Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration
 Chrystian J. Alves1
Chrystian J. Alves1  Rafael Dariolli2
Rafael Dariolli2  Frederico M. Jorge1 Matheus R. Monteiro1
Frederico M. Jorge1 Matheus R. Monteiro1  Jessica R. Maximino1 Roberto S. Martins3 Bryan E. Strauss4
Jessica R. Maximino1 Roberto S. Martins3 Bryan E. Strauss4  José E. Krieger2 Dagoberto Callegaro1
José E. Krieger2 Dagoberto Callegaro1  Gerson Chadi1*
Gerson Chadi1*- 1Department of Neurology, Neuroregeneration Center, University of São Paulo School of Medicine, University of São Paulo, São Paulo, Brazil
- 2Laboratory of Genetics and Molecular Cardiology/LIM13, Heart Institute, University of São Paulo School of Medicine, São Paulo, Brazil
- 3Department of Neurosurgery, Surgical Center of Functional Neurosurgery, Clinics Hospital of University of São Paulo, São Paulo, Brazil
- 4Viral Vector Laboratory, Center for Translational Investigation in Oncology/LIM24, Cancer Institute of São Paulo, University of São Paulo School of Medicine, São Paulo, Brazil
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease that leads to widespread motor neuron death, general palsy and respiratory failure. The most prevalent sporadic ALS form is not genetically inherited. Attempts to translate therapeutic strategies have failed because the described mechanisms of disease are based on animal models carrying specific gene mutations and thus do not address sporadic ALS. In order to achieve a better approach to study the human disease, human induced pluripotent stem cell (hiPSC)-differentiated motor neurons were obtained from motor nerve fibroblasts of sporadic ALS and non-ALS subjects using the STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus system and submitted to microarray analyses using a whole human genome platform. DAVID analyses of differentially expressed genes identified molecular function and biological process-related genes through Gene Ontology. REVIGO highlighted the related functions mRNA and DNA binding, GTP binding, transcription (co)-repressor activity, lipoprotein receptor binding, synapse organization, intracellular transport, mitotic cell cycle and cell death. KEGG showed pathways associated with Parkinson's disease and oxidative phosphorylation, highlighting iron homeostasis, neurotrophic functions, endosomal trafficking and ERK signaling. The analysis of most dysregulated genes and those representative of the majority of categorized genes indicates a strong association between mitochondrial function and cellular processes possibly related to motor neuron degeneration. In conclusion, iPSC-derived motor neurons from motor nerve fibroblasts of sporadic ALS patients may recapitulate key mechanisms of neurodegeneration and may offer an opportunity for translational investigation of sporadic ALS. Large gene profiling of differentiated motor neurons from sporadic ALS patients highlights mitochondrial participation in the establishment of autonomous mechanisms associated with sporadic ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that is characterized by a selective death of motor neurons in the brain and spinal cord of adults, leading to rapid respiratory muscle failure and patient death (Cleveland and Rothstein, 2001). Sporadic ALS is not genetically inherited and represents approximately 95% of all ALS cases. A minority of cases are familial forms and are associated with gene mutations (Gros-Louis et al., 2006; Turner and Talbot, 2008).
The generation of transgenic animals that carry mutated human genes associated with familial ALS, for instance superoxide dismutase (SOD1), tar DNA protein 43 (TDP-43), fused in sarcoma, and valosin-containing protein genes, has allowed the identification of basic mechanisms underlying neurodegeneration in the disease (McGoldrick et al., 2013). In fact, distinct processes of motor neuron death that are related to toxic glial paracrine (Nagai et al., 2007; Gowing et al., 2009) and autocrine (Ringer et al., 2012) signaling mechanisms have been described using animal models. Furthermore, the early peripheral pathological events of neuromuscular junction loss and motor axon retraction, which may start long before symptom onset, have also been described in these animal models (Rocha et al., 2013; Venkova et al., 2014). Indeed, recent evidence has pointed out the importance of close morphological and physiological relationships between peripheral non-neuronal Schwann cells (Keller et al., 2009; Chen et al., 2010; Maximino et al., 2014) and fibroblasts (Raman et al., 2015) with motor axons, as key participants in the early degeneration of motor neurons in ALS.
Despite significant progress, it is clear that shortcomings remain with regard to recapitulating the processes underlying the clinical form of sporadic ALS (Benatar, 2007b), based on mechanisms described in transgenic models carrying specific human gene mutations. Actually, these limitations inherent in animal models have been considered the major obstacle preventing the successful translation of results to clinical practice. No therapy has been effective in counteracting human disease progression or extending survival of ALS patients (Benatar, 2007a). Moreover, there is a general consensus that the development of therapeutic targets promoting positive clinical results have been hampered by lack of a relevant human disease model specific to sporadic ALS (Gordon and Meininger, 2011; Veyrat-Durebex et al., 2014).
One of the most promising approaches to provide a better understanding of the mechanisms underlying motor neuron degeneration in sporadic ALS is the generation of specific human-induced pluripotent stem cells (hiPSC) from fibroblasts of motor nerves from sporadic ALS patients. These hiPSCs can be differentiated into multiple cell types, including mature motor neurons which are normally inaccessible for in vitro studies (Veyrat-Durebex et al., 2014).
In fact, human motor neurons have been previously generated from human embryonic stem cells and hiPSCs carrying specific gene mutations, all related to the less prevalent, familial form of ALS (Di Giorgio et al., 2008; Marchetto et al., 2008; Mitne-Neto et al., 2011). Despite the substantial effort involved in generating iPSC-derived, differentiated neurons from familial ALS patients, such motor neurons developed from adult cells (i.e., skin fibroblasts and erythroblasts) harbor specific gene mutations, and thus may not represent an appropriate model from which to develop translational therapies specific to the more dominant, sporadic forms of ALS. Thus, this approach is presently restricted to the less prevalent form of ALS (Leblond et al., 2014).
In this study, hiPSC-derived motor neurons were obtained from sporadic ALS patients by reprogramming motor nerve-derived fibroblasts using a STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus System containing the four transcription factors OCT4, SOX2, KLF4, and CMYC (Somers et al., 2010). A large gene profiling analyses of these differentiated motor neurons was performed using a high-density oligonucleotide microarray linked to specific tools capable of identifying biological processes, molecular functions and pathways deregulated in the sporadic ALS form.
Our results demonstrate that the generation of hiPSCs by reprogramming motor nerve-derived fibroblasts from sporadic ALS patients, followed by their differentiation into adult motor neurons, is not only feasible, but more importantly, can be used to model key molecular mechanisms which may recapitulate those processes related to neurodegeneration in this disease state. This approach may thus provide one of the most promising platforms for the development of therapeutic targets specific to sporadic ALS.
Methods
Human Tissue Samples
Sporadic ALS patients accompanied at the ALS Ambulatory Unit of Department of Neurology of Clinics Hospital of University of São Paulo School of Medicine signed informed consent and were included in the present study after neurological evaluation. Inclusion criteria were less than 6 months of disease evolution after diagnosis according to EL ESCORIAL (Brooks et al., 2000) and motor impairments not affecting substantially one of the lower limbs. Sporadic ALS patients showed preserved movements of the ankle and big toe in the foot corresponding to the site of biopsy. Exclusion criteria were the presence of associated pathologies, breathing disorders, swallowing and cognitive disorders. Patients were submitted to surgical procedures in one of their feet by means of local anesthesia to collect a fragment of their extensor hallucis brevis nerve which is a motor nerve that innervates the extensor hallucis brevis muscle in the dorsal region of the foot. As control, biopsies were taken from the distal accessory nerve performed during reconstructive surgery of traumatic brachial plexus injuries of non-ALS patients at the Clinics Hospital of University of São Paulo School of Medicine. Non-ALS patients reported no history of familial ALS and clinical evaluation failed to show any signs of ALS. Both procedures were performed in the Surgical Center of Functional Neurosurgery of Clinics Hospital of University of São Paulo in accordance with protocol number 0187/11 approved by the Ethics Committee for Analysis of Research Projects of Clinics Hospital of University of São Paulo School of Medicine.
DNA Sequencing to Exclude Most Frequent Gene Mutations Associated with Familial ALS
SOD1 and TARDBP Analyses
The SOD1 and TARDBP genes were sequenced in ALS patients to verify the absence of DNA mutations in genes normally associated with familial ALS, thus providing further confirmation of the sporadic form of ALS in patients included in the study. Genomic DNA was extracted from peripheral blood using standard methods. For sequencing analysis, primers were designed so that they flanked all five exons of SOD1 and the first six exons of TARDBP, both within intronic regions (Hallewell et al., 1986; Xiong et al., 2010). Direct sequencing of amplified exons was performed using Big-dye® Terminator v3.1 sequencing (Applied Biosystems, USA). Reactions were performed by means of a ABI 3130 genetic analyzer (Applied Biosystems), and sequences were analyzed by using a Mutation Surveyor v5.0 (SoftGenetics, USA).
C9orf72 Repeat Expansions Analysis
Genomic DNA was isolated from peripheral blood according to standard protocols. The repeat number of the GGGGCC hexanucleotides was determined using genotyping primers. Repeat-primed polymerase chain reaction (PCR) was performed in order to provide a qualitative assessment of the presence of C9orf72 repeat expansions, as previously described (Dejesus-Hernandez et al., 2011). Briefly, 200 ng of genomic DNA were used as template in a final volume of 28 μl containing 12.5 μl of FastStart PCR Master Mix (Roche Applied Science, USA), and a final concentration of 0.25 mM 7-deaza-dGTP (Roche Applied Science), 5% dimetil sulfoxide (Sigma-Aldrich, USA), 1M betaine (Sigma-Aldrich) and 1 μM of each primer. Sample analyses were performed on an ABI 3500 genetic analyzer (Applied Biosystems), and data evaluated using the GeneMapper software. Repeat expansions are known to produce a characteristic sawtooth pattern with a 6-bp periodicity, as previously described (Dejesus-Hernandez et al., 2011).
hiPS Cell Generation
Fibroblasts were obtained from fragments of extensor hallucis brevis peripheral motor nerve biopsies from sporadic ALS patients and from distal accessory nerves collected from control non-ALS patients. Fibroblasts of sporadic ALS and non-ALS subjects were submitted to Sendai and STEMCCA methods of reprogramming as described below.
Sendai Reprogramming
After purification, fibroblasts were expanded in culture (Seluanov et al., 2010) and reprogrammed based on the protocol described by Macarthur et al. (2012). Briefly, approximately 105 human fibroblasts were seeded per well in a 6-well plate and incubated at 37°C and 5% CO2. The next day, cells were transduced with the CytoTune iPS Reprogramming Kit containing Sendai virus vectors (Life Technologies, USA; Cat. # A1378001) in a fibroblast growth medium at a multiplicity of infection of 3, as described in the manufacturer's protocols. The medium was replaced with fresh fibroblast growth medium 1 day after transduction and the cells were cultured for 7 days. Cells (2 × 105 cells per dish) were cultured on an inactivated mouse fibroblast feeder layer in 100 mm tissue culture dishes at day 8 of transduction. After an overnight period, the hiPSC medium was replaced daily. The hiPSC medium is composed of DMEM-F12 containing 20% knockout serum replacement (KSR), 1% MEM non-essential amino acids (NEAA) solution, 0.1% 2-mercaptoethanol. Medium was equilibrated at 37°C and added with 4 ng/mL FGF-2 just prior the use. Reagents were purchased from Life Technologies. The hiPSC colonies were picked for expansion and characterization from days 27 to 30 of reprogramming.
STEMCCA Reprogramming
Fibroblasts of sporadic ALS and non-ALS subject groups were expanded in culture as described above. The reprogramming was performed based on the protocol described by Somers et al. (2010). Briefly, 105 fibroblasts were plated onto a GelTrex (Life technologies) pretreated P35 mm dish in 10% fetal bovine serum and DMEM. The fibroblasts were subjected to reprogramming 24 h after plating by STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus (Millipore, USA) expressing the pluripotency related genes OCT4, SOX2, KLF4, and CMYC. DMEM media was replaced by E6 medium (Life Technologies) supplemented with 10 ng/ml human FGF-2 (Life Technologies) (E6F) and 0.5 mM NaB (Sigma-Aldrich) 24 h after transduction. The medium was changed by fresh E6F on days 3, 5, 7, and 9. Subsequently, the medium was changed daily from day 11 to 25 by the E8 medium (Life Technologies) supplemented with 0.25 mM of NaB (Sigma-Aldrich). The colonies were visualized with the aid of a phase contrast microscope at the end of that period and were collected manually. The colonies were then seeded in GelTrex pretreated dishes for the expansion in E8 medium. The hiPSCs-derived from sporadic ALS and non-ALS subjects were characterized by means of immunocytochemistry (Table 1), reverse transcriptase polymerase chain reaction (RT-PCR) and quantitative polymerase chain reaction (qPCR) (Table 2) and alkaline phosphatase live stain described below.
Table 1. Antibodies for characterization of hiPSC, embryoid body, differentiated motor neurons and cardiomyocytes.

Table 2. RT-PCR and qPCR primers for hiPSC and differentiated motor neuron gene markers.
Alkaline Phosphatase Live Staining
The culture media of hiPSCs from ALS and non-ALS subjects achieved with Sendai and STEMCCA methods of reprogramming were aspirated from colonies and were washed twice with DMEM/F-12 medium (Gibco). The colonies were incubated for 30 min at 37°C with alkaline phosphatase live stain (Life Technologies) diluted in DMEM/F-12 medium (1:500) and then washed 3x with the same medium. Positive stained colonies were visualized by means a FITC filter under fluorescent microscopy (EVOS XL Core Imaging System, Life Technologies).
Karyotyping
Eighty percent confluent hiPSC colonies were incubated for 2 h in the E8 medium with 0.5 μg/ml colchicine for karyotyping procedures. Cells were then harvested with TrypLE (Life Technologies) and incubated for 20 min at 37°C in 0.075 M KCl solution, fixed in methanol/acetic acid (3:1), placed on pre-wet chilled microscope slides, air-dried, and incubated for 2 days at 37°C. The microscope slides were stained for 90 s with Wright's stain in 2x buffer. Standard G-banding chromosome analysis was performed by Clinics Hospital Molecular Diagnostic Services (São Paulo, Brazil).
hiPSC Differentiation in vitro and Characterization
Differentiation of induced pluripotent cells was accomplished as described elsewhere (Evans and Kaufman, 1981; Martin, 1981).
Embryoid Body Differentiation
Briefly, hiPSCs were lifted using StemPro EZPassage (Life Technologies) and maintained for 4 days in suspension for embryoid body formation. The cells were cultured in embryoid body medium, composed of knockout DMEM/F12 (Life Technologies), supplemented with 20% KSR (Life Technologies), 2 mM GlutaMAX (Life Technologies), 100 μM NEAA (Life Technologies), 1% antibiotic-antimycotic (Sigma-Aldrich) and 100 μM 2-mercaptoethanol (Life Technologies). Addition of ROCK-inhibitor Y-27632 (Ascent Scientific, UK) for the first 24 h was used to improve the survival of hiPSC single cells (Lai et al., 2010) and to allow an adequate generation of embryoid bodies as well. The embryoid bodies were transferred to Geltrex-coated tissue-treated dishes to allow cell attachment and were maintained there for 1 week until their in vitro analysis of differentiation by means of immunocytochemistry with specific markers of three germ layers (Table 1).
Cardiomyocyte Differentiation
To further analyze their differentiation potential in vitro, hiPSCs were induced to directly differentiate into cardiomyocytes. The procedure was performed using a PSC cardiomyocyte differentiation Kit (Life Technologies), according to the manufacturer's protocol. Briefly, after splitting, cells were fed by cardiomyocyte differentiation medium A at day 4. Cells were then fed by cardiomyocyte differentiation medium B on day 6 and were maintained in a cardiomyocyte maintenance medium from day 8 to day 14. At that time point, cells were evaluated for their ability to show contraction as a normal feature of cardiomyocytes (Supplementary Video), characterized by immunocytochemistry for cardiomyocyte cell markers (Table 1) and, stained with fluorescent phalloidin actin binding (Life Technologies) performed according to the manufacture instructions.
hiPSC Differentiation In vivo
In order to evaluate the ability of hiPSC to develop teratomas, approximately 3x106 hiPSCs from ALS and non-ALS patients were injected subcutaneously into the dorsal flanks of nude rats anesthetized with ketamin (Cristália, Brazil)-xilazin (Vetbrands, Brazil; 62.5 mg/kg). Teratomas were allowed to expand for about 6 weeks. They were visualized and dissected by means of surgical procedure, fixed overnight in 4% paraformaldehyde and transferred to 70% ethanol until they were embedded in paraffin. Sections were stained with hematoxylin and eosin for microscopy analyses. Protocols for animal use were previously approved by the School of Medicine of the University of São Paulo Institutional Animal Care and Use Committee.
Motor Neuron Differentiation and Characterization
Motor neuron differentiation was performed according to a protocol described by Hu and Zhang (2009) and modified in our lab. Briefly, after reaching confluence, hiPSC colonies were cultured in suspension in the presence of embryoid body medium to achieve embryoid body formation as described above. The medium was replaced on day 4 by a neural differentiation medium containing DMEM/F12 (Life Technologies), N2-supplement (Invitrogen, USA), 100 μM NEAA (Life Technologies), 1% antibiotic-antimycotic (Sigma-Aldrich) and 2 μg/ml heparin (Cristália) to induce the formation of the neural progenitor cells. Clusters attached to laminin-coated dishes (20 μg/ml, Sigma-Aldrich) after 1 week in suspension. Primitive neuroepithelial cells were posteriorized by addition of 0.1 μM retinoic acid (Sigma-Aldrich) at day 10 and ventralizated by the addition of 100 ng/ml sonic hedgehog (Shh; Sigma-Aldrich) and B27 supplement (Gibco, USA) at day 14. The cells were collected at differentiation day 20 for microarray experiments. Samples of hiPSC-derived motor neurons from ALS and non-ALS subjects were also obtained on small laminin-coated coverslips (13 mm) and characterized by immunocytochemistry (Table 1), RT-PCR, qPCR (Table 2) and Hb9::GFP live reporter.
Hb9::GFP Live Reporter Technique for Motor Neuron Visualization
The construct containing Hb9-GFP (Hb9::GFP) reporter was transferred to the differentiated motor neurons using a lentivirus system (Marchetto et al., 2008). Hb9 is a motor neuron-specific transcription factor expressed in mature cells that binds to the promoter region of the green fluorescent protein (GFP) sequence in the construct. HEK 293T cells were employed for virus production. Briefly, triple transfection with calcium phosphate was performed 24 h after plating using the PSPAX2 plasmids, the pCMV-VSVg and the pLenti-Hb9-GFP (Addgene plasmid # 37080) or pEGIP (constitutive lentiviral vector for expression of GFP; Addgene plasmid # 26777) (Marchetto et al., 2008; Zou et al., 2009). The supernatant containing viral particles was collected 24 and 48 h after transfection. In parallel, differentiated motor neurons were plated at a concentration of 2 × 106 cells per dish on 60 mm plates. Transduction was achieved by adding the lentivirus to hiPSC-derived motor neuron cell cultures after the treatment with Shh. The differentiated motor neurons were maintained on small glass coverslips in 24-well plates and transduced using 200 μl viral supernatant plus 100 μl of neural differentiation medium containing 8 mg/ml polybrene. The medium was replaced 6 h after incubation at 37°C and 5% CO2. The cells were fixed 72 h after transduction and were evaluated for GFP expression using an Olympus AX-70 microscope (Olympus, JP).
Quantification of Differentiated Motor Neurons
The amount of differentiated motor neurons was quantified by the counting of ChAT immunoreactive profiles and also Hb9::GFP profiles counterstained with nuclear DAPI. Briefly, a sample of cells were seeded onto small laminin-coated (Sigma) glass coverslips and immunostained for ChAT (Millipore) or labeled with Hb9:GFP methodology (the construct Hb9::GFP as described above) followed by a DAPI nuclear labeling in order to determine the number of differentiated motor neurons in the sample fields. Evaluation of motor neuron vitality was performed by means of Fluoro-Jade C (FJC) analysis as described in the Figures S4E,F of the Supplementary Material (Schmued et al., 1997, 2005).
Immunocytochemical Characterization
The hiPSC colonies, the embryoid body differentiation in vitro and the differentiated motor neurons were characterized by indirect immunofluorescence, as described elsewhere (Maximino et al., 2014). Briefly, cells were fixed with 4% paraformaldehyde and incubated with primary antibodies shown in Table 1. Primary antibodies were detected using Alexa Fluor® 488 or 594-conjugated secondary antibodies specific for mouse, rabbit and goat (all from Invitrogen). Preparations were mounted on microscope slides and counterstained with nuclear 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI; Vector, USA). Digital images of immunofluorescence staining were obtained by means of an Olympus AX-70 microscope (Olympus).
RT-PCR and qPCR Characterization
Molecular characterizations of hiPSC and of differentiated motor neurons of the chamber culture were performed by means of RT-PCR and qPCR as described elsewhere (Maximino et al., 2014). Briefly, total RNA of cells contained in the 24-well plates was extracted using Trizol (Life Technologies) according to the manufacturer's protocol. The quantity (NanoDrop 1000 Spectrophotometer; Thermo Scientific, USA) and quality (Agilent 2100 bioanalyser, RNA 6000 Pico LabChip; Agilent Technologies, USA) of RNA were determined. cDNAs were amplified by PCR using the primers specific for hiPSC and differentiated motor neurons (Table 2). PCR products were submitted to electrophoresis in 2% agarosis gels containing ethidium bromide for 60 min at 100 volts, and then visualized under ultraviolet light exposure. qPCR reactions were performed as described below. hESC (Fraga et al., 2011) and neural progenitor cells were used as positive controls for hiPSC and negative controls for motor neuron characterization, respectively (Marti et al., 2013; Jha et al., 2015).
RNA Isolation and Microarray Experiments
The differentiated motor neurons that were achieved from a pool of neuronal progenitor lines of the two sporadic ALS and two non-ALS patients, this process were performed in three different times. A total of three ALS and three non-ALS (control) samples of differentiated motor neurons obtained were subjected to large gene profiling by means of microarray analysis. The same sampling procedure was employed in other quantification methods of the study. The procedures of RNA isolation and the methodology of microarray experiments were described in our previous publication (Maximino et al., 2014). Briefly, total RNA was extracted from differentiated motor neurons and linear amplification of RNA was performed using the RiboampHSplus kit (Arcturus) according to the manufacturer's protocol, control of RNA quality was them assessed, as described above. A representative electropherogram from a Bioanalyzer evaluation of RNA integrity of the differentiated motor neurons is shown in the Supplementary Material (Figure S4H). RNAs of samples (25 ng) and reference (100 ng) were reverse transcribed by the low-input RNA linear amplification kit and then transcribed to Cy5-labeled (samples) or Cy3-labeled (reference) according to the manufacturer's instructions (Agilent Technologies) and to our previous descriptions (de Oliveira et al., 2013). A total of 300 ng of Cy5-labeled cRNA was hybridized together with the same amount of Cy3-labeled reference to Whole Human Genome Oligo 8 × 60 K. All steps were performed according to the manufacturer's instructions (Agilent Technologies) and to our previous descriptions (de Oliveira et al., 2013; Maximino et al., 2014).
Microarray Analysis
The slide were scanned by a Microarray Scanner (Agilent) and the raw image data were converted to numerical data using the Agilent Feature Extraction Software, version 11.0.1.1 (Agilent Technologies), as described in our previous study (Maximino et al., 2014). Microarray raw data (.txt files) were imported into GeneSpring v.13 GX software package (Agilent Technologies). Raw signal intensities were normalized using this software. The probes were tested for differential expression using a moderated t-test for the comparisons between differentiated motor neurons that were achieved from a pool of neuronal progenitor lines of the two control patients and the two ALS patients, using GeneSpring v.13 GX Software. Genes with p < 0.05 were accepted as differentially expressed genes. The raw data from hybridizations are available on the Gene Expression Omnibus Database, and the GEO accession number is GSE68240. The data were registered in the GEO according to the following description: control samples (control sample 1, control sample 2, control sample 3) and ALS samples (ALS sample 1, ALS sample 2, ALS sample 3) of differentiated motor neurons that were achieved from a pool of neuronal progenitor lines of the two control patients and the two ALS patients, respectively.
Functional Enrichment Analysis
The functional annotation analysis of differentially transcribed genes was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) web-server v.6.7 (http://david.abcc.ncifcrf.gov) (Huang Da et al., 2009). Gene Ontology (GO) terms of Biological Process and Molecular Functions, and also Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways included in the DAVID knowledgebase were considered. We have also focused on the GO terms mitochondrion of the Cellular Component and their related genes. Furthermore, the mean of gene expression normalized signals related to the mitochondrion was compared between the two groups. GO terms were submitted to REVIGO, a web server that takes long lists of GO terms and summarizes them in categories and clusters of differentially expressed genes by removing redundant entries (Supek et al., 2011). The analysis was conducted on the list containing the up-regulated and down-regulated genes. High stringency (EASE score) parameters were selected to improve confidence in those values designated as enriched (Ashburner et al., 2000; Kanehisa et al., 2006).
Quantitative PCR
qPCR was carried out for the analysis the expression of pluripotency markers, NANOG, SOX2, OCT4, and CHAT in the hiPSCs and also the expression of the PAX6 (neural stem cell marker), OLIG2 (motor neuron precursor marker), and the CHAT, HB9 (mature motor neuron markers) in the differentiated motor neurons. The details of the normalization procedures were described in the legends of the Figures 2, 5 and Figure S4. Furthermore, a sample of differentially expressed genes described by microarray was selected for verification by qPCR, as described in our previous publication (de Oliveira et al., 2014). The genes were chosen for verification based on their high or low fold relative expression levels and possible involvement in ALS or other neurodegeneration related mechanisms. Thus, dual specificity phosphatase 6 (DUSP6), diacylglycerol o-acyltransferase 1 (DGAT1), potassium channel, subfamily K, member 12 (KCNK12), kinesin family member C1 (KIFC1); keratin associated protein 4-11 (KRTAP4-11); leucine zipper-EF-hand containing transmembrane protein 1 (LETM1); succinate dehydrogenase complex assembly factor 1 (SDHAF1); caspase 9, apoptosis-related cysteine peptidase (CASP9); vacuolar protein sorting 35 homolog (S. cerevisiae) (VPS35); and insulin-like growth factor 2 (IGF2) were verified by qPCR.
Briefly, cDNA of differentiated motor neurons, as described above was synthesized from 50 ng of total RNA by using a Maxima First Strand cDNA Synthesis Kit (ThermoScientific,USA) according to manufacturer. qPCR reactions were carried out in duplicate with 5 ng cDNA, the DyNAmo Color Flash SYBR Green qPCR kit (Thermo Scientific) and 400 nM of each primer in a final volume reaction of 20 μl, by using the Applied Biosystems 750 Real-Time PCR System (Applied Biosystems). The SYBR primers used in qPCR verification can be found in Table 3.
Table 3. Primers used in SYBR qPCR verification of sporadic ALS motor neuron deregulated genes.
The cycling in the SYBR reactions was composed of an initial denaturation at 95°C for 10 min. Templates were subsequently amplified by 40 cycles of 95°C for 15 s and 60°C for 30 s. A dissociation curve was then generated to ensure amplification of a single product, and the verification of no primer dimers formation. A standard curve was generated for each primer pair in order to determine the efficiency of the PCR reaction over a range of template concentrations from 0.032 ng/μl to 20 ng/μl, using cDNA synthesized from human differentiated motor neuron reference RNA. The efficiency for each set of primers was 100 ± 5%. Gene expression was normalized to GAPDH, and was determined using the ΔΔCt mathematical model (ABI PRISM 7700 Sequence Detection System protocol; Applied Biosystems). GAPDH was chosen as a housekeeping gene to normalize the qPCR values because the microarray analysis showed that its expression was stable across samples.
Statistical Analyses
The statistical method employed in the microarray analysis is described above. The One-Way ANOVA was applied with Tukey's multiple comparisons post-test to identify statistical significances between samples within groups. Furthermore, one-tailed unpaired t-test was used to determine the statistical significance of differences in qPCR experiments. All analyses were performed using Graphpad Prism 5 (GraphPad, USA). Data were presented as means ± SEM and significance level was set at p < 0.05.
Results
Clinical Evaluation of ALS Patients and DNA Sequencing
Sporadic ALS patients showed clinical and laboratory features of ALS according to EL SCORIAL (Table 4). The evolution of the disease did not exceed 29 months after initial symptoms and ALS patients showed preserved movements of the ankle and the big toe in the foot corresponding to the site of biopsy. The amplitude of the big toe movement diminished after the biopsy of the extensor hallucis brevis nerve, but they showed a progressive recovery after 4 weeks of surgical intervention. Unfortunately, due the natural evolution of the disease, ALS patients lost movements of the entire inferior limbs in the subsequent months. ALS patients did not present with any cognitive impairments or familial history of neurodegenerative disease.
Table 4. Patient's information.
Non-ALS patients were adult subjects that had suffered a unilateral injury of the brachial plexus months before the procedure of plexus surgery. The distal fragments of the preserved accessory nerve were obtained when the nerve was cut and transferred to one of the major nerves of the brachial plexus. That means the donor nerve was functional and did not show signs of local inflammation at the time of surgery.
Sporadic ALS patients showed no SOD1 and TARDBP gene mutations within the coding regions of those genes. Furthermore, C9orf72 repeat expansions were not present in the sporadic ALS patients (Figure S1).
Generation and Characterization of hiPSCs from Motor Nerve Fibroblasts
Integration-free iPS cell lines were generated using a Sendai virus -based on individual delivery of the 4 Yamanaka reprogramming factors, i.e., OCT4, SOX2, KLF4, and CMYC (Takahashi et al., 2007). Fibroblasts of all subjects showed successful reprogramming, with no morphological changes observed up to 12 days of reprogramming. The colonies of stem cells were tightly packed, showed a round morphology, and lacked well-defined sharp edges (Figures S2A,B). Unfortunately, several colonies regressed 3 weeks later and expansion of embryonic markers (see below) occurred in only a few colonies. The emerging colonies expressed the surface embryonic stem cell marker, SSEA4, and the pluripotency marker, alkaline phosphatase as shown in Supplementary Material (Figures S2C,D).
Based on the above results regarding the low level of reprogramming achieved by the Sendai method in our laboratory, we subsequently transduced fibroblasts using the STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus (Somers et al., 2010) in order to increase efficiency. In fact, morphological changes were observed as early as 6 days after reprogramming. Thus, 20 embryonic stem cell-like colonies were picked and expanded under feeder-free conditions 4 weeks after transduction. The best candidates for hiPSC clones from sporadic ALS and non-ALS subjects were selected based on their morphology and were employed in further characterization and motor neuron differentiation experiments.
All hiPSC colonies obtained from sporadic ALS and non-ALS control subjects showed classical morphology and their cells expressed the surface embryonic stem cell markers SSEA-4, TRA1-60, and TRA1-81, as well as the transcription factor OCT4 (Figures 1A–F) and alkaline phosphatase (Figure 1G). Karyotyping analysis of samples of hiPSC lines from all subjects showed an absence of alterations in the hiPSC chromosomal number and morphology (Figure 1H). Furthermore, colonies from ALS and non-ALS subjects showed NANOG, SOX2, OCT4, DPPA4, ESG/DPPA, DPPA2, REX1, GDF3, and CHAT signals which were similar to those of an hESC line and substantially different from the iPSC-differentiated motor neurons (Figure 2A). Furthermore, hiPSC colonies obtained from sporadic ALS and non-ALS control subjects expressed the higher levels of NANOG and OCT4 as evidenced by the qPCR experiments. Interestingly, the expression of SOX2, as seen in the hiPSCs and also in the differentiated motor neurons could be related to the fact that we have not performed the silencing of exogenous factors after lentiviral induction or to a natural time-dependent decrease of these pluripotency factors (Muratore et al., 2014; Raitano et al., 2015) (Figure 2B).
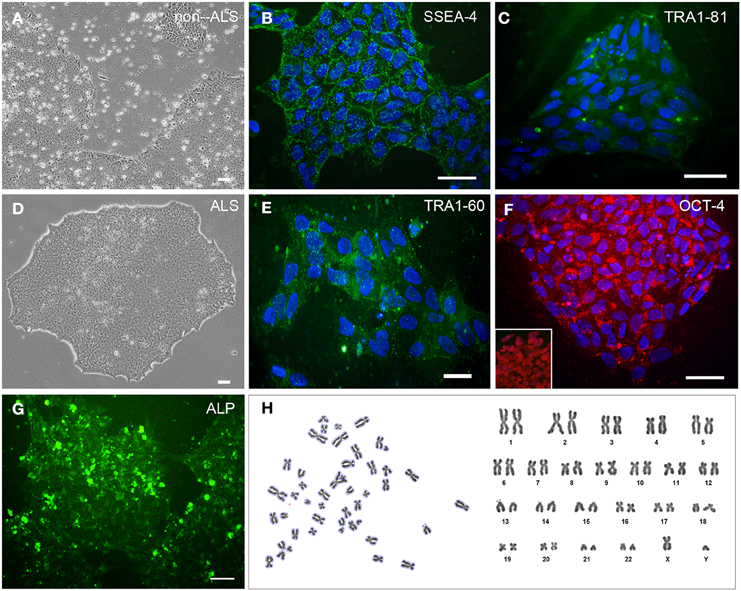
Figure 1. Generation and characterization of STEMCCA transduced hiPSC. Primary human induced pluripotent stem cells (hiPSC) colonies derived from non-ALS (A) and ALS patients of sporadic form (B–H) after transduction with STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus expressing the embrionary genes OCT4, SOX2, KLF4, and CMYC after several passages in E8 medium. hiPSCs were derived from fibroblasts of extensor hallucis brevis nerve obtained by biopsy from sporadic ALS patients and from distal fragments of the accessory nerve from a non-ALS subject. Immunostaining of cultured hiPSC for SSEA-4 (B), TRA1-60 (E), TRA1-81 (C), and OCT-4 (F). The presence of OCT-4 immunoreactivity in the nucleus of hiPSC samples that were not counterstained with nuclear DAPI is shown in the box inside of the (F). Of note, OCT-4 has been described in cytoplasmic vesicles (F), an event associated to regulation of pluripotency-associated protein homeostasis of pluripotent cells (Cho et al., 2014; Muratore et al., 2014). Cell nuclei were stained with DAPI (blue). hiPSC colonies were analyzed for alkaline phosphatase activity using the Alkaline Phosphatase Live Stain (G). Karyotyping sampling of hiPSC clone from an ALS patient, in which metaphase plates showed the normal male chromosomal content (H; 42,XY). The same pattern was observed for other subjects of the study, including the non-ALS patients (data not shown). Scale bars, 50 μm.
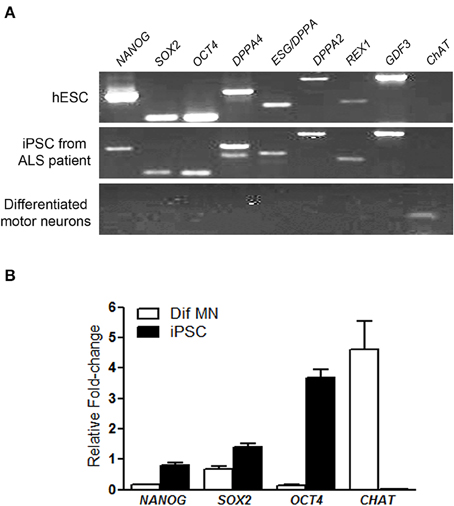
Figure 2. Pluripotency markers of generated hiPSC. RT-PCR analysis of pluripotency gene markers in human embryonic stem cells, in induced pluripotent stem cells (iPSC) from motor nerve fibroblasts of an ALS patient and in differentiated motor neurons after in vitro differentiation via embryoid body protocol. The pluripotency gene markers, NANOG, SOX2, OCT4, DPPA4, ESG/DPPA, DPPA2, REX1, GDF and also the gene choline acetyltransferase (CHAT), a marker of differentiated motor neurons were employed (A). The expression of NANOG, SOX2, OCT4, and CHAT by qPCR in the iPSC and differentiated motor neurons (Dif MN) was quantified and the graph shows relative fold change values; a pool of the hESC was used as reference samples with a reference value of 1 (B). iPSC and hESC show the pluripotency gene markers and differentiated motor neurons show only the CHAT marker (A,B). The hESCs were used as a reference parameter.
The pluripotency of the hiPSCs from ALS and non-ALS subjects was also evaluated in vitro through the formation of embryoid bodies. All hiPSC lines differentiated spontaneously into cell types of the three embryonic germ layers, as indicated by expression of the specific markers; Pck-26 (ectoderm), Myosin LC 2 (mesoderm), and EphA2 (endoderm) (Figures 3A–C).
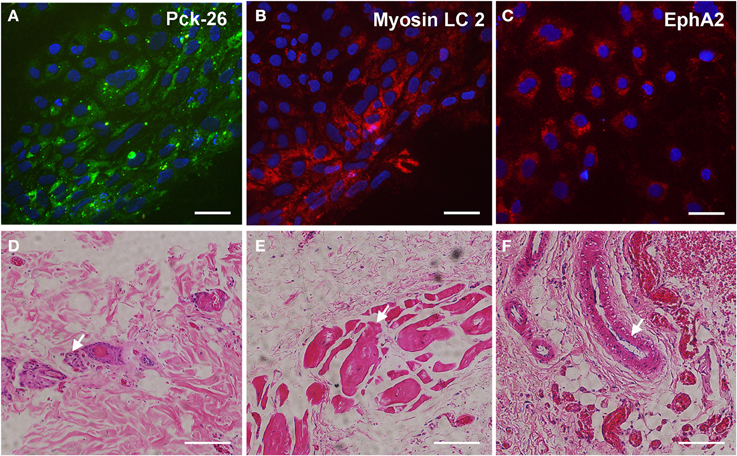
Figure 3. Spontaneous differentiation of hiPSC into three germ layers in vitro and in vivo. Immunocytochemical staining of anti-Pan cytokeratin (A; Pck-26, a marker of ectoderm layer), Myosin Light Chain (LC) 2 (B; a marker of mesoderm layer) and Ephrin A2 (C; a marker of endoderm layer) after spontaneous differentiation in vitro of human-induced pluripotent stem cells (hiPSC). hiPSC were derived from fibroblasts which were obtained from motor nerve of sporadic ALS patient. Cell nuclei were stained with DAPI (blue; A–C). Hematoxylin and eosin staining of sections from a teratoma formed after a subcutaneous injection of hiPSC into nude rats (D–F). Neural rosettes (arrow, D), muscle tissue (arrow; E), and gut-like epithelium (arrow, F) indicate tissues into the teratomas with ectoderm, mesoderm and endoderm features, respectively. Scale bars: 50 μm.
Furthermore, the pluripotency of hiPSC from ALS and non-ALS subjects was confirmed by the analysis of their ability to develop teratomas in vivo, after subcutaneous injection in nude rats. Histopathological analyses demonstrated tissues derived from the three germ layers, including neural rosettes from ectodermal origin, muscle tissue from mesodermal origin and gut-like cells from endodermal origin (Figures 3D–F) in the tumors 6 weeks after injection. These findings indicated a successful reprogramming of fibroblasts to a pluripotent state from sporadic ALS and non-ALS subjects.
Differentiation of hiPSC into Cardiomyocytes
hiPSCs were also differentiated into cardiomyocytes in order to further characterize their potential to generate additional mature cell types for further functional analyses. At 14 days, ALS differentiated cardiomyocytes were present as spontaneously contracting syncytia (Supplementary Video) expressing troponin T cardiac type 2 (cTnT) (Figures S3A,B), but were negative for CD-31 (an endothelial cell marker; Figure S3C), and TRA-1 (a stem cell marker, Figure S3D). hiPSC-differentiated cardiomyocytes expressed positive double-staining against myosin and actin to show the identity and the structural pattern of cardiac differentiated cells (Figures S3E–H).
Differentiation of hiPSCs into Motor Neurons
hiPSC lines from ALS and non-ALS subjects were also manipulated in order to generate mature motor neurons. hiPSC colonies (Figure 4A) first generated embryoid bodies in suspension (Figure 4B) and neuronal progenitor cells. Neural progenitor cells expressing the PAX6 marker (Figure 5K) were differentiated into post-mitotic motor neurons (Figures 4C,D) expressing MAP2 (Figure 5A) and ChAT (Figure 5B). Motor neurons were characterized further by means of a Hb9 (promoter/enhancer)-GFP (Hb9::GFP) reporter construct (Figure 5C), an early marker of these maturing motor neurons. Negative controls are shown in Figures 5D–F. There were no differences in the neuronal differentiation process between hiPSCs derived from sporadic ALS or non-ALS subjects. Samples of differentiated hiPSC-mature motor neurons from ALS patients (Figure 5G) and their non-ALS controls (Figure 5H) showed axonal projections. The results of the quantification of the number of differentiated motor neurons in the sample regions of the coverslips revealed the presence of 95.39 and 95.3% of ChAT positive and Hb9 positive profiles, respectively, in relation to the total amount of DAPI nuclear profiles in these regions (Figures 5I,J).
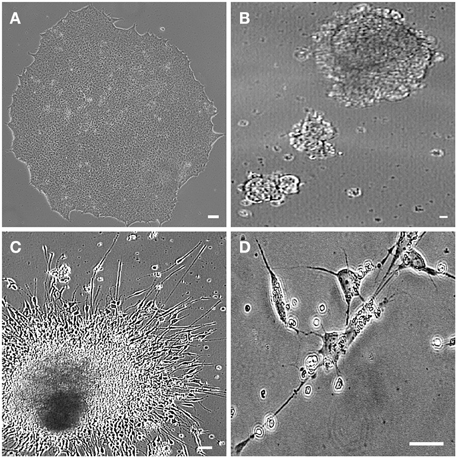
Figure 4. hiPSC-differentiated motor neurons. Human induced pluripotent stem cells (hiPSC) sample obtained from motor nerve fibroblasts of a sporadic ALS patient. hiPSC colony growing on feeder free medium forms uniform colonies (A). The motor neuron progenitors are cultured in suspension (embryoid bodies; B). Axonal lengths project from the clusters in an adherent culture (C). The hiPSC-derived motor neurons are showing classical morphology with axonal projections (D). Scale bars: 50 μm.
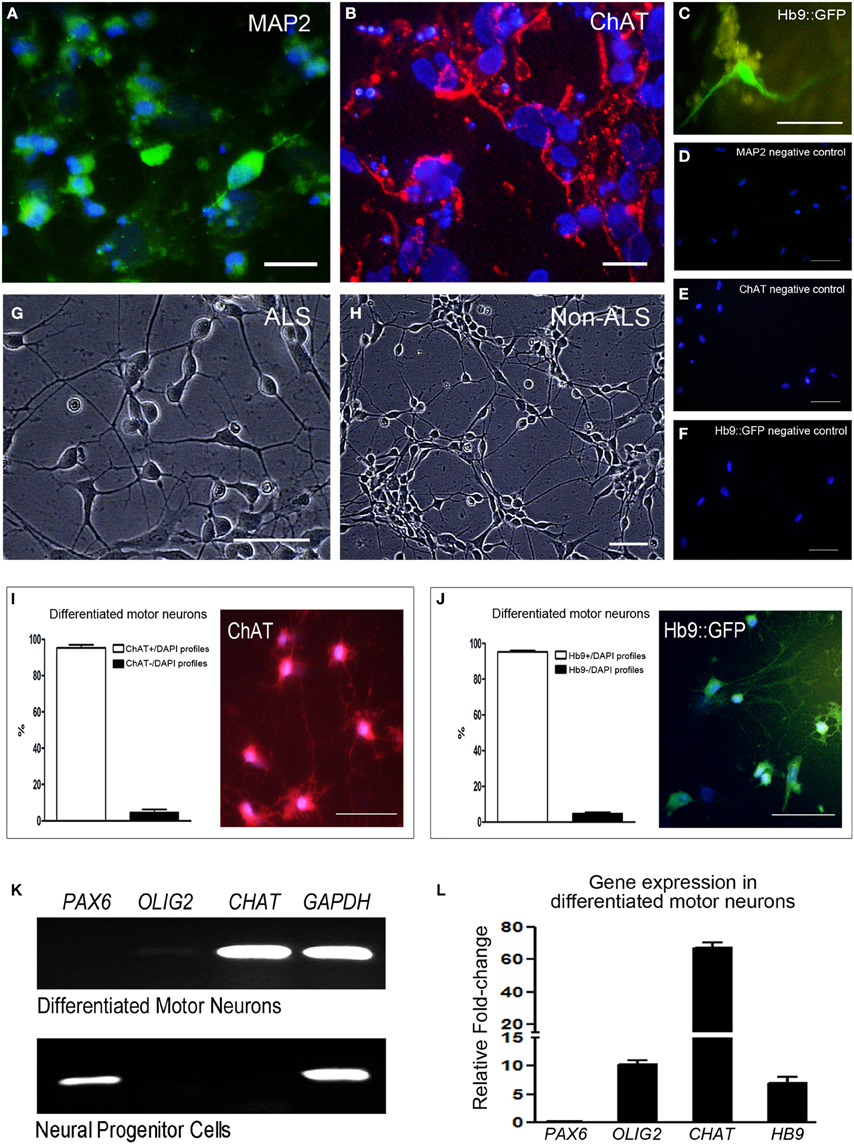
Figure 5. Specific labeling and qPCR differentiation gene markers of hiPSC-derived motor neurons. Sample of differentiated human induced pluripotent stem cells (hiPSC)-mature motor neurons are arranged in clusters and show axonal projections. Cells are positive for MAP2 (A; green), ChAT (B; red) and express GFP under control of motor neuron-specific gene Hb9 (C). The negative controls for each motor neuron marker mentioned above are seen in (D–F), respectively, and no specific labeling was found. Human induced pluripotent stem cells (hiPSC)-differentiated mature motor neurons from ALS patients (G) and their non-ALS controls (H) are arranged in clusters and show axonal projections. Number of differentiated motor neurons was quantified by the counting of ChAT+/DAPI profiles (I) and Hb9+/DAPI profiles (J) in sample regions of the coverslips and the results are expressed as percentage of total cells. Scale bars: 50 μm. RT-PCR analysis of neuronal gene markers of differentiated motor neurons and neural progenitor cells (K) contained in the 24-well plates. The markers of PAX6 (neural progenitor stage), OLIG2 (motor neuron progenitor stage), CHAT (mature motor neuron stage) and GAPDH (endogenous control gene) are evidenced. The expression of PAX6, OLIG2, CHAT, and HB9 was quantified in differentiated motor neurons contained in the 24-well plates by qPCR and the graphic shows the relative fold-change values; a pool of the neural progenitor cells was used as reference samples with a reference value of 1 (L).
RT-PCR and qPCR analyses of neuronal gene markers of neural progenitor cells and differentiated motor neurons contained in the 24-well plates are shown in the Figures 5K,L and Figure S4G of Supplementary Material. The marker PAX6 is present only in neural progenitor stage. Despite the very low OLIG2 signal seen by RT-PCR (Figure 5K), a higher level of signal amplification of OLIG2 was detected in differentiated motor neurons by qPCR (Figure 5L and Figure S4G of Supplementary Material). Furthermore, qPCR showed high expressions of CHAT and HB9 in the differentiated motor neurons compared to neural progenitor cells (Figure 5L and Figure S4G). Of note, an absence of Fluoro-Jade C staining was found in the 20 day-differentiated motor neurons, indicating that age as an adequate period for molecular study in order to avoid natural stress conditions of in vitro analysis.
Microarray Analysis
Microarray analysis demonstrated differentially expressed genes in the hiPSC-derived motor neurons from sporadic ALS and non-ALS subjects (Table S1). Statistical analyses have identified 1591 deregulated genes in differentiated motor neurons from sporadic ALS compared to control subjects. Of these 1591 genes, 526 were up-regulated and 1065 down-regulated in sporadic ALS compared to non-ALS subjects. The complete list of differentially expressed genes can be found in Supplementary Table S1.
Gene Ontology Terms Grouped by REVIGO
Differentially expressed genes based on GO and grouped by REVIGO could be characterized in clusters according to molecular function, e.g., mRNA binding, GTP binding, transcription (co)-repressor activities and low-density lipoprotein particle receptor binding (Figure S5); as well as gene clusters representing various biological processes such as macromolecule catabolism, synapse organization, aging, intracellular transport, negative regulation of DNA binding, death, cell death, mitotic cell cycle and response to inorganic substance (Figure S5). Detailed results for GO terms regarding the GO numbers and number of genes as well as the representative genes in each of these categories can be found in Tables S2, S3 in Supplementary Material. The Cellular Component indicated four GO term related with mitochondrion, specifically the mitochondrion (105 genes), mitochondrion part (65 genes), mitochondrion matrix (30 genes), and mitochondrion lumen (30 genes). The 105 genes of mitochondrion GO term are overlapped with all other GO terms (Table 5). The mean of gene expression normalized signal of 105 genes related to the mitochondrion of differentiated motor neurons from sporadic ALS and non-ALS samples are shown in Figure 6. Of note, the pattern of deregulated gene expression was consistent among the 3 lines of differentiated motor neurons of ALS and non-ALS groups, as exemplified by the absence of statistical differences of the mitochondrion gene expression within the each one of the two studied groups.
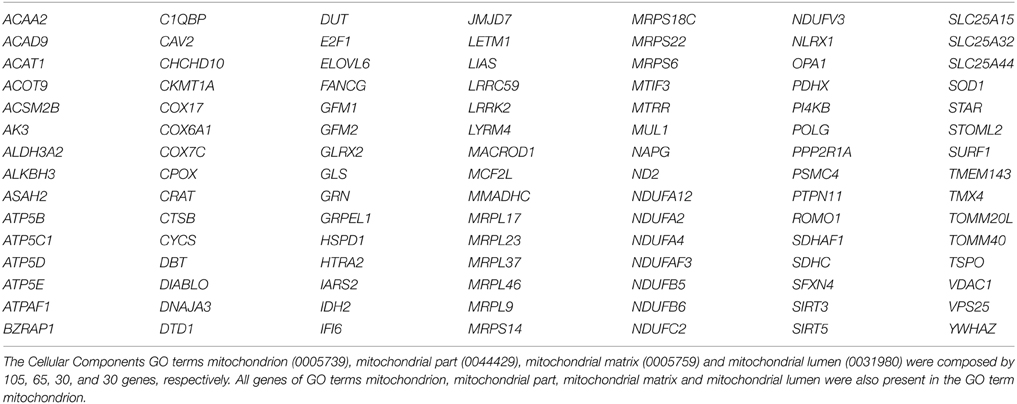
Table 5. Genes from GO terms related to the cellular component mitochondrion.
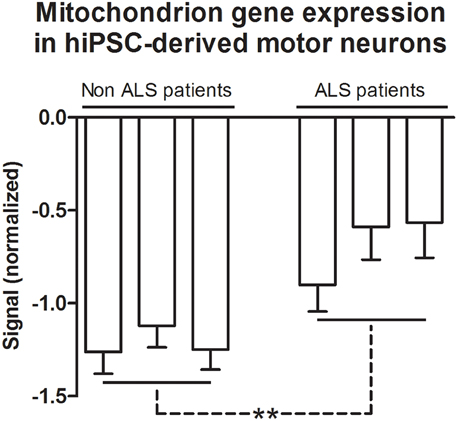
Figure 6. Mitochondrion gene expression in hiPSC-derived motor neurons. Gene expression normalized signals of 105 genes from GO terms of Cellular Components that are related to the mitochondrion. Bars represent the means ± SEM of the signals from three samples of differentiated motor neurons of non-ALS and ALS patients, as described in the text. The signal values (Cy5) were normalized by microarray reference (Cy3). The genes referred to GO terms mitochondrion (0005739), mitochondrial part (0044429), mitochondrial matrix (0005759), and mitochondrial lumen (0031980). See text and Table S4 for details. Differences within groups were analyzed by One-Way ANOVA followed by Tukey post-test, whereas differences between non-ALS and ALS groups were analyzed according to unpaired t-test **p < 0.01.
KEGG Analysis
Enriched KEGG terms were identified among differentially expressed genes (p < 0.05) in hiPSC-derived motor neurons of sporadic ALS and non-ALS subjects (Table 6). Importantly, differentially expressed genes associated with Parkinson's disease and oxidative phosphorylation are present as over-represented KEGG pathways. These pathways, and respective deregulated genes associated with them, are seen in Table 6. The prostate cancer pathway was omitted from the table because it was composed of genes unrelated to ALS.
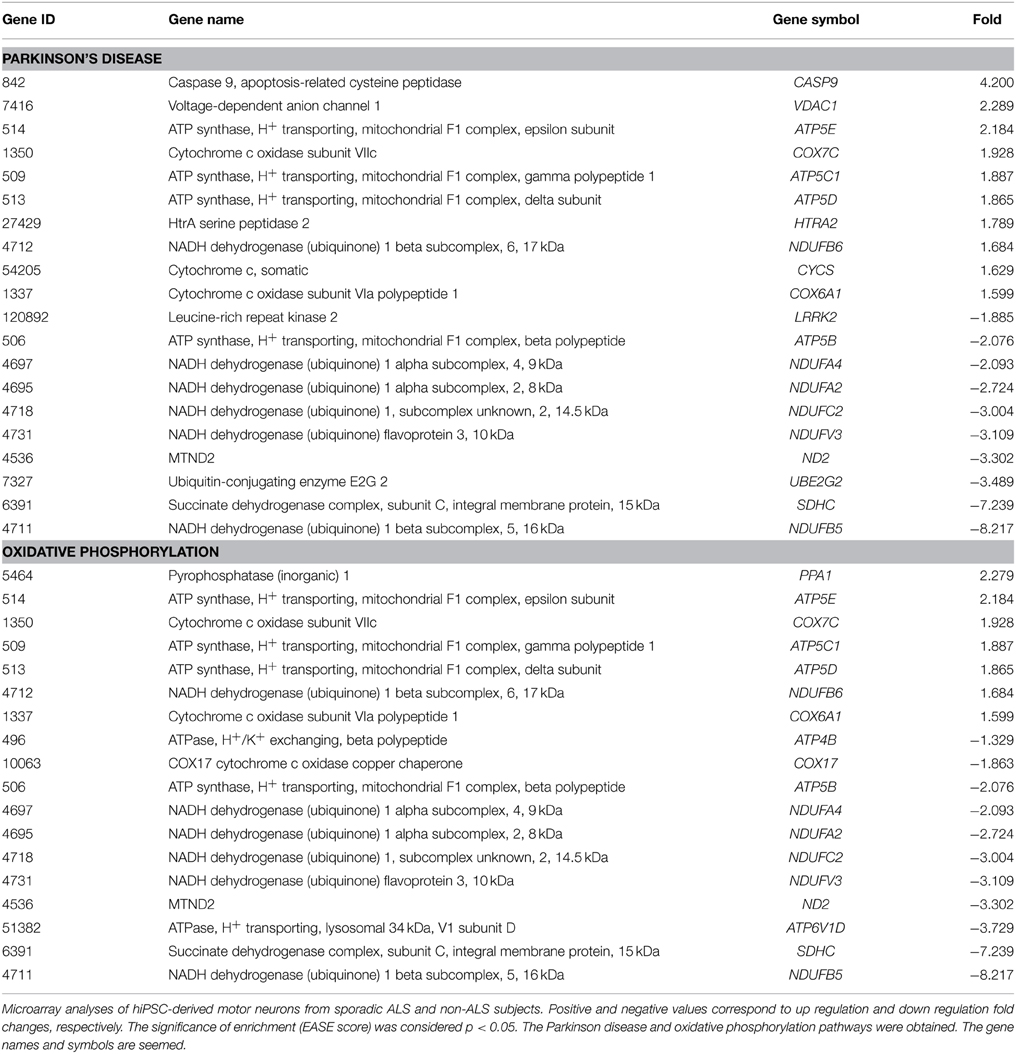
Table 6. KEGG pathways obtained from microarray analysis.
Verification of Microarray Results by qPCR
The results of qPCR for the 10 representative genes deregulated in differentiated motor neurons from sporadic ALS compared to non-ALS subjects are shown in Figure 7. Both the up (SDHAF1; 4.88-fold; CASP9; 7.64-fold; VPS35; 8.59-fold and IGF2; 13.52-fold) and down (DUSP6; –3.24-fold; DGAT1; –1.23-fold; KCNK12; –2.97-fold; KIFC1; –1.07-fold; KRTAP4-11; –2.46-fold; and LETM1; –3.03-fold) regulation observed for these selected genes, as determined by qPCR, were coincident with and supported our microarray results.
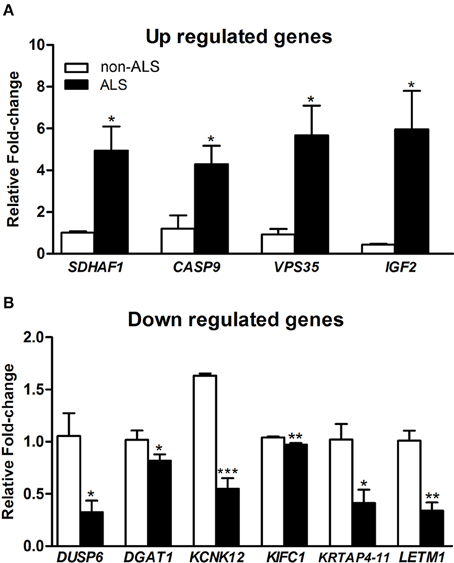
Figure 7. qPCR verification of deregulated genes. Graphs show relative fold change values of down regulated (A) and up regulated (B) of selected genes for verification in human-induced pluripotent stem cells (hiPSC)-differentiated motor neurons from sporadic ALS and non-ALS subjects. hiPSC were obtained from motor nerves of subjects. The selected genes were IGF2, VPS35, CASP9, HFE, and SDHF1 (A) and also DUSP6, DGAT1, KCNK12, KIFC1, KRTAP4-11, and LETM1 (B). A pool of the non-ALS group was used as reference samples with a reference value of 1. See text for details. Means ± SEM at least 3 replicates for each group. *, ** and *** p-values indicate the < 0.05, < 0.01 and < 0.001 levels of significance, respectively, according to unpaired t-test.
Discussion
Sporadic ALS is a complex disorder and patients present with a wide range of diverse clinical outcomes regarding disease onset, rate of progression and survival (Burkhardt et al., 2013). High throughput gene expression analysis using microarrays has not yet been described for iPSC-derived motor neurons from sporadic ALS patients, thus contributing to a lack of understanding of the mechanisms underlying neurodegeneration in this most common form of that human disease. The absence of clear knowledge regarding the etiopathogenesis and physiopathology of human sporadic ALS has been considered the major cause for failure to translate potential therapeutic strategies to the clinic. Indeed, most experimental studies resulting in therapeutic targets rely fundamentally on experiments using animal models carrying mutated genes of familial ALS (Beghi et al., 2007; Musarò, 2010). Of note, the patients of this study showed an early stage of ALS development, and the biopsy of the motor nerve was performed in a still functioning limb segment, thus the size of samples had to follow the roles of human ethical research. Despite that limitation, the results demonstrated the absence of internal variability.
Generation of hiPSC-derived Motor Neurons to Study Molecular Mechanisms in Sporadic ALS
Peripheral motor nerves of ALS patients were used for the first time to provide adult fibroblasts, in contrast to numerous previous studies using skin fibroblasts (Almeida et al., 2013; Zhang et al., 2013; Devlin et al., 2015), to be reprogrammed to hiPSCs. Motor nerves, as a source of adult cells for reprogramming, represent a logical choice with which to model ALS particularly since retraction of motor axons and denervation of the neuromuscular junction are the earliest events in the disease (Fischer et al., 2004; Schaefer et al., 2005; Pun et al., 2006). In fact, recent evidence describing neuronal dye-back mechanisms, combined with our own unpublished data, suggest that peripheral cells associated with motor axons, such as Schwann cells, may play a role in motor neuron degeneration in ALS (Keller et al., 2009; Maximino et al., 2014), particularly in the sporadic form. Furthermore, previous microarray studies also showed a differential pattern of gene expression in peripheral fibroblasts from ALS patients (Raman et al., 2015). These non-neuronal cells associated with peripheral motor nerves may provide additional information related to the pathogenic mechanisms of sporadic ALS once hiPSC-derived motor neurons could be produced from them. Of note, the growth and differentiation of peripheral motor nerve fibroblast-derived hiPSCs shown in this work seemed to be comparable to those hiPSCs derived from adult skin fibroblasts described elsewhere (Chestkov et al., 2014).
In support of this approach, in the present study, we initially generated integration-free iPS cell lines using Sendai virus -based individual delivery of each of the four Yamanaka reprogramming factors. However, a greater efficiency was achieved by using STEMCCA Cre-Excisable Constitutive Polycistronic Lentivirus. The combination of all factors in a single transcript using STEMCCA technology may be responsible for the increased efficiency of reprogramming (Awe et al., 2013; Kadari et al., 2014). In our study, this may have allowed the efficient production of hiPSCs from peripheral motor nerve fibroblasts and may have contributed to the subsequent success of motor neuron differentiation. Moreover, as far as we know, it is the first time that the STEMCCA methodology has been applied to generate hiPSC-derived motor neurons.
The potential for motor neuron differentiation achieved by means of the embryoid body method has been previously discussed by Hu and Zhang (2009). The neuronal differentiation process was initiated with the formation of cell aggregates in suspension (free-floating spheres). Furthermore, the subsequent stages involved in the acquisition of the precise motor neuron identities established by the neural progenitor cells, in a process that recapitulated those defined during normal mammalian embryonic development (Dasen and Jessell, 2009). The first step is the acquisition of a “spinal cord” character, i.e., caudalization, by means of extrinsic signals, i.e., retinoic acid. The cell specification along the dorsoventral axis, i.e., ventralization, occurs in response to Shh signaling (Jessell, 2000; Li et al., 2008). After that, differentiating neurons are maintained in the culture medium supplemented with specific neurotrophic factors to undergo further maturation, including the ability to produce specific neurotransmitters and to show specific morphological characteristics of adult motor neurons (Nizzardo et al., 2010). Because our objective was to assess early changes in the gene expression profile of differentiated motor neurons of sporadic ALS patients, microarray analyses were performed on day 20 of differentiation, a time point at which obtained motor neurons already expressed the specific markers ChAT and Hb9. Of note, we did not perform any electrophysiological tests related to mature neuron function on our iPSC-derived motor neurons. However, despite the OLIG2 expression shown by qPCR, the majority of differentiated motor neurons showed ChAT and also Hb9 markers at that culture age. Also, the cells possessed morphology of mature cells at that time. Thus, the results of CHAT and HB9 by qPCR together with high number of ChAT and Hb9 positive cells in our differentiated motor neuron culture conditions emphasize the state of early mature motor neurons in our system. Furthermore, the expression of OLIG2 seen in our system has been mentioned previously. In fact, expression of progenitress markers can be encountered in different types of transformed mature cells (Lian et al., 2012). The same should be true for IPSC-derived mature motor neurons because the co-expression of OLIG2, HB9, and CHAT were found at different stages of maturation (Hester et al., 2011; Jha et al., 2015), a process that may last for several weeks in culture. Nevertheless, the persistent presence of OLIG2 expression in motor neurons may not impair the differentiation process because generated motor neurons over-expressing Olig2, which were generated by a pLPCX-Olig2 retroviral transfection, were able to acquire morphological, biochemical and electrophysiological properties of mature cells (Lee et al., 2014). Nevertheless, it should be a matter of a next investigation whether a non-excisable system, as it is the case of the present study, may be responsible for the presence of OLIG2 in the mature differentiated motor neurons. Of note, despite the high number of differentiated motor neurons found in the coverslip, that sample analysis should not be taken as a parameter of reprograming/differentiation efficiency, which was not the scope of the present paper. A highly variable efficiency has been described in the literature, which is due to several factors related to differences of the protocols, iPSCs clones, substratum and also the limitation of the methodology to evaluate the final results (Hu and Zhang, 2009; Takazawa et al., 2012; Amoroso et al., 2013; Qu et al., 2014; Toli et al., 2015). Our qPCR analysis on regulation of transcripts of differentiated motor neuron markers may be a better method to document the type of cultured cell RNA that was evaluated in the microarray.
Furthermore, we decided to evaluate the gene profiling in the 20 day cultured ChAT and Hb9 expressed differentiated motor neurons in order to minimize the stress condition of in vitro assay and also because we were interested in the early molecular changes in the disease that could be related to the triggering of motor neuron death. Our decision to study molecular regulation on early mature differentiated motor neurons was also based on the fact that cultured motor neurons from ALS mouse start showing morphological alterations after 14 days in culture (Nagai et al., 2007), the expression of the mature marker may decrease in these cells from culture days 20–30 (Hester et al., 2011), and also because human derived motor neurons could show molecular instability in the process of maturation (Almeida et al., 2013). Despite the early age of differentiated mature motor neurons, that time point seems to be a safer stage to study molecular feature of the disease model in reprogrammed cells. Nevertheless, a detailed analysis of molecular changes at different phases of motor neuron maturation should be a matter of a future investigation.
The adequacy of sporadic ALS-derived iPSC-differentiated motor neurons for molecular studies is also highlighted by our results of the large gene profiling analysis, which is in line with profiles previously described using cells or tissues from post-mortem human ALS subjects or from animal models (Olsen et al., 2001; Dangond et al., 2004; Malaspina and De Belleroche, 2004; Jiang et al., 2005; Perrin et al., 2005; Ferraiuolo et al., 2007, 2011; Yamamoto et al., 2007; Offen et al., 2009; Brockington et al., 2010; D'Arrigo et al., 2010; Guipponi et al., 2010; Saris et al., 2013). Of note, we have not performed the excision of inserted cassette in our hiPSC cell lines to avoid an additional step of cell manipulation, what has not impaired the differentiation of mature Hb9 expressing motor neurons. There is a lack of detailed information regarding the presence and effects of residual reprogramming transgene expression the iPSCs-derived motor neurons. Based on the fact that residual reprogramming transgene expression from integrated viruses may alter the biological properties of iPSCs (Sommer et al., 2012), future work should analyze the gene profiling in hiPSC-derived motor neurons after silencing the exogenous factors. Despite the novelty of the methodological approach, the results of the present gene profiling in the differentiated motor neurons should be taken into attention before in vivo experiments because it is difficult to control all consequences of cell reprograming and also to avoid the stress conditions of any in vitro studies.
Importantly, the present gene profiling analysis of iPSC-derived motor neurons from sporadic ALS patients provides evidence of possible novel and autonomous mechanisms underlying neurodegeneration in the most abundant form of human ALS. It should be emphasized that previous descriptions on the regulation of gene expression in ALS are based solely on studies with animal models or human post-mortem tissue.
GO Molecular Function and Biological Processes from Sporadic ALS Motor Neuron Deregulated Genes
The microarray analyses identified 1591 deregulated genes in differentiated motor neurons from sporadic ALS patients compared to control subjects. The differentially expressed genes with a p-value lower than 0.05 were submitted to enrichment analyses based on GO and KEGG databases, according to our previous publications (de Oliveira et al., 2013; Maximino et al., 2014). Differentially expressed genes based on GO and grouped by REVIGO displayed molecular functions and biological processes consistent with mechanisms potentially contributing to the autonomous motor neuron cell death characteristic of sporadic ALS.
A wide range of terms of GO molecular function and biological processes grouped by REVIGO have already been implicated in ALS etiopathology. Moreover, their wide ranging and diverse activities underline the high degree of complexity of molecular and biochemical events taking place in the sporadic form of this disease. For instance, within the mRNA binding cluster (Takanashi and Yamaguchi, 2014; Štalekar et al., 2015), hnRNPA1, which codes for a heterogenous nuclear ribonucleoprotein A1, was up-regulated, and has been related to frontotemporal dementia and ALS (Seelen et al., 2014). The GTP binding cluster was previously correlated with ALS (Otomo et al., 2003; Droppelmann et al., 2014). However, the possible involvement of the down regulation of FKBP-4 in the cell-autonomous neurodegeneration characteristic of the sporadic form of ALS is an original contribution of this study. Furthermore, the deregulated FKBP-4, which belongs to the immunophilin chaperone protein family, may contribute to anti-aggregation processes of proteins widely described in ALS (Steiner et al., 1997; Gold et al., 1999; Manabe et al., 2002). Of note, the transcription (co)-repressor activity cluster (Tan et al., 2012; Dini Modigliani et al., 2014) revealed by REVIGO, identified NKX2 as an up-regulated gene product. NKX2 encodes a natural antisense RNA that modulates the expression of sense transcripts and mRNA processing, and might be involved in intracellular processes related to neuronal degeneration in sporadic ALS (Werner and Sayer, 2009). The synapse organization cluster (Shefner et al., 2006; Venkova et al., 2014) contains up-regulated MAP1B, which encodes a molecule linked to microtubule and synaptic stabilization and was found altered in spinal cords from ALS patients (Coyne et al., 2014).
Perhaps most significantly, REVIGO analysis has revealed a possible critical role for mitochondria in the diverse mechanisms contributing to neurodegeneration in ALS. Of note, the synapse organization cluster included the mitochondrion organization category in which the strongly deregulated genes, LETM1 and SDHF1, were identified (discussed below). Also, the intracellular transport cluster (Alami et al., 2014; Foran et al., 2014) contains the highly down-regulated gene, APOE, whose product is involved in mitochondrial oxidative stress in ALS, and its variants have been correlated to an increased risk of bulbar-onset of human ALS (Praline et al., 2011). Furthermore, the intracellular transport category also contains the significantly down-regulated KIF1A and KIFC1 genes (also included in several GO terms) whose protein products are responsible for anterograde axonal transport of mitochondria (De Vos et al., 2008; Hou and Yang, 2013), a process that is altered in ALS (De Vos et al., 2008). Additionally, the mitotic cell cycle cluster (Cova et al., 2010) revealed an up-regulation of DCTN1, encoding the p150 subunit of the axonal transport protein dynactin. Importantly, mutations in this gene have been associated with sporadic and familial ALS (Münch et al., 2004, 2005).
Significantly, REVIGO analysis provided direct evidence of mitochondrial involvement in the autonomous mechanisms underlying neurodegeneration in ALS with data associated with the cell death and death clusters (Martin et al., 2000; Tapia, 2014). These clusters contained the up-regulated gene, ATXN10, which has previously been associated with ALS (Figley et al., 2014), possibly involving mitochondria and activation of caspase 3 (White et al., 2010). Interestingly, the down-regulation of DYNLL1, also associated with this cluster, may be associated with impairment of dynein induced retrograde axonal transport of mitochondria in ALS (Chen et al., 2010).
Admittedly, it is difficult to separate the initial triggering from secondary events associated with stress of oncoming neurodegeneration based solely on results of the GO analysis of molecular function and biological process. Moreover, despite a huge diversity of events revealed by GO analysis, the general processes involving mitochondria may represent possible early autonomous mechanisms associated with sporadic ALS motor neurons. It should be stressed that mitochondrial functions also appear in several terms associated with cellular components of GO (data not shown).
KEGG Pathways from Sporadic ALS Motor Neuron Deregulated Genes
The DAVID analysis identified two specific KEGG pathways that might be related to ALS mechanisms of motor neuron degeneration in sporadic ALS, the Parkinson's disease and oxidative phosphorylation pathways. ALS and Parkinson's disease share some features and mechanisms of neuronal degeneration (Bosco et al., 2011; Shulman et al., 2011), thus emphasizing the relevance of the present molecular analysis employing human motor neurons. In fact, there is growing evidence from work of the past two decades suggesting that ALS may show a complex combination of events at both molecular and cellular levels that are common to Parkinsonism and frontotemporal dementia (Espay et al., 2011). For instance, TARDBP mutations (Mosca et al., 2012), TDP-43 proteinopathy (Mackenzie et al., 2010) and C9orf72 expansion (Lagier-Tourenne et al., 2013) are common to all three pathologies.
The oxidative phosphorylation pathway identified in the KEEG analysis is consistent with mitochondrial participation in sporadic ALS pathology (Ladd et al., 2014). For example, impairments of mitochondrial bioenergetics and energy metabolism, oxidative stress and apoptosis, among other features have been previously described (Cassina et al., 2008; Hedlund et al., 2010; Dupuis et al., 2011; Reddy et al., 2011; Cozzolino and Carrì, 2012; Federico et al., 2012; Brockington et al., 2013; Heath et al., 2013).
The importance of the above two KEEG pathways in the autonomous mechanisms related to neurodegeneration in sporadic ALS is highlighted by recent descriptions of the involvement of their genes in the degeneration of motor neurons in ALS and other neurodegenerative disorders related to ALS. For instance, LRRK2 mutations are associated with Parkinsonism, ALS and dementia (Wszolek et al., 1997; Zimprich et al., 2004), leading to mitochondrial fragmentation, mitochondrial dysfunction and increased reactive oxygen species (Whittle et al., 2007; Wang et al., 2012). Thus, the down regulation of LRRK2 in iPSC-derived motor neurons from sporadic ALS subjects in the present study provides further confirmation of the importance of mitochondrial dysfunction in this neurodegenerative disorder.
The down regulation of ATP5B we have identified in differentiated motor neurons from sporadic ALS patients in the present microarray analysis correlates with previous findings of reduced cytochrome oxidase and ATP synthase activities described in neurodegenerative disorders (Schägger and Ohm, 1995; Bosetti et al., 2002; Sergeant et al., 2003; Basso et al., 2004; Pamplona et al., 2005). For example, ATP5B deregulation was previously characterized in western Pacific ALS-parkinsonism-dementia complex (Shiraki, 1975).
Mitochondrial involvement in the autonomous motor neuron cell death in sporadic ALS is also supported by the up-regulation of VDAC1 seen in the microarray analyses. VDAC1 encodes for a voltage dependent anion channel in the outer mitochondrial membrane, and thus is able to regulate local ATP/ADP flux (Colombini, 2004; Lemasters and Holmuhamedov, 2006). Interestingly, VDAC1 is also a key player in mitochondria-mediated apoptosis and has been implicated in apoptotic-relevant mitochondrial events (Shimizu et al., 1999; Shoshan-Barmatz et al., 2006, 2008; Tajeddine et al., 2008; Abu-Hamad et al., 2009; Arbel and Shoshan-Barmatz, 2010). Misfolded mutant SOD1 directly binds and inhibits VDAC1 conductance, thus impacting the onset and survival in the ALS mouse model (Israelson et al., 2010). Furthermore, the deregulation of VDAC1 may also trigger neuronal damage by a direct interference in the regulation of mitochondrial reactive oxygen species (Liu et al., 2004; Vande Velde et al., 2004) or by a direct activation of mitochondrial apoptotic pathways (Tsujimoto and Shimizu, 2002; Shoshan-Barmatz et al., 2006; Yagoda et al., 2007; Abu-Hamad et al., 2008).
Additional evidence in support of mitochondrial dysfunction in autonomous sporadic ALS is provided by the demonstration that significant up-regulation of HFE may be associated with iron homeostasis. HFE mutations have been linked to human ALS (Rothstein, 2009; Kwan et al., 2012) and changes in iron metabolism may confer susceptibility to this disease in both humans (Li et al., 2014) and animals (Nandar et al., 2014). Because mitochondria regulate intracellular iron homeostasis (Horowitz and Greenamyre, 2010), HFE deregulation may represent an additional mitochondrial-based pathogenic mechanism in sporadic ALS. Mitochondrial dysfunction leading to cell autonomous neuronal damage in sporadic ALS is also supported by the significant deregulation of both TOMM20L (Ryan and Gogvadze, 1999) and HtrA2/Omi (Srinivasula et al., 2003), as seen in our microarray analysis.
Finally, many of the genes identified as being dysregulated in our microarray analysis have not yet been previously associated with ALS in any context. However, some (eg. ATP5D, ATP5E, NDUFA4, NDUFB5, NDUFB6) have been implicated in other neurodegenerative disorders (Chen et al., 2014; Suszyńska-Zajczyk et al., 2014). Additionally, NDUFC2, ND2, COX7C, NDUFV3, SDHC, ATP5C1, COX6A1, ATP4B, ATP6V1D, and COX17 are associated with mitochondrial function or impaired bioenergetic metabolism, and thus further support a role for mitochondrial dysfunction in the cell autonomous degeneration of motor neurons in sporadic ALS (Burman et al., 2014).
GO Mitochondrion Genes of the Cellular Component
Regulation of some of the mitochondrial genes and their related proteins of the cellular component mitochondrion described in the microarray analysis have been mentioned in the context of neuronal degeneration and ALS. For instance, C1QBP, CAV2, CTSB, DIABLO, GLRX2, NDUFA4, NLRX1, OPA1, SDHC, SIRT3, SURF1 have been correlated to neuronal death and to excitotoxic events (Manfredi and Beal, 2000; Weiergräber et al., 2007; Hayashi et al., 2009; Bräutigam et al., 2011; Gray et al., 2013; Tian et al., 2013; Imbeault et al., 2014). Some of them were related to complex cellular mechanisms of neurodegenerative disorders, also involving other cellular organelles as it is the case of CTSB in lysosomal function (Sevenich et al., 2006; Cho et al., 2013). Further indication of the importance of mitochondrion gene regulation in the mechanisms of ALS is highlighted with the descriptions of mitochondrial oxidative stress leading SDHC mutation-induced apoptosis (Ishii et al., 2011) and of neuroprotection after inhibition of mitochondrial release of the pro-apoptotic protein Smac/DIABLO (Soustiel and Larisch, 2010; Luan et al., 2012).
The genes ATP5B, E2F1, HTRA2, LETM1, LRRK2, POLG, SIRT3, SOD1, VDAC1 have been specifically described in the context of neuronal death in ALS. For instance, the gene polymerase gamma (POLG), which is related to mitochondrial biogenesis, was investigated in the context of ALS (Ladd et al., 2014). The mitochondrial protein ATP5B was also correlated to pathogenesis of neurodegenerative disorders (Yoon et al., 2009), actually the Western Pacific ALS-parkinsonism-dementia complex (Kisby et al., 2006). Mitochondrial proteins involved in cell-cycle and transcriptional regulation, i.e., E2F1, participate in the neuronal death pathways (Wu et al., 2012) and in ALS (Ranganathan and Bowser, 2003). Also, impairments of the apoptotic intermembrane mitochondrial serine protease HTRA2 lead to its accumulation in motor neuronal inclusions in ALS (Kawamoto et al., 2010) and neuronal death (Patterson et al., 2014). Importantly, the mitochondrial Ca(2+) transporter LETM1 is elevated in death resistant motor neurons of ALS mice. Conversely, LRRK2 was implicated in early neuronal death (Chen et al., 2012; Chou et al., 2014) while SIRT3 may exert neuroprotection by avoiding mitochondrial fragmentation in ALS mouse (Song et al., 2013), indicating the complex role of mitochondria in neurodegenerative diseases (Mühling et al., 2014). Furthermore, dysfunction of the mitochondrial voltage-dependent anion channel (VDAC) was associated with neuroprotection failure (Fernandez-Echevarria et al., 2014) and neurodegeneration in ALS (Israelson et al., 2010). Of note, a specific pathogenic LRRK2 mutation was described in ALS (Whittle et al., 2007). It should be noticed that the role of SOD1 in mitochondrial mechanism related to ALS has been largely described (Dupuis et al., 2004).
Finally, the PSMC4, GRN, and ND2 encoded genes of mitochondrial proteins have also been described in ALS, as it is the case of the 26S proteasome regulator PSMC4 (Trippier et al., 2014). Interestingly, a co-occurrence of GRN/C9ORF72 (Testi et al., 2015) and of NADH dehydrogenase subunit 2 (ND2) mutations were also detected in ALS patients (Lin et al., 1992).
It should be noticed that mutations of CHCHD10 have been described in ALS, a gene that encodes an intermembrane mitochondrial protein involved in mitochondrial genome stability and morphology (Bannwarth et al., 2014; Johnson et al., 2014).
It is well known that the regulation of mitochondrial gene expression is not restricted to pathological conditions but instead to a large range of cellular physiological conditions. However, we have demonstrated in our work no variations of mitochondrial gene expression within the groups but a significant difference between non-ALS and ALS subjects, emphasizing a possible participation of deregulated genes in the motor neurons disease. Actually, statistical significance between groups and small intragroup variability indicated an homogeneity of the samples.
qPCR Verification of Sporadic ALS Motor Neuron Deregulated Genes
The results of the qPCR and microarray analyses correlated well for all selected genes, confirming the efficacy of large gene profiling in the search for therapeutic targets in neurodegenerative disorders (de Oliveira et al., 2013; Maximino et al., 2014). Notably, a relationship between these deregulated genes with various molecular/biological processes related to intracellular trafficking, intercellular signaling, and neurotrophic functions (Vucic and Kiernan, 2009; Martin, 2012; Chaturvedi and Flint Beal, 2013) is supportive of a role for mitochondrial dysfunction in ALS, as previously postulated for other neurodegenerative diseases (Pagano et al., 2014) Furthermore, a critical analysis of the up and down regulated genes by qPCR supports their roles in both the events initiating neuronal damage and subsequent reactive events leading to neuroprotection.
Of note, LETM1, which encodes a mitochondrial Ca2+/H+ exchanger protein (Waldeck-Weiermair et al., 2011; Nowikovsky et al., 2012) was found to be up-regulated in iPSC-developed motor neurons of sporadic ALS patients. This mitochondrial ion exchange protein has been linked to protection of rescued neurons in the ALS mouse model (Mühling et al., 2014), possibly by its ability to regulate Ca2+ homeostasis. The high down regulation of LETM1 in sporadic ALS motor neurons supports a role for mitochondrial dysfunction in the cell-autonomous failure of neuroprotection. Moreover, DUSP6 was the most significantly down regulated gene identified by the microarray analyses. This gene encodes for a dual specificity phosphatase-6 protein, which is a critical determinant of ERK signaling (Bhalla et al., 2002), and is thus able to influence cell survival after neurotoxicity (Hetman and Gozdz, 2004). As ERK signaling has been implicated in ALS (Kim and Choi, 2010), a strong down-regulation of DUSP6 in sporadic ALS motor neurons is worthy of further investigation. Of note, despite of microarray and qPCR have shown a down regulation of DUSP6, the degree of regulation demonstrated by microarray was higher than that obtained by qPCR analysis The use of qPCR analysis to qualitatively verify the microarray results is largely accepted in the literature. However, small discrepancies in the degree of gene regulation between the two methods have been reported (Ferraiuolo et al., 2007). These methods have their specific quantitative differences (Chuaqui et al., 2002), which are thought to be related to the variation in the hybridization kinetics of the technologies, low fold changes or lack of concordance between transcripts accessed in each method (de Oliveira et al., 2013).
Dysfunction in lipid metabolism has also been associated with ALS (Schmitt et al., 2014; Palamiuc et al., 2015). The present study is the first to demonstrate a significant deregulation of DGAT1, a gene which encodes a multipass transmembrane protein that functions as a key metabolic enzyme in the conversion of diacylglycerol and fatty acetyl CoA to triacylglycerol, in the context of human ALS.
Also, specific processes involving potassium channels have been described in non-autonomous glial mechanisms of ALS (Bataveljić et al., 2012; Sato et al., 2014). However, this work contributed to the original description of this potassium channel's (subfamily K, member 12 gene, KCNK12) deregulation in differentiated human motor neurons from sporadic ALS patients.
The contribution of kinesins in the deregulated axonal transport (Landers et al., 2009; Shi et al., 2010; Kuzma-Kozakiewicz et al., 2013) as well as the participation of cytoskeleton components (Maximino et al., 2014) in ALS have been well documented. However, the significant down regulation in the genes, KIFC-1 and KRTAP4-11, point to the possibility of abnormalities in intracellular trafficking and cytoskeletal structural changes respectively, in the differentiated motor neurons from sporadic ALS patients.
The up-regulation of CASP9, which encodes for caspase-9 protein, might activate mitochondria-dependent apoptotic signaling, an event that has been described in neurodegenerative diseases (Andreoli et al., 2009; Darwish and Amiridze, 2010) including ALS (Pasinelli et al., 2000; Guégan et al., 2001, 2002; Inoue et al., 2003). Interestingly, caspase-9 levels have been correlated with clinical severity in ALS patients (Ilzecka, 2012). Considering the disease-specific phenotype of the iPSC-derived motor neurons in the present analysis, an early dysfunction of caspase-9 mediating motor neuron death may take place in sporadic ALS. Furthermore, the up regulation SDHAF1, which encodes for succinate dehydrogenase complex, an enzyme that participates in the respiratory chain and Krebs cycle (Rutter et al., 2010) further implicates mitochondrial participation in sporadic ALS.
Conversely, distinct from the putative roles of CASP9 and SDHAF-1, the up-regulation of IGF2 and VPS35 may indicate the occurrence of reactive autocrine neuroprotective responses in the sporadic ALS-differentiated motor neurons after an initial damage. Thus, it is likely that up-regulation of IGF2 may be involved in neuroprotection of motor neurons (Leventhal et al., 1999; Silva et al., 2009), in line to its previously suggested role in non-autonomous glial ALS mechanisms (Kihira et al., 2007; Dagvajantsan et al., 2008). The fact that IGF-1 treatment led to neuroprotection in an ALS animal model (Jablonka et al., 2011; Saenger et al., 2012) and to partial benefits in ALS clinical trials (Lai et al., 1997; Borasio et al., 1998), further supports our finding that up-regulation of IGF2 expression in the differentiated motor neurons from sporadic ALS patients highlights the importance of IGF family proteins as potential therapeutic targets for this disease. Accordingly, the regulation of VPS35 corroborates our findings with IGF2. The VPS35 gene product is involved in retrograde transport of proteins from endosomes to the Golgi complex (Zhang et al., 2001). VPS35 mutations and disruption of endosomal trafficking has previously been implicated in other neurodegenerative disorders (Small et al., 2005; Zimprich et al., 2011; Ando et al., 2012; Vilariño-Güell et al., 2014; Perrett et al., 2015). Thus, up-regulation of VPS35 might account for a neuroprotective response in ALS before degeneration of motor neurons begins.
In conclusion, the large gene profiling of early stage mature motor neurons differentiated from hiPSCs, which in turn, were derived from adult motor nerve associated fibroblasts from sporadic ALS subjects, has provided evidence for a wide range of molecular and cellular mechanisms possibly involved in the cell autonomous neuronal death in the disease. Of note, evidence supporting impairment of intracellular trafficking, intercellular signaling, oxidative phosphorylation and neurotrophic factor function among others, strongly implicate mitochondrial dysfunction as a key factor in this cell-autonomous neurodegeneration process. Interestingly, evidence is also provided that autocrine neuroprotective mechanisms coexist with those related to cell toxicity in these human sporadic ALS differentiated motor neurons. These results thus emphasize the importance of the use of human iPSC-derived motor neurons, obtained from motor nerve fibroblasts, as a reliable method to model disease mechanisms associated with neuronal degeneration in sporadic ALS. Importantly, the approach may also provide a viable opportunity to translate these results to the development of potential therapeutic targets specific to sporadic ALS.
Author Contributions
CA, RD, MM, and JM performed the experiments. FJ, GC, RM, and DC selected the patients and performed the biopsies. CA, JM, BS, JK, and GC designed the study, analyzed the results and wrote the manuscript. GC is responsible for the ALS Brazil Project of the University of São Paulo School of Medicine. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank professor Lygia da Veiga Pereira (Bioscience Institute of University of São Paulo) for providing hESC and valuable advice during the generation of hiPSCs. We thank professor Robert Carlone of Brock University for reading the manuscript and for his valuable suggestions and we also thank Leonardo Jensen and Graça Correa Rosas for helping with the preparation of iPSC cardiomyocytes. This work was supported by grants from the São Paulo Research Foundation (FAPESP; #2010/20457-7, #2010/20467-2) and National Council for Scientific and Technological Development (CNPq).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fncel.2015.00289
References
Abu-Hamad, S., Arbel, N., Calo, D., Arzoine, L., Israelson, A., Keinan, N., et al. (2009). The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 122, 1906–1916. doi: 10.1242/jcs.040188
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490. doi: 10.1074/jbc.M708216200
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Almeida, S., Gascon, E., Tran, H., Chou, H. J., Gendron, T. F., Degroot, S., et al. (2013). Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 126, 385–399. doi: 10.1007/s00401-013-1149-y
Amoroso, M. W., Croft, G. F., Williams, D. J., O'Keeffe, S., Carrasco, M. A., Davis, A. R., et al. (2013). Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci. 33, 574–586. doi: 10.1523/JNEUROSCI.0906-12.2013
Ando, M., Funayama, M., Li, Y., Kashihara, K., Murakami, Y., Ishizu, N., et al. (2012). VPS35 mutation in Japanese patients with typical Parkinson's disease. Mov. Disord. 27, 1413–1417. doi: 10.1002/mds.25145
Andreoli, V., Trecroci, F., La Russa, A., Valentino, P., Condino, F., Latorre, V., et al. (2009). CASP-9: a susceptibility locus for multiple sclerosis in Italy. J. Neuroimmunol. 210, 100–103. doi: 10.1016/j.jneuroim.2009.03.013
Arbel, N., and Shoshan-Barmatz, V. (2010). Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 285, 6053–6062. doi: 10.1074/jbc.M109.082990
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Awe, J. P., Lee, P. C., Ramathal, C., Vega-Crespo, A., Durruthy-Durruthy, J., Cooper, A., et al. (2013). Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res. Ther. 4, 87. doi: 10.1186/scrt246
Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. doi: 10.1093/brain/awu138
Basso, M., Giraudo, S., Corpillo, D., Bergamasco, B., Lopiano, L., and Fasano, M. (2004). Proteome analysis of human substantia nigra in Parkinson's disease. Proteomics 4, 3943–3952. doi: 10.1002/pmic.200400848
Bataveljić, D., Nikolić, L., Milosević, M., Todorović, N., and Andjus, P. R. (2012). Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia 60, 1991–2003. doi: 10.1002/glia.22414
Beghi, E., Mennini, T., Bendotti, C., Bigini, P., Logroscino, G., Chiò, A., et al. (2007). The heterogeneity of amyotrophic lateral sclerosis: a possible explanation of treatment failure. Curr. Med. Chem. 14, 3185–3200. doi: 10.2174/092986707782793862
Benatar, M. (2007a). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13. doi: 10.1016/j.nbd.2006.12.015
Benatar, M. (2007b). What zebras and mice can teach us about familial ALS. Neuromuscul. Disord. 17, 671–672. doi: 10.1016/j.nmd.2007.06.005
Bhalla, U. S., Ram, P. T., and Iyengar, R. (2002). MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297, 1018–1023. doi: 10.1126/science.1068873
Borasio, G. D., Robberecht, W., Leigh, P. N., Emile, J., Guiloff, R. J., Jerusalem, F., et al. (1998). A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 51, 583–586. doi: 10.1212/WNL.51.2.583
Bosco, D. A., LaVoie, M. J., Petsko, G. A., and Ringe, D. (2011). Proteostasis and movement disorders: Parkinson's disease and amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Biol. 3:a007500. doi: 10.1101/cshperspect.a007500
Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., et al. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol. Aging 23, 371–376. doi: 10.1016/S0197-4580(01)00314-1
Bräutigam, L., Schütte, L. D., Godoy, J. R., Prozorovski, T., Gellert, M., Hauptmann, G., et al. (2011). Vertebrate-specific glutaredoxin is essential for brain development. Proc. Natl. Acad. Sci. U.S.A. 108, 20532–20537. doi: 10.1073/pnas.1110085108
Brockington, A., Heath, P. R., Holden, H., Kasher, P., Bender, F. L., Claes, F., et al. (2010). Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis. BMC Genomics 11:203. doi: 10.1186/1471-2164-11-203
Brockington, A., Ning, K., Heath, P. R., Wood, E., Kirby, J., Fusi, N., et al. (2013). Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125, 95–109. doi: 10.1007/s00401-012-1058-5
Brooks, B. R., Miller, R. G., Swash, M., and Munsat, T. L. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi: 10.1016/j.mcn.2013.07.007
Burman, J. L., Itsara, L. S., Kayser, E. B., Suthammarak, W., Wang, A. M., Kaeberlein, M., et al. (2014). A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model. Mech. 7, 1165–1174. doi: 10.1242/dmm.015321
Cassina, P., Cassina, A., Pehar, M., Castellanos, R., Gandelman, M., De León, A., et al. (2008). Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J. Neurosci. 28, 4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008
Chaturvedi, R. K., and Flint Beal, M. (2013). Mitochondrial diseases of the brain. Free Radic. Biol. Med. 63, 1–29. doi: 10.1016/j.freeradbiomed.2013.03.018
Chen, C. M., Weng, Y. T., Chen, W. L., Lin, T. H., Chao, C. Y., Lin, C. H., et al. (2014). Aqueous extract of Glycyrrhiza inflata inhibits aggregation by upregulating PPARGC1A and NFE2L2-ARE pathways in cell models of spinocerebellar ataxia 3. Free Radic. Biol. Med. 71, 339–350. doi: 10.1016/j.freeradbiomed.2014.03.023
Chen, C. Y., Weng, Y. H., Chien, K. Y., Lin, K. J., Yeh, T. H., Cheng, Y. P., et al. (2012). (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 19, 1623–1633. doi: 10.1038/cdd.2012.42
Chen, K., Northington, F. J., and Martin, L. J. (2010). Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 214, 219–234. doi: 10.1007/s00429-009-0226-4
Chestkov, I. V., Vasilieva, E. A., Illarioshkin, S. N., Lagarkova, M. A., and Kiselev, S. L. (2014). Patient-specific induced pluripotent stem cells for SOD1-associated amyotrophic lateral sclerosis pathogenesis studies. Acta Naturae 6, 54–60.
Cho, K., Yoon, S. Y., Choi, J. E., Kang, H. J., Jang, H. Y., and Kim, D. H. (2013). CA-074Me, a cathepsin B inhibitor, decreases APP accumulation and protects primary rat cortical neurons treated with okadaic acid. Neurosci. Lett. 548, 222–227. doi: 10.1016/j.neulet.2013.05.056
Cho, Y. H., Han, K. M., Kim, D., Lee, J., Lee, S. H., Choi, K. W., et al. (2014). Autophagy regulates homeostasis of pluripotency-associated proteins in hESCs. Stem Cells 32, 424–435. doi: 10.1002/stem.1589
Chou, J. S., Chen, C. Y., Chen, Y. L., Weng, Y. H., Yeh, T. H., Lu, C. S., et al. (2014). (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol. Dis. 68, 190–199. doi: 10.1016/j.nbd.2014.04.021
Chuaqui, R. F., Bonner, R. F., Best, C. J., Gillespie, J. W., Flaig, M. J., Hewitt, S. M., et al. (2002). Post-analysis follow-up and validation of microarray experiments. Nat. Genet. 32(Suppl.), 509–514. doi: 10.1038/ng1034
Cleveland, D. W., and Rothstein, J. D. (2001). From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819. doi: 10.1038/35097565
Colombini, M. (2004). VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 256–257, 107–115. doi: 10.1023/B:MCBI.0000009862.17396.8d
Cova, E., Ghiroldi, A., Guareschi, S., Mazzini, G., Gagliardi, S., Davin, A., et al. (2010). G93A SOD1 alters cell cycle in a cellular model of amyotrophic lateral sclerosis. Cell. Signal. 22, 1477–1484. doi: 10.1016/j.cellsig.2010.05.016
Coyne, A. N., Siddegowda, B. B., Estes, P. S., Johannesmeyer, J., Kovalik, T., Daniel, S. G., et al. (2014). Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 34, 15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Dagvajantsan, B., Aoki, M., Warita, H., Suzuki, N., and Itoyama, Y. (2008). Up-regulation of insulin-like growth factor-II receptor in reactive astrocytes in the spinal cord of amyotrophic lateral sclerosis transgenic rats. Tohoku J. Exp. Med. 214, 303–310. doi: 10.1620/tjem.214.303
Dangond, F., Hwang, D., Camelo, S., Pasinelli, P., Frosch, M. P., Stephanopoulos, G., et al. (2004). Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genomics 16, 229–239. doi: 10.1152/physiolgenomics.00087.2001
D'Arrigo, A., Colavito, D., Peña-Altamira, E., Fabris, M., Dam, M., Contestabile, A., et al. (2010). Transcriptional profiling in the lumbar spinal cord of a mouse model of amyotrophic lateral sclerosis: a role for wild-type superoxide dismutase 1 in sporadic disease? J. Mol. Neurosci. 41, 404–415. doi: 10.1007/s12031-010-9332-2
Darwish, R. S., and Amiridze, N. S. (2010). Detectable levels of cytochrome C and activated caspase-9 in cerebrospinal fluid after human traumatic brain injury. Neurocrit. Care 12, 337–341. doi: 10.1007/s12028-009-9328-3
Dasen, J. S., and Jessell, T. M. (2009). Hox networks and the origins of motor neuron diversity. Curr. Top. Dev. Biol. 88, 169–200. doi: 10.1016/S0070-2153(09)88006-X
Dejesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
de Oliveira, G. P., Alves, C. J., and Chadi, G. (2013). Early gene expression changes in spinal cord from SOD1(G93A) amyotrophic lateral sclerosis animal model. Front. Cell. Neurosci. 7:216. doi: 10.3389/fncel.2013.00216
de Oliveira, G. P., Maximino, J. R., Maschietto, M., Zanoteli, E., Puga, R. D., Lima, L., et al. (2014). Early gene expression changes in skeletal muscle from SOD1(G93A) amyotrophic lateral sclerosis animal model. Cell. Mol. Neurobiol. 34, 451–462. doi: 10.1007/s10571-014-0029-x
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 5999. doi: 10.1038/ncomms6999
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173. doi: 10.1146/annurev.neuro.31.061307.090711
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3, 637–648. doi: 10.1016/j.stem.2008.09.017
Dini Modigliani, S., Morlando, M., Errichelli, L., Sabatelli, M., and Bozzoni, I. (2014). An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat. Commun. 5, 4335. doi: 10.1038/ncomms5335
Droppelmann, C. A., Campos-Melo, D., Volkening, K., and Strong, M. J. (2014). The emerging role of guanine nucleotide exchange factors in ALS and other neurodegenerative diseases. Front. Cell. Neurosci. 8:282. doi: 10.3389/fncel.2014.00282
Dupuis, L., Gonzalez De Aguilar, J. L., Oudart, H., De Tapia, M., Barbeito, L., and Loeffler, J. P. (2004). Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener. Dis. 1, 245–254. doi: 10.1159/000085063
Dupuis, L., Pradat, P. F., Ludolph, A. C., and Loeffler, J. P. (2011). Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 10, 75–82. doi: 10.1016/S1474-4422(10)70224-6
Espay, A. J., Spina, S., Houghton, D. J., Murrell, J. R., De Courten-Myers, G. M., Ghetti, B., et al. (2011). Rapidly progressive atypical parkinsonism associated with frontotemporal lobar degeneration and motor neuron disease. J. Neurol. Neurosurg. Psychiatry 82, 751–753. doi: 10.1136/jnnp.2009.201608
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. doi: 10.1038/292154a0
Federico, A., Cardaioli, E., Da Pozzo, P., Formichi, P., Gallus, G. N., and Radi, E. (2012). Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 322, 254–262. doi: 10.1016/j.jns.2012.05.030
Fernandez-Echevarria, C., Díaz, M., Ferrer, I., Canerina-Amaro, A., and Marin, R. (2014). Abeta promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer's disease. Neuroscience 278, 354–366. doi: 10.1016/j.neuroscience.2014.07.079
Ferraiuolo, L., Heath, P. R., Holden, H., Kasher, P., Kirby, J., and Shaw, P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641. doi: 10.1093/brain/awr193
Figley, M. D., Thomas, A., and Gitler, A. D. (2014). Evaluating noncoding nucleotide repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 35, 936.e931–e934. doi: 10.1016/j.neurobiolaging.2013.09.024
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Foran, E., Rosenblum, L., Bogush, A., Pasinelli, P., and Trotti, D. (2014). Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia 62, 1241–1253. doi: 10.1002/glia.22677
Fraga, A. M., Sukoyan, M., Rajan, P., Braga, D. P., Iaconelli, A. Jr., Franco, J. G. Jr., et al. (2011). Establishment of a Brazilian line of human embryonic stem cells in defined medium: implications for cell therapy in an ethnically diverse population. Cell Transplant. 20, 431–440. doi: 10.3727/096368910X522261
Gold, B. G., Densmore, V., Shou, W., Matzuk, M. M., and Gordon, H. S. (1999). Immunophilin FK506-binding protein 52 (not FK506-binding protein 12) mediates the neurotrophic action of FK506. J. Pharmacol. Exp. Ther. 289, 1202–1210.
Gordon, P. H., and Meininger, V. (2011). How can we improve clinical trials in amyotrophic lateral sclerosis? Nat. Rev. Neurol. 7, 650–654. doi: 10.1038/nrneurol.2011.147
Gowing, G., Lalancette-Hébert, M., Audet, J. N., Dequen, F., and Julien, J. P. (2009). Macrophage colony stimulating factor (M-CSF) exacerbates ALS disease in a mouse model through altered responses of microglia expressing mutant superoxide dismutase. Exp. Neurol. 220, 267–275. doi: 10.1016/j.expneurol.2009.08.021
Gray, J. J., Zommer, A. E., Bouchard, R. J., Duval, N., Blackstone, C., and Linseman, D. A. (2013). N-terminal cleavage of the mitochondrial fusion GTPase OPA1 occurs via a caspase-independent mechanism in cerebellar granule neurons exposed to oxidative or nitrosative stress. Brain Res. 1494, 28–43. doi: 10.1016/j.brainres.2012.12.001
Gros-Louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972. doi: 10.1016/j.bbadis.2006.01.004
Guégan, C., Vila, M., Rosoklija, G., Hays, A. P., and Przedborski, S. (2001). Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 21, 6569–6576.
Guégan, C., Vila, M., Teismann, P., Chen, C., Onteniente, B., Li, M., et al. (2002). Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol. Cell. Neurosci. 20, 553–562. doi: 10.1006/mcne.2002.1136
Guipponi, M., Li, Q. X., Hyde, L., Beissbarth, T., Smyth, G. K., Masters, C. L., et al. (2010). SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J. Mol. Neurosci. 41, 172–182. doi: 10.1007/s12031-009-9317-1
Hallewell, R. A., Mullenbach, G. T., Stempien, M. M., and Bell, G. I. (1986). Sequence of a cDNA coding for mouse manganese superoxide dismutase. Nucleic Acids Res. 14, 9539. doi: 10.1093/nar/14.23.9539
Hayashi, T., Ishimori, C., Takahashi-Niki, K., Taira, T., Kim, Y. C., Maita, H., et al. (2009). DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 390, 667–672. doi: 10.1016/j.bbrc.2009.10.025
Heath, P. R., Kirby, J., and Shaw, P. J. (2013). Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front. Cell. Neurosci. 7:259. doi: 10.3389/fncel.2013.00259
Hedlund, E., Karlsson, M., Osborn, T., Ludwig, W., and Isacson, O. (2010). Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 133, 2313–2330. doi: 10.1093/brain/awq167
Hester, M. E., Murtha, M. J., Song, S., Rao, M., Miranda, C. J., Meyer, K., et al. (2011). Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 19, 1905–1912. doi: 10.1038/mt.2011.135
Hetman, M., and Gozdz, A. (2004). Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 271, 2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x
Horowitz, M. P., and Greenamyre, J. T. (2010). Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimers. Dis. 20(Suppl. 2), S551–S568. doi: 10.3233/JAD-2010-100354
Hou, C. C., and Yang, W. X. (2013). Acroframosome-dependent KIFC1 facilitates acrosome formation during spermatogenesis in the caridean shrimp Exopalaemon modestus. PLoS ONE 8:e76065. doi: 10.1371/journal.pone.0076065
Hu, B. Y., and Zhang, S. C. (2009). Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 4, 1295–1304. doi: 10.1038/nprot.2009.127
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Ilzecka, J. (2012). Serum caspase-9 levels are increased in patients with amyotrophic lateral sclerosis. Neurol. Sci. 33, 825–829. doi: 10.1007/s10072-011-0837-4
Imbeault, E., Mahvelati, T. M., Braun, R., Gris, P., and Gris, D. (2014). Nlrx1 regulates neuronal cell death. Mol. Brain 7, 90. doi: 10.1186/s13041-014-0090-x
Inoue, H., Tsukita, K., Iwasato, T., Suzuki, Y., Tomioka, M., Tateno, M., et al. (2003). The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 22, 6665–6674. doi: 10.1093/emboj/cdg634
Ishii, T., Miyazawa, M., Hartman, P. S., and Ishii, N. (2011). Mitochondrial superoxide anion (O(2)(-)) inducible “mev-1” animal models for aging research. BMB Rep. 44, 298–305. doi: 10.5483/BMBRep.2011.44.5.298
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. doi: 10.1016/j.neuron.2010.07.019
Jablonka, S., Holtmann, B., Sendtner, M., and Metzger, F. (2011). Therapeutic effects of PEGylated insulin-like growth factor I in the pmn mouse model of motoneuron disease. Exp. Neurol. 232, 261–269. doi: 10.1016/j.expneurol.2011.09.015
Jessell, T. M. (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29. doi: 10.1038/35049541
Jha, B. S., Rao, M., and Malik, N. (2015). Motor neuron differentiation from pluripotent stem cells and other intermediate proliferative precursors that can be discriminated by lineage specific reporters. Stem Cell Rev. 11, 194–204. doi: 10.1007/s12015-014-9541-0
Jiang, Y. M., Yamamoto, M., Kobayashi, Y., Yoshihara, T., Liang, Y., Terao, S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. doi: 10.1002/ana.20379
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17, 664–666. doi: 10.1038/nn.3688
Kadari, A., Lu, M., Li, M., Sekaran, T., Thummer, R. P., Guyette, N., et al. (2014). Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Res. Ther. 5, 47. doi: 10.1186/scrt435
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., et al. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357. doi: 10.1093/nar/gkj102
Kawamoto, Y., Ito, H., Kobayashi, Y., Suzuki, Y., Akiguchi, I., Fujimura, H., et al. (2010). HtrA2/Omi-immunoreactive intraneuronal inclusions in the anterior horn of patients with sporadic and Cu/Zn superoxide dismutase (SOD1) mutant amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 36, 331–344. doi: 10.1111/j.1365-2990.2010.01075.x
Keller, A. F., Gravel, M., and Kriz, J. (2009). Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 57, 1130–1142. doi: 10.1002/glia.20836
Kihira, T., Suzuki, A., Kubo, T., Miwa, H., and Kondo, T. (2007). Expression of insulin-like growth factor-II and leukemia inhibitory factor antibody immunostaining on the ionized calcium-binding adaptor molecule 1-positive microglias in the spinal cord of amyotrophic lateral sclerosis patients. Neuropathology 27, 257–268. doi: 10.1111/j.1440-1789.2007.00776.x
Kim, E. K., and Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. doi: 10.1016/j.bbadis.2009.12.009
Kisby, G. E., Standley, M., Park, T., Olivas, A., Fei, S., Jacob, T., et al. (2006). Proteomic analysis of the genotoxicant methylazoxymethanol (MAM)-induced changes in the developing cerebellum. J. Proteome Res. 5, 2656–2665. doi: 10.1021/pr060126g
Kuzma-Kozakiewicz, M., Chudy, A., Gajewska, B., Dziewulska, D., Usarek, E., and Baranczyk-Kuzma, A. (2013). Kinesin expression in the central nervous system of humans and transgenic hSOD1G93A mice with amyotrophic lateral sclerosis. Neurodegener. Dis. 12, 71–80. doi: 10.1159/000339529
Kwan, J. Y., Jeong, S. Y., Van Gelderen, P., Deng, H. X., Quezado, M. M., Danielian, L. E., et al. (2012). Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS ONE 7:e35241. doi: 10.1371/journal.pone.0035241
Ladd, A. C., Keeney, P. M., Govind, M. M., and Bennett, J. P. Jr. (2014). Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromolecular Med. 16, 714–726. doi: 10.1007/s12017-014-8321-y
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Lai, E. C., Felice, K. J., Festoff, B. W., Gawel, M. J., Gelinas, D. F., Kratz, R., et al. (1997). Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 49, 1621–1630. doi: 10.1212/WNL.49.6.1621
Lai, W. H., Ho, J. C., Lee, Y. K., Ng, K. M., Au, K. W., Chan, Y. C., et al. (2010). ROCK inhibition facilitates the generation of human-induced pluripotent stem cells in a defined, feeder-, and serum-free system. Cell. Reprogram. 12, 641–653. doi: 10.1089/cell.2010.0051
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., Van Den Berg, L. H., Van Es, M. A., et al. (2009). Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 9004–9009. doi: 10.1073/pnas.0812937106
Leblond, C. S., Kaneb, H. M., Dion, P. A., and Rouleau, G. A. (2014). Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 262(Pt B), 91–101. doi: 10.1016/j.expneurol.2014.04.013
Lee, H. J., Kim, K. S., Ahn, J., Bae, H. M., Lim, I., and Kim, S. U. (2014). Human motor neurons generated from neural stem cells delay clinical onset and prolong life in ALS mouse model. PLoS ONE 9:e97518. doi: 10.1371/journal.pone.0097518
Lemasters, J. J., and Holmuhamedov, E. (2006). Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim. Biophys. Acta 1762, 181–190. doi: 10.1016/j.bbadis.2005.10.006
Leventhal, C., Rafii, S., Rafii, D., Shahar, A., and Goldman, S. A. (1999). Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol. Cell. Neurosci. 13, 450–464. doi: 10.1006/mcne.1999.0762
Li, M., Wang, L., Wang, W., Qi, X. L., and Tang, Z. Y. (2014). Mutations in the HFE gene and sporadic amyotrophic lateral sclerosis risk: a meta-analysis of observational studies. Braz. J. Med. Biol. Res. 47, 215–222. doi: 10.1590/1414-431X20133296
Li, X. J., Hu, B. Y., Jones, S. A., Zhang, Y. S., Lavaute, T., Du, Z. W., et al. (2008). Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells 26, 886–893. doi: 10.1634/stemcells.2007-0620
Lian, X., Hsiao, C., Wilson, G., Zhu, K., Hazeltine, L. B., Azarin, S. M., et al. (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 109, E1848–E1857. doi: 10.1073/pnas.1200250109
Lin, F. H., Lin, R., Wisniewski, H. M., Hwang, Y. W., Grundke-Iqbal, I., Healy-Louie, G., et al. (1992). Detection of point mutations in codon 331 of mitochondrial NADH dehydrogenase subunit 2 in Alzheimer's brains. Biochem. Biophys. Res. Commun. 182, 238–246. doi: 10.1016/S0006-291X(05)80136-6
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Luan, G., Zhao, Y., Zhai, F., Chen, Y., and Li, T. (2012). Ketogenic diet reduces Smac/Diablo and cytochrome c release and attenuates neuronal death in a mouse model of limbic epilepsy. Brain Res. Bull. 89, 79–85. doi: 10.1016/j.brainresbull.2012.07.002
Macarthur, C. C., Fontes, A., Ravinder, N., Kuninger, D., Kaur, J., Bailey, M., et al. (2012). Generation of human-induced pluripotent stem cells by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int. 2012:564612. doi: 10.1155/2012/564612
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Malaspina, A., and de Belleroche, J. (2004). Spinal cord molecular profiling provides a better understanding of amyotrophic lateral sclerosis pathogenesis. Brain Res. Brain Res. Rev. 45, 213–229. doi: 10.1016/j.brainresrev.2004.04.002
Manabe, Y., Warita, H., Murakami, T., Shiote, M., Hayashi, T., Omori, N., et al. (2002). Early decrease of the immunophilin FKBP 52 in the spinal cord of a transgenic model for amyotrophic lateral sclerosis. Brain Res. 935, 124–128. doi: 10.1016/S0006-8993(02)02466-6
Manfredi, G., and Beal, M. F. (2000). The role of mitochondria in the pathogenesis of neurodegenerative diseases. Brain Pathol. 10, 462–472. doi: 10.1111/j.1750-3639.2000.tb00278.x
Marchetto, M. C., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657. doi: 10.1016/j.stem.2008.10.001
Marti, M., Mulero, L., Pardo, C., Morera, C., Carrio, M., Laricchia-Robbio, L., et al. (2013). Characterization of pluripotent stem cells. Nat. Protoc. 8, 223–253. doi: 10.1038/nprot.2012.154
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U.S.A. 78, 7634–7638. doi: 10.1073/pnas.78.12.7634
Martin, L. J. (2012). Biology of mitochondria in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 107, 355–415. doi: 10.1016/B978-0-12-385883-2.00005-9
Martin, L. J., Price, A. C., Kaiser, A., Shaikh, A. Y., and Liu, Z. (2000). Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (Review). Int. J. Mol. Med. 5, 3–13. doi: 10.3892/ijmm.5.1.3
Maximino, J. R., de Oliveira, G. P., Alves, C. J., and Chadi, G. (2014). Deregulated expression of cytoskeleton related genes in the spinal cord and sciatic nerve of presymptomatic SOD1(G93A) Amyotrophic Lateral Sclerosis mouse model. Front. Cell. Neurosci. 8:148. doi: 10.3389/fncel.2014.00148
McGoldrick, P., Joyce, P. I., Fisher, E. M., and Greensmith, L. (2013). Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1832, 1421–1436. doi: 10.1016/j.bbadis.2013.03.012
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 20, 3642–3652. doi: 10.1093/hmg/ddr284
Mosca, L., Lunetta, C., Tarlarini, C., Avemaria, F., Maestri, E., Melazzini, M., et al. (2012). Wide phenotypic spectrum of the TARDBP gene: homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson's disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol. Aging 33, 1846.e1841–e1844. doi: 10.1016/j.neurobiolaging.2012.01.108
Mühling, T., Duda, J., Weishaupt, J. H., Ludolph, A. C., and Liss, B. (2014). Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca(2+) transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice. Front. Cell. Neurosci. 8:353. doi: 10.3389/fncel.2014.00353
Münch, C., Rosenbohm, A., Sperfeld, A. D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.WNL.0000134608.83927.B1
Muratore, C. R., Srikanth, P., Callahan, D. G., and Young-Pearse, T. L. (2014). Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS ONE 9:e105807. doi: 10.1371/journal.pone.0105807
Musarò, A. (2010). State of the art and the dark side of amyotrophic lateral sclerosis. World J. Biol. Chem. 1, 62–68. doi: 10.4331/wjbc.v1.i5.62
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nandar, W., Neely, E. B., Simmons, Z., and Connor, J. R. (2014). H63D HFE genotype accelerates disease progression in animal models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1842, 2413–2426. doi: 10.1016/j.bbadis.2014.09.016
Nizzardo, M., Simone, C., Falcone, M., Locatelli, F., Riboldi, G., Comi, G. P., et al. (2010). Human motor neuron generation from embryonic stem cells and induced pluripotent stem cells. Cell. Mol. Life Sci. 67, 3837–3847. doi: 10.1007/s00018-010-0463-y
Nowikovsky, K., Pozzan, T., Rizzuto, R., Scorrano, L., and Bernardi, P. (2012). Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454. doi: 10.1085/jgp.201110757
Offen, D., Barhum, Y., Melamed, E., Embacher, N., Schindler, C., and Ransmayr, G. (2009). Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 38, 85–93. doi: 10.1007/s12031-007-9004-z
Olsen, M. K., Roberds, S. L., Ellerbrock, B. R., Fleck, T. J., McKinley, D. K., and Gurney, M. E. (2001). Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann. Neurol. 50, 730–740. doi: 10.1002/ana.1252
Otomo, A., Hadano, S., Okada, T., Mizumura, H., Kunita, R., Nishijima, H., et al. (2003). ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 12, 1671–1687. doi: 10.1093/hmg/ddg184
Pagano, G., Talamanca, A. A., Castello, G., Cordero, M. D., d'Ischia, M., Gadaleta, M. N., et al. (2014). Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: toward mitochondria-targeted clinical strategies. Oxid. Med. Cell. Longev. 2014:541230. doi: 10.1155/2014/541230
Palamiuc, L., Schlagowski, A., Ngo, S. T., Vernay, A., Dirrig-Grosch, S., Henriques, A., et al. (2015). A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 7, 526–546. doi: 10.15252/emmm.201404433
Pamplona, R., Dalfó, E., Ayala, V., Bellmunt, M. J., Prat, J., Ferrer, I., et al. (2005). Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 280, 21522–21530. doi: 10.1074/jbc.M502255200
Pasinelli, P., Houseweart, M. K., Brown, R. H. Jr., and Cleveland, D. W. (2000). Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 13901–13906. doi: 10.1073/pnas.240305897
Patterson, V. L., Zullo, A. J., Koenig, C., Stoessel, S., Jo, H., Liu, X., et al. (2014). Neural-specific deletion of Htra2 causes cerebellar neurodegeneration and defective processing of mitochondrial OPA1. PLoS ONE 9:e115789. doi: 10.1371/journal.pone.0115789
Perrett, R. M., Alexopoulou, Z., and Tofaris, G. K. (2015). The endosomal pathway in Parkinson's disease. Mol. Cell Neurosci. 66(Pt A), 21–28. doi: 10.1016/j.mcn.2015.02.009
Perrin, F. E., Boisset, G., Docquier, M., Schaad, O., Descombes, P., and Kato, A. C. (2005). No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum. Mol. Genet. 14, 3309–3320. doi: 10.1093/hmg/ddi357
Praline, J., Blasco, H., Vourc'H, P., Garrigue, M. A., Gordon, P. H., Camu, W., et al. (2011). APOE epsilon4 allele is associated with an increased risk of bulbar-onset amyotrophic lateral sclerosis in men. Eur. J. Neurol. 18, 1046–1052. doi: 10.1111/j.1468-1331.2010.03330.x
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. doi: 10.1038/nn1653
Qu, Q., Li, D., Louis, K. R., Li, X., Yang, H., Sun, Q., et al. (2014). High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 5, 3449. doi: 10.1038/ncomms4449
Raitano, S., Ordovàs, L., De Muynck, L., Guo, W., Espuny-Camacho, I., Geraerts, M., et al. (2015). Restoration of progranulin expression rescues cortical neuron generation in an induced pluripotent stem cell model of frontotemporal dementia. Stem Cell Rep. 4, 16–24. doi: 10.1016/j.stemcr.2014.12.001
Raman, R., Allen, S. P., Goodall, E. F., Kramer, S., Ponger, L. L., Heath, P. R., et al. (2015). Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 41, 201–226. doi: 10.1111/nan.12147
Ranganathan, S., and Bowser, R. (2003). Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am. J. Pathol. 162, 823–835. doi: 10.1016/S0002-9440(10)63879-5
Reddy, P. H., Reddy, T. P., Manczak, M., Calkins, M. J., Shirendeb, U., and Mao, P. (2011). Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118. doi: 10.1016/j.brainresrev.2010.11.004
Ringer, C., Weihe, E., and Schütz, B. (2012). Calcitonin gene-related peptide expression levels predict motor neuron vulnerability in the superoxide dismutase 1-G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 45, 547–554. doi: 10.1016/j.nbd.2011.09.011
Rocha, M. C., Pousinha, P. A., Correia, A. M., Sebastião, A. M., and Ribeiro, J. A. (2013). Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS ONE 8:e73846. doi: 10.1371/journal.pone.0073846
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl. 1), S3–S9. doi: 10.1002/ana.21543
Rutter, J., Winge, D. R., and Schiffman, J. D. (2010). Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion 10, 393–401. doi: 10.1016/j.mito.2010.03.001
Ryan, S., and Gogvadze, V. (1999). Substrate-dependent effect of phenolic antioxidants on Ca2+ accumulation by rat liver mitochondria. Redox Rep. 4, 251–254. doi: 10.1179/135100099101534972
Saenger, S., Holtmann, B., Nilges, M. R., Schroeder, S., Hoeflich, A., Kletzl, H., et al. (2012). Functional improvement in mouse models of familial amyotrophic lateral sclerosis by PEGylated insulin-like growth factor I treatment depends on disease severity. Amyotroph. Lateral Scler. 13, 418–429. doi: 10.3109/17482968.2012.679944
Saris, C. G., Groen, E. J., Van Vught, P. W., Van Es, M. A., Blauw, H. M., Veldink, J. H., et al. (2013). Gene expression profile of SOD1-G93A mouse spinal cord, blood and muscle. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 190–198. doi: 10.3109/21678421.2012.749914
Sato, A., Sakai, N., Shinbo, J., Hashidate, H., Igarashi, S., Kakita, A., et al. (2014). An autopsy case of amyotrophic lateral sclerosis with prominent muscle cramps, fasciculation, and high titer of anti-voltage gated potassium channel (VGKC) complex antibody. Rinsho Shinkeigaku 54, 32–37. doi: 10.5692/clinicalneurol.54.32
Schaefer, A. M., Sanes, J. R., and Lichtman, J. W. (2005). A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 490, 209–219. doi: 10.1002/cne.20620
Schägger, H., and Ohm, T. G. (1995). Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 227, 916–921. doi: 10.1111/j.1432-1033.1995.tb20219.x
Schmitt, F., Hussain, G., Dupuis, L., Loeffler, J. P., and Henriques, A. (2014). A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front. Cell. Neurosci. 8:25. doi: 10.3389/fncel.2014.00025
Schmuck, G., and Kahl, R. (2009). The use of Fluoro-Jade in primary neuronal cell cultures. Arch. Toxicol. 83, 397–403. doi: 10.1007/s00204-008-0360-4
Schmued, L. C., Albertson, C., and Slikker, W. Jr. (1997). Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 751, 37–46. doi: 10.1016/S0006-8993(96)01387-X
Schmued, L. C., Stowers, C. C., Scallet, A. C., and Xu, L. (2005). Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 1035, 24–31. doi: 10.1016/j.brainres.2004.11.054
Seelen, M., Visser, A. E., Overste, D. J., Kim, H. J., Palud, A., Wong, T. H., et al. (2014). No mutations in hnRNPA1 and hnRNPA2B1 in Dutch patients with amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy. Neurobiol. Aging 35, 1956.e1959-1956.e1911. doi: 10.1016/j.neurobiolaging.2014.01.152
Seluanov, A., Vaidya, A., and Gorbunova, V. (2010). Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. doi: 10.3791/2033
Sergeant, N., Wattez, A., Galván-Valencia, M., Ghestem, A., David, J. P., Lemoine, J., et al. (2003). Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer's disease. Neuroscience 117, 293–303. doi: 10.1016/S0306-4522(02)00747-9
Sevenich, L., Pennacchio, L. A., Peters, C., and Reinheckel, T. (2006). Human cathepsin L rescues the neurodegeneration and lethality in cathepsin B/L double-deficient mice. Biol. Chem. 387, 885–891. doi: 10.1515/BC.2006.112
Shefner, J. M., Cudkowicz, M., and Brown, R. H. Jr. (2006). Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis. Muscle Nerve 34, 603–607. doi: 10.1002/mus.20628
Shi, P., Ström, A. L., Gal, J., and Zhu, H. (2010). Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 1802, 707–716. doi: 10.1016/j.bbadis.2010.05.008
Shimizu, S., Narita, M., and Tsujimoto, Y. (1999). Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487. doi: 10.1038/20959
Shiraki, H. Y. Y. (1975). “Amyotrophic lateral sclerosis in Japan,” in Handbook of Clinical Neurology, eds P. J. Vinken and G. W. Bruyn (Amsterdam; North-Holland: Elsevier), 353–419.
Shoshan-Barmatz, V., Israelson, A., Brdiczka, D., and Sheu, S. S. (2006). The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 12, 2249–2270. doi: 10.2174/138161206777585111
Shoshan-Barmatz, V., Keinan, N., and Zaid, H. (2008). Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191. doi: 10.1007/s10863-008-9147-9
Shulman, J. M., De Jager, P. L., and Feany, M. B. (2011). Parkinson's disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222. doi: 10.1146/annurev-pathol-011110-130242
Silva, D., Dikkes, P., Barnes, M., and Lopez, M. F. (2009). Decreased motoneuron survival in Igf2 null mice after sciatic nerve transection. Neuroreport 20, 1414–1418. doi: 10.1097/WNR.0b013e328330b735
Small, S. A., Kent, K., Pierce, A., Leung, C., Kang, M. S., Okada, H., et al. (2005). Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann. Neurol. 58, 909–919. doi: 10.1002/ana.20667
Somers, A., Jean, J. C., Sommer, C. A., Omari, A., Ford, C. C., Mills, J. A., et al. (2010). Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 28, 1728–1740. doi: 10.1002/stem.495
Sommer, C. A., Christodoulou, C., Gianotti-Sommer, A., Shen, S. S., Sailaja, B. S., Hezroni, H., et al. (2012). Residual expression of reprogramming factors affects the transcriptional program and epigenetic signatures of induced pluripotent stem cells. PLoS ONE 7:e51711. doi: 10.1371/journal.pone.0051711
Song, W., Song, Y., Kincaid, B., Bossy, B., and Bossy-Wetzel, E. (2013). Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiol. Dis. 51, 72–81. doi: 10.1016/j.nbd.2012.07.004
Soustiel, J. F., and Larisch, S. (2010). Mitochondrial damage: a target for new therapeutic horizons. Neurotherapeutics 7, 13–21. doi: 10.1016/j.nurt.2009.11.001
Srinivasula, S. M., Gupta, S., Datta, P., Zhang, Z., Hegde, R., Cheong, N., et al. (2003). Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2. J. Biol. Chem. 278, 31469–31472. doi: 10.1074/jbc.C300240200
Štalekar, M., Yin, X., Rebolj, K., Darovic, S., Troakes, C., Mayr, M., et al. (2015). Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 293, 157–170. doi: 10.1016/j.neuroscience.2015.02.046
Steiner, J. P., Hamilton, G. S., Ross, D. T., Valentine, H. L., Guo, H., Connolly, M. A., et al. (1997). Neurotrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proc. Natl. Acad. Sci. U.S.A. 94, 2019–2024. doi: 10.1073/pnas.94.5.2019
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800. doi: 10.1371/journal.pone.0021800
Suszyńska-Zajczyk, J., Luczak, M., Marczak, L., and Jakubowski, H. (2014). Hyperhomocysteinemia and bleomycin hydrolase modulate the expression of mouse brain proteins involved in neurodegeneration. J. Alzheimers. Dis. 40, 713–726. doi: 10.3233/JAD-132033
Tajeddine, N., Galluzzi, L., Kepp, O., Hangen, E., Morselli, E., Senovilla, L., et al. (2008). Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27, 4221–4232. doi: 10.1038/onc.2008.63
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takanashi, K., and Yamaguchi, A. (2014). Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem. Biophys. Res. Commun. 452, 600–607. doi: 10.1016/j.bbrc.2014.08.115
Takazawa, T., Croft, G. F., Amoroso, M. W., Studer, L., Wichterle, H., and Macdermott, A. B. (2012). Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS ONE 7:e40154. doi: 10.1371/journal.pone.0040154
Tan, A. Y., Riley, T. R., Coady, T., Bussemaker, H. J., and Manley, J. L. (2012). TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. U.S.A. 109, 6030–6035. doi: 10.1073/pnas.1203028109
Tapia, R. (2014). Cellular and molecular mechanisms of motor neuron death in amyotrophic lateral sclerosis: a perspective. Front. Cell. Neurosci. 8:241. doi: 10.3389/fncel.2014.00241
Testi, S., Tamburin, S., Zanette, G., and Fabrizi, G. M. (2015). Co-occurrence of the C9ORF72 expansion and a novel GRN mutation in a family with alternative expression of frontotemporal dementia and amyotrophic lateral sclerosis. J. Alzheimers. Dis. 44, 49–56. doi: 10.3233/JAD-141794
Tian, X., Zhou, Y., Gao, L., He, G., Jiang, W., Li, W., et al. (2013). Analysis of ischemic neuronal injury in Cav2.1 channel alpha1 subunit mutant mice. Biochem. Biophys. Res. Commun. 434, 60–64. doi: 10.1016/j.bbrc.2013.03.066
Toli, D., Buttigieg, D., Blanchard, S., Lemonnier, T., Lamotte d'Incamps, B., Bellouze, S., et al. (2015). Modeling amyotrophic lateral sclerosis in pure human iPSc-derived motor neurons isolated by a novel FACS double selection technique. Neurobiol. Dis. 82, 269–280. doi: 10.1016/j.nbd.2015.06.011
Trippier, P. C., Zhao, K. T., Fox, S. G., Schiefer, I. T., Benmohamed, R., Moran, J., et al. (2014). Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis. ACS Chem. Neurosci. 5, 823–829. doi: 10.1021/cn500147v
Tsujimoto, Y., and Shimizu, S. (2002). The voltage-dependent anion channel: an essential player in apoptosis. Biochimie 84, 187–193. doi: 10.1016/S0300-9084(02)01370-6
Turner, B. J., and Talbot, K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. doi: 10.1016/j.pneurobio.2008.01.001
Vande Velde, C., Garcia, M. L., Yin, X., Trapp, B. D., and Cleveland, D. W. (2004). The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med. 5, 193–203. doi: 10.1385/NMM:5:3:193
Venkova, K., Christov, A., Kamaluddin, Z., Kobalka, P., Siddiqui, S., and Hensley, K. (2014). Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 702–713. doi: 10.1097/NEN.0000000000000086
Veyrat-Durebex, C., Corcia, P., Dangoumau, A., Laumonnier, F., Piver, E., Gordon, P. H., et al. (2014). Advances in cellular models to explore the pathophysiology of amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 966–983. doi: 10.1007/s12035-013-8573-9
Vilariño-Güell, C., Rajput, A., Milnerwood, A. J., Shah, B., Szu-Tu, C., Trinh, J., et al. (2014). DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801. doi: 10.1093/hmg/ddt570
Vucic, S., and Kiernan, M. C. (2009). Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr. Mol. Med. 9, 255–272. doi: 10.2174/156652409787847173
Waldeck-Weiermair, M., Jean-Quartier, C., Rost, R., Khan, M. J., Vishnu, N., Bondarenko, A. I., et al. (2011). Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286, 28444–28455. doi: 10.1074/jbc.M111.244517
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944. doi: 10.1093/hmg/dds003
Weiergräber, M., Henry, M., Radhakrishnan, K., Hescheler, J., and Schneider, T. (2007). Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R-type voltage-gated calcium channel. J. Neurophysiol. 97, 3660–3669. doi: 10.1152/jn.01193.2006
Werner, A., and Sayer, J. A. (2009). Naturally occurring antisense RNA: function and mechanisms of action. Curr. Opin. Nephrol. Hypertens. 18, 343–349. doi: 10.1097/MNH.0b013e32832cb982
White, M. C., Gao, R., Xu, W., Mandal, S. M., Lim, J. G., Hazra, T. K., et al. (2010). Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 6:e1000984. doi: 10.1371/journal.pgen.1000984
Whittle, A. J., Ross, O. A., Naini, A., Gordon, P., Mistumoto, H., Dächsel, J. C., et al. (2007). Pathogenic Lrrk2 substitutions and Amyotrophic lateral sclerosis. J Neural Transm 114, 327–329. doi: 10.1007/s00702-006-0525-3
Wszolek, Z. K., Vieregge, P., Uitti, R. J., Gasser, T., Yasuhara, O., McGeer, P., et al. (1997). German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia - Longitudinal observations. Parkinsonism Relat. Disord. 3, 125–139. doi: 10.1016/S1353-8020(97)00013-8
Wu, J., Kharebava, G., Piao, C., Stoica, B. A., Dinizo, M., Sabirzhanov, B., et al. (2012). Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS ONE 7:e42129. doi: 10.1371/journal.pone.0042129
Xiong, H. L., Wang, J. Y., Sun, Y. M., Wu, J. J., Chen, Y., Qiao, K., et al. (2010). Association between novel TARDBP mutations and Chinese patients with amyotrophic lateral sclerosis. BMC Med. Genet. 11:8. doi: 10.1186/1471-2350-11-8
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yamamoto, M., Tanaka, F., and Sobue, G. (2007). [Gene expression profile of spinal ventral horn in ALS]. Brain Nerve 59, 1129–1139.
Yoon, S., Cong, W. T., Bang, Y., Lee, S. N., Yoon, C. S., Kwack, S. J., et al. (2009). Proteome response to ochratoxin A-induced apoptotic cell death in mouse hippocampal HT22 cells. Neurotoxicology 30, 666–676. doi: 10.1016/j.neuro.2009.04.013
Zhang, B., Chang, A., Kjeldsen, T. B., and Arvan, P. (2001). Intracellular retention of newly synthesized insulin in yeast is caused by endoproteolytic processing in the Golgi complex. J. Cell Biol. 153, 1187–1198. doi: 10.1083/jcb.153.6.1187
Zhang, Z., Almeida, S., Lu, Y., Nishimura, A. L., Peng, L., Sun, D., et al. (2013). Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS ONE 8:e76055. doi: 10.1371/journal.pone.0076055
Zimprich, A., Benet-Pagès, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. doi: 10.1016/j.ajhg.2011.06.008
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi: 10.1016/j.neuron.2004.11.005
Keywords: amyotrophic lateral sclerosis, sporadic ALS, induced pluripotent stem cells, motor neuron differentiation, microarray
Citation: Alves CJ, Dariolli R, Jorge FM, Monteiro MR, Maximino JR, Martins RS, Strauss BE, Krieger JE, Callegaro D and Chadi G (2015) Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 9:289. doi: 10.3389/fncel.2015.00289
Received: 07 May 2015; Accepted: 14 July 2015;
Published: 04 August 2015.
Edited by:
Manoj Kumar Jaiswal, Center for Neuroscience and Regenerative Medicine, USAReviewed by:
Stefan Hauser, Deutsches Zentrum für Neurodegenerative Erkrankungen, GermanyMu He, University of California, San Francisco, USA
Xiaogang Wu, Institute for Systems Biology, USA
Copyright © 2015 Alves, Dariolli, Jorge, Monteiro, Maximino, Martins, Strauss, Krieger, Callegaro and Chadi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerson Chadi, Department of Neurology, University of São Paulo, Av. Dr. Arnaldo, 455, 2nd Floor, Room 2119, 01246-903 São Paulo, Brazil, gerchadi@usp.br