 Changhua Shang
Changhua Shang Yujia Li1,2†
Yujia Li1,2†- 1College of Life Sciences, Guangxi Normal University, Guilin, China
- 2Key Laboratory of Ecology of Rare and Endangered Species and Environmental Protection (Guangxi Normal University), Ministry of Education, Guilin, China
- 3Guangxi Key Laboratory of Landscape Resources Conservation and Sustainable Utilization in Lijiang River Basin (Guangxi Normal University), Guilin, China
- 4School of Life Sciences, Sun Yat-sen University, Guangzhou, China
Bacterial communities in high-temperature Daqu and fermented grains are important for brewing Jiang-flavor Baijiu such as Danquan Baijiu. Daqu is a saccharifying and fermenting agent, which has a significant impact on the flavor of Baijiu. However, bacterial communities in three different types of samples from the Danquan distillery (dqjq_ck, dqjqcp, and dqjp3) were still unclear, which limited further development of Danquan Baijiu. “dqjq_ck” and “dqjqcp” indicate high-temperature Daqu at days 45 and 135, respectively. “dqjp3” indicates fermented grains. In this study, the bacterial communities of three samples were analyzed by Illumina Miseq high-throughput sequencing. The bacterial communities of three samples primarily composed of thermophilic bacteria and bacteria with stress resistance. The most abundant species in dqjq_ck, dqjqcp, and dqjp3 were Comamonas, Bacillus, and unclassified Lactobacillales, respectively. The main bacteria included Bacillus, Comamonas, Myroides, Paenibacillus, Acetobacter, Kroppenstedtia, Staphylococcus, Saccharopolyspora, Planifilum, Lactobacillus, Acinetobacter, Oceanobacillus, Enterococcus, Thermoactinomyces, Lactococcus, Streptomyces, Saccharomonospora, Tepidimicrobium, Anaerosalibacter, unclassified_Lactobacillales, unclassified_Thermoactinomycetaceae_1, unclassified_Bacillaceae_2, unclassified_Bacillales, unclassified_Microbacteriaceae, unclassified_Rhodobacteraceae, unclassified_Actinopolysporineae, and unclassified_Flavobacteriaceae in three samples (percentage was more than 1% in one of three samples). In our study, the succession of microbiota in three samples representing three important stages of Danquan Baijiu brewing was revealed. This article lays a good foundation for understanding the fermentation mechanism and screening some excellent indigenous bacteria to improve the quality of Danquan Baijiu in future.
Introduction
Traditional Chinese Baijiu has a variety of flavor styles such as Jiang flavor, light flavor, and Nong flavor, and a long history (Jia et al., 2020). Jiang-flavor Baijiu is one of the most popular and typical flavor styles in traditional Chinese Baijiu, which is represented by the famous Maotai from Huairen city, Guizhou Province, and Danquan Baijiu from Hechi city, Guangxi Autonomous Region.
In the past decades, there were some important discoveries about the origin of Chinese Baijiu based on the study of historical documents and the important physical evidence for Baijiu brewing (i.e., distillers). Different dynasties were suggested about the origin of Chinese Baijiu such as the Eastern Han Dynasty, Tang Dynasty, Song Dynasty, and Yuan Dynasty (Fu, 2005; Feng, 2015). However, regardless of scholars' perspectives, there is a completely consistent consensus. Chinese distilled liquor entered the consumer market and rapidly developed in the Yuan Dynasty (Mao, 2014). Chinese Baijiu, brandy, whisky, vodka, rum, and gin are known as the six major distilled spirits in the world, which highlighted the importance of Chinese Baijiu (Zheng et al., 2017). In 2020 and 2021, the export volumes of Chinese Baijiu were 14,246 and 16,023 kiloliters (with $459.912 and $564.827 million of sales), respectively, which accounted for only 0.19 and 0.22% of the domestic Chinese Baijiu yield. In 2020 and 2021, the total sales of Chinese Baijiu were 583.6 and 603.3 billion. The huge sales show the importance of Baijiu in the economy and society. However, the very low export proportion of Chinese Baijiu indicates that there is much room for expansion in the foreign market.
The production of Danquan Baijiu utilizes traditional and spontaneous multi-strain (including bacteria and fungi) solid-state fermentation (Liu et al., 2019; Jiang et al., 2020). The quality and yield of Baijiu are closely associated with microbes during the fermentation process (Yang et al., 2018; Jiang et al., 2020). The correlation between the microbes producing Taorong-type Baijiu and the major volatile substances in the pit mud was analyzed, which revealed the importance of the related microbes (Liu et al., 2022). Li et al. explored the effect of brewing ingredients in pit muds on the yield of Luzhou-flavor Baijiu and the specific effect of pit mud microbes in different locations on Baijiu production (Li et al., 2022). Luzhou-flavor baijiu is yielded by the combined activities of various microbes, and its flavor is affected by specific microbial community structures and the expression of specific genes (Zhou et al., 2022). The aforementioned studies fully clarified the importance of microorganisms in the Baijiu fermentation process. Chinese Baijiu is one of the world's oldest distilled liquors, which typically originated from the use of Daqu fermentation starters. Daqu is a saccharifying and fermenting agent, which has a significant effect on the flavor of Chinese Baijiu (Zheng et al., 2011). The microbes in Danquan Baijiu brewing primarily originates from high-temperature Daqu (HTD) which is responsible for saccharification and fermentation processes (Yang et al., 2018; Li et al., 2019). For Danquan Baijiu, HTD is made from wheat and old Daqu, then subjected to shaping, fermenting, and ripening, which involves numerous microbes and enzymes (Wang et al., 2022). HTD significantly affects the yield and quality of Baijiu (Liu et al., 2019). With the higher HTD-making temperature, most yeasts and molds are destroyed, and the microbial community of HTD primarily propagates bacteria, leading to the microbial composition dominated by thermophilic bacteria (Gan et al., 2019; Xie et al., 2020). Moreover, as an important functional community in HTD, bacteria can produce various enzymes and plentiful Baijiu aroma substances or their precursors, which give Baijiu a unique flavor (Zhao et al., 2020). Therefore, systematic research on the diversity of bacterial community in HTD is necessary for fully understanding the bacterial resources and fermentation mechanism of Baijiu such as Danquan Baijiu and improving the quality and yield of Baijiu.
At present, high-throughput sequencing technology has been widely utilized for the analysis of microbial community of diverse environmental samples (Dreier et al., 2022). Compared with traditional fingerprint technology such as restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), and random amplified polymorphic DNA (RAPD), high-throughput sequencing by Illumina MiSeq sequencing platform has many advantages such as high-throughput, fast rate, and accurate result (Hu et al., 2022). High-throughput sequencing has also been widely used to analyze the microbial community of various Daqu samples (Deng et al., 2020; Chen et al., 2021). Deng et al. investigated the differences of microbial communities and potential functions among four Jiang-flavor Daqu with different colors (named QW, QB, QY, and QR) by amplicon sequencing (Deng et al., 2020). Chen et al. used Illumina MiSeq sequencing to analyze the microbial community of special-flavor Baijiu Daqu in Jiangxi Province (Chen et al., 2021).
In this study, three samples (two HTDs and one fermented grains) were collected from the Danquan distillery. Through Illumina MiSeq high-throughput sequencing technology, the bacterial community in three samples was observed and compared. Then, the bacterial function was predicted by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSTs) (Douglas et al., 2018). Therefore, this study is aimed to (a) fully reveal the bacterial community of three samples, (b) identify the most abundant species of three samples, and (c) understand the bacterial function in three samples.
Materials and Methods
Sample Collection
Totally, three samples (dqjq_ck, dqjqcp, and dqjp3) were collected from the Danquan distillery in Nandan county, Hechi city, Guangxi Autonomous Region. Among them, dqjq_ck and dqjqcp were high-temperature Daqu (HTD) bricks (as saccharification fermentation agent for liquor fermentation) fermented for 45 d and 135 d, and dqjp3 was fermented grains (i.e., residues from the brewing process); three parallel materials were mixed in equal quantities for eachsample (dqjq_ck, dqjqcp, and dqjp3).
The preparation process of Daqu was as follows. First, raw materials (wheats) were broken and then mixed with water and crushing mother Daqu according to a certain proportion (wheat:water:crushing mother Daqu=100%: 40%: 7%, weight ratio). Second, the mixture was filled into brick-shaped molds (38 ×28 × 6 cm), and the workers stepped on it to obtain sufficient cohesion and proper air permeability. Third, the Daqu bricks were transferred into fermentation chambers, then layer-by-layer stacked up with space between each two, and covered with straw to obtain a proper solid-state fermentation environment (temperature: 65–70°C, humidity: 80–90%) for microbes. After 45 d of fermentation, Daqu bricks named dqjq_ck was obtained. At last, the Daqu bricks dqjq_ck were transferred into another fermentation chamber (temperature: about 40°C, humidity: natural humidity), then layer-by-layer stacked up in the space between each two for 90 d of fermentation sequentially to obtain ripe Daqu bricks (dqjqcp). Then ripe Daqu bricks were used for the brewing process. Therefore, there were two important Daqu fermentation stages (a total of 135 d): 45 d of the first fermentation and 90 d of the second fermentation, which was the standard operation of Daqu preparation in the Danquan distillery. This is the reason for choosing 45 and 135 days for Daqu sampling.
Metagenomic DNA Extraction
Metagenomic DNA was extracted from 0.5 g of each sample through an E.Z.N.A Mag-Bind Soil DNA Kit (Omega) in accordance with the manufacturer's instruction. DNA concentration was determined by using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific). Qualified DNA was stored at −80°C until required.
PCR Amplification and High-Throughput Sequencing
In this study, the V3-V4 region of 16S rDNA was amplified by sense primer 341F (CCTACGGGNGGCWGCAG) and antisense primer 805R (GACTACHVGGGTATCTAATCC). The PCR system was as follows: 15 μl 2×Hieff Robust PCR Master Mix, 1 μl 10 μM sense primer, 1 μl 10 μM antisense primer, 10 ng genomic DNA, and supplemented to 30 μl with ddH2O. The PCR condition was as follows: 94°C for 3 min, 5 cycles (94°C for 30 s, 45°C for 20 s, 65°C for 30 s), 20 cycles (94°C for 20 s, 55°C for 20 s, 72°C for 30 s), and 72°C for 5 min. Three DNA amplicons were sequenced by Miseq-Illumina sequencing platform in Sangon Biotech (China).
Bioinformatics Analysis
Raw reads were processed by three kinds of software (cutadapt, PEAR, and PRINSEQ) to remove adaptors, unknown nucleotides, and low-quality reads. Then clean reads were obtained. The clean reads were clustered by Usearch based on a 97% similarity level to generate operational taxonomic units (OTUs). OTUs were performed homology comparison by the RDP classifier with RDP database, then annotated taxonomic positions at different taxonomic levels (phylum, class, order, family, and genus). Bacterial abundance and diversity were assessed in three samples based on α-diversity indexes. The sequencing depth was assessed by a sparse curve in order to check compliance with requirement of bioinformatics analysis. The similarity among bacterial composition of three samples was determined by UniFrac analysis. Principal coordinates analysis (PCoA) was performed by unweighted and weighted UniFrac. PICRUSt software was used to predict the functional potential of bacterial communities in three samples.
Results
Analysis of Bacterial Diversity in Three Different Samples
The Illumina Miseq platform in the three samples (dqjq_ck, dqjqcp, and dqjp3) generated a total 60,079, 66,533, and 48,559 raw reads, with average lengths of 460 bp, 459 bp, and 460 bp, respectively. There were 58,783, 64,669, and 48,130 clean reads in the three samples (dqjq_ck, dqjqcp, and dqjp3), with average lengths of 426, 426, and 423 bp, respectively. According to a 97% similarity level, the clean reads were divided, and a total 266 OTUs were obtained in the three samples. The proportion of each taxonomic position is shown in Supplementary Table 1. Gene copy numbers of bacterial communities are shown in Supplementary Table 2 for three samples.
The sparse curve was used to evaluate the sequencing depth. From Figure 1, sparse curves were similar between the three samples, and the OTU number increased with the sequencing depth. With the gradual increase in sequencing depth, the OTU number of the three samples had an upward trend, which signified that the current sequencing depth was reasonable and sufficient to exhibit bacterial diversity in all samples. The α-diversity indexes in the three samples are shown in Table 1. The analysis of α-diversity showed that the highest and lowest richness (Chao index) were observed in dqjp3 and dqjq_ck, and the highest and lowest diversity (Shannon and Shannon even indexes) were observed in dqjqcp and dqjp3 (Table 1). The diversity indexes shown in Table 1 (including Shannon, Chao, Ace, Simpson, and Shannon even values) were utilized to evaluate the abundance and uniformity of the bacterial community. The richness estimator (Chao) was consistent with the number of OTUs. The bacterial OTUs and Chao were maximum in sample dqjp3, followed by sample dqjqcp. The Shannon index and Simpson index showed species diversity. The higher the Shannon index, the higher the community diversity. The bacterial Shannon value was the highest in sample dqjqcp and the lowest in sample dqjp3. The bacterial OTUs and Chao values of sample dqjp3 were the highest, while the Simpson value was the lowest.
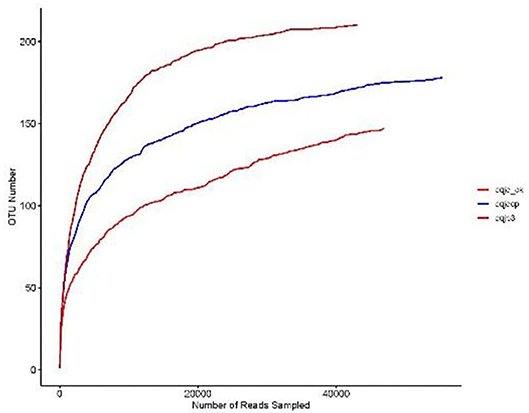
Figure 1. Sparse curves of three samples.
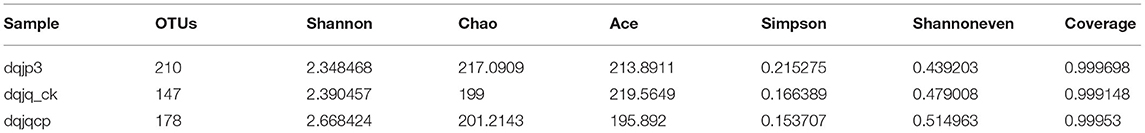
Table 1. α-Diversity indexes in three samples.
Analysis of Bacterial Community Structure in All Samples
In this study, phylum or genus (relative abundance>1.00% in one sample) was considered a dominant phylum or genus; other phyla or genera (relative abundance<1.00%) were classified as others. Sequences which were not be identified were classified as unclassified.
A total of 16 bacterial phyla were obtained from all samples, and four phyla were dominant among them, including Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes, which accounted for most of total sequences (Figure 2A). Firmicutes accounted for 38.26, 83.66, and 74.18% in dqjq_ck, dqjqcp, and dqjp3, respectively, which was an absolute dominant phylum. Proteobacteria were also dominant in the three samples, although its abundance in sample dqjqcp was relatively low (2.31%). At the genus level, a total of 167 bacterial genera were obtained from all samples, and 27 genera were dominant among them. Genera with a higher abundance included Bacillus, Comamonas, Myroides, Paenibacillus, Acetobacter, and unclassified_Lactobacillales (Figure 2B). The main bacteria included Bacillus, Comamonas, Myroides, Paenibacillus, Acetobacter, Kroppenstedtia, Staphylococcus, Saccharopolyspora, Planifilum, Lactobacillus, Acinetobacter, Oceanobacillus, Enterococcus, Thermoactinomyces, Lactococcus, Streptomyces, Saccharomonospora, Tepidimicrobium, Anaerosalibacter, unclassified_Lactobacillales, unclassified_Thermoactinomycetaceae_1, unclassified_Bacillaceae_2, unclassified_Bacillales, unclassified_Microbacteriaceae, unclassified_Rhodobacteraceae, unclassified_Actinopolysporineae, and unclassified_Flavobacteriaceae in the three samples (percentage was more than 1% in one of three samples). Deng et al. found that Kroppenstedtia and Bacillus were predominant in all Daqu samples with different colors (named QW, QB, QY, and QR). Saccharopolyspora was predominant in QB and QR (Deng et al., 2020). The predominant bacterial communities included Bacillales, Lactobacillales, and Rhodospirillales. Acetobacter was the predominant bacterial genus in the surface part of Daqu, whereas Kroppenstedtia, Oceanobacillus, and Bacillus were the predominant bacterial genera in the central part of Daqu (Chen et al., 2021). In our study, the predominant genera also included the aforementioned genera in the studies of Deng et al. and Chen et al. However, the relative abundance of each predominant genus varied widely between our study and these two studies.
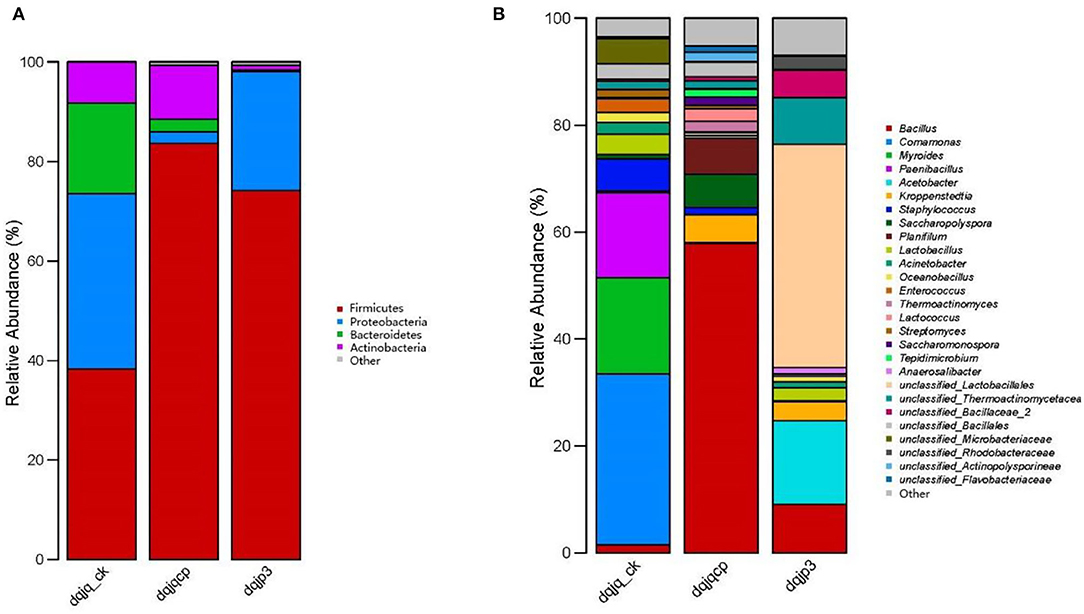
Figure 2. Taxonomic classification of bacterial reads retrieved from three samples at phylum and genus levels using RDP classifier. (A) Phylum level. (B) Genus level.
The bacterial community in the three samples was further characterized at the OTU level (Figure 3). In 266 OTUs found in all samples, there was one core OTU8, which was present in all samples with relative abundance (>1.00%). OTUs with higher abundance included OTU2, OTU3, OTU1, OTU7, OTU6, OTU4, OTU5, OTU8, OTU11, OTU17, OTU10, OTU9, OTU15, and OTU14.
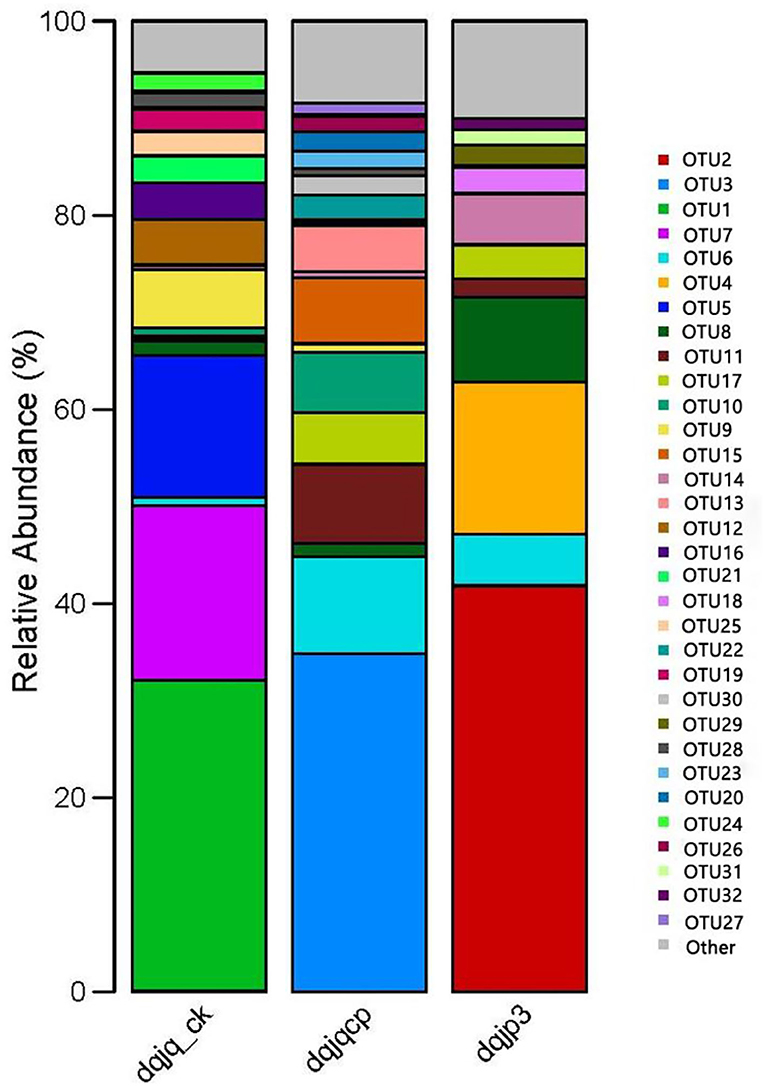
Figure 3. Bacterial community in three samples at the OTU level.
The main genera in our three samples were described as follows. Myroides sp. ZB35 was used to produce aroma volatiles (22 esters) by solid-state fermentation on a rice medium (Xiao et al., 2017). At 192 h, the total concentration of the esters reached 1774 μg/kg. In addition, six novel esterase genes were found in Myroides sp. ZB35. Aroma volatiles containing 2-methylbutyl isovalerate, isoamyl isovalerate, isoamyl 2-methylbutanoate, and 2-methylbutyl 2-methylbutanoate were identified from Myroides sp. ZB35 extracts (Xiao et al., 2014). Many species of Paenibacillus genus are well-known plant growth promoters, which can promote plant nutrient uptake, control phytopathogens, and produce phytohormones. In addition to agricultural applications, Paenibacillus can produce a diverse variety of antimicrobials, enzymes, and exopolysaccharides for bioremediation (Grady et al., 2016). Kroppenstedtia and Oceanobacillus can secret amylase and lipase, and metabolize acids, which can enhance the use of sorghum and raw materials in Daqu (Yu et al., 2014; Wang X. et al., 2018). Differences in the production of fermented sausage flavor were found among different strains of Staphylococcus xylosus and Staphylococcus carnosus in meat simulation medium (Ravyts et al., 2010). Staphylococcus gallinarum and Staphylococcus saprophyticus were found in southern Daqu and were absent in northern Daqu (Zheng et al., 2015). The representative dominant microbes in low-temperature Daqu Qingcha, such as Saccharopolyspora sp., showed a significant positive correlation with the starch content and a significant negative correlation with saccharifying power (Hou et al., 2022). Saccharopolyspora sp. is necessary for the generation of the flavor substance in Maotai Baijiu production, so it might also be a desirable genus in Jiang-flavor Danquan Baijiu production (Gan et al., 2019). Planifilum belongs to Thermoactinomycetaceae family, which comprises thermophilic organisms with Actinomyces-like mycelial growth (Logan and Halket, 2011). Planifilum can secrete xylanase to degrade xylan and fix nitrogen (Bjarnadóttir et al., 2018). Lactobacillus is frequently detected during the brewing process of different types of Baijiu in China, which plays a necessary role in yielding aroma components (He et al., 2019). Acinetobacter and Thermoactinomyces showed the strongest positive correlation with acetic acid synthesis; these two types of bacteria positively correlated with the synthesis of more than two organic acids (Zhao et al., 2020). As a lactic acid bacterium, Enterococcus can be isolated during the fermentation of HTD from Maotai liquor, and Enterococcus was also present in fermented grains (Wang et al., 2015). Lactococcus presented the strongest positive correlation with lactic acid synthesis (Zhao et al., 2020). Qiu et al. found that although Lactococcus garvieae LD3 had high arginine utilization in the production of medium temperature Daqu liquor, the strain could accumulate more citrulline to increase the ethyl carbamate content in fermented grains (Qiu et al., 2016). Previous studies have demonstrated that Streptomyces spp. in Daqu can introduce geosmin into the brewing process, resulting in the earthy-off flavor of the liquor (Du and Xu, 2012; Du et al., 2013). Saccharomonospora could hydrolyze phenolic compounds into a non-toxic form and reduce phytotoxicity (Shivlata and Satyanarayana, 2015). Tepidimicrobium belongs to Clostridiales, which played an important role in cellulose degradation (Koeck et al., 2013; Slobodkin et al., 2020). Anaerosalibacter is a halotolerant bacterium, which is associated with sludge and animal matter (Rezgui et al., 2012; Dione et al., 2016).
Differential Analysis of Bacterial Community in Three Samples
Principal coordinates analysis was used to visualize the difference of bacterial community in the three samples (Figure 4). PCoA based on the Bray–Curtis distance showed that three different samples have obvious difference in spatial arrangement, which was probably due to the large differences in fermentation time and type. It was worth noting that the spatial distance between dqjq_ck and dqjp3 was farthest, suggesting that dqjq_ck and dqjp3 differ more in bacterial community.
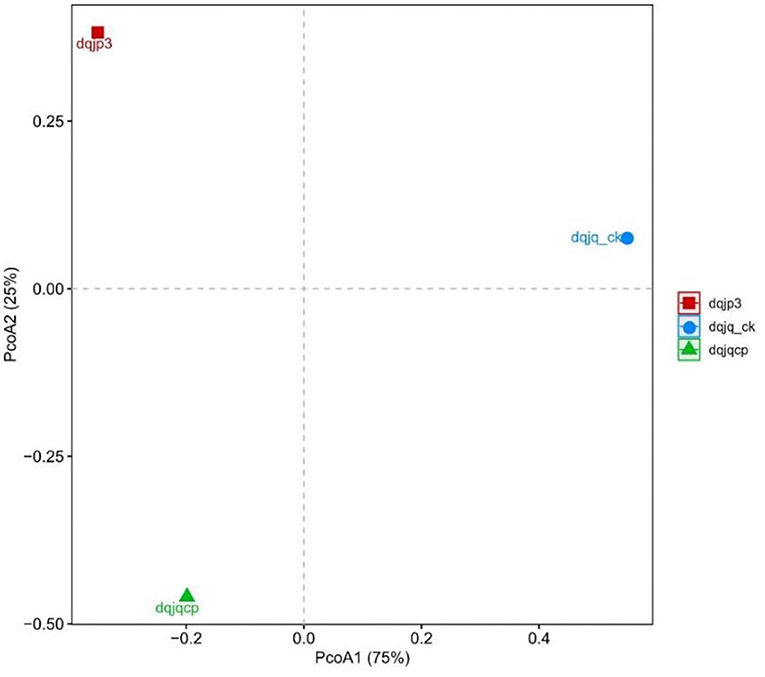
Figure 4. PCoA plot based on the 16S rDNA sequences from three samples.
PCoA showed significant differences among the bacterial communities of the three samples from the Danquan distillery (Figure 4). Although there were many identical genera in each sample, significant differences were apparent in the bacterial abundance among the three samples. In sample dqjq_ck, five genera such as Comamonas (32.00%), Myroides (18.03), Paenibacillus (15.92%), Staphylococcus (6.00%), and unclassified_Microbacteriaceae (4.70%) with high abundance accounted for 76.65% of the total abundance. In sample dqjqcp, five genera such as Bacillus (57.88%), Planifilum (6.76%), Saccharopolyspora (6.19), Kroppenstedtia (5.30%), and unclassified_Bacillales (2.78%) with high abundance accounted for 78.91% of the total abundance. In sample dqjp3, five genera such as unclassified_Lactobacillales (41.74%), Acetobacter (15.67%), Bacillus (9.04%), unclassified_Thermoactinomycetaceae_1 (8.72%), and unclassified_ Bacillaceae_2 (5.15%) with high abundance accounted for 80.32% of the total abundance. The significant differences of the bacterial communities occurred among the three samples, representing three important stages of Danquan Baijiu brewing, which were closely related to the environmental factors such as temperature, humidity, O2 content, and pH. Understanding the bacterial community succession in different stages of Baijiu production will help deepen the understanding of Baijiu fermentation mechanism and screen bacteria with excellent performance to prepare fortified Daqu for improving the quality of Baijiu. Some Bacillus strains with high yield of ethanol or flavor substance tetramethylpyrazine (TTMP) have been isolated and purified from three samples (dqjq_ck, dqjqcp, and dqjp3) based on our present study, and further functional research studies about these strains are ongoing.
Prediction of Bacterial Functional Potential
To profoundly recognize the role of bacteria in the three samples, based on 16S rDNA data, PICRUSt was used to predict bacterial functional potential in the three samples. Further analysis was performed in view of COG database. More than 4,000 functional proteins belonging to 24 COG categories were identified to obtain a bacterial COG profile. Enrichment of metabolic functions in the three samples is shown in Figure 5, which indicated that the bacterial metabolism was intense. It can be observed from Figure 5 that categories G (carbohydrate transport and metabolism), K (transcription), and T (signal transduction mechanisms) dominated in all samples, and the relative abundance of category K was highest among all categories.
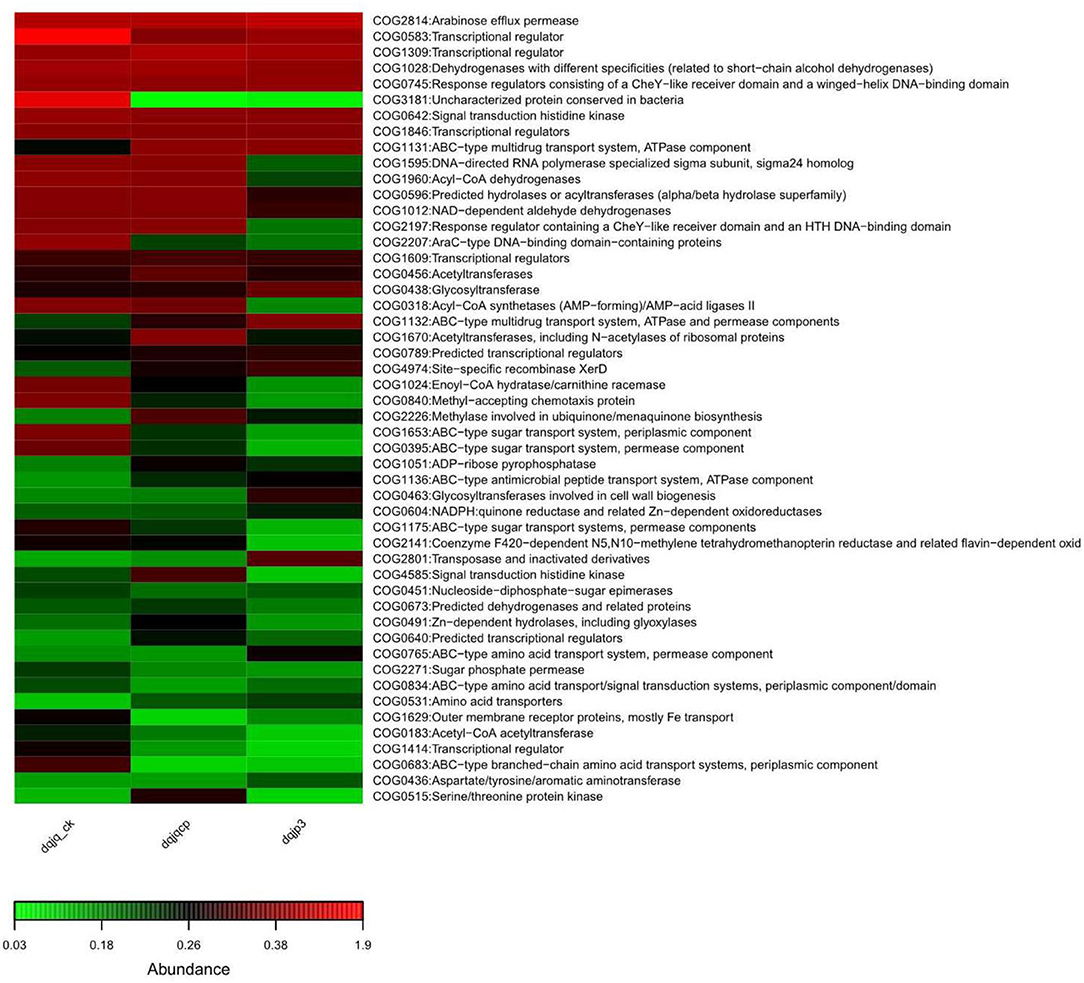
Figure 5. Overview of the bacterial COG profile in three samples.
The potential prediction of phenotypic functions in the three samples showed six dominant potential microbial phenotypes in all samples including chemoheterotrophy, aerobic chemoheterotrophy, fermentation, animal parasites or symbionts, human pathogens, and aromatic compound degradation (Figure 6).
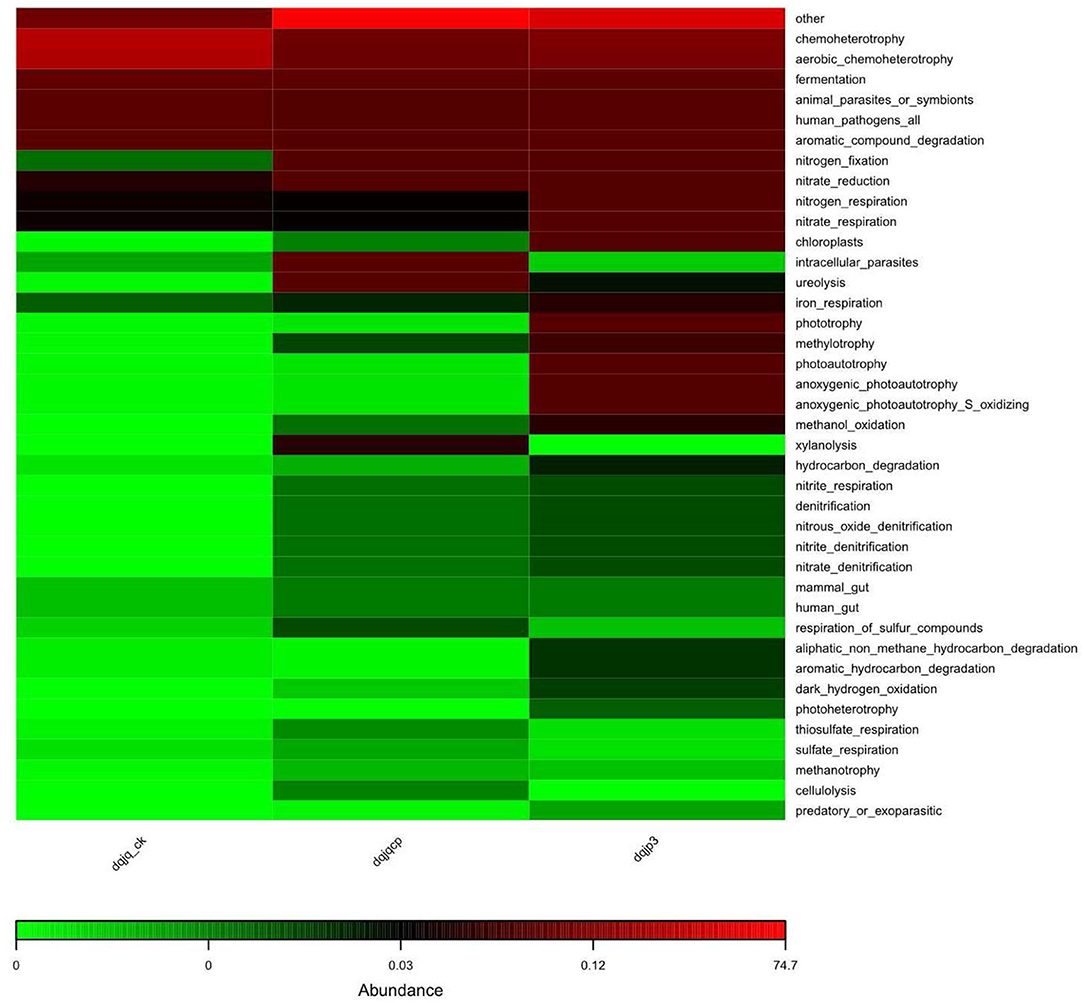
Figure 6. Prediction of bacterial functional potential.
Discussion
High-temperature Daqu is manufactured by traditional spontaneous solid-state fermentation in the open environment (Huang et al., 2017). The polymicrobial co-fermentation process under the high-temperature condition is a distinct character of HTD, which is obviously different from other types of Daqu such as low-temperature Daqu for light-flavor Baijiu (Cai et al., 2021). Bacteria are important microbes, which produce a variety of aroma substances and enzymes in HTD.
In this study, Illumina MiSeq high-throughput sequencing was used to examine bacterial diversity of two HTDs (dqjq_ck and dqjqcp) and one fermented grain (dqjp3), respectively. Figure 2B shows that the dominant genera in the three samples were Bacillus, Comamonas, Myroides, Paenibacillus, Acetobacter, and unclassified_Lactobacillales. Different from our results, Jin et al. (2019) revealed dominant bacterial genera in HTD from Moatai and Xijiu towns, Guizhou province, China, were Bacillales, Enterobacteriales, and Lactobacillales, and Wang Y. et al. (2020) found that dominant bacterial genera in HTD from a traditional Maotai-flavor liquor production factory in Xiangyang, Hubei province, China, were Thermoactinomyces, Staphylococcus, Lentibacillus, Kroppenstedtia, and Saccharopolyspora (Jin et al., 2019; Wang Y. et al., 2020). This phenomenon suggested that HTD from different regions had a distinct bacterial community due to different environmental conditions.
The result of Figure 2B indicated that the most abundant species in dqjq_ck was Comamonas, and its relative abundance was highest in sample dqjq_ck. As the denitrifying bacteria, Comamonas can use oxygen present in nitrate or nitrite to oxidize the carbon, which leads to the formation of nitrogen gas from harmful nitrate or nitrite for human health when there is no oxygen available (Chu et al., 2022). Comamonas could be used as the indole-degrading genus (Yang et al., 2022). The dominant microbe Comamonas sp. was linked to the yield of a unique PHA member poly(3-hydroxyisobutyrate) with an isobutyrate-fed enrichment culture (Vermeer et al., 2022). Comamonas played a dominant role in tetracycline hydrochloride degradation and power generation in a microbial fuel cell (Zhang et al., 2021). Comamonas testosteroni YAZ2 could be used for the cleanup of polychlorinated biphenyl water pollution (Hara et al., 2021). The laccase enzymatic traits and delignification procedures of rice straw by Comamonas testosteroni FJ17 were investigated (Wang et al., 2021). Comamonas thiooxydans CG-1 had the ability for biodegradation of polyethylene terephthalate (a general plastic with non-degradable property) (Huang et al., 2022). Comamonas genus can degrade the raw materials in fermented grains, provide carbon source for the growth of other microorganisms, and participate in the formation of organic acids (Li and Qiu, 2021). Comamonas acidovorans has the ability of inhibiting spoilage mold. The existence of these bacteria can reduce the side effects caused by the invasion of spoilage mold in the brewing system (Yu et al., 2016). In brief, genus Comamonas could degrade a variety of pollutants in liquor production process.
The result of Figure 2B indicated that the most abundant species in dqjqcp was Bacillus, and its relative abundance was highest in sample dqjqcp. Genus Bacillus had been previously thought as a most represented bacterium with importance in a variety of Daqu (Zhang et al., 2014; Zheng et al., 2014; Hu et al., 2017). Genus Bacillus had been demonstrated to be the major Jiang flavor-producing bacterium for the yield of Jiang-flavor Baijiu, which could produce a variety of degrading enzymes such as protease, cellulase, amylase, and glucoamylase. These enzymes could hydrolyze macromolecules (protein, starch, etc.) to produce many flavor compounds during the production of Jiang-flavor Baijiu (Cai et al., 2021; Wu et al., 2021). Specific species of genus Bacillus and its quantity obviously affect the quality of Daqu and liquor style (Fan et al., 2018). For example, previous studies indicated that Bacillus subtilis and Bacillus sarcina could produce a red pigment, which might play an important role in Baijiu production (Zhao et al., 2009; Wei et al., 2014; Deng et al., 2020). Flavor metabolism of Saccharomyces cerevisiae by mixed culture with Bacillus licheniformis was improved during Chinese Maotai-flavor Baijiu fermentation, and the content of 12 flavor compounds, containing fatty acids and their esters, terpene, and aromatic compounds, was improved (Meng et al., 2015). Bacillus licheniformis stains MT-6 and MT-15 could produce the volatile compounds including C4 compounds, volatile acids, pyrazines, and aromatic and phenolic compounds, which could improve aroma concentration during the fermentation of Moutai-flavor Baijiu (Zhang et al., 2013). Bacillus cereus could make flavor compound ethyl hexanoate during the production process of Baijiu (Zhao et al., 2017). B. cereus and B. licheniformis were the most abundant bacteria to produce α-amylase in Jiuqu (Wang Z. M. et al., 2020). As the flavor-producing strains, Bacillus amyloliquefaciens and Bacillus subtilis could secrete α-amylase to enhance the saccharification process of raw materials and improve the contents of alcohols, esters, and diverse organic acids (Li et al., 2014; Yao et al., 2015). The relative abundance of genus Bacillus was higher at sites rich in oxygen in Daqu (He et al., 2019). Therefore, the most abundant species in sample dqjqcp, which was distributed in a well-ventilated area with more oxygen, is genus Bacillus. However, sample dqjq_ck was covered with thick rice straw with the least oxygen concentration, so relative abundance of genus Bacillus was the lowest. In addition, Bacillus could adapt to various unfavorable conditions such as heat and acid owing to the formation of spores (Wang W. et al., 2018; Mirza et al., 2022). In addition, genus Bacillus was the dominant genus in wheat (Zheng et al., 2015). Therefore, wheat as one of raw materials of two HTD samples (dqjqcp and dqjq_ck) was an important source of genus Bacillus (Du et al., 2019).
The result of Figure 2B indicated that the most abundant species in dqjp3 was unclassified Lactobacillales, and its relative abundance was highest in sample dqjp3. Lactobacillales is in the order of lactic acid bacteria. Lactobacillales was the dominant bacterium in Daqu owing to its extraordinary ability to adapt to various temperature and moisture, which helped it survive in high-temperature environments (Zhang et al., 2014; Wang and Xu, 2015). Lactobacillales can produce organic acids which may affect the growth of other microbes. For example, acetic acid can inhibit the growth of microbes and affect the structure of microbial community during the process of high-temperature fermentation (Nie et al., 2013). Furthermore, most genera of lactic acid-producing bacteria such as Lactobacillales could produce antibiotics such as bacteriocin, tetracycline, erythromycin, aminoglycoside, and vancomycin (Wu et al., 2017; Anisimova and Yarullina, 2019; Campedelli et al., 2019; Todorov et al., 2019; Arellano et al., 2020; Colautti et al., 2022). Lactobacillales and Acetobacter were found to affect the biomarkers through producing organic acids and bacteriocins and advance the occurrence of microbial community succession (Ma et al., 2022). In summary, Lactobacillales played an important role in flavor formation, microorganism antagonism, and microbial community succession.
Danquan Baijiu production involved three stages: the first stage (45 d of the first Daqu fermentation), the second stage (90 d of the second Daqu fermentation for the mature Daqu), and the third stage (mixing the crushed mature Daqu with the steamed sorghum for Baijiu brewing). After the end of each stage, the samples were collected, which in turn were dqjq_ck, dqjqcp, and dqjp3. Numerous studies had shown that Bacillus genus had the potential to produce alcohol. Bacillus subtilis can survive in different substrates because of its metabolic diversity and robust systems for the yield and secretion of a diversity of enzymes (Stülke and Hillen, 2000; Zhang and Zhang, 2010; Gu et al., 2018; Banerjee et al., 2019). Bacillus subtilis with innate ability to ferment a diversity of carbohydrates from mono-, di-, oligo-, and polysaccharides (such as starch, xylan, galactan, pullulan, arabinan, rhamnogalacturonan, and pectin) seems promising for inexpensive consolidated bioprocessing of bioethanol production using renewable plant biomass and wastes (Maleki et al., 2021). In addition, the ethanologenic Bacillus subtilis AP was estimated without and with Xyn-2 for bioethanol production from wheat bran. The strain Bacillus subtilis AP could produce 5.5 g/L ethanol, with a yield of 22.6% in consolidated bioprocessing (Rajabi et al., 2022). The proportions of Bacillus genus in dqjq_ck, dqjqcp, and dqjp3 were 1.43, 57.88, and 9.04%, respectively. With the higher HTD-making temperature, most yeasts and molds are destroyed, and the microbial community of HTD primarily propagates thermophilic bacteria (Gan et al., 2019; Xie et al., 2020). Therefore, we inferred that as a kind of thermophilic bacteria, Bacillus genus might play an important role in the bioethanol production for Danquan Baijiu brewing. Through our study, the succession of microbiota in the three samples representing the three important stages of Danquan Baijiu brewing were revealed. Our results will provide an important theoretical basis for enhancing the brewing process and the quality of Jiang-flavor Danquan Baijiu.
The result of functional potential and microbiome phenotype prediction is shown in Figures 5, 6. The predicted COG functional profile by PICRUSt showed that four functional categories with higher relative abundance in the three samples included K (transcription), E (amino acid transport and metabolism), G (carbohydrate transport and metabolism), and C (energy production and conversion) among all functional categories. In the meantime, amino acid transport and metabolism also require energy. In HTD, amino acids took part in Maillard reaction at high temperature, which gave HTD unique Jiang flavor (Gan et al., 2019; Feng et al., 2020). The noticeable expression of the functional category G (carbohydrate transport and metabolism) could provide a lot of energy for material transport, the reproduction and growth, and respiration in microbes. Carbohydrate metabolism and amino acid metabolism are very vital for microbes, which play an important role in the survival of microbes and the implementation of related functions. The KEGG-based analysis indicated that Baijiu Daqu and huangjiu wheat Qu microbiota were enriched with genes related to carbohydrate metabolism, amino acid metabolism, and energy metabolism. This result suggested that Daqu and wheat Qu microbiota had huge potential for the degradation of raw materials (such as wheat, rice, and barley) and flavor compound metabolism, which was consistent with our study (Zhang et al., 2022). Cai et al. suggested that the dominant microbial functions (such as E, G, and C) were the most vital functions in low-temperature Daqu (LTD), which indicated that the fermentation of LTD depended on the transport and metabolism of carbohydrates and amino acids; meanwhile, the life activity of microbes relied on energy metabolism (Cai et al., 2021). Given all that, categories E, G, and C were vital for different types of Qu.
Simultaneously, all samples also had a higher relative abundance in categories K (transcription), J (translation, ribosomal structure, and biogenesis), L (replication, recombination, and repair), and M (cell wall/membrane/envelope biogenesis), which indicated that bacteria in all samples maintained the frequent metabolism and had a high ability of growth and reproduction. Furthermore, all samples also had a high relative abundance in categories P (inorganic ion transport and metabolism) and H (coenzyme transport and metabolism). The transport of different inorganic ions is essential for bacterial metabolism, which can be utilized as cofactors of a variety of enzymes to advance different biochemical reactions. About coenzyme transport and metabolism, enzymes in HTD could catalyze the metabolism of amino acids to enhance the fermentation rate and provide substrates for Maillard reaction under high-temperature conditions, which caused Jiang flavor of HTD [7, 8] (Gan et al., 2019; Xie et al., 2020).
Traditional fermented foods such as Baijiu are frequently produced by spontaneous fermentation with multiple microbes. Environmental factors play important roles in microbial succession. Models were constructed to predict the population of core microbiota by processing parameters (room humidity and room temperature) (Ban et al., 2022). This research will be helpful for regulating microbes via controlling processing parameters in spontaneous fermentation such as Daqu fermentation (Ban et al., 2022). The microbial community succession (MCS) was driven by environmental factors such as temperature, humidity, CO2, O2, acidity, and moisture during fermentation of Nongxiangxing Daqu, and each environmental factor drove the occurrence of MCS by different mechanisms (Ma et al., 2022). The primary driving factors of MCS were acidity, moisture, and temperature for fermentation of Nongxiangxing Daqu (Ma et al., 2022). In our study, the fermentation temperature and humidity of dqjq_ck and dqjqcp were 65–70°C/80–90% and about 40°C/natural humidity, respectively. We inferred that the significant differences in environmental factors such as temperature and humidity affected MCS in samples dqjq_ck and dqjqcp. Traditional spontaneous fermentation with MCS and the changes of environmental factors result in an unstable quality of fermented foods in the microecosystem (He et al., 2020). He et al. found that a higher abundance affected the yield of flavor metabolites and changed the interspecies interactions of microbial community (He et al., 2020). Then He et al. investigated the bioturbation effect of fortified Daqu inoculating Bacillus strains on microbial composition and flavor metabolite and found that fortified Daqu could reduce the yield of lactic acid (He et al., 2020). More studies had explained that the acidification of fermentation cellar was primarily derived from the accumulation of lactic acid bacteria. In our study, as lactic acid bacterium, unclassified Lactobacillales had the highest abundance in fermented grains (i.e., sample dqjp3), which perhaps affected the flavor and quality of Baijiu. The fortified Daqu could help improve the flavor and quality of Baijiu. The indigenous microorganisms can better adapt to the local environment. Therefore, we isolated the suitable microorganisms from fermented grains such as Bacillus strains to improve the flavor and quality of Danquan Baijiu.
Conclusion
In summary, the bacterial communities of the three samples from the Danquan distillery were analyzed. This article discussed the dominant genera with the potential for the improvement of the quality of Danquan Baijiu in the three samples. The most abundant species in dqjq_ck, dqjqcp, and dqjp3 were Comamonas, Bacillus, and unclassified Lactobacillales, respectively. Genus Comamonas could degrade a variety of pollutants in the fermentation process. Genus Bacillus could adapt to various unfavorable conditions, which was the major Jiang flavor-producing bacterium for the yield of Jiang-flavor Baijiu. The unclassified Lactobacillales played an important role in flavor formation, microorganism antagonism, and microbial community succession. With the changes in environmental conditions (fermentation temperature and humidity) and sample types, the microflora also underwent the corresponding adaptive changes. The results would lay the foundation for the isolation of some excellent indigenous bacteria and the construction of their genetic engineering modification to prepare fortified Daqu and improve the quality of Danquan Baijiu in future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioSample - SAMN26292234, SAMN26292235, and SAMN26292236.
Author Contributions
CS: conceptualization, writing-review and editing, supervision, project administration, funding acquisition, and methodology. YL: software. YL and JZ: validation. CS, YL, and JZ: formal analysis. YL, JZ, and SG: investigation and data curation and writing-original draft preparation. JZ and SG: visualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by Guangxi Key Research and Development Program (AB21220057 and 2021AB27009), Research Funds of the Guangxi Key Laboratory of Landscape Resources Conservation and Sustainable Utilization in Lijiang River Basin, Guangxi Normal University (LRCSU21Z0207), Research Funds of Key Laboratory of Ecology of Rare and Endangered Species and Environmental Protection (Guangxi Normal University), Ministry of Education, China (ERESEP2022Z11), National Training Program of Innovation and Entrepreneurship for Undergraduates (202210602064), and Innovation Project of Guangxi Graduate Education (YCSW2022178 and XJCY2022011).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.883122/full#supplementary-material
Supplementary Table 1. The proportion of OTUs in three samples.
Supplementary Table 2. Gene copy numbers of bacterial communities in three samples.
References
Anisimova, E. A., and Yarullina, D. R. (2019). Antibiotic resistance of LACTOBACILLUS strains. Curr. Microbiol. 76, 1407–1416. doi: 10.1007/s00284-019-01769-7
Arellano, K., Vazquez, J., Park, H., Lim, J., Ji, Y., Kang, H. J., et al. (2020). Safety evaluation and whole-genome annotation of lactobacillus plantarum strains from different sources with special focus on isolates from green tea. Probiotics. Antimicrob. Prot. 12, 1057–1070. doi: 10.1007/s12602-019-09620-y
Ban, S., Chen, L., Fu, S., Wu, Q., and Xu, Y. (2022). Modelling and predicting population of core fungi through processing parameters in spontaneous starter (Daqu) fermentation. Int. J. Food Microbiol. 363, 109493. doi: 10.1016/j.ijfoodmicro.2021.109493
Banerjee, S., Mishra, G., and Roy, A. (2019). Metabolic engineering of bacteria for renewable bioethanol production from cellulosic biomass. Biotechnol. Bioprocess. Eng. 24, 713–733. doi: 10.1007/s12257-019-0134-2
Bjarnadóttir, M., Að*albjörnsson, B. V., Nilsson, A., Slizyte, R., Roleda, M. Y., Hreggvið*sson, G. Ó., et al. (2018). Palmaria palmata as an alternative protein source: enzymatic protein extraction, amino acid composition, and nitrogen-to-protein conversion factor. J. Appl. Phycol. 30, 2061–2070. doi: 10.1007/s10811-017-1351-8
Cai, W., Wang, Y., Ni, H., Liu, Z., Liu, J., Zhong, J., et al. (2021). Diversity of microbiota, microbial functions, and flavor in different types of low-temperature Daqu. Food Res. Int. 150, 110734. doi: 10.1016/j.foodres.2021.110734
Campedelli, I., Mathur, H., Salvetti, E., Clarke, S., Rea, M. C., Torriani, S., et al. (2019). Genus-wide assessment of antibiotic resistance in Lactobacillus spp. Appl. Enveron. Microbiol. 85, 1–21. doi: 10.1128/AEM.01738-18
Chen, Y., Li, K., Liu, T., Li, R., Fu, G., Wan, Y., et al. (2021). Analysis of difference in microbial community and physicochemical indices between surface and central parts of Chinese special-flavor Baijiu Daqu. Front. Microbiol. 11, 592421. doi: 10.3389/fmicb.2020.592421
Chu, Y. X., Wang, J., Jiang, L., Tian, G., and He, R. (2022). Intermittent aeration reducing N2O emissions from bioreactor landfills with gas-water joint regulation. Waste Manag. 139, 309–320. doi: 10.1016/j.wasman.2021.12.041
Colautti, A., Arnoldi, M., Comi, G., and Iacumin, L. (2022). Antibiotic resistance and virulence factors in lactobacilli: something to carefully consider. Food Microbiol. 103, 103934. doi: 10.1016/j.fm.2021.103934
Deng, L., Mao, X., Liu, D., Ning, X. Q., Shen, Y., Chen, B., et al. (2020). Comparative analysis of physicochemical properties and microbial composition in high-temperature Daqu with different colors. Front. Microbiol. 11, 588117. doi: 10.3389/fmicb.2020.588117
Dione, N., Sankar, S. A., Lagier, J. C., Khelaifia, S., Michele, C., Armstrong, N., et al. (2016). Genome sequence and description of Anaerosalibacter massiliensis sp. nov. New Microbes New Infect. 10, 66–76. doi: 10.1016/j.nmni.2016.01.002
Douglas, G. M., Beiko, R. G., and Langille, M. G. (2018). Predicting the functional potential of the microbiome from marker genes using PICRUSt. Methods Mol. Biol. 1849, 169–177. doi: 10.1007/978-1-4939-8728-3_11
Dreier, M., Meola, M., Berthoud, H., Shani, N., Wechsler, D., and Junier, P. (2022). High-throughput qPCR and 16S rRNA gene amplicon sequencing as complementary methods for the investigation of the cheese microbiota. BMC Microbiol. 22, 48. doi: 10.1186/s12866-022-02451-y
Du, H., Lu, H., Xu, Y., and Du, X. (2013). Community of environmental streptomyces related to geosmin development in Chinese liquors. J. Agric. Food Chem. 61, 1343–1348. doi: 10.1021/jf3040513
Du, H., Wang, X., Zhang, Y., and Xu, Y. (2019). Exploring the impacts of raw materials and environments on the microbiota in Chinese Daqu starter. Int. J. Food Microbiol. 297, 32–40. doi: 10.1016/j.ijfoodmicro.2019.02.020
Du, H., and Xu, Y. (2012). Determination of the microbial origin of geosmin in Chinese liquor. J. Agric. Food Chem. 60, 2288–2292. doi: 10.1021/jf204648e
Fan, G., Sun, B., Fu, Z., Xia, Y., Huang, M., Xu, C., et al. (2018). Analysis of physicochemical indices, volatile flavor components, and microbial community of a light-flavor Daqu. J. Am. Soc. Brew. Chem. 76, 209–218. doi: 10.1080/03610470.2018.1424402
Feng, E. (2015). Preliminary analysis on origin of distilled spirit in China. Jilin Univ. J. Soc. Sci. Edit. 55, 163–170. doi: 10.15939/j.jujsse.2015.01.018
Feng, N., Wu, H., Xie, Y., and Wu, Q. (2020). A novel drug delivery system obtained from hydrophobic modified amphiphilic polymers by Maillard reaction. Int. J. Biol. Macromol. 157, 146–150. doi: 10.1016/j.ijbiomac.2020.04.218
Fu, J. (2005). Study on the origin of liquor-making by <<Picture Illustration of China General History>>. Liquor-Mak. Sci. Technol. 3, 92–93. doi: 10.13746/j.njkj.2005.03.030
Gan, S. H., Yang, F., Sahu, S. K., Luo, R. Y., Liao, S. L., Wang, H. Y., et al. (2019). Deciphering the composition and functional profile of the microbial communities in Chinese Moutai liquor starters. Front. Microbiol. 10, 1540. doi: 10.3389/fmicb.2019.01540
Grady, E. N., MacDonald, J., Liu, L., Richman, A., and Yuan, Z. C. (2016). Current knowledge and perspectives of Paenibacillus: a review. Microb. Cell Fact. 15, 203. doi: 10.1186/s12934-016-0603-7
Gu, Y., Xu, X., Wu, Y., Niu, T., Liu, Y., Li, J., et al. (2018). Advances and prospects of Bacillus subtilis cellular factories: From rational design to industrial applications. Metab. Eng. 50, 109–121. doi: 10.1016/j.ymben.2018.05.006
Hara, T., Takatsuka, Y., Nakata, E., and Morii, T. (2021). Augmentation of an engineered bacterial strain potentially improves the cleanup of PCB water pollution. Microbiol. Spectr. 9, e0192621. doi: 10.1128/spectrum.01926-21
He, G., Huang, J., Wu, C., Jin, Y., and Zhou, R. (2020). Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res. Int. 129, 108851. doi: 10.1016/j.foodres.2019.108851
He, G., Huang, J., Zhou, R., Wu, C., and Jin, Y. (2019). Effect of fortified Daqu on the microbial community and flavor in Chinese strong-flavor liquor brewing process. Front. Microbiol. 10, 56. doi: 10.3389/fmicb.2019.00056
Hou, Q., Wang, Y., Cai,W., Ni, H., and Zhao,H., Zhang, Z., et al. (2022). Metagenomic and physicochemical analyses reveal microbial community and functional differences between three types of low-temperature Daqu. Food Res. Int. 156, 111167. doi: 10.1016/j.foodres.2022.111167
Hu, Y., Chen, M., Yang, Z., Cong, M., Zhu, X., and Jia, H. (2022). Soil microbial community response to nitrogen application on a swamp meadow in the arid region of central Asia. Front. Microbiol. 12, 797306. doi: 10.3389/fmicb.2021.797306
Hu, Y., Dun, Y., Li, S., Fu, B., Xiong, X., Peng, N., et al. (2017). Changes in microbial community during fermentation of high-temperature Daqu used in the production of Chinese ‘Baiyunbian’ liquor. J. Inst. Brew. 123, 594–599. doi: 10.1002/jib.455
Huang, Q. S., Yan, Z. F., Chen, X. Q., Du, Y. Y., Li, J., Liu, Z. Z., et al. (2022). Accelerated biodegradation of polyethylene terephthalate by Thermobifida fusca cutinase mediated by Stenotrophomonas pavanii. Sci. Total Environ. 808, 152107. doi: 10.1016/j.scitotenv.2021.152107
Huang, Y., Yi, Z., Jin, Y., Huang, M., He, K., Liu, D., et al. (2017). Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese nong-flavor liquor starter. Front. Microbiol. 8, 1747. doi: 10.3389/fmicb.2017.01747
Jia, W., Fan, Z. B., Du, A., Li, Y. L., Zhang, R., Shi, Q. Y., et al. (2020). Recent advances in Baijiu analysis by chromatography based technology–a review. Food Chem. 324, 126899. doi: 10.1016/j.foodchem.2020.126899
Jiang, L., Su, W., Mu, Y., and Mu, Y. (2020). Major metabolites and microbial community of fermented black glutinous rice wine with different starters. Front. Microbiol. 11, 593. doi: 10.3389/fmicb.2020.00593
Jin, Y., Li, D., Ai, M., Tang, Q., Huang, J., Ding, X., et al. (2019). Correlation between volatile profiles and microbial communities: a metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 121, 422–432. doi: 10.1016/j.foodres.2019.03.021
Koeck, D. E., Wibberg, D., Koellmeier, T., Blom, J., Jaenicke, S., Winkler, A., et al. (2013). Draft genome sequence of the cellulolytic Clostridium thermocellum wild-type strain BC1 playing a role in cellulosic biomass degradation. J. Biotechnol. 168, 62–63. doi: 10.1016/j.jbiotec.2013.08.011
Li, D. N., Huang, W., and Qiu, S. Y. (2019). Thermoflavimicrobium daqui sp. nov., a thermophilic microbe isolated from Moutai-flavour Daqu. Int. J. Syst. Evol. Microbiol. 69, 2709–2716. doi: 10.1099/ijsem.0.003528
Li, J., Sun, H., Wang, Q., Cai, Y., Shi, Z., Jia, J., et al. (2022). Microbial community spatial structures in Luzhou-flavored liquor pit muds with different brewing materials. PeerJ. 10, e12987. doi: 10.7717/peerj.12987
Li, Z., Bai, Z., Wang, D., Zhang, W., Zhang, M., Lin, F., et al. (2014). Cultivable bacterial diversity and amylase production in three typical Daqus of Chinese spirits. Int. J. Food Sci. Tech. 49, 776–786. doi: 10.1111/ijfs.12365
Li, Z., and Qiu, L. (2021). Change of prokaryotic community succession process of fermented grains during strong-flavor baijiu fermentation. J. Qilu Univ. Technol. 35, 26–31. doi: 10.16442/j.cnki.qlgydxxb.2021.04.005
Liu, P., Zhang, L., Du, X., Zhao, J., Gao, G., and Zhang, X. (2019). Dynamic analysis of physicochemical and biochemical indices and microbial communities of light-flavor Daqu during storage. J. Am. Soc. Brew. Chem. 77, 287–294. doi: 10.1080/03610470.2019.1629238
Liu, Y., Sun, M., Hou, P., Wang, W., Shen, X., Zhang, L., et al. (2022). Analysis of microbial community structure and volatile compounds in pit mud used for manufacturing Taorong-type Baijiu based on high-throughput sequencing. Sci. Rep. 12, 7347. doi: 10.1038/s41598-022-10412-8
Logan, N. A., and Halket, G. (2011). “Developments in the taxonomy of aerobic, endospore-forming bacteria,” in Endospore-Forming Soil Bacteria, eds Logan, N.A., Vos, P. (Berlin, Heidelberg: Springer Berlin Heidelberg), pp.1–29.
Ma, S., Luo, H., Zhao, D., Qiao, Z., Zheng, J., An, M., et al. (2022). Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing daqu. Bioresour. Technol. 345, 126549. doi: 10.1016/j.biortech.2021.126549
Maleki, F., Changizian, M., Zolfaghari, N., Rajaei, S., Noghabi, K. A., and Zahiri, H. S. (2021). Consolidated bioprocessing for bioethanol production by metabolically engineered Bacillus subtilis strains. Sci. Rep. 11, 13731. doi: 10.1038/s41598-021-92627-9
Mao, Q. (2014). ZaoShao Baijiu is the origin of Chinese Baijiu. Liquor-Mak. Sci. Technol. 10, 143. doi: 10.3969/j.issn.1001-9286.2014.10.057
Meng, X., Wu, Q., Wang, L., Wang, D., Chen, L., and Xu, Y. (2015). Improving flavor metabolism of Saccharomyces cerevisiae by mixed culture with Bacillus licheniformis for Chinese Maotai-flavor liquor making. J. Ind. Microbiol. Biotechnol. 42, 1601–1608. doi: 10.1007/s10295-015-1647-0
Mirza, M. A., Aruna, D., and Irukulla, M. (2022). Efficacy of Bacillus clausii UBBC-07 spores in the amelioration of oral mucositis in head and neck cancer patients undergoing radiation therapy. Cancer Treat Res. Commun. 31, 100523. doi: 10.1016/j.ctarc.2022.100523
Nie, Z., Zheng, Y., Wang, M., Han, Y., Wang, Y., Luo, J., et al. (2013). Exploring microbial succession and diversity during solid-state fermentation of Tianjin duliu mature vinegar. Bioresour. Technol. 148, 325–333. doi: 10.1016/j.biortech.2013.08.152
Qiu, Y., Fang, F., Zhou, X., Chen, X., Zhang, L., Du, G., et al. (2016). Characterization of arginine utilization strains from fermented grains and evaluation of their contribution to citrulline accumulation in Chinese Luzhou- flavor spirits, Acta. Microbiologica. Sinica. 56, 1638–1646. doi: 10.13343/j.cnki.wsxb.20160007
Rajabi, M., Nourisanami, F., Ghadikolaei, K. K., Changizian, M., Noghabi, K. A., and Zahiri, H. S. (2022). Metagenomic psychrohalophilic xylanase from camel rumen investigated for bioethanol production from wheat bran using Bacillus subtilis AP. Sci. Rep. 12, 8152. doi: 10.1038/s41598-022-11412-4
Ravyts, F., Steen, L., Goemaere, O., Paelinck, H., De Vuyst, L., and Leroy, F. (2010). The application of staphylococci with flavour-generating potential is affected by acidification in fermented dry sausages. Food Microbiol. 27, 945–954. doi: 10.1016/j.fm.2010.05.030
Rezgui, R., Maaroufi, A., Fardeau, M. L., Ben Ali Gam, Z., Cayol, J. L., Ben Hamed, S., et al. (2012). Anaerosalibacter bizertensis gen. nov., sp. nov., a halotolerant bacterium isolated from sludge. Int. J. Syst. Evol. Microbiol. 62, 2469–2474. doi: 10.1099/ijs.0.036566-0
Shivlata, L., and Satyanarayana, T. (2015). Thermophilic and alkaliphilic Actinobacteria: biology and potential applications. Front. Microbiol. 6, 1014. doi: 10.3389/fmicb.2015.01014
Slobodkin, A. I., Tourova, T. P., Kostrikina, N. A., Lysenko, A. M., German, K. E., Bonch-Osmolovskaya, E. A., et al. (2020). Tepidimicrobium ferriphilum gen. nov. sp. nov. a novel moderately thermophilic, Fe(III)-reducing bacterium of the order Clostridiales. Int. J. Syst. Evol. Microbiol. 56, 369–372. doi: 10.1099/ijs.0.63694-0
Stülke, J., and Hillen, W. (2000). Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 54, 849–880. doi: 10.1146/annurev.micro.54.1.849
Todorov, S. D., Tome, E., and Nero, L. A. (2019). Not everything is a question of reputation: safety of bacteriocinogenic lab isolated from smoked salmon. Chem. Eng. Trans. 75, 451–456. doi: 10.3303/CET1975076
Vermeer, C. M., Bons, L. J., and Kleerebezem, R. (2022). Production of a newly discovered PHA family member with an isobutyrate-fed enrichment culture. Appl. Microbiol. Biotechnol. 106, 605–618. doi: 10.1007/s00253-021-11742-9
Wang, H. Y., and Xu, Y. (2015). Effect of temperature on microbial composition of starter culture for Chinese light aroma style liquor fermentation. Lett. Appl. Microbiol. 60, 85–91. doi: 10.1111/lam.12344
Wang, L., Wang, Y. Y., Wang, D. Q., Xu, J., Yang, F., Liu, G., et al. (2015). Dynamic changes in the bacterial community in Moutai liquor fermentation process characterized by deep sequencing. J. Inst. Brew. 121, 603–608. doi: 10.1002/jib.259
Wang, L., Xue, C., Owens, G., and Chen, Z. (2021). Artificial intelligence modeling and molecular docking to analyze the laccase delignification process of rice straw by Comamonas testosteroni FJ17. Bioresour. Technol. 345, 126565. doi: 10.1016/j.biortech.2021.126565
Wang, W., Liu, R., Shen, Y., and Lian, B. (2018). The potential correlation between bacterial sporulation and the characteristic flavor of Chinese maotai liquor. Front. Microbiol. 9, 1435. doi: 10.3389/fmicb.2018.01435
Wang, X., Du, H., Zhang, Y., and Xu, Y. (2018). Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl. Environ. Microb. 84, e02369–e02317. doi: 10.1128/AEM.02369-17
Wang, Y., Cai, W., Wang, W., Shu, N., Zhang, Z., Hou, Q., et al. (2020). Analysis of microbial diversity and functional differences in different types of high-temperature Daqu. Food Sci. Nutr. 9, 1003–1016. doi: 10.1002/fsn3.2068
Wang, Z., Wang, S., Liao, P., Chen, L., Sun, J., Sun, B., et al. (2022). HS-SPME combined with GC-MS/O to analyze the flavor of Strong aroma baijiu Daqu. Foods 11, 116. doi: 10.3390/foods11010116
Wang, Z. M., Wang, C. T., Shen, C. H., Wang, S. T., Mao, J. Q., Li, Z., et al. (2020). Microbiota stratification and succession of amylase-producing Bacillus in traditional Chinese Jiuqu (fermentation starters). J. Sci. Food Agric. 100, 3544–3553. doi: 10.1002/jsfa.10405
Wei, P. W., Wang, L. Y., Luo, H. B., Rao, J. Q., Cao, Z. H., and Xu, D. C. (2014). Study on the metabolism and ecological conditions of two pigment-producing strains from Daqu. Liquor Mak. Sci. Technol. 242, 12–15. doi: 10.13746/j.njkj.2014.0126
Wu, C., Huang, J., and Zhou, R. (2017). Genomics of lactic acid bacteria: Current status and potential applications. Crit. Rev. Microbiol. 43, 393–404. doi: 10.1080/1040841X.2016.1179623
Wu, X., Jiang, Q., Wang, Z., Xu, Y., Chen, W., Sun, J., et al. (2021). Diversity, enzyme production and antibacterial activity of Bacillus strains isolated from sesame-flavored liquor Daqu. Arch. Microbiol. 203, 5831–5839. doi: 10.1007/s00203-021-02552-8
Xiao, Z., Liang, W., Zhu, X., and Zhao, J. Y. (2017). Solid-state fermentative production of aroma esters by Myroides sp. ZB35 and its complete genome sequence. J. Biotechnol. 245, 28–33. doi: 10.1016/j.jbiotec.2017.02.003
Xiao, Z., Zhu, X., Xi, L., Hou, X., Fang, L., and Lu, J. R. (2014). Biodegradation of C5-C8 fatty acids and production of aroma volatiles by Myroides sp. ZB35 isolated from activated sludge. J. Microbiol. 52, 407–412. doi: 10.1007/s12275-014-4109-x
Xie, M., Lv, F., Ma, G., Farooq, A., Li, H., Du, Y., et al. (2020). High throughput sequencing of the bacterial composition and dynamic succession in Daqu for Chinese sesame flavour liquor. J. Inst. Brew. 126, 98–104. doi: 10.1002/jib.592
Yang, J., Dou, X., and Ma, Y. (2018). Diversity and dynamic succession of microorganisms during Daqu preparation for Luzhou-flavour liquor using second-generation sequencing technology. J. Inst. Brew. 124, 498–507. doi: 10.1002/jib.528
Yang, J., Ma, F., Dai, C., Wu, W., Fan, S., Lian, S., et al. (2022). Indole metabolism by phenol-stimulated activated sludges: Performance, microbial communities and network analysis. Environ. Res. 207, 112660. doi: 10.1016/j.envres.2021.112660
Yao, S., Liu, Y., Li, H., Ge, Y. Y., Zhang, M. J., Xin, C. H., et al. (2015). Bacterial communities during the process of high-temperature Daqu production of roasted sesame-like flavour liquor. J. Inst. Brew. 121, 440–448. doi: 10.1002/jib.235
Yu, C., Xue, D., Zhou, M., and Wang, J. (2016). Analysis of diversity and brewing characteristics of bacterium in mechanization process of Fen-flavor Xiaoqu liquor. Hubei Agricult. Sci. 55, 2860–2863.
Yu, C., Yu, S., Zhang, Z., Li, Z., and Zhang, X. (2014). Oceanobacillus pacificus sp. nov., isolated from a deep-sea sediment. Int. J. Syst. Evol. Micr. 64, 1278–1283. doi: 10.1099/ijs.0.056481-0
Zhang, G., Liang, D., Zhao, Z., Qi, J., and Huang, L. (2021). Enhanced performance of microbial fuel cell with electron mediators from tetracycline hydrochloride degradation. Environ. Res. 206, 112605. doi: 10.1016/j.envres.2021.112605
Zhang, J., Liu, S., Sun, H., Jiang, Z., Xu, Y., Mao, J., et al. (2022). Metagenomics-based insights into the microbial community profiling and flavor development potentiality of baijiu Daqu and huangjiu wheat Qu. Food Res. Int. 152, 110707. doi: 10.1016/j.foodres.2021.110707
Zhang, L., Wu, C., Ding, X., Zheng, J., and Zhou, R. (2014). Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World J. Microbiol. Biotechnol. 30, 3055–3063. doi: 10.1007/s11274-014-1732-y
Zhang, R., Wu, Q., and Xu, Y. (2013). Aroma characteristics of Moutai-flavour liquor produced with Bacillus licheniformis by solid-state fermentation. Lett. Appl. Microbiol. 57, 11–18. doi: 10.1111/lam.12087
Zhang, X. Z., and Zhang, Y. H. P. (2010). One-step production of biocommodities from lignocellulosic biomass by recombinant cellulolytic Bacillus subtilis: Opportunities and challenges. Eng. Life Sci. 10, 398–406. doi: 10.1002/elsc.201000011
Zhao, C., Yan, X., Yang, S., and Chen, F. (2017). Screening of Bacillus strains from Luzhou-flavor liquor making for high-yield ethyl hexanoate and low-yield propanol. LWT-Food Sci. Technol. 77, 60–66. doi: 10.1016/j.lwt.2016.11.035
Zhao, D., Niu, G. J., Peng, Z. Y., Yang, R., and Chen, L. (2009). Isolation and identification of a red pigment-producing bacteria strain from luzhouflavor Daqu. Liquor Making Sci. Technol. 185, 53–57. doi: 10.13746/j.njkj.2009.11.003
Zhao, Q. S., Yang, J. G., Zhang, K. Z., Wang, M. Y., Zhao, X. X., Su, C., et al. (2020). Lactic acid bacteria in the brewing of traditional Daqu liquor. J. Inst. Brew. 126, 14–23. doi: 10.1002/jib.593
Zheng, F., Ma, Y., Hou, M., Sun, J., Sun, X., Huang, M., et al. (2017). Progress and prospect in aroma components in top six distilled spirits. Journal of Food Science and Technology 35, 1–12. doi: 10.3969/j.issn.2095-6002.2017.02.001
Zheng, X., Yan, Z., Nout, M. J. R., Smid, E. J., Zwietering, M. H., Boekhout, T., et al. (2014). Microbiota dynamics related to environmental conditions during the fermentative production of FenDaqu, a Chinese industrial fermentation starter. Int. J. Food Microbiol. 182, 57–62. doi: 10.1016/j.ijfoodmicro.2014.05.008
Zheng, X. W., Tabrizi, M. R., Nout, M. J. R., and Han, B. Z. (2011). Daqu—A traditional Chinese liquor fermentation starter. J. Inst. Brew. 117, 82–90. doi: 10.1002/j.2050-0416.2011.tb00447.x
Zheng, X. W., Yan, Z., Nout, M. J., Boekhout, T., Han, B. Z., Zwietering, M. H., et al. (2015). Characterization of the microbial community in different types of Daqu samples as revealed by 16S rRNA and 26S rRNA gene clone libraries. World J. Microbiol. Biotechnol. 31, 199–208. doi: 10.1007/s11274-014-1776-z
Keywords: bacterial diversity, Jiang-flavor Baijiu, Daqu, fermented grains, high-throughput sequencing
Citation: Shang C, Li Y, Zhang J and Gan S (2022) Analysis of Bacterial Diversity in Different Types of Daqu and Fermented Grains From Danquan Distillery. Front. Microbiol. 13:883122. doi: 10.3389/fmicb.2022.883122
Received: 24 February 2022; Accepted: 07 June 2022;
Published: 04 July 2022.
Edited by:
Nathalia Silva, State University of Campinas, BrazilReviewed by:
Huaxi Yi, Ocean University of China, ChinaLiliana Rocha, State University of Campinas, Brazil
Guangsen Fan, Beijing Technology and Business University, China
Copyright © 2022 Shang, Li, Zhang and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changhua Shang, shangchanghua@mailbox.gxnu.edu.cn
†These authors share first authorship