 Rayana Katylin Mendes Da Silva1
Rayana Katylin Mendes Da Silva1 Joana Morais2,3
Joana Morais2,3 Brian Thomas Foley4
Brian Thomas Foley4 Gonzalo Bello1
Gonzalo Bello1 Mariza Gonçalves Morgado1
Mariza Gonçalves Morgado1 Monick Lindenmeyer Guimarães1*
Monick Lindenmeyer Guimarães1*- 1Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil
- 2Laboratório de Biologia Molecular, Instituto Nacional de Investigação em Saúde, Ministério da Saúde de Angola, Luanda, Angola
- 3Departamento de Bioquímica, Faculdade de Medicina, Universidade Agostinho Neto, Luanda, Angola
- 4Theoretical Biology and Biophysics, Los Alamos National Laboratory, Los Alamos, NM, United States
Angola, located in Central Africa, has around 320,000 (270,000–380,000) people living with human immunodeficiency virus (HIV)/AIDS, equivalent to 1% of the country’s population at the end of 2021. A previous study conducted in 2012, using Angolan samples collected between 2008 and 2010 revealed a high prevalence of HIV-1 recombinants, around 42% of sequences, with 21% showing the same UH profile in partial pol region which were grouped into a monophyletic cluster with high bootstrap support. Thus, the objective of the present work was to obtain complete genomes of those sequences and characterize them, aiming at a description of a new circulating recombinant form (CRF). Whole blood from nine HIV-1 UH pol-infected individuals had their genomic DNA extracted, and nested PCR was used to amplify seven overlapping fragments targeting the full-length HIV-1 genome. The final classification was based on maximum likelihood trees, and recombination analyses were performed using a bootscan from the Simplot program. BLAST and Los Alamos Database inspections were used to search other similar H-like pol sequences. Complete genome amplification was possible for three samples, partial genomes were obtained for the other three, and only pol was available for the remaining three sequences. Bootscan analysis of the two whole-genome and three partial genome sequences retrieved from people living with HIV/AIDS (PLHIVA) without epidemiological linkage showed the same complex recombination profile involving HIV-1 subtypes A/G/H/CRF27_cpx, with a total of six recombinant breakpoints, aiming to classify a new HIV-1 CRF124_cpx. We found no other full-length HIV-1 genomes with the same mosaic profile; however, we identified 33 partial pol sequences, mainly sampled from Angola between 2001 to 2019, with the same H-like profile. Bayesian analysis of H and H-like pol sequences indicates that CRF124_cpx probably originated in Angola at mid-1970s, indicating that this CRF has been circulating in the country for a long time. In summary, our study describes a new CRF circulating principally in Angola and highlights the importance of continuing molecular surveillance studies, especially in countries with high molecular diversity of HIV.
Introduction
About 40 years after its discovery, the human immunodeficiency virus (HIV) remains a global public health challenge. By the end of 2021, 38.4 million people were living with HIV/AIDS (PLWHA) worldwide (UNAIDS, 2022).
Human immunodeficiency virus is classified into two types (type 1 and type 2). HIV-1 is made up of four groups (M, N, O, and P), while HIV-2 is made up of eight groups (A–H; Salvi et al., 1998; Simon et al., 1998; Heyndrickx et al., 2000; Robertson, 2000; Guimarães et al., 2002; Roques et al., 2004; Plantier et al., 2009). HIV-1 group M is responsible for the HIV/AIDS pandemic. Full-genome phylogenetic analyzes have revealed that this group is subdivided into 10 subtypes (A–D, F–H, and J–L) and nine sub-subtypes (A1–A4, A6–A8, F1, and F2). In addition to more than 100 CRFs and uncounted unique recombinant forms (URFs; Désiré et al., 2018; Yamaguchi et al., 2020; Mendes Da Silva et al., 2021; Los Alamos database, 2022).
Overall, on the African continent, the most remarkable diversity of HIV is found in the Central African region, where HIV-1 originated, and all subtypes and many CRFs and URFs are verified (Hemelaar et al., 2019). Like other countries in this region, Angola has high genetic variability of HIV-1 in its population, with an estimated 320,000 (270,000–380,000) PLHIVA, equivalent to 1% of the country’s population until the end of 2021 (UNAIDS, 2021). In a previous study of HIV samples from Angola, molecular epidemiological data of the pol region revealed a high prevalence of HIV-1 recombinant forms (42%), subtypes C (16%) and F1 (14%), followed by G (6%), A, D, and H (5% each), and K (1%) (Delwart et al., 1993; Delatorre et al., 2017). Among the 42% of the sequences classified as recombinants, 21% had the same UH pol profile and were grouped in a monophyletic cluster with high support (Passaes et al., 2009; Afonso et al., 2012a). Thus, the present work aimed to obtain and characterize the HIV-1 full-length genome sequences, aiming to describe a new CRF.
Materials and methods
Study population
Previous studies from our group recruited 101 PLHIVA in Angola, and none of them were under the regular antiretroviral therapy. The study was approved by the CE-FMUAN 027/08 Ethical Committee. After signing the informed consent, patient’s blood collections were carried out in three phases from July 2008 to November 2010 (August 2008, July 2009, and November 2010) at São Lucas Medical Center (CSSL), one of the National Referral Centers for HIV diagnoses, in Kifangondo village, located in the border between Luanda and Bengo provinces. HIV-1 pol sequences [covering the protease (PR) and partial reverse-transcriptase (RT), positions 2,313–3,272 relative to the HXB2 genome] were generated. A highly significant supported UH pol cluster of nine sequences, that corresponded to 8.9% of the total HIV sequences analyzed (Qu et al., 2010; Afonso et al., 2012a), were the focus of the present study.
Amplification of HIV-1 full-length genomes and phylogenetic analyses
Biological samples were stored at −20°C since collection in 2008/2010. DNA was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany) or QIAmp Viral RNA (QIAGEN, Germany). The double-stranded proviral DNA was amplified using nested-PCR employing Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA, United States) into seven overlapping fragments using HIV-1 specific primers, as presented in Supplementary Table S1 (Zazzi et al., 1993).
The amplified products were purified using the Illustra GFX PCR DNA and gel Purification Kit (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) and sequenced on an ABI 3130 Genetic Analyzer using the ABI BigDye Terminator v.3.1 Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, United States). The chromatograms were analyzed and edited using the Seqman software package DNASTAR Lasergene (DNAStar, Madison, WI, United States).
The phylogenetic trees of maximum likelihood (ML) were reconstructed with PhyML version 3.0 (Guindon et al., 2010) using the general time-reversible (GTR) model of nucleotide substitutions. The approximate likelihood ratio test (aLRT) was used to estimate the confidence of the branch in the tree. The phylogenetic trees reconstructed were visualized and edited using the Figtree software version 1.4.4 [13]. Reference sequences of HIV-1 group M subtypes (A–D, F–H, and J–L), sub-subtypes (A1–A4, A6–A8, F1, and F2), and all CRF (until CRF118) sequences were obtained from the Los Alamos HIV database (Available from: http://www.hiv.lanl.gov/).
Recombination analyses were performed using a Bootscan implemented in SimPlot v3.5.1 software with the following parameters: 400 nucleotides (nt) window, 20 nt increments, and NJ method under Kimura’s two-parameter correction with 100 bootstrap replicates (Lole et al., 1999). For better characterization of recombination breakpoints, the midpoints were mapped as suggested in previous analyzes performed by Sierra et al. (2005).
A basic local alignment search tool (BLAST, Available at: https://blast.ncbi.nlm.nih.gov) was performed to identify sequences with high similarity to the studied sequences. The retrieved sequences, together with those from HIV-1 subtype H or UH from Los Alamos Database, were used to search other H-like pol sequences and were included in phylogenetic analyses. (available from: https://blast.ncbi.nlm.nih.gov; http://www.hiv.lanl.gov).
Sequence availability
All HIV-1 sequences generated in this study were deposited in the GenBank database (accession numbers ON962802–ON962807).
Analysis of spatiotemporal dispersion pattern and demographic history
The evolutionary rate (μ, nucleotide substitutions per site per year, subst./site/year), the time of the most recent common ancestor (TMRCA, years), and the ancestral geographic movements of HIV-1 H and H-like pol sequences were jointly estimated using the Bayesian Markov Chain Monte Carlo (MCMC) approach as implemented in BEAST v1.10 (Drummond et al., 2002; Drummond and Rambaut, 2007) with BEAGLE to improve run-time (Suchard and Rambaut, 2009). Analyses were performed under a GTR + I + G nucleotide substitution model, a relaxed uncorrelated lognormal molecular clock model with a uniform prior on clock rate (1.0–2.0 × 10−3 subst/site/year), and a Bayesian Skyline coalescent model (Drummond et al., 2005). Migration events throughout the phylogenetic history were inferred using a reversible discrete Bayesian phylogeographic model (Lemey et al., 2009) and a CTMC rate reference prior (Ferreira and Suchard, 2008). MCMC chain was run for 10 × 106 generations and adequate chain mixing (Effective Sample Size > 200) and uncertainty in parameter estimates [95% Highest Probability Density (HPD) interval] were assessed using the TRACER v1.6 program (Rambaut and Drummond, 2009). Maximum clade credibility (MCC) tree was summarized from the posterior distribution of trees with Tree Annotator and visualized with FigTree v1.4.0 (Available from: http://tree.bio.ed.ac.uk/software/figtree/).
Results
Sociodemographic and clinical data
Among the nine sequences previously classified as HIV-1 UH investigated in PLWHA from Angola, seven were from provinces bordering the CSSL, and two were from the province of Cabinda and Uíge (Northern Angola). Seven were female, and two were male. Sociodemographic and clinical data of our study group are presented in Table 1. The mean age of these individuals was 34 (24–36) years at the time of sample collection. Only two patients had a known epidemiological linkage to sexual transmission (ANG. 107e ANG.113). None of the patients were under antiretroviral therapy (ART). However, some patients reported being part of a spiritual group where the chief prescribed some natural herbs and medicines for treatment (Afonso et al., 2012a).
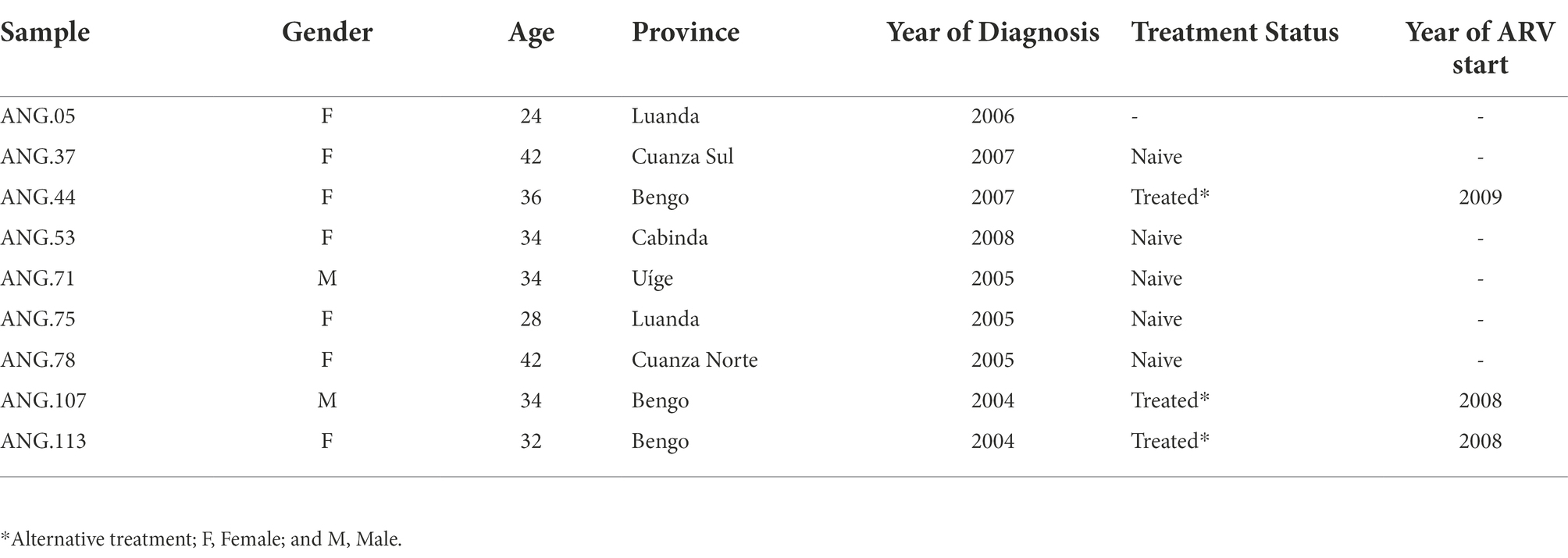
Table 1. Sociodemographic and clinical data of the studied individuals.
Genome amplification and sequence analysis
From these previously classified HIV-1 UH pol samples, it was possible to amplify and sequence the complete genome of three of them (ANG.37, ANG.44, and ANG.78), obtained from PLWHA without epidemiological linkage. Due to the low amount of the biological material available, only partial genome sequences could be obtained for three individuals (ANG.05, ANG.71, and ANG.113), and, for the three remaining ones, only the initial fragment of PR/RT was obtained from the original study and investigated (Table 2).
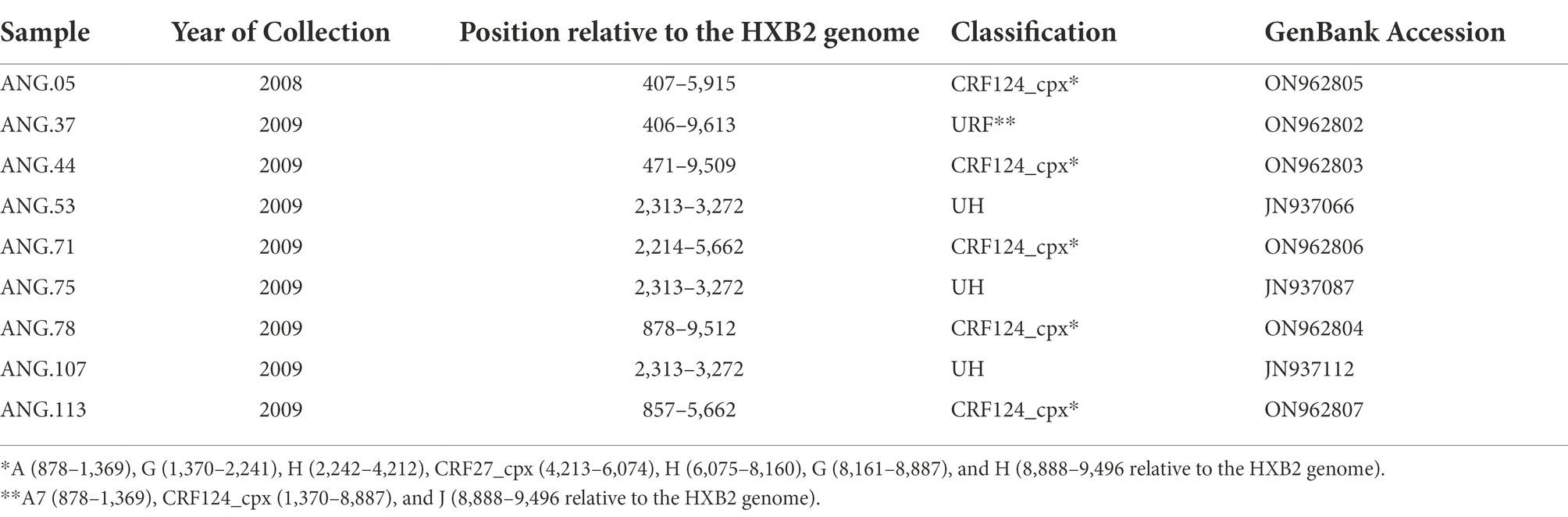
Table 2. Genomic data related to the studied HIV Angolan sequences.
The ML tree performed confirmed that the sequences ANG.37, ANG.44, and ANG.78 grouped apart of the remaining HIV-1 subtypes and CRFs, suggesting a new subtype or CRF (Supplementary Figure 1). Bootscan analysis, including all HIV-1 group M subtypes and A and F sub-subtypes was conducted for the full-length genome sequences (Figure 1A) and partial genome sequences (Figure 1B). This analysis revealed that five of the six sequences (Figures 1A,B) showed the same recombination profile, involving HIV-1 subtypes A, G, H, and J, with eight recombinant breakpoints. In order to confirm these breakpoints and the HIV-1 subtype of each fragment, partial ML phylogenetic trees were performed (Supplementary Figure 2). The sequence ANG.37 showed the same recombinant pattern as ANG.44 and ANG.78, from the fragment positions 1,370 to 8,887, depicting a distinct recombination profile outside this fragment (Figure 1C), showing a URF profile.
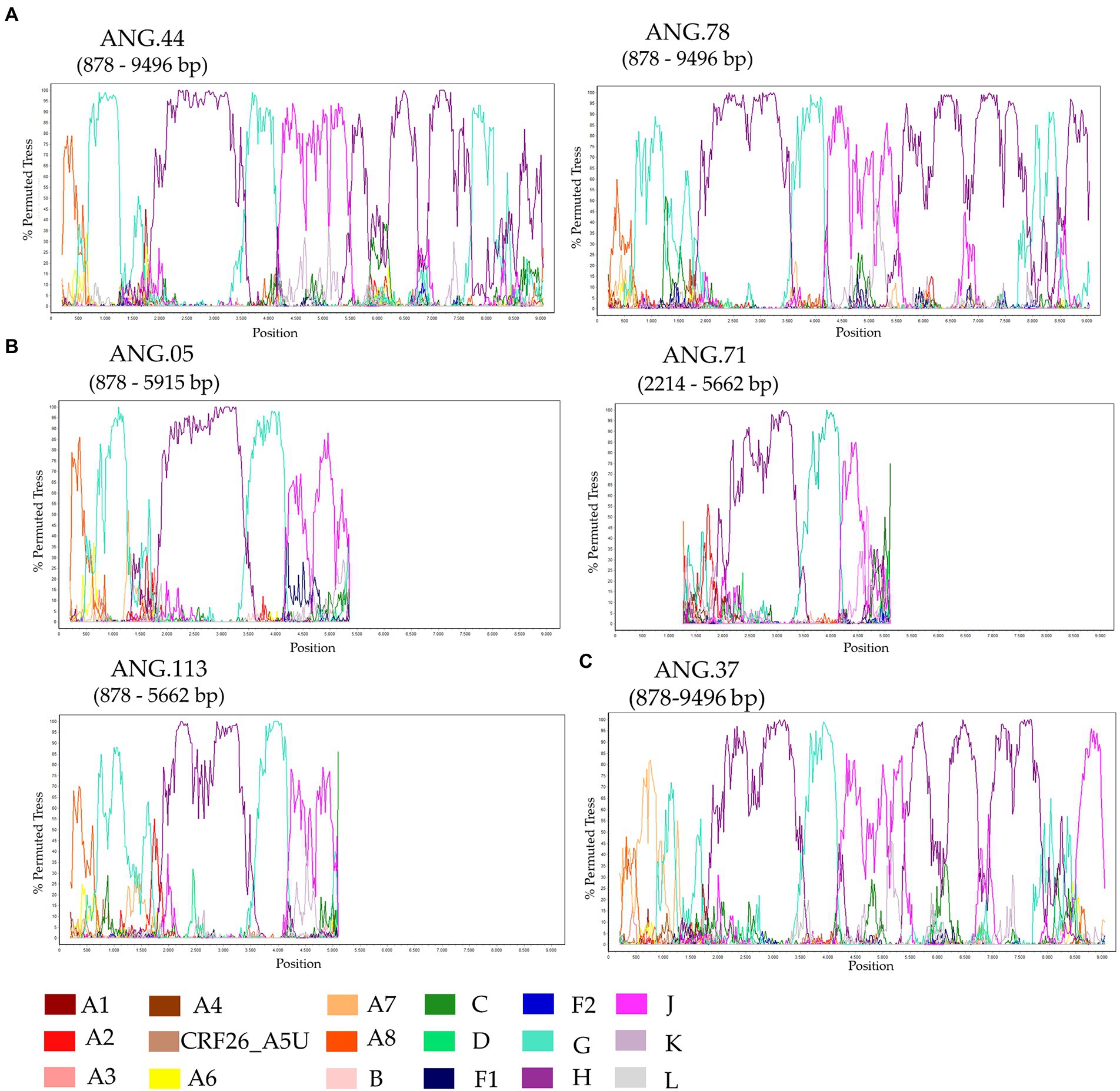
Figure 1. Investigation of human immunodeficiency virus (HIV) genome recombination in HIV-1 samples from Angola. Bootscan analysis was performed in SimPlot software using a 400 nt sliding window and 20 nt increments. (A) Complete genome (ANG.44 and ANG.78), (B) Partial genome (ANG.05, ANG.71, and ANG.113), and (C) ANG.37. The legend in the lower corner indicates the colors that represent the different clades of HIV-1.
After observing that ANG.44 and ANG.78 sequences cluster together with the CRF13_cpx, CRF18_cpx, and CRF27_cpx, all of them involved the HIV-1 subtypes A, G, J, and H (except CRF13_cpx), we have included their full-length genome sequences to perform a new bootscan analysis. Through bootscanning it was possible to verify that the fragments IV (subtype G) and V (subtype J), represented at Supplementary Figure 2, should be reclassified in CRF27_cpx (4213–6,075 relative position to HXB2 genome; Figure 2). Based on these analyzes and in the lack of epidemiological linked among this PLHIVA, we could describe a new CRF, designated as CRF124_cpx, depicting six recombinant breakpoints and involving HIV-1 subtypes A, G, H, and CRF27_cpx (Table 2).
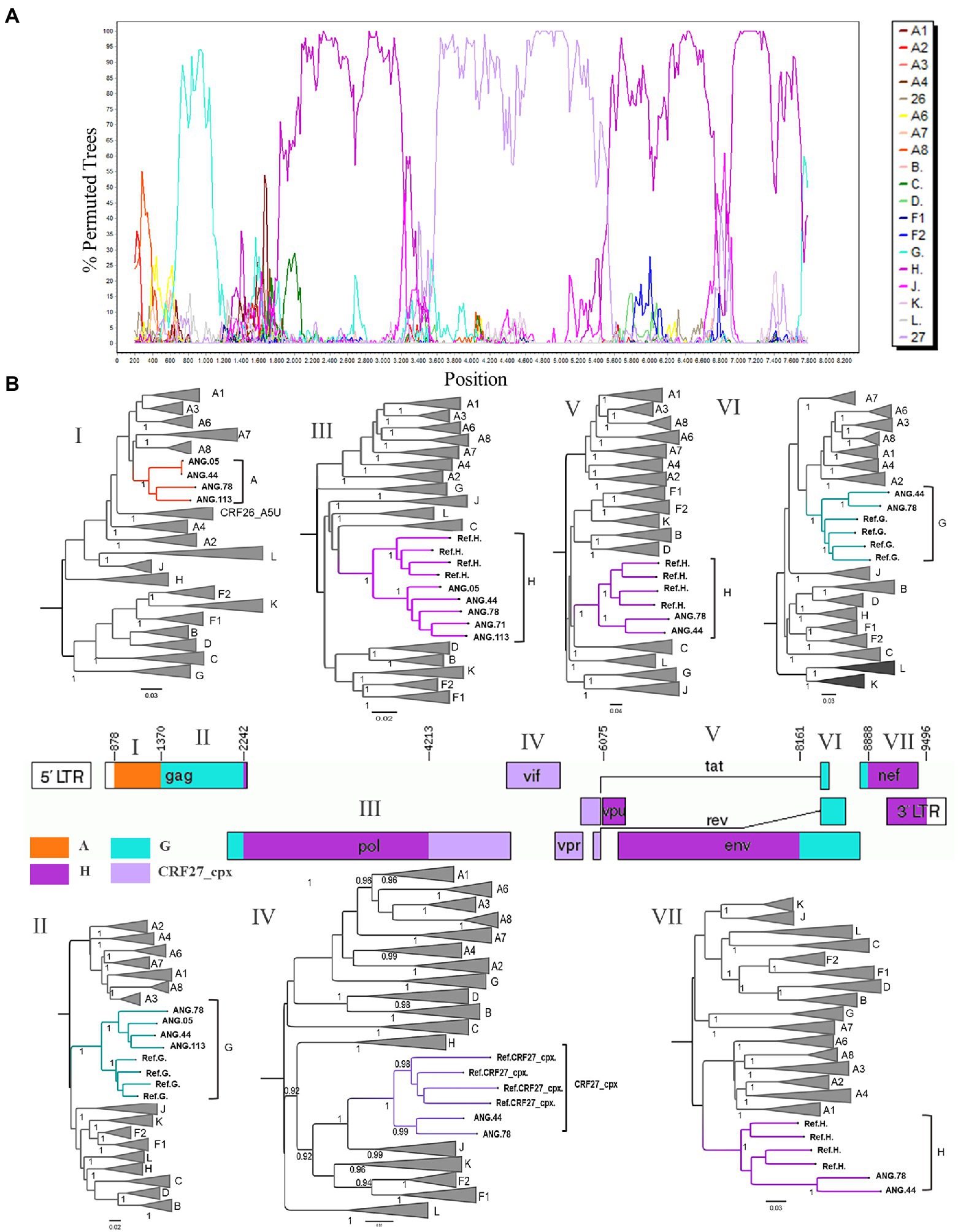
Figure 2. Determination of the new circulating recombinant form (CRF) among HIV-1 samples circulating in Angola. (A) Bootscan analysis of the sequence ANG.44 was performed in SimPlot software using a 400 nt sliding window and 20 nt increments. (B) Genomic structure of CRF124_cpx colored according to the HIV-1 subtyping. The mosaic map was generated using the Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html). The maximum likelihood tree (ML) was performed to confirmation of the HIV-1 subtype of each fragment. ML tree implementing nucleotide substitution model General Time Reversible (GTR), indicating the phylogenetic relationships between pure HIV-1 subtypes and CRF124_cpx sequences. ALRT values were represented only if >0.90.
Complete genome sequences and all seven fragments that compose the CRF124_cpx were submitted to BLAST to scan for related sequences and did not recover significantly related sequences. Thus, we performed BLAST analyses of partial pol (2,261–3,274) and retrieved 100 sequences with homology higher than 91.4%. These retrieved sequences and those subtyped as H, previously published and available in the database at Los Alamos HIV database were included in this alignment, and the ML tree was performed. This analysis allowed us to identify another 24 sequences that presented the same H-like profile defined by bootscan analysis, showing part of the segment with threshold below 70 and another part well defined as H, and all these sequences clustered together with high support (aLTR 0.99) with CRF124_cpx in this fragment, as highlighted in purple in Figure 3.
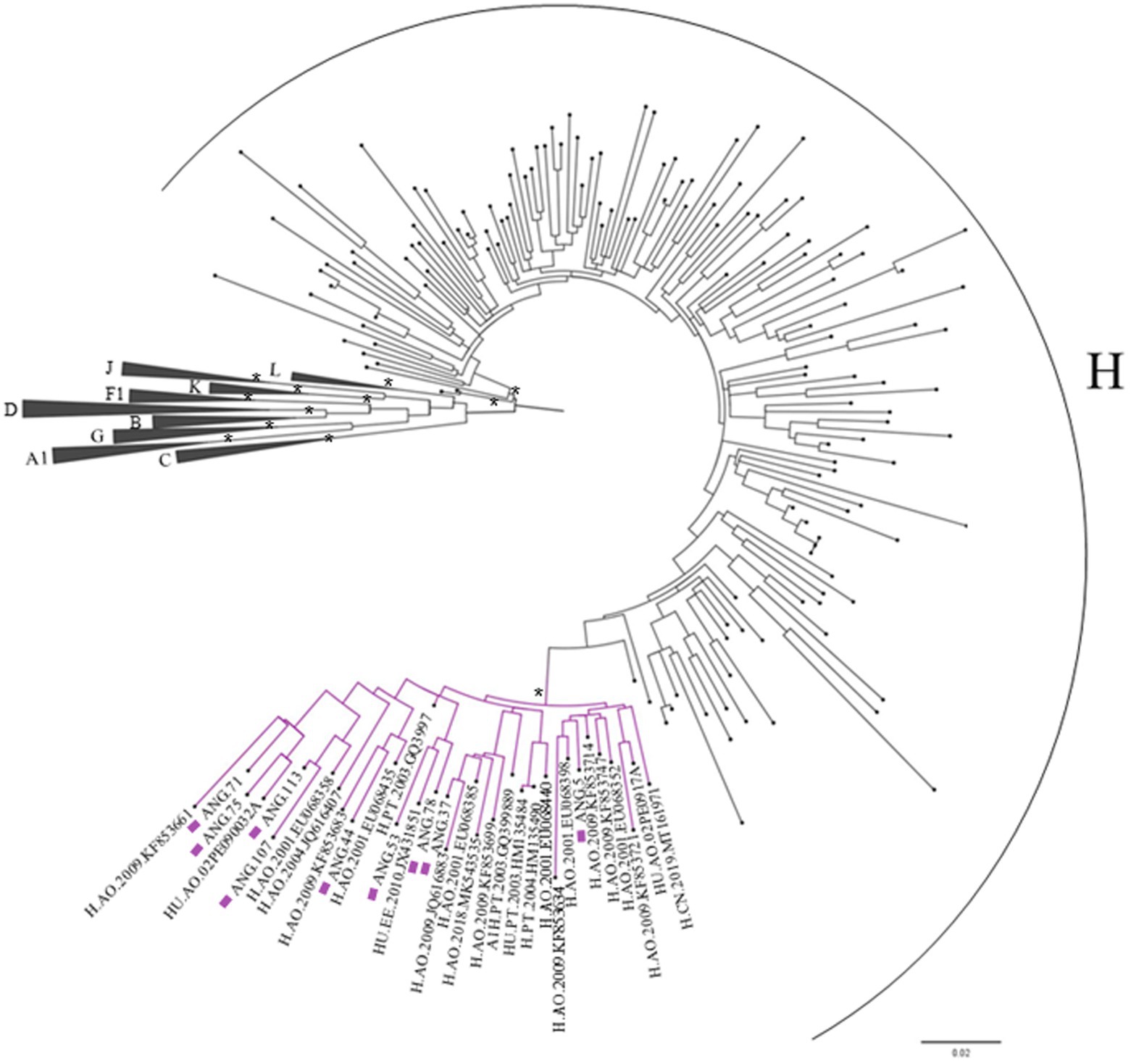
Figure 3. Maximum likelihood (ML) tree of Pol fragment (2,261–3,274 relative to the HXB2 reference) implementing nucleotide substitution model General Time Reversible (GTR), indicating the phylogenetic relationships between the studied sequences and those retrieved from BLAST. Approximate likelihood ratio test (ALRT) values are represented only if >0.90 with asterisk. The sequences belonging to the CRF124_cpx are depicted with purple squares and their cluster are highlighted in purple.
Analysis of spatiotemporal dispersion pattern and demographic history
To reconstruct the origin of the HIV-1 CRF124_cpx, the two full-length and three partial CRF124_cpx sequences here characterized were combined with 28 H-like and 27 subtype H pol sequences sampled between 2000 and 2019, retrieved from Los Alamos HIV database, and subjected to Bayesian phylogeographic analysis. Our analysis indicates that the CRF124_cpx/H-like and subtype H pol sequences branched in two highly supported (posterior probability, PP = 1 and PP = 0.74, respectively) reciprocally monophyletic sister clades, with full-length CRF124_cpx sequences widely intermixed among the H-like pol sequences (Figure 4). The ancestor of the H and CRF124_cpx/H-like clades was placed in the Democratic Republic of Congo (posterior state probability, PSP = 0.60) at 1963 (95% HPD, 1951–1976; Figure 4). The most probable root location of the CRF124_cpx/H-like clade was placed in Angola (posterior state probability, PSP = 1) and the TMRCA was estimated at 1975 (95% HPD, 1967–1984), while the subtype H ancestor was placed in the Democratic Republic of Congo (posterior state probability, PSP = 0.79) at 1970 (95% HPD, 1958–1980; Figure 4). The subtype H was disseminated from the Democratic Republic of Congo to other Central African countries (Angola, Cameroon, and Central African Republic) and to other countries around the world, while the CRF124_cpx/H-like clade migrates from Angola to Portugal, Estonia and China at multiple independent times.
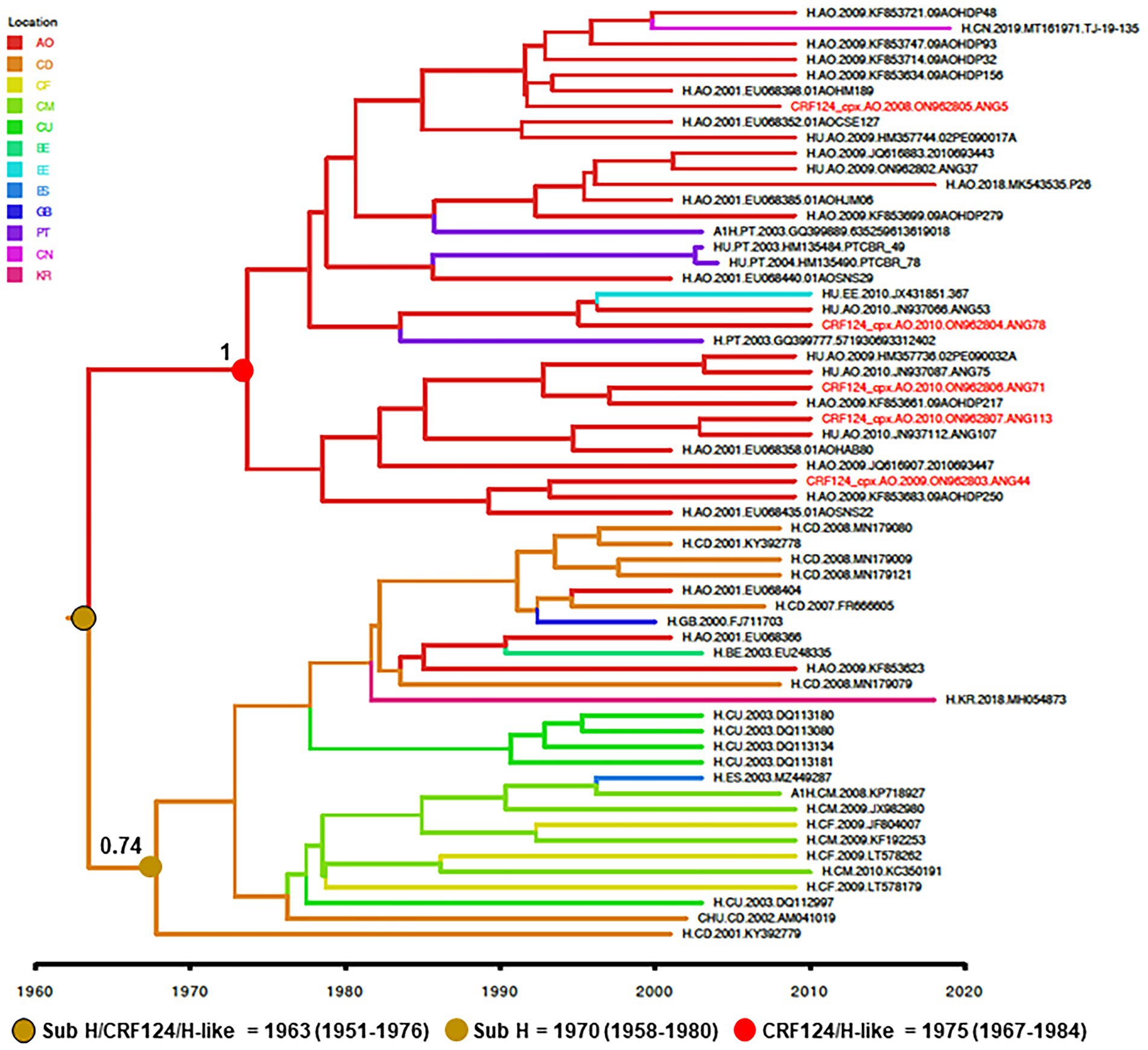
Figure 4. Time-scaled Bayesian MCC tree of HIV-1 CRF124_cpx, H and H-like pol sequences (~700 nt). Branches are colored according to the most probable location state of their descendent nodes as indicated in the legend (upper left). Red tips indicate the position of full-length CRF124_cpx sequences. PP support is shown at nodes of subtype H and clade CRF124_cpx/H-like. Branch lengths are drawn to a scale of years. The tree was automatically rooted under the assumption of a relaxed molecular clock. AO, Angola; CD, Democratic Republic of Congo; CF, Central African Republic; CM, Cameroon; CU, Cuba; BE, Belgium; EE, Estonia; ES, Spain; GB, Great Britain; PT, Portugal CN, China; and KR, South Korea.
Discussion
Angola is a Central Africa country that borders the Republic of Congo, Democratic Republic of Congo, Zambia, and Namibia. As previously described the HIV-1 epidemic in Angola is highly complex; all pure non-B pure subtypes belonging to group M have already been identified, in addition to the high proportion of URFs and CRFs (Bártolo et al., 2009; Afonso et al., 2012a; Sebastião et al., 2020). As compiled by Hemelaar et al. in 2019, the prevalence of recombinant forms increased from 16.5% (1990–1999) to 46.8% (2010–2015) in this African region (Hemelaar et al., 2019). Through HIV molecular epidemiology studies carried out between 2000–2009 and 2010–2019, it was also possible to observe an increasing number of recombinant sequences from 23.6 to 41.4% in Angola in these periods. Among the sequences with a H-like profile in the pol- PR/RT region, it was possible to stand out that sequences presenting the same profile have already been reported in five of eight published studies, totalizing 20 sequences. Based on the 448 HIV-1 Angolan sequences (positions 2,661 –3,274 relative to HXB2), this profile, H-like detected in 28 Angolan sequences corresponds to 6.2% of the deposited sequences (Bártolo et al., 2009, 2014; Castelbranco et al., 2010; Clemente, 2011; Afonso et al., 2012a; Sebastião et al., 2020; Los Alamos database, 2022).
Recombinants are composed of segments of different combinations between subtypes or CRFs, potentially giving viruses an evolutionary advantage as enhanced transmission, pathogenesis, or drug resistance. Some recombinants are so successful in spreading that they become more prevalent than some pure HIV-1 subtypes, such as the worldwide prevalence of CRF02_AG compared to subtype K, 7.7 and 0.1%, respectively (Hemelaar et al., 2020; Los Alamos database, 2022). In the present study, it was possible to characterize a new circulating recombinant form, called CRF124_cpx, composed of fragments of subtypes A, G, H, and CRF27_cpx. After the change in the nomenclature proposed to the HIV classification based on genomes which included the criterion adopted for the description of new CRFs, proposed for Robertson in 2000, over the years, more than 100 CRFs have been identified. CRFs involving subtypes A, G, H, and J do not appear to be uncommon; about 12 CRFs already been described were composed by the association of at least two of the subtypes involved in this newly described (Los Alamos database, 2022). By inspecting complete genomic sequences in the Los Alamos Database, it was possible to identify that HIV-1 subtype A is mainly found in Rwanda and Russian Federation; subtype G in Nigeria and Cameroon; subtype H in the Democratic Republic of Congo and Belgium; and subtype J is more detected in sequences from Sweden and Democratic Republic of Congo, being the Democratic Republic of Congo one of the countries that border Angola (Supplementary Table S2). Interestingly, the HIV-1 sub-subtype A detected in fragment I (878–1,370 from HXB2 genome) from the CRF124_cpx is distinct from the other A sub-subtype, suggesting that it took part in a new one not already described.
Different studies have been carried out to analyze the molecular epidemiology of HIV-1 in Angola, based in the partial pol region, and the HIV-1 subtypes A, G, H, and J, which comprise CRF124_cpx, were identified in their pure forms. In these studies, the HIV-1 subtype A prevalence ranged from 5 to 13.8%, subtype G from 5.7 to 10.8%, subtype H from 3.8 to 5.7% and J was not found in one study and was found 3.2% in another (Bártolo et al., 2009, 2014; Afonso et al., 2012a; Sebastião et al., 2019). The co-circulation of these subtypes in Angola may have favored the occurrence of recombination events that gave rise to the CRF124_cpx. It is also possible that the CRF124_cpx ancestor was disseminated from the neighboring country of Democratic Republic of Congo to Angola. Our Bayesian phylogeographic analysis supports that the CRF124_cpx/H-like pol sequences branched as a sister clade of subtype H and that the ancestor of both lineages probably arose in the Democratic Republic of Congo in the early 1960s. The subtype H probably started to spread in the Democratic Republic of Congo at the early 1970s and was disseminated to neighboring Central African countries (including Angola, Cameroon, and the Central African Republic). The CRF124_cpx/H-like lineage started to spread in Angola around the mid-1970s and was not detected in other Central African countries. Of note, the CRF27_cpx that comprise fragment 4,213–6,075 (relative to the HXB2 genome) of the CRF124_cpx genomes is detected at low prevalence (0.75%) in the Democratic Republic of Congo (Vidal et al., 2008). We may thus speculate that the H-like ancestor may represents a H sub-subtype that circulated in the Democratic Republic of Congo between 1960 and 1980 and that became extinct as a pure sub-subtype, but recombined with other HIV-1 group M subtypes to originate the CRF124_cpx and others URFs.
The inferred TMRCA of the CRF124_cpx clade overlap with the estimated TMRCA of the subtype F1 clade (Bello et al., 2012) and of some local subtype C clades (Afonso et al., 2012b) in Angola. Of note, the concurrent onset date of HIV-1 clades C, F1, and CRF124_cpx in Angola between the mid-1970s at the early 1980s coincides with important socio-political changes that occurred in the country after the beginning of the civil war in 1975 and also coincides with a period of positive migration influx for Angola according to the estimates of the United Nations World Population Prospects (available from: http://esa.un.org/unpd/wpp/Excel-Data/migration.htm). Those changing migratory patterns between mid-1970s and early 1980s may have fueled the introduction, recombination, and local dissemination of different HIV-1 viruses in Angola. The search for sequences presenting the same pol profile of this new CRF124_cpx demonstrates that it is not restricted to Angola, but have been probably disseminated to Portugal, China, and Estonia. Some studies have shown a substantial similarity in the HIV-1 molecular epidemiology profile between Angola and Portugal. Their hypothesis is that socio-historical ties and the intense human migration between the 1970s and 1980s, added to the current intense migratory flow between Portugal and Sub-Saharan African countries, mainly those of the Portuguese-speaking African countries, such as Angola, contributed to the introduction of non-B HIV-1 subtypes in Portugal (Bártolo et al., 2009; Niyasom et al., 2009; Sebastião et al., 2020).
The diversity of HIV can impact diagnosis, viral load measurement, drug resistance, responses to antiretroviral treatment, pathogenesis, vaccine design, immune response, and viral escape (Mackay et al., 2004; Hemelaar, 2013), which demonstrates the need for continuous monitoring of the molecular epidemiology of HIV worldwide.
Data availability statement
The data presented in the study are deposited in the GenBank database repository, accession number ON962802–ON962807.
Ethics statement
The studies involving human participants were reviewed and approved by CE-FMUAN 027/08 Ethical Committee. The patients/participants provided their written informed consent to participate in this study.
Author contributions
MG, MM, and JM conceived and designed the study. RS, JM, GB, and MG performed the experiments. JM participated in patient recruitment. RS, MG, GB, and BF analyzed the data. MG and RS drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We received financial support from Conselho Nacional de Desenvolvimento Científico e Tecnológico/CNPq (Grant number: 307972/2014-3) and from the Laboratory of AIDS and Molecular Immunology, Oswaldo Cruz Institute, FIOCRUZ. RS, MM, and MG are recipients of fellowships from CNPq, numbers 132017/2020-2, 314064/2018-4, and 305919/2018-0, respectively. GB is funded by fellowships of CNPq (Grant number: 304883/2020-4) and FAPERJ (Grant number: E-26/202.896/2018). This study was partially supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES - Finance Code 001.
Acknowledgments
The authors are grateful for genomic platform-DNA sequencing—RPT01A (Rede de Plataformas Tecnológicas Fiocruz).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.992640/full#supplementary-material
References
Afonso, J. M., Bello, G., Guimarães, M. L., Sojka, M., and Morgado, M. G. (2012a). HIV-1 genetic diversity and transmitted drug resistance mutations among patients from the north, central and south regions of Angola. PLoS One 7:e42996. doi: 10.1371/journal.pone.0042996
Afonso, J. M., Morgado, M. G., and Bello, G. (2012b). Evidence of multiple introductions of HIV-1 subtype C in Angola. Infect. Genet. Evol. 12, 1458–1465. doi: 10.1016/j.meegid.2012.05.005
Bártolo, I., Rocha, C., Bartolomeu, J., Gama, A., Marcelino, R., Fonseca, M., et al. (2009). Highly divergent subtypes and new recombinant forms prevail in the HIV/AIDS epidemic in Angola: new insights into the origins of the AIDS pandemic. Infect. Genet. Evol. 9, 672–682. doi: 10.1016/j.meegid.2008.05.003
Bártolo, I., Zakovic, S., Martin, F., Palladino, C., Carvalho, P., Camacho, R., et al. (2014). HIV-1 diversity, transmission dynamics and primary drug resistance in Angola. PLoS One 9, 1–17. doi: 10.1371/journal.pone.0113626
Bello, G., Afonso, J. M., and Morgado, M. G. (2012). Phylodynamics of HIV-1 subtype F1 in Angola, Brazil and Romania. Infect. Genet. Evol. 12, 1079–1086. doi: 10.1016/j.meegid.2012.03.014
Castelbranco, E. P. A. F., Da Silva Souza, E., Cavalcanti, A. M. S., Martins, A. N., De Alencar, L. C. A., and Tanuri, A. (2010). Frequency of primary resistance to antiretroviral drugs and genetic variability of HIV-1 among infected pregnant women recently diagnosed in Luanda-Angola. AIDS Res. Hum. Retrovir. 26, 1313–1316. doi: 10.1089/aid.2010.0111
Clemente, S. (2011). Epidemiologia Molecular da Infecção por VIH/SIDA, em Angola. Faculdade de Medicina, Universidade de Lisboa.
Delatorre, E., Silva-De-Jesus, C., Couto-Fernandez, J. C., Pilotto, J. H., and Morgado, M. G. (2017). High HIV-1 diversity and prevalence of transmitted drug resistance among antiretroviral-naive HIV-infected pregnant women from Rio de Janeiro, Brazil. AIDS Res. Hum. Retrovir. 33, 68–73. doi: 10.1089/aid.2016.0159
Delwart, E. L., Shpaer, E. G., Louwagie, J., McCutchan, F. E., Grez, M., Rübsamen-Waigmann, H., et al. (1993). Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science 262, 1257–1261. doi: 10.1126/science.8235655
Désiré, N., Cerutti, L., Le Hingrat, Q., Perrier, M., Emler, S., Calvez, V., et al. (2018). Characterization update of HIV-1 M subtypes diversity and proposal for subtypes a and D sub-subtypes reclassification. Retrovirology 15, 80–87. doi: 10.1186/s12977-018-0461-y
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G., and Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161, 1307–1320. doi: 10.1093/genetics/161.3.1307
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214–218. doi: 10.1186/1471-2148-7-214
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Ferreira, M. A. R., and Suchard, M. A. (2008). Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 36, 355–368. doi: 10.1002/cjs.5550360302
Guimarães, M. L., Dos Santos Moreira, A., Loureiro, R., Galvão-Castro, B., and Morgado, M. G. (2002). High frequency of recombinant genomes in HIV type 1 samples from Brazilian southeastern and southern regions. AIDS Res. Hum. Retrovir. 18, 1261–1269. doi: 10.1089/088922202320886307
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hemelaar, J. (2013). Implications of HIV diversity for the HIV-1 pandemic. J. Inf. Secur. 66, 391–400. doi: 10.1016/j.jinf.2012.10.026
Hemelaar, J., Elangovan, R., Yun, J., Dickson-Tetteh, L., Fleminger, I., Kirtley, S., et al. (2019). Global and regional molecular epidemiology of HIV-1, 1990–2015: a systematic review, global survey, and trend analysis. Lancet Infect. Dis. 19, 143–155. doi: 10.1016/S1473-3099(18)30647-9
Hemelaar, J., Elangovan, R., Yun, J., Dickson-Tetteh, L., Kirtley, S., Gouws-Williams, E., et al. (2020). Global and regional epidemiology of HIV-1 recombinants in 1990–2015: a systematic review and global survey. Lancet HIV 7, e772–e781. doi: 10.1016/S2352-3018(20)30252-6
Heyndrickx, L., Janssens, W., Zekeng, L., Musonda, R., Anagonou, S., Van der Auwera, G., et al. (2000). Simplified strategy for detection of recombinant human immunodeficiency virus type 1 group M isolates by gag/env Heteroduplex mobility assay. J. Virol. 74, 363–370. doi: 10.1128/jvi.74.1.363-370.2000
Lemey, P., Rambaut, A., Drummond, A. J., and Suchard, M. A. (2009). Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5:e1000520. doi: 10.1371/journal.pcbi.1000520
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected Seroconverters in India, with evidence of Intersubtype recombination. J. Virol. 73, 152–160. doi: 10.1128/JVI.73.1.152-160.1999
Los Alamos database (2022). Available at: http://www.hiv.lanl.gov/
Mackay, G. A., Liu, Z., Singh, D. K., Smith, M. S., Mukherjee, S., Sheffer, D., et al. (2004). Protection against late-onset AIDS in macaques prophylactically immunized with a live simian HIV vaccine was dependent on persistence of the vaccine virus. J. Immunol. 173, 4100–4107. doi: 10.4049/jimmunol.173.6.4100
Mendes Da Silva, R. K., de Pina, M., Araujo, I. I., Venegas Maciera, K., Gonçalves Morgado, M., and Lindenmeyer Guimarães, M. (2021). Genetic characterization of a new hiv-1 sub-subtype a in Cabo Verde, denominated a8. Viruses 13, 2–10. doi: 10.3390/v13061093
Niyasom, C., Horthongkham, N., Sreephiang, A., Kantakamalakul, W., Louisirirotchanakul, S., Chuenchitra, T., et al. (2009). HIV-l subtype b tat gene activities and disease progression in hiv-l CRF01-AE infection. Southeast Asian. J. Trop. Med. Public Health 40, 748–758.
Passaes, C. P. B., Bello, G., Lorete, R. S., Matos Almeida, S. E., Junqueira, D. M., Veloso, V. G., et al. (2009). Genetic characterization of HIV-1 BC recombinants and evolutionary history of the CRF31_BC in southern Brazil. Infect. Genet. Evol. 9, 474–482. doi: 10.1016/j.meegid.2009.01.008
Plantier, J. C., Leoz, M., Dickerson, J. E., De Oliveira, F., Cordonnier, F., Lemée, V., et al. (2009). A new human immunodeficiency virus derived from gorillas. Nat. Med. 15, 871–872. doi: 10.1038/nm.2016
Qu, S., Ma, L., Yuan, L., Xu, W., Hong, K., Xing, H., et al. (2010). Co-receptor usage and prediction of v3 genotyping algorithms in hiv-1 subtype b' from paid blood donors experienced antiretroviral therapy in chinese central province. Virol. J. 7, 1–7. doi: 10.1186/1743-422X-7-280
Rambaut, A., and Drummond, A. (2009). Trace v1.6. Available at: http://treebioedacuk/software/tracer/
Robertson, D. L. (2000). HIV-1 Nomenclature Proposal. Science 288, 55–56. doi: 10.1126/science.288.5463.55d
Roques, P., Robertson, D. L., Souquière, S., Apetrei, C., Nerrienet, E., Barré-Sinoussi, F., et al. (2004). Phylogenetic characteristics of three new HIV-1 N strains and implications for the origin of group N. AIDS 18, 1371–1381. doi: 10.1097/01.aids.0000125990.86904.28
Salvi, R., Garbuglia, A. R., Di Caro, A., Pulciani, S., Montella, F., and Benedetto, A. (1998). Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term Nonprogressor. J. Virol. 72, 3646–3657. doi: 10.1128/jvi.72.5.3646-3657.1998
Sebastião, C. S., Morais, J., and Brito, M. (2020). Clinical and public health implications of hiv-1 genetic diversity and drug resistance mutations in Angola: a systematic review. AIDS Rev. 23, 48–56. doi: 10.24875/AIDSRev.20000057
Sebastião, C. S., Neto, Z., de Jesus, C. S., Mirandela, M., Jandondo, D., Couto-Fernandez, J. C., et al. (2019). Genetic diversity and drug resistance of HIV-1 among infected pregnant women newly diagnosed in Luanda, Angola. PLoS One 14:e0225251. doi: 10.1371/journal.pone.0225251
Sierra, M., Thomson, M. M., Ríos, M., Casado, G., Ojea-de Castro, R., Delgado, E., et al. (2005). The analysis of near full-length genome sequences of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Chile, Venezuela and Spain reveals their relationship to diverse lineages of recombinant viruses related to CRF12_BF. Infect. Genet. Evol. 5, 209–217. doi: 10.1016/j.meegid.2004.07.010
Simon, F., Mauclère, P., Roques, P., Loussert-Ajaka, I., Müller-Trutwin, M. C., Saragosti, S., et al. (1998). Identification of a new human immunodeficiency virus type 1 distinct from group M and group O. Nat. Med. 4, 1032–1037. doi: 10.1038/2017
Suchard, M. A., and Rambaut, A. (2009). Many-core algorithms for statistical phylogenetics. Bioinformatics 25, 1370–1376. doi: 10.1093/bioinformatics/btp244
UNAIDS (2021). Factsheets Angola 2021. UNAIDS. Available at: https://aidsinfo.unaids.org
UNAIDS (2022). Global HIV & AIDS statistics — Fact sheet. Available at: https://www.unaids.org/en/resources/fact-sheet
Vidal, N., Frange, P., Chaix, M. L., Mulanga, C., Lepira, F., Bazepeo, S. E., et al. (2008). Characterization of an old complex circulating recombinant form, CRF27_cpx, originating from the Democratic Republic of Congo (DRC) and circulating in France. AIDS Res. Hum. Retrovir. 24, 315–321. doi: 10.1089/aid.2007.0241
Yamaguchi, J., Vallari, A., McArthur, C., Sthreshley, L., Cloherty, G. A., Berg, M. G., et al. (2020). Brief report: complete genome sequence of CG-0018a-01 establishes HIV-1 subtype L. J. Acquir. Immune Defic. Syndr. 83, 319–322. doi: 10.1097/QAI.0000000000002246
Keywords: HIV-1, diversity, CRF124_cpx, surveillance, Angola
Citation: Da Silva RM, Morais J, Foley BT, Bello G, Morgado MG and Guimarães ML (2022) Identification of a new circulating recombinant form of human immunodeficiency virus type 1, CRF124_cpx involving subtypes A, G, H, and CRF27_cpx in Angola. Front. Microbiol. 13:992640. doi: 10.3389/fmicb.2022.992640
Edited by:
Mohammed Rohaim, Lancaster University, United KingdomReviewed by:
Sudhanshu Shekhar Pandey, Trivitron healthcare Ltd., IndiaEdson Delatorre, Federal University of Espirito Santo, Brazil
Copyright © 2022 Da Silva, Morais, Foley, Bello, Morgado and Guimarães. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monick Lindenmeyer Guimarães, monicklg@ioc.fiocruz.br