A Systematic Strategy to Find Potential Therapeutic Targets for Pseudomonas aeruginosa Using Integrated Computational Models
 Fernando Medeiros Filho1†*
Fernando Medeiros Filho1†*  Ana Paula Barbosa do Nascimento1†
Ana Paula Barbosa do Nascimento1†  Maiana de Oliveira Cerqueira e Costa2
Maiana de Oliveira Cerqueira e Costa2  Thiago Castanheira Merigueti1
Thiago Castanheira Merigueti1  Marcio Argollo de Menezes3
Marcio Argollo de Menezes3  Marisa Fabiana Nicolás2
Marisa Fabiana Nicolás2  Marcelo Trindade dos Santos2
Marcelo Trindade dos Santos2  Ana Paula D’Alincourt Carvalho-Assef4
Ana Paula D’Alincourt Carvalho-Assef4  Fabrício Alves Barbosa da Silva1*
Fabrício Alves Barbosa da Silva1*- 1Programa de Computação Científica, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil
- 2Laboratório Nacional de Computação Científica, Petrópolis, Brazil
- 3Instituto de Física, Universidade Federal Fluminense, Niterói, Brazil
- 4Laboratório de Pesquisa Em Infecção Hospitalar, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil
Pseudomonas aeruginosa is an opportunistic human pathogen that has been a constant global health problem due to its ability to cause infection at different body sites and its resistance to a broad spectrum of clinically available antibiotics. The World Health Organization classified multidrug-resistant Pseudomonas aeruginosa among the top-ranked organisms that require urgent research and development of effective therapeutic options. Several approaches have been taken to achieve these goals, but they all depend on discovering potential drug targets. The large amount of data obtained from sequencing technologies has been used to create computational models of organisms, which provide a powerful tool for better understanding their biological behavior. In the present work, we applied a method to integrate transcriptome data with genome-scale metabolic networks of Pseudomonas aeruginosa. We submitted both metabolic and integrated models to dynamic simulations and compared their performance with published in vitro growth curves. In addition, we used these models to identify potential therapeutic targets and compared the results to analyze the assumption that computational models enriched with biological measurements can provide more selective and (or) specific predictions. Our results demonstrate that dynamic simulations from integrated models result in more accurate growth curves and flux distribution more coherent with biological observations. Moreover, identifying drug targets from integrated models is more selective as the predicted genes were a subset of those found in the metabolic models. Our analysis resulted in the identification of 26 non-host homologous targets. Among them, we highlighted five top-ranked genes based on lesser conservation with the human microbiome. Overall, some of the genes identified in this work have already been proposed by different approaches and (or) are already investigated as targets to antimicrobial compounds, reinforcing the benefit of using integrated models as a starting point to selecting biologically relevant therapeutic targets.
Introduction
Infectious diseases are a concerning public health problem worldwide. Among the most life-threatening infectious diseases are the bacterial infections caused by the “ESKAPE” pathogens, an acronym for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp., which are known for their ability to escape the action of multiple drugs (Pachori, Gothalwal, and Gandhi 2019). As result, the World Health Organization classified some of these multidrug-resistant pathogens like P. aeruginosa as a critical priority on the pathogens list for new antibiotics research and development (World Health Organization 2017). P. aeruginosa is an opportunistic human pathogen known for its metabolic versatility, virulence factor diversity, and great intrinsic and acquired antibiotic resistance. These traits allow the bacterium to cause infections in different areas, e.g., the lower respiratory tract, skin, urinary tract, eyes, leading to bacteremia, endocarditis, and other complications (Lister, Wolter, and Hanson 2009; Silby et al., 2011).
Identifying potential targets for new drug discovery or drug repurposing is achieved when crucial biological processes are well-characterized. The complete genome of P. aeruginosa PAO1, widely used as a reference strain, was sequenced two decades ago by Stover et al. (2000). Furthermore, the ongoing advance of “omics” technologies had provided even more details to unveil the functioning of P. aeruginosa. From a systems biology perspective, these data are the fundamentals of different computational approaches such as the reconstruction of biological networks, a mathematical representation of cell molecules and their interactions. The most common types of biological networks are the metabolic, gene regulatory, and signaling networks (Koutrouli et al., 2020). Genome-scale metabolic networks (GEMs) encompass a set of known biochemical reactions of an organism using gene-protein-reaction (GPR) associations, constrained in some models by thermodynamic directionality, transcription factor activity, gene expression level, and others. The growth rate of an organism in a given condition or the production rate of a metabolite of interest can be predicted from a GEM using optimization methods such as flux balance analysis (FBA) (Ruppin et al., 2010; Hyduke, Lewis, and Palsson 2013).
In addition, we can integrate different types of networks or incorporate additional biological measurements into a single network to provide more robust computational models. Despite the challenge of integrating gene expression data with GEMs, there are several methods proposed to achieve this task. The majority is based on FBA-driven algorithms considering experimentally measured RNA levels to turn off or to constrain the reactions, such as PROM, E-Flux, CoRegFlux, TRFBA, and others (Blazier and Papin 2012; Banos, Trébulle, and Elati 2017; Cruz et al., 2020). PROM is one of the first methods developed to be applied to genome-scale networks in an automated manner. PROM uses transcriptome data to define gene activation or repression, and interactions between regulators and targets. However, it requires a large amount of data (Chandrasekaran and Price 2010). E-Flux defines the reaction maximum flux to the gene expression level, while CoRegFlux applies a statistical approach to infer the gene regulatory network from transcriptome data (Colijn et al., 2009; Banos, Trébulle, and Elati 2017). TRFBA uses the gene expression level converted by the constant parameter C to constrain the reaction upper bound. TRFBA does not require a large amount of data nor previous knowledge of the regulator-target relationship (Motamedian et al., 2017; Malek Shahkouhi and Motamedian 2020).
The growth rate assessment using biological networks enables the prediction of drug targets since gene knockout can result in growth arrest or growth loss. Likewise, the cell response to antimicrobial compounds can be predicted (Chavali et al., 2012; Chung et al., 2021). Indeed, this approach was applied to several pathogens, including ESKAPE Gram-negative bacteria as A. baumannii, K. pneumoniae, and P. aeruginosa, to identify novel targets and to evaluate the impact of last-resort antibiotics on the metabolism (Presta et al., 2017; Ramos et al., 2018; Zhu et al., 2018; Norsigian et al., 2019). Moreover, it is possible to simulate the temporal and spatial dynamics of the growth process, i.e., in the first step of the simulation, biomass and metabolite production rates calculated using FBA update the extracellular concentrations. In the next step, uptake rates of compounds required for FBA calculation are subjected to the previous updated extracellular concentrations and could lead to environmental changes for the following time step. The process may continue until there are no more substrates available in the extracellular space (Scott et al., 2018).
In this work, we applied the TRFBA method for integrating transcriptome data with two metabolic reconstructions of PAO1. Next, we used the ACBM, an agent and constraint-based modeling approach, to simulate the temporal dynamics of the growth process. The primary goals were: 1) to generate an integrated computational model of P. aeruginosa, which incorporates gene expression data in the GEM; and 2) compare the dynamics of metabolic and integrated models to analyze if the progressive inclusion of biological data results in more reliable computational models capable of simulating the biological growth of P. aeruginosa. Then, we investigated the hypothesis that identifying potential targets from integrated models is more accurate than from metabolic models that do not consider information from other cellular processes. We used the algorithm FindTargetsWEB (Merigueti et al., 2019) to find these targets from both models and compare them to discuss their accuracy. The goal is to show improved selectivity and (or) specificity of target prediction from integrated models compared to metabolic models.
Materials and Methods
Data Selection
The genome-scale metabolic models of P. aeruginosa PAO1 used in this work were iMO1056 and iPAO1 (Oberhardt et al., 2008; Zhu et al., 2018). The iMO1056 model contains 992 reactions, 858 metabolites, and 1,042 genes encompassed in the cytoplasmic and extracellular compartments. This metabolic model is the first genome-scale metabolic model of P. aeruginosa, also used extensively in the literature. The iPAO1 model contains 4,365 reactions, 3,022 metabolites, and 1,458 genes encompassed in the cytoplasmic, periplasmic, and extracellular compartments. It is the only model of P. aeruginosa that includes the periplasmic space. We edited the iMO1056 model by adding calcium and chloride ions into the biomass reaction, and their corresponding exchange and transport reactions, because they are essential molecules to the cellular homeostasis and components of the growth media used in our work. Based on biological measurements obtained from Pseudomonas genus, we also adjusted the non-growth-associated maintenance value to 3.96 mmol ATP gDW−1 h−1 in both models (van Duuren et al., 2013). The transcriptome data used in this work are available under the access number E-MTAB-8374 at Array Express database and was performed by Dolan et al. (2020), where total RNA was isolated from cells grown in MOPS minimal medium supplemented with glycerol or acetate as the carbon source. Furthermore, to reproduce these same biological conditions, we properly adjusted the lower and upper bounds of exchange reactions in both computational models.
Transcriptome Data Analysis
First, we assessed the raw reads of RNA-sequencing experiment in fastq format using FastQC (Andrews 2010). Then, we performed quality and adapter filtering using Trimmomatic with default parameters when necessary (Bolger, Lohse, and Usadel 2014). For the alignment of the processed reads to the PAO1 genome (available at GenBank database under the accession number NC_002516), we used HISAT2 (version 2.2.0) (Kim et al., 2019). We used the featureCounts program to count the number of reads mapped to each coding sequence of the PAO1 genome (Liao, Smyth, and Shi 2014) and we normalized the transcript abundance by applying the transcripts per million measure (Wagner, Kin, and Lynch 2012).
Integration of Metabolic Network and Transcriptome Data
We used the TRFBA algorithm in its linear form, i.e. the version which integrates a metabolic network with expression data to model a specific condition, to construct the integrated models of PAO1 from iMO1056 and iPAO1 models, and the chosen transcriptome data (Motamedian et al., 2017; Malek Shahkouhi and Motamedian 2020). First, the TRFBA converts all reactions in the model to their irreversible form. Given a reversible reaction
Flux Balance and Variability Analysis
FBA is a constraint-based mathematical approach used to calculate the fluxes of a metabolic network under the steady-state by optimizing an objective function through linear programming (Orth, Thiele, and Palsson 2010). In this work, we set growth rate as the objective function to be maximized. As the optimal growth rate is calculated, FBA returns a single flux distribution. However, different flux distributions are possible for the same maximal growth. Flux variability analysis (FVA) is a mathematical approach used to determine the minimum and maximum flux value for each reaction in a given model obeying the same constraints and the same objective value as FBA within the solution space (Mahadevan and Schilling 2003; Schellenberger et al., 2011). We used the functions optimizeCbModel (FBA) e fluxVariability (FVA) from COBRA Toolbox to perform these analyses.
Dynamic Simulations of Metabolic and Integrated Models
We used the user-friendly ACBM framework to simulate the growth of P. aeruginosa PAO1 over time using both metabolic and integrated models (Karimian and Motamedian 2020). Briefly, it uses agent and constraint-based modeling to apply intracellular and extracellular restrictions to the cell population in a three-dimensional space, where each cell, metabolite (carbon source), and environment are modeled as an agent. ACBM requires the input of several parameters such as initial cell amount, radius, length, mass, initial metabolite amount, and volume of the simulated environment. The movement of metabolites and cells in the environment is predicted based on stochastic simulations and FBA or TRFBA are used to predict growth. In each time step, if there is a metabolite close to the cell, it consumes the metabolite, moves at random, and the metabolite is removed from the environment. If there is no metabolite, the cell moves at random. All possible events, such as biomass generation, production, or consumption of metabolites are calculated and updated to the next step. If the cell mass doubles, a new cell object is included in the environment (cell division). Cells are removed from the environment when they do not find metabolites during their survival time (cell death). This process continues until the simulation time ends or until all cells die. The computational models and parameters given as input to ACBM (Supplementary Table 1: https://github.com/medeirosfilho1/Integration-paeruginosa/blob/main/Supplementary_table_1.xlsx) were based on experimental data obtained in previous studies (Dolan et al., 2020; H. ; Zhang et al., 2016).
Identification of Potential Therapeutic Targets
We used the FindTargetsWEB online application to identify potential therapeutic targets in PAO1 metabolic and integrated models (Merigueti et al., 2019). The same models used to perform dynamic simulations were converted from MAT to SBML level 3 format using the writeSBML function from COBRA Toolbox, which is the file format required by FindTargetsWEB (all files are available at: https://github.com/medeirosfilho1/Integration-paeruginosa). The application is composed of nine steps. The first step evaluates if the model generates a biomass value greater than zero. As a second step, FindTargetsWEB uses FVA to filter reactions whose flux range is equal to zero. Since those reactions do not accept any variation, they may be more susceptible to perturbations (Oberhardt et al., 2010). However, the results of this step are only maintained if the user chooses to perform the FBA + FVA analysis. Therefore, we used the FBA + FVA option for identifying potential targets. The following steps comprise the knockout of single reactions followed by gene knockouts if GPR associations are available. When knockout simulation results in a biomass value of zero, the reaction GPR and (or) gene information is stored. If the knocked-out gene is included in the stored reaction GPR, this gene is considered essential. When GPR associations are not provided, FindTargetsWEB retrieves EC numbers from the KEGG database through the reaction compounds. Otherwise, EC numbers are retrieved through gene ID. Then, EC numbers are used to query the DrugBank database to obtain protein name, organism, and UniProt ID. The UniProt ID is used to perform blastp searches against the genome of the model organism. Proteins with identity ≥30% are kept. Finally, FindtargetsWEB uses the recovered UniProt IDs to search for inhibitors in the DrugBank database. After obtaining the results from FindTargetsWEB, we filtered the application output to keep only hits with identity ≥60% and coverage ≥70% and used these proteins to carry out the analysis to prioritize targets according to non-host homology and microbiome conservation. First, we performed a blastp search against the human proteome (GRCh38. p13 release available at RefSeq database under the accession number GCF_000001405.39) using E value ≤ 1e-5 and coverage ≥70% as parameters. Next, hits with identity ≥40% were filtered out. Likewise, the remaining proteins were compared to the proteome of 454 organisms (Supplementary Table 2: https://github.com/medeirosfilho1/Integration-paeruginosa/blob/main/Supplementary_table_2.xlsx) from the gastrointestinal tract of NIH Human Microbiome Project (Human Microbiome Project Consortium 2012a; 2012b). Alignments with identity ≥40% were considered as a hit.
Technical Specifications
TRFBA algorithm, ACBM framework, FBA, and FVA analysis were performed using the MATLAB program (R2020b version 9.9) with the COBRA Toolbox (version 2.0.0) and the gplk solver (version 2.7) in the Java runtime environment (version 1.8.0_281).
Results
Dynamic Simulations of Metabolic and Integrated Models
The iMO1056 and iPAO1 metabolic models were modified to reproduce the experimental environment used by Dolan et al. (2020). First, we adjusted the lower and upper bounds of all exchange reactions to zero except those related to the MOPS minimal medium compounds and carbon source, which were set to −1,000 (lower bound) and 1,000 (upper bound). Then, we performed the FBA analysis of metabolic models. The iMO1056 model generated a biomass reaction flux of 11.26 h−1 for acetate as a carbon source and 21.90 h−1 for glycerol as a carbon source. The iPAO1 model generated a biomass reaction flux of 8.68 h−1 and 17.32 h−1 for acetate and glycerol, respectively. After, the ACBM framework was used to dynamically simulate P. aeruginosa PAO1 growth in MOPS minimal medium with acetate (Figures 1A,B) or glycerol (Figures 1C,D) as a carbon source. Since ACBM executions are computationally expensive, the environment volume simulated is only 0.16 μL. Therefore, due to the randomness and discretization effects, it would be improper to directly compare experimental results generated in a counting unit (CFU/mL) and computational results generated in a concentration unit (g/L), whereas the numbers obtained during simulations are relatively low. Instead, we choose to compare the exponential phase duration, entry points in the stationary phase, and growth curve shapes. Simulations revealed that the growth curve reached the stationary phase after 1 hour. In the same way, the carbon sources were consumed in less than 1 hour.
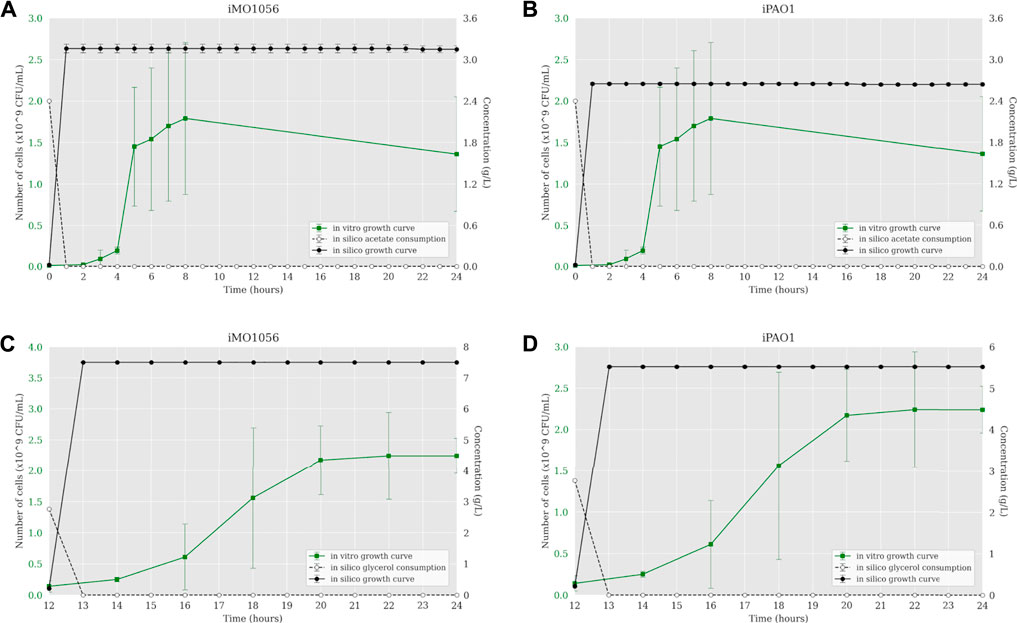
FIGURE 1. Comparison between P. aeruginosa growth curves predicted by ACBM from the metabolic models and growth curves measured by Dolan et al. (2020). (A,B) MOPS minimal medium with acetate as carbon source. (C,D) MOPS minimal medium with glycerol as carbon source. The graphics include the carbon source consumption over time (dashed lines). All measurements were made at least in triplicates, and curves represent the mean of all replicates. Error bars are the standard deviation of the mean.
The next step was to insert experimentally measured uptake rates for both carbon sources as input to the ACBM framework. The default uptake upper bound value is 1,000 mmol gDW−1 h−1. In line with Dolan et al. (2020), we adjusted this parameter to 30.4 mmol gDW−1 h−1 for acetate and 9.2 mmol∙gDW−1 h−1 for glycerol. Figure 2 shows a predicted behavior closer to the observed biologically depicting a sigmoidal curve typical of bacterial growth. When acetate was the carbon source, cells reached the stationary phase after 7 and 8 h of simulation from the iMO1056 and iPAO1 models, respectively, almost depleting acetate concentration (Figures 2A,B). When glycerol was the carbon source, cells reached the stationary phase at 21 and 22 h of simulation from the iMO1056 and iPAO1 models respectively, almost depleting glycerol concentration (Figures 2C,D).
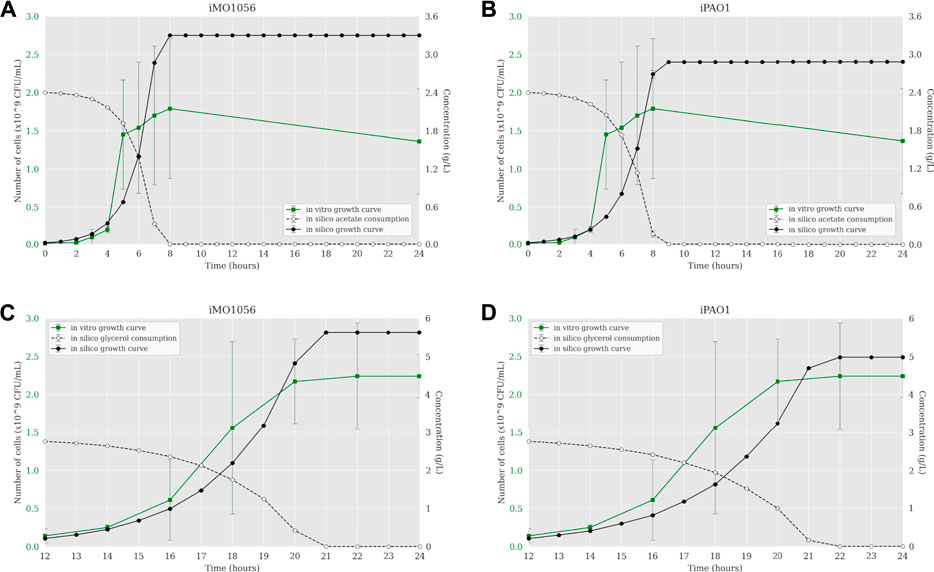
FIGURE 2. Comparison between P. aeruginosa growth curves predicted by ACBM from the metabolic models with adjusted carbon source uptake rates and growth curves measured by Dolan et al. (2020). (A,B) MOPS minimal medium with acetate as carbon source. (C,D) MOPS minimal medium with glycerol as carbon source. The graphics include the carbon source consumption over time (dashed lines). All measurements were made at least in triplicates, and curves represent the mean of all replicates. Error bars are the standard deviation of the mean.
According to Motamedian et al. (2017), we calculated the C constant value for both metabolic models in each growth condition using the transcriptome data of P. aeruginosa PAO1. The resulting C values were 0.605 and 0.172 mmol gDW−1 h−1 in acetate to iMO1056 and iPAO1 models, and 0.063 and 0.043 mmol gDW−1 h−1 in glycerol to iMO1056 and iPAO1 models respectively. We used these values to perform the integration using the TRFBA algorithm adjusting the growth rates to approximately 0.80 h−1 (acetate) and 0.37 h−1 (glycerol) as experimentally measured by Dolan et al. (2020). The dynamic simulations with the integrated models predicted growth curves with a shape more similar to the in vitro growth curve (Figure 3). In acetate, cells entered the stationary phase after 6 h for both integrated models (Figures 3A,B). Likewise, in glycerol, cells reached the stationary phase at 19 h of simulation (Figures 3C,D).
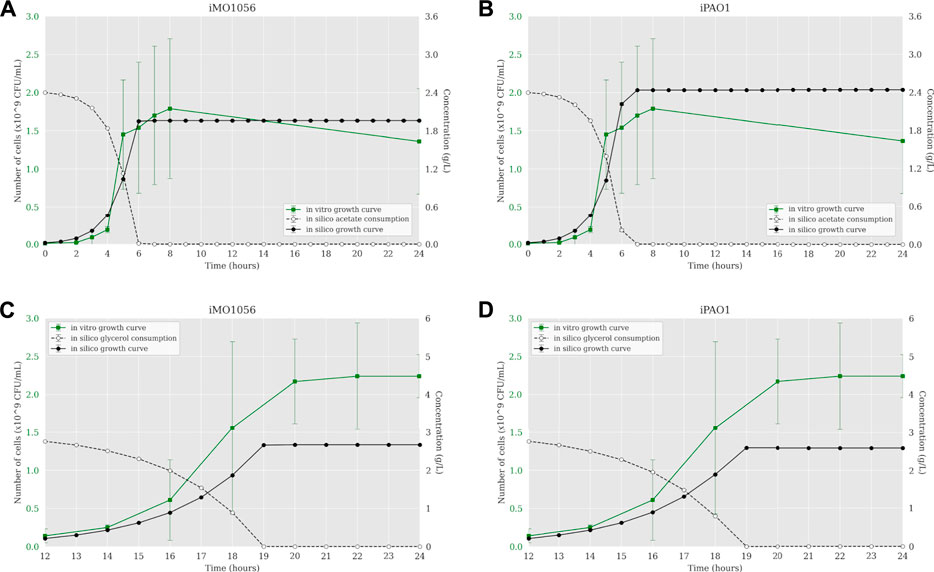
FIGURE 3. Comparison between P. aeruginosa growth curves predicted by ACBM from the integrated models and growth curves measured by Dolan et al. (2020). (A,B) MOPS minimal medium with acetate as carbon source. (C,D) MOPS minimal medium with glycerol as carbon source. The graphics include the carbon source consumption over time (dashed lines). All measurements were made at least in triplicates and curves represent the mean of all replicates. Error bars are the standard deviation of the mean.
Flux Balance and Variability Analysis
The flux distribution in the central metabolism of P. aeruginosa PAO1 during growth in acetate or glycerol was experimentally measured by Dolan et al. (2020). Based on the results, we analyzed the predicted flux flow through the network upon carbon source uptake for both models in all growth conditions using FBA and FVA. The analysis revealed that the fluxes predicted from the iMO1056 and iPAO1 models with acetate as carbon source agreed with the biological pathway (Figure 4 and Supplementary Table 3: https://github.com/medeirosfilho1/Integration-paeruginosa/blob/main/Supplementary_table_3.xlsx). It is noteworthy that the known utilization of isocitrate by tricarboxylic acid cycle and glyoxylate shunt was computationally reproduced by all models (Figure 4, reaction 15 to reactions 16 and 22). However, the reaction numbered as 21 in Figure 4, although catalyzed by the same enzyme, the malate dehydrogenase, showed a slight difference between metabolic and integrated models. The flux in the metabolic adjusted models was mainly going through the reaction using ubiquinone as a cofactor, while in the integrated models, the flux passed through the reaction with nicotinamide adenine dinucleotide as a cofactor. Regarding the flux values, the predicted acetate uptake rates ranged from 460 to 600 mmol gDW−1 h−1; consequently, the enchained reactions also had high flux values. FBA analysis from integrated models revealed acetate uptake rates coherent with those experimentally measured ranging from 45 to 52 mmol gDW−1 h−1.
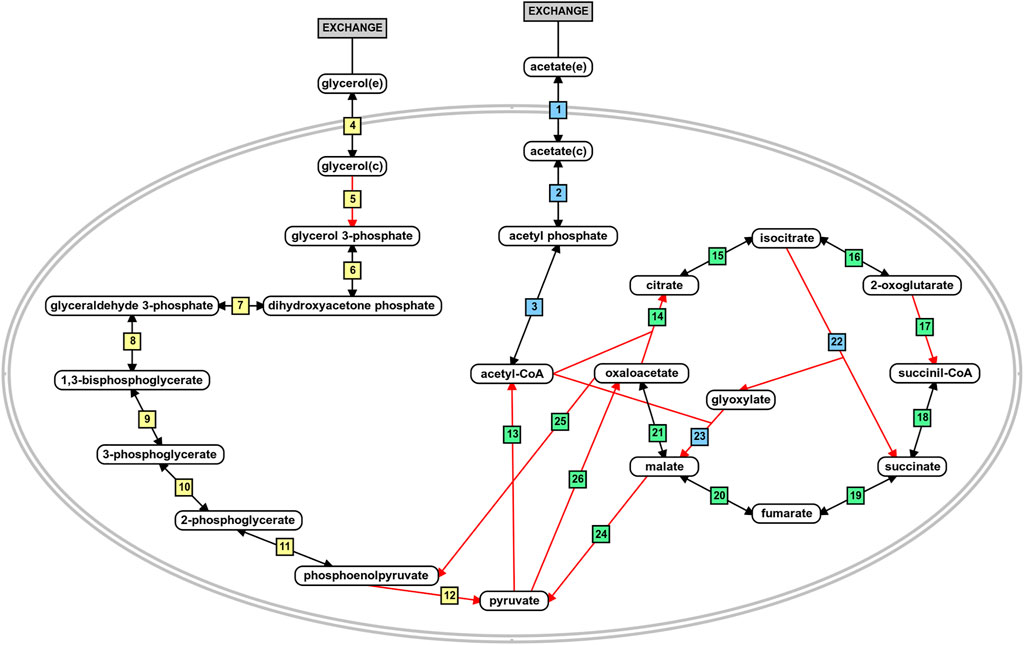
FIGURE 4. Schematic representation of acetate and glycerol central metabolism in P. aeruginosa PAO1. The blue boxes depict reactions related to acetate metabolism. The yellow boxes depict reactions related to glycerol metabolism, and the green boxes are the reactions shared by the metabolism of both carbon sources. The red arrows indicate irreversible reactions. This figure was generated using the PathVisio software (Kutmon et al., 2015).
Likewise, during the simulated growth in glycerol, the uptake rate ranged from 360 to 400 mmol gDW−1 h−1 in the metabolic models and 13–14 mmol gDW−1 h−1 in the integrated models. However, the reaction numbered as 5 in Figure 4 of the iPAO1 metabolic models, an essential step to glycerol entrance in the central metabolism, had no flux suggesting the flow was shifted through reaction steps not observed biologically. After integration, the flux through reaction 5 was restored. The flux distribution calculated for all models is available in the Supplementary Material.
Identification of Potential Therapeutic Targets
We used the FindTargetsWEB application (Merigueti et al., 2019) to analyze the selectivity of integrated models in identifying therapeutic targets compared to metabolic models with adjusted carbon source uptake rates, which do not consider gene expression data. Among the eight models analyzed, FindTargetsWEB identified a total of 68 different drug targets. However, the application applies an identity cutoff of 30% to search for similar DrugBank proteins in the target organism. We choose to apply a more conservative filter using an identity greater than 60% and coverage greater than 70%. The number of targets decreased to 32. In addition, to avoid undesirable host-drug interactions, we filtered out P. aeruginosa proteins homologous to any human proteins resulting in a final list of 26 targets. Considering both models and no growth condition, 18 of the 26 targets were also predicted from the integrated models, while 8 were unique from adjusted metabolic models (Figure 5 and Table 1).
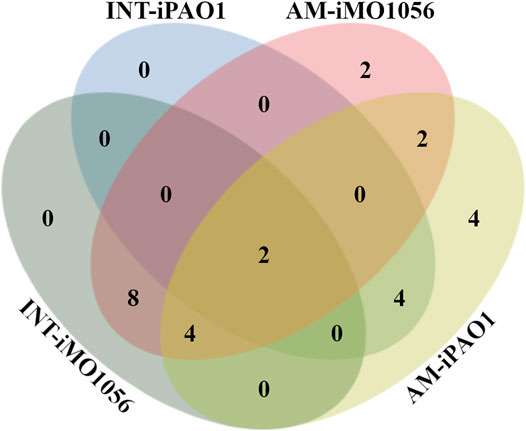
FIGURE 5. Venn diagram illustrating the number of targets shared by the metabolic models with adjusted carbon source uptake rates (AM) and integrated models (INT).
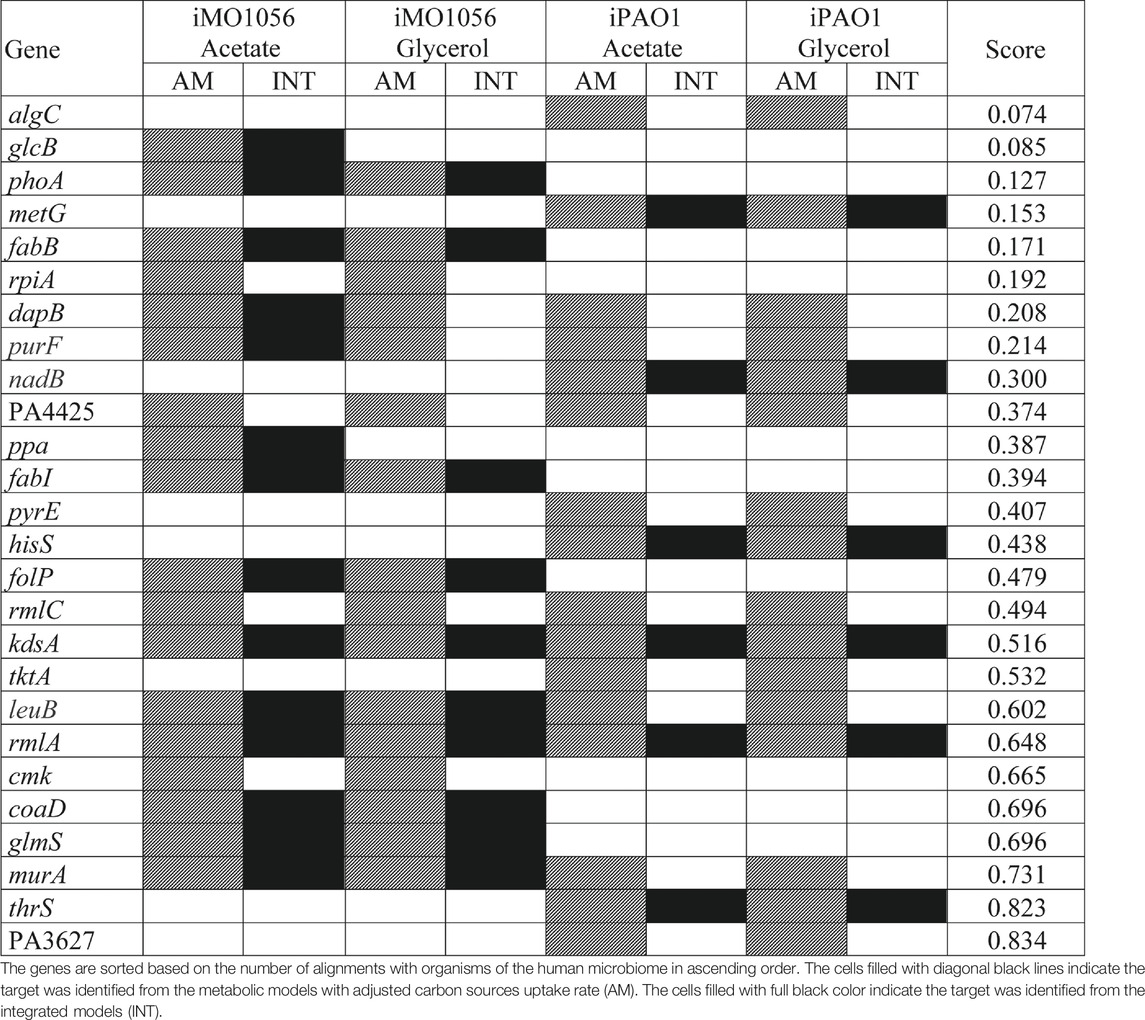
TABLE 1. List of potential therapeutic targets identified by the FindTargetWEB application from the metabolic models with adjusted carbon source uptake rates and integrated models.
Several organisms inhabit the host gastrointestinal tract, and antimicrobial effects upon the normal microbiota could result in adverse effects. Although we do not consider homology an excluding factor, we used blastp results of the predicted targets against the human microbiome to compute a score and suggest a prioritization. The score was the ratio between the number of organisms that presented at least one hit with the target protein sequence and the total number of organisms from the gastrointestinal tract (Table 1). Table 1 emphasizes the selectivity of the integrated models showing the targets predicted by each model in the different growth conditions. It is noteworthy that targets found in integrated models are proper subsets of the targets found in the corresponding metabolic adjusted models. Except for specific genes of the iMO1056 model, there are no differences in the prediction between growth conditions.
Discussion
Evolving antibiotic resistance profiles emphasize the need to research and develop drug targets and effective therapies against infections caused by P. aeruginosa. In the last few decades, computational approaches have become essential tools to help researchers screen new drug targets and hasten drug discovery and design. Reconstruction of biological networks from “omics” data is one of these tools. Moreover, integrating different networks (e.g., metabolic, gene regulatory, signaling networks) is expected to yield more comprehensive computational models that allow a more accurate prediction of any condition of interest. This work aimed to integrate GEMs of P. aeruginosa PAO1 with publicly available gene expression data to better reproduce the biological behavior and identify potential therapeutic targets.
The dynamic simulations performed from both metabolic and integrated models have demonstrated that as we include more layers of biological information in the computational model, the more precise the predictions are. According to Dolan et al. (2020), cells grown in MOPS minimal medium with acetate as carbon source show a growth rate of 0.80 h−1 and an exponential phase that ranges from 2 up to 8 h. However, the cells grew slower in glycerol (0.37 h−1), starting the exponential phase at 12 h of growth and reaching the stationary phase at 20 h. The predictions based on metabolic models show cells reaching the stationary phase before the first hour of simulation, also depleting all carbon sources available (Figure 1). This growth rate is not consistent with biological behavior. Once we adjust carbon source uptake rates to values measured experimentally, the growth curve based on metabolic models showed a more suitable shape. Indeed, in acetate, cells reached stationary phase at closer times than in vitro growth, showing a growth rate of 0.82 h−1 to iMO1056 and 0.71 h−1 to iPAO1 model (Figures 2A,B). Likewise, in glycerol, the shape of the growth curves is less incoherent, showing a growth rate of 0.55 h−1 to iMO1056 and 0.48 h−1 to the iPAO1 model (Figures 2C,D). These results show that the addition of a single biological measure could improve the accuracy of simulations. The next step was to analyze the dynamics of models after the integration process. Both growth curves had a shape closer to the observed experimentally.
In acetate, cells reached the stationary phase after 6 h of simulation from both models (Figures 3A,B). In glycerol, cells reached the stationary phase at 7 h of simulation from both models, considering the starting point of 12 h, it was equal to 19 h of growth (Figures 3C,D). As part of the integration process, the growth rates of integrated models were equal to those measured by Dolan et al. (2020). However, it is noteworthy that simulations from integrated models showed carbon source uptake rates close to 30.4 (acetate) and 9.2 (glycerol) mmol gDW−1 h−1. In addition, we analyzed the internal flux distribution. FBA uses linear programming to find reaction fluxes based on maximizing an objective function. In this work, the objective function is biomass production. An optimal solution can obtain non-zero flux values to reactions that are not part of a known biological pathway but are still correct from a mathematical perspective. We observed that sometimes the optimal solution found included fluxes not observed experimentally. However, we could observe that the integration of metabolic networks with transcriptome data allowed the flux to pass through the correct biological pathways. Integrated models have a reduced solution space compared to non-integrated models, which may be related to the flux reorientations observed in the former. These results indicate that the definition of constraints based on expression gene levels allowed the system to approximate to better approximate the experimental growth curve dynamics compared to the previous approaches.
Identification of potential drug targets from metabolic networks is commonly employed. There are different approaches to identify targets from genomic-scale metabolic networks, with several levels of automation. In this work, we used FindTargetsWEB, which is an online application developed to identify targets based on gene essentiality under a given condition. Although metabolic networks are a valuable resource, we could observe that as we include more biological measurements in a computational model, the predictions based on this model are more accurate. Furthermore, since we observed better growth dynamics with integrated models, we suggest identifying targets from these models could also be more reliable. In order to analyze this hypothesis, we submitted both metabolic and integrated models to FindTargetsWEB and compared the results. We found that 18 of the genes identified from the integrated models as potential drug targets were a subset of the 26 genes identified from the metabolic models with adjusted carbon source uptake rates. This observation is a consequence of the solution space reduction due to more strict constraints imposed by the gene expression data. Our intention is not to point out that the genes found from non-adjusted metabolic models are not reliable targets, but the integration is a method to improve selectivity and narrow the screening process.
According to the target scores (Table 1), we highlight the five top-ranked genes selected from the integrated models, glcB, phoA, metG, fabB, and dapB.Poulsen et al. (2019) pointed out 321 genes of P. aeruginosa as high-priority drug targets based on the definition of an essential core genome. The essentiality analysis was performed with different strains carrying transposon insertions grown in different media. We observed that the five top-ranked genes are included in the essential core genome described by Poulsen et al. (2019) except for glcB and phoA. It is noteworthy that the minimal medium used was M9 and the reference strain was PA14 (Poulsen et al., 2019). The fact that the genes listed in Table 1 have already been identified in the literature as potential targets reinforces the adequacy of the method described in our work for selecting biologically relevant targets.
The first placed gene among the five top-ranked is glcB, which encodes a malate synthase (MALS). MALS catalyzes the condensation of acetyl-CoA to glyoxylate to form malate and coenzyme A. This reaction is an essential step of the glyoxylate cycle (Figure 4, reaction 23), an anaplerotic pathway providing intermediates for the tricarboxylic acid cycle or precursors for amino acid synthesis. It acts as a modified version of the tricarboxylic acid cycle, bypassing the carbon dioxide-producing steps to conserve carbon atoms for gluconeogenesis (Beeckmans 2009; Kornberg 1966). This glyoxylate shunt is involved in metabolic adaptation to environmental changes, and it is essential for bacterial growth in acetate, ethanol, fatty acids, or any substrate whose acetyl-CoA is a direct product of the pathway. Besides enabling the use of different carbon sources, the glyoxylate shunt plays an important role in virulence, oxidative stress defense, and antibiotic resistance in several clinically relevant pathogens (Maloy, Bohlander, and Nunn 1980; Renilla et al., 2012; Dunn, Ramírez-Trujillo, and Hernández-Lucas 2009; Lorenz and Fink 2002; Meylan et al., 2017). The chronic P. aeruginosa infections in cystic fibrosis patients showed MALS upregulation. A double mutant of MALS and isocitrate lyase (another enzyme of the glyoxylate cycle) was avirulent in a mouse pulmonary infection model, which emphasizes MALS as an attractive target for drug development. In addition, MALS has great potential as a broad-spectrum target because it is conserved in pathogenic species, selective, and has a narrow distribution in the gastrointestinal microbiota (Table 1) (Hagins et al., 2010; Fahnoe et al., 2012; Myler and Stacy 2012; Murima, McKinney, and Pethe 2014; McVey et al., 2017). A class of Mg2+ chelators compounds called phenyl-diketoacid were described as MALS inhibitors in Mycobacterium tuberculosis (Krieger et al., 2012; Shukla, Shukla, and Tripathi 2021). In P. aeruginosa, Fahnoe et al. (2012) identified eight different new compounds with inhibitory activities against MALS. Furthermore, these compounds impaired both MALS and isocitrate lyase enzymes within the glyoxylate shunt pathway, an advantageous property to prevent the rapid development of resistance against new antimicrobial agents.
Inorganic phosphate is an essential component of nucleotides, membrane phospholipids, and phosphorylated proteins. In bacteria, phosphonates and organophosphates are viable sources of inorganic phosphate upon the enzymatic activity of alkaline phosphatases (AP). The gene phoA encodes a periplasmic AP, and it is highly expressed under phosphate-limiting conditions, e.g., human airway epithelial infections such as cystic fibrosis (Chekabab, Harel, and Dozois 2014; Jones et al., 2021). In addition, AP seems to contribute to the cell division cycle in a low-phosphate environment, possibly a consequence of its role in the inorganic phosphate scavenge (Bhatti, DeVoe, and Ingram 1976). To the best of our knowledge, there is no recent scientific literature reporting effective inhibitors to AP of organisms phylogenetically close to P. aeruginosa. In contrast, several inhibitors are described to mammalian AP since they have an important role in bone formation and prevention of intestinal inflammation. Bacterial and mammalian AP have significant differences regarding their catalytic sites (number and type of metal ions, amino acid residues), molecular weight, and kinetics (Rashida and Iqbal 2015). Indeed, Chakraborty et al. (2012) described the inhibition of Vibrio AP by imipenem, a β-lactam antibiotic, but the same effect was not observed in the Escherichia coli AP. Even inside the same domain, the AP of these phylogenetically distant organisms is not affected by the same compounds. Thus, it is theoretically possible to discover a drug capable of inhibiting the P. aeruginosa AP without an undesired effect on the host.
The gene metG encodes the enzyme methionyl-tRNA synthetase (MetRS), which belongs to the same class of two other targets identified in our work: hisS (histidyl-tRNA synthetase, HisRS) and thrS (threonyl-tRNA synthetase, ThrRS) (Table 1). Overall, aminoacyl-tRNA synthetases (AaRSs) constitute a class of 20 enzymes essential for protein biosynthesis, corresponding to each canonical amino acid. AaRSs catalyze a specific amino acid attachment to their cognate tRNAs, playing a crucial role during the initiation and elongation phase of protein biosynthesis. Due to their primordial function, AaRSs are present in all three kingdoms of life. Despite their similarity among organisms, structural differences between prokaryotic and eukaryotic AaRSs are sufficient to select pathogen-specific inhibitors (Kwon, Fox, and Kim 2019; Pang, Weeks, and Van Aerschot 2021). There are two known AaRSs inhibitors approved for clinical use, but none are designed for Gram-negative pathogens. Most bacteria contain one of the two forms of a MetRS, where MetRS1 is found in Gram-positive bacteria, protozoa, and mitochondria, and MetRS2 is found in archaea, the cytosol of eukaryotic cells, and Gram-negative bacteria, including P. aeruginosa (Nakama, Nureki, and Yokoyama 2001; Gentry et al., 2003; Rock et al., 2007). According to Mercaldi et al. (2021), the auxiliary pockets of MetRS1 and MetRS2 differ in their amino acid composition, leading to less or no effect of known MetRS1 inhibitors upon MetRS2. In P. aeruginosa, Robles et al. (2017) found one candidate among 1,690 compounds with satisfactory inhibition results, the isopomiferin. However, isopomiferin did not show broad-spectrum activity, in addition to high-level toxicity in human cells when compared to other antibiotics of common use. Instead, promising compounds have been found for the other two targets, HisRS and ThrRS. A screening assay analyzing nearly 1700 compounds selected 15 with activity against HisRs of P. aeruginosa. Among them, four (BT02C02, BT02D04, BT08E04, and BT09C11) were highlighted for presenting effective inhibition results associated with a broad-spectrum activity. Furthermore, the compounds bound to other places besides the active site of aminoacylation, which is advantageous to avoid resistance mechanisms, having low-level toxicity in eukaryotic cells. An interesting feature of BT09C11 is the presence of a sulfonamide group, which has antimicrobial activity against other enzymes, e.g., acting as a competitive inhibitor of dihydropteroate synthase (an enzyme encoded by another potential target, the gene folP). This fact could imply more than one form of inhibition (Henry 1943; Hu et al., 2018). Regarding ThrRS, Scott et al. (2019) described obafluorin, a natural compound produced by Pseudomonas fluorescens. Obafluorin is active against Gram-positive and Gram-negative bacteria, including P. aeruginosa. Interestingly, P. fluorescens has a homolog to its ThrRS called ObaO, which confers immunity to obafluorin, and it is not present in the P. aeruginosa chromosome.
The gene fabB encodes the cytoplasmic enzyme 3-oxoacyl-[acyl-carrier-protein] synthase 1 or β-ketoacyl-ACP synthase (KAS) I (Feng and Cronan 2009). In P. aeruginosa, fabB is co-transcribed with fabA establishing the fabAB operon that plays a crucial role in unsaturated fatty acid (UFA) biosynthesis via the anaerobic type II biosynthetic pathway (Hoang and Schweizer 1997; Subramanian, Rock, and Zhang 2010). The joint function of both enzymes FabA and FabB impact the membrane fluidity under different growth conditions contributing to the dominant UFA synthetic pathway in P. aeruginosa. Indeed, the composition of P. aeruginosa membrane contains more UFAs than saturated fatty acids, whose balance depends on a coordinated regulation at the transcriptional level in response to changes in the environment (Zhang et al., 2007). Two major inhibitors of KAS were described to date, cerulenin and thiolactomycin. Although fabB was not considered homologous to human proteins in our analysis, cerulenin was not selective also inhibiting eukaryotic KAS. In contrast, thiolactomycin interacts with FabB preventing the elongation of UFAs. It has a broad spectrum against several pathogens and is selective (Jackowski et al., 2002; Khandekar, Daines, and Lonsdale 2003). In P. aeruginosa, Schweizer (1998) demonstrates intrinsic resistance to thiolactomycin conferred by efflux pump systems. However, the critical role of FabB in the composition of cellular fatty acids and membrane fluidity of P. aeruginosa emphasizes its importance as a drug target, and the combination of existing drugs with antimicrobial adjuvants like efflux pump inhibitors could lead to effective therapeutic options (Peraman et al., 2021).
The gene dapB encodes the enzyme dihydrodipiconilate reductase (DHDPR). DHDPR catalyzes an intermediate reaction in the diaminopimelate (DAP) pathway responsible for the biosynthesis of two essential compounds, meso-diaminopimelate (meso-DAP) and lysine. Lysine is important for protein synthesis and meso-DAP is a cell wall component in Gram-negative bacteria, such as P. aeruginosa. In mammalians, lysine is an essential amino acid, i.e., it is not synthesized and must be acquired from the diet. DAP pathway is only present in bacteria and plants. These points reinforce the potential of DapB inhibitors as antimicrobial agents and minimize the possibility of toxicity in human cells (Hutton, Perugini, and Gerrard 2007; Impey et al., 2020). The efforts towards the identification of DHDPR inhibitors are focused on Mycobacterium tuberculosis. Some effective candidates were found, including sulfonamides. Among them, one has a sulfonamide group replaced by a sulfone, showing an increased potency against DHDPR of M. tuberculosis in addition to DHDPR of E. coli (Paiva et al., 2001). There are no reports in the scientific literature on investigating dapB for drug discovery in P. aeruginosa.
Although low-ranking targets, the genes kdsA and rmlA also stand out in Table 1 because they were identified as potential drug targets in all models. It is noteworthy that both genes are involved in the biosynthesis of lipopolysaccharide (LPS) constituents. LPS is a major component of P. aeruginosa outer membrane. It is an important virulence factor and an efficient permeability barrier. The kdsA gene encodes a key enzyme 2-dehydro-3-deoxyphosphooctonate aldolase that catalyzes the production of 2-keto-3-deoxy-D-manno-octulosonate-8-phosphate, an essential compound for the assembly of LPS (Nelson et al., 2013; Valvano 2015). The kdsA gene is part of the essential core genes described by Poulsen et al. (2019), and previous works corroborate its essentiality through experimental techniques and different media (Skurnik et al., 2013; Lee et al., 2015; Turner et al., 2015). The inhibition of KdsA leads to cell growth arrest by limiting replication (Xu et al., 2003; Ahmad et al., 2019). There are several inhibitors of KdsA described with potent in vitro activity, including the {[(2,2-Dihydroxy-Ethyl)-(2,3,4,5-Tetrahydroxy-6-Phosphonooxy-Hexyl)-Amino]-Methyl}-Phosphonic acid (DB02433) present in DrugBank as an experimental drug (Grison et al., 2005; Harrison, Reichau, and Parker 2012; Ahmad et al., 2019). The gene rmlA encodes the enzyme glucose-1-phosphate thymidylyltransferase (G1PTMT), which catalyzes the first step in the biosynthesis of rhamnose, a homopolymer component of P. aeruginosa LPS (King et al., 2009; Alphey et al., 2013). Poulsen et al. (2019) did not mention rmlA in their work, but a knockout mutant of the rmlA gene in PAO1 could not grow in M9 minimal medium. In addition, the mutant released very low extracellular DNA, which is related to biofilm formation and induction of antibiotic resistance in biofilm (Elamin et al., 2017). The substrates of G1PTMT are glucose-1-phosphate and deoxy-thymidine triphosphate. Smithen et al. (2015) demonstrate that bisubstrate analogs, i.e., a molecule that resembles both substrates in a transient state, are potent inhibitors of G1PTMT of Streptococcus pneumoniae. In P. aeruginosa, Alphey et al. (2013) show small thymidine-containing molecules that inhibit G1PTMT through binding the allosteric site. Allosteric inhibitors are considered more promising drugs because they are more specific thus less toxic. The reason is that allosteric sites often are more selective due to lower amino acid residue conservation among protein families when compared to active sites. Despite recent advances, the discovery of G1PTMT inhibitors to be used as antimicrobial agents remains a challenge. The genes kdsA and rmlA have numerous essential criteria for prioritization, such as essentiality, absence of homologs in humans, broad-spectrum target, and druggability. However, they did not obtain a good classification because we used the microbiome conservation criterion to rank the identified targets, minimizing adverse effects caused by the elimination of intestinal flora. However, this is not an exclusion criterion, only the parameter of prioritization adopted in our work.
The main goal in building computational models based on different layers of biological data is to improve the accuracy of in silico simulations. We demonstrate that the integration of transcriptome data to metabolic networks described in our work successfully achieved this objective, resulting in growth curves and flux distributions in line with biological observations. In addition, the identification of potential drug targets from integrated computational models is more selective and points out genes with reported biological relevance. Indeed, some targets identified in our work have already been proposed as drug targets through distinct methodologies. Others are already known drug targets. These observations corroborate that further investigating unexploited targets is a promising approach. A noteworthy remark is that the reference organism used in this work is not a multidrug-resistant strain. However, the methodology applied here can be extended to other strains, other genera, and other conditions since “omics” data are available for several organisms. Finally, we advocate integrating multiple layers of omics data for accurate phenotype prediction and therapeutic target identification, enabling new drug discovery through advanced systems biology approaches instead of time-consuming and expensive conventional screening.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, and available at https://github.com/medeirosfilho1/Integrationpaeruginosa. Further inquiries can be directed to the corresponding authors.
Author Contributions
MC and MM performed the SBML improvements. FF, AN, MC performed the file conversions, integration processes, and dynamic simulations. AN and MC performed the flux variability analysis. FF, TM, and FS performed the target identification analysis. Targets filtering and scoring were performed by AN and MC. MN and AC provided support on the biological interpretation of results. FS, AC, and MT supervised the work. AN wrote the manuscript with inputs and comments from FF, MC, MM, MN, MT, AC, and FS. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Programa Fiocruz de Fomento à Inovação—Inova Fiocruz (Grant Number VPPCB-007-FIO-18-2-29) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil—CAPES (Grant Number 88882.442959/2019-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Dr. Stephen K. Dolan for kindly providing additional data and enlightening information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.728129/full#supplementary-material
Abbreviations
AaRS, aminoacyl-tRNA synthetase; AM, metabolic model with adjusted carbon source uptake rate; AP, alkaline phosphatase; DAP, diaminopimelate; DHDPR, dihydrodipiconilate reductase; FBA, flux balance analysis; FVA, flux variability analysis; G1PTMT, glucose-1-phosphate thymidylyltransferase; GEM, genome-scale metabolic model; GPR, gene-protein-reaction; HisRS, histidyl-tRNA synthetase; INT, integrated model; KAS, ketoacyl-ACP synthase; LPS, lipopolysaccharide; MALS, malate synthase; MetRS, methionyl-tRNA synthetase; ThrRS, threonyl-tRNA synthetase; UFA, unsaturated fatty acid.
References
Ahmad, S., Raza, S., Abro, A., Liedl, K. R., and Azam, S. S. (2019). Toward Novel Inhibitors against KdsB: A Highly Specific and Selective Broad-Spectrum Bacterial Enzyme. J. Biomol. Struct. Dyn. 37 (5), 1326–1345. doi:10.1080/07391102.2018.1459318
Alphey, M. S., Pirrie, L., Torrie, L. S., Boulkeroua, W. A., Gardiner, M., Sarkar, A., et al. (2013). Allosteric Competitive Inhibitors of the Glucose-1-Phosphate Thymidylyltransferase (RmlA) from Pseudomonas Aeruginosa. ACS Chem. Biol. 8 (2), 387–396. doi:10.1021/cb300426u
Andrews, Simon. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Banos, D. T., Trébulle, P., and Elati, M. (2017). Integrating Transcriptional Activity in Genome-Scale Models of Metabolism. Bmc Syst. Biol. 11 (S7), 134. doi:10.1186/s12918-017-0507-0
Beeckmans, S. (2009). Glyoxylate Cycle. Encyclopedia Microbiol. 159–79, 159–179. Elsevier. doi:10.1016/B978-012373944-5.00075-4
Bhatti, A. R., DeVoe, I. W., and Ingram, J. M. (1976). Cell Division in Pseudomonas aeruginosa: Participation of Alkaline Phosphatase. J. Bacteriol. 126 (1), 400–409. doi:10.1128/jb.126.1.400-409.1976
Blazier, A. S., and Papin, J. A. (2012). Integration of Expression Data in Genome-Scale Metabolic Network Reconstructions. Front. Physio. 3, 299. doi:10.3389/fphys.2012.00299
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Chakraborty, S., Ásgeirsson, B., Minda, R., Salaye, L., Frère, J.-M., and Rao, B. J. (2012). Inhibition of a cold-active alkaline phosphatase by imipenem revealed by in silico modeling of metallo-β-lactamase active sites. FEBS Lett. 586 (20), 3710–3715. doi:10.1016/j.febslet.2012.08.030
Chandrasekaran, S., and Price, N. D. (2010). Probabilistic integrative modeling of genome-scale metabolic and regulatory networks inEscherichia coliandMycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 107 (41), 17845–17850. doi:10.1073/pnas.1005139107
Chavali, A. K., D’Auria, K. M., Hewlett, E. L., Pearson, R. D., and Papin, J. A. (2012). A Metabolic Network Approach for the Identification and Prioritization of Antimicrobial Drug Targets. Trends Microbiol. 20 (3), 113–123. doi:10.1016/j.tim.2011.12.004Pearson
Chekabab, S. M., Harel, J., and Dozois, C. M. (2014). Interplay between Genetic Regulation of Phosphate Homeostasis and Bacterial Virulence. Virulence 5 (8), 786–793. doi:10.4161/viru.29307Dozois
Chung, W. Y., Zhu, Y., Mahamad Maifiah, M. H., Shivashekaregowda, N. K. H., Wong, E. H., and Abdul Rahim, N. (2021). Novel Antimicrobial Development Using Genome-Scale Metabolic Model of Gram-Negative Pathogens: A Review. J. Antibiot. 74 (2), 95–104. doi:10.1038/s41429-020-00366-2
Colijn, C., Brandes, A., Zucker, J., Lun, D. S., Weiner, B., Farhat, M. R., et al. (2009). Interpreting Expression Data with Metabolic Flux Models: Predicting Mycobacterium tuberculosis Mycolic Acid Production. Plos Comput. Biol. 5 (8), e1000489. doi:10.1371/journal.pcbi.1000489
Cruz, F., Faria, J. P., Rocha, M., Rocha, I., and Dias, O. (2020). A Review of Methods for the Reconstruction and Analysis of Integrated Genome-Scale Models of Metabolism and Regulation. Biochem. Soc. Trans. 48 (5), 1889–1903. doi:10.1042/BST20190840
Dolan, S. K., Kohlstedt, M., Trigg, S., Vallejo Ramirez, P., Kaminski, C. F., Wittmann, C., et al. (2020). Contextual Flexibility in Pseudomonas aeruginosa Central Carbon Metabolism during Growth in Single Carbon Sources. MBio 11 (2), e02684. doi:10.1128/mBio.02684-19
Dunn, M. F., Ramírez-Trujillo, J. A., and Hernández-Lucas, I. (2009). Major Roles of Isocitrate Lyase and Malate Synthase in Bacterial and Fungal Pathogenesis. Microbiology 155 (10), 3166–3175. doi:10.1099/mic.0.030858-0
Elamin, A. A., Steinicke, S., Oehlmann, W., Braun, Y., Wanas, H., Shuralev, E. A., et al. (2017). Novel drug targets in cell wall biosynthesis exploited by gene disruption in Pseudomonas aeruginosa. PLOS ONE 12 (10), e0186801. doi:10.1371/journal.pone.0186801
Fahnoe, K. C., Flanagan, M. E., Gibson, G., Shanmugasundaram, V., Che, Y., and Tomaras, A. P. (2012). Non-Traditional Antibacterial Screening Approaches for the Identification of Novel Inhibitors of the Glyoxylate Shunt in Gram-Negative Pathogens. PLoS ONE 7 (12), e51732. doi:10.1371/journal.pone.0051732
Feng, Y., and Cronan, J. E. (2009). Escherichia coli Unsaturated Fatty Acid Synthesis. J. Biol. Chem. 284 (43), 29526–29535. doi:10.1074/jbc.M109.023440
Gentry, D. R., Ingraham, K. A., Stanhope, M. J., Rittenhouse, S., Jarvest, R. L., O'Hanlon, P. J., et al. (2003). Variable Sensitivity to Bacterial Methionyl-TRNA Synthetase Inhibitors Reveals Subpopulations of Streptococcus pneumoniae with Two Distinct Methionyl-TRNA Synthetase Genes. Antimicrob. Agents Chemother. 47 (6), 1784–1789. doi:10.1128/AAC.47.6.1784-1789.2003
Grison, C., Petek, S., Finance, C., and Coutrot, P. (2005). Synthesis and Antibacterial Activity of Mechanism-Based Inhibitors of KDO8P Synthase and DAH7P Synthase. Carbohydr. Res. 340 (4), 529–537. doi:10.1016/j.carres.2004.11.019
Hagins, J. M., Scoffield, J. A., Suh, S.-J., and Silo-Suh, L. (2010). Influence of RpoN on Isocitrate Lyase Activity in Pseudomonas aeruginosa. Microbiology 156 (4), 1201–1210. doi:10.1099/mic.0.033381-0
Harrison, A. N., Reichau, S., and Parker, E. J. (2012). Synthesis and Evaluation of Tetrahedral Intermediate Mimic Inhibitors of 3-Deoxy-d-Manno-Octulosonate 8-Phosphate Synthase. Bioorg. Med. Chem. Lett. 22 (2), 907–911. doi:10.1016/j.bmcl.2011.12.025
Henry, R. J. (1943). The Mode of Action of Sulfonamides. Bacteriol. Rev. 7 (4), 175–262. doi:10.1128/br.7.4.175-262.1943
Hoang, T. T., and Schweizer, H. P. (1997). Fatty Acid Biosynthesis in Pseudomonas aeruginosa: Cloning and Characterization of the FabAB Operon Encoding Beta-Hydroxyacyl-Acyl Carrier Protein Dehydratase (FabA) and Beta-Ketoacyl-Acyl Carrier Protein Synthase I (FabB). J. Bacteriol. 179 (17), 5326–5332. doi:10.1128/jb.179.17.5326-5332.1997
Hu, Y., Palmer, S. O., Robles, S. T., Resto, T., Dean, F. B., and Bullard, J. M. (2018). Identification of Chemical Compounds That Inhibit the Function of Histidyl-TRNA Synthetase from Pseudomonas aeruginosa. SLAS DISCOVERY: Advancing Sci. Drug Discov. 23 (1), 65–75. doi:10.1177/2472555217722016
Human Microbiome Project Consortium (2012a). A Framework for Human Microbiome Research. Nature 486 (7402), 215–221. doi:10.1038/nature11209
Human Microbiome Project Consortium (2012b). Structure, Function and Diversity of the Healthy Human Microbiome. Nature 486 (7402), 207–214. doi:10.1038/nature11234
Hutton, C. A., Perugini, M. A., and Gerrard, J. A. (2007). Inhibition of Lysine Biosynthesis: An Evolving Antibiotic Strategy. Mol. Biosyst. 3 (7), 458. doi:10.1039/b705624a
Hyduke, D. R., Lewis, N. E., and Palsson, B. Ø. (2013). Analysis of Omics Data with Genome-Scale Models of Metabolism. Mol. Biosyst. 9 (2), 167–174. doi:10.1039/c2mb25453k
Impey, R. E., Panjikar, S., Hall, C. J., Bock, L. J., Sutton, J. M., Perugini, M. A., et al. (2020). Identification of two dihydrodipicolinate synthase isoforms fromPseudomonas aeruginosathat differ in allosteric regulation. Febs J. 287 (2), 386–400. doi:10.1111/febs.15014
Jackowski, S., Zhang, Y.-M., Price, A. C., White, S. W., and Rock, C. O. (2002). A Missense Mutation in the fabB (β-Ketoacyl-Acyl Carrier Protein Synthase I) Gene Confers Thiolactomycin Resistance to Escherichia coli. Antimicrob. Agents Chemother. 46 (5), 1246–1252. doi:10.1128/AAC.46.5.1246-1252.2002
Jones, R. A., Shropshire, H., Zhao, C., Murphy, A., Lidbury, I., Wei, T., et al. (2021). Phosphorus Stress Induces the Synthesis of Novel Glycolipids in Pseudomonas aeruginosa That Confer Protection against a Last-Resort Antibiotic. Isme J. doi:10.1038/s41396-021-01008-7
Karimian, E., and Motamedian, E. (2020). ACBM: An Integrated Agent and Constraint Based Modeling Framework for Simulation of Microbial Communities. Sci. Rep. 10 (1), 8695. doi:10.1038/s41598-020-65659-w
Khandekar, S., Daines, R., and Lonsdale, J. (2003). Bacterial β-Ketoacyl-Acyl Carrier Protein Synthases as Targets for Antibacterial Agents. Cpps 4 (1), 21–29. doi:10.2174/1389203033380377
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 37 (8), 907–915. doi:10.1038/s41587-019-0201-4
King, J. D., Kocíncová, D., Westman, E. L., and Lam, J. S. (2009). Review: Lipopolysaccharide Biosynthesis in Pseudomonas aeruginosa. Innate Immun. 15 (5), 261–312. doi:10.1177/1753425909106436
Kornberg, H. (1966). The Role and Control of the Glyoxylate Cycle in Escherichia coli. Biochem. J. 99 (1), 1–11. doi:10.1042/bj0990001
Koutrouli, M., Karatzas, E., Paez-Espino, D., and Pavlopoulos, G. A. (2020). A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 88 (January), 34. doi:10.3389/fbioe.2020.00034
Krieger, I. V., Freundlich, J. S., Roberts, J. P., Gawandi, V. B., Sun, Q., Owen, J. L., et al. (2012). Structure-Guided Discovery of Phenyl-Diketo Acids as Potent Inhibitors of M. tuberculosis Malate Synthase. Chem. Biol. 19 (12), 1556–1567. doi:10.1016/j.chembiol.2012.09.018FreundlichGawandi
Kutmon, M., van Iersel, M. P., Bohler, A., Kelder, T., Nunes, N, Pico, A. R., et al. (2019). “PathVisio 3: An Extendable Pathway Analysis Toolbox”. PLoS Comput. Biol. Editors R. F. Murphy11 (2), e1004085. doi:10.1371/journal.pcbi.1004085
Kwon, N. H., Fox, P. L., and Kim, S. (2019). Aminoacyl-TRNA Synthetases as Therapeutic Targets. Nat. Rev. Drug Discov. 18 (8), 629–650. doi:10.1038/s41573-019-0026-3
Lee, S. A., Gallagher, L. A., Thongdee, M., Staudinger, B. J., Lippman, S., Singh, P. K., et al. (2015). General and condition-specific essential functions ofPseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 112 (16), 5189–5194. doi:10.1073/pnas.1422186112
Liao, Y., Smyth, G. K., and Shi, W. (2014). FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 30 (7), 923–930. doi:10.1093/bioinformatics/btt656
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-Resistant Pseudomonas aeruginosa : Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 22 (4), 582–610. doi:10.1128/CMR.00040-09
Lorenz, M. C., and Fink, G. R. (2002). Life and Death in a Macrophage: Role of the Glyoxylate Cycle in Virulence. Eukaryot. Cel 1 (5), 657–662. doi:10.1128/EC.1.5.657-662.2002
Mahadevan, R., and Schilling, C. H. (2003). The Effects of Alternate Optimal Solutions in Constraint-Based Genome-Scale Metabolic Models. Metab. Eng. 5 (4), 264–276. doi:10.1016/j.ymben.2003.09.002
Malek Shahkouhi, A., and Motamedian, E. (2020). Reconstruction of a Regulated Two-Cell Metabolic Model to Study Biohydrogen Production in a Diazotrophic Cyanobacterium Anabaena variabilis ATCC 29413. PLoS ONE 15 (1), e0227977. doi:10.1371/journal.pone.0227977
Maloy, S. R., Bohlander, M., and Nunn, W. D. (1980). Elevated Levels of Glyoxylate Shunt Enzymes in Escherichia coli Strains Constitutive for Fatty Acid Degradation. J. Bacteriol. 143 (2), 720–725. doi:10.1128/jb.143.2.720-725.1980
McVey, A. C., Medarametla, P., Chee, X., Bartlett, S., Poso, A., Spring, D. R., et al. (2017). Structural and Functional Characterization of Malate Synthase G from Opportunistic Pathogen Pseudomonas aeruginosa. Biochemistry 56 (41), 5539–5549. doi:10.1021/acs.biochem.7b00852
Mercaldi, G. F., Andrade, M. d. O., Zanella, J. d. L., Cordeiro, A. T., and Benedetti, C. E. (2021). Molecular Basis for Diaryldiamine Selectivity and Competition with TRNA in a Type 2 Methionyl-TRNA Synthetase from a Gram-Negative Bacterium. J. Biol. Chem. 296, 100658. doi:10.1016/j.jbc.2021.100658
Merigueti, T. C., Carneiro, M. W., Carvalho-Assef, A. P. D. A., Silva-Jr, F. P., and Silva, F. A. B. d. (2019). FindTargetsWEB: A User-Friendly Tool for Identification of Potential Therapeutic Targets in Metabolic Networks of Bacteria. Front. Genet. 1010, 633. doi:10.3389/fgene.2019.00633
Meylan, S., Porter, C. B. M., Yang, J. H., Belenky, P., Gutierrez, A., Lobritz, M. A., et al. (2017). Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas aeruginosa via Tricarboxylic Acid Cycle Control. Cel Chem. Biol. 24 (2), 195–206. doi:10.1016/j.chembiol.2016.12.015
Motamedian, E., Mohammadi, M., Shojaosadati, S. A., and Heydari, M. (2017). TRFBA: An Algorithm to Integrate Genome-Scale Metabolic and Transcriptional Regulatory Networks with Incorporation of Expression Data. Bioinformatics 33 (7), btw772–63. doi:10.1093/bioinformatics/btw772
Murima, P., McKinney, J. D., and Pethe, K. (2014). Targeting Bacterial Central Metabolism for Drug Development. Chem. Biol. 21 (11), 1423–1432. doi:10.1016/j.chembiol.2014.08.020
Myler, P. J., and Stacy., R. (2012). A New Drug for an Old Bug. Chem. Biol. 19 (12), 1499–1500. doi:10.1016/j.chembiol.2012.12.003
Nakama, T., Nureki, O., and Yokoyama, S. (2001). Structural Basis for the Recognition of Isoleucyl-Adenylate and an Antibiotic, Mupirocin, by Isoleucyl-TRNA Synthetase. J. Biol. Chem. 276 (50), 47387–47393. doi:10.1074/jbc.M109089200
Nelson, S. K., Kelleher, A., Robinson, G., Reiling, S., and Asojo, O. A. (2013). Structure of 2-keto-3-deoxy-D-manno-octulosonate-8-phosphate synthase fromPseudomonas aeruginosa. Acta Cryst. Sect F 69 (10), 1084–1088. doi:10.1107/S1744309113023993
Norsigian, C. J., Attia, H., Szubin, R., YassinYassin, A. S., Palsson, B. Ø., Aziz, R. K., and Monk, J. M. (2019). Comparative Genome-Scale Metabolic Modeling of Metallo-Beta-Lactamase-Producing Multidrug-Resistant Klebsiella pneumoniae Clinical Isolates. Front. Cel. Infect. Microbiol. 9 (May), 161. doi:10.3389/fcimb.2019.00161
Oberhardt, M. A., Goldberg, J. B., Hogardt, M., and Papin, J. A. (2010). Metabolic Network Analysis of Pseudomonas aeruginosa during Chronic Cystic Fibrosis Lung Infection. J. Bacteriol. 192 (20), 5534–5548. doi:10.1128/JB.00900-10
Oberhardt, M. A., Puchałka, J., Fryer, K. E., Martins dos Santos, V. A. P., and Papin, J. A. (2008). Genome-Scale Metabolic Network Analysis of the Opportunistic Pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 190 (8), 2790–2803. doi:10.1128/JB.01583-07
Orth, J. D., Thiele, I., and Palsson, B. Ø. (2010). What Is Flux Balance Analysis? Nat. Biotechnol. 28 (3), 245–248. doi:10.1038/nbt.1614
Pachori, P., Gothalwal, R., and Gandhi, P. (2019). Emergence of Antibiotic Resistance Pseudomonas aeruginosa in Intensive Care Unit; a Critical Review. Genes Dis. 6 (2), 109–119. doi:10.1016/j.gendis.2019.04.001
Paiva, A. M., Vanderwall, D. E., Vanderwall, J. S., Kozarich, J. W., Williamson, J. M., and Kelly, T. M. (2001). Inhibitors of Dihydrodipicolinate Reductase, a Key Enzyme of the Diaminopimelate Pathway of Mycobacterium tuberculosis. Biochim. Biophys. Acta 1545 (1–2), 67–77. doi:10.1016/S0167-4838(00)00262-4
Pang, L., Weeks, S. D., and Van Aerschot, A. (2021). Aminoacyl-TRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Ijms 22 (4), 1750. doi:10.3390/ijms22041750
Peraman, R., Sure, S. K., Dusthackeer, V. N. A., Chilamakuru, N. B., Yiragamreddy, P. R., Pokuri, C., et al. (2021). Insights on Recent Approaches in Drug Discovery Strategies and Untapped Drug Targets against Drug Resistance. Futur J. Pharm. Sci. 7 (1), 56. doi:10.1186/s43094-021-00196-5
Poulsen, B. E., Yang, R., Clatworthy, A. E., White, T., Osmulski, S. J., Li, L., et al. (2019). Defining the core essential genome ofPseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 116 (20), 10072–10080. doi:10.1073/pnas.1900570116
Presta, L., Bosi, E., Mansouri, L., Dijkshoorn, L., Fani, R., and Fondi, M. (2017). Constraint-Based Modeling Identifies New Putative Targets to Fight Colistin-Resistant A. baumannii Infections. Sci. Rep. 7 (1), 3706. doi:10.1038/s41598-017-03416-2
Ramos, P. I. P., Fernández Do Porto, D., Lanzarotti, E., Sosa, E. J., Burguener, G., Pardo, A. M., et al. (2018). An Integrative, Multi-Omics Approach towards the Prioritization of Klebsiella pneumoniae Drug Targets. Sci. Rep. 8 (1), 10755. doi:10.1038/s41598-018-28916-7
Rashida, M., and Iqbal, J. (2015). Inhibition of Alkaline Phosphatase: An Emerging New Drug Target. Mrmc 15 (1), 41–51. doi:10.2174/1389557515666150219113205
Renilla, S., Bernal, V., Fuhrer, T., Castaño-Cerezo, S., Pastor, J. M., Iborra, J. L., et al. (2012). Acetate scavenging activity in Escherichia coli: interplay of acetyl-CoA synthetase and the PEP-glyoxylate cycle in chemostat cultures. Appl. Microbiol. Biotechnol. 93 (5), 2109–2124. doi:10.1007/s00253-011-3536-4
Robles, S., Hu, Y., Resto, T., Dean, F., and Bullard, J. M. (2017). Identification and Characterization of a Chemical Compound That Inhibits Methionyl-TRNA Synthetase from Pseudomonas aeruginosa. Cddt 14 (3), 156–168. doi:10.2174/1570163814666170330100238
Rock, F. L., Mao, W., Yaremchuk, A., Tukalo, M., Crepin, T., Zhou, H., et al. (2007). An Antifungal Agent Inhibits an Aminoacyl-TRNA Synthetase by Trapping TRNA in the Editing Site. Science 316 (5832), 1759–1761. doi:10.1126/science.1142189
Ruppin, E., Papin, J. A., de Figueiredo, L. F., and Schuster, S. (2010). Metabolic Reconstruction, Constraint-Based Analysis and Game Theory to Probe Genome-Scale Metabolic Networks. Curr. Opin. Biotechnol. 21 (4), 502–510. doi:10.1016/j.copbio.2010.07.002
Schellenberger, J., Que, R., Fleming, R. M. T., Thiele, I., Orth, J. D., Feist, A. M., et al. (2011). Quantitative Prediction of Cellular Metabolism with Constraint-Based Models: The COBRA Toolbox v2.0. Nat. Protoc. 6 (9), 1290–1307. doi:10.1038/nprot.2011.308
Schweizer, H. P. (1998). Intrinsic Resistance to Inhibitors of Fatty Acid Biosynthesis in Pseudomonas Aeruginosa Is Due to Efflux: Application of a Novel Technique for Generation of Unmarked Chromosomal Mutations for the Study of Efflux Systems. Antimicrob. Agents Chemother. 42 (2), 394–398. doi:10.1128/aac.42.2.394
Scott, F., Wilson, P., Conejeros, R., and Vassiliadis, V. S. (2018). Simulation and Optimization of Dynamic Flux Balance Analysis Models Using an Interior Point Method Reformulation. Comput. Chem. Eng. 119 (November), 152–170. doi:10.1016/j.compchemeng.2018.08.041
Scott, T. A., Batey, S. F. D., Wiencek, P., Wiencek, G., Alt, S., Francklyn, C. S., et al. (2019). Immunity-Guided Identification of Threonyl-tRNA Synthetase as the Molecular Target of Obafluorin, a β-Lactone Antibiotic. ACS Chem. Biol. 14 (12), 2663–2671. doi:10.1021/acschembio.9b00590
Shukla, R., Shukla, H., and Tripathi, T. (2021). Structure-Based Discovery of Phenyl-Diketo Acids Derivatives as Mycobacterium tuberculosis Malate Synthase Inhibitors. J. Biomol. Struct. Dyn. 39 (8), 2945–2958. doi:10.1080/07391102.2020.1758787
Silby, M. W., Winstanley, C., Godfrey, S. A. C., Levy, S. B., and Jackson, R. W. (2011). Pseudomonasgenomes: diverse and adaptable. FEMS Microbiol. Rev. 35 (4), 652–680. doi:10.1111/j.1574-6976.2011.00269.x
Skurnik, D., Roux, D., Aschard, H., Cattoir, V., Yoder-Himes, D., Lory, S., et al. (2013). A Comprehensive Analysis of In Vitro and In Vivo Genetic Fitness of Pseudomonas aeruginosa Using High-Throughput Sequencing of Transposon Libraries. Plos Pathog. 9 (9), e1003582. doi:10.1371/journal.ppat.1003582
Smithen, D. A., Forget, S. M., McCormick, N. E., Syvitski, R. T., and Jakeman, D. L. (2015). Polyphosphate-Containing Bisubstrate Analogues as Inhibitors of a Bacterial Cell Wall Thymidylyltransferase. Org. Biomol. Chem. 13 (11), 3347–3350. doi:10.1039/C4OB02583K
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete Genome Sequence of Pseudomonas aeruginosa PAO1, an Opportunistic Pathogen. Nature 406 (6799), 959–964. doi:10.1038/35023079
Subramanian, C., Rock, C. O., and Zhang, Y.-M. (2010). DesT Coordinates the Expression of Anaerobic and Aerobic Pathways for Unsaturated Fatty Acid Biosynthesis in Pseudomonas aeruginosa. J. Bacteriol. 192 (1), 280–285. doi:10.1128/JB.00404-09
Turner, K. H., Wessel, A. K., WesselMurray, G. C., MurrayPalmer, J. L., and Whiteley, M. (2015). Essential genome ofPseudomonas aeruginosain cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 112 (13), 4110–4115. doi:10.1073/pnas.1419677112
Valvano, M. A. (2015). Genetics and Biosynthesis of LipopolysaccharideMol. Med. Microbiol. 55–89. Elsevier, 55–89. doi:10.1016/B978-0-12-397169-2.00004-4
van Duuren, J. B., Puchałka, J., Mars, A. E., Bücker, R., Eggink, G., Wittmann, C., et al. (2013). Reconciling in Vivo and in Silico Key Biological Parameters of Pseudomonas putida KT2440 during Growth on Glucose under Carbon-Limited Condition. BMC Biotechnol. 13 (October), 93. doi:10.1186/1472-6750-13-93
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of MRNA Abundance Using RNA-Seq Data: RPKM Measure Is Inconsistent among Samples. Theor. Biosci. 131 (4), 281–285. doi:10.1007/s12064-012-0162-3
World Health Organization (2017). “Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis.” World Health Organization. https://bit.ly/2RQUG71 September 4.2017
Xu, X., Wang, J., Grison, C., Petek, S., Coutrot, P., Birck, M., et al. (2003). Structure-Based Design of Novel Inhibitors of 3-Deoxy-d-Manno-Octulosonate 8-Phosphate Synthase. Drug Des. Discov. 18 (2–3), 91–99. doi:10.3109/10559610290271787
Zhang, H., Zeng, H., Ulrich, A. C., and Liu, Y. (2016). Comparison of the transport and deposition ofPseudomonas aeruginosaunder aerobic and anaerobic conditions. Water Resour. Res. 52 (2), 1127–1139. doi:10.1002/2015WR017821
Zhang, Y.-M., Zhu, K., Frank, M. W., and Rock, C. O. (2007). A Pseudomonas aeruginosa Transcription Factor That Senses Fatty Acid Structure. Mol. Microbiol. 66 (3), 622–632. doi:10.1111/j.1365-2958.2007.05934.x
Keywords: Pseudomonas aeruginosa, metabolic network, transcriptome data, integrated model, therapeutic target
Citation: Medeiros Filho F, Nascimento APBd, Costa MdOCe, Merigueti TC, Menezes MAd, Nicolás MF, Santos MTd, Carvalho-Assef APD and Silva FABd (2021) A Systematic Strategy to Find Potential Therapeutic Targets for Pseudomonas aeruginosa Using Integrated Computational Models. Front. Mol. Biosci. 8:728129. doi: 10.3389/fmolb.2021.728129
Received: 29 June 2021; Accepted: 31 August 2021;
Published: 20 September 2021.
Edited by:
Emmanuel Mikros, National and Kapodistrian University of Athens, GreeceReviewed by:
Miguel Rocha, University of Minho, PortugalChang-Jer Wu, National Taiwan Ocean University, Taiwan
Copyright © 2021 Medeiros Filho, Nascimento, Costa, Merigueti, Menezes, Nicolás, Santos, Carvalho-Assef and Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernando Medeiros Filho, medeiros.fiocruz@gmail.com; Fabrício Alves Barbosa da Silva, fabricio.silva@fiocruz.br
†These authors have contributed equally to this work and share first authorship