 Yaovi M. G. Hounmanou1*
Yaovi M. G. Hounmanou1* Anders Dalsgaard1,2
Anders Dalsgaard1,2 Tirzania Frannetta Sopacua1Gazi Md. Noor Uddin1
Tirzania Frannetta Sopacua1Gazi Md. Noor Uddin1 Pimlapas Leekitcharoenphon3
Pimlapas Leekitcharoenphon3 Rene S. Hendriksen3
Rene S. Hendriksen3 John E. Olsen1
John E. Olsen1 Marianne Halberg Larsen1
Marianne Halberg Larsen1- 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 2School of Chemical and Biological Engineering, Nanyang Technological University, Singapore, Singapore
- 3Research Group for Genomic Epidemiology, National Food Institute, Technical University of Denmark, Kongens Lyngby, Denmark
Salmonella Weltevreden is increasingly reported from aquatic environments, seafood, and patients in several Southeast Asian countries. Using genome-wide analysis, we characterized S. Weltevreden isolated from cultured shrimp and tilapia from Vietnam and China to study their genetic characteristics and relatedness to clinical isolates of S. Weltevreden ST-365. The phylogenetic analysis revealed up to 312 single-nucleotide polymorphism (SNP) difference between tilapia isolates, whereas isolates from shrimp were genetically more closely related. Epidemiologically unrelated isolates from Vietnam were closely related to isolates from China, e.g., 20 SNPs differences between strains 28V and 75C. In comparison with strains from other parts of the world, our environmental isolates predominantly clustered within the continental South Asia lineage, constituted mostly of strains from human stool with as low as seven SNPs difference, e.g., 30V versus Cont_ERR495254. All sequenced isolates were MLST type ST-365 and contained the major virulence-related genes encoded by the Salmonella Pathogenicity Islands 1–5. Ten of the isolates harbored the IncFII(S) plasmid similar to the virulence genes-mediated plasmid pSPCV of S. Paratyphi C, and one isolate had the IncQ1 plasmid on the same contig with strA/B, sul2, and tetA resistance genes similar to the IncQ1 type, pNUC of S. Typhimurium. A pangenomic analysis yielded 7891 genes including a core genome of 4892 genes, with a closely related accessory genome content between clinical and environmental isolates (Benjamini p > 0.05). In a search for differences that could explain the higher prevalence of S. Weltevreden in aquatic samples, genomes were compared with those of other Salmonella enterica serovars. S. Weltevreden revealed specific regions harboring glpX (Fructose-1;6-bisphosphatase; class II), rfbC (dTDP-4-dehydrorhamnose 3;5-epimerase), and cmtB (PTS Mannitol-specific cryptic phosphotransferase enzyme IIA component) involved in carbohydrate biosynthesis pathways. Our study builds grounds for future experiments to determine genes or pathways that are essential when S. Weltevreden are in aquatic environments and microbial interactions providing survival advantages to S. Weltevreden in such environments.
Introduction
Salmonella enterica subsp. enterica serovar Weltevreden is a common cause of human salmonellosis in Asian countries like China, Singapore, Thailand, and Vietnam (Aarestrup, 2003; Makendi et al., 2016; Manipura, 2016; Ferrari et al., 2019; Lin et al., 2019). S. Weltevreden is frequently found in seafood and aquatic environments in Asia (Bangtrakulnonth et al., 2004; Ponce et al., 2008; Noor Uddin et al., 2015; Li et al., 2017; Ferrari et al., 2019). It is also found in other food sources including broilers in China (Ren et al., 2016), poultry and pigs in Vietnam (Lettini et al., 2016; Trung et al., 2017), as well as in vegetables in Malaysia (Awang Salleh et al., 2003). The source and transmission routes of S. Weltevreden, when found in meat products, are unknown, but fish meal used as a protein source in livestock feed may be an important source, as S. Weltevreden is often found in fish and seafood (Kumar et al., 2015; Makendi et al., 2016). This underlines that S. Weltevreden, like Vibrio cholerae (Hounmanou et al., 2019a, b), might have a preference to the aquatic environment, from where it spreads to humans, domestic animals, and food products. We previously reported a close genetic relatedness of epidemiological unrelated S. Weltevreden isolated from cultured shrimp in Vietnam and cultured tilapia in China using pulsed field gel electrophoresis genotyping (Noor Uddin et al., 2015; Li et al., 2017) suggesting occurrence and spread of clonal strains of S. Weltevreden in aquatic environments.
The increasing association with S. Weltevreden and human salmonellosis in Southeast Asia (Aarestrup, 2003; Makendi et al., 2016; Manipura, 2016; Lin et al., 2019) correlates with its high occurrence in seafood in the region (Ferrari et al., 2019). However, little is currently known about genetic differences between S. Weltevreden and other serovars that favor its frequent occurrence in the aquatic environment. Comparison of S. Weltevreden 2007-60-3289-1, a plant isolate, with the genome of the seafood isolate SL484 showed that the genomes of both strains are very similar and no genetic marker was found to justify the aquatic life of the seafood isolate (Brankatschk et al., 2012). An increased expression of the enterotoxin stn gene in S. Weltevreden was reported when they were grown in seafood (Kumar et al., 2015); however, this expression was also observed in S. Typhi. Therefore, the genetic determinants of S. Weltevreden that favors its frequent occurrence in the aquatic environment compared with other non-typhoidal Salmonella remain to be described.
The present study aimed to determine the genetic characteristics and diversity of S. Weltevreden originating from shrimp and tilapia in Vietnam and China. We also performed a pangenomic analysis to understand the genetic relationship between environmental and clinical S. Weltevreden and to identify potential genetic traits associated with an aquatic occurrence of the serovar. Finally, a genome-wide comparison was performed between our S. Weltevreden strains and selected broad host range and host-adapted Salmonella enterica serovars.
Materials and Methods
Strain Collection
Six strains of S. Weltevreden (95V, 74V, 30V, 28V, 13V, 3V) recovered in 2013 from cultured shrimp in Vietnam (Noor Uddin et al., 2015) and six strains (85C, 75C, 62C, 30C, 28C, 24C) isolated in 2013 in cultured tilapia obtained from China (Li et al., 2017) were characterized by whole genome sequencing.
DNA Extraction and Whole Genome Sequencing
DNA was extracted using the DNeasy Blood and tissue kit following the manufacturer’s protocol for Gram-negative bacteria (Qiagen, Hilden, Germany). DNA concentrations were determined using the Qubit dsDNA BR assay kit (Invitrogen, Carlsbad, CA, United States). Genome sequencing was performed using the MiSeq instrument (Illumina, San Diego, CA, United States) at a 300 bp paired-end-read format. Raw reads were de novo assembled using the SPAdes v. 3.9.0 (Bankevich et al., 2012). The raw reads from the 12 S. Weltevreden strains were submitted to the European Nucleotide Archive under the project accession number PRJEB37452.
Characterization and Phylogenetic and Comparative Genomics of S. Weltevreden
Assembled genomes of the 12 isolates were analyzed using the Salmonella In Silico Typing Resource (SISTR) (Yoshida et al., 2016) for confirmation of serovar. In silico MLST typing was performed with MLST 2.0 (Larsen et al., 2012). The sequences were further analyzed to identify acquired resistance genes by ResFinder v.3.2 (Zankari et al., 2012) and presence of known point mutations with 80% threshold for identity and 60% minimum alignment length. PlasmidFinder 2.1 (Carattoli et al., 2014) was used to identify plasmid replicons that are present in the genomes using default settings. Moreover, the assembled genomes were analyzed for identification of the main pathogenic markers on the Salmonella Pathogenicity Islands (SPIs) using SPI databases from CGE1 and the Pathogenicity Islands Database PAIDB v.2.0 (Yoon et al., 2015). The genomes were all analyzed for presence of prophages in PHASTER2 (Arndt et al., 2016), where the intact phage regions were reported as SWΦ.
For phylogenetic analysis, the raw reads of the 12 strains were analyzed using CSI-Phylogeny (Kaas et al., 2014) where single-nucleotide polymorphism (SNP) analysis was performed using the already described S. Weltevreden 10259 (Acc: SAMEA1904377) as reference (Makendi et al., 2016). To analyze the S. Weltevreden strains from China and Vietnam in a global context, the raw reads were further analyzed along with a collection of 60 S. Weltevreden strains. The global strains selected were all of the same sequence type ST-365 including 30 from the continental lineage and 30 from the island clonal line (Makendi et al., 2016) isolated from human, duck, pig, fish, poultry, seafood, environment, and vegetables (Supplementary Table S1). The obtained trees were annotated in iTOL (Letunic and Bork, 2016) and rooted with the stated reference strain.
To describe the genetic relationship between the environmental and clinical isolates, we performed a pangenomic analysis. In this analysis, 17 environmental strains (including our 12 strains and 5 additional strains isolated from seafood) and 17 clinical strains were used. The selected genomes for the pangenomic analysis were publicly available and part of the initial phylogenetic analysis and located on the continental clade, where our strains predominantly clustered (see “Results” and “Discussion” sections). All the 34 genomes analyzed were annotated using Prokka (Seemann, 2014) and the gff files were used as an input to the Roary (v.3.7.0) (Page et al., 2015) pangenome analysis tool in a Linux interface. The binary presence/absence data of accessory genes produced in Roary was used to calculate the associations between all genes in the accessory genome and the sample types of the isolates by employing the Scoary (v.1.6.11) tool (Brynildsrud et al., 2016). The accessory genome tree was visualized in phandango (Hadfield et al., 2018).
To decipher the potential genetic markers associated with the aquatic occurrence of S. Weltevreden, a number of known genes associated with aquatic bacteria were selected to make a database for local nucleotide BLAST using MyDbFinder 2.0. These include the salt resistance plasmids described in marine microorganisms (pSR1KM603475, pSR2KM603476, pSR4KM603478, pSR3KM 603477, pSR5KM603479, pSR6KM603480) (Mirete et al., 2015), as well as the SPI-2 encoded T3SS genes sseC and ssaU described to support survival of S. Typhi in Acanthamoeba in the aquatic environment (Bleasdale et al., 2009). Moreover, due to its previously reported role in the survival of S. Weltevreden in seafood, the chromosomally located stn gene (Salmonella enterotoxin) (Kumar et al., 2015; Ammar et al., 2016) was included in a locally built database. Using the SEED genome viewer from RAST (Overbeek et al., 2014), a functional gene comparison was made between clinical and environmental strains of S. Weltevreden. This included genes involved in carbohydrate biosynthesis, stress response as well as regulation and cell signaling established in the SEED subsystem annotation and previously reported to play a role in the survival of Flavobacterium spp. and V. cholerae in the aquatic environment (Kolton et al., 2013; Hounmanou et al., 2019a).
In addition, another genome-wide comparison was performed between our 12 S. Weltevreden strains and seven broad host range and host-adapted Salmonella enterica serovars including S. Typhi CT18, S. Typhimurium LT2, S. Enteritidis 92-0392, S. Dublin USMARC-69838, S. Pullorum QJ-2D-Sal, S. Gallinarum 287/91, and S. Choleraesuis SCSA50. The reference S. Weltevreden 10259 (SAMEA1904377), which is a clinical strain, served as reference to compare the genome content of S. Weltevreden with these other serovars using Blast Atlas in GView3. The functions of the obtained S. Weltevreden specific gene products were determined in the QuickGO Molecular function4 through UniProt (The UniProt Consortium., 2019).
Results and Discussion
Genetic Variations and Relatedness Among S. Weltevreden From Shrimp and Tilapia
The strains characterized in this study were all of ST-365 and were previously isolated from tilapia aquaculture in China and shrimp culture sites in Vietnam without any known epidemiological association between strains and sites of isolation (Noor Uddin et al., 2015; Li et al., 2017). By PFGE fingerprinting, it was previously found that the strains isolated from the same source were closely related. We made a genomic comparison to get a better view of the relatedness of the isolates (Figure 1). This comparison showed that some strains isolated from shrimp in Vietnam are clonal with zero SNPs, e.g., 74V and 30V, as well as 3V and 13V (Figure 1). Other strains such as 24C, 30C, and 95V were located on the continental cluster and differed by up to 100 SNPs belonging to separate sub-clades as previously reported by Makendi et al. (2016) based on intrinsic genomic variations. As expected due to the higher discriminatory power of WGS compared with PFGE (Bayliss et al., 2017), other isolates with similar PFGE fingerprints were more diverse and had up to 312 SPNs within isolates from tilapia in China (Figure 1). Surprisingly, some strains isolated in shrimp from Vietnam were genetically related to strains recovered from tilapia in China, i.e., for instance 28V and 75C (20 SNPs), 30V and 28C (31 SNPs), and 13V and 85C (34 SNPs) (Supplementary Table S1). When analyzed with selected S. Weltevreden isolates from different parts of the world, the isolates predominantly clustered within a so-called continental lineage which constitutes mostly of isolates of clinical origin, with as low as seven SNPs difference, e.g., 30V versus Cont_ERR495254 (Figure 2 and Supplementary Table S1). The similarity among genomes of epidemiologically unrelated strains from shrimp and tilapia recovered in Vietnam and China suggests that the S. Weltevreden circulating in Southeast Asia may have a common source and recent spread, or they have a conserved genome. The observation corroborates their main clustering with other strains found predominantly in the Southeast Asian lineage of the continental cluster (Makendi et al., 2016). It should be noted that we did not select all available S. Weltevreden genomes, but a representative number of strains (selected across all countries, years of isolation, hosts, and sources of isolation) representing the continental and island clusters (Supplementary Table S1) from the previous study of Makendi et al. (2016) for the global localization of the genomes of our environmental strains from shrimp and tilapia.
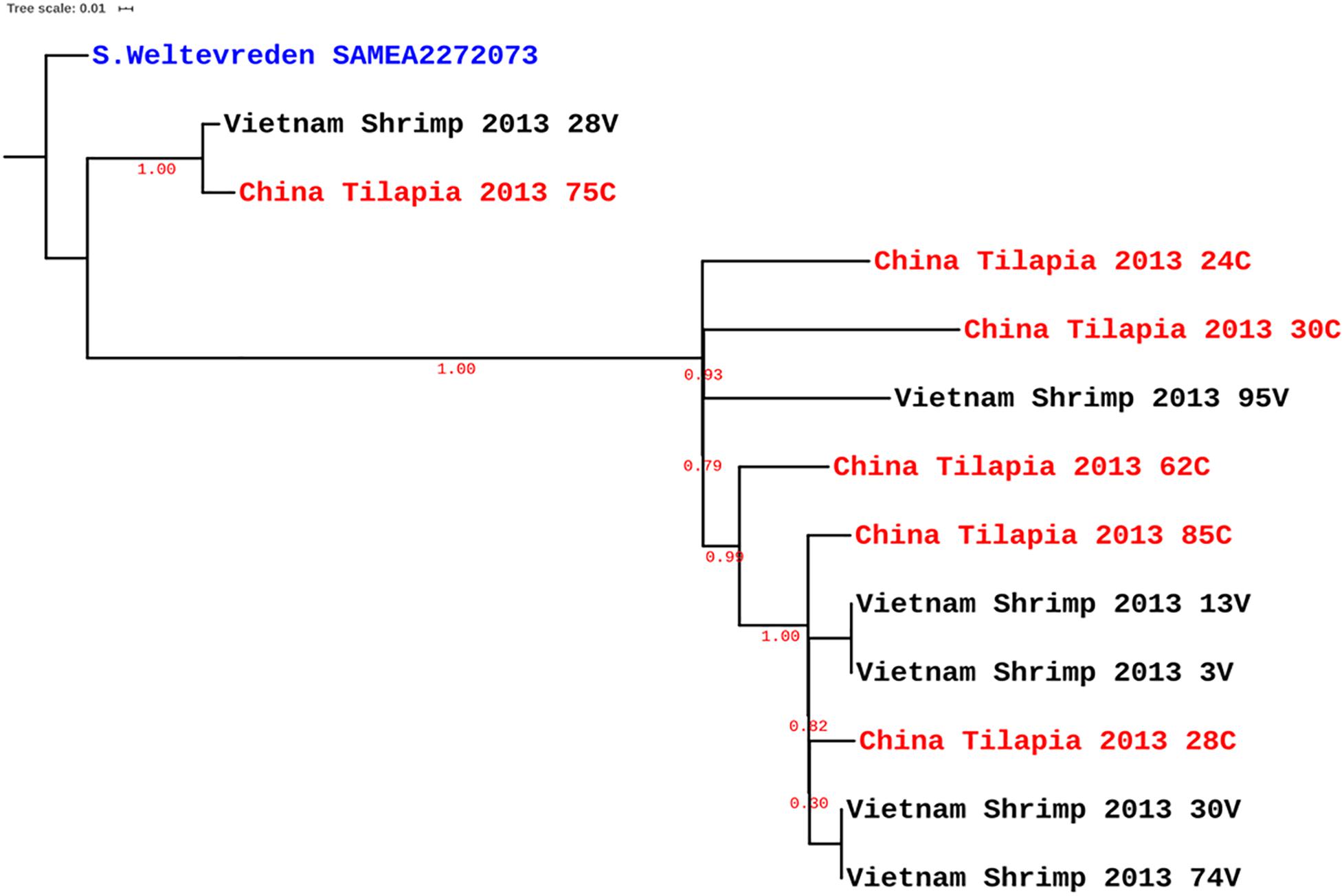
Figure 1. Maximum-likelihood SNP tree of Salmonella Weltevreden isolated from shrimp (black) in Vietnam and tilapia (red) in China. S. Weltevreden 10259 (SAMEA1904377; blue) served as reference to root the tree.
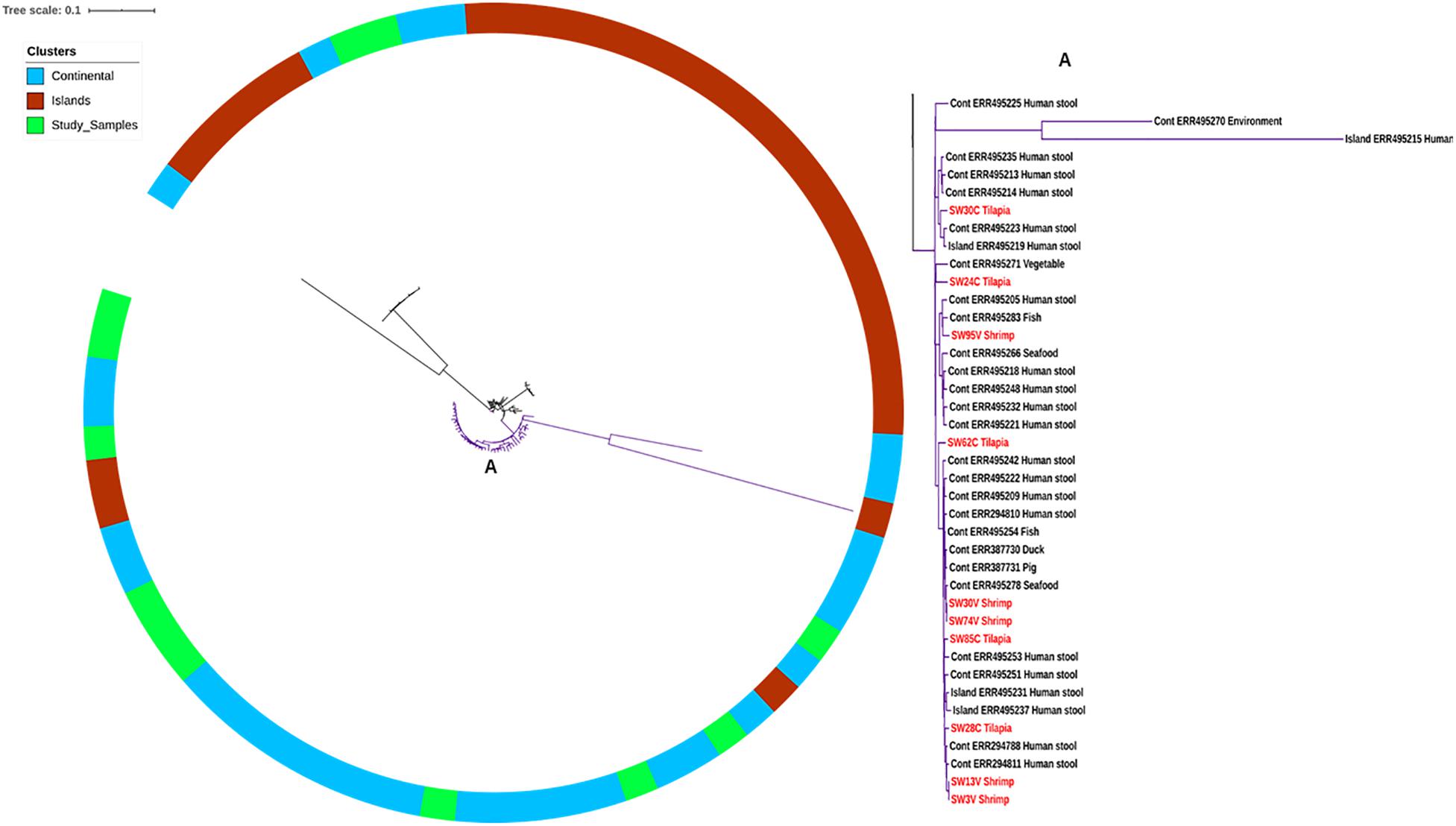
Figure 2. SNP tree showing Salmonella Weltevreden from this study (red font types in A) within a global context of the continental and island clusters (Makendi et al., 2016). (A) Depicts the continental clade where 10 of our 12 strains are clustered.
Genomic Characteristics of S. Weltevreden From Shrimp and Tilapia
The observed phylogenetic similarity between our environmental isolates and clinical strains from the continental Southeast Asian cluster is not surprising and indicates that our strains from shrimp and tilapia have the potential to cause disease in humans. This is further substantiated by the presence in our strains of main virulence-associated genes on the Salmonella Pathogenicity Islands (SPIs) 1–5, as well as a number of other pathogenicity islands (data not shown), which have been associated with virulence in humans and animals for Salmonella Typhimurium (Marcus et al., 2000). The presence of SPI1-5 known to be important for intestinal invasion indicates that the environmental isolates are capable of causing infection (Marcus et al., 2000; Lou et al., 2019) and invading the intestine. However, isolates of S. Weltevreden were previously reported to be significantly less invasive in Hep-2 cells but caused inflammatory reactions in mice (Makendi et al., 2016).
The IncFII plasmid replicon is present in most of our strains (Table 1). This plasmid is part of a large family of IncF plasmids that have been associated with Salmonella virulence and play a role during infection under low-iron conditions in host cells (Khajanchi et al., 2017). The closest reference to the IncFII plasmid identified in our genomes is the highly transmissible plasmid pSPCV (CP000858) reported in the human pathogen Salmonella Paratyphi C encoding virulence genes such as the SPI2 effector genes of the spv gene clusters (Liu et al., 2009); however, the spv genes were not present in the genomes of S. Weltevreden.
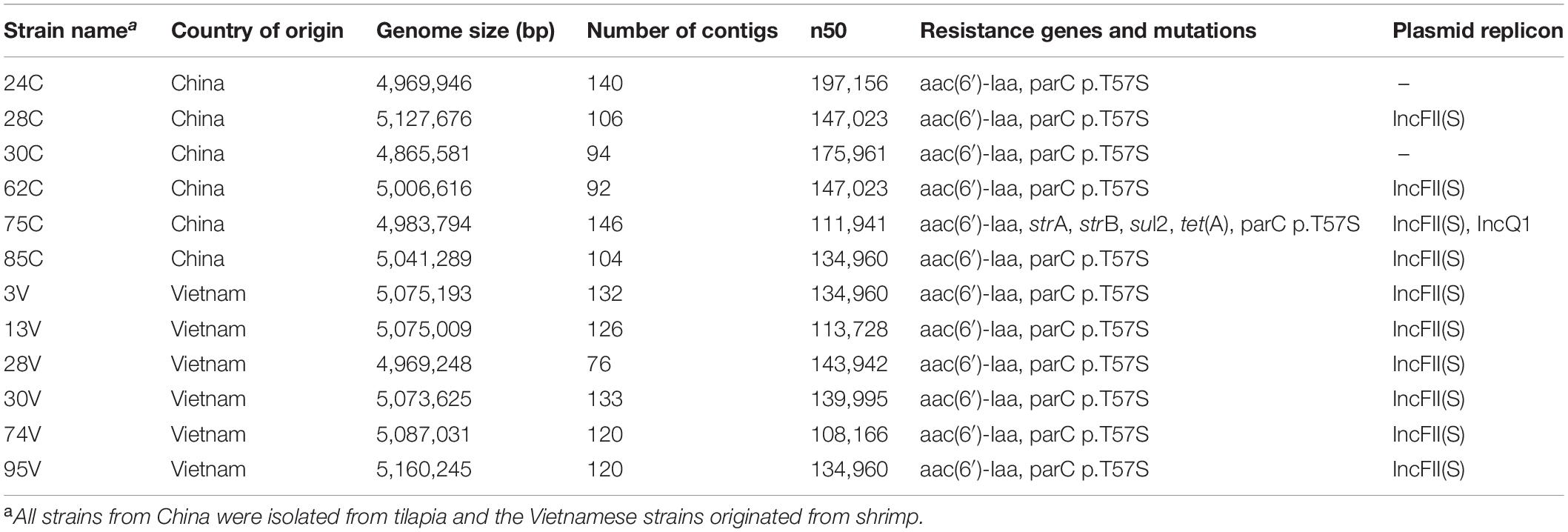
Table 1. General properties and genetic characteristics of S. Weltewreden strains isolated from shrimp and tilapia.
Altogether, our data indicate that the S. Weltevreden strains isolated in shrimp and tilapia are closely related to clinical strains with pathogenic potentials to cause infection in mammals. Moreover, our strains do not carry many antimicrobial resistance genes. Except for strain 75 C from tilapia, all the remaining strains only carried the aminoglycoside resistance gene aac(6′)-Iaa and presented a point mutation in parC p.T57S encoding resistance to quinolones, although they were originally reported susceptible to nalidixic acid and ciprofloxacin (Noor Uddin et al., 2015; Li et al., 2017). The discrepancy between susceptibility to gentamicin and the presence of the aac(6′)-Iaa gene is because the aac(6′)-Iaa enzyme does not acetylate gentamicin (Salipante and Hall, 2003). However, a previous study showed the presence of plasmid-located aph(3′)-Ia and strA/B genes conferring aminoglycoside resistance in S. Weltevreden from food products (Makendi et al., 2016); genes not found in our strains. Phenotypic resistance to quinolones seems increasingly to require both plasmid-mediated quinolone resistance genes like qnrA, qnrC, and two point mutations in the quinolone resistance-determining region such as parC and gyrA (Kotb et al., 2019). Point mutations in the parC gene alone are usually associated with a low increase in the minimum inhibitory concentrations to quinolones, so the discrepant results shown between our genomic and phenotypic observations are not surprising. Overall, the general susceptibility of S. Weltevreden to different antimicrobials suggests that it has a habitat and ecology with little if any exposure to antimicrobials, e.g., an aquatic habitat (Noor Uddin et al., 2015; Li et al., 2017).
The strain 75 C carried, in addition to the parC p.T57S mutation, strA, strB, sul2, and tetA resistance genes along with an IncQ1 type plasmid on the same contig suggesting a plasmid-mediated resistance in this strain confirmed after blasting the contig. The identified IncQ1 plasmid is similar to the pRSF1010 (M28829) of E. coli and the pNUC isolated in clinical S. Typhimurium conferring resistance to sulfamethoxazole, streptomycin, and tetracycline through the presence of sul2, strAB, and tetA genes (Oliva et al., 2017). Strain 75 C clusters with strains showing similar levels of antimicrobial resistance as shown for strains previously characterized and located in a continental sub-clade named “Vietnam Antimicrobial resistant” (Makendi et al., 2016). Nevertheless, in our study, this sub-clade of strains grouped on the island cluster, a discrepancy that we also found in a recent study which is probably caused by minor differences in the SNP calling methods (FastTree versus mpileup) (Minh et al., 2020).
Sequences of the phage VibrioΦ_X29 was found in strain 30C and the EnterobacterΦ_Tyrion phage in 95V, and the strains carried at least one of the common Salmonella prophages such as Gifsy_1 and 2, phiV10, sal3, Fels, PsP3, g341c, and SEN34 (Supplementary Table S2). The presence of the lysogenic phage VibrioΦ_X29 (Bhandare et al., 2017) in S. Weltevreden is interesting as it has not been reported previously in S. Weltevreden (Makendi et al., 2016) and deserves further attention. The finding suggests that S. Weltevreden has acquired the phage from Vibrio spp. which suggests that marine environments may be important ecological niches for this serovar.
It was also investigated if our environmental S. Weltevreden strains possess other genetic markers supporting their presence in marine environments including seafood such as salt tolerance. However, none of the strains carried genes and plasmids reported to encode salt resistance in marine microorganisms which may be due to the fact that the reference genes used for this analysis emanated from marine bacteria (Mirete et al., 2015), whereas S. Weltevreden may be less halophilic as it has also been isolated in freshwater aquaculture (Li et al., 2017). Thus, the absence of the high salt tolerance genes in our strains should be interpreted with caution because S. Weltevreden is often isolated in seafood.
Our strains possessed the SPI-2 encoded T3SS genes sseC and ssaU described to support survival of S. Typhi in Acanthamoeba in the aquatic environment (Bleasdale et al., 2009). Also, all our S. Weltevreden isolates harbor the chromosomally located gene stn which encodes the Salmonella enterotoxin Stn and has been shown to be upregulated during growth in seafood (Kumar et al., 2015); a gene which was found in all our strains. These genes are however not restricted to S. Weltevreden but are present in most Salmonella enterica serovars (Marcus et al., 2000; Ammar et al., 2016). The association of S. Weltevreden with aquatic environments is therefore difficult to document by comparative genomics alone because the strains causing disease in human are genetically similar to those in water which probably serves as their ecological niche. This observation is similar to the case of V. cholerae O1 found in aquatic environments where there was no genetic distinction between clinical and environmental strains isolated from Lake Victoria, although V. cholerae is known to have a tropism for the aquatic environment (Hounmanou et al., 2019a, b). Future laboratory experiments could assess the growth of S. Weltevreden in water and use proteomics to detect all differential expressed genes providing survival advantages to the bacterium. Similar to studies in V. cholerae and S. Typhi, the persistence of S. Weltevreden in the aquatic environment could also be assessed through various microbial interaction studies such as relationships with protozoa and other aquatic organisms like phytoplankton, zooplankton, and fish (Tezcan-Merdol et al., 2004; Douesnard-Malo and Daigle, 2011; Hounmanou et al., 2019c).
The Accessory Genome of Isolated Strains in Comparison With Clinical Strains
The genetic relatedness between our environmental and clinical S. Weltevreden strains revealed by the core genome–based phylogeny was also confirmed by the pangenome analysis. Roary alignment of the genomes in the clinical and environmental strains located in the continental clade where 10 of our 12 strains clustered revealed a pangenome size of 7891 genes including a core genome of 4892 genes which is similar to the number of core genes in most Salmonella enterica (Stevens et al., 2018). In a pangenome analysis, the accessory genome serves to reveal significantly unique genes for each clade. However, when scored and analyzed with Scoary commands, it was shown that the accessory genome content of the environmental strains was not significantly different from clinical strains with a Benjamini p > 0.05 between strains from the two origins (Supplementary Table S3 and Figure 3). Nevertheless, there was a unique genetic region in strain 24C compared with all the strains included in the analysis (Figure 3). This region was visualized using CLC genomic workbench and by the over 500 annotated proteins that are mostly additional copies of the already present genes in all the strains from the region (sheet 2 of Supplementary Table S3).
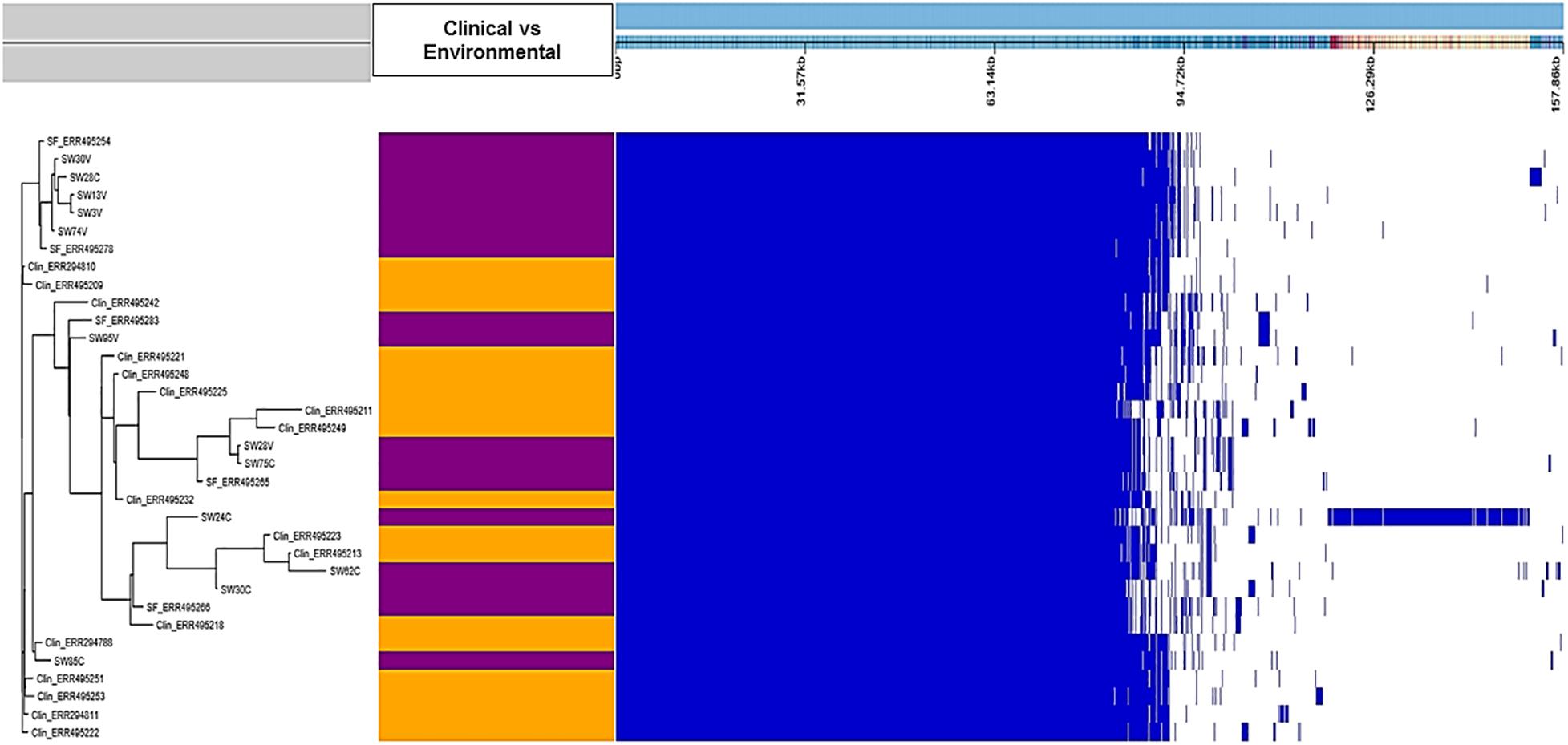
Figure 3. Presence–absence map of the genes in the pangenome of clinical and environmental S. Weltevreden. The tree to the left is the accessory binary tree where environmental strains are marked with purple whereas clinical strains are shown in orange color. The blue box marks the presence of a region whereas the white gaps represent the absent regions.
Following methodologies used to distinguish terrestrial and aquatic Flavobacterium spp. (Kolton et al., 2013), we performed a functional gene comparative genomic analysis with the SEED viewer in RAST (see “Materials and Methods” section). The findings of this analysis corroborated our pangenomic analysis demonstrating that all the SEED’s functional subsystems present in environmental strains were also found in clinical strains with similar proportions of gene content. For instance, functional genes involved in “virulence, disease, and defense”; “regulation and cell signaling”; “stress response”; and “carbohydrate biosynthesis” were evenly distributed in genomes from both sources. This further confirms the zoonotic potential of S. Weltevreden isolated in aquatic environments and seafood.
Content Comparison of the Genome of S. Weltevreden to Seven Other Salmonella enterica
In a further pangenomic analysis, we compared S. Weltevreden with genomes of seven other Salmonella enterica serovars, i.e., S. Typhi, S. Typhimurium, S. Enteritidis, S. Dublin, S. Pullorum, S. Gallinarum, and S. Choleraesuis. The objective was to detect genetic regions that are unique to S. Weltevreden and assess if the genes in such regions may play a role in aquatic niche adaptation. Various genetic elements were detected as specific to S. Weltevreden including glpX (Fructose-1;6-bisphosphatase; class II), rfbC (dTDP-4-dehydrorhamnose 3;5-epimerase), different PTS mannitol transporters, and sorE (L-sorbose 1-phosphate reductase) predominantly involved in the carbohydrate biosynthesis pathways (Figure 4 and Supplementary Table S4). Other unique elements were the O-antigen flippase Wzx which has an antiporter activity and xenobiotic transmembrane transporter activity present on the cell membrane (The UniProt Consortium., 2019). This analysis also detected DEAD/DEAH box helicases involved in various aspects of RNA metabolism, including nuclear transcription, pre-mRNA splicing, ribosome biogenesis, nucleocytoplasmic transport, translation, RNA decay, and organellar gene expression (The UniProt Consortium., 2019). Although these elements can potentially distinguish S. Weltevreden from other serovars, their role in aquatic niche adaption remains to be established. Furthermore, when blasted, the detected regions are also present in other bacteria such as E. coli, Shigella spp., Mycobacterium leprae, and Pseudomonas spp. (The UniProt Consortium., 2019). The identified S. Weltevreden specific elements are similar to the genetic markers found in the six genomic islands reported when S. Weltevreden was compared with other serovars including the DEAD-box ATP-dependent RNA helicase (ydbR), the mannose-specific phosphotransferase system (PTS), and the plasmid pSW82 (Acc. FR775255) (Brankatschk et al., 2012).
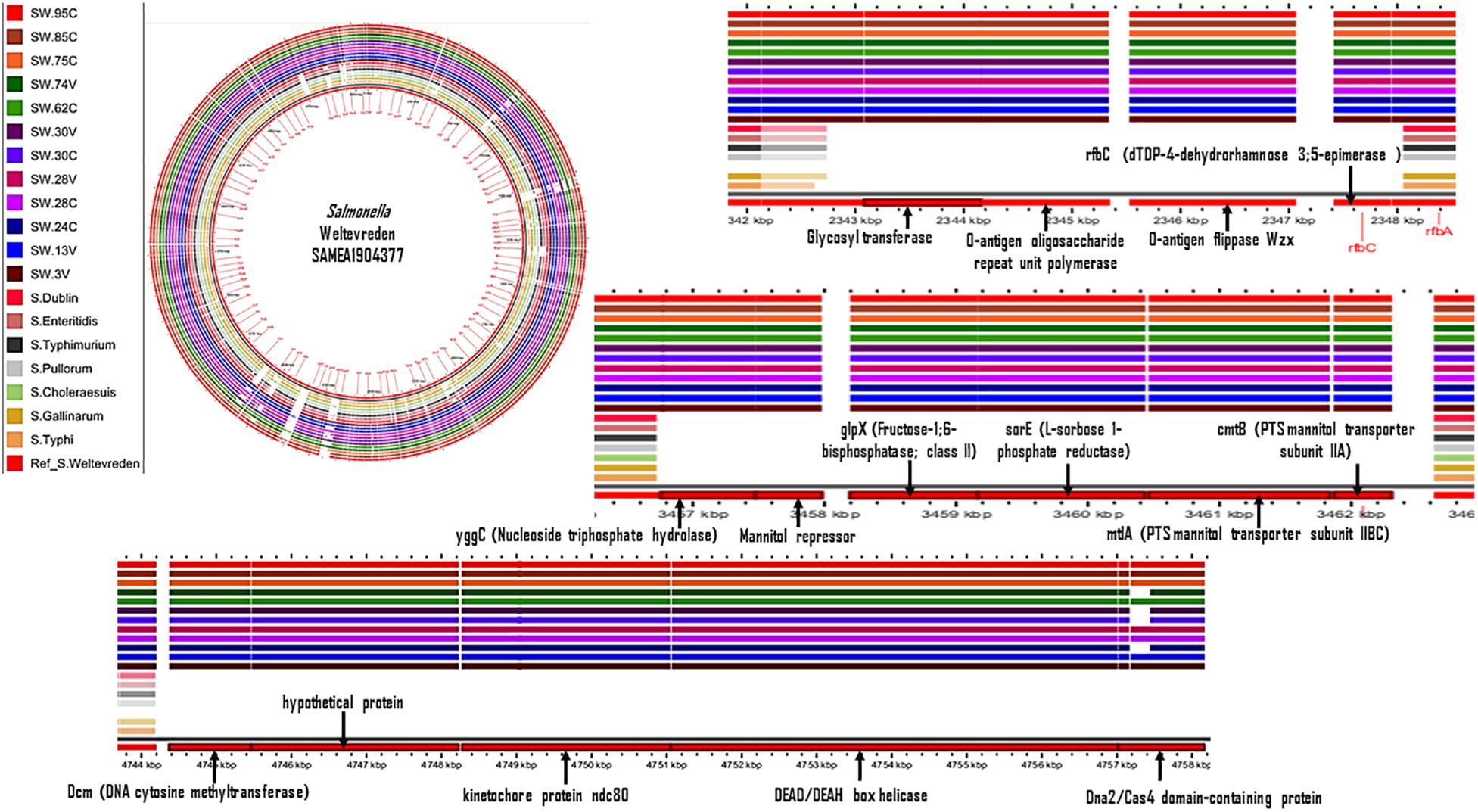
Figure 4. Genome content comparison of S. Weltevreden with S. Typhi, S. Typhimurium, S. Enteritidis, S. Dublin, S. Pullorum, S. Gallinarum, and S. Choleraesuis. Gaps in the genome show genetic regions potentially specific to S. Weltevreden.
We are currently conducting growth experiments to verify findings in the genomic study. Preliminary data from these experiments show no differences between S. Weltevreden and S. Typhimurium LT2 when grown in media with different salt concentrations representing different aquatic environments from fresh to brackish water (unpublished data). However, increasing NaCl concentrations (0.5–6%) induced different growth responses within the S. Weltevreden populations. Moreover, because the genomic study revealed the presence of specific PTS systems in S. Weltevreden, we performed growth experiments with different concentrations of carbohydrates. The growth of S. Weltevreden in the minimal media M9 with glucose, mannitol, galactose, glycerol, and in combination glycerol + mannitol was compared with growth of S. Typhimurium TL2, S. Agona, and S. Seftenberg. The bioscreen showed that all tested strains were able to grow with the different tested carbon sources from 0.001 to 0.2% of carbohydrate with no difference between the growth rates (unpublished data). These preliminary results show no growth differences between S. Weltevreden and other Salmonella serotypes as opposed to the expectations from the genomic data and therefore points to the need for further growth experiments of other more specific carbon sources for PTS-specific system found in S. Weltevreden. This study revealed that the S. Weltevreden isolated from shrimp and tilapia in Vietnam and China possess pathogenic characteristics and are genetically closely related to clinical strains found in Southeast Asia. Antimicrobial resistance genes were uncommon. Comparative genomics indicate that the sources of human infections are likely fish, shrimp, and possibly other types of seafood. Various genomic regions were detected in the strains that differentiate them from other serovars and need to be further studied in experimental studies for their functional roles in the adaptation of S. Weltevreden to the aquatic environment. Moreover, the present study shows the need for new experiments describing aquatic reservoirs and microbial interactions that provide survival advantages to S. Weltevreden in aquatic environments. Genes that are overexpressed when S. Weltevreden is in water may also be determined. Growth experiments under different environmental conditions should also be considered.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
GU isolated the Vietnamese strains. AD, ML, TS, and GU wrote the first draft of the manuscript. YH performed the bioinformatics analyses with some guidance from PL and RH and revised and finalized the manuscript. AD, PL, RH, JO, and ML critically revised the manuscript to reach a final version. AD and ML conceived the study and provided funding. All authors revised and approved the final version of the manuscript.
Funding
Costs for genomic analyses were covered by the Department of Veterinary and Animal Sciences, University of Copenhagen. Salary for YH was covered by the Danish International Development Assistance (Danida) through the project “Innovations and Markets for Lake Victoria Fisheries” (IMLAF); Grant No. DFC File No. 14-PO1-TAN.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Li Kang, Shanghai Ocean University, China for doing the initial field work and isolation of Salmonella Weltevreden in tilapia aquaculture and visiting fellow Jingyuan Wang for initiating the whole genome sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01985/full#supplementary-material
TABLE S1 | Public strains used and pairwise SNPs between genomes (Excel file).
TABLE S2 | Prophages identified in the genomes of S. Weltevreden isolated in shrimp and tilapia from Vietnam and China, respectively.
TABLE S3 | Content of the accessory genome in clinical and environmental strains (Excel file).
TABLE S4 | Additional S. Weltevreden-specific genes when compared with S. Typhi, S. Typhimurium, S. Enteritidis, S. Dublin, S. Pollorum, S. Gallinarum, and S. Choleraesuis.
Footnotes
- ^ https://cge.cbs.dtu.dk/services/SPIFinder/
- ^ http://phaster.ca/
- ^ https://server.gview.ca/
- ^ https://www.ebi.ac.uk/QuickGO/
References
Aarestrup, F. M. (2003). Antimicrobial susceptibility and occurrence of resistance genes among Salmonella enterica serovar Weltevreden from different countries. J. Antimicrob. Chemother. 52, 715–718. doi: 10.1093/jac/dkg426
Ammar, A. M., Mohamed, A. A., Abd El-Hamid, M. I., and El-Azzouny, M. M. (2016). Virulence genotypes of clinical SalmonellaSerovars from broilers in Egypt. J. Infect. Dev. Countries 10, 337–346. doi: 10.3855/jidc.7437
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Awang Salleh, N., Rusul, G., Hassan, Z., Reezal, A., Hajar Isa, S., Nishibuchi, M., et al. (2003). Incidence of Salmonella spp. in raw vegetables in Selangor, Malaysia. Food Control 14, 475–479. doi: 10.1016/S0956-7135(02)00105-6
Bangtrakulnonth, A., Pornreongwong, S., Pulsrikarn, C., Sawanpanyalert, P., Hendriksen, R. S., Lo Fo Wong, D. M. A., et al. (2004). Salmonella serovars from humans and other sources in Thailand, 1993–2002. Emerg. Infect. Dis. 10, 131–136. doi: 10.3201/eid1001.02-0781
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bayliss, S. C., Verner-Jeffreys, D. W., Bartie, K. L., Aanensen, D. M., Sheppard, S. K., Adams, A., et al. (2017). The promise of whole genome pathogen sequencing for the molecular epidemiology of emerging aquaculture pathogens. Front. Microbiol. 8:121. doi: 10.3389/fmicb.2017.00121
Bhandare, S. G., Warry, A., Emes, R. D., Hooton, S. P. T., Barrow, P. A., and Atterbury, R. J. (2017). Complete genome sequences of Vibrio cholerae-specific bacteriophages 24 and X29. Genome Announc. 5, e1013-17. doi: 10.1128/genomeA.01013-17
Bleasdale, B., Lott, P. J., Jagannathan, A., Stevens, M. P., Birtles, R. J., and Wigley, P. (2009). The Salmonella pathogenicity Island 2-encoded type iii secretion system is essential for the survival of Salmonella enterica serovar Typhimurium in free-living amoebae. Appl. Environ. Microbiol. 75, 1793–1795. doi: 10.1128/AEM.02033-08
Brankatschk, K., Blom, J., Goesmann, A., Smits, T. H. M., and Duffy, B. (2012). Comparative genomic analysis of Salmonella enterica subsp. enterica serovar Weltevreden foodborne strains with other serovars. Int. J. Food Microbiol. 155, 247–256. doi: 10.1016/j.ijfoodmicro.2012.01.024
Brynildsrud, O., Bohlin, J., Scheffer, L., and Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17:238. doi: 10.1186/s13059-016-1108-8
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Douesnard-Malo, F., and Daigle, F. (2011). Increased persistence of Salmonella enterica Serovar Typhi in the presence of Acanthamoeba castellanii. Appl. Environ. Microbiol. 77, 7640–7646. doi: 10.1128/AEM.00699-11
Ferrari, R. G., Rosario, D. K. A., Cunha-Neto, A., Mano, S. B., Figueiredo, E. E. S., and Conte-Junior, C. A. (2019). Worldwide epidemiology of Salmonella serovars in animal-based foods: a meta-analysis. Appl. Environ. Microbiol. 85, e00591-19. doi: 10.1128/AEM.00591-19
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Hounmanou, Y. M. G., Leekitcharoenphon, P., Hendriksen, R. S., Dougnon, T. V., Mdegela, R. H., Olsen, J. E., et al. (2019a). Surveillance and genomics of toxigenic Vibrio cholerae O1 from fish, phytoplankton and water in Lake Victoria, Tanzania. Front. Microbiol. 10:901. doi: 10.3389/fmicb.2019.00901
Hounmanou, Y. M. G., Leekitcharoenphon, P., Kudirkiene, E., Mdegela, R. H., Hendriksen, R. S., Olsen, J. E., et al. (2019b). Genomic insights into Vibrio cholerae O1 responsible for cholera epidemics in Tanzania between 1993 and 2017. PLoS Negl. Trop. Dis. 13:e0007934. doi: 10.1371/journal.pntd.0007934
Hounmanou, Y. M. G., Mdegela, R. H., Dougnon, T. V., Madsen, H., Withey, J. H., Olsen, J. E., et al. (2019c). Tilapia (Oreochromis niloticus) as a putative reservoir host for survival and transmission of Vibrio cholerae O1 biotype El Tor in the aquatic environment. Front. Microbiol. 10:1215. doi: 10.3389/fmicb.2019.01215
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. doi: 10.1371/journal.pone.0104984
Khajanchi, B. K., Hasan, N. A., Choi, S. Y., Han, J., Zhao, S., Colwell, R. R., et al. (2017). Comparative genomic analysis and characterization of incompatibility group FIB plasmid encoded virulence factors of Salmonella enterica isolated from food sources. BMC Genomics 18:570. doi: 10.1186/s12864-017-3954-5
Kolton, M., Sela, N., Elad, Y., and Cytryn, E. (2013). Comparative genomic analysis indicates that niche adaptation of terrestrial flavobacteria is strongly linked to plant glycan metabolism. PLoS One 8:e76704. doi: 10.1371/journal.pone.0076704
Kotb, D. N., Mahdy, W. K., Mahmoud, M. S., and Khairy, R. M. M. (2019). Impact of co-existence of PMQR genes and QRDR mutations on fluoroquinolones resistance in Enterobacteriaceae strains isolated from community and hospital acquired UTIs. BMC Infect. Dis. 19:979. doi: 10.1186/s12879-019-4606-y
Kumar, R., Datta, T. K., and Lalitha, K. V. (2015). Salmonella grows vigorously on seafood and expresses its virulence and stress genes at different temperature exposure. BMC Microbiol. 15:254. doi: 10.1186/s12866-015-0579-1
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Lettini, A. A., Vo Than, T., Marafin, E., Longo, A., Antonello, K., Zavagnin, P., et al. (2016). Distribution of Salmonella serovars and antimicrobial susceptibility from poultry and swine farms in central Vietnam. Zoonoses Public Health 63, 569–576. doi: 10.1111/zph.12265
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, K., Petersen, G., Barco, L., Hvidtfeldt, K., Liu, L., and Dalsgaard, A. (2017). Salmonella Weltevreden in integrated and non-integrated tilapia aquaculture systems in Guangdong, China. Food Microbiol. 65, 19–24. doi: 10.1016/j.fm.2017.01.014
Lin, Y. N., Fong, R., Leo, J., Kwan, W. W., Ye, A., Chan, P. P., et al. (2019). Distribution of Salmonella spp. along the food chain in Singapore, 2008-2016. Epidemiol. News Bull. 45, 44–54.
Liu, W.-Q., Feng, Y., Wang, Y., Zou, Q.-H., Chen, F., Guo, J.-T., et al. (2009). Salmonella paratyphi C: genetic divergence from Salmonella choleraesuis and pathogenic convergence with Salmonella typhi. PLoS One 4:e4510. doi: 10.1371/journal.pone.0004510
Lou, L., Zhang, P., Piao, R., and Wang, Y. (2019). Salmonella pathogenicity Island 1 (SPI-1) and its complex regulatory network. Front. Cell. Infect. Microbiol. 9:270. doi: 10.3389/fcimb.2019.00270
Makendi, C., Page, A. J., Wren, B. W., Le Thi Phuong, T., Clare, S., Hale, C., et al. (2016). A phylogenetic and phenotypic analysis of Salmonella enterica serovar weltevreden, an emerging agent of diarrheal disease in tropical regions. PLoS Negl. Trop. Dis. 10:e0004446. doi: 10.1371/journal.pntd.0004446
Manipura, R. (2016). A rare serotype Salmonella weltevreden causing enteric fever in an HIV positive patient in Mangalore. Asian J. Pharm. Clin. Res. 9, 14–15. doi: 10.22159/ajpcr.2016.v9i6.14144
Marcus, S. L., Brumell, J. H., Pfeifer, C. G., and Finlay, B. B. (2000). Salmonella pathogenicity Islands: big virulence in small packages. Microbes Infect. 2, 145–156. doi: 10.1016/S1286-4579(00)00273-2
Minh, D. K., Hounmanou, Y. M. G., Mai, H. B. T., Olsen, J. E., and Dalsgaard, A. (2020). Prevalence and genomic characterization of Salmonella Weltevreden in commercial pig feed. Vet. Microbiol. 246:108725. doi: 10.1016/j.vetmic.2020.108725
Mirete, S., Mora-Ruiz, M. R., Lamprecht-Grandío, M., de Figueras, C. G., Rosselló-Móra, R., and González-Pastor, J. E. (2015). Salt resistance genes revealed by functional metagenomics from brines and moderate-salinity rhizosphere within a hypersaline environment. Front. Microbiol. 6:1121. doi: 10.3389/fmicb.2015.01121
Noor Uddin, G., Larsen, M. H., Barco, L., Minh Phu, T., and Dalsgaard, A. (2015). Clonal occurrence of Salmonella Weltevreden in cultured shrimp in the Mekong Delta, Vietnam. PLoS One 10:e0134252. doi: 10.1371/journal.pone.0134252
Oliva, M., Monno, R., D’Addabbo, P., Pesole, G., Dionisi, A. M., Scrascia, M., et al. (2017). A novel group of IncQ1 plasmids conferring multidrug resistance. Plasmid 89, 22–26. doi: 10.1016/j.plasmid.2016.11.005
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Ponce, E., Khan, A. A., Cheng, C. M., Summage-West, C., and Cerniglia, C. E. (2008). Prevalence and characterization of Salmonella enterica serovar Weltevreden from imported seafood. Food Microbiol. 25, 29–35. doi: 10.1016/j.fm.2007.09.001
Ren, X., Li, M., Xu, C., Cui, K., Feng, Z., Fu, Y., et al. (2016). Prevalence and molecular characterization of Salmonella enterica isolates throughout an integrated broiler supply chain in China. Epidemiol. Infect. 144, 2989–2999. doi: 10.1017/S0950268816001515
Salipante, S. J., and Hall, B. G. (2003). Determining the limits of the evolutionary potential of an antibiotic resistance gene. Mol. Biol. Evol. 20, 653–659. doi: 10.1093/molbev/msg074
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Stevens, M. J. A., Zurfluh, K., Althaus, D., Corti, S., Lehner, A., and Stephan, R. (2018). Complete and assembled genome sequence of Salmonella enterica subsp. enterica serotype Senftenberg N17-509, a strain lacking Salmonella pathogen Island 1. Genome Announc. 6, e00156-18. doi: 10.1128/genomeA.00156-18
Tezcan-Merdol, D., Ljungström, M., Winiecka-Krusnell, J., Linder, E., Engstrand, L., and Rhen, M. (2004). Uptake and replication of Salmonella enterica in Acanthamoeba rhysodes. Appl. Environ. Microbiol. 70, 3706–3714. doi: 10.1128/AEM.70.6.3706-3714.2004
The UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. doi: 10.1093/nar/gky1049
Trung, N. V., Carrique-Mas, J. J., Nghia, N. H., Tu, L. T. P., Mai, H. H., Tuyen, H. T., et al. (2017). Non-typhoidal Salmonella colonization in chickens and humans in the Mekong delta of Vietnam. Zoonoses Public Health 64, 94–99. doi: 10.1111/zph.12270
Yoon, S. H., Park, Y.-K., and Kim, J. F. (2015). PAIDB v2.0: exploration and analysis of pathogenicity and resistance Islands. Nucleic Acids Res. 43, D624–D630. doi: 10.1093/nar/gku985
Yoshida, C. E., Kruczkiewicz, P., Laing, C. R., Lingohr, E. J., Gannon, V. P. J., Nash, J. H. E., et al. (2016). The Salmonella In Silico Typing Resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS One 11:e0147101. doi: 10.1371/journal.pone.0147101
Keywords: Salmonella Weltevreden, genome comparison, non-typhoidal Salmonella, microbial ecology, whole genome sequencing
Citation: Hounmanou YMG, Dalsgaard A, Sopacua TF, Uddin GMN, Leekitcharoenphon P, Hendriksen RS, Olsen JE and Larsen MH (2020) Molecular Characteristics and Zoonotic Potential of Salmonella Weltevreden From Cultured Shrimp and Tilapia in Vietnam and China. Front. Microbiol. 11:1985. doi: 10.3389/fmicb.2020.01985
Received: 23 March 2020; Accepted: 27 July 2020;
Published: 25 August 2020.
Edited by:
Zhi Ruan, Zhejiang University, ChinaReviewed by:
Vitali Sintchenko, The University of Sydney, AustraliaJeannette Barba-León, University of Guadalajara, Mexico
Copyright © 2020 Hounmanou, Dalsgaard, Sopacua, Uddin, Leekitcharoenphon, Hendriksen, Olsen and Larsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaovi M. G. Hounmanou, gil@sund.ku.dk