 Ajeng K. Pramono
Ajeng K. Pramono Ardhiani K. Hidayanti
Ardhiani K. Hidayanti Yohsuke Tagami
Yohsuke Tagami Hiroki Ando
Hiroki Ando- 1Laboratory of Phage Biologics, Graduate School of Medicine, Gifu University, Gifu, Japan
- 2School of Biological Environment, The United Graduate School of Agricultural Science, Gifu University, Gifu, Japan
- 3School of Life Sciences and Technology, Institut Teknologi Bandung (ITB), Bandung, Indonesia
- 4Laboratory of Applied Entomology, Faculty of Agriculture, Shizuoka University, Shizuoka, Japan
- 5Center for One Medicine Innovative Translational Research (COMIT), Gifu University, Gifu, Japan
Liriomyza trifolii, an agricultural pest, is occasionally infected by Wolbachia. A Wolbachia strain present in Liriomyza trifolii is associated with cytoplasmic incompatibility (CI) effects, leading to the death of embryos resulting from incompatible crosses between antibiotic-treated or naturally Wolbachia-free strain females and Wolbachia-infected males. In this study, high-throughput sequencing of hypervariable rRNA genes was employed to characterize the bacterial community in Wolbachia-infected L. trifolii without antibiotic treatment. The analysis revealed that Wolbachia dominates the bacterial community in L. trifolii, with minor presence of Acinetobacter, Pseudomonas, and Limnobacter. To elucidate the genetic basis of the CI phenotype, metagenomic sequencing was also conducted to assemble the genome of the Wolbachia strain. The draft-genome of the Wolbachia strain wLtri was 1.35 Mbp with 34% GC content and contained 1,487 predicted genes. Notably, within the wLtri genome, there are three distinct types of cytoplasmic incompatibility factor (cif) genes: Type I, Type III, and Type V cifA;B. These genes are likely responsible for inducing the strong cytoplasmic incompatibility observed in L. trifolii.
1 Introduction
Wolbachia are intracellular symbiont bacteria (Phylum: Pseudomonadota, Class: Alphaproteobacteria) found in various terrestrial arthropods and nematodes. About 20–66% of both animal taxa are infected by Wolbachia (Hilgenboecker et al., 2008; Gomes et al., 2022). The maternally inherited endosymbiotic Wolbachia can manipulate the reproduction of their hosts, using several mechanisms, including cytoplasmic incompatibility (CI), male killing, parthenogenesis, and feminization (Correa and Ballard, 2016). CI causes offspring death when an infected male mates with an uninfected female, while the mating of infected females with the same Wolbachia strain produces viable offspring either way (Yen and Barr, 1973; Shropshire et al., 2020). The degree of CI induction can vary significantly between different Wolbachia strains, with some strains causing no reproductive manipulation or CI, such as wAu in Drosophila simulans and wMau in D. mauritiana, while others can cause weak CI as in wYak of D. yakuba, or complete CI that affects all embryos in wPip of Culex pipiens complex species (Laven, 1967; Hoffmann et al., 1996; Meany et al., 2019; Beckmann et al., 2021). CI induced by Wolbachia can be a useful technique for controlling insect populations. The use of CI-Wolbachia has been effective in controlling the mosquito vector-borne diseases population, reducing the transmission of disease in the health sector, and can be considered in the agricultural sector through incompatible insect technique (IIT) to control insect pests (Laven, 1967; Hidayanti et al., 2022).
Cytoplasmic incompatibility is a two-sided phenomenon, involving a form of “modification” in sperm and a corresponding “rescue” mechanism occurring within the eggs (Hurst, 1991). The CI effect in Wolbachia is mainly attributed to a class of genes known as cytoplasmic incompatibility factor (cif) genes, which forms the molecular basis of CI (Beckmann et al., 2017, 2019; Le Page et al., 2017). The cif genes, cifA and cifB, usually occur in an operon, but unpaired and fragmented cif genes are also found in some strains (Martinez et al., 2021). In addition, multiple pair’s amplification and diversification of cif genes have been reported to contribute to CI diversity in wPip from the mosquito Culex pipiens (Bonneau et al., 2018a,b). The cif gene products are categorized into Type I–V based on their protein domain similarity (Lindsey et al., 2018; Martinez et al., 2021). Cif proteins with deubiquitinase activity are sometimes referred to as Cid, while Cif proteins with DNase activity are sometimes referred to as Cin (Beckmann et al., 2017). The affinity between CidA–CidB, and CinA–CinB, have been confirmed (Beckmann et al., 2017; Chen et al., 2019). Furthermore, co-expressing cifA/cifB transgenes in Drosophila melanogaster also mimics the embryonic defects, a feature of CI that results in embryo death (Le Page et al., 2017; Chen et al., 2019), while transgenic expression of cifB alone induces CI in Anopheles gambiae (Adams et al., 2021). Besides, transgenic expression of a single cifA gene can rescue defects in egg-hatch rates and growth defects induced by CifB in yeast (Shropshire et al., 2018; Adams et al., 2021).
Liriomyza trifolii (Diptera: Agromyzidae), a polyphagous leafminer insect is a significant invasive pest of agricultural vegetable and ornamental plants (Kang et al., 2009; Zhang et al., 2017). Wolbachia infection is prevalent in Liriomyza species in Japan and the Indo-Pacific region, with 30–80% of the population being positive for the infection (Xu et al., 2021). Wolbachia-infected L. trifolii exhibited strong CI phenotype, resulting in very few eggs hatching from the crossing between infected males with naturally Wolbachia-free or antibiotic-treated females (Tagami et al., 2006a). The Wolbachia strain found in L. trifolii has been assigned to Supergroup B, in contrast to the majority of Wolbachia strains identified in Diptera, which belong to Supergroup A (Scholz et al., 2020). Unfortunately, the low completeness of the initial genome has hindered the identification of cif genes and other important gene markers, such as the Wolbachia surface protein gene wsp (Braig and Zhou, 1998) and the five housekeeping genes (gatB, coxA, hcpA, ftsZ, and fbpA) used in the Wolbachia multilocus strain typing (MLST) methodology (Baldo et al., 2006).
In this study, we surveyed the bacterial community in L. trifolii to explore its microbiome, and sequenced, assembled, and analyzed the genome of its Wolbachia to investigate the putative genetic basis of the strong CI effect on its insect host. We additionally provided a detailed description of the newly assembled genome through phylogenetic and comparative genome analysis. This involved the identification of cif genes and the prophage region to further characterize the strains in comparison to other closely related Wolbachia genomes.
2 Materials and methods
2.1 Insect rearing, sample collection, and DNA extraction
Liriomyza trifolii were provided by Applied Entomology Laboratory, Faculty of Agriculture, Shizuoka University. The flies were isolated in Hamamatsu, Shizuoka, Japan in 1991, and maintained on the leaves of kidney bean plants in a 34 cm (width) × 35 cm (length) × 34 cm (height) cage with light–dark regime (16:8) at 23°C (Tagami et al., 2006a; Hidayanti et al., 2022).
Total genomic DNA was extracted from L. trifolii using Qiagen DNeasy Blood and Tissue Kit, following the manufacturer’s instruction (Qiagen, Hilden, Germany), with slight modifications. Adults insects (n = 30–50) were crushed in 50 μL ATL buffer with a motorized pestle (Power Masher II; Nippi, Tokyo, Japan); 130 μL of ATL buffer, and 20 μL proteinase-K (20 mg/mL) were added to the homogenate and incubated in a 56°C dry bath incubator (Major Science, Taiwan) for 2–3 h; 200 μL of buffer AL and 200 μL of 99.5% ethanol were added following incubation at 56°C; to maximize DNA yield, the DNA was eluted in 50 μL of buffer AE. DNA yield and purity were verified with a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). The extracted DNA was aliquoted for sequencing and cloning of 16S rRNA gene and the Wolbachia surface protein (wsp) gene, and metagenomic library construction for next-generation sequencing (NGS).
2.2 High-throughput and sanger amplicon sequencing
2.2.1 High-throughput 16S rRNA amplicon sequencing
The total DNA from L. trifolii was amplified using primers to target the hypervariable V3–V4 regions of the 16S rRNA gene, which includes Illumina adaptor sequences (in triplicate), using these primers: Forward Primer (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and Reverse Primer (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′). The amplicon classification was also confirmed with the near-full-length 16S rRNA gene clones sequencing, amplified using primers 27F-mix (5’-AGRGTTTGATYMTGGCTCAG-3′) and 1492R (5’-GGHTACCTTGTTACGACTT-3′) (Hongoh et al., 2007).
The high-throughput amplicon sequencing analysis was performed using the QIIME2-2021.4 platform (Bolyen et al., 2019). Adapter and primer sequences were removed using the following options: --p-front-f CCTACGGGNGGCWGCAG --p-front-r GACTACHVGGGTATCTAATCC. The reads were trimmed, denoised, paired, and dereplicated with the built-in dada2 algorithm (Callahan et al., 2016). The amplicon sequence variants (ASVs) were taxonomically classified using the q2-feature-classifier, which had been trained for V3–V4 regions of 16S rRNA (Bokulich et al., 2018).
2.2.2 Wolbachia amplicon sequencing
The presence of Wolbachia was detected using Wolbachia surface protein (wsp) gene primers (wsp81F: 5’-TGGTCCAATAAGTGATGAAGAAAC-3′ and wsp691R: 5’-AAAAATTAAACGCTACTCCA-3′) (Braig and Zhou, 1998). The wsp amplicons were cloned and sequenced to confirm whether the Wolbachia strain in the current sample was identical to the previously detected Wolbachia associated with strong cytoplasmic incompatibility in L. trifolii (Tagami et al., 2006a). The alignment of the wsp sequences was also performed with the previously sequenced wsp from the Wolbachia survey in Japan and Indo-pacific region (Xu et al., 2021).
2.3 Wolbachia shotgun sequencing, genome assembly and bioinformatics analyses
The Wolbachia draft genome was obtained by preparing a shotgun metagenome library with an Illumina DNA prep kit, which was then sequenced using the MiSeq Reagent Kit v2 and v3. The metagenomic sequencing reads were filtered based on base quality (Q20) and length (>50 bp). Next, the filtered reads were paired and assembled using MEGAHIT (Li et al., 2015) and only contigs longer than 1,000 bp were retained for further analysis. A custom database of Wolbachia genomes was utilized to identify Wolbachia contigs based on nucleotide similarity using blastn (Altschul et al., 1990). Scaffolds were created using Codon Code Aligner (v. 9.0.1, Codon Code Corporation) and Mauve Contig Mover (MCM) (Rissman et al., 2009), followed by manual inspection and visualization in Geneious Prime 2022.1.1. Genome completeness was determined by the presence of single-copy genes of Proteobacteria (proteobacteria_odb10) using BUSCO 5.2.2 (Manni et al., 2021). Average Nucleotide Identity (ANI) was calculated using FastANI tool (Jain et al., 2018). Annotation of coding regions, RNA genes, and other genomic features was done through the RAST-tk pipeline (Aziz et al., 2008). The prophage region was automatically detected using PHASTER (Arndt et al., 2016) and subsequently refined manually based on similarity searches against prophage region in the Wolbachia of Ischnura elegans, the largest Wolbachia genome assembled from the Darwin Tree of Life biodiversity genomics project (Vancaester and Blaxter, 2023). Identification of Gene Transfer Agents (GTAs) was also performed through sequence similarity searched against RcGTA in Rhodobacter capsulatus and a putative GTA in wMel. The sequence similarity searches were conducted using the BLAST suite against the nr database (https://www.ncbi.nlm.nih.gov/), a custom Wolbachia genome database (Supplementary Table S1; Pascar and Chandler, 2018), or specific genes as otherwise mentioned.
2.4 Phylogenomic analysis
A phylogenetic tree of Wolbachia was created from concatenated 40 single copy orthologous genes, aligned with MAFFT 7.402 (Katoh and Standley, 2013). The trees were generated with 1,000 bootstraps using Maximum Likelihood method and JTT matrix-based model in MEGAX (Kumar et al., 2018). For the CI genes phylogeny, the cifA and cifB homologs were identified and constructed as previously described (Martinez et al., 2021). To clarify the origin of the large terminase (terL) in wMeg, an unrooted terL phylogenetic tree was constructed using identified homologs from reference genomes. These homologs were retrieved through nucleotide similarity searches, using the following sequences as queries: the terL sequences downstream of the Type III cifA;B in wMeg (CP021120.1), and the previously identified terL sequences in WORiC (CP001391.1; sr1WO), WORiA (CP001391.1; sr2WO), WOMelB (AE017196.1; sr3WO), as well as WOFol2 (CP015510.2; sr4WO).
2.5 Protein domain prediction
The cif genes were translated into amino acid then queried individually using HHpred (https://toolkit.tuebingen.mpg.de/hhpred/; Söding et al., 2005) with defaults parameters against the following databases: PDB_mmCIF70_24_Oct, SCOPe70_2.08, Pfam-A_v36, SMART_v6.0, PHROGs_v4, TIGRFAMs_v15.0, and COG_KOG_v1.0.
3 Results
3.1 Microbiome of Liriomyza trifolii
The Wolbachia strains in the current study was identical to the strain associated with strong CI in L. trifolii in the previous study (Tagami et al., 2006a). The identity was confirmed using a partial wsp gene which was amplified, cloned, and Sanger-sequenced (n = 24). The wsp gene is commonly used as an indicator of Wolbachia infection in insects. There are at least three wsp alleles have been identified in Wolbachia in L. trifolii, wLtriA, wLsatA, and wLsatD. All clones from the sample were identical to the wLsatD-type allele, hereafter referred to as wLtri.
High-throughput sequencing of the hypervariable region of the 16S rRNA gene was also performed to gain insights into the bacterial community present in L. trifolii without the antibiotic treatment. A total of 533,982 V3–V4 amplicon reads were quality-filtered, de-noised, and merged into 176,493 functional sequences. These sequences were de-replicated into 161 ASVs, in which 28 ASVs accounted for more than 80% of the total reads. Each of these ASVs were classified into taxa; in contrast to operational taxonomic unit (OTU)-based clustering, different ASVs could be classified into the same taxon.
The bacterial community in L. trifolii was largely dominated by Pseudomonadota (synonym, Proteobacteria) members, accounting for 96% of the total ASVs, with a minor presence of Actinobacteria, Bacteroidota, and Firmicutes. Among the identified genera, Wolbachia was the most abundant taxon (Figure 1A), with the representative ASV exhibiting an identical sequence to that of CI-inducing Wolbachia strains which infect a rice moth, Corcyra cephalonica (wCcep), as well as Wolbachia strains derived from other moths, such as Agriphila tristella, Erebia cassioides, and Operophtera brumata. The cloning and sequencing of an almost full-length 16S gene confirmed this finding. Likewise, ASVs from Acinetobacter and Pseudomonas were detected and matched to the 16S rRNA sequences recovered from metagenomic assembled contigs. These genera have been consistently reported to co-exist with Wolbachia in both wild and laboratory-reared mosquitoes, including Culex and Aedes (Beier et al., 1996; Pidiyar et al., 2002, 2004; Lindh et al., 2005; Zouache et al., 2009a,b; Minard et al., 2013; Schrieke et al., 2021; Rau et al., 2022).
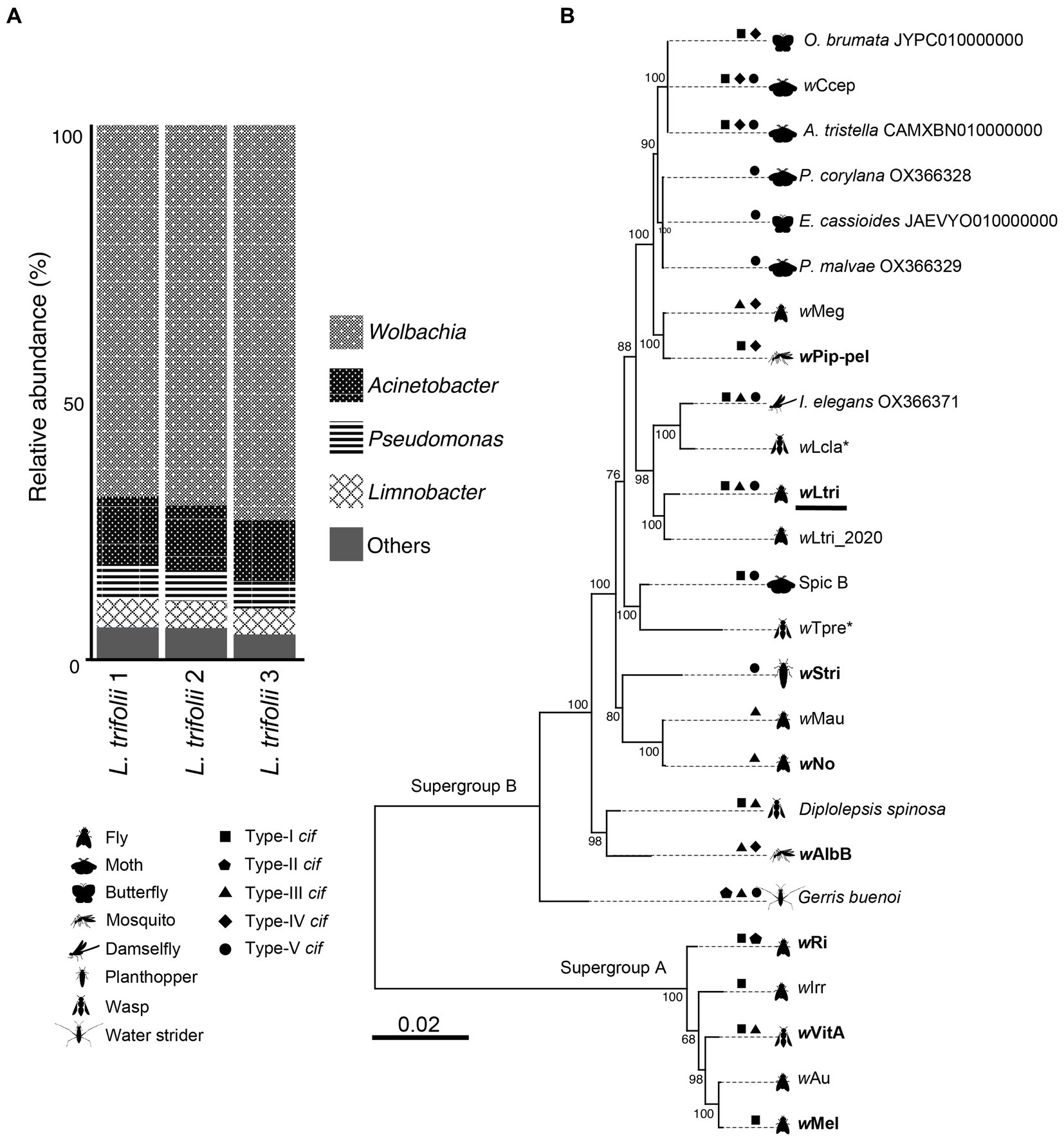
Figure 1. (A) Prokaryote composition in Liriomyza trifolii at the genus level in triplicate. (B) Phylogeny of 25 strains of Wolbachia. The tree was constructed based on nucleotide sequences of 40 single-copy orthologous genes. The Wolbachia strains that are reported to cause CI are presented in boldface, while the parthenogenesis-inducing strains are marked with an asterisk. The wLtri from this current study is underlined.
3.2 The genome of wLtri, the Wolbachia strain in Liriomyza trifolii
A total of 15,421,862,646 high-quality bases were assembled using MEGAHIT into contigs. A contig covering the entire L. trifolii mitochondrial sequence was identified, and it exhibited 99.1% nucleotide similarity to L. trifolii GU327644. A total of 435 contigs originating from Wolbachia were identified using BLASTN searches against a custom database (Supplementary Table S1) and further validated via manual inspection. The wLtri contigs were reordered using Mauve Contig Mover (MCM) to create a draft genome (Figure 2). The genome comparison analysis also included a previously sequenced L. trifolii, which contains 443 contigs of Wolbachia, assembled using a similar short-read sequencing technology and genome assembler, referred to as wLtri_2020 hereinafter (Vicoso and Bachtrog, 2015; Scholz et al., 2020). The Average Nucleotide Identity score between wLtri draft genome and wLtri_NCBI was 98.1%. The completeness of the wLtri genome based on BUSCO (proteobacteria_odb10) was 80.3%, which represents a typical value for complete Wolbachia genomes (Sinha et al., 2019), while the completeness of wLtri_2020 was only 31.6%. The assembly size of wLtri was 1,358,284 bp, which was longer than that of wLtri_2020 (879,722 bp). The genome of wLtri contained 1,487 coding sequences (CDSs), along with 5S, 23S, and 16S rRNA, and 34 tRNA genes, while wLtri_2020 had fewer CDSs (n = 1,206) and an incomplete set of rRNA genes (Table 1).
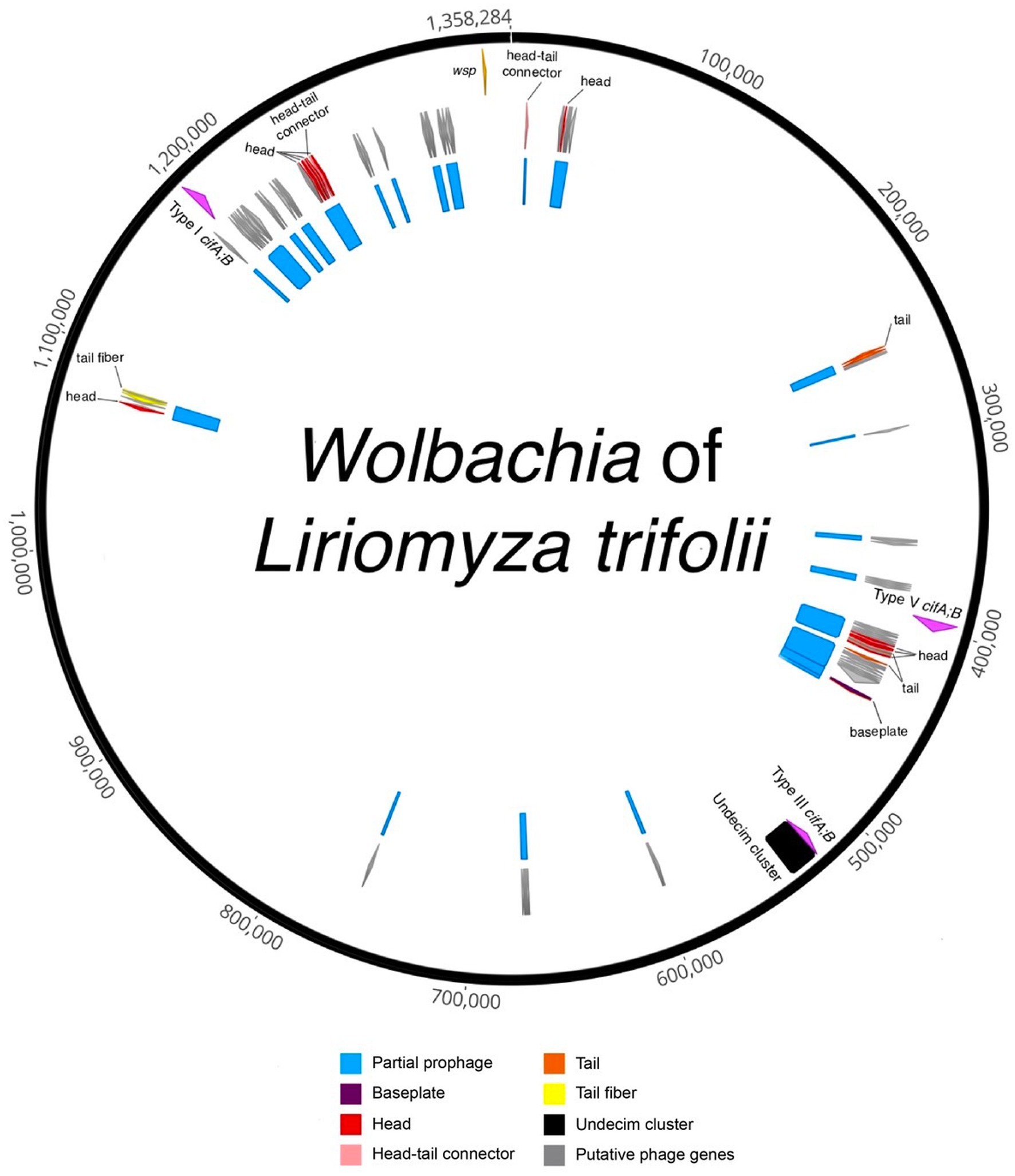
Figure 2. Circularized draft genome annotation of the Wolbachia of Liriomyza trifolii. Partial prophage regions are indicated in blue, representing regions with incomplete prophage regions predicted by PHASTER and/or parts of prophage WO in the Wolbachia of Ischnura elegans. Structural genes are denoted in red (head), pink (head-tail connector), purple (baseplate), orange (tail), and yellow (tail fiber), while gray signifies putative phage genes, encompassing genes found in phages but with unknown functions. The Undecim cluster is depicted in black. The cif genes are highlighted in fuchsia, and the Wolbachia surface protein (wsp) gene is represented in light orange.
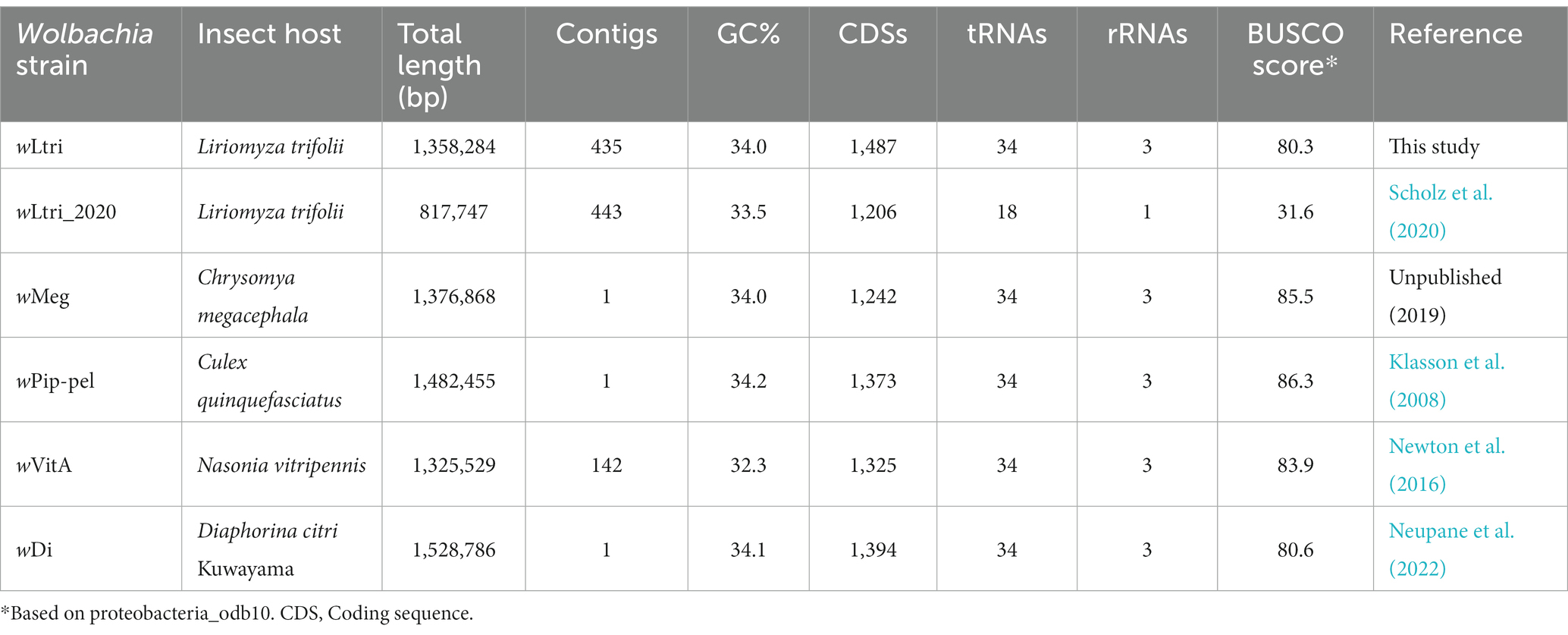
Table 1. Comparison of assembly status and genome characteristics among Wolbachia strains.
Based on the maximum likelihood phylogenetic tree of single-copy genes, the newly sequenced wLtri belongs to the Supergroup B and it clustered with wLtri_2020, which infects the same host species, L. trifolii (Figure 1B). Wolbachia of Supergroup B is commonly found in Lepidopteran hosts, with only a few instances identified in Dipteran hosts (Meany et al., 2019; Scholz et al., 2020; Vancaester and Blaxter, 2023). Within the Supergroup B clade, wLtri formed a monophyletic group with Wolbachia strains that infect other arthropods, such as damselflies (I. elegans) and wasps (Leptopilina clavipes), rather than with strains infecting flies, such as wMau of Drosophila mauritiana and wNo of Drosophila simulans (Figure 1B). To further investigate the relationship between wLtri and Wolbachia strains in wasps, a phylogenetic tree was constructed using wsp genes (Supplementary Figure S1). The analysis included a parasitoid wasp, Hemiptarsenus varicornis, found near the initial L. trifolii sampling location (Tagami et al., 2006b). Although the wsp gene undergoes rapid evolution, the wsp gene derived from wLtri consistently clustered with the wsp amplified from wasps H. varicornis and Trichogramma pretiosum (wTpre).
3.3 Prophage regions in wLtri
In the case of wLtri, prophage sequences were analyzed using the PHASTER tool and sequence homology to previously known prophage WO to identify incomplete prophage regions with a combined size of 98.9 Kb (Figure 2). Prophage regions were also identified using nucleotide similarity searches against Wovirus, which includes WO phages, from a newly proposed family, Symbioviridae. The Wovirus was further subclassified into four groups, sr1WO, sr2WO, sr3WO, and sr4WO, based on gene synteny in the phage core module and serine-recombinase nucleotide identity (Bordenstein and Bordenstein, 2022). A BLASTN search using these recombinases revealed fragments in wLtri with 84 and 88% nucleotide similarity to sr1WO and sr3WO, respectively.
Our analysis also revealed the presence of a set of 11 conserved genes, known as Undecim Cluster, in wLtri genome. This cluster is part of the EAM of Phage WO, which is commonly found in sr3WO and is occasionally present in sr4WO and WO-like islands (Bordenstein and Bordenstein, 2022). The EAM in wLtri displays a module synteny similar to that of the WO-like island in WOAlbB3, wNo, WOMau2, and wVitA, where it also comprises cifA and cifB genes (Bordenstein and Bordenstein, 2022). Notably, the cif genes found in these WO-like islands belong to Type III cifA;B, unlike in sr3WO, which mostly includes Type I cifA;B.
Interestingly, in wMeg, downstream of Undecim cluster—Type III cifA;B, a large terminase gene (terL) was identified (Supplementary Figure S2A). The gene is commonly used as a prophage marker due to its high degree of conservation and ubiquity across phage genomes. To determine whether the terL was originated from a Phage WO carrying the Type III cifA;B, the intergenic region between the cif genes and terL was aligned with other Wolbachia which possessed same type of cif genes in the reference genomes, including wLtri. The intergenic region (1,113 bp) displayed potential shared synteny (Supplementary Figure S2B), revealing distinct partitioning into two segments: a left portion (L; 554 bp) downstream of the Undecim cluster—cif genes, and a right portion (R; 559 bp) upstream of terL. While both segments were occasionally repeated in some of the reference genomes, they were never co-occurred, except in wMeg (Supplementary Figure S2C; Supplementary Tables S2, S3).
The L was found adjacent to cif genes and/or the Undecim Cluster in the genomes that possess it. In the Wolbachia of I. elegans, the L was identified in two locations: the first near a partial Undecim Cluster—cif genes, and the second near an incomplete Undecim Cluster without cif genes in a different location (Supplementary Table S2). In wIrr, the L was associated with IS256, while in the Wolbachia of Erebia cassioides and Leptopilina clavipes, wLcla, it was associated with IS110 family transposase (Supplementary Table S2). Conversely, the R was found in more location within the genomes, often at breakpoints, and was associated with IS982, IS5, and IS630 (Supplementary Figure S3; Supplementary Table S3).
In the Wolbachia genome, a terminase gene may also have been derived from Gene Transfer Agents (GTAs). BLAST searches (TBLASTN, BLASTN) using the amino acid and nucleotide sequences of a terminase-like protein (RCAP_rcc01683) from RcGTA, a well-studied GTA in Rhodobacter capsulatus, as queries against the Wolbachia reference genomes and wLtri, did not yield any significant matches. However, when a terminase from the putative GTA in wMel (WD_1016; AE017196) was used as a query, it revealed matches within the reference genomes (Supplementary Table S4). Notably, these findings included the terL which was located downstream of the WO-like island in wMeg, revealing 82% nucleotide and 83% amino acid similarities, respectively. In addition, a phylogenetic tree constructed using homologs of terL from wMeg and prophage WO revealed that they belong to two distinct clades, which further divided into a sub-clade that separates Supergroup A from Supergroup B (Supplementary Figure S2C).
3.4 Cytoplasmic incompatibility genes
wLtri has three phylogenetically distinct cifA–cifB gene pairs which belong to Type I, III, and V (Figure 3A). The wLtri Type I showed moderate to low amino acid similarity to the CifA (63–68%) and CifB (48–51%) of the experimentally validated CI-inducing Wolbachia strains wMel and wPip, respectively. The Type V also showed moderate and low amino acid similarity of 57 and 20% to the CifA–CifB proteins of wStri, respectively. On the other hand, wLtri Type III showed high amino acid similarity (98%) to CifA and CifB of CI-inducing wNo. Furthermore, the wLtri Type III was the only gene pair located within the EAM of WO-like islands, adjacent to an Undecim cluster.
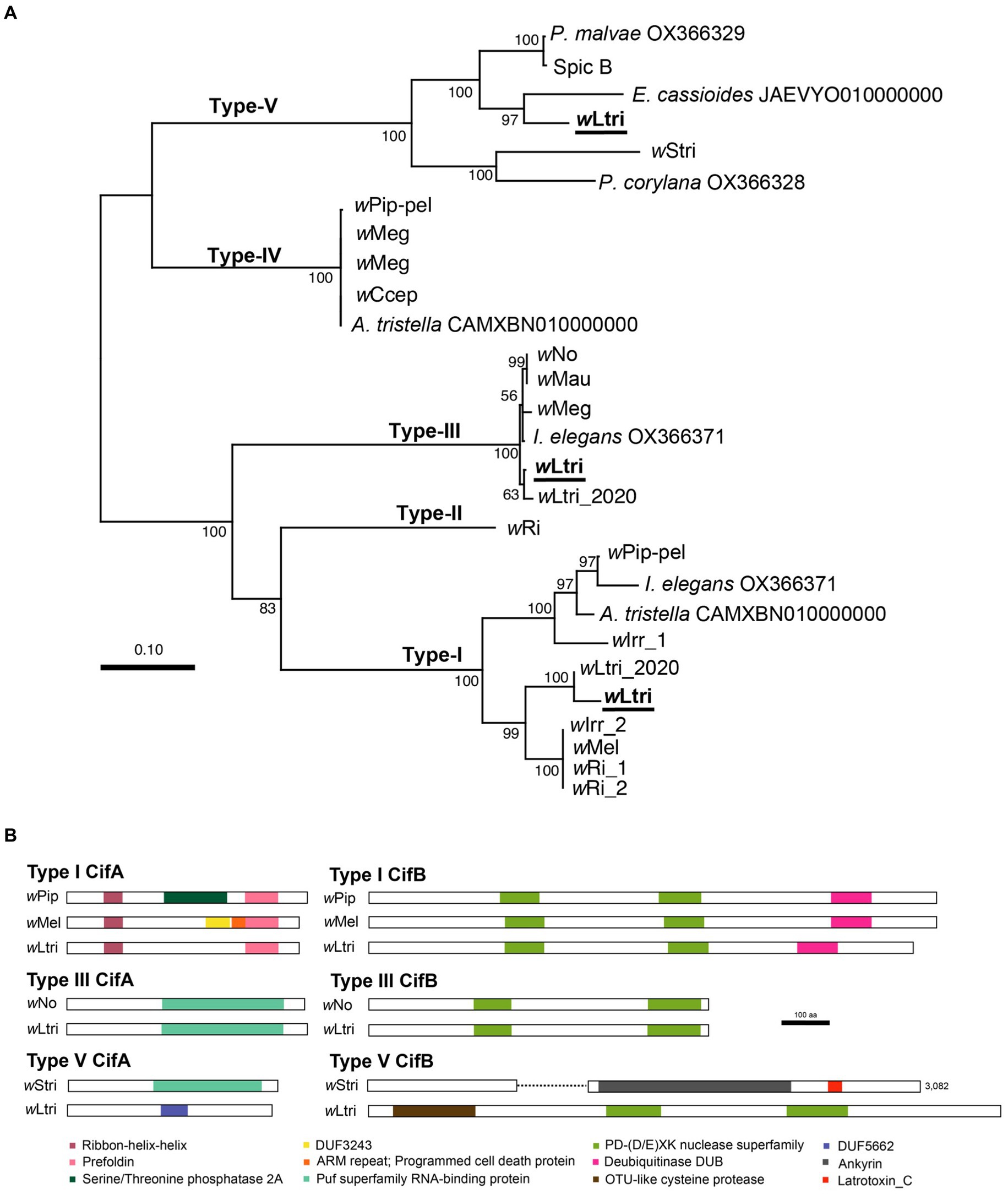
Figure 3. (A) Maximum likelihood tree of concatenated cifA and cifB nucleotide sequences. Partially sequenced cif homologs were excluded. Bootstrap values were estimated from 1,000 replicates. (B) Representative structures of Cif proteins with predicted domains. The wStri type V CifB has a length of 3,082 amino acids. To accommodate presentation constraints, it was shortened without eliminating any identifiable domains.
In wLtri CifA, four protein domains were identified (Figure 3B). The first two were the Ribon-helix–helix Protein (RHH domain) and Prefoldin, found in the Type I CifA of wLtri. Despite a low probability of homology (probability 22–31%), these domains were also found in functional CifAs (CidA). However, the CifA of wLtri lacks Serine/Threonine phosphatase 2A and DUF3243, which are present in wPip and wMel, respectively. Additionally, the Puf superfamily RNA-binding protein (Type III) was identified in both wLtri and wNo. Lastly, the DUF 5662 (Type V) was found in wLtri but not in wStri Type V CifA, which contains the Puf superfamily. In wLtri CifB, three protein domains were identified (Figure 3B). The PD-(D/E)XK nuclease superfamily was consistently found in all wLtri CifB with a high probability (>96%). Furthermore, the Type I exhibited the presence of a deubiquitinase domain DUB (probability 96%), while the Type V contained an OTU-like cysteine protease (probability 99%).
4 Discussion
This study represents the first survey of the bacterial community of the American Serpentine Leafminer fly, L. trifolii, in Japan. Using 16S rRNA high-throughput amplicon sequencing, it revealed Wolbachia as the most abundant bacterium in L. trifolii. The genome of the Wolbachia strain, wLtri, had never been assembled with high completeness before. This wLtri assembly represents the most complete genome sequence of a cytoplasmic-inducing Wolbachia of L. trifolii. Another available Wolbachia genome from L. trifolii, wLtri_2020, is a binning of 443 contigs with only 31.6% BUSCO completeness (Vicoso and Bachtrog, 2015; Scholz et al., 2020). wLtri_2020 was not included in some of the analyses in this study as it contains only partial genome information. We have also determined that wLtri was the main endosymbiont of L. trifolii, using16S rRNA gene amplicon sequencing and metagenomic data.
Unlike the majority of Wolbachia strains found in Dipteran hosts, which belong to Supergroup A, wLtri is classified under Supergroup B. The phylogeny of supergroup A and B have been found to be incongruent with those of their hosts, due to frequent horizontal transmission of Wolbachia strains across diverse host species (Raychoudhury et al., 2009; Wang et al., 2020). Insect parasitoids have been proposed as a means of facilitating this horizontal transmission of endosymbionts when infected and uninfected parasitoid wasps develop within the same host insect (Huigens et al., 2000, 2004; Ahmed et al., 2015). Liriomyza trifolii is also susceptible to parasitoid wasps, with approximately 24 species of leafminer parasitoids identified in Japan (Arakaki and Kinjo, 1998). Among these wasps, a wsp sequence from H. varicornis, found near the original sampling location of L. trifolii, formed a clade with that of wLtri. However, due to the limited availability of wsp sequences from other parasitoids, it was the only sequence included in the analysis. This limitation is attributed to the lower prevalence of Wolbachia infection in leafminer parasitoids compared to Liriomyza, in which among the surveyed 15 leafminer parasitoid species, only H. varicornis was infected with Wolbachia (Tagami et al., 2006b). Besides, the migration of leafminers Liriomyza from another country (Abe, 2017) may potentially introduce endosymbiont transfer between the established Liriomyza and the invasive species. Therefore, additional research is needed to comprehensively understand the potential role of Liriomyza parasitoids and the impact of invasive Liriomyza on the horizontal transmission of Wolbachia strains.
Wolbachia is well known for inducing CI in many insects, including L. trifolii (Tagami et al., 2006a). Recent studies have shown that the proteins which are responsible for CI, CifA and CifB, can be classified into Types I–V (Martinez et al., 2021). In the genome of wLtri, three sets of cifA;B genes—Types I, III, and V—were identified. Notably, the genes encoding Type I and Type V CifB in wLtri were shorter, exhibiting low protein similarity to functional Cif in wPip, wMel, wNo, and wStri. Typically, cifB often accumulates more mutations before cifA, rendering the gene non-functional before being eliminated from the genome (Martinez et al., 2021). However, in wLtri, although the genes were shorter, the predicted gene products containing domains commonly found in Type I CifB, such as PD-(D/E)XK nuclease superfamily and DUB, remained recognizable. The Type V CifB in wLtri was also considerably shorter than that of wStri, a Wolbachia strain in Laodelphax striatellus. This difference is not unexpected due to the greater diversity of protein domains in Type V CifB compared to other types, encompassing domains such as the C-terminal domain of Latrotoxin, those involved in protein–protein interactions (tetratricopeptide and ankyrin repeats), and a protease domain (OTU-like cysteine protease) (Martinez et al., 2021). However, in wLtri, only the latter and a PD-(D/E)XK were present. In contrast, the Type III cifA;B of wLtri appeared highly conserved, sharing adjacent gene synteny with CI-inducing Wolbachia strains like wNo, which exclusively contains Type III cifA;B genes.
The amplification-diversification of functional cif genes and the cumulative presence of these genes have been associated with cytoplasmic incompatibility (CI) strength (Le Page et al., 2017; Bonneau et al., 2018b). In the genome of Wolbachia in Culex pipiens wPip, variations and copy numbers of cif genes (cidA, cidB) are identified and the expression of these multiple cid gene variants in males may account for differences in CI cellular phenotypes (Bonneau et al., 2018a,b). Furthermore, wMel, a Wolbachia strain with only one copy of these genes exhibits a weak CI phenotype, whereas strains with two or three copies of the genes, such as wRi and wHa, showed a strong CI effects (Le Page et al., 2017). The facts that wLtri causes strong CI in L. trifolii (Tagami et al., 2006a) and its genome harbored three sets of cifA–cifB genes, might supported this correlation. Ultimately it is necessary to confirm that these cif gene products interact with each other through performing an in vitro pull-down study, which demonstrates the specific binding of functional cognate protein pairs (CifA and CifB). Afterward, to determine the individual gene activity, transgenic expression of a single cif gene can be conducted, allowing for the assessment of whether a gene alone can induce CI or whether other cif genes or factors are necessary for the CI to occur (Beckmann et al., 2017; Le Page et al., 2017; Adams et al., 2021; Horard et al., 2022).
The wLtri Type III cifA;B are located adjacent to a conserved set of 11 genes, collectively known as the Undecim Cluster, which constitutes a eukaryotic association module (EAM) within phage WO. Phage WO, a bacteriophage that infects intracellular Wolbachia, faces the challenge of 2-fold barriers: the eukaryotic cell membrane and the intracellular bacterial cell membranes. Consequently, it frequently carries an EAM containing genes that exhibit eukaryotic-like functions and origins, which have the potential to influence host-Wolbachia interactions (Bordenstein and Bordenstein, 2022). After infection, the phage WO that integrates its genetic material into the Wolbachia genome refer as putative prophage WO. Although it is thought that no complete prophage WO has been identified in wLtri, the genes surrounding the cifs exhibit module synteny akin to that of the WO-like island found in wNo, wMau, and wAlbB. These WO-like Islands are considered defective prophages, likely stemming from an ancestral prophage WO genome, which has since undergone domestication by the bacterial host or is undergoing degradation and elimination from the chromosome (Bordenstein and Bordenstein, 2022).
The WO-like island of wLtri also exhibits module synteny with that of wMeg, a Wolbachia strain found in the blowfly Chrysomya megacephala, which is commonly associated with carrion and other decaying materials in human environments (Badenhorst and Villet, 2018). This similarity extends to the intergenic region between their Type III cifA;B and terL. However, the terL was distinct from the known terL genes of phage WO. Within Wolbachia genomes, terL is not exclusively associated with prophage WO but is also linked to Gene Transfer Agents (GTAs), which are virus-like structures responsible for packaging and transferring prokaryotic DNA between donor and recipient prokaryotic cells (Lang and Beatty, 2000; Lang et al., 2017; Bordenstein and Bordenstein, 2022). Although the terL in wMeg exhibited low nucleotide sequence similarity to a terminase-like gene in RcGTA of R. capsulatus, it demonstrated higher similarities to a terminase found in a putative GTA from wMel (AE017196). This observation suggests that the terL in wMeg might be a component of GTAs within Wolbachia genomes. Furthermore, the terL homologs formed a distinct clade, separating them from other terL genes within the prophage WO region, suggests that the terL genes in wMeg and prophage WO have different evolutionary origins. This clade further branched into sub-clades that distinguished Supergroups A and B, consistent with previous finding that terL genes within putative GTAs in Wolbachia genomes can effectively differentiate between these Supergroups (Bordenstein and Bordenstein, 2022).
Regarding the potential synteny of the intergenic region between the cif genes and terL in the Wolbachia genomes, it appears to involve at least two “genomic scars” resulting from ancestral transposition events associated with IS256, IS110, IS982, IS5, and IS630. In wMeg, an ancient phage WO carrying Type III cifA;B might had integrated its genome into or near a GTA sequence. Subsequently, the GTA and the prophage WO may have deteriorated over time, or transposition events could have joined the breakpoints near the cif genes and terL, ultimately leading to the genetic remnants that are presently observed. Given that the intergenic sequences were frequently located in the vicinity of breakpoints in wLtri and other Wolbachia genomes, the observed similarities in this region are likely a consequence of the latter phenomenon.
In summary, our bacterial community survey indicates that Wolbachia is the main endosymbiont in L. trifolii, alongside minor occurrences of Acinetobacter, Pseudomonas, and Limnobacter. The Wolbachia strain in L. trifolii, wLtri, possesses three distinct types of cytoplasmic incompatibility factor (cif) genes: Type I, Type III, and Type V cifA;B. The diversification and cumulative presence of these genes may contribute to the strong CI effects observed in L. trifolii.
Data availability statement
The data presented in the study are deposited in the DNA Data Bank of Japan (DDBJ) repository, accession number DRA017213 (https://ddbj.nig.ac.jp/resource/bioproject/PRJDB16160).
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
AP: Conceptualization, Data curation, Investigation, Validation, Writing – original draft, Writing – review & editing. AH: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. YT: Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing. HA: Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was partly funded by a grant from the Japan Society for the Promotion of Science (JSPS) KAKENHI to HA (Grant Numbers: 20H03723, 15K21770) and YT (Grant Number: JP419K06069).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1304401/full#supplementary-material
References
Abe, Y. (2017). Invasion of Japan by exotic leafminers Liriomyza spp. (Diptera: Agromyzidae) and its consequences. Appl. Entomol. Zool. 52, 175–182. doi: 10.1007/s13355-017-0486-z
Adams, K. L., Abernathy, D. G., Willett, B. C., Selland, E. K., Itoe, M. A., and Catteruccia, F. (2021). Wolbachia cifB induces cytoplasmic incompatibility in the malaria mosquito vector. Nat. Microbiol. 6, 1575–1582. doi: 10.1038/s41564-021-00998-6
Ahmed, M. Z., Li, S. J., Xue, X., Yin, X. J., Ren, S. X., Jiggins, F. M., et al. (2015). The intracellular bacterium Wolbachia uses parasitoid wasps as phoretic vectors for efficient horizontal transmission. PLoS Pathog. 10:e1004672. doi: 10.1371/journal.ppat.1004672
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arakaki, N., and Kinjo, K. (1998). Notes on the parasitoid fauna of the serpentine leafminer Liriomyza trifolii (burgess) (Diptera: Agromyzidae) in Okinawa, southern Japan. Appl. Entomol. Zool. 33, 577–581. doi: 10.1303/aez.33.577
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Badenhorst, R., and Villet, M. H. (2018). The uses of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae) in forensic entomology. Forens. Sci. Res. 3, 2–15. doi: 10.1080/20961790.2018.1426136
Baldo, L., Dunning Hotopp, J. C., Jolley, K. A., Bordenstein, S. R., Biber, S. A., Choudhury, R. R., et al. (2006). Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 72, 7098–7110. doi: 10.1128/AEM.00731-06
Beckmann, J. F., Bonneau, M., Chen, H., Hochstrasser, M., Poinsot, D., Merçot, H., et al. (2019). The toxin–antidote model of cytoplasmic incompatibility: genetics and evolutionary implications. Trends Genet. 35, 175–185. doi: 10.1016/j.tig.2018.12.004
Beckmann, J. F., Ronau, J. A., and Hochstrasser, M. (2017). A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat. Microbiol. 2:17007. doi: 10.1038/nmicrobiol.2017.7
Beckmann, J. F., Van Vaerenberghe, K., Akwa, D. E., and Cooper, B. S. (2021). A single mutation weakens symbiont-induced reproductive manipulation through reductions in deubiquitylation efficiency. Proc. Natl. Acad. Sci. 118:e2113271118. doi: 10.1073/pnas.2113271118
Beier, J. C., Pumpuni, C. B., Demaio, J., and Kent, M. (1996). The midgut bacterial Flora of wild Aedes triseriatus, Culex pipiens, and Psorophora columbiae mosquitoes. Am. J. Trop. Med. Hyg. 54, 219–223. doi: 10.4269/ajtmh.1996.54.219
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonneau, M., Atyame, C., Beji, M., Justy, F., Cohen-Gonsaud, M., Sicard, M., et al. (2018a). Culex pipiens crossing type diversity is governed by an amplified and polymorphic operon of Wolbachia. Nat. Commun. 9:319. doi: 10.1038/s41467-017-02749-w
Bonneau, M., Landmann, F., Labbé, P., Justy, F., Weill, M., and Sicard, M. (2018b). The cellular phenotype of cytoplasmic incompatibility in Culex pipiens in the light of cidB diversity. PLoS Pathog. 14:e1007364. doi: 10.1371/journal.ppat.1007364
Bordenstein, S. R., and Bordenstein, S. R. (2022). Widespread phages of endosymbionts: phage WO genomics and the proposed taxonomic classification of Symbioviridae. PLoS Genet. 18:e1010227. doi: 10.1371/journal.pgen.1010227
Braig, H. R., and Zhou, W., Dobson, S.’L. & O'Neill, S. L. (1998). Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J. Bacteriol. 180:2373–2378. doi: 10.1128/JB
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, H., Ronau, J. A., Beckmann, J. F., and Hochstrasser, M. (2019). A Wolbachia nuclease and its binding partner provide a distinct mechanism for cytoplasmic incompatibility. Proc. Natl. Acad. Sci. U. S. A. 116, 22314–22321. doi: 10.1073/pnas.1914571116
Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing against a master manipulator. Front. Ecol. Evol. 3:153. doi: 10.3389/fevo.2015.00153
Gomes, T. M. F. F., Wallau, G. L., and Loreto, E. L. S. (2022). Multiple long-range host shifts of major Wolbachia supergroups infecting arthropods. Sci. Rep. 12:8131. doi: 10.1038/s41598-022-12299-x
Hidayanti, A. K., Gazali, A., and Tagami, Y. (2022). Effect of quorum sensing inducers and inhibitors on cytoplasmic incompatibility induced by Wolbachia (Rickettsiales: Anaplasmataceae) in American serpentine Leafminer (Diptera: Agromyzidae): potential tool for the incompatible insect technique. J. Insect Sci. 22:8. doi: 10.1093/jisesa/ieab106
Hilgenboecker, K., Hammerstein, P., Schlattmann, P., Telschow, A., and Werren, J. H. (2008). How many species are infected with Wolb–chia?—a statistical analysis of current data. FEMS Microbiol. Lett. 281, 215–220. doi: 10.1111/j.1574-6968.2008.01110.x
Hoffmann, A. A., Clancy, D., and Duncan, J. (1996). Naturally-occurring Wolbachia infection in Drosophila simulans that does not cause cytoplasmic incompatibility. Heredity 76, 1–8. doi: 10.1038/hdy.1996.1
Hongoh, Y., Sato, T., Dolan, M. F., Noda, S., Ui, S., Kudo, T., et al. (2007). The motility symbiont of the termite gut flagellate Caduceia versatilis is a member of the “Synergistes” group. Appl. Environ. Microbiol. 73, 6270–6276. doi: 10.1128/AEM.00750-07
Horard, B., Terretaz, K., Gosselin-Grenet, A. S., Sobry, H., Sicard, M., Landmann, F., et al. (2022). Paternal transmission of the Wolbachia CidB toxin underlies cytoplasmic incompatibility. Curr. Biol. 32, 1319–1331.e5. doi: 10.1016/j.cub.2022.01.052
Huigens, M. E., de Almeida, R. P., Boons, P. A., Luck, R. F., and Stouthamer, R. (2004). Natural interspecific and intraspecific horizontal transfer of parthenogenesis-inducing Wolbachia in Trichogramma wasps. Proc. R. Soc. B Biol. Sci. 271, 509–515. doi: 10.1098/rspb.2003.2640
Huigens, M. E., Luck, R. F., Klaassen, R. H., Maas, M. F., Timmermans, M. J., and Stouthamer, R. (2000). Infectious parthenogenesis. Nature 405, 178–179. doi: 10.1038/35012066
Hurst, L. D. (1991). The evolution of cytoplasmic incompatibility or when spite can be successful. J. Theor. Biol. 148, 269–277. doi: 10.1016/s0022-5193(05)80344-3
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Kang, L., Chen, B., Wei, J.-N., and Liu, T.-X. (2009). Roles of thermal adaptation and chemical ecology in Liriomyza distribution and control. Annu. Rev. Entomol. 54, 127–145. doi: 10.1146/annurev.ento.54.110807.090507
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klasson, L., Walker, T., Sebaihia, M., Sanders, M. J., Quail, M. A., and Lord, A. (2008). Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Molecular biology and evolution, 25, 1877–1887. doi: 10.1093/molbev/msn133
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lang, A. S., and Beatty, J. T. (2000). Genetic analysis of a bacterial genetic exchange element: the gene transfer agent of Rhodobacter capsulatus. Proc. Natl. Acad. Sci. U. S. A. 97, 859–864. doi: 10.1073/pnas.97.2.859
Lang, A. S., Westbye, A. B., and Beatty, J. T. (2017). The distribution, evolution, and roles of gene transfer agents in prokaryotic genetic exchange. Annu. Rev. Virol. 4, 87–104. doi: 10.1146/annurev-virology-101416-041624
Laven, H. (1967). Eradication of Culex pipiens fatigans through cytoplasmic incompatibility. Nature 216, 383–384. doi: 10.1038/216383a0
Le Page, D. P., Metcalf, J. A., Bordenstein, S. R., On, J., Perlmutter, J. I., Shropshire, J. D., et al. (2017). Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543, 243–247. doi: 10.1038/nature21391
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Lindh, J. M., Terenius, O., and Faye, I. (2005). 16S rRNA gene-based identification of midgut Bacteria from field-caught Anopheles gambiae Sensu Lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl. Environ. Microbiol. 71, 7217–7223. doi: 10.1128/AEM.71.11.7217-7223.2005
Lindsey, A. R. I., Rice, D. W., Bordenstein, S. R., Brooks, A. W., Bordenstein, S. R., and Newton, I. L. G. (2018). Evolutionary genetics of cytoplasmic incompatibility genes cifA and cifB in prophage WO of Wolbachia. Genome Biol. Evol. 10, 434–451. doi: 10.1093/gbe/evy012
Manni, M., Berkeley, M. R., Seppey, M., and Zdobnov, E. M. (2021). BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1:e323. doi: 10.1002/cpz1.323
Martinez, J., Klasson, L., Welch, J. J., and Jiggins, F. M. (2021). Life and death of selfish genes: comparative genomics reveals the dynamic evolution of cytoplasmic incompatibility. Mol. Biol. Evol. 38, 2–15. doi: 10.1093/molbev/msaa209
Meany, M. K., Conner, W. R., Richter, S. V., Bailey, J. A., Turelli, M., and Cooper, B. S. (2019). Loss of cytoplasmic incompatibility and minimal fecundity effects explain relatively low Wolbachia frequencies in Drosophila mauritiana. Evolution 73, 1278–1295. doi: 10.1111/evo.13745
Minard, G., Tran, F. H., Raharimalala, F. N., Hellard, E., Ravelonandro, P., Mavingui, P., et al. (2013). Prevalence, genomic and metabolic profiles of Acinetobacter and Asaia associated with field-caught Aedes albopictus from Madagascar. FEMS Microbiol. Ecol. 83, 63–73. doi: 10.1111/j.1574-6941.2012.01455.x
Neupane, S., Bonilla, S. I., Manalo, A. M., and Pelz-Stelinski, K. S. (2022). Complete de novo assembly of Wolbachia endosymbiont of Diaphorina citri Kuwayama (Hemiptera: Liviidae) using long-read genome sequencing. Sci. Rep. 16:125. doi: 10.1038/s41598-021-03184-0
Newton, I. L., Clark, M. E., Kent, B. N., Bordenstein, S. R., Qu, J., Richards, S., et al. (2016). Comparative genomics of two closely related Wolbachia with different reproductive effects on hosts. Genome Biol. Evol. 8, 1526–1542. doi: 10.1093/gbe/evw096
Pascar, J., and Chandler, C. H. (2018). A bioinformatics approach to identifying Wolbachia infections in arthropods. PeerJ 6:e5486. doi: 10.7717/peerj.5486
Pidiyar, V. J., Jangid, K., Patole, M. S., and Shouche, Y. S. (2004). Studies on cultured and uncultured microbiota of wild Culex quinquefasciatus mosquito midgut based on 16s ribosomal RNA gene analysis. Am. J. Trop. Med. Hyg. 70, 597–603. doi: 10.4269/ajtmh.2004.70.597
Pidiyar, V., Kaznowski, A., Narayan, N. B., Patole, M., and Shouche, Y. S. (2002). Aeromonas culicicola sp. nov., from the midgut of Culex quinquefasciatus. Int. J. Syst. Evol. Microbiol. 52, 1723–1728. doi: 10.1099/00207713-52-5-1723
Rau, J., Werner, D., Beer, M., Höper, D., and Kampen, H. (2022). The microbial RNA metagenome of Aedes albopictus (Diptera: Culicidae) from Germany. Parasitol. Res. 121, 2587–2599. doi: 10.1007/s00436-022-07576-7
Raychoudhury, R., Baldo, L., Oliveira, D. C. S. G., and Werren, J. H. (2009). Modes of acquisition of Wolbachia: horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution 63, 165–183. doi: 10.1111/j.1558-5646.2008.00533.x
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the mauve aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Scholz, M., Albanese, D., Tuohy, K., Donati, C., Segata, N., and Rota-Stabelli, O. (2020). Large scale genome reconstructions illuminate Wolbachia evolution. Nat. Commun. 11:5235. doi: 10.1038/s41467-020-19016-0
Schrieke, H., Maignien, L., Constancias, F., Trigodet, F., Chakloute, S., Rakotoarivony, I., et al. (2021). The mosquito microbiome includes habitat-specific but rare symbionts. Comput. Struct. Biotechnol. J. 20, 410–420. doi: 10.1016/j.csbj.2021.12.019
Shropshire, J. D., Leigh, B., and Bordenstein, S. R. (2020). Symbiont-mediated cytoplasmic incompatibility: what have we learned in 50 years? elife 9, 1–36. doi: 10.7554/ELIFE.61989
Shropshire, J. D., On, J., Layton, E. M., Zhou, H., and Bordenstein, S. R. (2018). One prophage WO gene rescues cytoplasmic incompatibility in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 115, 4987–4991. doi: 10.1073/pnas.1800650115
Sinha, A., Li, Z., Sun, L., Carlow, C. K. S., and Cordaux, R. (2019). Complete genome sequence of the Wolbachia wAlbB endosymbiont of Aedes albopictus. Genome Biol. Evol. 11, 706–720. doi: 10.1093/gbe/evz025
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248. doi: 10.1093/nar/gki408
Tagami, Y., Doi, M., Sugiyama, K., Tatara, A., and Saito, T. (2006a). Wolbachia-induced cytoplasmic incompatibility in Liriomyza trifolii and its possible use as a tool in insect pest control. Biol. Control 38, 205–209. doi: 10.1016/j.biocontrol.2006.03.008
Tagami, Y., Doi, M., Sugiyama, K., Tatara, A., and Saito, T. (2006b). Survey of leafminers and their parasitoids to find endosymbionts for improvement of biological control. Biol. Control 38, 210–216. doi: 10.1016/j.biocontrol.2006.01.015
Vancaester, E., and Blaxter, M. (2023). Phylogenomic analysis of Wolbachia genomes from the Darwin tree of life biodiversity genomics project. PLoS Biol. 21. doi: 10.1371/journal.pbio.3001972
Vicoso, B., and Bachtrog, D. (2015). Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 13:e1002078. doi: 10.1371/journal.pbio.1002078
Wang, X., Xiong, X., Cao, W., Zhang, C., Werren, J. H., and Wang, X. (2020). Phylogenomic analysis of Wolbachia strains reveals patterns of genome evolution and recombination. Genome Biol. Evol. 12, 2508–2520. doi: 10.1093/GBE/EVAA219
Xu, X., Ridland, P. M., Umina, P. A., Gill, A., Ross, P. A., Pirtle, E., et al. (2021). High incidence of related Wolbachia across unrelated leaf-mining diptera. Insects 12:788. doi: 10.3390/insects12090788
Yen, J. H., and Barr, A. R. (1973). The etiological agent of cytoplasmic incompatibility in Culex pipiens. J. Invertebr. Pathol. 22, 242–250. doi: 10.1016/0022-2011(73)90141-9
Zhang, X. R., Xing, Z. L., Lei, Z. R., and Gao, Y. L. (2017). Recent status of the invasive Leafminer Liriomyza trifolii in China. Southwest. Entomol 42, 301–304. doi: 10.3958/059.042.0130
Zouache, K., Voronin, D., Tran-Van, V., and Mavingui, P. (2009a). Composition of bacterial communities associated with natural and laboratory populations of Asobara tabida infected with Wolbachia. Appl. Environ. Microbiol. 75, 3755–3764. doi: 10.1128/AEM.02964-08
Keywords: Wolbachia , Liriomyza trifolii , CIF, cytoplasmic incompatibility factor, phage WO
Citation: Pramono AK, Hidayanti AK, Tagami Y and Ando H (2024) Bacterial community and genome analysis of cytoplasmic incompatibility-inducing Wolbachia in American serpentine leafminer, Liriomyza trifolii. Front. Microbiol. 15:1304401. doi: 10.3389/fmicb.2024.1304401
Edited by:
Didier Bouchon, University of Poitiers, FranceReviewed by:
Sarah Bordenstein, The Pennsylvania State University (PSU), United StatesMathieu Sicard, Université Montpellier 2, France
Copyright © 2024 Pramono, Hidayanti, Tagami and Ando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroki Ando, ando.hiroki.m4@f.gifu-u.ac.jp