 Qun Lan1
Qun Lan1 Yuju Lian1Peiya Peng1Long Yang1Heng Zhao2
Yuju Lian1Peiya Peng1Long Yang1Heng Zhao2 Peng Huang1
Peng Huang1 Haiming Ma1
Haiming Ma1 Hongjiang Wei2
Hongjiang Wei2 Yulong Yin1,3*
Yulong Yin1,3* Mei Liu1,4*
Mei Liu1,4*- 1College of Animal Science and Technology, Hunan Agricultural University, Changsha, China
- 2State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan Agricultural University, Kunming, China
- 3Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Hunan Provincial Engineering Research Center for Healthy Livestock and Poultry Production, Scientific Observing and Experimental Station of Animal Nutrition and Feed Science in South-Central, Ministry of Agriculture, Changsha, China
- 4Kunpeng Institute of Modern Agriculture at Foshan, Foshan, China
Finishing weight is a key economic trait in the domestic pig industry. Evidence has linked the gut microbiota and SCFAs to health and production performance in pigs. Nevertheless, for Diannan small ear (DSE) pigs, a specific pig breed in China, the potential effect of gut microbiota and SCFAs on their finishing weight remains unclear. Herein, based on the data of the 16S ribosomal RNA gene and metagenomic sequencing analysis, we found that 13 OTUs could be potential biomarkers and 19 microbial species were associated with finishing weight. Among these, carbohydrate-decomposing bacteria of the families Streptococcaceae, Lactobacillaceae, and Prevotellaceae were positively related to finishing weight, whereas the microbial taxa associated with intestinal inflammation and damage exhibited opposite effects. In addition, interactions of these microbial species were found to be linked with finishing weight for the first time. Gut microbial functional annotation analysis indicated that CAZymes, such as glucosidase and glucanase could significantly affect finishing weight, given their roles in increasing nutrient absorption efficiency. Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologies (KOs) and KEGG pathways analysis indicated that glycolysis/gluconeogenesis, phosphotransferase system (PTS), secondary bile acid biosynthesis, ABC transporters, sulfur metabolism, and one carbon pool by folate could act as key factors in regulating finishing weight. Additionally, SCFA levels, especially acetate and butyrate, had pivotal impacts on finishing weight. Finishing weight-associated species Prevotella sp. RS2, Ruminococcus sp. AF31-14BH and Lactobacillus pontis showed positive associations with butyrate concentration, and Paraprevotella xylaniphila and Bacteroides sp. OF04-15BH were positively related to acetate level. Taken together, our study provides essential knowledge for manipulating gut microbiomes to improve finishing weight. The underlying mechanisms of how gut microbiome and SCFAs modulate pigs’ finishing weight required further elucidation.
Introduction
Domesticated pigs account for a high proportion of meat production in the world and also serve as an important animal model for basic biomedical research studies (Lunney, 2007). The swine intestinal tract harbors trillions of diverse microorganisms, which get involved in regulating host nutrient digestion, energy absorption, and disease development (Zmora et al., 2019). Short-chain fatty acids (SCFAs), mainly comprised of propionate, butyrate, and acetate, are regarded as important metabolites derived from bacteria-dependent hydrolysis of indigestible dietary fibers, which play crucial roles in energy metabolism modulation and improving intestinal homeostasis (Priyadarshini et al., 2018).
Recently, emerging evidence suggests that SCFAs and gut microbiomes exert positive effects in regulating the growth performances of livestock animals. For instance, the increased abundance of bacteria belonging to families Ruminococcaceae and Lachnospiraceae could generate a high concentration of butyrate, which is responsible for the higher feed intake and body weight of broilers (Yacoubi et al., 2018). The genus Prevotella, genus Faecalibacterium, and family Ruminococcaceae are participated in digesting dietary fibers which subsequently influence SCFA production, which played vital roles in modulating body weight and feed intake of pigs (Wang J. et al., 2020). Moreover, research pointed out that the significant improvement in feed intake and average daily gain (ADG) of fattening cattle were closely related to levels of acetic acid and propionic acid, and their producing-bacteria Acetobacterium and Fournierella, respectively (Zhuang et al., 2021). Additionally, the increased abundance of Bifidobacterium and Lactobacillus in the gut of broiler chickens has contributed to the generation of lactate and SCFAs, which possess positive effects on ADG and food conservation rate (Yu et al., 2019).
Finishing weight is considered to be a critical growth trait of pigs, which directly reflects meat production. Nevertheless, only few works have unraveled the potential relationship between the intestinal microbiome and the finishing weight of pigs. For example, research indicated that the abundance of Parabacteroides, Bacteroides, and ClostridiumXI showed positive associations with the final body weight of pigs (Yang et al., 2021b). The body weight of Enshi pigs at the finishing stage was positively correlated with Ruminococcaceae UCG-005 and Alloprevotella (Tang et al., 2020). In addition, genera Prevotella, unclassified Bacteroidales, and unclassified Veillonellaceae in multi-intestinal segments of pigs were positively associated with finishing weight (De Rodas et al., 2018). In contrast, genera Stenotrophomonas, Sphaerochaeta, and Desulfovibrio in the gut microbial communities of pigs were negatively associated with finishing weight (Tang et al., 2020). Another study reported that genera Parabacteroides, Desulfovibrio, and Treponema had negative correlations with the body weight of pigs at the finisher stage (Oh et al., 2020). Moreover, the final body weight of the three-way hybrid pig was negatively correlated with the genera Roseburia, Prevotella, and Anaerovibrio (Yang et al., 2021b). However, these above-mentioned studies are mainly based on 16S rRNA gene sequencing analysis, which cannot identify the specific species and functional capacities that affect finishing weight. As a specific pig breed of China, the Diannan small-ear (DSE) pigs are primarily raised in the southern region of Yunnan province with a subtropical climate, and are characterized by more fat deposition, lower growth rate, better meat quality, and strong resistance to adverse surroundings (Wang et al., 2015). The potential characters of the gut microbiome and SCFAs in regulating the finishing weight of DSE pigs remain largely unknown. Hence, we devised the present study aimed to investigate the effects of gut microbiome and SCFAs on body weight of DSE pigs in finishing phase using data of 16S rRNA gene sequencing, metagenomic sequencing, and fecal SCFAs. We finally identified some microbial species and metabolic functions related to finishing weight. Also, the correlations between SCFAs concentration and finishing weight were unraveled. Our study helps to uncover the effects of gut microbiome and SCFAs on modulating finishing weight and hence provide certain reference significances for improving production performance of DSE pigs and others.
Materials and methods
Experimental pigs and sample collection
One hundred and two Diannan small-ear (DSE) pigs (40 ± 3 days, 48 females and 54 males) with similar weaning weights were grouped into different commercial pens and bred under the same nutritional and management conditions until the finishing stage (345 ± 3 days). At the finishing stage, all DSE pigs were fed with same diet two times 1 day using the compound feeds containing 14–16% of crude protein, 0.25–0.60% of sodium chloride, 0.60–1.50% of calcium, 8% of coarse fiber, 3,100–3,200 kJ of digestible energy, and 0.70% of lysine, and offered an ad libitum water. The finishing body weight of all DSE pigs was measured and recorded. Then, finishing weights were sorted to select the bottom six pigs with the lowest body weight (low group) and the top six pigs with the highest body weight (high group) for both 16S rRNA gene sequencing and metagenomic sequencing. The normal distribution of finishing weight for all DSE pigs was displayed in Supplementary Figure S1A. There exists a significant difference in finishing weight among low and high groups (Supplementary Figure S1B, FDR adjusted p < 0.01). All DSE pigs were healthy and not fed with probiotics, prebiotics, antibiotics, and anti-coccidial drugs throughout the whole experimental period. The fresh feces of the DSE population were collected directly from the rectum. All samples were stored in the 2-ml sterilized plastic sampling tubes and then dipped into liquid nitrogen immediately. After long-distance transportation, samples were stored in a refrigerator (−80°C) until processing.
DNA extraction and 16S rRNA gene sequencing
The QIAamp fast DNA stool Mini Kit (QIAGEN, Germany) was selected to extract microbial DNA based on the manufacturer’s guidelines. The concentration and quality of stool DNA were assessed by spectrophotometer Nanodrop 2000 (Thermo Scientific, United States) and 1.5% agarose gel electrophoresis, respectively. The specific primer 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) was used to amply the hypervariable regions (V3–V4) for 16S rRNA gene. The PCR amplification was accomplished by following steps: initial 94°C denaturation step for 5 min, subsequently, 30 cycles of 95°C for 30 s, 55°C for 25 s, and 72°C for 25 s followed by a final extension step for 10 min at 72°C. All amplicons were sequenced using the paired-end method on a MiSeq platform (Illumina, United States) following the standard protocols. The quality control process of raw data was performed by using QIIME (v.1.9.1), including removing primers, barcodes, and low-quality sequences (Caporaso et al., 2010). FLASH (v.1.2.11) was used for merging high-quality paired-end reads into tags (Magoc and Salzberg, 2011). Before further analysis, the library size of sequences was rarefied to 40,000 tags for each sample to normalize the sequencing depth. USEARCH (v.11.0) was used for matching tags into operational taxonomic units (OTUs) based on 97% sequence identity (Edgar, 2010). We filtered out those OTUs as following criteria: relative abundance was less than 0.05% and OTUs were presented in 1% of the experimental pigs. OTU taxonomic category assignments were finished by using the SILVA database (v.138.1; Quast et al., 2013). The MOTHUR (v.1.28.1; Chappidi et al., 2019) and QIIME were used for calculating the alpha and beta diversity indices, respectively.
Biomarker discovery in 16S rRNA data
To identify the biomarker characteristics among low and high finishing weight groups, linear discriminant analysis effect size (LEfSe; Segata et al., 2011) was performed based on a normalized OTU relative abundance matrix. As a tool for high-dimensional biomarker identification and explanation, the LEfSe algorithm uses the non-parametric factoria Kruskal-Wallis test to identify the features with significant differences and performs LDA to estimate the effect size of each feature. The LDA threshold score and significant level in the current study were set at 2 and 0.05, respectively.
Metagenomic sequencing
Following the instruction of the manufacturer, a paired-end (PE) DNA library was established for each sample, and sequencing was implemented on the Illumina HiSeq 4000 platform. Quality control, adapter trimming, and low-quality read removal of sequenced data were proceeded by software fastp (v.0.23.2; Chen et al., 2018) to achieve clean data, and an average of 59,008,155 high-quality reads was obtained for each sample. The MEGAHIT (v.1.2.9; Li et al., 2015) was then used for assembling high quality reads into contigs, and the length of contigs with more than 200 bp was used to perform open reading frames (ORF) prediction using MetaGeneMark (v.3.25; Zhu et al., 2010). We used Cd-hit (v.4.6.8; Fu et al., 2012) to discard the redundant genes derived from the predicted ORFs for constructing the non-redundant gene catalog. The gene abundance content was obtained by mapping the high-quality reads against the non-redundant gene catalog via MOCAT (v2.0; Kultima et al., 2016). The program of hmmscan in HMMER (v.3.0; Eddy, 2011) was used to annotate Carbohydrate-Active enZYmes (CAZymes). Profiles of KEGG pathway annotation information and KEGG Orthologies (KOs) were extracted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using GhostKOALA (Kanehisa et al., 2016).
Determinations of SCFA in fecal samples
The measurement of SCFAs concentrations in stool samples was using gas chromatography on a Shimadzu GC-2010 plus system with a flame ionization detector (Shimadzu, Japan) according to the approaches previously described with small modifications (Xu et al., 2021). A total of 1,000 mg fresh feces sample was vortex-mixed with 5 ml ultrapure water and centrifuged (1,000 rpm) at 4°C for 30 min. Subsequently, the supernatant (900 μl) which was mixed with 100 μl metaphosphoric acid solution (25%; Macklin, United States) for 3 or 4 h was used for filtering by a 0.45 μm filter membrane. Aliquots of the supernatants (600 μl) were pipetted into a glass gas chromatography vial to make preparations for the following GC instrument analysis. Briefly, the temperature of the flame ionization detector was set at 280°C. The carrier gas was highly purified N2 (99.99%) with a flow rate of 0.8 ml/min. The concentration of SCFAs in fecal samples was determined by comparing them with the standard curve. We used Mantel tests based on the Spearman method to perform the correlation analysis between SCFAs levels and finishing weight-associated species.
Interaction network analysis
To construct the interaction network of all finishing weight associated species, the pair-wised Spearman correlations analysis was performed between these species. Spearman’s correlation coefficient of less than 0.65 and adjust p value of more than 0.05 were excluded, which allows us to only focus on those species that strongly co-occurred and were more likely to affect each other. The major modules in the interaction network were calculated and visualized using Gephi software (v.0.9.2; https://gephi.org/). After that, Spearman’s correlation analysis was used to test for the correlations between finishing weight phenotypic value and modules.
Basic statistical analysis
The differential metagenomic species and function capacities for the gut microbiome between low and high finishing pigs were detected by the Wilcoxon test with FDR correction. Statistical comparisons of SCFAs in stool samples were done using the two-sided Student’s t-test (unpaired) with FDR correction between low and high finishing weight pigs. Interactive Tree Of Life (iTOL) was used to visualize the differential metagenomic species, and the other charts were finished by R software.
Results
Comparison of bacterial diversity and microbial composition
The bacterial diversity (PD and Shannon) and richness (Chao) indices for alpha-diversity were compared to discover the difference for the gut microbial community. As shown in Figure 1A, the PD index in high finishing weight pigs was significantly decreased in comparison with low finishing weight pigs (FDR adjusted p < 0.01). In addition, the same tendency was also observed in the Chao index (FDR adjusted p < 0.05; Figure 1B). The Shannon index between the two groups did not exhibit a statistical difference while the value in the high finishing weight group was lower than the low finishing weight group (Supplementary Table S1).

Figure 1. Alpha, beta-diversity comparison, and microbial composition among low and high finishing weight groups. (A) Comparison of PD index for two groups. (B) Comparison of Chao index between two groups. (C) Principal Coordinate Analysis (PCoA) analysis for gut microbial community structures based on Unweighted Unifrac distance. (D) Unweighted Unifrac distance metric. (E,F) The gut microbial composition for low and high finishing weight DSE pigs in phylum (E) and genus (F) levels. The box means the interquartile range, and the box with the midline represented the median. “L” and “H” represent low and high finishing weight, respectively; “*” FDR adjusted p < 0.05, “**” FDR adjusted p < 0.01.
Principal Coordinate Analysis (PCoA) was performed to observe the change of gut microbial community structures between two experimental populations. The distribution of microbiota between the two groups was clustered separately along principal coordinates (Figure 1C). The analysis of unweighted UniFrac distance metric comparison indicated that low finishing weight pigs had higher dissimilarity among intestinal microbiota than high finishing weight pigs (Figure 1D, FDR adjusted p < 0.01).
For gut microbial composition, Firmicutes, Bacteroidetes, and Proteobacteria were the top three dominant phyla (Figure 1E) and represented more than 95.10% of all sequence reads. At the genus level, Streptococcus, Lachnospiraceae, Oscillospiraceae_UCG-002, and Lachnospiraceae_XPB1014_group were the first four predominant genera (Figure 1F). Approximately 0.03% of sequences in fecal samples cannot be assigned to a certain rank (unclassified; Supplementary Table S2).
Differential microbiota compositions in low and high finishing weight groups
The fecal microbiota from low and high finishing weight individuals was analyzed by LEfSe multilevel species discrimination. A total of 13 OTUs were identified (Figure 2). Nine OTUs were specifically found in low finishing weight group, namely, genera Piscicoccus, Corynebacterium, Dietzia, dgA_11_gut_group, Acidaminococcus, Dialister, families Rikenellaceae and Desulfovibrionaceae, and class Bacteroidales. Whereas for the high finishing weight group, three genera Streptococcus, Prevotellaceae UCG-003, Frisingicoccus, and one family Bacteroidales_RF16_group could be the potential biomarkers.
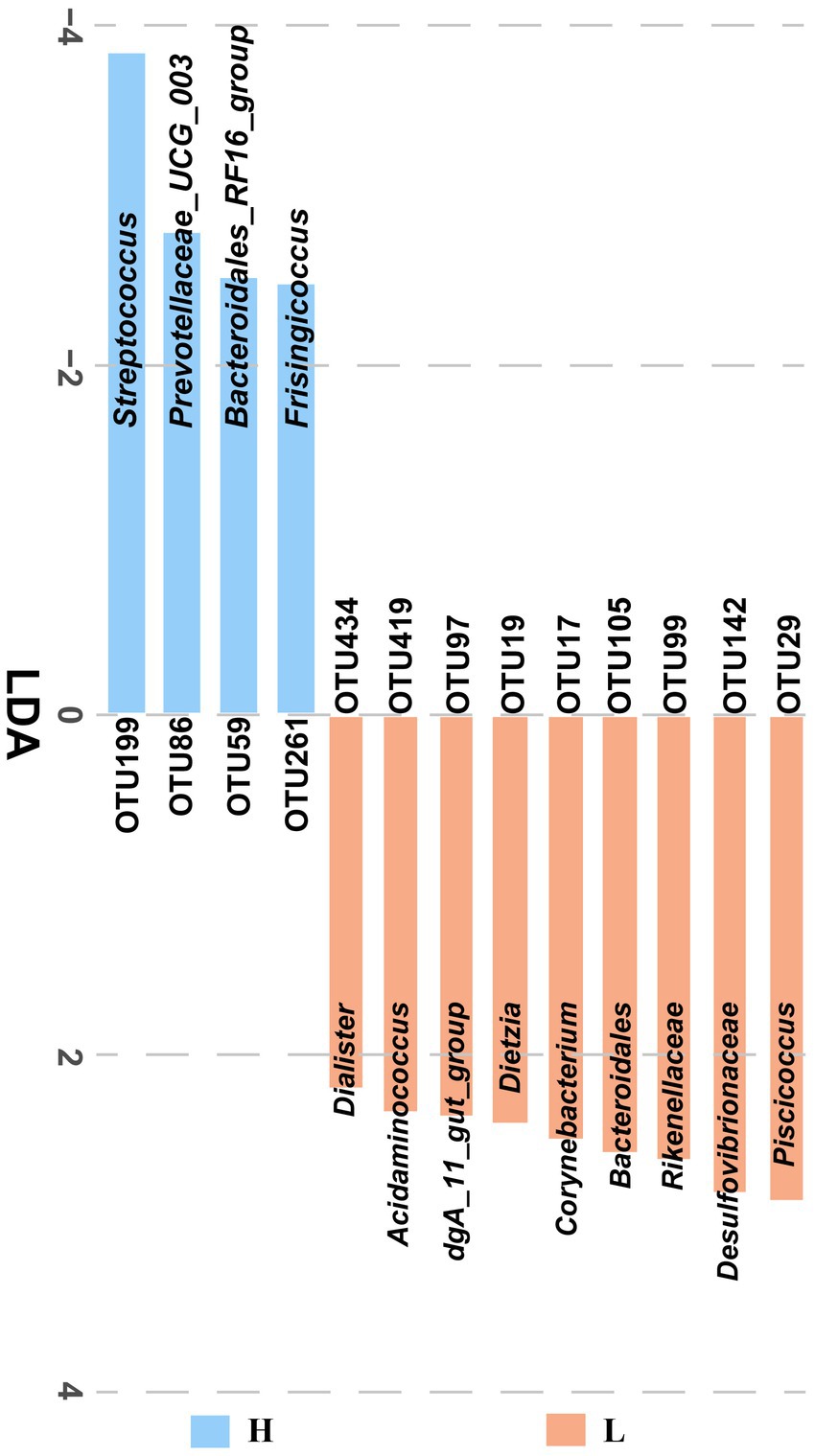
Figure 2. The identified significantly different OTUs in low and high finishing weight pigs. The “H” under the blue bar represents the high finishing weight group, and the “L” under the orange bar represents the low finishing weight group.
Microbial species associated with finishing weight
At the species level, 16S rRNA gene sequencing possesses large limitations in both accuracy and resolution. Herein, we performed metagenomic sequencing for 12 individuals to further identify the differential species between low and high finishing weight groups. A total of 19 species showed different enrichments among two groups identified by the Wilcoxon rank-sum test with FDR correction using metagenomic sequencing data (Figure 3 and Supplementary Table S3, FDR adjusted p < 0.05). For low finishing weight group, Desulfovibrio piger and Desulfovibrio sp. An276 from the family Desulfovibrionaceae were the most abundant species. In addition, three species belong to the family Clostridiaceae, including Clostridium perfringens, Clostridium sp. CAG:349, and Clostridium paraputrificum were abundant in pigs with low finishing weight. Three species from the member of family Corynebacteriaceae, named Corynebacterium freneyi, Corynebacterium halotolerans, and Corynebacterium humireducens were also enriched in the low finishing weight group. Conversely, we found that Streptococcus equinus, Streptococcus salivarius, and Streptococcus lutetiensis (members of the family Streptococcaceae) were augmented in high finishing weight group, followed by Lactobacillus reuteri, Lactobacillus pontis, Lactobacillus plantarum, and Lactobacillus equigenerosi. Moreover, Prevotella sp. RS2, Ruminococcus sp. AF31-14BH, Paraprevotella xylaniphila, and Bacteroides sp. OF04-15BH were also abundant in the high finishing weight group.
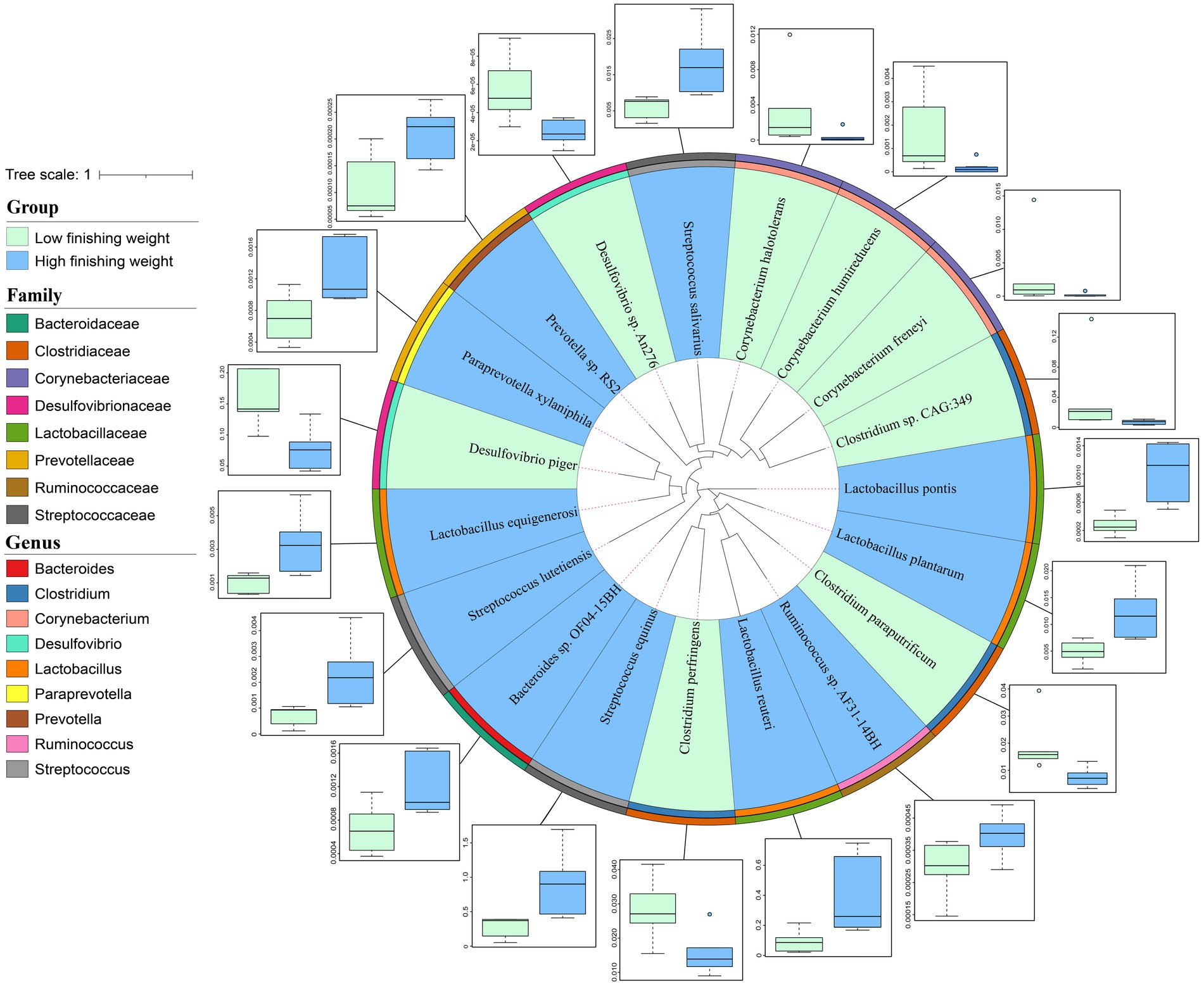
Figure 3. The phylogenetic relationships and abundance comparison of metagenomic species in low and high finishing weight pigs. The phylogenetic tree of species is displayed as the innermost layers. Labels with green and blue colors represent low and high finishing weight groups, respectively. The different color strips in the third and four layers, respectively, correspond to different genera and families as indicated by the color codes on the left. The outermost layer was the boxplot representing the abundance comparison of finishing weight associated species. The dots in the bar chart represent species abundance.
Interactions of microbial species associated with finishing weight
To determine whether these significantly enriched species play a vital role in affecting the finishing weight by their interactions, they were used to estimate the correlation coefficient using the Spearman method. The interaction network consisted of two modules (Figure 4A). We found that dominant species from family Desulfovibrionaceae, Corynebacteriaceae, and Clostridiaceae were clustered into Module 1 through intra-positive interactions, whereas Module 2 was constituted by intra-positive interactions among the species mainly from families Streptococcaceae, Lactobacillaceae, and Prevotellaceae, which were positively related to finishing weight. In addition, as shown in Figure 4B, we found that Module 1 was positively associated with finishing weight, but Module 2 was negatively correlated with finishing weight.
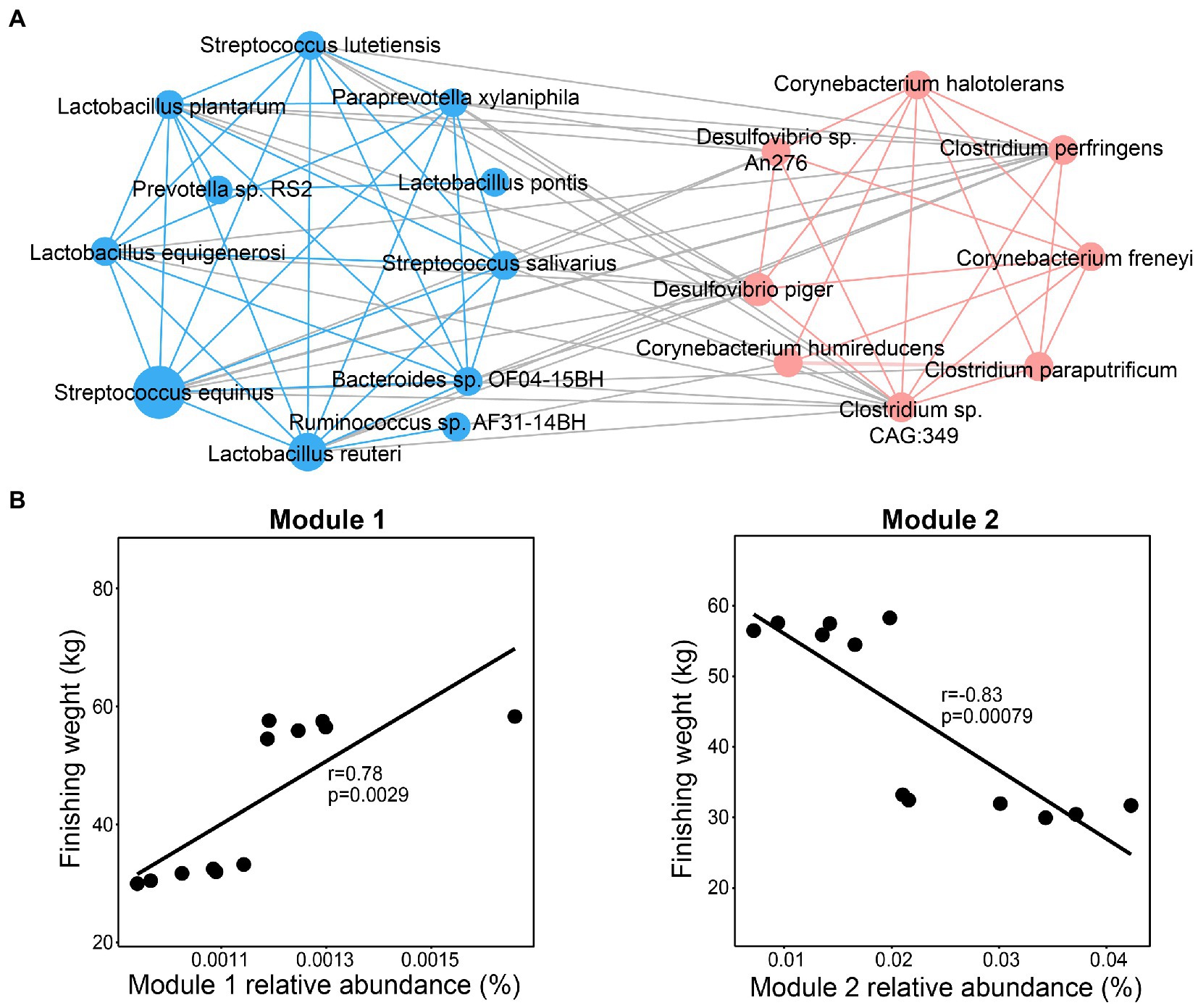
Figure 4. Interactions among the species responding for finishing weight. (A) The interaction network among finishing weight-related species. The blue nodes and lines, respectively, denote positively associated species and positive correlations among them. The coral nodes and lines, respectively, represent negatively associated species and positive correlations among them. Lines with gray color indicate a negative correlation. The node size was drawn according to the average relative abundance of different species. Each line linked to nodes represents significant correlations (FDR adjusted p < 0.05, |r| > 0.65). (B) The associations of the abundance of Module 1 and Module 2 with finishing weight.
Functional compositions of the gut microbiome related to finishing weight
Functionalities of the gut microbiome associated with finishing weight were analyzed by comparing the abundances of KEGG items and CAZymes among low and high finishing individuals.
Twenty-one CAZymes with significantly different abundances were detected (Figure 5A, FDR-adjusted p < 0.05). Among these, 11 CAZymes especially glucosidase and glucanase (e.g., GH1, GH128, and GH6), lytic cellulose monooxygenase (e.g., AA9 and AA10), and Glucosyltransferase (e.g., GT113 and GT26) were significantly enriched in high finishing weight group. While regarding low finishing weight pigs, we found that mannosyltransferase (e.g., GT39 and GT62), peptidoglycan lytic transglycosylase, and levansucrase had significantly higher abundances.
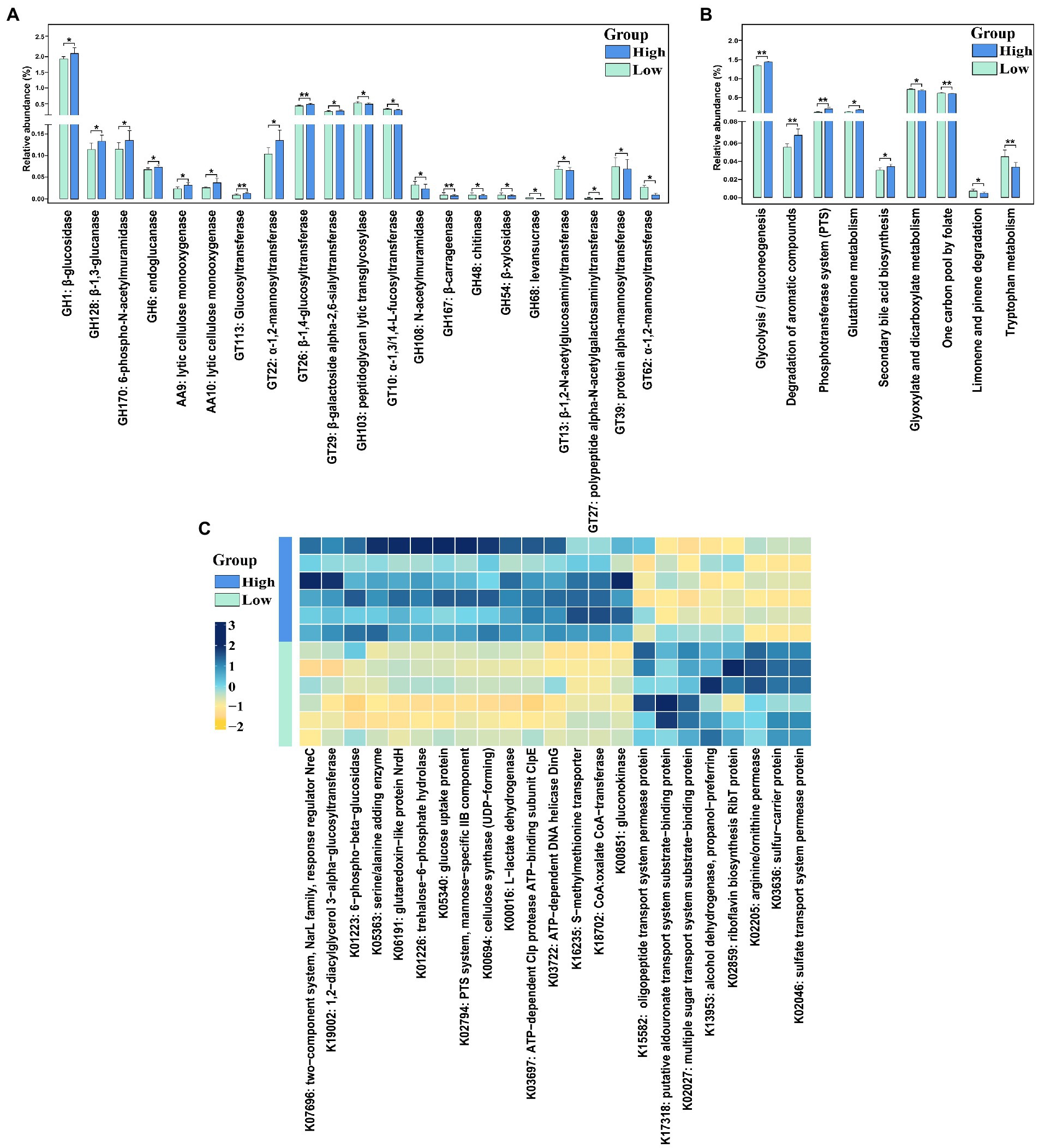
Figure 5. The CAzymes and KEGG function terms exhibiting different enrichments between low and high finishing weight pigs. (A) The differential CAzymes are detected to relate with finishing weight. (B) The differential KEGG pathways are identified to associate with finishing weight. (C) Heat map of KOs displaying different enrichments between low and high finishing weight pigs. “*” and “**” represents for FDR adjusted p < 0.05 and p < 0.01, respectively. The rectangles with green and blue colors represent low and high finishing weight groups, respectively.
A total of nine differential enriched KEGG pathways were detected in the gut microbiome of pigs with distinct finishing weights (Figure 5B, FDR-adjusted p < 0.05). We found that one carbon pool by folate, limonene, and pinene degradation, and tryptophan metabolism were more active in the gut microbial communities of the low finishing weight group. Meanwhile, in pigs with high finishing weight, gut microbiota was more capable of operating glycolysis/gluconeogenesis, degradation of aromatic compounds, phosphotransferase system (PTS), glutathione metabolism, and secondary bile acid biosynthesis.
On the other hand, 23 KOs showed distinct abundances in pigs with low and high finishing weights (Figure 5C, FDR-adjusted p < 0.05). Eight KOs mainly related to ABC transporters (e.g., K15582 and K17318), Fatty acid degradation (e.g., K13953), and Sulfur metabolism (e.g., K02046) were plentiful in low finishing weight group. The other 15 KOs were more abundant in high finishing weight group, most of which were associated with glycolysis/gluconeogenesis (e.g., K00016 and K01223), carbohydrate metabolism (e.g., K01226, K02794, and K00694), lipid metabolism:(e.g., K19002), and the biosynthesis of secondary metabolites (e.g., K00851).
Changes in fecal SCFAs linked to finishing weight
Owing to the SCFAs produced by intestinal microbiota playing vital roles in regulating intestinal health status and host energy metabolism, we detected the concentrations of butyric acid, propionic acid, and acetic acid in the feces. Our results showed that both the concentrations of acetate and butyrate for the high finishing weight group were significantly greater than the low finishing weight group (FDR-adjusted p < 0.05 and p < 0.01). No significant change was found in the levels of propionic acid between the two groups (Figure 6A). By further correlation analysis, we observed that the levels of acetic acid and butyric acid displayed a positive association with finishing weight, but the levels of propionic acid only exhibited a tendency of positive association with finishing weight (Figure 6B).
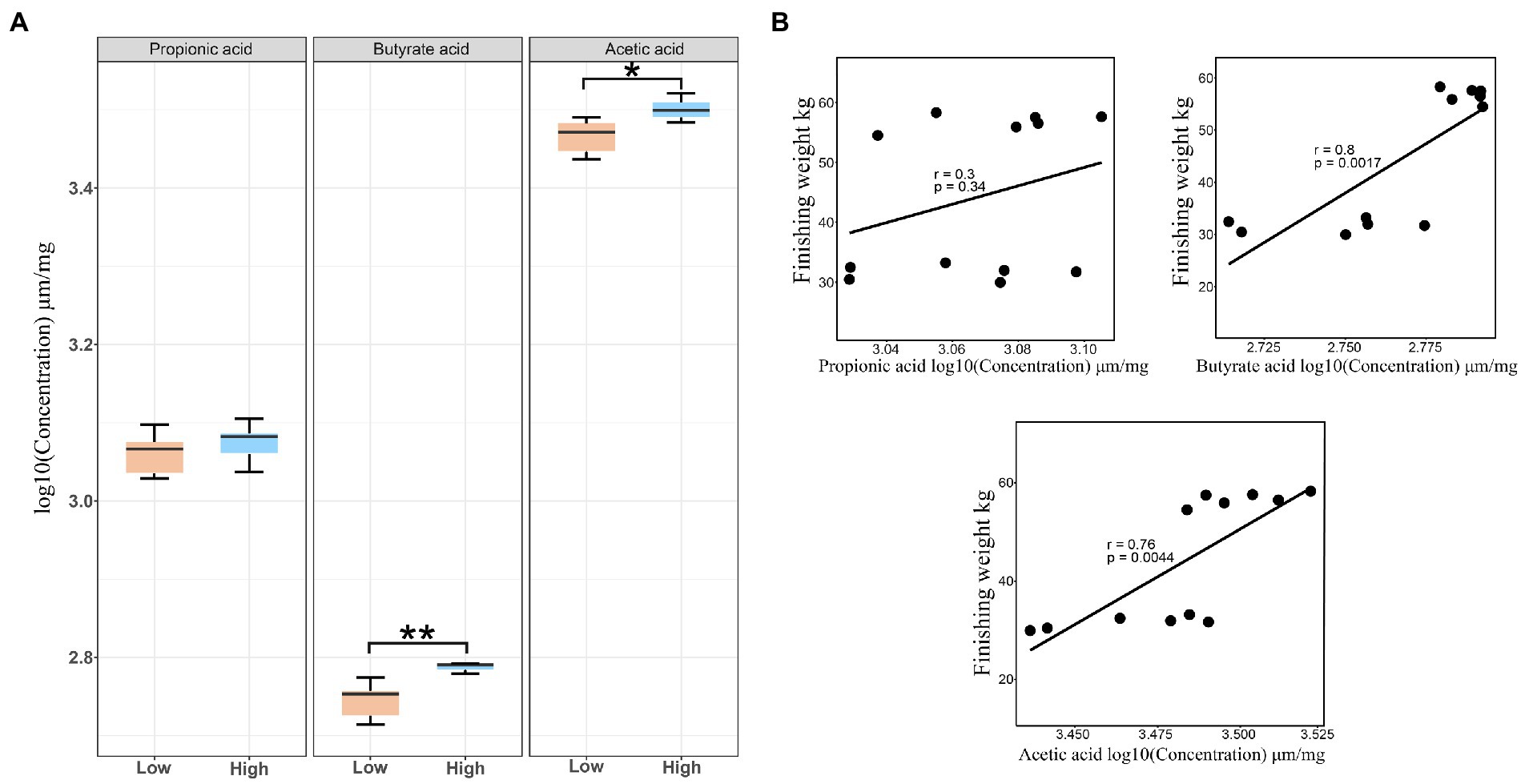
Figure 6. Stool short-chain fatty acid concentrations in low and high finishing weight pigs and their correlations with finishing weight. (A) The comparisons of fecal short-chain fatty acid levels between low and high finishing weight pigs. (B) The correlations between stool short-chain fatty acid levels and finishing weight. “*” and “**” represents for FDR adjusted p < 0.05 and p < 0.01, respectively.
Finishing weight associated species-SCFAs correlation
Mantel tests were performed to analyze the correlations between SCFAs level and finishing weight-associated species. As exhibited in Figure 7, we found that three species had positive relationships with the level of butyric acid, they were Prevotella sp. RS2, Ruminococcus sp. AF31-14BH, and Lactobacillus pontis, the species Desulfovibrio sp. An276, Desulfovibrio piger, and Corynebacterium humireducens showed negative correlations. Moreover, the level of acetic acid was positively related to Paraprevotella xylaniphila and Bacteroides sp. OF04.15BH, respectively. Nevertheless, no significant correlations were found between the level of propionic acid and finishing weight-associated species.
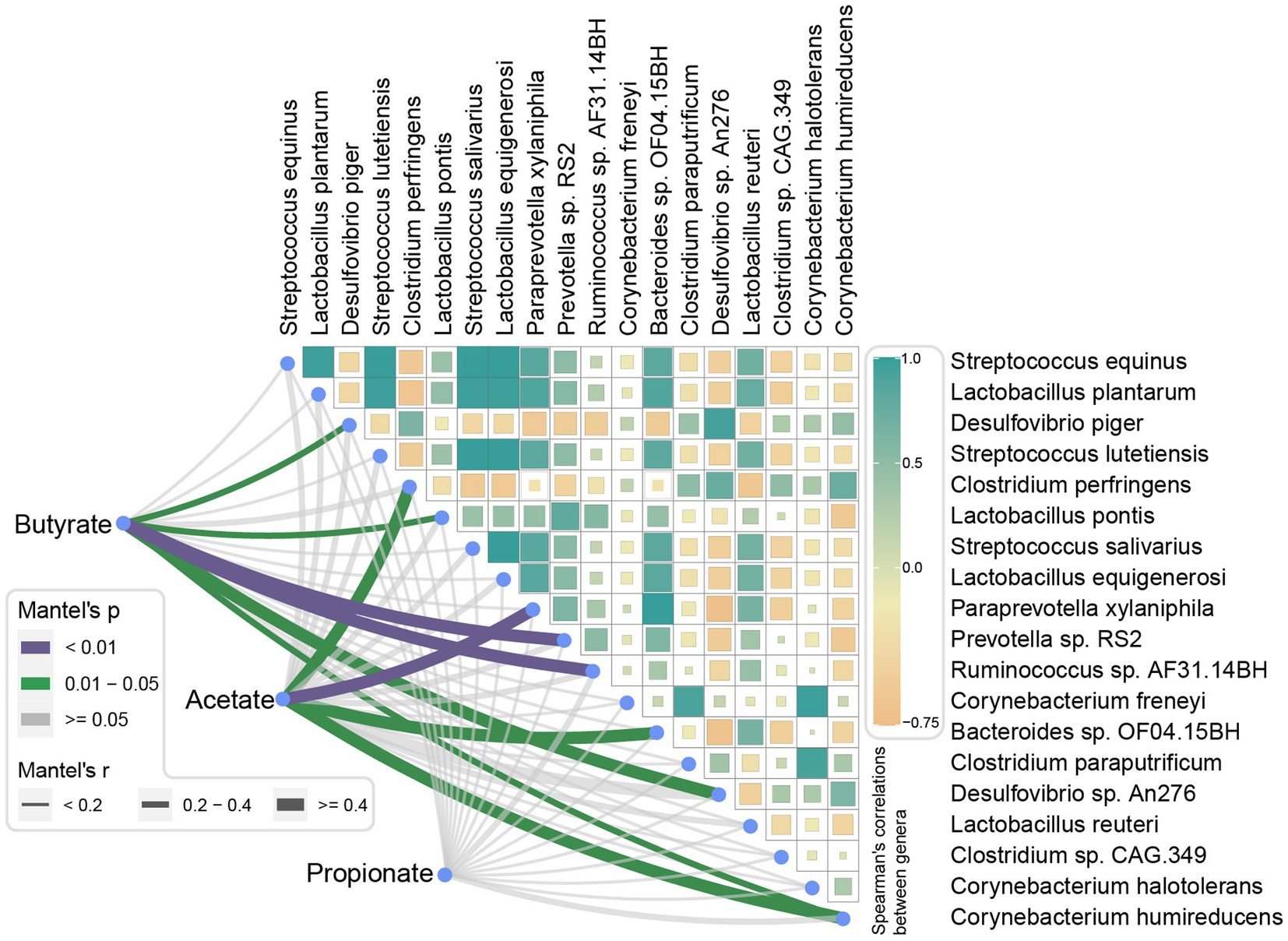
Figure 7. Correlations between short-chain fatty acid levels and finishing weight-associated species. Pairwise comparisons of finishing weight related-species are shown with a color gradient denoting Spearman’s correlation coefficient. Butyrate, acetate, and propionate were related to each microbial species by partial Spearman tests. Edge width corresponds to the Partial Spearman’s r statistic for the corresponding distance correlations, and edge color represents the statistical significance.
Discussion
The pivotal roles of the gut microbiome in domestic animals have been continuously demonstrated, especially in aspects of nutrient metabolism, energy harvesting, and health maintenance (O'Callaghan et al., 2016). Accordingly, the gut microbiome is regarded as an important player in affecting the growth of livestock animals. Hence, identifying core gut microbiota is an important cut-in point for improving livestock production performances (Maltecca et al., 2020). SCFAs are essential metabolites generated by large intestinal anaerobic bacteria by fermenting the complex carbohydrates, which have emerged as an important modulator in gut health status and energy metabolism regulation (Koh et al., 2016). However, the effects of gut microbiota and SCFAs on the finishing weight of DSE pigs have received little attention. We, here, systematically and exhaustively assessed how the gut microbiome and SCFAs affect the finishing weight of DSE pigs.
A previous study indicated that overweight pigs have markedly lower species richness in alpha diversity and high dissimilarities in beta diversity (Panasevich et al., 2018). Another study also found that the higher body weight of Wuzhishan minipigs exhibited lower alpha diversity (Xu et al., 2022). We here confirmed these previously reported outcomes demonstrating that pigs with high body weight have lower gut microbial diversity and richness. In addition, in consistent with previous studies on the gut microbiota of swine (Long et al., 2021; Shang et al., 2022), we found that the gut microbiota of DSE pigs in both phylum and genus levels were dominated by Firmicutes and Bacteroides, which have a better capacity in producing various enzymes that are responsible for carbohydrate degradation and fermentation (Stojanov et al., 2020).
In total, 13 OTUs were specifically found in low and high finishing weight groups. Among these, four OTUs were detected in the high finishing weight group. Prevotellaceae UCG-003 is a member of acetate-producing bacteria capable of degrading structural carbohydrates (cellulose and hemicellulose) in feeds and possesses beneficial roles in intestinal health by regulating the immune-inflammatory system (Li et al., 2019; Duan et al., 2021). Regarding the genus Streptococcus, which was subordinate to the order of Lactobacillales and the dominant lactate-producing genus in different segments of the intestine and also the main utilizers of simple carbohydrates (Chen et al., 2016). In contrast, Desulfovibrionaceae and Corynebacterium were identified in the low finishing weight group. Studies have shown that a high abundance of Desulfovibrio and Corynebacterium were detected in diarrhea-related cases (Zhu et al., 2018; Jo et al., 2021; Wang et al., 2021), suggesting that they might be the pathogenic bacteria that exert harmful effects on DSE pig’s growth and development.
Using metagenomic sequencing technology, we obtain more information about bacterial strains associated with finishing weight. A particularly notable finding is the larger proportion of Streptococcus strains in high finishing weight pigs, including S. equinus, S. salivarius, and S. lutetiensis. As the lactate-producing bacteria, Bifidobacterium and Lactobacillus are among the better described probiotics, the effect of Streptococci on the host intestine is less studied. A recent study demonstrated that S. salivarius exhibited an anti-inflammatory effect when co-incubated with gut epithelial cells (Li et al., 2022). Researchers found a remarkable increase of S. lutetiensis in the intestine after supplementing fructan (Vanhoutte et al., 2005). Furthermore, β-glucanases, α-galactosidase, and endoglucanases of S. equinus play a special role in degrading complex carbohydrates (Jans and Boleij, 2018). Commonly, Streptococcus mainly colonizes the surfaces of the intestinal mucosa. Studies provided supportive evidence that this genus could actively regulate peroxisome proliferator-activated receptors (PPARs; Are et al., 2008; Couvigny et al., 2015). These receptors, which were subordinate to the nuclear receptor superfamily of regulatory factors, are a class of ligand-activated transcription factors (Mirza et al., 2019). Their primary responsibility in the gut mucosa is to modulate genes related to lipid metabolism (Tenenbaum et al., 2003). They stimulate cell differentiation, especially as an essential inducer of adipocyte differentiation and growth (Brun et al., 1997; Lefterova and Lazar, 2009). Importantly, in human intestinal epithelial cells, S. salivarius has been demonstrated that significantly decreased the PPARγ-targeted metabolic genes (e.g., I-FABP and Angptl4), which were involved mainly in glucose metabolism and lipid accumulation (Couvigny et al., 2015). Consequently, down-regulating the level of PPARγ might lead to the effect of metabolic complications such as rapid weight gain. Here, we observe that the relative abundance of Streptococcus strains increased dramatically in the high finishing weight individuals. We hypothesize that the greater abundance of Streptococcus could fundamentally promote the enhancement of finishing weight by modulating energy harvesting and intake.
Furthermore, some species from Lactobacillus spp. could significantly affect finishing weight, such as L. reuteri, L. plantarum, L. equigenerosi, and L. pontis. In recent years, researchers unraveled the beneficial roles of Lactobacillus spp. on the growth performance of pigs. Supplementing extra L. plantarum in the pig diet could significantly increase both ADG and body weight (Yang et al., 2014). Another study has shown a significant elevation in feed conversion and ADG by dietary supplementation of L. reuteri (Hou et al., 2015). Additionally, the compound strains of L. reuteri and L. plantarum could decrease pig diarrhea rate (Dell'Anno et al., 2021). More importantly, the study pointed out that Lactobacillus spp. colonized the intestinal tract could elevate porcine growth hormone levels, antioxidation, and immunity, ultimately helping to fight stress (Yang et al., 2020). These investigations help explain why Lactobacillus spp. significantly enriched in high finishing weight pigs. We, therefore, speculated that L. equigenerosi and L. pontis may also serve as beneficial strains which play positive roles in pig growth performance.
We also observed that Paraprevotella xylaniphila was positively associated with finishing weight. A positive association between P. xylaniphila and obesity indices (body mass index, hip circumference, and waist circumference) for children who consumed high carbohydrate and saturated fat diets was found in a recent investigation (Orbe-Orihuela et al., 2022). Moreover, previous studies indicated that P. xylaniphila was a main succinate producing bacterium, it was positively associated with insulin, BMI, glucose, and triglycerides (Morotomi et al., 2009; Serena et al., 2018). On the contrary, certain species from Clostridium spp. and Desulfovibrio spp. showed negative correlations with finishing weight. Clostridium perfringens is a popular opportunistic pathogen inhabiting pig intestines (Zhou et al., 2021). The classical symptom of C. perfringens in pigs includes severe diarrhea, accompanied by intestinal villi damage and necrotic mucosa (Kiu and Hall, 2018). Desulfovibrio piger, which was a sulfate-reducing bacterium mainly degrades both sulfates and sulfites from the diet and generates hydrogen sulfide (H2S) as a product (Gibson, 1990). H2S can cause damage to the intestinal epithelium, triggering systemic and chronic inflammation (Attene-Ramos et al., 2007). More importantly, a newly published study indicated that Desulfovibrio piger was positively related to sarcopenia, which was typically characterized by weight loss (Wang et al., 2022). In short, these species augmenting the gut microbiome of DSE pigs could be the potential precipitating factors for the reduction of finishing weight.
The interaction network exhibited the co-exclusion and co-occurrence relationships among finishing weight-related species. Their interactions which could affect the finishing weight have a closer relationship with their diverse functions in host energy metabolism and intestinal health modulation. Similarly, earlier research indicated that the co-occurrence network embraced core genera including Streptococcus, Lactobacillus, Bacteroides, and Prevotellaceae were positively associated with porcine daily weight gains and body weight (Yang et al., 2021a). Moreover, the porcine body weight gain might be affected by the strong co-occurrence relationship between lactic acid producers (e.g., Lactobacillus) and butyrate-producer (e.g., Ruminococcaceae) through regulating host appetite and feeding behavior (Yang et al., 2018). Researchers also emphasized the essential role of gut microbial interactions in improving the growth and development of broiler chickens (Ma et al., 2018).
Compositions in metagenomic functional capacities were different between low and high finishing weight pigs. Both KOs and KEGG comparison analysis demonstrated that the glycolysis/gluconeogenesis, PTS, lipid metabolism, and secondary bile acid biosynthesis were more active in high finishing weight pigs, while the pathways of ABC transporters, sulfur metabolism, and one carbon pool by folate were overrepresented in low finishing weight group. The glycolysis/gluconeogenesis and PTS pathway were for the majority associated with being overweight (Graessler et al., 2013; Del Chierico et al., 2018). PTS exists only in bacteria where it gets involved in catalyzing the transportation and phosphorylation of carbohydrates (e.g., polysaccharides, monosaccharides, polyols, and amino sugars; Kashani et al., 2019). An earlier study indicated that both Lactobacillus and Streptococcus could ferment carbohydrates into lactate by their highly expressed PTS and carbohydrate metabolic genes (Shen et al., 2020). These two genera were overrepresented in high finishing weight pigs. This might be the main reason why these two pathways were active. Furthermore, the second bile acid (BA) biosynthesis, which is important for lipid and glucose metabolism regulation in the intestine (Zeng et al., 2019), was also active. Under the function of bile salt hydrolase (BSH), primary BA can undergo biotransformation to the secondary BA in the colon (Chiang, 2009). Intriguingly, previous studies have found that both Lactobacillus and Streptococcus were bile salt hydrolase (BSH) bacteria (Degirolamo et al., 2014), and Streptococcus was associated with lipid and bile acid homeostasis in the gut (Ren et al., 2017). It is possible that a high proportion of these two genera might contribute to increased concentrations of secondary BA and subsequently enhanced intestinal energy harvest and lipid metabolism to affect finishing weight. On the other hand, ABC transports are responsible for transporting various lipids, sterols, and drugs (Hou et al., 2017) and have recently been associated with multi-drug resistance and colonic inflammation (Andersen et al., 2015; Fang et al., 2017), which may be detrimental to growth performances in animals. Sulfur metabolism activated by sulfate-reducing bacteria (e.g., Desulfovibrio) has been demonstrated to be associated with inflammatory bowel diseases in mice (Metwaly et al., 2020). In addition, the biosynthesis pathway of folate seems to have a positive role in weight gain (Del Chierico et al., 2018). Recently, the research highlighted that one carbon pool by folate was enriched in Saba pigs with low body weight (Gao et al., 2020), which was in line with our study.
The CAZyme comparison analysis reflected that glucanase and glucosidase were more active in pigs with high finishing weights. Previous research has shown that glucanase supplementation could elevate the ratio of villi height to crypt depth, improve the absorption of nutrients, and finally promote body weight gain (Karunaratne et al., 2021). Furthermore, a greater abundance of glucosidase in the intestinal microbiome resulted in increased body weight gain in broilers (De Cesare et al., 2017). This could be used to explain why DSE pigs with a higher concentration of these two enzymes displayed greater finishing weight.
Our results also showed that both the levels of butyric and acetic acid were significantly increased in pigs with high finishing weights compared to pigs with low finishing weights, and these two SCFAs showed positive correlations with finishing weight. Butyrate possesses wide energy and metabolic modulation capacities and has strong anti-inflammatory effects that positively affect animal’s body weight gain (Bedford and Gong, 2018). On the other hand, research showed that an increased proportion of fecal and serum acetate contribute to the activation of the parasympathetic nervous system which reversely promotes elevated glucose-stimulated insulin secretion (GSIS), hyperphagia, and finally causes weight gain (Perry et al., 2016). Correlation analysis revealed positive associations between butyric level and Prevotella sp. RS2, Ruminococcus sp. AF31-14BH, and Lactobacillus pontis. As reported in previous literature, members from Prevotella possess butyrate kinase and phosphotransbutyrylase, which are essential for the biosynthesis of butyrate via the succinic pathway (Wang L. et al., 2020). Furthermore, Ruminococcus sp. AF31-14BH is a member that belongs to the family Ruminococcaceae, some species from it were well-known butyrate producers. However, it is necessary to perform future investigations for these two species and butyrate formation to support our findings. Additionally, research demonstrated a high abundance of intestinal Lactobacillus spp. contributed to the elevation of butyric acid levels (Le Roy et al., 2015), it makes sense to explain why there was a positive association between Lactobacillus pontis and butyric acid. Particularly, we observed that P. xylaniphila showed a positive association with acetic acids. According to the previous findings, P. xylaniphila was a bacterium that participated in glucose metabolism to produce acetic acids and succinic as the end products (Morotomi et al., 2009). Of note, evidence showed a strong association between levels of succinate/acetate and body weight (Serena et al., 2018; Sakamoto et al., 2022). Therefore, we suggested that P. xylaniphila might be a potential microbial marker for improving the growth performance of pigs.
Our study, although limited by a small pig population, offers important information that upholds the potential of intestinal flora manipulation in improving the finishing weight of DSE pigs. Nevertheless, future studies aiming to validate the positive or negative role of certain bacterial species in regulating finishing weight via specific pathogen-free (SPF) mice/minipigs’ intervention experiment and to obtain more mechanistic understandings into the cross-talk between gut microbes and host and its effects on growth performance of DSE pigs by multi-omics sequencing analysis are needed. In addition, supplementing the microbial species from Lactobacillus spp., Streptococcus spp., and Prevotella spp., or SCFAs (e.g., butyric and acetic acids) into the basal diet might be a valuable strategy to improve the finishing weight of DSE pigs.
Conclusion
In summary, the present study detected some key microbial taxa related to finishing weight. We also performed functional annotation analysis and identified several finishing weight-associated CAZymes, KOs, and KEGG pathways. Furthermore, we highlighted the important role of SCFAs in finishing weight, especially acetate and butyrate. Hence, our investigation provides essential insights into the microbe-SCFAs interplay in affecting finishing weight and hints that modulating the intestinal bacterial communities could be an effective scheme to ameliorate finishing weight for DSE pigs and possibly other pig breeds.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI BioProject—PRJNA910336.
Ethics statement
The animal study was reviewed and approved by Animal Care and Use Committee (ACUC) in Hunan Agricultural University (Changsha, China; Permit Number: CACAHU 20210701).
Author contributions
ML and YY conceived and designed the experiments, supervised the experiment progress, and revised the manuscript. YL, PP, LY, and HZ helped to collect the samples. QL performed the experiments, analyzed the data, and wrote the manuscript. PH, HM, and HW revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Natural Science Foundation of Hunan Province (No. 2022JJ40171), National Natural Science Foundation of China (No. 32002166), Breeding and Promotion of Lancang black pig (2021kjc-js072), Key Science and Technology Project of Yunnan Province (202202AE090032), Consulting research program of Yunnan Province pig industry development strategy (2022YNZH7-2), and the Science and technology innovation Program of Hunan Province (2021RC3091).
Acknowledgments
We are grateful to HW’s group for their help in feeding the experimental Diannan small-ear (DSE) pigs population and collecting samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1117965/full#supplementary-material
SUPPLEMENTARY MATERIAL FIGURE S1
Finishing weight phenotypic values of all pigs (A) and low and high individuals (B). “**” represents FDR adjusted p < 0.01. “L” and “H” in the boxplot and PCoA plot represent low and high finishing weight, respectively.
SUPPLEMENTARY MATERIAL TABLE S1
Estimation of richness and diversity indices in gut microbiota among low and high finishing weight groups.
SUPPLEMENTARY MATERIAL TABLE S2
The relative abundances of OTUs in the phylum and genus level.
SUPPLEMENTARY MATERIAL TABLE S3
Differential microbial species between low and high finishing weight pigs.
References
Andersen, V., Svenningsen, K., Knudsen, L. A., Hansen, A. K., Holmskov, U., Stensballe, A., et al. (2015). Novel understanding of ABC transporters ABCB1/MDR/P-glycoprotein, ABCC2/MRP2, and ABCG2/BCRP in colorectal pathophysiology. World J. Gastroenterol. 21, 11862–11876. doi: 10.3748/wjg.v21.i41.11862
Are, A., Aronsson, L., Wang, S., Greicius, G., Lee, Y. K., Gustafsson, J. A., et al. (2008). Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 105, 1943–1948. doi: 10.1073/pnas.0711734105
Attene-Ramos, M. S., Wagner, E. D., Gaskins, H. R., and Plewa, M. J. (2007). Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 5, 455–459. doi: 10.1158/1541-7786.MCR-06-0439
Bedford, A., and Gong, J. (2018). Implications of butyrate and its derivatives for gut health and animal production. Anim. Nutr. 4, 151–159. doi: 10.1016/j.aninu.2017.08.010
Brun, R. P., Kim, J. B., Hu, E., and Spiegelman, B. M. (1997). Peroxisome proliferator-activated receptor gamma and the control of adipogenesis. Curr. Opin. Lipidol. 8, 212–218. doi: 10.1097/00041433-199708000-00004
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chappidi, S., Villa, E. C., and Cantarel, B. L. (2019). Using Mothur to determine bacterial community composition and structure in 16S ribosomal RNA datasets. Curr. Protoc. Bioinformatics 67:e83. doi: 10.1002/cpbi.83
Chen, L., Luo, Y., Wang, H., Liu, S., Shen, Y., and Wang, M. (2016). Effects of glucose and starch on lactate production by newly isolated Streptococcus bovis S1 from Saanen goats. Appl. Environ. Microbiol. 82, 5982–5989. doi: 10.1128/AEM.01994-16
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chiang, J. Y. (2009). Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966. doi: 10.1194/jlr.R900010-JLR200
Couvigny, B., de Wouters, T., Kaci, G., Jacouton, E., Delorme, C., Dore, J., et al. (2015). Commensal streptococcus salivarius modulates PPARgamma transcriptional activity in human intestinal epithelial cells. PLoS One 10:e0125371. doi: 10.1371/journal.pone.0125371
De Cesare, A., Sirri, F., Manfreda, G., Moniaci, P., Giardini, A., Zampiga, M., et al. (2017). Effect of dietary supplementation with lactobacillus acidophilus D2/CSL (CECT 4529) on caecum microbioma and productive performance in broiler chickens. PLoS One 12:e0176309. doi: 10.1371/journal.pone.0176309
De Rodas, B., Youmans, B. P., Danzeisen, J. L., Tran, H., and Johnson, T. J. (2018). Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 96, 1778–1794. doi: 10.1093/jas/sky109
Degirolamo, C., Rainaldi, S., Bovenga, F., Murzilli, S., and Moschetta, A. (2014). Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 7, 12–18. doi: 10.1016/j.celrep.2014.02.032
Del Chierico, F., Abbatini, F., Russo, A., Quagliariello, A., Reddel, S., Capoccia, D., et al. (2018). Gut microbiota markers in obese adolescent and adult patients: age-dependent differential patterns. Front. Microbiol. 9:1210. doi: 10.3389/fmicb.2018.01210
Dell'Anno, M., Callegari, M. L., Reggi, S., Caprarulo, V., Giromini, C., Spalletta, A., et al. (2021). Lactobacillus plantarum and lactobacillus reuteri as functional feed additives to prevent Diarrhoea in weaned piglets. Animals 11:1766. doi: 10.3390/ani11061766
Duan, J., Huang, Y., Tan, X., Chai, T., Wu, J., Zhang, H., et al. (2021). Characterization of gut microbiome in mice model of depression with divergent response to escitalopram treatment. Transl. Psychiatry 11:303. doi: 10.1038/s41398-021-01428-1
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Fang, S., Xiong, X., Su, Y., Huang, L., and Chen, C. (2017). 16S rRNA gene-based association study identified microbial taxa associated with pork intramuscular fat content in feces and cecum lumen. BMC Microbiol. 17:162. doi: 10.1186/s12866-017-1055-x
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, H., Yang, Y., Cao, Z., Ran, J., Zhang, C., Huang, Y., et al. (2020). Characteristics of the Jejunal microbiota in 35-day-old Saba and Landrace piglets. Pol. J. Microbiol. 69, 367–378. doi: 10.33073/pjm-2020-041
Gibson, G. R. (1990). Physiology and ecology of the sulphate-reducing bacteria. J. Appl. Bacteriol. 69, 769–797. doi: 10.1111/j.1365-2672.1990.tb01575.x
Graessler, J., Qin, Y., Zhong, H., Zhang, J., Licinio, J., Wong, M. L., et al. (2013). Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: correlation with inflammatory and metabolic parameters. Pharm. J. 13, 514–522. doi: 10.1038/tpj.2012.43
Hou, Y. P., He, Q. Q., Ouyang, H. M., Peng, H. S., Wang, Q., Li, J., et al. (2017). Human gut microbiota associated with obesity in Chinese children and adolescents. Biomed. Res. Int. 2017, 1–8. doi: 10.1155/2017/7585989
Hou, C., Zeng, X., Yang, F., Liu, H., and Qiao, S. (2015). Study and use of the probiotic lactobacillus reuteri in pigs: a review. J. Anim. Sci. Biotechnol. 6:14. doi: 10.1186/s40104-015-0014-3
Jans, C., and Boleij, A. (2018). The road to infection: host-microbe interactions defining the pathogenicity of Streptococcus bovis/Streptococcus equinus complex members. Front. Microbiol. 9:603. doi: 10.3389/fmicb.2018.00603
Jo, H. E., Kwon, M. S., Whon, T. W., Kim, D. W., Yun, M., Lee, J., et al. (2021). Alteration of gut microbiota after antibiotic exposure in finishing swine. Front. Microbiol. 12:596002. doi: 10.3389/fmicb.2021.596002
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Karunaratne, N. D., Newkirk, R. W., Ames, N. P., Van Kessel, A. G., Bedford, M. R., and Classen, H. L. (2021). Effects of exogenous beta-glucanase on ileal digesta soluble beta-glucan molecular weight, digestive tract characteristics, and performance of coccidiosis vaccinated broiler chickens fed hulless barley-based diets with and without medication. PLoS One 16:e0236231. doi: 10.1371/journal.pone.0236231
Kashani, A., Brejnrod, A. D., Jin, C., Kern, T., Madsen, A. N., Holm, L. A., et al. (2019). Impaired glucose metabolism and altered gut microbiome despite calorie restriction of Ob/Ob mice. Anim. Microb. 1:11. doi: 10.1186/s42523-019-0007-1
Kiu, R., and Hall, L. J. (2018). An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 7:141. doi: 10.1038/s41426-018-0144-8
Koh, A., De Vadder, F., Kovatcheva-Datchary, P., and Backhed, F. (2016). From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cells 165, 1332–1345. doi: 10.1016/j.cell.2016.05.041
Kultima, J. R., Coelho, L. P., Forslund, K., Huerta-Cepas, J., Li, S. S., Driessen, M., et al. (2016). MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics 32, 2520–2523. doi: 10.1093/bioinformatics/btw183
Le Roy, C. I., Stsepetova, J., Sepp, E., Songisepp, E., Claus, S. P., and Mikelsaar, M. (2015). New insights into the impact of lactobacillus population on host-bacteria metabolic interplay. Oncotarget 6, 30545–30556. doi: 10.18632/oncotarget.5906
Lefterova, M. I., and Lazar, M. A. (2009). New developments in adipogenesis. Trends Endocrinol. Metab. 20, 107–114. doi: 10.1016/j.tem.2008.11.005
Li, S., Li, N., Wang, C., Zhao, Y., Cao, J., Li, X., et al. (2022). Gut microbiota and immune modulatory properties of human breast Milk streptococcus salivarius and S. parasanguinis strains. Front. Nutr. 9:798403. doi: 10.3389/fnut.2022.798403
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, L., Qu, M., Liu, C., Pan, K., Xu, L., OuYang, K., et al. (2019). Expression of a recombinant Lentinula edodes cellobiohydrolase by Pichia pastoris and its effects on in vitro ruminal fermentation of agricultural straws. Int. J. Biol. Macromol. 134, 146–155. doi: 10.1016/j.ijbiomac.2019.05.043
Long, C. X., Wu, J. Q., and Tan, Z. J. (2021). Intestinal microbiota disturbance affects the occurrence of African swine fever. Anim. Biotechnol., 1–10. doi: 10.1080/10495398.2021.2010089
Lunney, J. K. (2007). Advances in swine biomedical model genomics. Int. J. Biol. Sci. 3, 179–184. doi: 10.7150/ijbs.3.179
Ma, Y., Wang, W., Zhang, H., Wang, J., Zhang, W., Gao, J., et al. (2018). Supplemental Bacillus subtilis DSM 32315 manipulates intestinal structure and microbial composition in broiler chickens. Sci. Rep. 8:15358. doi: 10.1038/s41598-018-33762-8
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maltecca, C., Bergamaschi, M., and Tiezzi, F. (2020). The interaction between microbiome and pig efficiency: a review. J. Anim. Breed. Genet. 137, 4–13. doi: 10.1111/jbg.12443
Metwaly, A., Dunkel, A., Waldschmitt, N., Raj, A. C. D., Lagkouvardos, I., Corraliza, A. M., et al. (2020). Integrated microbiota and metabolite profiles link Crohn's disease to sulfur metabolism. Nat. Commun. 11:4322. doi: 10.1038/s41467-020-17956-1
Mirza, A. Z., Althagafi, I. I., and Shamshad, H. (2019). Role of PPAR receptor in different diseases and their ligands: physiological importance and clinical implications. Eur. J. Med. Chem. 166, 502–513. doi: 10.1016/j.ejmech.2019.01.067
Morotomi, M., Nagai, F., Sakon, H., and Tanaka, R. (2009). Paraprevotella clara gen. Nov., sp. nov. and Paraprevotella xylaniphila sp. nov., members of the family 'Prevotellaceae' isolated from human faeces. Int. J. Syst. Evol. Microbiol. 59, 1895–1900. doi: 10.1099/ijs.0.008169-0
O'Callaghan, T. F., Ross, R. P., Stanton, C., and Clarke, G. (2016). The gut microbiome as a virtual endocrine organ with implications for farm and domestic animal endocrinology. Domest. Anim. Endocrinol. 56, S44–S55. doi: 10.1016/j.domaniend.2016.05.003
Oh, J. K., Chae, J. P., Pajarillo, E. A. B., Kim, S. H., Kwak, M. J., Eun, J. S., et al. (2020). Association between the body weight of growing pigs and the functional capacity of their gut microbiota. Anim. Sci. J. 91:e13418. doi: 10.1111/asj.13418
Orbe-Orihuela, Y. C., Godoy-Lozano, E. E., Lagunas-Martinez, A., Castaneda-Marquez, A. C., Murga-Garrido, S., Diaz-Benitez, C. E., et al. (2022). Association of gut Microbiota with dietary-dependent childhood obesity. Arch. Med. Res. 53, 407–415. doi: 10.1016/j.arcmed.2022.03.007
Panasevich, M. R., Wankhade, U. D., Chintapalli, S. V., Shankar, K., and Rector, R. S. (2018). Cecal versus fecal microbiota in Ossabaw swine and implications for obesity. Physiol. Genomics 50, 355–368. doi: 10.1152/physiolgenomics.00110.2017
Perry, R. J., Peng, L., Barry, N. A., Cline, G. W., Zhang, D., Cardone, R. L., et al. (2016). Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 534, 213–217. doi: 10.1038/nature18309
Priyadarshini, M., Kotlo, K. U., Dudeja, P. K., and Layden, B. T. (2018). Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr. Physiol. 8, 1091–1115. doi: 10.1002/cphy.c170050
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ren, Z., Jiang, J., Xie, H., Li, A., Lu, H., Xu, S., et al. (2017). Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 8, 95176–95191. doi: 10.18632/oncotarget.18820
Sakamoto, M., Ikeyama, N., Iino, T., and Ohkuma, M. (2022). Growth of succinate consumer Dialister hominis is supported by Bacteroides thetaiotaomicron. Anaerobe 77:102642. doi: 10.1016/j.anaerobe.2022.102642
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Serena, C., Ceperuelo-Mallafre, V., Keiran, N., Queipo-Ortuno, M. I., Bernal, R., Gomez-Huelgas, R., et al. (2018). Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 12, 1642–1657. doi: 10.1038/s41396-018-0068-2
Shang, P., Wei, M., Duan, M., Yan, F., and Chamba, Y. (2022). Healthy gut microbiome composition enhances disease resistance and fat deposition in Tibetan pigs. Front. Microbiol. 13:965292. doi: 10.3389/fmicb.2022.965292
Shen, J., Mu, C., Wang, H., Huang, Z., Yu, K., Zoetendal, E. G., et al. (2020). Stimulation of gastric transit function driven by hydrolyzed casein increases small intestinal carbohydrate availability and its microbial metabolism. Mol. Nutr. Food Res. 64:e2000250. doi: 10.1002/mnfr.202000250
Stojanov, S., Berlec, A., and Strukelj, B. (2020). The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 8:1715. doi: 10.3390/microorganisms8111715
Tang, S., Xin, Y., Ma, Y., Xu, X., Zhao, S., and Cao, J. (2020). Screening of microbes associated with swine growth and fat deposition traits across the intestinal tract. Front. Microbiol. 11:586776. doi: 10.3389/fmicb.2020.586776
Tenenbaum, A., Fisman, E. Z., and Motro, M. (2003). Metabolic syndrome and type 2 diabetes mellitus: focus on peroxisome proliferator activated receptors (PPAR). Cardiovasc. Diabetol. 2:4. doi: 10.1186/1475-2840-2-4
Vanhoutte, T., Huys, G., De Brandt, E., Fahey, G. C. Jr., and Swings, J. (2005). Molecular monitoring and characterization of the faecal microbiota of healthy dogs during fructan supplementation. FEMS Microbiol. Lett. 249, 65–71. doi: 10.1016/j.femsle.2005.06.003
Wang, Z., Li, Q., Chamba, Y., Zhang, B., Shang, P., Zhang, H., et al. (2015). Identification of genes related to growth and lipid deposition from transcriptome profiles of pig muscle tissue. PLoS One 10:e0141138. doi: 10.1371/journal.pone.0141138
Wang, J., Liu, Y., Yang, Y., Bao, C., and Cao, Y. (2020). High-level expression of an acidic thermostable xylanase in Pichia pastoris and its application in weaned piglets. J. Anim. Sci. 98:skz364. doi: 10.1093/jas/skz364
Wang, X., Wang, Z., Pan, H., Qi, J., Li, D., Zhang, L., et al. (2021). Captivity influences the gut microbiome of Rhinopithecus roxellana. Front. Microbiol. 12:763022. doi: 10.3389/fmicb.2021.763022
Wang, Y., Zhang, Y., Lane, N. E., Wu, J., Yang, T., Li, J., et al. (2022). Population-based metagenomics analysis reveals altered gut microbiome in sarcopenia: data from the Xiangya sarcopenia study. J. Cachexia. Sarcopenia Muscle 13, 2340–2351. doi: 10.1002/jcsm.13037
Wang, L., Zhang, G., Li, Y., and Zhang, Y. (2020). Effects of high forage/concentrate diet on volatile fatty acid production and the microorganisms involved in VFA production in cow rumen. Animals 10:223. doi: 10.3390/ani10020223
Xu, S. S., Wang, N., Huang, L., Zhang, X. L., Feng, S. T., Liu, S. S., et al. (2022). Changes in the mucosa-associated microbiome and transcriptome across gut segments Are associated with obesity in a metabolic syndrome porcine model. Microbiol. Spectr. 10:e0071722. doi: 10.1128/spectrum.00717-22
Xu, E., Yang, H., Ren, M., Wang, Y., Xiao, M., Tang, Q., et al. (2021). Identification of Enterotype and its effects on intestinal butyrate production in pigs. Animals 11:730. doi: 10.3390/ani11030730
Yacoubi, N., Saulnier, L., Bonnin, E., Devillard, E., Eeckhaut, V., Rhayat, L., et al. (2018). Short-chain arabinoxylans prepared from enzymatically treated wheat grain exert prebiotic effects during the broiler starter period. Poult. Sci. 97, 412–424. doi: 10.3382/ps/pex297
Yang, K. M., Jiang, Z. Y., Zheng, C. T., Wang, L., and Yang, X. F. (2014). Effect of lactobacillus plantarum on diarrhea and intestinal barrier function of young piglets challenged with enterotoxigenic Escherichia coli K88. J. Anim. Sci. 92, 1496–1503. doi: 10.2527/jas.2013-6619
Yang, Y., Liu, Y., Liu, J., Wang, H., Guo, Y., Du, M., et al. (2021a). Composition of the fecal microbiota of piglets at various growth stages. Front. Vet. Sci. 8:661671. doi: 10.3389/fvets.2021.661671
Yang, Y., Shen, L., Gao, H., Ran, J., Li, X., Jiang, H., et al. (2021b). Comparison of cecal microbiota composition in hybrid pigs from two separate three-way crosses. Anim. Biosci. 34, 1202–1209. doi: 10.5713/ab.20.0681
Yang, J., Wang, C., Huang, K., Zhang, M., Wang, J., and Pan, X. (2020). Compound lactobacillus sp. administration ameliorates stress and body growth through gut microbiota optimization on weaning piglets. Appl. Microbiol. Biotechnol. 104, 6749–6765. doi: 10.1007/s00253-020-10727-4
Yang, H., Yang, M., Fang, S., Huang, X., He, M., Ke, S., et al. (2018). Evaluating the profound effect of gut microbiome on host appetite in pigs. BMC Microbiol. 18:215. doi: 10.1186/s12866-018-1364-8
Yu, M., Li, Z., Chen, W., Wang, G., Cui, Y., and Ma, X. (2019). Dietary supplementation with citrus extract altered the intestinal microbiota and microbial metabolite profiles and enhanced the mucosal immune homeostasis in yellow-feathered broilers. Front. Microbiol. 10:2662. doi: 10.3389/fmicb.2019.02662
Zeng, H., Umar, S., Rust, B., Lazarova, D., and Bordonaro, M. (2019). Secondary bile acids and short chain fatty acids in the colon: a focus on colonic microbiome, cell proliferation, inflammation, and cancer. Int. J. Mol. Sci. 20:1214. doi: 10.3390/ijms20051214
Zhou, Y., Wang, B., Wang, Q., Tang, L., Zou, P., Zeng, Z., et al. (2021). Protective effects of lactobacillus plantarum Lac16 on Clostridium perfringens infection-associated injury in IPEC-J2 cells. Int. J. Mol. Sci. 22:12388. doi: 10.3390/ijms222212388
Zhu, W., Lomsadze, A., and Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38:e132. doi: 10.1093/nar/gkq275
Zhu, H., Zeng, D., Wang, Q., Wang, N., Zeng, B., Niu, L., et al. (2018). Diarrhea-associated intestinal microbiota in captive Sichuan Golden snub-nosed monkeys (Rhinopithecus roxellana). Microbes Environ. 33, 249–256. doi: 10.1264/jsme2.ME17163
Zhuang, X., Chen, Z., Sun, X., Li, F., Luo, J., Chen, T., et al. (2021). Fermentation quality of herbal tea residue and its application in fattening cattle under heat stress. BMC Vet. Res. 17:348. doi: 10.1186/s12917-021-03061-y
Keywords: Diannan small-ear pigs, finishing weight, 16S rRNA gene, metagenomic, SCFAs, gut microbiome
Citation: Lan Q, Lian Y, Peng P, Yang L, Zhao H, Huang P, Ma H, Wei H, Yin Y and Liu M (2023) Association of gut microbiota and SCFAs with finishing weight of Diannan small ear pigs. Front. Microbiol. 14:1117965. doi: 10.3389/fmicb.2023.1117965
Edited by:
Weiwei Wang, South China Agricultural University, ChinaReviewed by:
Liang Chen, State Key Laboratory of Animal Nutrition, Institute of Animal Sciences (CAAS), ChinaShaoming Fang, Fujian Agriculture and Forestry University, China
Copyright © 2023 Lan, Lian, Peng, Yang, Zhao, Huang, Ma, Wei, Yin and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulong Yin, ✉ yinyulong@isa.ac.cn; Mei Liu, ✉ Mei.Liu@hunau.edu.cn