6:122–4. doi: 10.1080/14660820510034451
14. Tan C-F, Piao Y-S, Hayashi S, Obata H, Umeda Y, Sato M, et al. Familial amyotrophic lateral sclerosis with bulbar onset and a novel Asp101Tyr Cu/Zn superoxide dismutase gene mutation. Acta Neuropathol. (2004) 108:332–6. doi: 10.1007/s00401-004-0893-4
15. Salameh JS, Atassi N, David WS. SOD1 (A4V)-mediated ALS presenting with lower motor neuron facial diplegia and unilateral vocal cord paralysis. Muscle Nerve. (2009) 40:880–2. doi: 10.1002/mus.21321
16. Kostrzewa M, Damian MS, Müller U. Superoxide dismutase 1: identification of a novel mutation in a case of familial amyotrophic lateral sclerosis. Hum Genet. (1996) 98:48–50. doi: 10.1007/s004390050157
17. Hermann A, Reuner U, Ziethe G, Bräuer A, Gölnitz U, Rolfs A, et al. Vocal cord paralysis and rapid progressive motor neuron disease by the I113F mutation in SOD1 gene. Amyotroph Lateral Scler. (2011) 12:382–4. doi: 10.3109/17482968.2011.565775
18. Origone P, Caponnetto C, Mantero V, Cichero E, Fossa P, Geroldi A, et al. Fast course ALS presenting with vocal cord paralysis: clinical features, bioinformatic and modelling analysis of the novel SOD1 Gly147Ser mutation. Amyotroph Lateral Scler. (2012) 13:144–8. doi: 10.3109/17482968.2011.614254
19. Kühnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M, et al. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. (2008) 4:366–74. doi: 10.1038/ncpneuro0853
20. Sperfeld A-D, Hanemann CO, Ludolph AC, Kassubek J. Laryngospasm: an underdiagnosed symptom of X-linked spinobulbar muscular atrophy. Neurology. (2005) 64:753–4. doi: 10.1212/01.WNL.0000151978.74467.E7
Keywords: Amyotrophic Lateral Sclerosis, SOD1, p.D90A, laryngospasm, case report
Citation: Capece G, Ceroni M, Alfonsi E, Palmieri I, Cereda C and Diamanti L (2021) Case Report: Laryngospasm as Initial Manifestation of Amyotrophic Lateral Sclerosis in a Long-Survival Patient With Heterozygous p.D90A – SOD1 Mutation. Front. Neurol. 12:708885. doi: 10.3389/fneur.2021.708885
Received: 12 May 2021; Accepted: 30 August 2021;
Published: 30 September 2021.
Edited by:
Xin-Ming Shen, Mayo Clinic, United StatesReviewed by:
Zhang-Yu Zou, Fujian Medical University Union Hospital, ChinaMarianne De Visser, University of Amsterdam, Netherlands
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, Iran
Wladimir Bocca Vieira De Rezende Pinto, Federal University of São Paulo, Brazil
Copyright © 2021 Capece, Ceroni, Alfonsi, Palmieri, Cereda and Diamanti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Cereda, cristina.cereda@mondino.it