 Qian Wang
Qian Wang Lei Zhang
Lei Zhang Yiju Zhang
Yiju Zhang Huamin Chen
Huamin Chen Jianghua Song
Jianghua Song Mingjie Lyu
Mingjie Lyu Rui Chen
Rui Chen Lixin Zhang
Lixin Zhang- 1Anhui Province Key Laboratory of Integrated Pest Management on Crops, College of Plant Protection, Anhui Agricultural University, Hefei, China
- 2Institute of Crop Germplasm and Biotechnology, Tianjin Academy of Agricultural Sciences, Tianjin, China
- 3State Key Laboratory for Biology of Plant Disease and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 4College of Horticulture, Anhui Agricultural University, Hefei, China
Bacillus pumilus plays an important role in industrial application and biocontrol activities, as well as causing humans and plants disease, leading to economic losses and biosafety concerns. However, until now, the pathogenesis and underlying mechanisms of B. pumilus strains remain unclear. In our previous study, one representative isolate of B. pumilus named HM-7 has been recovered and proved to be the causal agent of fruit rot on muskmelon (Cucumis melo). Herein, we present a complete and annotated genome sequence of HM-7 that contains 4,111 coding genes in a single 3,951,520 bp chromosome with 41.04% GC content. A total of 3,481 genes were functionally annotated with the GO, COG, and KEGG databases. Pan-core genome analysis of HM-7 and 20 representative B. pumilus strains, as well as six closely related Bacillus species, discovered 740 core genes and 15,205 genes in the pan-genome of 21 B. pumilus strains, in which 485 specific-genes were identified in HM-7 genome. The average nucleotide identity (ANI), and whole-genome-based phylogenetic analysis revealed that HM-7 was most closely related to the C4, GR8, MTCC-B6033, TUAT1 and SH-B11 strains, but evolutionarily distinct from other strains in B. pumilus. Collinearity analysis of the six similar B. pumilus strains showed high levels of synteny but also several divergent regions for each strains. In the HM-7 genome, we identified 484 genes in the carbohydrate-active enzymes (CAZyme) class, 650 genes encoding virulence factors, and 1,115 genes associated with pathogen-host interactions. Moreover, three HM-7-specific regions were determined, which contained 424 protein-coding genes. Further investigation of these genes showed that 19 pathogenesis-related genes were mainly associated with flagella formation and secretion of toxic products, which might be involved in the virulence of strain HM-7. Our results provided detailed genomic and taxonomic information for the HM-7 strain, and discovered its potential pathogenic mechanism, which lay a foundation for developing effective prevention and control strategies against this pathogen in the future.
Introduction
Bacillus pumilus, an endospore-forming Gram-positive bacterium, residing in stratospheric air, soil, deep-sea sediments, and some extreme environments, is one of the best-known Bacillus species (Logan et al., 2009; Stepanov et al., 2016; Zhou et al., 2022). B. pumilus plays a crucial role in industry for producing abundant extracellular enzymes and secondary metabolites, and shows high adaptability and stress resistance (Bonifer et al., 2019; Zuo et al., 2022). This species is commonly utilized as probiotics in animal (Sanders et al., 2003; Hong et al., 2005). B. pumilus has also been widely used in agriculture for their beneficial actions on plants, such as plant-growth-promoting effect by facilitating plant nutrient uptake, phytohormone synthesis, phosphate solubilization, and biological nitrogen fixation (Hernandez et al., 2009), as well as antagonistic activities by producing antimicrobial agents (Huang et al., 1992; Agarwal et al., 2017; Dai et al., 2021). Meanwhile, increasing research has revealed that some B. pumilus strains were determined as pathogenic due to causing diseases in human and plants. For instance, foodborne illness (Logan, 2012) and cutaneous infection (Tena et al., 2007; Johnson et al., 2008) in human was associated with toxicity of B. pumilus strains. Several previous reports confirmed that B. pumilus caused diseases in a variety of forest trees (Kovaleva et al., 2015; Mazlan et al., 2019), fruits (Saleh et al., 1997; Galal et al., 2006; Li et al., 2009; Song et al., 2018), and vegetables (Peng et al., 2013), including the staple-food potato (Bathily et al., 2010), resulting in enormous economic losses and potential biological risk. However, to date, the pathogenic mechanism of these B. pumilus strains remain unclear.
Advances in sequencing technologies and the rapid development of genomic tools facilitate researchers to gain essential insights into the molecular basis of the strains at the genome level. So far, 177 genome assemblies of B. pumilus strains have been deposited into the National Center for Biotechnology Information (NCBI) public database, including 15 complete genomes, 4 chromosomes, and 158 draft genomes, most of which have been sequenced in the past 3 years. Comparative genomic approaches have been proven to be extremely valuable for functional characterization and classification of bacteria and fungi (Jothi et al., 2006). Recently, non-pathogenic B. pumilus have been investigated for their genomic features, phylogenetic relationships and evolutionary mechanisms by genomic analysis. Comparative genomic analysis of B. pumilus strains 7P and 3–19, revealed that nucleotide variants affected the streptomycin resistance and overproduction of extracellular hydrolases in B. pumilus 3–19 (Pudova et al., 2022). The niche-specific differences in genome expansion of antibacterial B. pumilus SF-4 and other eleven strains were revealed by genome mining and comparative genome analysis (Iqbal et al., 2021). Comparative analysis of marine-derived and the terrestrial B. pumilus strains revealed the evolutionary relationships, differentiation, and environmental adaptation (Fu et al., 2021). Yuan and Gao (2015) conducted genomic analysis of the ginger pathogen B. pumilus strain GR8, which revealed plant candidate pathogenic genes. Taken together, previous reports focused on the non-pathogenic strains of B. pumilus, whereas the pathogenic strains of B. pumilus and the underlying pathogenic mechanisms are remain elusive.
In our previous work, we isolated the pathogenic strain HM-7 from symptomatic fruit, which causes bacterial soft rot in melon. This strain was recognized as B. pumilus by sequence analysis of the 16S rRNA and the b-subunit of DNA gyrase (gyrB) gene (Song et al., 2018). In this study, we sequenced and assembled the genome of B. pumilus HM-7 and carried out comparative genomic analysis based on pan-core genome analysis, average nucleotide identity, phylogenetic and genomic collinearity analysis of all the available complete genomes or chromosomes of B. pumilus strains, as well as six closely related species (Bacillus anthracis, Bacillus cereus, Bacillus licheniformis, Bacillus safensis, Bacillus subtilis, and Bacillus thuringiensis). Genome annotation and analysis of pathogenic-associated genes revealed the genome characteristics, phylogenetic and taxonomic status of HM-7. Our results provide new insights into the pathogenic properties of B. pumilus and lay a solid foundation for controlling bacterial diseases caused by B. pumilus HM-7.
Materials and methods
The bacterial strain and DNA extraction
The representative isolate B. pumilus HM-7 used in this study was previously recovered from diseased tissues of muskmelon fruit (cv. “Xingtian20”) in Huainan, Anhui Province, China. The strain HM-7 was shake-cultured (220 rpm) in Nutrient Agar (NA) liquid medium (0.3% beef extract, 1.0% glucose, 0.5% peptone, 0.05% yeast extract) at 28°C for 24 h, and bacterial cells were harvested by centrifugation at 5,000 × g for 10 min at 4°C. Genomic DNA was extracted from the bacterial pellet using the TAKARA Bacterial DNA Kit (TaKaRa, Japan) according to the manufacturer’s instructions. The concentration and quality of genomic DNA was determined by NanoDrop (Thermo Fisher Scientific, Loughborough, UK) and agarose gel electrophoresis. B. pumilus HM-7 was preserved in the China Center for Type Culture Collection as CCTCC AB 2019388.
Genome sequencing, assembly and annotation
The whole genome sequencing of B. pumilus HM-7 was performed using the PacBio RS II single-molecule real-time (SMRT) sequencing platform. De novo assembly of high-quality long reads was carried out using HGAP (Chin et al., 2013) and further polished by Quiver (Chin et al., 2013). The resulting sequence data was assembled into a single contiguous genome. Genome annotation was performed using Prokka version 1.14 (Seemann, 2014). Open reading frames (ORFs) were predicted by Prodigal (Hyatt et al., 2010). Ribosomal RNA (rRNA) genes and transfer RNA (tRNA) genes was identified by the RNAmmer V1.2 (Lagesen et al., 2007) and tRNAscan-SE V2.0 (Lowe and Eddy, 1997), respectively. The interspersed repetitive sequences and tandem repeats were detected by RepeatMasker V4.1 (Chen, 2004) and the Tandem Repeats Finder (Benson, 1999). Genome visualization was carried out using the CGView Comparison Tool (Grant et al., 2012).
Functional analysis of the protein-coding genes was performed using Clusters of Orthologous Groups (COG) (Tatusov et al., 1997), Gene Ontology (GO) (Levasseur et al., 2013; Jones et al., 2014), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2016) databases. Pathogenicity analysis was conducted via a whole genome Blast search of the Carbohydrate-Active enZYmes (CAZy) database (Levasseur et al., 2013), the Virulence Factors Database (VFDB) (Chen et al., 2012), and the Pathogen Host Interactions (PHI) database (Winnenburg, 2006). An E-value cut-off of 1e-10 was set for the BLAST analysis.
Pan-genome analysis
A total of 20 complete genomes or chromosomes of B. pumilus strains and six type strains of closely related species were retrieved from the NCBI ftp site1 and listed in Supplementary Table 1. All genomes were re-annotated using Prokka (version 1.14) (Seemann, 2014) with identical default parameters. The pan-genome analysis of these genomes was evaluated, using the Roary program (Page et al., 2015) with a blast identity cutoff of 97% for comparison between B. pumilus strains, and a 40% cutoff for comparison between Bacillus species. Furthermore, we used R to map the petal plot to visualize the number of unique genes in each strain derived from the pan-genome analysis, as well as an UpSet plot to visualize the intersecting gene sets between B. pumilus HM-7 and five closely B. pumilus strains.
Average nucleotide identity and phylogenetic analysis
ANI were calculated by using JSpecies software (Richter and Rosselló-Móra, 2009) with default parameters to elucidate the interspecific relationship of these strains. A Pearson correlation matrix was generated, and correlation analysis ordered by hierarchical clustering was performed according to the procedures of Espariz et al. (2016).
Gene clusters that were shared among all strains and contained only single gene copies from each strain were referred to as single-copy genes (Zhong et al., 2018). We constructed phylogenetic tree based on single copies from the clustering result of OrthoFinder version 2.5 (Emms and Kelly, 2015) with default parameters. These protein sequences of each strains obtained from Prokka were selected and rooted using MUSCLE V3.8 (Edgar, 2004) with default parameters to perform multiple sequence alignment. The alignment was trimmed with GBlocks 0.91b (Gblocks $i -b4 = 5 -b5 = h -t = p -e = 0.2) (Talavera and Castresana, 2007) and used to infer the evolutionary history of strains with Randomized Axelerated Maximum Likelihood Algorithm (RAxML) (Stamatakis et al., 2008), based on the GAMMAJTT model for proteins (raxmlHPC-PTHREADS-SSE3 -T 64 -f a -x 123 -p 123 -N 1000 -m PROTGAMMAJTT -k -O -n $output_tag.tre -s $input_tag.phy). The reliability of the inferred tree was tested by bootstrapping with 1,000 replicates. The online tool iTOL2 was used for visualization.
Genomic collinearity analysis and three HM-7-specific regions
We analyzed the genomic architectures and syntenic relationships among the genomes clustered together with B. pumilus HM-7 using the Mauve Alignment System (Darling et al., 2004). Genomic islands (Gls) were determined with the IslandViewer 4 using four independent methods IslandPick, SIGI-HMM, IslandPath-DIMOB, and Islander. Prophages were detected using the PHASTER (PHAge Search Tool Enhanced Release).3 Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) elements were identified using CRISPR Finder software (Grissa et al., 2007). The output file “gene_presence_absence.csv” (Supplementary Table 2) was analyzed using Microsoft Excel to identify strain-specific genes present in HM-7 and absent in other strains based on the results of Roary analysis.
Results
Genomic features and annotation of B. pumilus HM-7
In order to obtain a high-quality genome sequence of B. pumilus HM-7, 5.27 Gb raw reads were produced from the PacBio platform, with an N50 length of 13,912 bp and an average length of 8,640 bp, accounting for approximately 1,340-fold genome coverage. De novo assembly of high-quality long reads generated a single contiguous sequence (contig) with a size of 3,951,520 bp and 41.04% GC content (Table 1). In total, 4,111 genes, three sets of 16S, 23S, and 5S rRNA operons, 24 rRNA genes, 82 tRNA genes, and 1 tmRNA were identified. Moreover, 136 interspersed repetitive sequences (IRSs) and 118 tandem repeats (TRs) were also discovered in this assembled genome (Table 1). The genomic loci of these protein-coding, rRNA, and tRNA genes were unevenly distributed across the genome (Figure 1A).

Table 1. General features of the B. pumilus HM-7 genome.
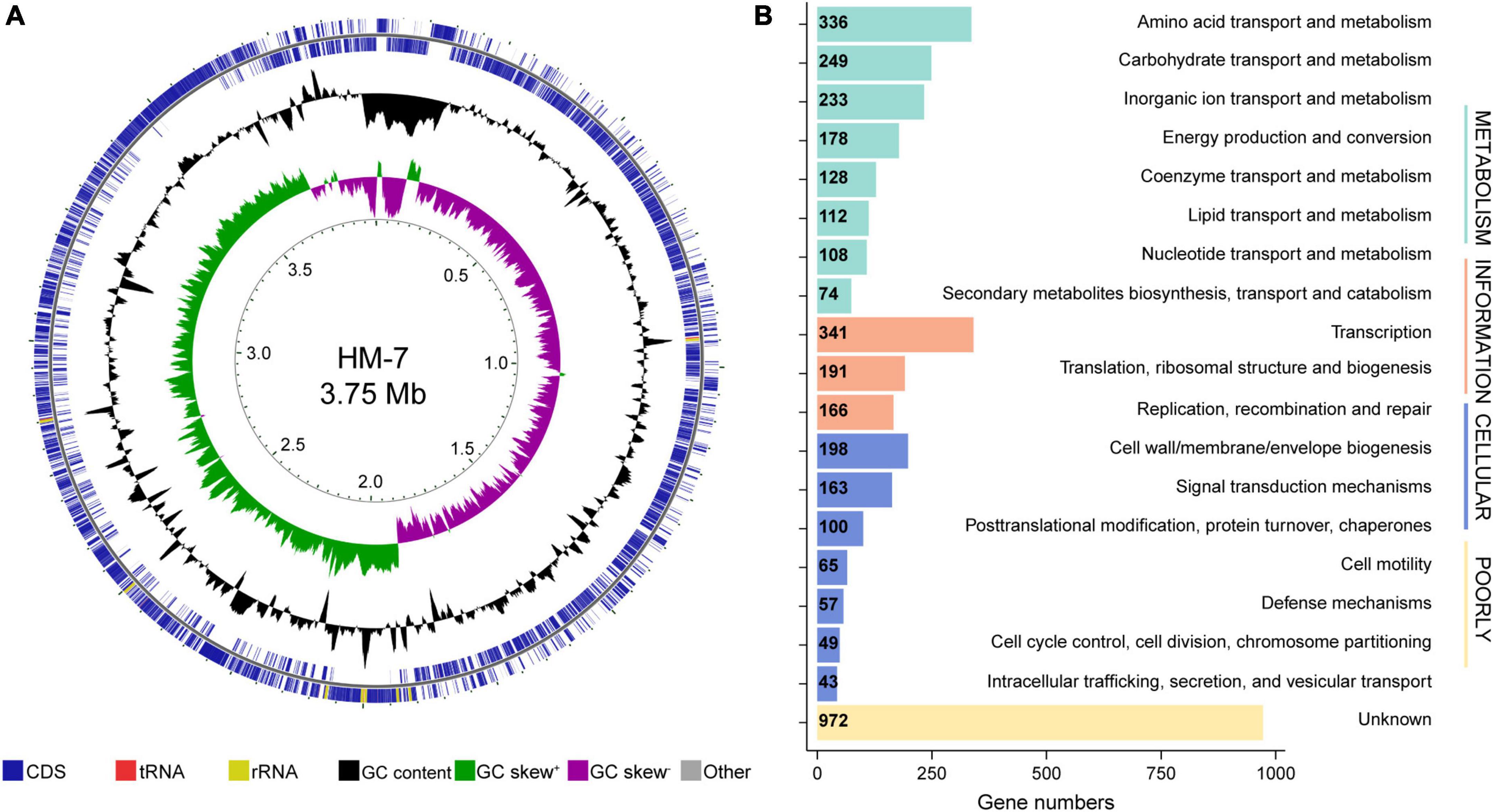
Figure 1. Genome analysis of B. pumilus strain HM-7. (A) From the inner to the outer circle: Circle 1, genomic position; Circle 2, GC skew; Circle 3, GC content; Circle 4 and 5, predicted protein-coding sequences (CDS) transcribed anticlockwise (inner part) and clockwise (outer part), respectively. (B) COG function classification of genes in HM-7, grouped into four main parts: Metabolism, cellular processes, information, and poorly.
To further determine the functions of the 4,111 annotated genes, we performed BLAST searches against the COG, GO, and KEGG databases. Of these, 3,479 (84.6%) predicted genes were assigned to the COG categories (Figure 1B): 1,418 of the genes were related to metabolism, 675 to cellular processes, 698 genes to information, and 972 genes to poorly characterized. A total of 919 (22.4%) genes could be assigned with certain GO definitions, including 677 genes in biological processes, 577 genes in cellular components, and 654 genes in molecular functions. Enriched GO terms focused on cellular process (608), metabolic process (528), cellular anatomical entity (567), obsolete cell part (534), and catalytic activity (454) (Supplementary Figure 1A). The KEGG annotation led to the identification of 1,268 genes with definite functions. Among the categories, metabolism was the largest group, containing metabolic pathway (613 genes), biosynthesis of secondary metabolites (294 genes), microbial metabolism in diverse environments (187 genes), biosynthesis of amino acids (131 genes), and others. The second largest group was the environmental information processing, mainly composed of ABC transporters (155 genes) and two-component system (109 genes) (Supplementary Figure 1B).
Potential pathogenesis-related genes in HM-7 genome
To investigate the pathogenic mechanisms of B. pumilus HM-7, we carried out the homology screening of potential virulence factors based on the CAZymes, VFDB, and PHI databases. CAZymes is an important virulence factors that involved in metabolism of host cell wall and required for invasion into host tissue (Lee et al., 2014). As a result, 484 (11.77%) CAZymes genes were identified in HM-7 genome, which were classified into six classes, encompassing auxiliary activities (AAs), glycosyltransferase (GT), glycoside hydrolases (GHs), carbohydrate-binding molecules (CBMs), carbohydrate esterases (CEs), and polysaccharide lyases (PLs). Of these, the most abundant enzymatic family was GT with 233 CAZyme encoding genes that could be further divided into 19 different families (Supplementary Table 3). The second most frequent enzyme subfamily was GHs (137 genes), followed by CBMs (58 genes), CEs (45 genes), AAs (8 genes), and PLs (3 genes) (Figure 2A and Supplementary Table 3).
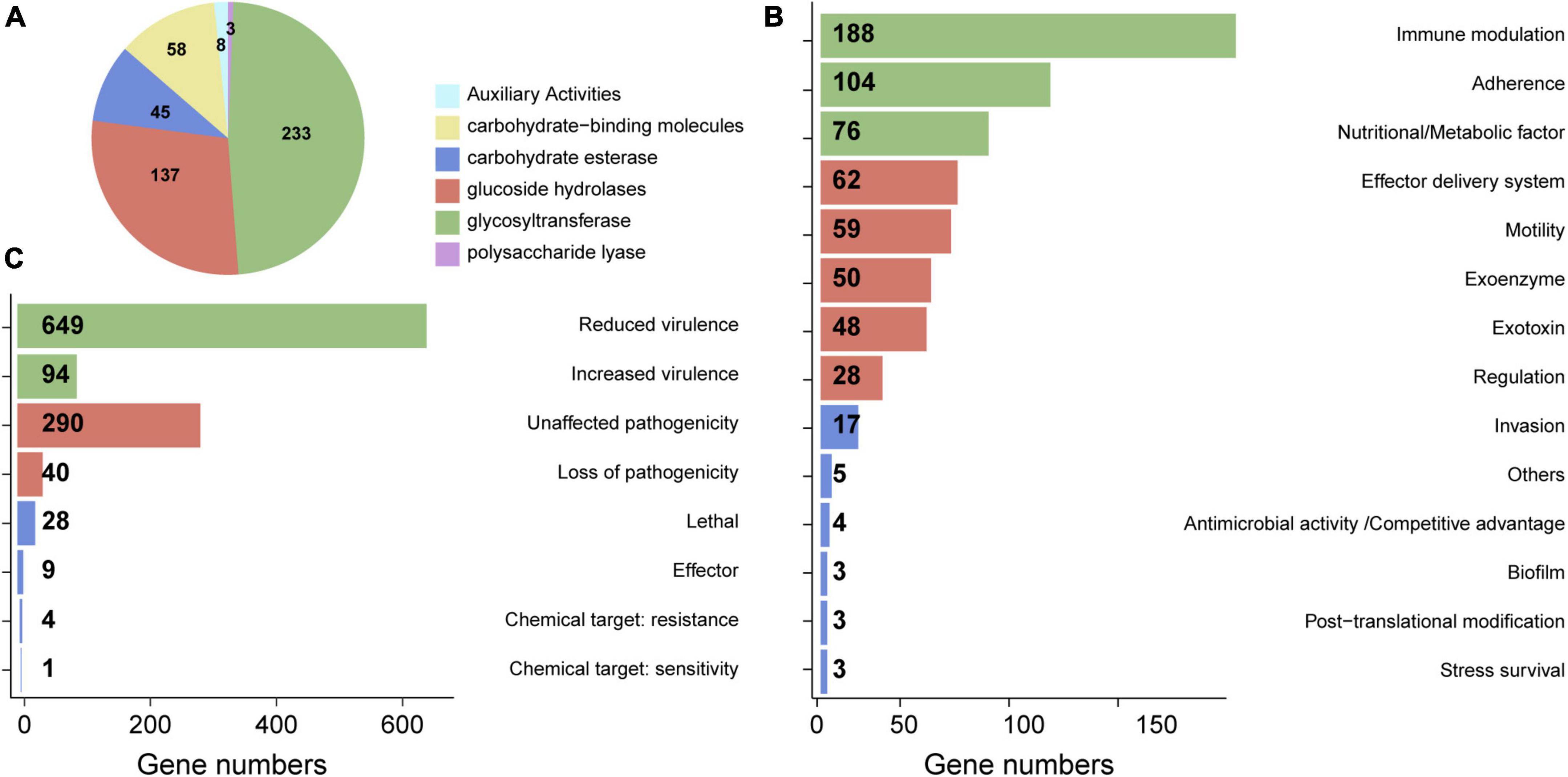
Figure 2. The annotation of CAZymes, VFDB, and PHI in B. pumilus HM-7 genome. (A) Distribution of CAZymes categories and gene number. (B) The functional categories of the B. pumilus HM-7 virulence genes according to the classification in the VFDB. (C) PHI classification and the genes number in HM-7. The list of genes are presented in Supplementary Tables 3–5.
The VFDB provides up-to-date knowledge of the virulence factors (VFs) of various bacterial pathogens (Yao et al., 2020). As a result, 650 (15.81%) virulence genes were identified, which could be assigned to 14 basal categories of VFDB. The largest identified category was immune modulation (188 genes), which facilitates the survival of pathogenic bacteria by controlling the host immune system, including anti-phagocytosis, disruption, and depletion of the complement system (Liu et al., 2022). pdgA, the most abundant gene in HM-7 genome, is involved in PG N-deacetylation to evade the host innate immune system (Supplementary Table 4; Coullon et al., 2020). Adherence, as the primary step in bacterial pathogenesis (Hori and Matsumoto, 2010), was detected as the second category, and consisted of 104 genes, such as chaperonin GroEL (groEL), immunogenic lipoprotein (IlpA), pili (pilD, pilJ, pilR), and EF-Tu (tuf) (Figure 2B and Supplementary Table 4). A previous study indicated that GroEL, pili, and EF-Tu may facilitate the invasion of the pathogenic B. pumilus strain GR-8 through motility and adhesion (Yuan and Gao, 2015). This suggests that these genes might play crucial roles for the pathogenesis of B. pumilus HM-7. In addition, distributions and abundances of nutritional/metabolic factors (metal uptake and metabolic adaptation), effector delivery systems (e.g., type II/III/IV/VII secretion system), motility, exoenzyme, exotoxins, and other categories were also identified in the genome of HM-7 (Figure 2B and Supplementary Table 4).
The PHI database contains experimentally verified pathogenicity, virulence, and effector genes from bacterial, fungal and protist pathogens (Urban et al., 2020). We predicted a total of 1,115 (27.12%) PHI genes, which were classified into five categories: virulence, pathogenicity, chemical susceptibility, effector, and lethal. Most of the PHI genes were assigned to the “virulence” class (743 genes), followed by the “pathogenicity” class (330 genes). Furthermore, 28, 9, and 5 genes were annotated as “lethal,” “effector,” and “chemical target” (Figure 2C and Supplementary Table 5). Taken together, these pathogenesis-related candidate genes offered important clues for understanding the pathogenic mechanisms of B. pumilus HM-7.
Pan-core genome analysis
To comprehensively determine the diversity and strain-specific characteristics among Bacillus species, pan-core analysis was performed using 21 strains of B. pumilus and six closely related Bacillus species. As expected, evidence from phylogenetic tree and core-pan gene numbers indicated that the percentage of unique genes was divergent in 27 strains of Bacillus spp. (Supplementary Figure 2A). The number of unique genes in B. subtilis 168 (1,002 genes), B. licheniformis SCDB-14 (1,226 genes), B. thuringiensis ATCC-10792 (1,231 genes), B. anthracis Ames-Ancestor-A2084 (877 genes), and B. cereus BC33 (466 genes) strains was much higher than those in the B. pumilus strains (Supplementary Figure 2B).
In terms of 21 strains of B. pumilus, five strains including MTCC-B6033, TUAT1, C4, SH-B11, and GR8 showed closer relationships to HM-7, which were clustered in the same branch (Figure 3A). The pangenome comprised 15,945 genes, and 740 genes shared by all B. pumilus strains (Figure 3B). The 8,631 genes present in more than one strain formed the accessory genome, and 6,574 genes specific to one single strain formed the unique genome ranging from 83 (B. pumilus 3–19) to 1,863 (B. pumilus 145) genes (Figures 3A,B). B. pumilus strain 145 also contained the highest percentage of unique genes (192 genes) within the Bacillus species (Supplementary Figure 2B). This was consistent with the previous study that strain 145 was evolutionarily distant from the other B. pumilus strains (Figure 3A; Iqbal et al., 2021). The soft core, shell, and cloud contained 406, 5,036, and 9,763 genes, respectively (Figure 3D).
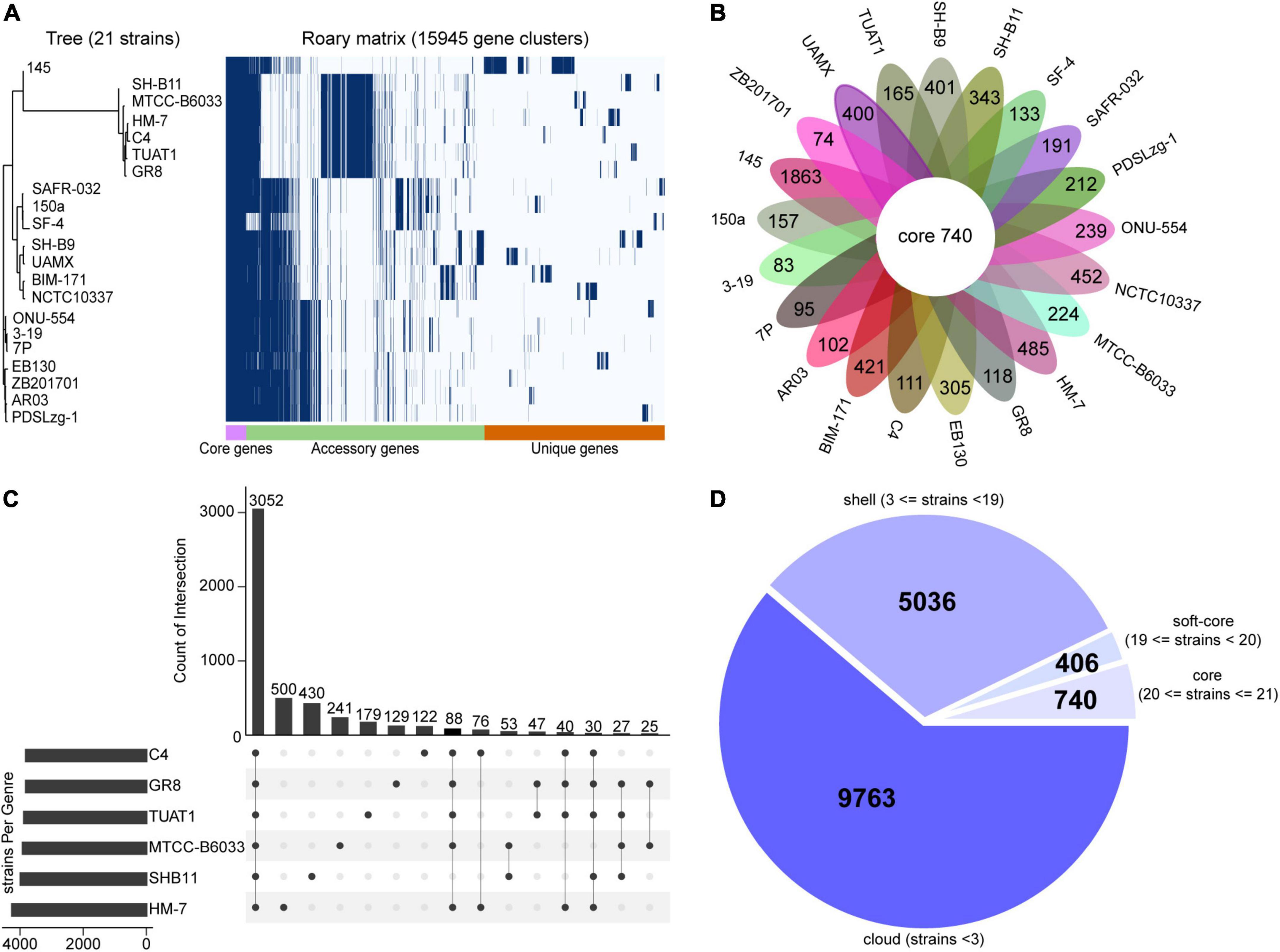
Figure 3. Pan-core genome analysis of 21 strains of B. pumilus. (A) Gene distribution of 21 B. pumilus strains based on the gene presence–absence matrix generated from Roary. A purple box, a green box, and an orange box to represent the core gene, accessory genes and specific genes, respectively. The phylogenetic tree on the left represents the phylogenetic relationships among the strains involved in the corresponding pan-genome. (B) Flower petal plot of 21 strains. Pan-genome analysis of 21 strains yielded unique genes for each strain, the numbers of which are shown on each petal plot. The center Circos shows that the number of core gene obtained from 21 strains with hard core threshold. (C) Upset plot show the gene set derived from the six strains. Bar numbers are sorted in descending order. Each bar represents the corresponding overlap core genes between two strains. Information provided on the left represent corresponding strain set. (D) Pie plot of 21 strains. Pan-genome analysis of 21 strains yielded accessory genes, including shell and cloud genes.
Further analysis of the six closely related B. pumilus strains led to the identification of 3,052 core genes, 1,601 unique genes, and 917 accessory genes (Figure 3C and Supplementary Table 2). Notably, HM-7 possessed the most abundant unique genes (500), which was more than the other five strains (Figure 3C).
Average nucleotide identity and phylogenetic analysis
To confirm the taxonomic identity and explore the phylogenetic relationship of these B. pumilus strains, pairwise ANI values were calculated, and a phylogenetic tree was constructed based on single-copies shared by all genomes. As a result, MTCC-B6033, TUAT1, C4, SH-B11, GR8, and HM-7 were clustered together, sharing more than 98% ANI with each other and 89% ANI with the other 15 strains of B. pumilus (Figure 4A). According to the ANI matrix and phylogenetic tree, 21 strains of B. pumilus were classified into four distinct clades (Figures 4A,B). HM-7 was clustered with MTCC-B6033, TUAT1, C4, SH-B11, and GR-8 (Figures 4A,B). Clade B comprised of seven strains: including ONU-554, 7P, 3-19, EB130, ZB201701, PDSLzg-1, and AR03. Clade D comprised of the other seven strains: SF-4, 150a, SAFR-032, SH-B9, UAMX, BIM-171, and NCTC10337 (Figure 4A). However, B. pumilus 145 was evolutionarily distant from the other strains and regarded as a separate clade C (Figure 4A and Supplementary Figure 3A). The ANI matrix and phylogenetic tree of all 27 Bacillus species supported this classification, in which B. pumilus 145 was obviously divergent from other B. pumilus strains (Supplementary Figure 3).
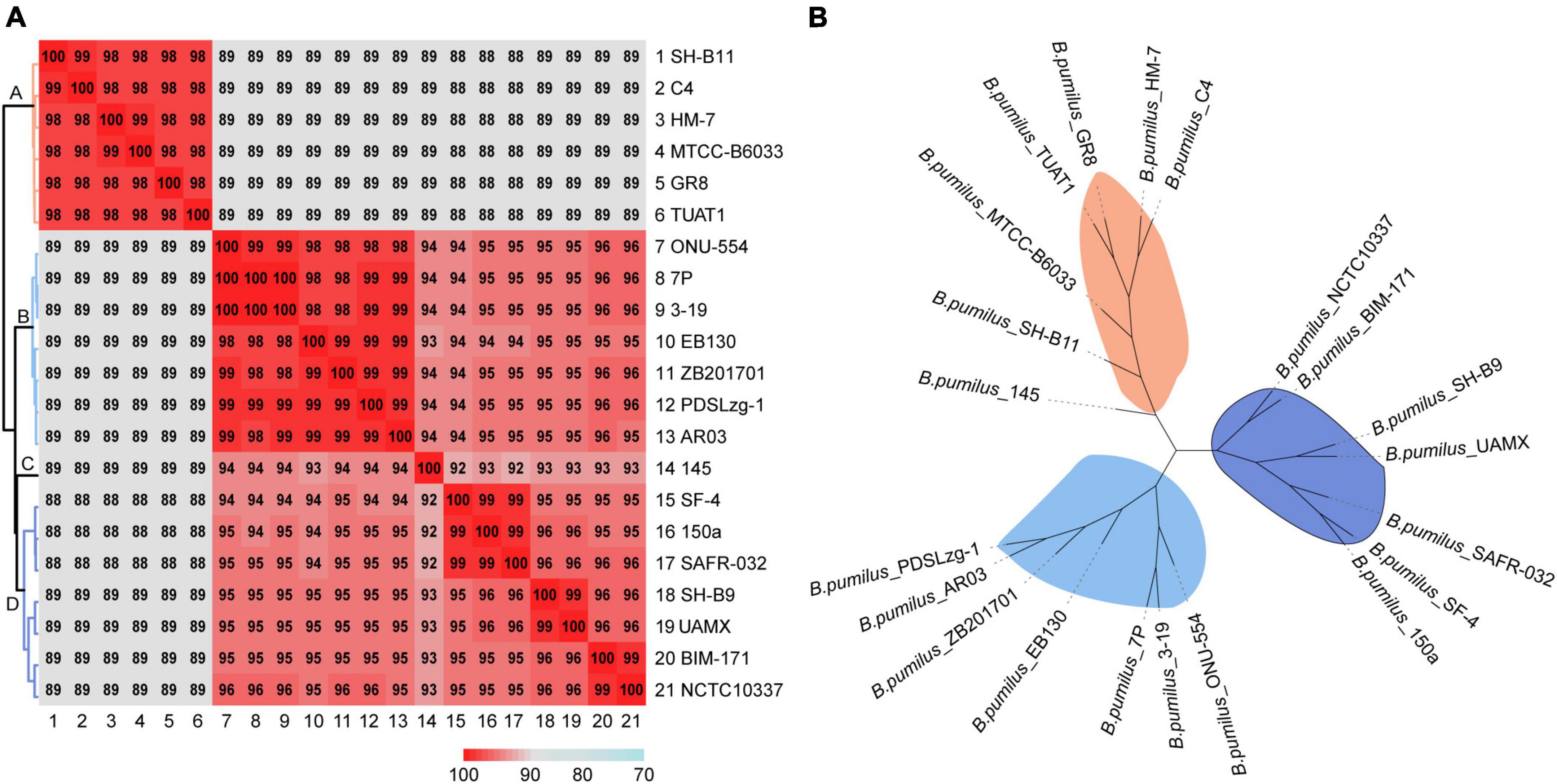
Figure 4. Evolutionary relationships of 21 strains of B. pumilus. (A) Heatmap and dendrogram of average nucleotide identity among different strains. (B) Phylogenetic tree based on total single-copy orthologous.
In addition, these 27 Bacillus strains were clearly clustered into five clades (Supplementary Figure 3A). B. safensis PgKB20 shared more than 89% ANI with 21 strains of B. pumilus, and 72–73% with other strains of Bacillus species (Supplementary Figure 3A). B. safensis PgKB20 was also clustered together with B. pumilus strains, but clearly separated from other Bacillus species (B. anthracis Ames-Ancestor-A2084, B. cereus BC33, B. licheniformis SCDB-14, B. subtilis 168, and B. thuringiensis ATCC-10792) (Supplementary Figure 3B). These results suggested that B. safensis PgKB20 was more closely related to B. pumilus than the other five strains of Bacillus species, confirming previous observations (Fu et al., 2021).
Genomic collinearity analysis and specific-regions of HM-7
Collinearity analysis can further identify the uniformity and variability among bacterial species at the genome-level, reflecting the common origin and specific features of target genome (Datta et al., 2020). The complete genome of HM-7 was compared with the other five closely releated B. pumilus strains (GR8, MTCC-B6033, TUAT1, C4, and SH-B11) to investigate the collinear relationship and orthologous distribution of genes. As a result, a total of 9 homologous regions were detected among the six genomes (Figure 5A). On the whole, a high degree of synteny was demonstrated among the six strains (regions with the same color). As for HM-7, three genomic regions with 107.3 kb (X), 153.2 kb (Y), and 36.8 kb (Z) were detected, which are obviously different from other B. pumilus strains.
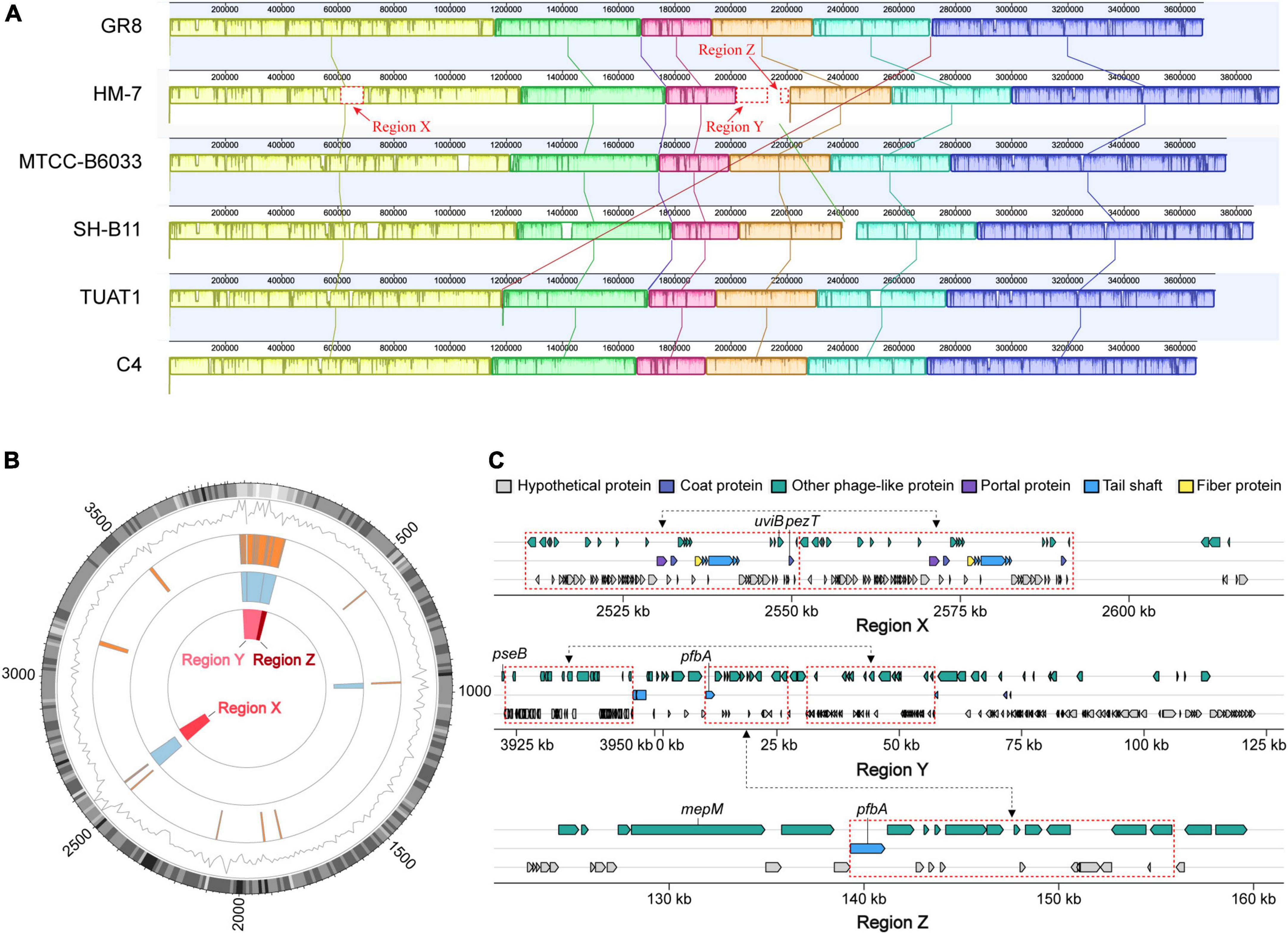
Figure 5. Comparative analysis and mobile genetic elements in the B. pumilus strains. (A) MAUVE alignment of the genome sequences of B. pumilus MTCC-B6033, TUAT1, C4, SH-B11, HM-7 and GR-8. Boxes in the same color represent homologous regions [local colinear blocks (LCBs)] between B. pumilus genomes. Uncolored regions within the LCBs or in-between LCBs indicate the presence of specific sequences in that strain. (B) Circular genome maps showing the locations of mobile genetic elements and three unique genomic regions (X, Y, Z) in B. pumilus strain HM-7. From the inner to the outer circle: Three unique genomic regions position (red boxes); prophages position (light blue boxes); GEIs position (orange boxes); black lines and gray heatmap indicate gene density and GC content, respectively. (C) Detailed information and predicted functions of genes located in the regions of X, Y, Z. Important virulence factors were marked with gene ID. Three pairs of blocks (dashed red) linked with black arrows represent repeat fragments.
Before dissecting the genes located in the specific-regions of HM-7, mobile genetic elements (MGEs) were analyzed, resulting in the identification of 21 genomic islands (GIs) including 5 putative prophages in the HM-7 genome (Figure 5B). Intriguingly, the X, Y, and Z specific-regions of HM-7 were overlapped with four prophages (177-100,490, 93,656-161,316, 2,508,533-2,591,517, and 3,920,319-3,951,028) (Figure 5B and Supplementary Table 6). The GC content of these specific-regions was relatively low in contrast to the average level of the genome (Figure 5B). In addition, three large repeat fragments were detected in the three regions (Figure 5C).
In total, we identified 424 strain-specific genes in these three specific-regions, accounting for 84.8% of the unique genes in HM-7 (Supplementary Table 7). Most of these genes were distributed in phage regions, suggesting that they were derived from phage-mediated lateral gene transfer (Figure 5C). In the case of pathogenic bacteria, strain-specific genes frequently encoded important virulence factors such as bacterial toxins (Fortier and Sekulovic, 2013). Herein, 19 candidate genes were concerned that might contribute to its pathogenesis (Table 2). Among them, the uviB and pezT genes were found in the region X that are associated with the export of bacteriocin and toxin, respectively. The pseB gene was predicted as VFDB factor ABZJ_00087, and encodes the UDP-N-acetylglucosamine- 4% 2C6-dehydratase, which is a sialic-acid-like sugar involved in flagellin modification and capsule formation (Hinderlich et al., 2013). It has been reported that phage tail genes seem to have developed dual functions and also serve as adhesion proteins for bacterial host attachment (Brüssow et al., 2004). Phage tail gene pfbA was predicted to encode the plasmin fibronectin-binding protein A, which binding to fibronectin and plasmin, working as virulence factors like adhesins/invasins for the LPXTG motif of the cell-anchoring sequence in Gram-positive bacteria (Yamaguchi et al., 2008). The mepM gene families were predicted to be the Murein DD-endopeptidase MepM, and functionally affected membrane invagination and cytokinesis (Babu et al., 2011). These candidate strain-specific genes located in the specific regions provided important information for parsing the pathogenesis of B. pumilus HM-7.

Table 2. The annotation of 19 specific-genes in HM-7 against the VFDB, PHI, and CAZYmes databases.
Discussion
B. pumilus has been widely applied in industry and agriculture for its ability to produce substances with biocatalysis, antimicrobial, and plant growth promoting activities (Gutierrez-Manero et al., 2001; Saggese et al., 2018; Hayat et al., 2020). However, increasing studies have revealed some B. pumilus strains can cause opportunistic infections and elicit biosecurity concerns (Johnson et al., 2008; Logan, 2012; Peng et al., 2013; Song et al., 2018; Mazlan et al., 2019), although its pathogenesis is still vague. In recent years, genome sequencing approaches and genomic analysis strategies have provided powerful tools to facilitate researchers distinguishing pathogenic from non-pathogenic microbe and dissecting the underlying mechanisms of bacterial pathogens (Thomson et al., 2006; Panthee et al., 2021). In our previous report, B. pumilus HM-7 was identified as a pathogen of muskmelon, causing the bacterial soft rot (Song et al., 2018). Herein, we performed the genome assembly and comparative genomic analysis to clarify genomic features and determine the virulence genes for B. pumilus HM-7, offering valuable information for better utilization of B. pumilus strains in the future.
The ANI analysis with genomic information is commonly used for evaluating the genomic distance and establishing species boundaries, which overcomes the challenges caused by evolutionary mutation rates and HGT events (Konstantinidis and Tiedje, 2007). An accurate phylogenetic tree supports our understanding of the major transitions in evolution (Emms and Kelly, 2015; Kapli et al., 2020), the phylogenetic tree based on single-copies shared by all genomes is more accurate than a tree constructed from only the 16S rRNA gene (Hahnke et al., 2016). In our present study, core and pan-genome analysis of 27 Bacillus strains revealed extensive genetic diversity among Bacillus species and B. pumilus strains. Notably, B. subtilis, B. licheniformis, and B. safensis, are much closer to B. pumilus than other members of the Bacillus species (Supplementary Figures 2, 3), corroborating previous findings (Zhao et al., 2012; Fu et al., 2021). Additionally, tightly clustered SF-4, SAFR-032, and 150a, as well as the highly divergent of strain 145, were consistent with a previously reported study (Iqbal et al., 2021). As a result, we found that MTCC-B6033, TUAT1, C4, SH-B11, and GR8 were highly similar to HM-7 based on the evidence from ANI analysis and phylogenetic trees using single-copy genes. Intriguingly, HM-7 had a high proportion of strain-specific genes, which provided an important clue to investigate pathogenic genes. According to previous reports, MTCC-B6033, SH-B11, TUAT1, and C4 were identified as biocatalyst, antifungal, biofertilizer, and keratin-degrading (Villanueva et al., 2014; Fellahi et al., 2016; Zam et al., 2016; Okazaki et al., 2019). While GR8 and HM-7 strains exhibited pathogenic effects to ginger and muskmelon, respectively (Yuan and Gao, 2015; Song et al., 2018). It was supposed that GR8 and HM-7 might be more similar in terms of virulence.
Previous studies have been elaborated the capacities and processes of bacterial pathogens, including adherence to host tissues, invasion, modulation of host inflammatory responses, and secretion of toxic products (Ramarao and Sanchis, 2013; Do Vale et al., 2016; Gu et al., 2019). In this study, candidate pathogenic genes associated with adhesion, immune escape and toxicity were found and potentially responsible for the virulence of strain HM-7 (Supplementary Table 4). It was reported that biofilm formation and bacterial adherence of B. amyloliquefaciens were disrupted when the genes of collagen-like proteins (CLPs, ClpA, ClpB, ClpC, and ClpD) were inactivated (Zhao et al., 2015). Simultaneously, the flagellar biosynthesis-related genes (fliD, fliE, flgD, flgE, flhA) were expressed in pathogenicity-activated Xanthomonas oryzae pv. oryzae cells at the transcriptome and proteome levels (Kim et al., 2021). These adhesion-related genes were detected in HM-7 genome, which might play important roles during invasion in this bacterium. The bacterial pathogen needs to escape the immune assaults of host in various forms such as phagocytosis and complement-mediated killing. Previous reports revealed that anti-phagocytosis of immune escape were associated with capsules genes in B. subtilis and B. anthracis (Makino et al., 2002; Gu et al., 2019). Here, cap genes (capB, capC, and capA), cps genes (cpsF, cpsG, cpsJ), and the genes of LplA1, LspA, PanD, PanC, SodA, and SodB related to capsule biosynthesis in Bacillus species (Koehler, 2002; Gu et al., 2019), were also found in the HM-7 genome. Moreover, genes involved in nutritional/metabolic system, such as metal (e.g., zinc, iron and magnesium) uptake and adaptation, could partially explain the virulence of strain HM-7, since they play the crucial role in proliferation and pathogenicity of bacterial pathogens (Liu et al., 2022).
Until now, B. pumilus has caused a variety of symptoms on muskmelon, ginger, peach, pine, and Asian pear (Saleh et al., 1997; Kotan et al., 2006; Li et al., 2009; Peng et al., 2013; Kovaleva et al., 2015; Song et al., 2018), while the virulence factors involved in the pathogenic mechanism in B. pumilus still remain unclear. It is well known that bacterial soft rot results from the general disorganization of plant tissues following the degradation of the major component of primary cell walls (Bauer et al., 1994). Yuan and Gao (2015) confirmed that B. pumilus GR8 could cause ginger rhizome rot by producing plant cell wall-degrading enzymes to destroy ginger cells. It has also reported that hydrolytic enzymes could decompose plant cells and tissues without wounds by pathogenic B. altitudinis (Lemjiber et al., 2021). Phytopathogenic CAZymes, such as cellulases, pectinases, xylanases, and proteases, play a central role in plant cell wall degradation and facilitate bacterial colonization and nutrient acquisition (Cantarel et al., 2009; He et al., 2021). The candidate genes encoding CAZymes for vegetal tissue degradation may support the phytopathogenicity. The pectate lyase genes pelA and pelD in B. subtilis could degrade gum from the plant cell wall (Zou et al., 2013). The deletion of the endoglucanase gene celA or the pectinase gene pelA1 in virulent strains of Clavibacter michiganensis can lead to significantly decreased pathogenicity and reduced canker symptom (Wang et al., 2022). The homologous genes celY and pelA, encoding glucanase and pectate trisaccharide lyase, respectively, were found in HM-7 genome.
Interestingly, we found that the presence of several genomic islands (GEIs) and prophages with large (>10 kb) integrative elements and repeat fragments in HM-7 genome, indicating that horizontal gene transfer (HGT) occurred during evolution. Virulence factors and pathogenicity determinants can spread via HGT through mobile genetic elements (MGEs) such as plasmids, bacteriophages, and genetic islands (GIs) (Dobrindt et al.,, 2004; Sobecky and Hazen, 2009). Phages play an important role in the evolution and virulence of bacterial pathogens for carrying key virulence factors (Brüssow et al., 2004). In HM-7 genome, varied GC-content around prophages suggested a phage-mediated gene transfer from a rare heterologous host differing in GC content. The molecular mechanisms underlying pathogenicity in bacteria might be related with the acquisition of new DNA sequences through HGT (Bartoli et al., 2016). Flagella are essential membrane structures that contribute to bacterial virulence and mediate the secretion of extracellular toxins (Sperandio et al., 2002). Toxins were capable of manipulating host cell functions and vital processes of living organisms to favor microbial infection (Do Vale et al., 2016). In this study, functional annotation of strain-specific genes in HM-7 led to the characterization of 19 candidate genes involved in flagella formation and toxin production, which may assist the activation of virulence in HM-7. Future work is necessary to reveal the functions of these candidate genes potentially involved in the pathogenicity of the bacterium associated with muskmelon fruit rot.
As previously reported, phages could effectively control the ginger rhizome rot caused by B. pumilus GR8 (Yuan et al., 2015; Yuan and Gao, 2016). Bacteriophages could be a promising antimicrobial approach as biocontrol agents against pathogens in animals, food products and plants (Goodridge and Bisha, 2011; Yuan et al., 2015; Gambino and Brøndsted, 2021). Considering that the CRISPR/Cas system was absent in HM-7, phage therapy could be a practical strategy for managing bacterial soft rot of muskmelon caused by strain HM-7 in the future.
In this study, the genome sequencing and comparative genomic analyses facilitated the determination of genomic features and virulence factors in the pathogenic strain B. pumilus HM-7. Our results explored to the current understanding of pathogenesis involved in the aspects of adhesion, invasion, intracellular survival, and evasion of host defenses, which would be applied in preventions of bacterial attack and infection in future. Admittedly, more efforts are required to validate the functions of these candidate genes and figure out the pathogenic models as well as involved physiological and metabolic processes.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA857747.
Author contributions
QW and LXZ contributed to conception and design of the study. QW performed the bioinformatic data analyses and wrote the manuscript draft. LZ carried out collection and organized the data. YZ, HC, JS, ML, and RC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by the National Natural Science Foundation of China (32072378), Innovation Research and Experiment Program for Youth Scholar (2021023), the Research Foundation of State Key Laboratory for Biology of Plant Diseases and Insects Pest (SKLOF201611), and the Key Research and Development Projects in Anhui Province (202104b11020006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1008648/full#supplementary-material
Supplementary Figure 1 | Gene annotation by GO, and KEGG for B. pumilus HM-7. (A) GO function classification of genes in HM-7. GO analysis was performed for three main categories: cellular components, molecular function, and biological processes. (B) The KEGG pathway classification of genes in HM-7 contains six groups: cellular processes, environmental, genetic, human diseases, metabolism, and organismal systems.
Supplementary Figure 2 | Pan-core genome analysis of 27 strains of Bacillus species. (A) Gene distribution of 27 strains based on the gene presence-absence matrix generated from Roary. A purple box, green box and an orange box to represent the core gene, accessory genes and specific genes, respectively. The phylogenetic tree on the left represents the phylogenetic relationships among the strains that make up the corresponding pan-genome; (B) Flower petal plot of 27 strains. Pan-genome analysis of 27 strains yielded unique genes for each strain, the numbers of which are shown on each petal plot. The center Circos shows that the number of core gene obtained from 27 strains.
Supplementary Figure 3 | Evolutionary relationships of 27 strains of Bacillus species. (A) Heatmap and dendrogram of average nucleotide identity among different strains. (B) Phylogenetic tree based on total single-copy orthologous.
Supplementary Table 1 | Characteristics of the strains used in this study.
Supplementary Table 2 | Genes presence and absence in six strains of B. pumilus. The presence of each CDS in the respective genome is listed below each labeled strain column. If a cell is blank the respective strain does not have the gene.
Supplementary Table 3 | CAZymes categories identification in HM-7 using the CAZymes database.
Supplementary Table 4 | Virulence-related factor identification in HM-7 using the VFDB database.
Supplementary Table 5 | Gene identification in HM-7 using the PHI database.
Supplementary Table 6 | Prophage regions identification in HM-7 using PHASTER.
Supplementary Table 7 | Genes in three unique regions of HM-7.
Footnotes
References
Agarwal, M., Dheeman, S., Dubey, R. C., Kumar, P., Maheshwari, D. K., and Bajpai, V. K. (2017). Differential antagonistic responses of Bacillus pumilus MSUA3 against Rhizoctonia solani and Fusarium oxysporum causing fungal diseases in Fagopyrum esculentum Moench. Microbiol. Res. 205, 40–47. doi: 10.1016/j.micres.2017.08.012
Babu, M., Dıaz-Mejıa, J. J., Vlasblom, J., Gagarinova, A., Phanse, S., Graham, C., et al. (2011). Genetic interaction maps in escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 7:e1002377. doi: 10.1371/journal.pgen.1002377
Bartoli, C., Roux, F., and Lamichhane, J. R. (2016). Molecular mechanisms underlying the emergence of bacterial pathogens: An ecological perspective. Mol. Plant Pathol. 17, 303–310. doi: 10.1111/mpp.12284
Bathily, H., Babana, A. H., and Samaké, F. (2010). Bacillus pumilus, a new pathogen on potato tubers in storage in Mali. Afr. J. Microbiol. Res. 4, 2067–2071.
Bauer, D. W., Bogdanove, A. J., Beer, S. V., and Collmer, A. (1994). Erwinia chrysanthemi hrp genes and their involvement in soft rot pathogenesis and elicitation of the hypersensitive response. MPMI-Mol. Plant Microbe Interact. 7, 573–581. doi: 10.1094/mpmi-7-0573
Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bonifer, K. S., Wen, X., Hasim, S., Phillips, E. K., Dunlap, R. N., Gann, E. R., et al. (2019). Bacillus pumilus B12 degrades polylactic acid and degradation is affected by changing nutrient conditions. Front. Microbiol. 10:2548. doi: 10.3389/fmicb.2019.02548
Brüssow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. R. 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Research 37:D233–D238. doi: 10.1093/nar/gkn663
Chen, L., Xiong, Z., Sun, L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40:D641–D645. doi: 10.1093/nar/gkr989
Chen, N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 5, 4–10. doi: 10.1002/0471250953.bi0410s05
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Coullon, H., Rifflet, A., Wheeler, R., Janoir, C., Boneca, I. G., and Candela, T. (2020). Peptidoglycan analysis reveals that synergistic deacetylase activity in vegetative Clostridium difficile impacts the host response. J. Biol. Chem. 295, 16785–16796. doi: 10.1074/jbc.RA119.012442
Dai, Y., Wu, X. Q., Wang, Y. H., and Zhu, M. L. (2021). Biocontrol potential of Bacillus pumilus HR10 against Sphaeropsis shoot blight disease of pine. Biol. Control 152:104458. doi: 10.1016/j.biocontrol.2020.104458
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Datta, S., Saha, D., Chattopadhyay, L., and Majumdar, B. (2020). Genome comparison identifies different bacillus species in a bast fibre-retting bacterial consortium and provides insights into pectin degrading genes. Sci. Rep. 10:8169. doi: 10.1038/s41598-020-65228-1
Do Vale, A., Cabanes, D., and Sousa, S. (2016). Bacterial toxins as pathogen weapons against phagocytes. Front. Microbiol. 7:42. doi: 10.3389/fmicb.2016.00042
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Espariz, M., Zuljan, F. A., Esteban, L., and Magni, C. (2016). Taxonomic identity resolution of highly phylogenetically related strains and selection of phylogenetic markers by using genome-scale methods: the bacillus pumilus group case. PLoS One 11:e0163098. doi: 10.1371/journal.pone.0163098
Fellahi, S., Chibani, A., Feuk-Lagerstedt, E., and Taherzadeh, M. J. (2016). Identification of two new keratinolytic proteases from a Bacillus pumilus strain using protein analysis and gene sequencing. AMB Exp. 6:42. doi: 10.1186/s13568-016-0213-0
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Fu, X., Gong, L., Liu, Y., Lai, Q., Li, G., and Shao, Z. (2021). Bacillus pumilus group comparative genomics: toward pangenome features, diversity, and marine environmental adaptation. Front. Microbiol. 12:571212. doi: 10.3389/fmicb.2021.571212
Galal, A. A., El-Bana, A. A., and Janse, J. (2006). Bacillus pumilus, a new pathogen on mango plants. Egypt. J. Phytopathol. 34, 17–29.
Gambino, M., and Brøndsted, L. (2021). Looking into the future of phage-based control of zoonotic pathogens in food and animal production. Curr. Opin. Biotech. 68, 96–103. doi: 10.1016/j.copbio.2020.10.003
Goodridge, L. D., and Bisha, B. (2011). Phage-based biocontrol strategies to reduce foodborne pathogens in foods. Bacteriophage 1, 130–137. doi: 10.4161/bact.1.3.17629
Grant, J. R., Arantes, A. S., and Stothard, P. (2012). Comparing thousands of circular genomes using the CG view comparison tool. BMC Genomics 13:202. doi: 10.1186/1471-2164-13-202
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57. doi: 10.1093/nar/gkm360
Gu, H. J., Sun, Q. L., Luo, J. C., Zhang, J., and Sun, L. (2019). A first study of the virulence potential of a Bacillus subtilis isolate from deep-sea hydrothermal vent. Front. Cell. Infect. Mi. 9:183. doi: 10.3389/fcimb.2019.00183
Gutierrez-Manero, F. J., Ramos-Solano, B., Probanza, A., Mehouachi, J., Tadeo, F. R., and Talon, M. (2001). The plant-growth-promoting rhizobacteria Bacillus pumilus and Bacillus licheniformis produce high amounts of physiologically active gibberellins. Physiol. Plantarum 111, 206–211. doi: 10.1034/j.1399-3054.2001.1110211.x
Hahnke, R. L., Meier-Kolthoff, J. P., García-López, M., Mukherjee, S., Huntemann, M., Ivanova, N. N., et al. (2016). Genome-based taxonomic classification of bacteroidetes. Front Microbiol. 7:2003. doi: 10.3389/fmicb.2016.02003
Hayat, K., Menhas, S., Bundschuh, J., Zhou, P., Niazi, N. K., Hussain, A., et al. (2020). Plant growth promotion and enhanced uptake of Cd by combinatorial application of Bacillus pumilus and EDTA on Zea mays L. Int. J. Phytorem. 22, 1372–1384. doi: 10.1080/15226514.2020.1780410
He, X., Lu, T., and Zhou, X. (2021). Whole genome sequencing and comparative genomics analysis of Pectobacterium carotovorum identifies key pathogenic genes. Mol. Phylogenet. Evol. 162:107114. doi: 10.1016/j.ympev.2021.107114
Hernandez, J.-P., de-Bashan, L. E., Rodriguez, D. J., Rodriguez, Y., and Bashan, Y. (2009). Growth promotion of the freshwater microalga Chlorella vulgaris by the nitrogen-fixing, plant growth-promoting bacterium Bacillus pumilus from arid zone soils. Eur. J. Soil Biol. 45, 88–93. doi: 10.1016/j.ejsobi.2008.08.004
Hinderlich, S., Weidemann, W., Yardeni, T., Horstkorte, R., and Huizing, M. (2013). UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A master regulator of sialic acid synthesis. SialoGlyco Chem. Biol. 366, 97–137. doi: 10.1007/128_2013_464
Hong, H. A., Duc, L. H., and Cutting, S. M. (2005). The use of bacterial spore formers as probiotics. FEMS Microbiol. Rev. 29, 813–835. doi: 10.1016/j.femsre.2004.12.001
Hori, K., and Matsumoto, S. (2010). Bacterial adhesion: From mechanism to control. Biochem. Eng. J. 48, 424–434. doi: 10.1016/j.bej.2009.11.014
Huang, Y., Wild, B. L., and Morris, S. C. (1992). Postharvest biological control of Penicillium digitatum decay on citrus fruit by Bacillus pumilus. Ann. Appl. Biol. 120, 367–372. doi: 10.1111/j.1744-7348.1992.tb03433.x
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11:119. doi: 10.1186/1471-2105-11-119
Iqbal, S., Vollmers, J., and Janjua, H. A. (2021). Genome mining and comparative genome analysis revealed niche-specific genome expansion in antibacterial bacillus pumilus strain SF-4. Genes 12:1060. doi: 10.3390/genes12071060
Johnson, B. T., Shaw, L. N., Nelson, D. C., and Mayo, J. A. (2008). Extracellular proteolytic activities expressed by Bacillus pumilus isolated from endodontic and periodontal lesions. J. Med. Microbiol. 57, 643–651. doi: 10.1099/jmm.0.47754-0
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jothi, R., Zotenko, E., Tasneem, A., and Przytycka, T. M. (2006). COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics 22, 779–788. doi: 10.1093/bioinformatics/btl009
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44:D457–D462. doi: 10.1093/nar/gkv1070
Kapli, P., Yang, Z., and Telford, M. J. (2020). Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21, 428–444. doi: 10.1038/s41576-020-0233-0
Kim, S., Jang, W. E., Park, J., Kim, M. S., Kim, J. G., and Kang, L. W. (2021). Combined analysis of the time-resolved transcriptome and proteome of plant pathogen Xanthomonas oryzae pv. oryzae. Front. Microbiol. 12:664857. doi: 10.3389/fmicb.2021.664857
Koehler, T. M. (2002). Bacillus anthracis genetics and virulence gene regulation. Curr. Top. Microbiol. Immunol. 271, 143–164. doi: 10.1007/978-3-662-05767-4_7
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Kotan, R., Sahin, F., and Ala, A. (2006). Identification and pathogenicity of bacteria isolated from pome fruit trees in the Eastern Anatolia region of Turkey/Identifizierung und Pathogenität von Bakterien aus ostanatolischen Kernobstbäumen. J. Plant Dis. Prot. 113, 8–13.
Kovaleva, V. A., Shalovylo, Y. I., Gorovik, Y. N., Lagonenko, A. L., Evtushenkov, A. N., and Gout, R. T. (2015). Bacillus pumilus-a new phytopathogen of Scots pine. J. For. Sci. 61, 131–137. doi: 10.17221/16/2014-JFS
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lee, D. H., Kim, J. B., Lim, J. A., Han, S. W., and Heu, S. (2014). Genetic Diversity of Pectobacterium carotovorum subsp. brasiliensis Isolated in Korea. Plant Pathol. J. 30, 117–124. doi: 10.5423/PPJ.OA.12.2013.0117
Lemjiber, N., Naamani, K., Merieau, A., Dihazi, A., Zhar, N., Jediyi, H., et al. (2021). Identification and genomic characterization of pathogenic bacillus altitudinis from common pear trees in Morocco. Agronomy 11:1344.
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6:41. doi: 10.1186/1754-6834-6-41
Li, B., Qiu, W., Tan, Q. M., Su, T., Fang, Y., and Xie, G. L. (2009). Association of a bacillus species with leaf and twig dieback of asian pear (Pyrus Pyrifolia) in China. J. Plant Pathol. 91, 705–708. doi: 10.4454/jpp.v91i3.565
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50:D912–D917. doi: 10.1093/nar/gkab1107
Logan, N. A. (2012). Bacillus and relatives in foodborne illness: Bacillus in foodborne illness. J. Appl. Microbiol. 112, 417–429. doi: 10.1111/j.1365-2672.2011.05204.x
Logan, N. A., Berge, O., Bishop, A. H., Busse, H. J., De Vos, P., Fritze, D., et al. (2009). Proposed minimal standards for describing new taxa of aerobic, endospore-forming bacteria. Int. J. Syst. Evol. Microbiol. 59, 2114–2121. doi: 10.1099/ijs.0.013649-0
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Makino, S., Watarai, M., Cheun, H. I., Shirahata, T., and Uchida, I. (2002). Effect of the lower molecular capsule released from the cell surface of Bacillus anthracis on the pathogenesis of anthrax. J. Infect. Dis. 186, 227–233. doi: 10.1086/341299
Mazlan, S., Zulperi, D., Wahab, A., Jaafar, N. M., Sulaiman, Z., and Rajandas, H. (2019). First report of Bacillus pumilus causing trunk bulges of rubber tree (Hevea brasiliensis) in Malaysia. Plant Dis. 103, 1016. doi: 10.1094/PDIS-08-18-1409-PDN
Okazaki, S., Sano, N., Yamada, T., Ishii, K., Kojima, K., Djedidi, S., et al. (2019). Complete genome sequence of plant growth-promoting bacillus pumilus TUAT1. Microbiol. Resour. Announc. 8:e76–e19. doi: 10.1128/MRA.00076-19
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Panthee, S., Paudel, A., Hamamoto, H., Ogasawara, A. A., Iwasa, T., Blom, J., et al. (2021). Complete genome sequence and comparative genomic analysis of Enterococcus faecalis EF-2001, a probiotic bacterium. Genomics 113, 1534–1542. doi: 10.1016/j.ygeno.2021.03.021
Peng, Q., Yuan, Y., and Gao, M. (2013). Bacillus pumilus, a novel ginger rhizome rot pathogen in China. Plant Dis. 97, 1308–1315. doi: 10.1094/PDIS-12-12-1178-RE
Pudova, D. S., Toymentseva, A. A., Gogoleva, N. E., Shagimardanova, E. I., Mardanova, A. M., and Sharipova, M. R. (2022). Comparative genome analysis of two bacillus pumilus strains producing high level of extracellular hydrolases. Genes 13:409. doi: 10.3390/genes13030409
Ramarao, N., and Sanchis, V. (2013). The pore-forming haemolysins of Bacillus cereus: a review. Toxins 5, 1119–1139. doi: 10.3390/toxins5061119
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Saggese, A., Culurciello, R., Casillo, A., Corsaro, M., Ricca, E., and Baccigalupi, L. (2018). A marine isolate of bacillus pumilus secretes a pumilacidin active against staphylococcus aureus. Mar. Drugs 16, 180. doi: 10.3390/md16060180
Saleh, O. I., Huang, P.-Y., and Huang, J.-S. (1997). Bacillus pumilus, the cause of bacterial blotch of immature balady peach in egypt. J. Phytopathol. 145, 447–453. doi: 10.1111/j.1439-0434.1997.tb00348.x
Sanders, M. E., Morelli, L., and Tompkins, T. A. (2003). Sporeformers as human probiotics: bacillus, sporolactobacillus, and brevibacillus. Compr. Rev. Food Sci. F. 2, 101–110. doi: 10.1111/j.1541-4337.2003.tb00017.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sobecky, P. A., and Hazen, T. H. (2009). Horizontal gene transfer and mobile genetic elements in marine systems. Methods Mol. Biol. 435, 435–453. doi: 10.1007/978-1-60327-853-9_25
Song, J. H., Wu, Z. R., Zhang, L. X., Tan, G. J., Wang, S., and Wang, J. J. (2018). First report of bacillus pumilus causing fruit rot on muskmelon (cucumis melo) in China. Plant Dis. 102, 439–439. doi: 10.1094/PDIS-08-17-1169-PDN
Sperandio, V., Torres, A. G., and Kaper, J. B. (2002). Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol. Microbiol. 43, 809–821. doi: 10.1046/j.1365-2958.2002.02803.x
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stepanov, V. G., Tirumalai, M. R., Montazari, S., Checinska, A., Venkateswaran, K., and Fox, G. E. (2016). Bacillus pumilus SAFR-032 genome revisited: sequence update and re-annotation. PLoS One 11:e0157331. doi: 10.1371/journal.pone.0157331
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tatusov, R. L., Koonin, E. V., and Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Tena, D., Martinez-Torres, J. A., Perez-Pomata, M. T., Saez-Nieto, J. A., Rubio, V., and Bisquert, J. (2007). Cutaneous infection due to bacillus pumilus: report of 3 cases. Clin. Infect. Dis. 44:e40–e42. doi: 10.1086/511077
Thomson, N. R., Howard, S., Wren, B. W., Holden, M. T. G., Crossman, L., Challis, G. L., et al. (2006). The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLoS Genet. 2:e206. doi: 10.1371/journal.pgen.0020206
Urban, M., Cuzick, A., Seager, J., Wood, V., Rutherford, K., Venkatesh, S. Y., et al. (2020). PHI-base: the pathogen–host interactions database. Nucleic Acids Res. 48:D613–D620. doi: 10.1093/nar/gkz904
Villanueva, J., Switala, J., Ivancich, A., and Loewen, P. C. (2014). Genome sequence of bacillus pumilus MTCC B6033. Genome Announc. 2:e327–e314. doi: 10.1128/genomeA.00327-14
Wang, Y., Deng, S., Li, Z., and Yang, W. (2022). Advances in the characterization of the mechanism underlying bacterial canker development and tomato plant resistance. Horticulturae 8:209. doi: 10.3390/horticulturae8030209
Winnenburg, R. (2006). PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 34:D459–D464. doi: 10.1093/nar/gkj047
Yamaguchi, M., Terao, Y., Mori, Y., Hamada, S., and Kawabata, S. (2008). PfbA, a novel plasmin- and fibronectin-binding protein of streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J. Biol. Chem. 283, 36272–36279. doi: 10.1074/jbc.M807087200
Yao, G., Zhang, W., Yang, M., Yang, H., Wang, J., Zhang, H., et al. (2020). MicroPhenoDB associates metagenomic data with pathogenic microbes, microbial core genes, and human disease phenotypes. Genom. Proteom. Bioinf. 18, 760–772. doi: 10.1016/j.gpb.2020.11.001
Yuan, Y., and Gao, M. (2015). Genomic analysis of a ginger pathogen Bacillus pumilus providing the understanding to the pathogenesis and the novel control strategy. Sci. Rep. 5:10259. doi: 10.1038/srep10259
Yuan, Y., and Gao, M. (2016). Characteristics and complete genome analysis of a novel jumbo phage infecting pathogenic Bacillus pumilus causing ginger rhizome rot disease. Arch. Virol. 161, 3597–3600. doi: 10.1007/s00705-016-3053-y
Yuan, Y., Peng, Q., Wu, D., Kou, Z., Wu, Y., Liu, P., et al. (2015). Effects of actin-like proteins encoded by two Bacillus pumilus phages on unstable lysogeny, revealed by genomic analysis. Appl. Environ. Microb. 81, 339–350. doi: 10.1128/AEM.02889-14
Zam, S. I., Agustien, A., Jannah, M., Aldi, Y., and Djamaan, A. (2016). Isolation, characterization of endophytic bacteria from Citrus aurantifolia swingle leaves and testing of antifungal activity towards Fusarium oxysporum. Der Pharm. Lett. 8, 83–89.
Zhao, C. W., Wang, H. Y., Zhang, Y. Z., and Feng, H. (2012). Draft genome sequence of Bacillus pumilus BA06, a producer of alkaline serine protease with leather-dehairing function. J. Bacteriol 194, 6668–6669. doi: 10.1128/JB.01694-12
Zhao, X., Wang, Y., Shang, Q., Li, Y., Hao, H., and Zhang, Y. (2015). Collagen-like proteins (ClpA, ClpB, ClpC, and ClpD) are required for biofilm formation and adhesion to plant roots by Bacillus amyloliquefaciens FZB42. PloS One 10:e0117414. doi: 10.1371/journal.pone.0117414
Zhong, C., Han, M., Yu, S., Yang, P., Li, H., and Ning, K. (2018). Pan-genome analyses of 24 Shewanella strains re-emphasize the diversification of their functions yet evolutionary dynamics of metal-reducing pathway. Biotechnol. Biofuels 11:193. doi: 10.1186/s13068-018-1201-1
Zhou, Z., Chang, N., Lv, Y., Jiang, H., Yao, C., Wan, X., et al. (2022). K solubilizing bacteria (Bacillus) promote theanine synthesis in tea roots (Camellia sinensis) by activating CsTSI activity. Tree Physiol 42, 1613–1627. doi: 10.1093/treephys/tpac027
Zou, M., Li, X., Zhao, J., and Qu, Y. (2013). Characteristics of polygalacturonate lyase C from Bacillus subtilis 7-3-3 and its synergistic action with PelA in enzymatic degumming. PLoS One 8:e79357. doi: 10.1371/journal.pone.0079357
Keywords: Bacillus pumilus, complete genome sequence, pan-core genome, comparative genomic analysis, pathogenic gene
Citation: Wang Q, Zhang L, Zhang Y, Chen H, Song J, Lyu M, Chen R and Zhang L (2022) Comparative genomic analyses reveal genetic characteristics and pathogenic factors of Bacillus pumilus HM-7. Front. Microbiol. 13:1008648. doi: 10.3389/fmicb.2022.1008648
Received: 01 August 2022; Accepted: 10 October 2022;
Published: 07 November 2022.
Edited by:
Yue Xie, Sichuan Agricultural University, ChinaReviewed by:
Yihui Yuan, Hainan University, ChinaRavi Shah, Medical College of Wisconsin, United States
Saumya Patel, Gujarat University, India
Archna Suman, Indian Agricultural Research Institute (ICAR), India
Mario Alberto Martínez Núñez, Universidad Nacional Autónoma de México, Mexico
Copyright © 2022 Wang, Zhang, Zhang, Chen, Song, Lyu, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixin Zhang, lxzhang@ahau.edu.cn
†These authors have contributed equally to this work