PSEN1 c.1292C<A Variant and Early-Onset Alzheimer’s Disease: A Scoping Review
 Maribel Orozco-Barajas1,2
Maribel Orozco-Barajas1,2  Yulisa Oropeza-Ruvalcaba2
Yulisa Oropeza-Ruvalcaba2  Alejandro A. Canales-Aguirre3
Alejandro A. Canales-Aguirre3  Victor J. Sánchez-González1,4*
Victor J. Sánchez-González1,4*- 1Doctorado en Biociencias, Centro Universitario de los Altos, Universidad de Guadalajara, Guadalajara, Mexico
- 2Centro de Atención Psicológica, Tepatitlán de Morelos, Mexico
- 3Departamento de Biotecnología Médica y Farmacéutica, Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco A. C. (CIATEJ), Guadalajara, Mexico
- 4Departamento de Clínicas, Centro Universitario de los Altos, Universidad de Guadalajara, Guadalajara, Mexico
Alzheimer’s disease (AD) is the most common cause of dementia, characterized by progressive loss of cognitive function, with β-amyloid plaques and neurofibrillary tangles being its major pathological findings. Although the disease mainly affects the elderly, c. 5–10% of the cases are due to PSEN1, PSEN2, and APP mutations, principally associated with an early onset of the disease. The A413E (rs63750083) PSEN1 variant, identified in 2001, is associated with early-onset Alzheimer’s disease (EOAD). Although there is scant knowledge about the disease’s clinical manifestations and particular features, significant clinical heterogeneity was reported, with a high incidence of spastic paraparesis (SP), language impairments, and psychiatric and motor manifestations. This scoping review aims to synthesize findings related to the A431E variant of PSEN1. In the search, we followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement and the guidelines proposed by Arksey and O’Malley. We searched and identified 247 studies including the A431E variant of PSEN1 from 2001 to 2021 in five databases and one search engine. After the removal of duplicates, and apply inclusion criteria, 42 studies were finally included. We considered a narrative synthesis with a qualitative approach for the analysis of the data. Given the study sample conformation, we divided the results into those carried out only with participants carrying A431E (seven studies), subjects with PSEN variants (11 studies), and variants associated with EOAD in PSEN1, PSEN2, and APP (24 studies). The resulting synthesis indicates most studies involve Mexican and Mexican-American participants in preclinical stages. The articles analyzed included carrier characteristics in categories such as genetics, clinical, imaging techniques, neuropsychology, neuropathology, and biomarkers. Some studies also considered family members’ beliefs and caregivers’ experiences. Heterogeneity in both the studies found and carrier samples of EOAD-related gene variants does not allow for the generalization of the findings. Future research should focus on reporting data on the progression of carrier characteristics through time and reporting results independently or comparing them across variants.
Introduction
Alzheimer’s disease causes c. 70% of dementia cases (World Health Organization, 2020), which is a neurodegenerative disease resulting in progressive cognitive deficits due to plaques and tangles accumulation leading to inflammation and oxidative stress responses (Amponsah et al., 2021).
Most cases are related to susceptibility genes and risk factors such as age, obesity, hypertension, diabetes, depression, and others (Livingston et al., 2020). Variants in the causality genes, including PSEN1, PSEN2, and APP (identified with high penetrance), are responsible for only 5–10% of the cases (Hoogmartens et al., 2021). With 326 reported, among them the A431E, PSEN1 has the most pathogenic variants associated with EOAD (Alzforum, 2021).
With a 1292c> a, rs63750083 nomenclature, A431E is in the exon 12 of PSEN1, and in the transmembrane region nine, it changes an alanine by glutamic acid and alters its physical–chemical interaction (Landrum et al., 2018; Alzforum, 2021). A431E has the OMIM code 104311.0033 and is associated with EOAD type 3 (McKusick-Nathans Institute of Genetic Medicine JHU, 2019). A431E has complete penetrance (Bateman et al., 2012). According to the guidelines of the American College of Medical Genetics and Genomics and Association of Molecular Pathology (ACMG/AMP), it is classified as pathogenic due to studies indicating, in a moderate range, an effect on Aβ42/Aβ40 levels and a decreased ratio (PS3-M classification criteria), critical functional location (PM1-M) and low frequency or no control (PM2-M), and strong co-segregation (PP1-S) (Alzforum, 2022).
Rogaeva et al. (2001), made the first description of the variant, which was found in five unrelated cases with a family history of AD and onset before 65 years of age. Later, Yescas et al. (2006) identified 12 families and hypothesized a founder effect of A431E. At the same time, Murrell et al. (2006) added an extra 15 independent families with an A431E history.
A431E is one of the three variants in PSEN1 with the highest number of affected individuals in Latin America (Dumois-Petersen et al., 2020; Llibre-Guerra et al., 2021). The estimated population varies from 381 (Llibre-Guerra et al., 2021) to 301 (Dumois-Petersen et al., 2020), while the number of people at risk ranges from 463 (Llibre-Guerra et al., 2021) to 560 (Dumois-Petersen et al., 2020). Therefore, an increase in A431E carriers is expected, as diagnostic studies and evaluations are still being held mostly by our group.
The distinctive phenotypic feature of A431E is the high frequency of SP (Santos-Mandujano et al., 2020; Llibre-Guerra et al., 2021). A431E is associated with generalized white matter abnormalities which precede SP (Soosman et al., 2016). In addition, Yescas et al. (2006) identified an exclusive motor presentation along with pyramidal signs, myoclonus, and seizures in cases where the onset of EOAD was before the average age of onset. Llibre-Guerra et al. (2021) reported such findings as uncommon in other PSEN1 variants.
Cases such as the A431E offer the possibility of understanding the disease’s genetic basis and pathology from the early stages (Fuller et al., 2019), which has triggered interest in genotypic and phenotypic characterization of its carriers. As studies involving individuals with a history of A431E continue to expand, it is necessary to have a reference framework regarding the findings of the phenotypic characteristics of this variant.
Previous literature reviews have focused on genetic aspects of EOAD caused by PSEN1, PSEN2, and APP variants (Tanzi, 2012; Ringman and Coppola, 2013; Ringman et al., 2014; Hoogmartens et al., 2021), and on the study of biomarkers in both cerebrospinal fluid (Ghidoni et al., 2011a,b; Rostgaard et al., 2015; Schindler et al., 2019) and plasma (Blennow et al., 2012). In addition, reviews focused on the clinical heterogeneity of PSEN1 variants were also published (Larner and Doran, 2006, 2008; Larner, 2013). However, there are no articles specifically focused on reviewing the findings of A431E, which represents a gap in the literature.
In this article, we sought to review studies that include carriers of the A431E variant to describe and characterize findings (clinical, neuroanatomical, neuropathological, neuropsychological, and possible biomarkers) associated with EOAD through a narrative review synthesis with a qualitative approach. We choose an exploratory review to focus on providing scanning of existing knowledge evidence in response to a specific objective (Arksey and O’Malley, 2005).
This scoping review aims to identify and synthesize the characteristics of the A431E variant of PSEN1 presented in the findings of EOAD-related studies.
Methods
Study Design
An exploratory review was chosen to achieve the aim of this study. The search was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) (Page et al., 2021) statement and the guidelines proposed by Arksey and O’Malley (2005). The authors suggest that this type of study aims to quickly provide a general frame of reference for key aspects such as concepts, sources, and types of evidence. It was chosen to “evaluate the extent, range, and nature of the research activity” regarding the A431E variant associated with EOAD, which corresponds to the first of the aims of exploratory studies proposed by the authors (Arksey and O’Malley, 2005, p. 21).
Search Strategy
In order to identify the articles to be included, the search was conducted in medicine, biomedical, and multiple search field databases: MEDLINE, PubMed, Scopus, WOS (Web of Science), Ovid, BMC (BioMed Central) databases, and the search engine Google Scholar. Research articles were considered if they included the following keywords: A431E AND PSEN1 OR PS1 AND ALZHEIMER; although the following search codes were also used A431E AND PSEN1 OR PS1 AND ALZHEIMER NOT SPORADIC NOT LATE ONSET. Articles from all databases published from 2001 to September 2021 were considered. Subsequently, duplicates were removed.
Study Selection/Screening
An initial screening of the research papers’ abstracts was independently conducted by two reviewers (MO-B and YO-R) considering the aim and the inclusion and exclusion criteria of the study.
Research papers included were those fulfilling the following inclusion criteria: (a) articles that report both the presence of the variant and disease; (b) scientific papers with an original contribution; (c) peer-reviewed publications; and (d) papers available in Spanish or English.
Articles were excluded when they met at least one of the following criteria: (a) a review paper; (b) abstracts of posters, conferences, or academic paper works where no access to the full text was available; (c) independent reviews or no peer-reviewed paper; and (d) experimental studies in cellular models. Full texts were also reviewed by the researchers to determine their eligibility by two reviewers (MO-B and YO-R). During this process, disagreements about the inclusion or exclusion of articles were discussed with third and fourth researchers (VJSG and AACA).
As a final criterion, we considered excluding experimental model studies. Articles were identified in which carriers and individuals with a history of the A431E were included along with participants with a history of other variants and even sporadic AD (SAD). Although in many of these papers the authors did not report the results separately or compared between groups, we highlight that in many of these studies the participant samples consisted mainly of people with a history of EOAD or A431E carriers. Therefore, we considered it appropriate to include the studies and, to facilitate the reading of results, separate them into those that included only people with a history of EOAD or A431E carriers, those including, in addition to the variant of interest, (a) other variants in PSEN1, (b) PSEN1 and PSEN2, and (c) PSEN and APP.
Data Extraction and Analysis
General data extracted from the studies include the following: authors, year of publication, country, study group, type of sample, and stage/type of participant. In addition, information about techniques and instruments used, study aim, and key findings were also extracted.
Data for each article were extracted without distinguishing between (1) study aim, (2) discipline in which the study is framed, (3) significant findings in results, (4) size sample, (5) country of origin of author or group of authors, and (6) authors discipline.
Data extraction was carried out by two of the authors (MO-B and YO-R). The following types of findings were included in this review: beliefs, caregivers, clinical, genetics, neuroanatomical, neuropathology, neuropsychological, biochemical, electrophysiological, and cerebrospinal fluid (CSF) biomarkers.
Analysis and synthesis of the data were performed using a narrative review approach to capture the diversity of findings related to A431E. We consider this method to be the most appropriate given the heterogeneous characteristics of the results. Gaps in the literature were also identified.
Results
Selection of Studies
The search has a date range from 2001 to September 2021. We identified a total of 484 articles in five electronic databases and one search engine. After duplicates were removed, a total of 247 articles were left. Following the inclusion criteria, 42 articles were finally considered to be included in this review. The process of identification, screening, eligibility, and inclusion as well as the articles identified in each phase are shown in Figure 1.
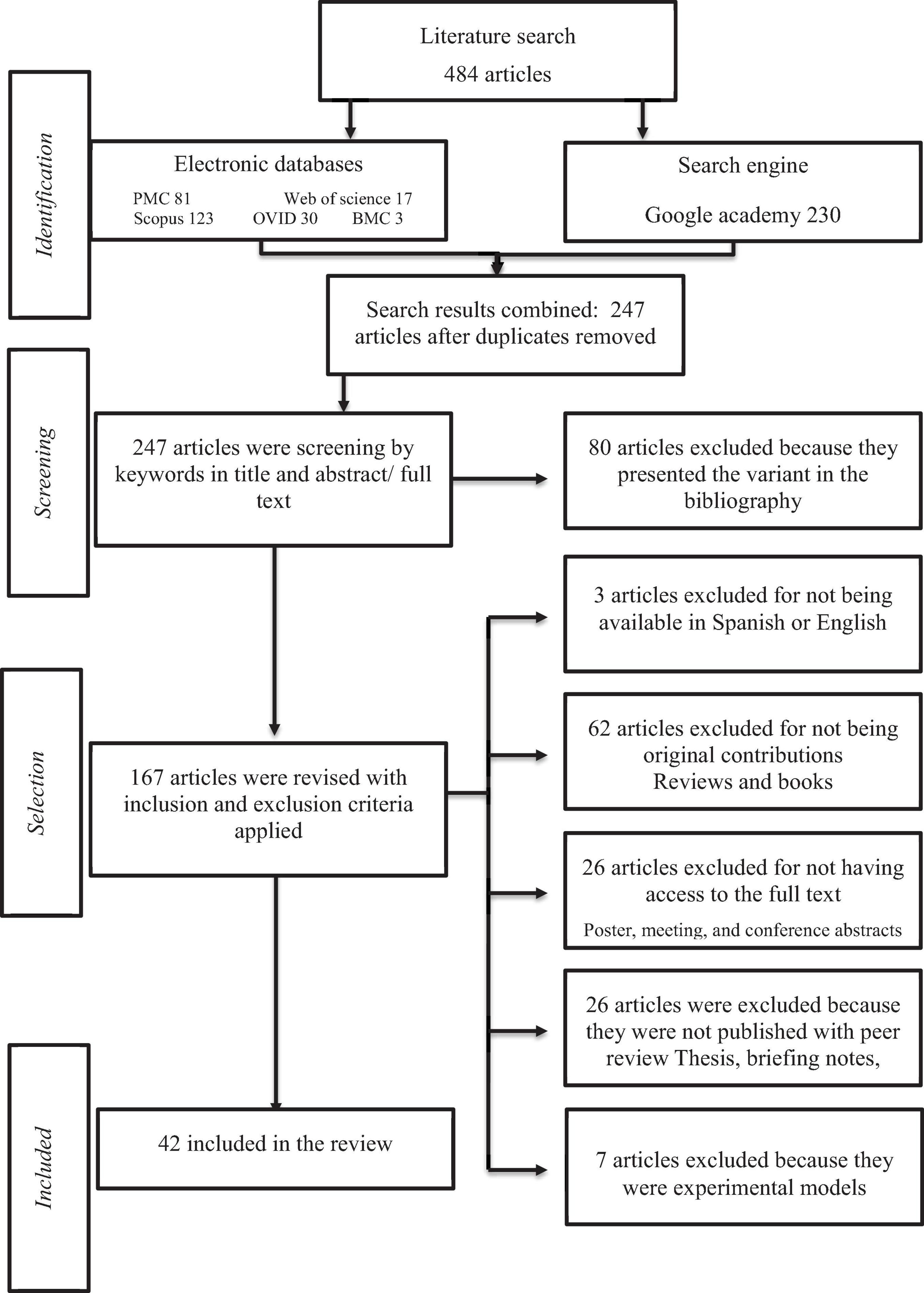
Figure 1. Screening process.
General Characteristics
Since 2001, there has been a gradual increase in the number of publications that consider the A431E PSEN1 variant (see Table 1).
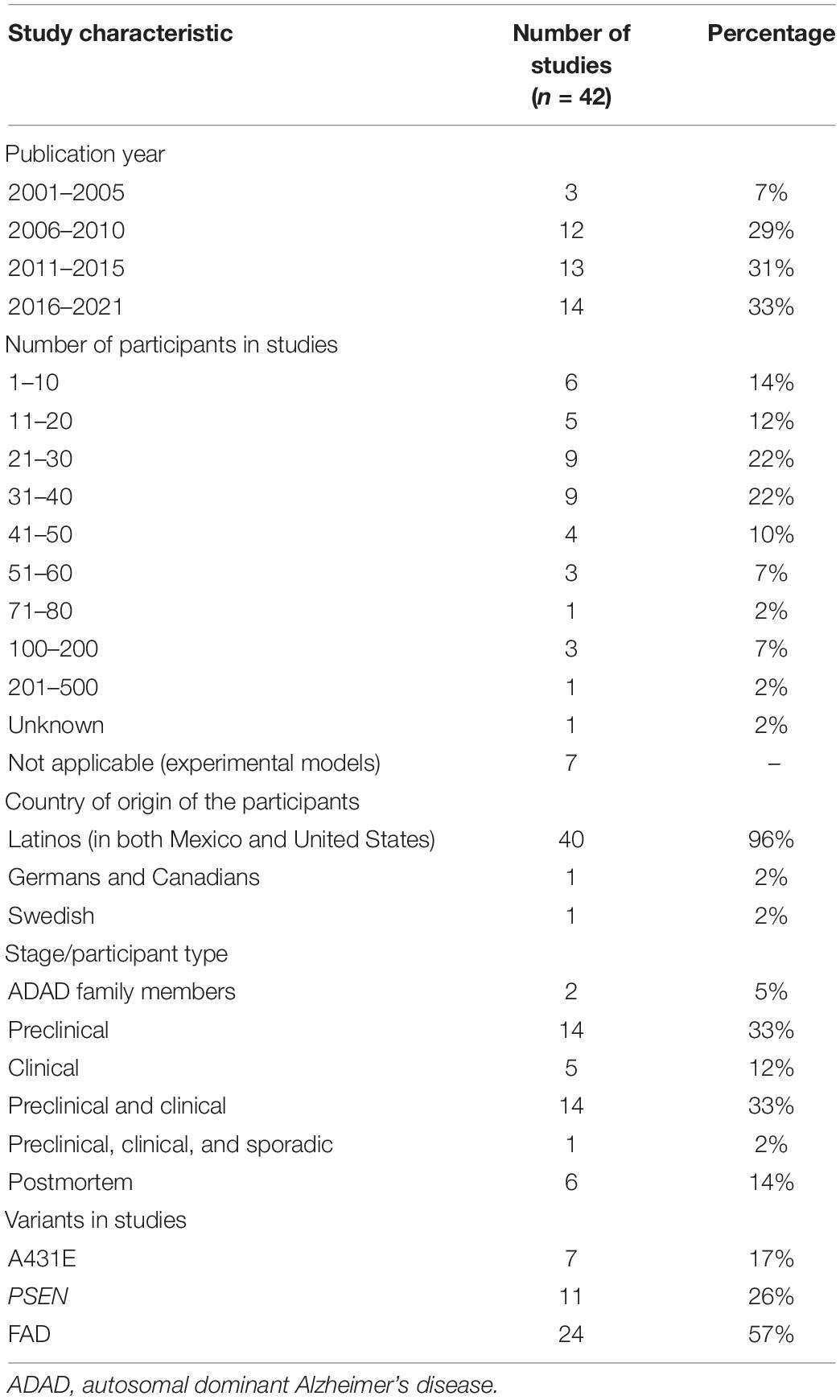
Table 1. Summary of general study characteristics.
The number of participants in the studies is also variable, being the studies with the highest percentage of those evaluating c. 21–30 and 31–40 participants. Most of the studies are related to the Latino population, although studies conducted in Germany, Canada, and Sweden have also been performed.
Of the 42 studies, 14 were conducted in a preclinical phase, another 14 in both a preclinical and clinical phase, five were limited to the clinical phase, six of them were postmortem studies, two were in family members at risk for AD, and one included preclinical and clinical participants and individuals with SAD.
Variants in Studies
Seven of the studies were only on A431E, 11 studies with PSEN variants, and the remaining 24 articles are about EOAD [these studies also use the abbreviations FAD (familial Alzheimer’s disease) and autosomal dominant Alzheimer’s disease (ADAD)].
General details of the studies included in this review are presented in Supplementary Table 1, with a detailed study group sample and country, presented accordingly, based on the year of publication. All the studies were conducted in North America, with an exception of two articles in a collaboration with the United States, Canada, and Germany and another in a Swedish publication.
Category Combinations
The identified 42 studies are all cross-sectional and were classified into several categories according to what was analyzed. Among these, the main ones were beliefs, caregivers, clinical, genetics, studies using imaging techniques, neuropathology, and CSF biomarkers. Such categories are combined between them and with others including neuropsychological, biochemical, and electrophysiological. This classification is presented in Table 2.
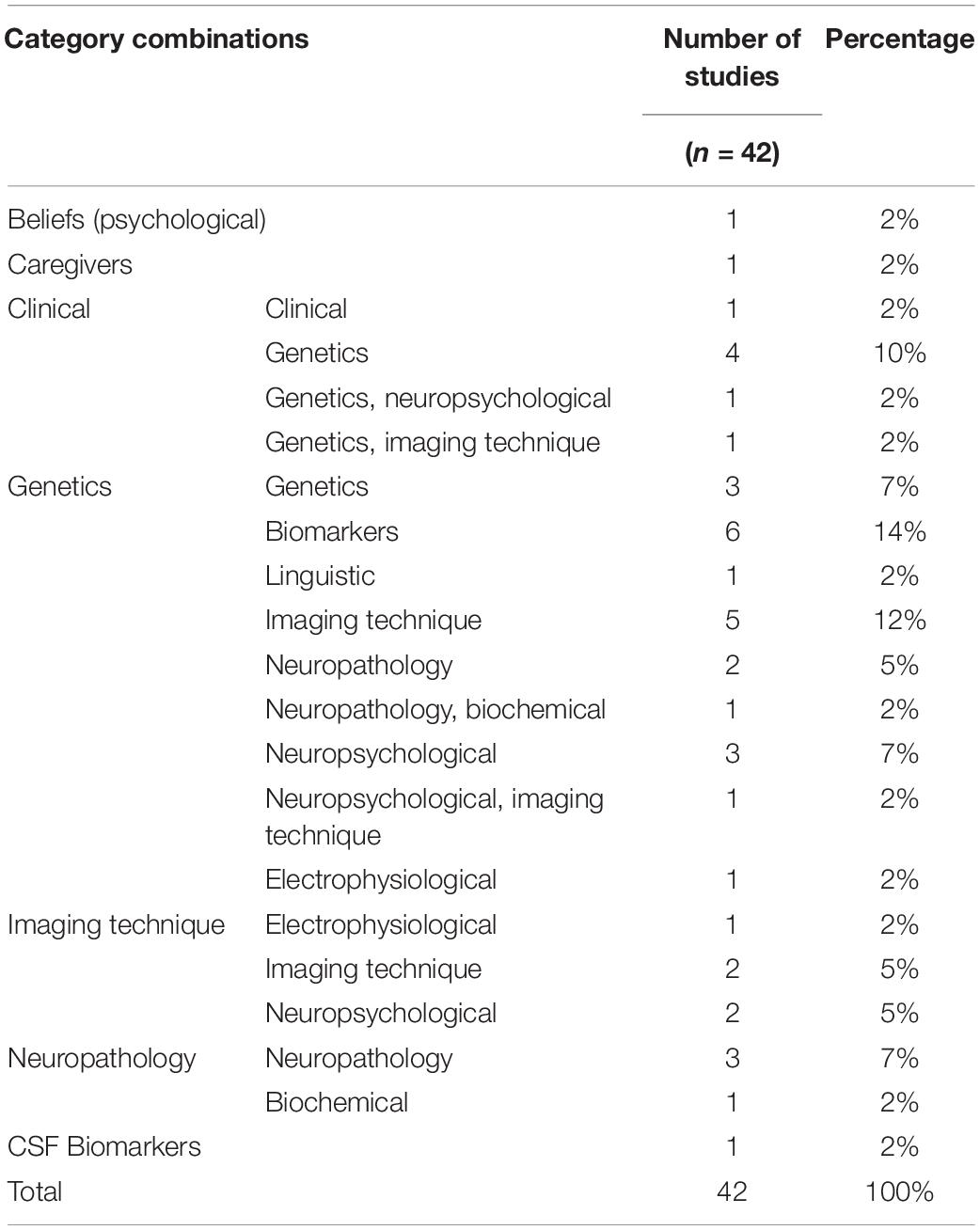
Table 2. Category combinations.
Participant Type
Of the 42 studies, two were conducted on members of families with a history of EOAD (Withers et al., 2019, 2021). In the preclinical stage, two of the studies were conducted on PSEN variant carriers (Ringman et al., 2004, 2007a) and 12 on subjects with a history or carriers of EOAD-associated variants Ringman et al., 2008a,b, 2011, 2012b,d,e; Golob et al., 2009; Medina et al., 2011, 2021; Braskie et al., 2013; Petok et al., 2018; Joe et al., 2019). In the clinical phase, three of the studies were conducted in A431E carriers (Parker et al., 2019; Alakkas et al., 2020; Dumois-Petersen et al., 2020), and two of the studies were conducted in PSEN variant carriers (Joshi et al., 2012; Soosman et al., 2016). Three studies were conducted on A431E carriers in both preclinical and clinical stages (Murrell et al., 2006; Yescas et al., 2006; Santos-Mandujano et al., 2020), “three in PSEN variant carriers (Rogaeva et al., 2001; Ringman et al., 2005; Leverenz et al., 2006), and eight in subjects with a history or carriers of EOAD-associated variants (Ringman et al., 2007b,2010, 2012a,c; Apostolova et al., 2011; Braskie et al., 2012; Lee et al., 2013; Singer et al., 2021). One study included preclinical and clinical phase participants and individuals with SAD (Portelius et al., 2010). This is classified as part of the studies conducted in A431E carriers as the results were reported per participant. Of the studies conducted on brain tissue, four were performed in PSEN variant carriers (Maarouf et al., 2008; Roher et al., 2013; Beck et al., 2016; Gefen et al., 2020) and two in EOAD cases (Albrecht et al., 2009; Ringman et al., 2016).
Variants Presented in the Studies
A431E PSEN1 Variant
Seven studies are focused on A431E (see Table 3). In 2006, a study hypothesized the founding effect of this variant in the Altos de Jalisco area (Yescas et al., 2006), a letter to the editor, further added 15 independent families to this finding (Murrell et al., 2006). Phenotypic variability reported in the studies, such as SP (Murrell et al., 2006; Yescas et al., 2006; Parker et al., 2019; Dumois-Petersen et al., 2020), motor impairment, visuospatial deficits, olfactory dysfunctions, such as hyposmia and anosmia, as well as respiratory difficulties and visual impairment (Santos-Mandujano et al., 2020), language disorders (Dumois-Petersen et al., 2020), neuropsychiatric symptoms (Alakkas et al., 2020; Dumois-Petersen et al., 2020), atrophy disproportionate to age (Parker et al., 2019; Alakkas et al., 2020; Santos-Mandujano et al., 2020), chronic microhemorrhages within bilateral occipital, temporal, and right frontal lobes and pseudobulbar affect (Parker et al., 2019), and periventricular white matter hyperintensities (Santos-Mandujano et al., 2020), were reported. Two of these studies are case reports (Parker et al., 2019; Alakkas et al., 2020), one of them describing a case of a homozygous person (Parker et al., 2019). In two studies, the average age of dementia onset is mentioned as 42.5 ± 3.9 years (Dumois-Petersen et al., 2020) and 40 years (Yescas et al., 2006). One article identifies low levels of Aβ1–37, Aβ1–38, and Aβ1–39 in cerebrospinal fluid in carriers of this variant, compared to people with SAD (Portelius et al., 2010).
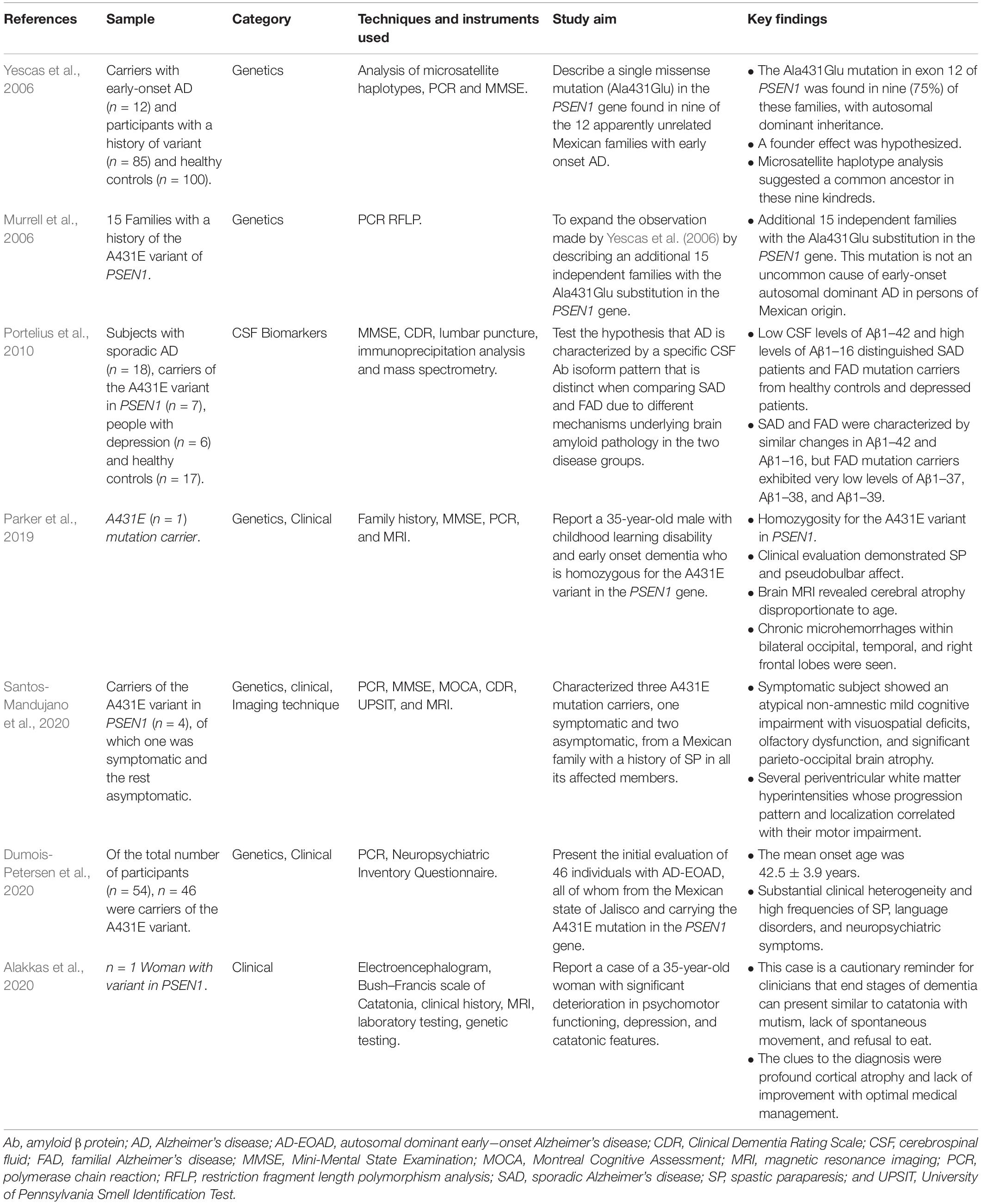
Table 3. A431E of PSEN1 studies.
We found A431E in studies mixed with other genetic variants; in Table 4, we classified the articles analyzing PSEN variants. Eight studies focused on PSEN1 variants (Rogaeva et al., 2001; Ringman et al., 2004, 2005, 2007a; Joshi et al., 2012; Beck et al., 2016; Soosman et al., 2016; Gefen et al., 2020) and, in three more, variants in PSEN1 and PSEN2 are analyzed (Leverenz et al., 2006; Maarouf et al., 2008; Roher et al., 2013).
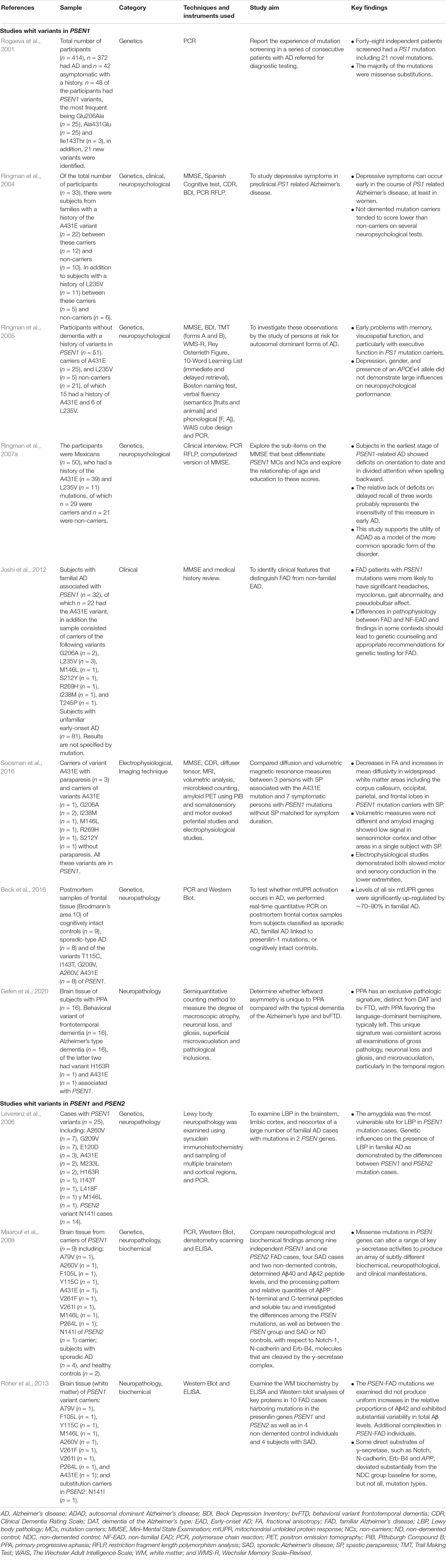
Table 4. PSEN studies.
PSEN1
One article in the Genetics category described novel missense variant substitutions (Rogaeva et al., 2001), among them, is the Ala431Glu. In four studies, the authors identified clinical and neuropsychological characteristics in the participants with variants in PSEN1. In the early stages, memory, visuospatial, and executive function deficits were found (Ringman et al., 2005). In a study using the Mini-Mental State Examination (MMSE), a decreased temporal orientation performance and divided attention were observed, whereas the three-word list subtest has no sensitivity in identifying changes in memory in the preclinical stage (Ringman et al., 2007a). In the case of women, depressive symptoms were identified (Ringman et al., 2004), and carriers of PSEN1 mutations in the clinical stage more significantly presented headaches, myoclonus, gait abnormality, and pseudobulbar affect (Joshi et al., 2012). In these articles, the sample is composed primarily of people with the A431E variant. One article reported findings related to electrophysiological and imaging techniques in carriers with and without SP, where SP carriers showed decreased fractional anisotropy, increased mean diffusivity in widespread white matter areas, and slow motor and sensory conduction in the inferior extremities (Soosman et al., 2016).
Two articles study brain tissue of carriers of PSEN1 variants, and one presents a significant upregulation of six genes related to mitochondrial unfolded protein response (Beck et al., 2016) with primarily differences between primary progressive aphasia (PPA), AD, and a behavioral variant of frontotemporal dementia (Gefen et al., 2020).
PSEN1 and PSEN2
Three studies presented neuropathological features; differences among these genes were found in one article: in PSEN1 mutation cases, the amygdala was more vulnerable to Lewy body pathology than in PSEN2 (Leverenz et al., 2006). The other two analyze the biochemical and neuropathological implications (Maarouf et al., 2008; Roher et al., 2013), variation in levels of beta-amyloid, and differences in some substrates of gamma secretase (Roher et al., 2013) along with its dysfunction (Maarouf et al., 2008).
After presenting the studies exclusively to PSEN, we categorized those who reported in combination with APP. In Table 5, we present these articles in three categories: first, articles with PSEN1 and APP (Ringman et al., 2007b, 2008a,b, 2010, 2011, 2012a,b,c,d,e; Golob et al., 2009; Apostolova et al., 2011; Braskie et al., 2012, 2013; Lee et al., 2013; Joe et al., 2019; Medina et al., 2021; Singer et al., 2021); second, articles with PSEN1, PSEN2, and APP variants (Albrecht et al., 2009; Medina et al., 2011; Ringman et al., 2016; Petok et al., 2018), and finally, those articles who reported families with a history of EOAD (Withers et al., 2019) and another with A431E PSEN1 and an unknown mutation (Withers et al., 2021).
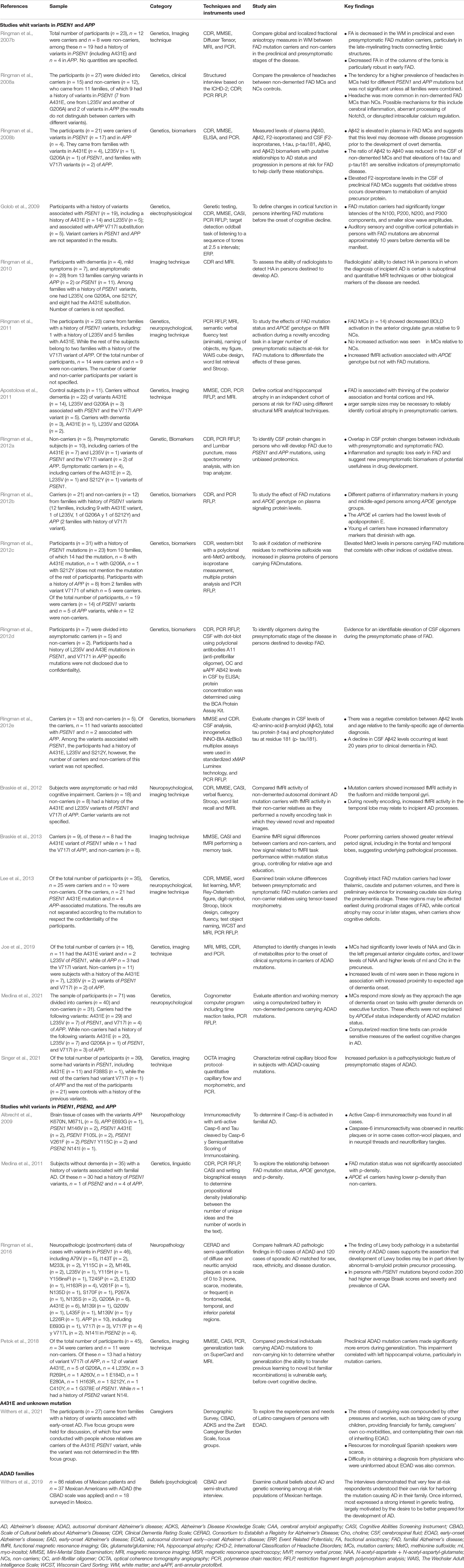
Table 5. PSEN and APP studies.
PSEN1 and APP
Six of the studies included genetic analysis in combination with biomarkers (Ringman et al., 2008b, 2012a,b,c,d,e): four CSF quantifications, two of which identified Aβ42 depletion (Ringman et al., 2008b, 2012e), one finds oligomers elevation (Ringman et al., 2012d), and one identified overlapped protein changes (Ringman et al., 2012a). Three articles measured plasma levels; one found elevated Aβ42 (Ringman et al., 2008b), other inflammatory markers (Ringman et al., 2012b), and other elevated MetO (Ringman et al., 2012c).
In imaging studies (Ringman et al., 2007b,2010, 2011; Apostolova et al., 2011; Braskie et al., 2012, 2013; Lee et al., 2013; Joe et al., 2019; Singer et al., 2021), Ringman et al. (2007b) reported a decreased FA in white matter in the preclinical stage. Singer et al. (2021) found increased retinal perfusion also in presymptomatic carriers, and Joe et al. (2019) revealed that carriers had significantly lower levels of NAA and glutamine in the left pregenual anterior cingulate cortex, and lower levels of NAA and higher levels of myoinositol and choline in the precuneus, and a thinning of the posterior association and frontal cortices with hippocampal atrophy (Apostolova et al., 2011), lower thalamic, caudate, putamen volumes (Lee et al., 2013), and decreased BOLD activation in the anterior cingulate gyrus (Ringman et al., 2011). One article examined the ability of radiologists in the diagnosis of early AD stages in people in early stages of EOAD and found it to be suboptimal. Therefore, another marker is considered necessary for diagnosis (Ringman et al., 2010). Braskie et al. (2012) identified mutation carriers showing an increased fMRI activity in the fusiform and middle temporal gyri and a greater retrieval period signal, including in the frontal and temporal lobes (Braskie et al., 2013). These findings were related to the predementia phase.
Three of the studies with neuropsychological findings (Ringman et al., 2011; Braskie et al., 2012; Lee et al., 2013; Medina et al., 2021) reported no differences in cognitive tests between preclinical carriers of the mutations and non-carriers (Ringman et al., 2011; Braskie et al., 2012; Lee et al., 2013). However, in a memory retrieval task, a lower fMRI activity in the hippocampus was observed (Braskie et al., 2012) while for the executive function, the response gets slower as they approach the age onset of dementia (Medina et al., 2021). In carriers with mild cognitive impairment, lower memory, language, and visuospatial, executive functioning scores, were observed compared to preclinical carriers and controls (Lee et al., 2013).
In the clinical, a higher prevalence of headaches in MCs is held for different PSEN1 and APP mutations (Ringman et al., 2008a) and electrophysiological features related to longer latencies of the N100, P200, N200, and P300 components, and smaller slow wave amplitudes (Golob et al., 2009).
PSEN1, PSEN2, and APP
Two of the studies analyzed brain tissue; Albrecht et al. (2009) identified that Casp-6 immunoreactivity was active in every participant, while Ringman et al. (2016) found Lewy body pathology in 27.1% of the ADAD cases and a higher Braak scores and cerebral amyloid angiopathy (CAA) prevalence. One study focused on linguistic aspects of the carriers (Medina et al., 2011), in which p-density was neither related to the status of participants with a history of EOAD nor with years to clinical onset of the disease, but it was associated with the presence of the APOEe4 allele. The last article reported cognitive data in relation to neuroanatomical findings (Petok et al., 2018), in which more errors in generalization tasks were associated with smaller left hippocampal volume in carriers.
History of Early-Onset Alzheimer’s Disease
Two studies were conducted on participants with a history of EOAD. One of the articles assessed cultural beliefs related to AD and genetic testing (Withers et al., 2019); in this study, some of the participants with a history of ADAD associated with genetic variants know their own risk of developing the disease. The authors found that providing information about the genetic bases of AD increased the interest of people with a history of ADAD for genetic testing. The other article assessed the experiences and needs of caregivers (Withers et al., 2021) and found the stress of informal caregivers comes from different sources beyond caregiving, such as knowing their own risk, caring for other family members including children, providing financially, their own health, and diagnosis access, and their main need is to access information in their own language.
Discussion
The most common cause of hereditary EOAD is PSEN1 mutations followed by PSEN2 and APP mutations (Ramos et al., 2020). While the pathophysiology is similar, there are differences in the AD phenotype (Ringman et al., 2014). Among the variants in PSEN1, A431E is one of the primary three due to the number of carriers, which varies from 381 to 301 (Dumois-Petersen et al., 2020; Llibre-Guerra et al., 2021), and more descendants have been reported to be at risk. Therefore, the incidence is currently open (Llibre-Guerra et al., 2021).
This scoping review aims to synthesize the findings related to the characteristics of the PSEN1 A431E variant associated with EOAD. The results of this review integrated a few studies focused only on the variant of our interest; most studies included other PSEN1, PSEN2, and APP variants in addition to A431E.
In Mexico, a founder effect (Ala431Glu in PSEN1) was hypothesized in the Altos de Jalisco area by Yescas et al. (2006). This could explain why the carriers were mainly Mexican and Mexican Americans both in Mexico and in the United States.
The founder effect hypothesis and the identification of more families could be associated with the growing interest in the variant reflected in the considerable increase in studies between 2006 and 2010 and it has been rising since then.
Analyzing the pathophysiology of EOAD-associated variants, such as A431E, from the early stages of the disease, provides comprehensive knowledge especially to identify biomarkers and interventions with therapeutic potential (Pilotto et al., 2013; Russell et al., 2014). We consider this could be the reason why most of the identified studies have been performed at the preclinical stage.
Synthesizing the findings of the A431E variant is crucial, for it allows comparison of phenotypic features between this and other variants and with SAD cases as well, and provides a framework for clinicians working with individuals with such a history of AD. Therefore, we highlight the importance of constructing knowledge by means of independently reporting results or comparing variants in studies that involve people with a history of two or more variants.
A431E in PSEN
The average age of dementia onset is 40 years with a range of either 34–48 years (Yescas et al., 2006) or 42.5 ± 3.9 years of age (Dumois-Petersen et al., 2020). In both studies, the onset of symptoms was established based on reports from family members. Since families usually seek medical attention in advanced stages, we consider these reports may be biased. There were no longitudinal studies among the articles included in this review, only one study reported cross-sectional data over the course of 6 years of disease evolution (Dumois-Petersen et al., 2020). This lack of longitudinal studies may be due to the relatively recent identification of A431E, barriers that delay diagnosis such as the cost and time implications of assessments, and the attitudes of people with a history of the variant toward genetic analysis and research.
Evidence regarding phenotypic variability is not conclusive. Joshi et al. (2012) identified atypical features in carriers of variants associated with FAD, and these features were also reported by other authors: Pseudobulbar effect was identified also in the case of Parker et al. (2019); myoclonus was presented in one of the families identified by Yescas et al. (2006); gait abnormality was identified as a first symptom (Dumois-Petersen et al., 2020); and headaches were reported in the case of an A431E carrier (Alakkas et al., 2020), but the prevalence and intensity of the headaches could not be inquired; in addition, headaches were present in 67% of carriers in the study by Ringman et al. (2008a) and allowed for differentiating carriers and non-carriers of variants in both PSEN1 and APP in a sample composed mainly of families with a history of A431E. Could these symptoms be part of a continuum or manifest at any moment is a topic of study and could be addressed in upcoming longitudinal studies. Spastic paraparesis is one of the main clinical features associated with A431E (Parker et al., 2019; Dumois-Petersen et al., 2020; Santos-Mandujano et al., 2020). In addition, pure motor presentations have been frequently identified in carriers of this variant (Llibre-Guerra et al., 2021). A431E has been associated with white matter abnormalities which correlate with motor impairments (Santos-Mandujano et al., 2020) which in some cases even preceded and exceeded cognitive symptoms.
Yescas et al., 2006, reported that one of the families in their study had a history of partial seizures 20 years prior to the clinical onset of AD, which they did not consider to be related to the A431E phenotype. However, seizures have been reported to occur in 15% of A431E carriers (Dumois-Petersen et al., 2020). The small population in the Yescas study may have been responsible for that finding and EEG evaluation in the early stages of the disease or a specific questioning for epileptic activity may be considered in EOAD cases, especially in those harboring the A431E variant.
Although evidence is still inconclusive, this type of manifestation could indicate A431E leads to an atypical presentation of EOAD whose early manifestations in the preclinical stage are not amnestic; therefore, this symptomatology should be explored as part of the clinical practice in this disease.
Given the heterogeneous characteristics of the studies involving carriers and non-carriers of different variants in the three principal genes associated with EOAD, we considered discussing the findings of A431E, PSEN1, PSEN2, and APP in a unified manner to contrast characteristic types among carriers of different variants.
Neuropsychological reports are inconsistent. Although memory deficits have been reported as the first cognitive symptom (Dumois-Petersen et al., 2020), verbal memory test scores did not allow differentiation between carriers and non-carriers at the preclinical stage (Ringman et al., 2004, 2011). In language, findings varied between those in which performance on language tests allowed differentiation (Lee et al., 2013) or not (Ringman et al., 2005; Medina et al., 2011) between carriers and non-carriers, and language disorders were observed as an initial symptom only in a minority (Dumois-Petersen et al., 2020). Visuospatial deficits are present since the early stages of the disease (Ringman et al., 2005; Lee et al., 2013; Santos-Mandujano et al., 2020). As for executive functioning, in mild cognitive impairment, both the carriers (Lee et al., 2013) and non-demented (Ringman et al., 2005) had lower and slower performance (Medina et al., 2021). These changes have been associated with Tau neurofibrillary tangles in the prefrontal areas (Weintraub et al., 2012) and may reflect dual protein participation in the early stages of the disease.
Neuropsychiatric manifestations complicate the diagnosis and even disguise some of the symptoms of the clinical onset of EOAD, as in the case reported by Alakkas et al. (2020). Thus, we highlight the importance of exploring a family history of AD in the neuropsychiatric clinical practice and following up over time. Depressive symptoms are common during EOAD associated with A431E in 53% of the cases (Dumois-Petersen et al., 2020). In the case of female carriers, depressive symptoms usually appear in the first stages of the disease and could be associated with the neuropathology of AD (Ringman et al., 2004). Other neuropsychiatric symptoms were reported infrequently in the studies reviewed, including hallucinations in 11.8% of the A431E carriers (Dumois-Petersen et al., 2020), catatonia, mutism, lack of spontaneous movement, and refusal to eat (Alakkas et al., 2020).
The identified neuropathological and biomarker findings and distinctive clinical features, such as SP, support the classification of the A431E variant as pathogenic, as described by the ACMG/AMP guidelines (Alzforum, 2022).
A neuropathological characterization of a brain of an A431E carrier showed severe frontal atrophy, neuronal loss, and gliosis from moderate to severe, and a predominance of neurofibrillary tangles followed by cotton-wool plaques, with greater accumulation of the beta-amyloid 40. The type and concentration per area of amyloid deposits and neurofibrillary tangles have been reported to differ widely among carriers of PSEN1 and PSEN2 variants and SAD (Maarouf et al., 2008).
Lewy body pathology (LBP) was more common in the amygdala of carriers of variants in PSEN1 (found in 96% of carriers) than in PSEN2 (Leverenz et al., 2006). Ringman et al. (2016), when analyzing middle frontal, superior temporal, and inferior parietal regions, reported LBP in only 21.1% of the ADAD cases due to variants in PSEN1, PSEN2, and APP. In these regions, this pathology type was significantly higher in SAD.
In contrast, CAA scores were higher in ADAD due to variants in PSEN1 beyond codon 200 (in 63.3% of the cases analyzed) than in SAD (39.2%) (Ringman et al., 2016). Cases with ADAD had a CAA mean score situated in the mild range with a trend toward the moderate (Ringman et al., 2016). Similarly, the case described by Maarouf et al. (2008) of A431E had a moderate CAA score.
Activation levels of mtUPR genes in the frontal cortex were significantly higher in PSEN1 variant carriers (70–90%) compared with levels in those with SAD (40–60%) (Beck et al., 2016). This may result in increased vulnerability to pathological processes associated with this response in carriers of PSEN1 variants.
Active Casp-6 immunoreactivity is present in cases of EOAD due to variants in PSEN1, PSEN2, and APP, and in SAD (Albrecht et al., 2009). The two A431E carriers in this study presented a neuritic plaque and neurofibrillary tangle in densities ranging from moderate to severe and mild to moderate neuropil threads in the superior/medial temporal gyrus, hippocampus, and entorhinal cortex (Albrecht et al., 2009).
Although the sample size of these studies was small and the minority were carriers of A431E, it is important to highlight that the neuropathological findings in postmortem studies allow in the first instance the differentiation between EOAD due to A431E variant or others in PSEN1, and variants in PSEN2 or APP, SAD, and dementias caused by other conditions such as PPA and bvFTD (Gefen et al., 2020), and may ultimately be of help for the differential diagnosis (Braak and Braak, 1991; McKhann et al., 2011).
Case reports identified atrophy disproportionate to age (Parker et al., 2019; Alakkas et al., 2020; Santos-Mandujano et al., 2020) in structures such as thalamic, caudate, the putamen (Lee et al., 2013), hippocampus, and in posterior association and frontal cortices (Apostolova et al., 2011). As in the case of Alakkas et al. (2020), levels of cortical and subcortical atrophy are key in the differential diagnosis of the disease, but there are also other potential measures in the identification of EOAD, for example, decreased BOLD activation in the cingulate gyrus (Ringman et al., 2011), hyperactivity in the fusiform gyrus and medial temporal gyrus (Braskie et al., 2012), and lower levels of NAA and higher levels of myoinositol and choline in the precuneus (Joe et al., 2019).
Biomarkers are useful, especially CSF tau/ratio for differential diagnosis, therapeutic targets, and even to measure the progression of the disease (Pilotto et al., 2013; Russell et al., 2014). In CSF, toxic effects of Aβ42 oligomerization at synapses independent of amyloid plaque formation have been studied (Ringman et al., 2012d), and Ringman et al. (2012e) found elevated oligomers and low levels of Aβ42 in asymptomatic individuals with a history of variants in PSEN1 (including A431E) and APP. Of the seven participants, five were carriers, with a significant elevation of ring protofibrils with a progressive reduction identified 20 years before to age of onset of dementia. In carriers, the Aβ42/Aβ40 ratio was lower (Ringman et al., 2008b) whereas SAD participants and A431E carriers had similar changes in Aβ42 and Aβ16, but symptomatic and asymptomatic carriers show a downward trending pattern of Aβ37, Aβ38, and Aβ39 isoform levels, suggesting that this variant determines the cleavage site of γ-secretase which is associated with disease manifestation (Portelius et al., 2010).
Plasma Aβ42 levels have been identified as elevated in carriers of variants associated with FAD and may decrease with disease progression prior to the development of dementia (Ringman et al., 2008b). An association was found between methionine sulfoxide levels with the amount of plasma F2-isoprostane and superoxide dismutase-1 (Ringman et al., 2012c); in addition, inflammatory markers and synaptic degeneration in presymptomatic carriers have been found, which has been indicated as a potential therapeutic target (Ringman et al., 2012b).
Providing information about the genetic basis of AD may increase the interest of people with a history of variants associated with EOAD to participate in clinical research and act focused on staying informed about the implications of this disease (Withers et al., 2019). However, it is important to consider that few studies have been conducted in this specific area. Other authors identified the factors leading to genetic testing in individuals with a history of variants associated with FAD were worry about the clinical onset of the disease and if they were carriers or not, and family and financial planning (Steinbart et al., 2001). The financial aspect was also an important stress source for caregivers of variant carriers associated with EOAD since some of them were also the main financial providers of their families (Withers et al., 2021).
An expansion of the findings regarding A431E is to be expected because a current area of research in our University is focused on studying the clinical, neuropsychological, social, and pathological hallmarks of these variants and has started collaborations with groups devoted to the ADAD. We expect that broader, longitudinal studies in both the preclinical and clinical stages will also help in a better understanding of the clinical course and the physiopathology of EOAD with the A431E variant.
Limitations
The limitations in the methodology consist of having only considered studies in humans or brain tissue. The exclusion of experimental studies could result in relevant information on pathophysiology, which was omitted from this review.
This article has described the extent, type, and research findings related to A431E in PSEN1. However, quality criteria were not applied to assess the methods of the included studies or the validity of their results.
Although among many of the studies identified, participants were carriers of other variants of PSEN1 or associated with FAD, and most of the participants carried the A431E represents a limitation when characterizing these carriers. We recommend avoiding the generalization of the different findings.
In addition, the number of participants in each study and the design are variable.
Future systematic reviews should integrate more information about other variants mostly studied to contrast the information found.
Conclusion
This scoping review summarizes research associated with the A431E variant of PSEN1 associated with EOAD.
In total, we reviewed 42 studies, all of them cross-sectional, seven focused on A431E, 11 analyzed that variant and others in the PSEN1 and PSEN2 genes, and 24 whose samples are composed of cases in the genes PSEN and APP.
The included studies indicate that A431E has been studied in several categories, such as genetics, clinical, imaging, neuropsychology, neuropathology, and biomarkers in carriers or participants with a history of the variant in the preclinical and clinical phases.
The key findings in these studies identify several changes that occur years before the age of onset of dementia. Further studies can be designed to monitor through time the early changes associated with the disease, allowing the establishment of programs to attend to the needs of these families.
The clinical heterogeneity found in the different studies is diagnosis challenging, which in clinical practice represents a burden for health professionals and public health measures, where families are affected by late diagnosis, delaying their intervention and support.
This study provides a helpful synthesis for researchers and clinicians who work with AD-related gene variants carriers, mostly with early-onset familial AD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
MO-B made the conception and study design. MO-B, YO-R, AC-A, and VS-G performed the database search. MO-B and YO-R performed data analysis. MO-B, YO-R, VS-G, and AC-A wrote the manuscript screening. All authors contributed with feedback and edited the manuscript.
Funding
Funding was given by the PROINPEP fund of the University of Guadalajara.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the support of the 727497 CONACYT grant.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.860529/full#supplementary-material
References
Alakkas, A., Meyer, A., Debbold, E., Yagudayeva, R., and Bui, J. (2020). Early-Onset Alzheimer’s Disease Masquerading as Catatonia. Case. Rep. Neurol. Med. 2020:1493481. doi: 10.1155/2020/149348
Albrecht, S., Bogdanovic, N., Ghetti, B., Winblad, B., and LeBlanc, A. C. (2009). Caspase-6 Activation in Familial Alzheimer Disease Brains Carrying Amyloid Precursor Protein or Presenilin I or Presenilin II Mutations. J. Neuropathol. Exp. Neurol. 68:12. doi: 10.1097/NEN.0b013e3181c1da10
Alzforum. (2021). Mutations: PSEN-1. Available online at https://www.alzforum.org/mutations/psen-1 (accessed Jun 28, 2021)
Alzforum. (2022). Mutations; PSEN1 A431E. Available online at https://www.alzforum.org/mutations/psen1-a431e (accessed on Apr 28, 2022)
Amponsah, A. E., Guo, R., Kong, D., Feng, B., He, J., Zhang, W., et al. (2021). Patient-derived iPSCs, a reliable in vitro model for the investigation of Alzheimer’s disease. Rev. Neurosci. 32, 379–402. doi: 10.1515/revneuro-2020-0065
Apostolova, L. G., Hwang, K. S., Medina, L. D., Green, A. E., Braskie, M. N., Dutton, R. A., et al. (2011). Cortical and Hippocampal Atrophy in Patients with Autosomal Dominant Familial Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 32, 118–25. doi: 10.1159/000330471
Arksey, H., and O’Malley, L. (2005). Scoping studies: towards a methodological framework. Int. J. Soc. Res. Methodol. 8, 19–32. doi: 10.1080/1364557032000119616
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. NEJM 367, 795–804. doi: 10.1056/NEJMoa1202753
Beck, J. S., Mufson, E. J., and Counts, S. E. (2016). Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer. Res. 13, :610–4. doi: 10.2174/1567205013666151221145445
Blennow, K., Zetterberg, H., and Fagan, A. M. (2012). Fluid Biomarkers in Alzheimer Disease. Cold. Spring. Harb. Perspect. Med. 2:a006221. doi: 10.1101/cshperspect.a006221
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta. Neuropathol. 82, 239–59. doi: 10.1007/BF00308809
Braskie, M. N., Medina, L. D., Rodriguez-Agudelo, Y., Geschwind, D. H., Macias-Islas, M. A., Cummings, J. L., et al. (2012). Increased fMRI signal with age in familial Alzheimer’s disease mutation carriers. Neurobiol. Aging. 33:424.e11–21. doi: 10.1016/j.neurobiolaging.2010.09.028
Braskie, M. N., Medina, L. D., Rodriguez-Agudelo, Y., Geschwind, D. H., Macias-Islas, M. A., Thompson, P. M., et al. (2013). Memory performance and fMRI signal in presymptomatic familial Alzheimer’s disease: memory Performance and fMRI in Familial AD. Hum. Brain. Mapp. 34:3308–19. doi: 10.1002/hbm.22141
Dumois-Petersen, S., Gallegos-Arreola, M. P., Magaña-Torres, M. T., Perea-Díaz, F. J., Ringman, J. M., and Figuera, L. E. (2020). Autosomal dominant early onset Alzheimer’s disease in the Mexican state of Jalisco: high frequency of the mutation PSEN1 c. 1292C> A and phenotypic profile of patients. Am J Med. Genet. C. Semin. Med. Genet. 184, 1023–1029. doi: 10.1002/ajmg.c.31865
Fuller, J. T., Cronin-Golomb, A., Gatchel, J. R., Norton, D. J., Guzmán-Vélez, E., Jacobs, H. I. L., et al. (2019). Biological and Cognitive Markers of Presenilin1 E280A Autosomal Dominant Alzheimer’s Disease: a Comprehensive Review of the Colombian Kindred. J. Prev. Alzheimers. Dis. 6, 112–120. doi: 10.14283/jpad.2019.6
Gefen, T., Mao, Q., Kohler, M., Moeller, S., Kawles, A., Coventry, C., et al. (2020). Primary Progressive Aphasia has a Unique Signature Distinct from Dementia of the Alzheimer’s Type and Behavioral Variant Frontotemporal Dementia Regardless of Pathology. J. Neuropathol. Exp. Neurol. 79, 1379–1381. doi: 10.1093/jnen/nlaa080
Ghidoni, R., Benussi, L., Paterlini, A., Albertini, V., Binetti, G., and Emanuele, E. (2011a). Cerebrospinal Fluid Biomarkers for Alzheimer’s Disease: the Present and the Future. J. Neurodegener. Dis. 8, 413–20. doi: 10.1159/000327756
Ghidoni, R., Paterlini, A., Albertini, V., Stoppani, E., Binetti, G., Fuxe, K., et al. (2011b). A Window into the Heterogeneity of Human Cerebrospinal Fluid Aβ Peptides. J. Biomed. Biotechnol 2011:697036. doi: 10.1155/2011/697036
Golob, E. J., Ringman, J. M., Irimajiri, R., Bright, S., Schaffer, B., Medina, L. D., et al. (2009). Cortical event-related potentials in preclinical familial Alzheimer disease. Neurology 73, 1649–55. doi: 10.1212/WNL.0b013e3181c1de77
Hoogmartens, J., Cacace, R., and Van Broeckhoven, C. (2021). Insight into the genetic etiology of Alzheimer’s disease: a comprehensive review of the role of rare variants. Alzheimers. Dement. 13:e12155. doi: 10.1002/dad2.12155
Joe, E., Medina, L. D., Ringman, J. M., and O’Neill, J. (2019). 1H MRS spectroscopy in preclinical autosomal dominant Alzheimer disease. Brain Imaging and Behav. 13, 925–932. doi: 10.1007/s11682-018-9913-1
Joshi, A., Ringman, J. M., Lee, A. S., Juarez, K. O., and Mendez, M. F. (2012). Comparison of clinical characteristics between familial and non-familial early onset Alzheimer’s disease. J. Neurol. 259, 2182–8. doi: 10.1007/s00415-012-6481-y
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic. Acids. Res 46, D1062–D1067. doi: 10.1093/nar/gkx1153
Larner, A. J. (2013). Presenilin-1 Mutations in Alzheimer’s Disease: an Update on Genotype-Phenotype Relationships. J. Alzheimers. Dis. 37, 653–9. doi: 10.3233/JAD-130746
Larner, A. J., and Doran, M. (2006). Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin–1 gene. J. Neurol. 253, 139–58. doi: 10.1007/s00415-005-0019-5
Larner, A. J., and Doran, M. (2008). Genotype-Phenotype Relationships of Presenilin-1 Mutations in Alzheimer’s Disease: an Update. J. Alzheimers. Dis. 17, 259–65. doi: 10.3233/JAD-2009-1042
Lee, G. J., Lu, P. H., Medina, L. D., Rodriguez-Agudelo, Y., Melchor, S., Coppola, G., et al. (2013). Regional brain volume differences in symptomatic and presymptomatic carriers of familial Alzheimer’s disease mutations. J. Neurol. Neurosurg. Psychiatry. 84, 154–62. doi: 10.1136/jnnp-2011-302087
Leverenz, J. B., Fishel, M. A., Peskind, E. R., Montine, T. J., Nochlin, D., Steinbart, E., et al. (2006). Lewy Body Pathology in Familial Alzheimer Disease: evidence for Disease- and Mutation-Specific Pathologic Phenotype. Arch. Neurol. 63, 370–6. doi: 10.1001/archneur.63.3.370
Livingston, G., Huntley, J., Sommerlad, A., Ames, D., Ballard, C., Banerjee, S., et al. (2020). Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396:10248. doi: 10.1016/S0140-6736(20)30367-6
Llibre-Guerra, J. J., Li, Y., Allegri, R. F., Mendez, P. C., Surace, E. I, Llibre-Rodriguez, J. J., et al. (2021). Dominantly inherited Alzheimer’s disease in Latin America: genetic heterogeneity and clinical phenotypes. Alzheimers. Dement. 17, 653–664. doi: 10.1002/alz.12227
Maarouf, C. L., Daugs, I. D., Spina, S., Vidal, R., Kokjohn, T. A., Patton, R. L., et al. (2008). Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol. Neurodegener. 3:20. doi: 10.1186/1750-1326-3-20
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers. Dement. 7, 263–9. doi: 10.1016/j.jalz.2011.03.005
McKusick-Nathans Institute of Genetic Medicine JHU. (2019). Online Mendelian Inheritance in Man, OMIM®. Available online at http://www.omim.org/entry/104311#0033 (accessed on Feb 02, 2021)
Medina, L. D., Rodriguez-Agudelo, Y., Geschwind, D. H., Gilbert, P. E., Liang, L. J., Cummings, J. L., et al. (2011). Propositional Density and Apolipoprotein E Genotype among Persons at Risk for Familial Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 32, 188–92. doi: 10.1159/000333023
Medina, L. D., Woo, E., Rodriguez-Agudelo, Y., Chaparro Maldonado, H., Yi, D., Coppola, G., et al. (2021). Reaction time and response inhibition in autosomal dominant Alzheimer’s disease. Brain Cogn. 147:105656. doi: 10.1016/j.bandc.2020.105656
Murrell, J., Ghetti, B., Cochran, E., Macias-Islas, M. A., Medina, L., Varpetian, A., et al. (2006). The A431E mutation in PSEN1 causing Familial Alzheimer’s Disease originating in Jalisco State. Mexico: an additional fifteen families. Neurogenetics 7, 277–9. doi: 10.1007/s10048-006-0053-1
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 372:71. doi: 10.1136/bmj.n71
Parker, J., Mozaffar, T., Messmore, A., Deignan, J. L., Kimonis, V. E., and Ringman, J. M. (2019). Homozygosity for the A431E mutation in PSEN1 presenting with a relatively aggressive phenotype. Neurosci. Lett. 699, 195–198. doi: 10.1016/j.neulet.2019.01.047
Petok, J. R., Myers, C. E., Pa, J., Hobel, Z., Wharton, D. M., Medina, L. D., et al. (2018). Impairment of memory generalization in preclinical autosomal dominant Alzheimer’s disease mutation carriers. Neurobiol. Aging 65, 149–157. doi: 10.1016/j.neurobiolaging.2018.01.022
Pilotto, A., Padovani, A., and Borroni, B. (2013). Clinical, Biological, and Imaging Features of Monogenic Alzheimer’s Disease. Biomed. Res. Int. 2013:689591. doi: 10.1155/2013/689591
Portelius, E., Andreasson, U., Ringman, J. M., Buerger, K., Daborg, J., Buchhave, P., et al. (2010). Distinct cerebrospinal fluid amyloid β peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Mol. Neurodegener. 5:1. doi: 10.1186/1750-1326-5-2
Ramos, C., Aguillon, D., Cordano, C., and Lopera, F. (2020). Genetics of dementia: insights from Latin America. Dement. Neuropsychol 14, 223–236. doi: 10.1590/1980-57642020dn14-030004
Ringman, J. M., and Coppola, G. (2013). New Genes and New Insights from Old Genes: update on Alzheimer Disease. Continuum. 19, 358–71. doi: 10.1212/01.CON.0000429179.21977.a1
Ringman, J. M., Diaz-Olavarrieta, C., Rodriguez, Y., Chavez, M., Fairbanks, L., Paz, F., et al. (2005). Neuropsychological function in nondemented carriers of presenilin-1 mutations. Neurology 65, 552–8. doi: 10.1212/01.wnl.0000172919.50001.d6
Ringman, J. M., Diaz-Olavarrieta, C., Rodriguez, Y., Chavez, M., Paz, F., Murrell, J., et al. (2004). Female preclinical presenilin-1 mutation carriers unaware of their genetic status have higher levels of depression than their non-mutation carrying kin. J. Neurol. Neurosurg. Psychiatry. 75, 500–2. doi: 10.1136/jnnp.2002.005025
Ringman, J. M., Goate, A., Masters, C. L., Cairns, N. J., Danek, A., Graff-Radford, N., et al. (2014). Genetic Heterogeneity in Alzheimer Disease and Implications for Treatment Strategies. Curr. Neurol. Neurosci. Rep. 14:499. doi: 10.1007/s11910-014-0499-8
Ringman, J. M., Medina, L. D., Braskie, M., Rodriguez-Agudelo, Y., Geschwind, D. H., Macias-Islas, M. A., et al. (2011). Effects of Risk Genes on BOLD Activation in Presymptomatic Carriers of Familial Alzheimer’s Disease Mutations during a Novelty Encoding Task. Cereb. Cortex 21, 877–83. doi: 10.1093/cercor/bhq158
Ringman, J. M., Monsell, S., Ng, D. W., Zhou, Y., Nguyen, A., Coppola, G., et al. (2016). Neuropathology of Autosomal Dominant Alzheimer Disease in the National Alzheimer Coordinating Center Database. J. Neuropathol. Exp. Neurol. 75, 284–90. doi: 10.1093/jnen/nlv028
Ringman, J. M., O’Neill, J., Geschwind, D., Medina, L., Apostolova, L. G., Rodriguez, Y., et al. (2007a). Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain 130, 1767–76. doi: 10.1093/brain/awm102
Ringman, J. M., Rodriguez, Y., Diaz-Olavarrieta, C., Chavez, M., Thompson, M., Fairbanks, L., et al. (2007b). Performance on MMSE sub-items and education level in presenilin-1 mutation carriers without dementia. Int. Psychogeriatr. 19, 323–32. doi: 10.1017/S1041610206003772
Ringman, J. M., Pope, W., and Salamon, N. (2010). Insensitivity of visual assessment of hippocampal atrophy in familial Alzheimer’s disease. J. Neurol. 257, 839–42. doi: 10.1007/s00415-009-5436-4
Ringman, J. M., Elashoff, D., Geschwind, D. H., Welsh, B. T., Gylys, K. H., Lee, C., et al. (2012b). Plasma Signaling Proteins in Persons at Genetic Risk for Alzheimer Disease: influence of APOE Genotype. Arch. Neurol. 69, 757–64. doi: 10.1001/archneurol.2012.277
Ringman, J. M., Tomic, J. L., Coppola, G., Elashoff, D., Gylys, K. H., and Glabe, C. G. (2012d). Conformation-dependent oligomers in cerebrospinal fluid of presymptomatic familial Alzheimer’s disease mutation carriers. Dement. Geriatr. Cogn. Disord. 2, 652–7. doi: 10.1159/000345771
Ringman, J. M., Coppola, G., Elashoff, D., Rodriguez-Agudelo, Y., Medina, L. D., Gylys, K., et al. (2012e). Cerebrospinal Fluid Biomarkers and Proximity to Diagnosis in Preclinical Familial Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 33, 1–5. doi: 10.1159/000335729
Ringman, J. M., Schulman, H., Becker, C., Jones, T., Bai, Y., Immermann, F., et al. (2012a). Proteomic Changes in Cerebrospinal Fluid of Presymptomatic and Affected Persons Carrying Familial Alzheimer Disease Mutations. Arch. Neurol. 69, 96–104. doi: 10.1001/archneurol.2011.642
Ringman, J. M., Fithian, A. T., Gylys, K., Cummings, J. L., Coppola, G., Elashoff, D., et al. (2012c). Plasma Methionine Sulfoxide in Persons with Familial Alzheimer’s Disease Mutations. Dement. Geriatr. Cogn. Disord. 33, 219–25. doi: 10.1159/000338546
Ringman, J. M., Romano, J. D., Medina, L. D., Rodriguez-Agudelo, Y., Schaffer, B., Varpetian, A., et al. (2008a). Increased Prevalence of Significant Recurrent Headache in Preclinical Familial Alzheimer’s Disease Mutation Carriers. Dement. Geriatr. Cogn. Disord. 25, 380–4. doi: 10.1159/000121986
Ringman, J. M., Younkin, S. G., Pratico, D., Seltzer, W., Cole, G. M., Geschwind, D. H., et al. (2008b). Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 71, 85–92. doi: 10.1212/01.wnl.0000303973.71803.81
Rogaeva, E. A., Fafel, K. C., Song, Y. Q., Medeiros, H., Sato, C., Liang, Y., et al. (2001). Screening for PS1 Mutations in a Referral-Based Series of AD Cases: 21 Novel Mutations. Neurology. 57, 621–5. doi: 10.1212/WNL.57.4.621
Roher, A. E., Maarouf, C. L., Malek-Ahmadi, M., Wilson, J., Kokjohn, T. A., Daugs, I. D., et al. (2013). Subjects harboring presenilin familial Alzheimer’s disease mutations exhibit diverse white matter biochemistry alterations. Am. J. Neurodegener. Dis. 2, 187–207.
Rostgaard, N., Waldemar, G., Nielsen, J. E., and Simonsen, A. H. (2015). Cerebrospinal Fluid Biomarkers in Familial Forms of Alzheimer’s Disease and Frontotemporal Dementia. Dement. Geriatr. Cogn. Dis. Extra. 40, 54–62. doi: 10.1159/000381828
Russell, C. L., Koncarevic, S., and Ward, M. A. (2014). Post-Translational Modifications in Alzheimer’s Disease and the Potential for New Biomarkers. J. Alzheimers. Dis. 41, 345–64. doi: 10.3233/JAD-132312
Santos-Mandujano, R. A., Ryan, N. S., Chávez-Gutiérrez, L., Sánchez-Torres, C., and Meraz-Ríos, M. A. (2020). Clinical Association of White Matter Hyperintensities Localization in a Mexican Family with Spastic Paraparesis Carrying the PSEN1 A431E Mutation. J. Alzheimers. Dis. 73, 1075–1083. doi: 10.3233/JAD-190978
Schindler, S. E., Bollinger, J. G., Ovod, V., Mawuenyega, K. G., Li, Y., Gordon, B. A., et al. (2019). High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. J. Neurol. 93, e1647–e1659. doi: 10.1212/WNL.0000000000008081
Singer, M. B., Ringman, J. M., Chu, Z., Zhou, X., Jiang, X., Shahidzadeh, A., et al. (2021). Abnormal retinal capillary blood flow in autosomal dominant Alzheimer’s disease. Alzheimer’s Dement. 13:e12162. doi: 10.1002/dad2.12162
Soosman, S. K., Joseph-Mathurin, N., Braskie, M. N., Bordelon, Y. M., Wharton, D., Casado, M., et al. (2016). Widespread white matter and conduction defects in PSEN1-related spastic paraparesis. Neurobiol. Aging 47, 201–209. doi: 10.1016/j.neurobiolaging.2016.07.030
Steinbart, E. J., Smith, C. O., Poorkaj, P., and Bird, T. D. (2001). Impact of DNA Testing for Early-Onset Familial Alzheimer Disease and Frontotemporal Dementia. Arch. Neurol. 58, 1828–31. doi: 10.1001/archneur.58.11.1828
Tanzi, R. E. (2012). The Genetics of Alzheimer Disease. Cold. Spring. Harb. Perspect. Med. 2, 10. doi: 10.1101/cshperspect.a006296
Weintraub, S., Wicklund, A. H., and Salmon, D. P. (2012). The neuropsychological profile of Alzheimer disease. Cold. Spring. Harb. Perspect Med. 2:a006171. doi: 10.1101/cshperspect.a006171
Withers, M., Cortez-Sanchez, K., Herrera, J., Ringman, J. M., and Segal-Gidan, F. (2021). “My backpack is so heavy”: experiences of Latino caregivers of family with early-onset Alzheimer’s. J. Am. Geriatr. Soc. 69, 1539–1547. doi: 10.1111/jgs.17091
Withers, M., Sayegh, P., Rodriguez-Agudelo, Y., Ernstrom, K., Raman, R., Montoya, L., et al. (2019). A mixed-methods study of cultural beliefs about dementia and genetic testing among Mexicans and Mexican-Americans at-risk for autosomal dominant Alzheimer’s disease. J. Genet. Couns. 28, 921–932. doi: 10.1002/jgc4.1133
World Health Organization (2020). Dementia. Available online at: https://www.who.int/es/news-room/fact-sheets/detail/dementia (accessed June 28, 2021).
Yescas, P., Huertas-Vazquez, A., Villarreal-Molina, M. T., Rasmussen, A., Tusié-Luna, M. T., López, M., et al. (2006). Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early-onset Alzheimer’s disease in Mexican families. Neurogenetics. 7, 195–200. doi: 10.1007/s10048-006-0043-3
Keywords: A431E, c.1292C<A, PSEN1, EOAD, founder effect, dementia, ADAD, scoping review
Citation: Orozco-Barajas M, Oropeza-Ruvalcaba Y, Canales-Aguirre AA and Sánchez-González VJ (2022) PSEN1 c.1292C<A Variant and Early-Onset Alzheimer’s Disease: A Scoping Review. Front. Aging Neurosci. 14:860529. doi: 10.3389/fnagi.2022.860529
Received: 23 January 2022; Accepted: 02 June 2022;
Published: 22 July 2022.
Edited by:
Jun Xu, Capital Medical University, ChinaReviewed by:
Mitsuru Shinohara, National Center for Geriatrics and Gerontology (NCGG), JapanSeong An, Gachon University, South Korea
Copyright © 2022 Orozco-Barajas, Oropeza-Ruvalcaba, Canales-Aguirre and Sánchez-González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor J. Sánchez-González, victor.sanchez@academicos.udg.mx