 Phillip D. Fromm
Phillip D. Fromm Jessica C. Kling
Jessica C. Kling Annika Remke2
Annika Remke2 Christian Bogdan
Christian Bogdan Heinrich Körner
Heinrich Körner- 1Comparative Genomics Centre, James Cook University, Townsville, QN, Australia
- 2Menzies Institute for Medical Research Tasmania, Hobart, TAS, Australia
- 3Blumenthal Group, The University of Queensland Diamantina Institute, Translational Research Institute, Woolloongabba, QLD, Australia
- 4Mikrobiologisches Institut – Klinische Mikrobiologie, Immunologie und Hygiene, Friederich-Alexander-Universität Erlangen-Nürnberg, Universitätsklinikum Erlangen, Erlangen, Germany
Induction of inducible nitric oxide synthase in mononuclear phagocytes by IFN-γ and innate tumor necrosis factor (TNF) provide the basis for an effective immune response to the intracellular parasite Leishmania (L.) major. In previous experiments, we observed a fatal visceral form of leishmaniasis in L. major-infected C57BL/6 TNF-/- mice. To further delineate the protective function of TNF and its receptor requirements, we comparatively assessed L. major-infected C57BL/6 mice that were either deficient for membrane and soluble TNF (Tnf-/-), for soluble TNF alone (memTnfΔ/Δ), or the TNF receptors type 1 (Tnfr1-/-) or type 2 (Tnfr2-/-). We detected locally and systemically increased levels of the cytokine IFN-γ in the absence of the TNF-TNFR1-signaling pathway. An analysis of transcription factors and cytokines revealed that activated Tnf-/- CD4+ T cells displayed a highly active Th1 phenotype with a strong usage of the T cell receptor Vβ5.1/2. From these data we conclude that the fatal outcome of L. major infection in Tnf-/- mice does not result from a skewed or deficient Th1 differentiation.
Introduction
Cutaneous leishmaniasis is caused by different species of the protozoan parasite genus Leishmania (L.) such as the “old world” species of Leishmania major (Reithinger et al., 2007; Schonian et al., 2008), which are transferred by the bite of a sand fly (Sacks and Perkins, 1985). In the case of a L. major infection, the clinical manifestation is limited to a local skin lesion that heals without further treatment. However, depending on the host immune response and the parasite species and strain, chronic, non-healing skin ulcers and widespread tissue destruction or even systemic parasite dissemination have also been observed (Reithinger et al., 2007).
The immune response to L. major in the skin and draining LNs has been analyzed extensively using high- or low-dose mouse infection models (Sacks and Noben-Trauth, 2002; Bogdan, 2008; Liese et al., 2008), but is still only partly understood. In genetically self-healing C57BL/6 mice a protective immune response against L. major occurs that is characterized by an initial boost of interferon (IFN)-α/β (Diefenbach et al., 1998), the rapid induction of IL-12 (Scharton-Kersten et al., 1995), the production of IFN-γ by NK cells and the differentiation of Th1 cells (Liese et al., 2007; Lykens et al., 2010; Prajeeth et al., 2011). IFN-γ drives macrophages to upregulate the enzyme inducible nitric oxide synthase (iNOS, NOS2; Ding et al., 1998) to produce large amounts of the leishmanicidal effector molecule nitric oxide (NO) from L-arginine (Deng et al., 1993), a process synergistically supported by tumor necrosis factor (TNF; Ding et al., 1998). Synthesis of NO is essential for the resolution of L. major infections in vivo (Liew et al., 1990; Stenger et al., 1994, 1996; Diefenbach et al., 1998; Prajeeth et al., 2011). This well established chain of immunological events in the resistant C57BL/6 mouse contrasts with the immune response to L. major in the genetically susceptible (i.e., non-healing) BALB/c mouse which shows an early and sustained IL-4 expression that results in a progressive infection (Sacks and Noben-Trauth, 2002). The observed dichotomy in the cytokine response led to the development of the Th1–Th2 model of T cell differentiation, which interprets genetic differences of cytokine expression as causal for the disparate clinical outcomes observed in experimental cutaneous Leishmaniasis (Mosmann and Coffman, 1989). However, more recent work has demonstrated that this classical model is too simplistic, as newly characterized T cell populations (e.g., Th17 cells, regulatory T cells) (Alexander and Brombacher, 2012), early chemokine expression (Roebrock et al., 2014) and differential wound healing mechanisms (Baldwin et al., 2007) need to be taken into account when discussing genetic and immunological reasons for susceptibility to L. major. In addition, in TNF-deficient mice L. major-specific T cells displayed strong in vitro IFN-γ expression, but in vivo failed to achieve control of L. major as TNF-/- mice succumbed to the infection within 6–7 weeks (Wilhelm et al., 2001). These and other results, which contradicted the classical Th1/Th2 dichotomy (Belosevic et al., 1989; Anderson et al., 2005), suggested that the presence of IFN-γ is necessary, but not sufficient for the control of L. major in vivo.
To further address the role of TNF for the innate and adaptive response to L. major and to reconcile previous discrepant results obtained with TNF- or TNF-receptor-deficient mouse strains of different genetic origins (Vieira et al., 1996; Nashleanas et al., 1998; Chakour et al., 2003), we infected mice deficient for membrane and soluble TNF (Tnf-/-), for soluble TNF alone (memTnfΔ/Δ), for TNF receptor type 1 (Tnfr1-/-), or type 2 (Tnfr2-/-) on a pure C57BL/6 background to comprehensively assess the course of leishmaniasis. Subsequently, we compared the expression of IFN-γ and the underlying T cell response in Wt and Tnf-/- mice to analyze and quantify the adaptive immune response in the absence of TNF in more detail.
Materials and Methods
Mice
The gene-targeted C57BL/6 mouse strains deficient for both soluble and membrane TNF (Tnf-/-) or for soluble TNF only (memTnfΔ/Δ) were generated on a genetically pure C57BL/6 (Wt) background (Körner et al., 1997; Ruuls et al., 2001). The Tnfr1-/- (Jackson stock number: 003242) and Tnfr2-/- mice (Jackson stock number: 002620) were obtained from Jackson Laboratories and had been backcrossed more than 10 times or had been established on a C57BL/6 background, respectively (Peschon et al., 1998). The screening procedure followed the protocols published previously (Körner et al., 1997; Peschon et al., 1998). All animals were kept under specific pathogen-free conditions at the Animal Research Facilities of the University of Tasmania, Australia, the Comparative Genomics Centre, James Cook University, Australia, and the Institute for Clinical Microbiology, Immunology and Hygiene at the University Hospital of Erlangen, Germany. All experiments followed protocols approved by the animal ethics committees of James Cook University, Townsville, the University of Tasmania, Hobart, Tasmania and the Government of Mittelfranken, Germany. Mice of an age of 8–12 weeks were used in all experiments.
Parasites and Infection
The virulent L. major isolate MHOM/IL/81/FE/BNI (Solbach et al., 1986; Stenger et al., 1996) was maintained through serial passage in BALB/c mice in vivo and cultured in vitro in Novy-Nicolle-MacNeal blood agar slants in RPMI1640 medium supplemented with 10% new born calf serum, penicillin/streptomycin, non-essential amino acids and 10 mM HEPES, all supplied by Invitrogen Life Technologies (Mount Waverly, Australia). For infection, stationary phase L. major promastigotes were used between passage 2 and 6 and 3 × 106 parasites were injected in a volume of 40 μl into one hind footpad. The infection site was monitored daily and the increase in lesion size was determined weekly by measuring the footpad thickness with a metric caliper (Kroeplin Schnelltaster, Schluechtern, Germany). The percentage of increase in footpad thickness was determined by the formula (thickness of infected footpad minus thickness of non-infected footpad/mean thickness of non-infected footpad) × 100. The parasite burden (per gram of tissue) was determined at day 28 post infection (p. i.) using a limiting dilution method and L-Calc software version 1.1 (Stem Cell Technologies, www.Stemcell.com) which performs a generalized Pearson Chi-squared test (Wilhelm et al., 2001).
Flow Cytometry
Draining popliteal (p) lymph nodes (LN) or footpad lesions were incubated with collagenase D (1 mg/ml, Roche Products Australia, Brisbane, QLD, Australia) and DNAse 1 (100 U/ml, Sigma–Aldrich) for 30 min at 37°C and disrupted between frosted glass slides yielding single cell suspensions. Cells were filtered through 60 μm nylon meshes or 40 μm cell strainers (BD Biosciences, Sydney, NSW, Australia) to remove tissue debris. Prior to FACS staining the cells were blocked with either anti-CD16/32 antibody (clone 2.4G2, eBioscience, San Diego, CA, USA) or 10% rat serum (IMVS, Adelaide, SA, Australia). Cells were stained with rat-anti-mouse antibodies specific for B220 (RA3-6B2, Pacific Blue, or APC-Cy7), CD4 (RM4-5, PerCP-Cy5.5, or Pacific Blue), CD8 (53–6.7, Pacific Blue), CD25 (PC61, APC; 7D4, FITC), CD44 (IM7, PeCy7), CD62L (MEL-14, APC), GITR (DTA-1, PeCY7), Vß4 TCR (KT4, PE), Vß5.1/5.2 TCR (MR9-4, FITC) CD3𝜀 (145-2C11, PE-Cy7), IL-17 (TC11-18H10, PE), IL-4 (11B11, PE), IFN-γ (XMG1.2, Alexa Fluor-488) and with Armenian hamster-anti-mouse antibodies specific for TCR β-chain (H57-597, APC or biotin/Streptavidin Pacific Orange). Multicolour staining of single cells for surface antigens was performed as published (Wilhelm et al., 2001). Data were acquired using a Cyan ADP (Beckman Coulter, Fullerton, CA, USA). Analyses were performed using FlowJo® version 8.86 (Tree Star Inc.).
Cell Isolation
CD4+ T cells were isolated from draining LN using antibody labeled magnetic beads (Miltenyi Biotec Australia, Sydney, NSW, Australia). Briefly, the LN were disrupted between frosted glass slides yielding single cell suspensions. The cells were washed with PBS/0.1% BSA and incubated with anti-CD4 beads (Miltenyi Biotec) for 15 min on ice. After labeling, all CD4+ cells (naïve as well as activated) were isolated using a standard lymphocyte protocol on a fully automated autoMacs Pro Separator (Miltenyi Biotec) which was provided by the manufacturer.
Cytokine Analysis
Intracellular cytokine staining was performed on antigen-stimulated T cells. Cells that included CD4+ T cells and antigen-presenting cells were re-stimulated in 96 well plates (Sarstedt Australia, Mawson Lakes, SA, Australia) at a density of 5 × 105 cells using freeze-thawed L. major antigen (MOI equivalent = three) for 72 h in RPMI1640 medium supplemented with 10% new born calf serum, penicillin/streptomycin, non-essential amino acids, and 10 mM HEPES, all supplied by Invitrogen Life Technologies (Mount Waverly, Australia). For the last 6 h of culture the cells were kept in the presence of PMA (20 ng/ml)/ionomycin (1 μg/ml) and Golgi-StopTM (BD Biosciences, 4 μg/ml). Subsequently, the cells were stained for surface antigens, fixed, permeabilised using FoxP3-FixPerm buffer (Biolegend, Perth, Australia) and stained for IFN-γ, IL-4, and IL-17 following the manufacturers’ instructions.
Serum was collected from experimental animals weekly during the course of infection. Cytokine levels were measured in mouse serum and tissue culture supernatants using mouse inflammation cytokine bead array (CBA, BD Biosciences). CBA were acquired on the ARIA II and FCS 2.0 files were analyzed using FCAP Array (Soft Flow Inc, Burnsville, MN, USA).
Real-time RT-PCR was performed with total RNA extracted from footpad lesions using Trizol (Invitrogen). Following DNase treatment of RNA (using RQ1 RNase free DNase; Promega, Sydney Australia) cDNA synthesis was performed with Superscript III reverse transcriptase (Invitrogen). Gene expression analysis was performed using SYBR-green-ER qPCR kit (Invitrogen) or Brilliant II SYBR-green (Agilent Technologies, Integrated Sciences, Willoughby, NSW, Australia), run on a Corbett Rotor Gene 6000 (Qiagen, Doncaster, Australia) and analyzed using REST 2009 gene expression software (Qiagen) to determine relative expression of genes. PCR primers were designed using Vector NTI (Invitrogen): β-actin-forward: AAT CCT GTG GCA TCC ATG AAA C, β-actin-reverse: CGC AGC TCA GTA ACA GTC CG; GAPDH- forward: GTG AAG GTC GGT GTG AAC GG, GAPDH-reverse: ATG TTA GTG GGG TCT CGC TCC; Gata3-forward: GAG GTG GAC GTA CTT TTT AAC AT, Gata3-reverse: GGC ATA CCT GGC TCC CGT; Hprt-forward: GTT GGT TAC AGG CCA GAC TTT GTT G, Hprt-reverse: GAG GGT AGG CTG GCC TAT AGG CT; Il-10-forward: GGT TGC CAA GCC TTA TCG GA, Il-10-reverse: ACC TGC TCC ACT GCC TTG CT; Il-17A-forward: TCT GTG TCT CTG ATG CTG TTT GC, Il17A-reverse: ACG GTT GAG GTA GTC TGA GGG C; Ifn-γ-forward: AGA GCC AGA TTA TCT CTT TCT AC, Ifn-γ-reverse: CTT TTT TCG CCT TGC TGC TG; Rorγt-forward: CCG CTG AGA GGG CTT CAC, Rorγt-reverse: TGC AGG AGT AGG CCA CAT TAC A; Tbx21-forward: CAA CAA CCC CTT TGC CAA AG, Tbx21-reverse: TCC CCAA GCA AGT TGA CAGT.
Statistics
Statistical analysis was performed using a non-parametric Mann Whitney U-test, a Kruskall–Wallis with Dunn’s multiple comparison test or a two-way ANOVA test with Bonferroni correction to test for multiple hypotheses. Mean values (±SEM) are shown. Analysis was performed using GraphPad Prism 5.0 for MacIntosh (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Each experimental group was compared to Wt controls. Statistical p values of p < 0.05 were considered to be significant (with ∗p < 0.05 and ∗∗p < 0.01, respectively).
Results
Clinical Course of L. major Infection in the Absence of TNF or its Receptors
The published clinical outcomes of L. major infection in Tnfr1-/- and Tnfr2-/- mice (Vieira et al., 1996; Nashleanas et al., 1998) were significantly different from the infection of Tnf-/- mice (Wilhelm et al., 2001). It has been shown earlier using bone marrow reconstitution that changes in the lymphoid organs due to the absence of TNF signaling only have a minimal influence on the clinical outcome of the infection (Wilhelm et al., 2001). Therefore, this could reflect the use of different L. major strains (Ritter et al., 2004) or the genetic heterogeneity of the mouse strains. Therefore, we infected Tnf-/-, Tnfr1-/-, Tnfr2-/-, and memTnfΔ/Δ strains (Körner et al., 1997; Peschon et al., 1998; Ruuls et al., 2001) with the pathogenic L. major isolate BNI and monitored the course of disease (Figure 1). In accordance with previously described results obtained with a different strain of L. major (Allenbach et al., 2008), infected memTNFΔ/Δ mice, which express the membrane form but lack the ability to release soluble TNF, and Wt control mice showed a comparable course of infection (Figure 1) and were able to control the infection. In contrast, L. major-infected Tnf-/-, Tnfr1-/-, and Tnfr2-/- mice all developed similar, large skin lesions strikingly different from the Wt control mice. At day 56 all control mice had resolved their lesions, whereas infected Tnf- and Tnfr1-deficient mice had to be euthanized in accordance with animal ethics considerations because the animals developed signs of systemic distress. L. major-infected Tnfr2-deficient mice developed large lesions, which were comparable in size to those in TNF or TNFR1 mice, but ultimately controlled the infection (Figure 1 and data not shown). The wildtype controls [C57BL/6, shown as B6.WT (Figure 1) and BALB/c (data not shown)] exhibited the expected symptoms with B6.Wt controlling the infection whereas BALB/c succumbing to a progressive infection. From these data we conclude that TNF and TNFR1 are both essential for controlling an infection with L. major, whereas TNFR2 only plays a contributory role. Furthermore, membrane TNF alone is sufficient to convey protection.
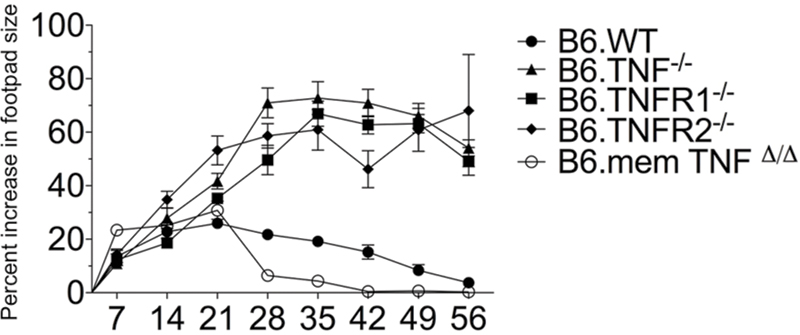
FIGURE 1. Course of infection in Tnf- and Tnfr-negative mouse strains. The lesion sizes of Tnf-/- (n = 34), memTnfΔ/Δ (n = 5), Tnfr1-/- (n = 27), and Tnfr2-/- (n = 27) mice, which had been infected subcutaneously with Leishmania major in one hind footpad were determined and compared to Wt (n = 36). The data are presented as percent increase in footpad lesion size. The combined results of four experiments are shown as mean (±SEM) and the number of animals stated above is the maximal number at the beginning of the experiment. The differences in clinical outcome were combined for analysis using a two-way ANOVA with Bonferroni correction to test for multiple hypotheses.
Induction of IFN-γ Response in Tnf-/- Mice During L. major Infection
The cytokine milieu is essential for induction of a leishmanicidal response in macrophages and for the establishment of protective immunity. To determine the impact of a TNF or TNF receptor deficiency on the development of the adaptive immune response, we first infected Wt, Tnf-/- and BALB/c mice with L. major and analyzed CD4+ T cells from popliteal draining lymph nodes (pLN) for their expression of IL-4, IFN-γ, IL-17, and IL-10. We observed a relatively early, strong IL-4 expression in all mouse strains on day 7 p. i., which had largely disappeared on day 21 p. i. (Figure 2A). IFN-γ was present in all three genotypes with a significantly elevated expression in TNF-/- (see Figure 2B), while another pro-inflammatory cytokine, IL-17, was hardly detectable in CD4+ T cells of any strain at the analyzed time-points (Figure 2A). The expression of IL-10 was determined after 7, 21, and 28 days and was very similar to IL-4 expression with a strong presence at day 7 p. i., and fast down-regulation thereafter (Figure 2A). Both IL-4 and IL-10 were co-expressed with IFN-γ in 1 – 2% of all cells (Figure 2A). This finding is likely to reflect a random heterogeneity during the differentiation of Th1 and Th2 cells at the beginning of the adaptive immune response (Morris et al., 1992), as the IL-4-, IL-10-, and IFN-γ-co-expressing cells were only detectable at day 7 p. i.
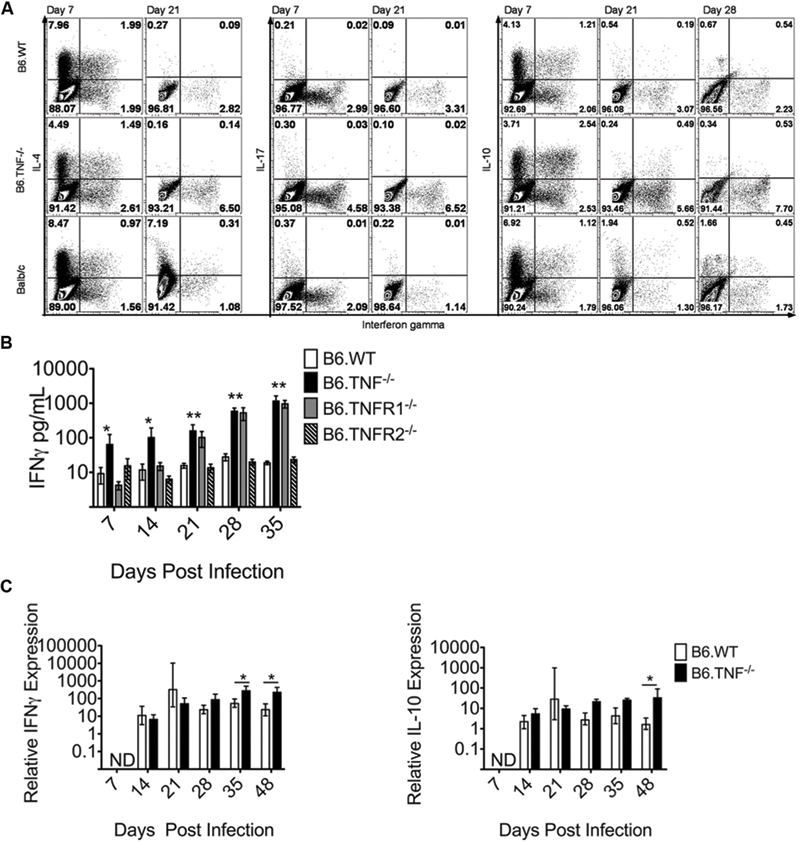
FIGURE 2. TNF-TNFR1 signaling pathway deficiency results in elevated IFN-γ expression after L. major infection. (A) Cytokine expression in CD4+ T cells from popliteal draining LNs of L. major infected Wt, Tnf-/- and BALB/c mice was analyzed. The use of the Tnf-/- mice constitutes the disruption of the complete TNF-TNFR1 signaling pathway. Intracellular flow cytometry was used to determine the proportional expression of IFN-γ, IL-4, IL-17, and IL-10 in CD4+ T cells at day 7 and 21 (and in the case of IL-10 also day 28) p. i. The experiment was performed two times independently and a representative staining is shown. (B) The concentration of IFN-γ in the serum of L. major infected Wt, Tnf-/-, Tnfr1-/-, and Tnfr2-/- was determined. The data are presented as mean (±SEM; n = 5–6 per genotype). Results are representative of at least 3 independent experiments. (C) The relative expression of IFN-γ and IL-10 in the footpad lesion of infected Wt or Tnf-/- mice was compared to uninfected controls. Relative expression was calculated relative to β-actin as described (Meissner et al., 2003). The data are presented as median (±SEM; n = 3–5 mice, representative of two independent experiments). The white bar represents Wt, the black bar Tnf-/- mice. The non-parametric Mann–Whitney U-test was used for analysis of experiments displayed in (B,C) to test for statistical differences (∗p < 0.05 ∗∗p < 0.01).
A quantification of the IFN-γ expression in the serum of all genotypes at days 7, 14, 21, 28, and 35 p. i. showed a significantly increased concentration of IFN-γ throughout the course of infection exclusively in both Tnf-/- and Tnfr1-/- mice (Figure 2B). The expression level of IFN-γ mRNA in the footpad lesions was initially comparable between infected wt and Tnf-/- mice, but was significantly increased in Tnf-/- mice after day 35 of infection (Figure 2C). IL-10 expression relative to the housekeeping gene was also increased significantly at day 35 p. i. (Figure 2C). However, as demonstrated by flow cytometry, the protein expression of IL-10 was substantially lower than of IFN-γ which points to an unexpectedly stable and even enhanced Th1-type response in the absence of TNF (Figure 2C).
Expression Analysis of Transcription Factors and Cytokines in Activated CD4+ T cells in L. major-Infected Wt and Tnf-/- Mice
The polarization of T cell responses is strongly regulated by the balance of a number of transcription factors such as Tbx21 (Tbet) and Gata3 that regulate the expression of IFN-γ and IL-4, respectively (Zheng and Flavell, 1997; Szabo et al., 2000, 2002). To investigate a potential cause for the increased IFN-γ expression in the absence of the TNF-TNFR1 signaling pathway during the course of L. major infection, we isolated populations of both activated (CD62L- CD44+) and naïve CD4+ T cells (CD62L+ CD44-) from the spleens of Wt and Tnf-/- mice at day 50 p. i. and analyzed the expression level of several genes. The relative increase in Tbx21 expression in activated T cells of both genotypes (Wt: mean = 134 [range 111.43–170.87]; Tnf-/-: mean = 34 [range 23.7–41.56]) was in agreement with the observed increase in Ifn-γ expression (497-fold upregulation in wt mice and 324-fold upregulation in Tnf-/- mice) (Figure 3B). In contrast, the expression of Gata3, which directs both IL-4 and IL-10 production (Zheng and Flavell, 1997; Shoemaker et al., 2006), was only weakly induced (1.6-fold) in activated wt CD4+ T cells and decreased (1.42-fold) in activated Tnf-/- CD4+ T cells as compared to the respective naive CD4+ T-cell populations (Figure 3A). This was associated with a dramatic reduction of the IL-10 mRNA expression (Figure 3B). Additionally, we analyzed the expression of the transcription factor Rorγt (Rorc) which is characteristic for the pro-inflammatory Th17 T cell subset (Yang et al., 2008). At this late stage of infection it was up-regulated in activated CD4+ T cells of both wt (mean = 67.9-fold) and Tnf-/- (mean = 48.6-fold) mice to a similar extent (Figure 3A), although expression of IL-17A was generally low and barely detectable either by real-time PCR (Figure 3B) or by intracellular cytokine staining (Figure 2A).
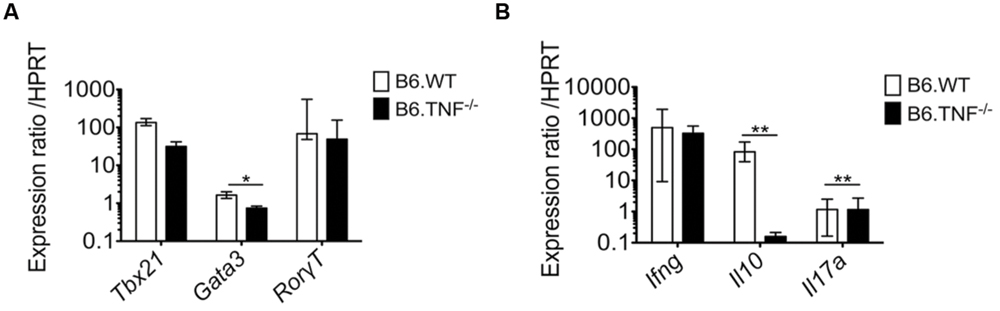
FIGURE 3. Expression of Tbx21 is unchanged while expression of Gata3 is reduced in activated Tnf-deficient CD4+ T cells. Naïve (CD62L+ CD44-) as well as activated (CD62L- CD44+) CD4+ T cells from Wt or Tnf-/- mice were isolated from the spleens of individual mice at day 50 after L. major infection. Quantitative real time RT-PCR was performed to analyze the relative mRNA expression of transcription factors (A) and cytokines (B). The expression of the different genes was normalized with the house keeping gene hypoxanthine guanine phosphoribosyl transferase (HPRT). Data are presented as median (±SEM) of the mRNA expression ratio of activated vs. naive CD4+ T cells (n = 5–6 mice, one representative of two independent experiments is shown) (∗p < 0.05, ∗∗p < 0.01).
Thus, only the Gata3 gene was expressed differently between the genotypes and consequently IL-10 expression was dramatically reduced in infected Tnf-/- mice. These data further corroborate that mice lacking the TNF-TNFR1 signaling pathway show an exaggerated Th1 response but nevertheless are unable to resolve the infection.
Analysis of CD4+ T cell Activation During Leishmaniasis
Both susceptible and resistant strains of mice were reported to develop strong antigen-specific responses to epitopes of the Leishmania homolog of receptors for activated C-kinase antigen (LACK) with a predominant stimulation of Vα8 Vβ4-positive T cells, whereas the Vβ5-expressing T cell population did not expand (Launois et al., 2007). Therefore, we followed the expansion of Vβ4 TCR+ CD4+ T cells and concurrently analyzed the Vβ5.1/5.2 TCR+ CD4+ T cells (Launois et al., 1997) in the draining pLNs of Wt, Tnf-, Tnfr1-, or Tnfr2-deficient mice. The activation of these T cells was studied using two classical activation markers of peripheral CD4+ T cells, CD62L and CD44 (Tough et al., 1996). Within the draining LN isolated from Wt, Tnf-/-, Tnfr1-/-, and Tnfr2-/- mice, a comparable proportion of T cells was Vβ4 TCR+ CD4+ (Figure 4A). However, the population of Vβ4 TCR+ CD4+ T cells from Tnf-/- and Tnfr1-/- mice contained a significantly larger proportion of activated (CD62L- CD44+) T cells (Figure 4A) and showed also an increase in the absolute numbers of activated Vβ4 TCR+ CD4+ T cells within the draining pLN (Figure 4B). Interestingly, Vβ5.1/5.2 TCR+ CD4+ T cells from Tnf-/- and Tnfr1-/- mice showed a similar increase in activation as shown for Vβ4 TCR+ CD4+ T cells, pointing to a strong non-specific activation of the T cell compartment in these genotypes (Figure 4B). In Wt as well as Tnfr2-deficient mice neither Vβ4 TCR+ T cells nor Vβ5 TCR+ T cells displayed significant activation at this time-point of analysis. By day 50 p. i. the T cell compartment of Tnf-/- mice had collapsed and both Vβ4 and Vβ5.1/5.2 TCR+ CD4+ T cell subsets were reduced within the pLN (data not shown).
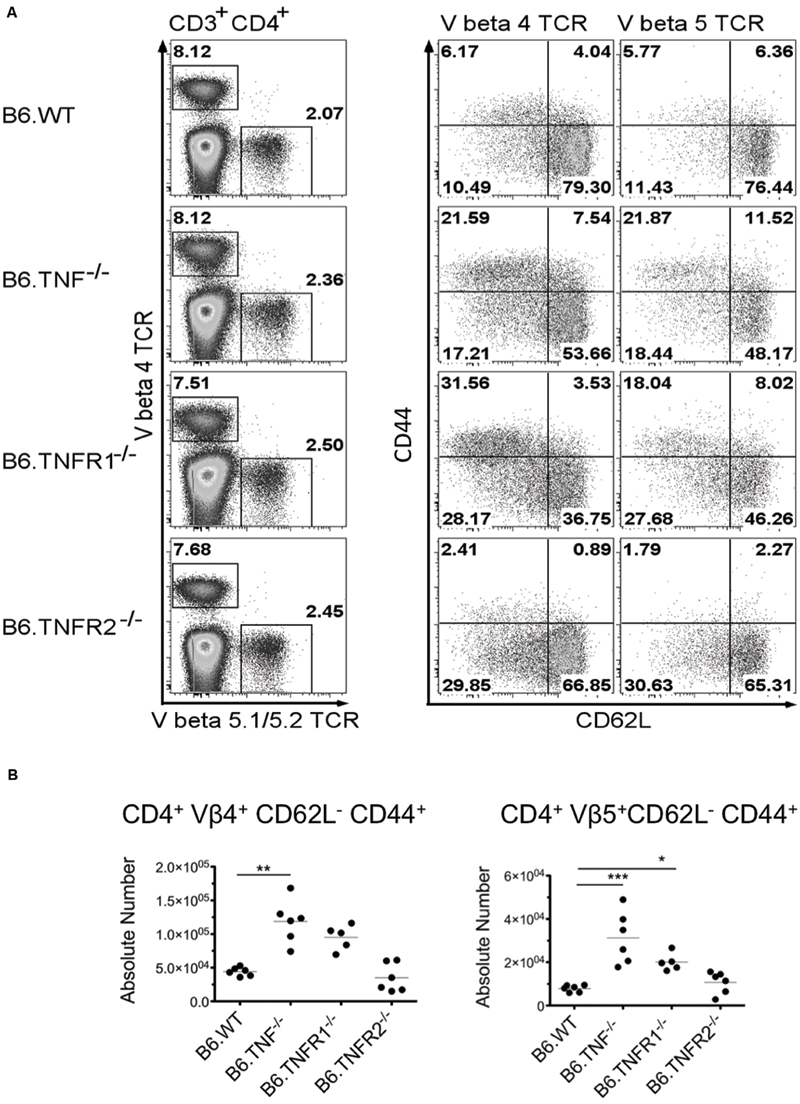
FIGURE 4. Expansion and activation of Vβ4 and Vβ5.1/5.2 TCR repertoire occurs in the absence of TNF or TNFR1 but not TNFR2. Wt mice or mice lacking TNF, TNFR1, or TNFR2 were infected with 3 × 106 L. major promastigotes and draining pLN were harvested at day 21 p. i. (A) Tnf- and Tnfr1-deficient mice showed increased activation of CD4+ T cells expressing either Vβ4 or Vβ5.1/5.2 TCR chains. One of two representative experiments is shown. (B) Increased activation was correlated with increased absolute numbers of both Vβ4 or 5 restricted activated (CD62L- CD44+) CD4+ T cells in the draining pLN by a comparable size of the organs. Statistical comparisons were performed using a Kruskal–Wallis Test with Dunns correction; n = 5–6 per genotyope ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p < 0.001.
Discussion
The TNF signaling pathways, especially the TNF-TNFR1 pathway, are essential in generating an inflammatory response and mediating resistance to infection by intracellular pathogens such as L. major (Wilhelm et al., 2001; Körner et al., 2010). In the absence of the pro-inflammatory cytokine TNF the protection against L. major is severely impaired resulting in a progressive infection and eventually a fatal outcome. Interestingly, using reciprocal bone marrow chimeras we could show that the ability of hemopoietic cells to express TNF confers protection (Wilhelm et al., 2001). The relatively subtle structural changes of lymphoid organs in Tnf- and Tnfr1-gene deficient mice were not relevant with regard to protection against L. major (Wilhelm et al., 2001).
In the present study we have analyzed the clinical course of L. major infection in C57BL/6 mouse strains deficient for TNF, soluble TNF (memTnfΔ/Δ), TNFR1 or TNFR2 and could show that the outcome was fatal in the absence of TNF and TNFR1. A deficiency of soluble TNF did not impair the healing phenotype associated with the C57BL/6 background as long as the membrane-bound cytokine was still expressed. This is in agreement with earlier studies that showed that transmembrane TNF expressed on CD4+ T cells was sufficient to induce a protective anti-Leishmania immune response (Birkland et al., 1992; Allenbach et al., 2008). A Tnfr2-deficiency resulted in the development of large skin lesions, which, however, ultimately was resolved. The fatal outcome observed in L. major infected Tnf- or Tnfr1-deficient mice occurred despite a seemingly intact adaptive immune response and an over-expression of IFN-γ.
Our results describing a fatal course of L. major infection in TNF-/- (Wilhelm et al., 2001) contrast with two other studies published in experimental cutaneous leishmaniasis, which either used anti-TNF antibodies in C3H/HeN mice (Titus et al., 1989) or a Tnf-/- strain on a different mouse background (Chakour et al., 2003). In both cases an aggravated clincial course of infection was observed, but the mice resolved the lesions and survived. Similarly, our findings on the contribution of TNFR1 or TNFR2 to the protective response to L. major also differ from several earlier reports. Vieira et al. (1996) used a Tnfr1-/- strain generated on a 129Sv background and showed that these mice survived the infection and eliminated the parasites but did not completely resolve the skin swelling and pathology. Infection of Tnfr2-/- mice on a mixed 129Sv × C57BL/6 background showed that this receptor was dispensable for the control of L. major (Nashleanas et al., 1998) and, finally, an infection of a Tnfr1/2 double-deficient 129Sv × C57BL/6 mouse followed the Tnfr1-/- phenotype (Nashleanas et al., 1998). In the present study, we compared memTnfΔ/Δ, Tnf-/-, Tnfr1-/-, and Tnfr2-/- mice on an identical C57BL/6 genetic background using the L. major parasite strain BNI to account for differences caused by the genetic variability of the infected mouse strain and the parasite strain used (Ritter et al., 2004). In our experiments the previously observed phenotypes could not be replicated with the exception of the resistance of memTnfΔ/Δ to the infection. Both Tnfr1-/- and Tnfr2-/- mice displayed a lesion development comparable to Tnf-/- mice suggesting overlapping roles for the two receptors in the pathogenesis of leishmaniasis. Interestingly, Tnfr2-/- mice ultimately survived, whereas both Tnf-/- and Tnfr1-/- mice developed signs of severe systemic disease and succumbed to the infection.
Experimental cutaneous leishmaniasis in genetically inbred mice was the first model which showed a mouse strain-dependent polarization of IL-4 and IFN-γ production by CD4+ T cells correlating with either disease susceptibility or resistance (Heinzel et al., 1989) and therefore formed the basis of the Th1 and Th2 paradigm (Mosmann and Coffman, 1989). In our experiments, the expression of IL-10 paralleled the expression of IL-4. Additionally, we detected a small percentage of both IFN-γ/IL-10 and IFN-γ/IL-4 double positive T cells at day 7 p. i. which disappeared soon after. These cells could be part of a regulatory subpopulation or represent random heterogeneity during T cell differentiation at the beginning of the antigen-specific immune response (Morris et al., 1992). The major argument for a central role of IFN-γ in resistance to L. major infection has been its polarizing influence on CD4+ T cell differentiation (Heinzel et al., 1989; Laouar et al., 2005) and its activating effect on macrophages with the induction of leishmanicidal NO in vitro (Liew et al., 1990) and in vivo (Stenger et al., 1994, 1996; Diefenbach et al., 1998). This has been tested by administration of anti-IFN-γ mAbs to C3H/HeN mice which prevented the development of natural resistance (Belosevic et al., 1989; Scott, 1991). However, several studies have called into question this fundamental role for IFN-γ in the sequence of events resulting in protection. First, while the administration of anti-IL-4 mAb to susceptible BALB/c mice prevented progressive uncontrolled infection of L. major (Sadick et al., 1990) and was paralleled by an up-regulation of IFN-γ production, the concurrent neutralization of this elevated IFN-γ did not abrogate or change the resistance phenotype. Second, mice congenic for known resistance loci (Lmr1, Lmr2, Lmr3) derived from either resistant C57BL/6 or susceptible BALB/c mice did not display the expected susceptible/resistant phenotype despite expressing either IL-4 or IFN-γ suggesting that the IFN-γ/IL-4 cytokine profile alone is not a sufficient determinant of disease resistance (Elso et al., 2004a,b; Sakthianandeswaren et al., 2010). Resistant C57BL/6 mice carrying a BALB/c congenic region for lmr1 displayed increased susceptibility while the reciprocal congenic strain (BALB/c) containing the C57BL/6 allele showed an intermediate phenotype (Elso et al., 2004a). Third, following infection with a L. major strain isolated from patient with chronic cutaneous leishmaniasis a non-healing course of disease was observed in C57BL/6 mice despite efficient Th1 polarization (Anderson et al., 2005). Fourth, in our present study we showed that mice deficient for TNF or TNFR1 overexpressed IFN-γ and displayed intact, relatively strong expression of iNOS in the draining LN (Wilhelm et al., 2001), yet, counterintuitively, developed progressive, and ultimately fatal leishmaniasis. While a synergy between IFN-γ and pro-inflammatory mediators (e.g., TNF) based on cooperative signaling through STAT1, IRF-1 and NFκB (Drapier et al., 1988; Ohmori et al., 1997; Saura et al., 1999; Schroder et al., 2004; Farlik et al., 2010) has been described in vitro for the production of NO by macrophages (Bogdan et al., 1990; Green et al., 1990; Ding et al., 1998), there appears to be a high level of redundancy in vivo. Nevertheless, despite intact induction of iNOS which is associated with parasite clearance (Stenger et al., 1994, 1996; Diefenbach et al., 1998; Blos et al., 2003), Tnf-/- mice still failed to control parasite growth and dissemination (Wilhelm et al., 2001).
Taken together, the lack of resistance to L. major infection in Tnf-/- and Tnfr1-/- mice despite an increased production of IFN-γ points to a central role for TNF/TNFR1 signaling in linking innate leishmanicidal effector mechanisms with the adaptive immune response. Since TNF has been shown to modulate the expansion of regulatory T cell networks (Chen et al., 2007), the absence of TNF signaling may modify the local immune response in a way that it becomes refractory to the activity of IFN-γ and iNOS and therefore unable to prevent progressive infection. In addition, the sustained presence of large amounts of systemic IFN-γ throughout the course of leishmaniasis in Tnf-negative mice may be the consequence of ongoing infection and parasite dissemination. Instead of promoting protection by activating macrophages the overproduction of IFN-γ could result in immunopathology that contributes to a fatal outcome in response to infection with L. major.
Funding
This study was supported by a travel grant of the European Macrophage and Dendritic Cell Society and the Logan Travel Award of the School of Pharmacy and Molecular Sciences (to PF), the Research Advancement program of JCU and the NHMRC (485807 to HK), the German Research Foundation (grant SFB643, project A6, and SFB1181, project C4, to CB) and the Interdisciplinary Center for Clinical Research (IZKF) of the University Hospital of Erlangen (project grant A61 to CB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Ulrike Schleicher and Rene Gollan for their technical help. Furthermore, we thank Chris Engwerda for providing memTNFΔ/Δ mice and Nicole Fraser and Kylie Robertson as well as the animal facility of the University of Tasmania for expert animal husbandry.
References
Alexander, J., and Brombacher, F. (2012). T helper1/T helper2 cells and resistance/susceptibility to Leishmania infection: is this paradigm still relevant? Front. Immunol. 3:80. doi: 10.3389/fimmu.2012.00080
Allenbach, C., Launois, P., Mueller, C., and Tacchini-Cottier, F. (2008). An essential role for transmembrane TNF in the resolution of the inflammatory lesion induced by Leishmania major infection. Eur. J. Immunol. 38, 720–731. doi: 10.1002/eji.200737662
Anderson, C. F., Mendez, S., and Sacks, D. L. (2005). Nonhealing infection despite th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J. Immunol. 174, 2934–2941. doi: 10.4049/jimmunol.174.5.2934
Baldwin, T., Sakthianandeswaren, A., Curtis, J. M., Kumar, B., Smyth, G. K., Foote, S. J., et al. (2007). Wound healing response is a major contributor to the severity of cutaneous Leishmaniasis in the ear model of infection. Parasite Immunol. 29, 501–513. doi: 10.1111/j.1365-3024.2007.00969.x
Belosevic, M., Finbloom, D., Van Der Meide, P., Slayter, M., and Nacy, C. (1989). Administration of monoclonal anti-IFN-gamma antibodies in vivo abrogates natural resistance of C3H/HeN mice to infection with Leishmania major. J. Immunol. 143, 266–274.
Birkland, T. P., Sypek, J. P., and Wyler, D. J. (1992). Soluble TNF and membrane TNF expressed on CD4+ T lymphocytes differ in their ability to activate macrophage antileishmanial defense. J. Leukoc. Biol. 51, 296–299.
Blos, M., Schleicher, U., Soares Rocha, F. J., Meissner, U., Rollinghoff, M., and Bogdan, C. (2003). Organ-specific and stage-dependent control of Leishmania major infection by inducible nitric oxide synthase and phagocyte NADPH oxidase. Eur. J. Immunol. 33, 1224–1234. doi: 10.1002/eji.200323825
Bogdan, C. (2008). Mechanisms and consequences of persistence of intracellular pathogens: leishmaniasis as an example. Cell. Microbiol. 10, 1221–1234. doi: 10.1111/j.1462-5822.2008.01146.x
Bogdan, C., Moll, H., Solbach, W., and Rollinghoff, M. (1990). Tumor necrosis factor-alpha in combination with interferon-gamma, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur. J. Immunol. 20, 1131–1135. doi: 10.1002/eji.1830200528
Chakour, R., Guler, R., Bugnon, M., Allenbach, C., Garcia, I., Mauel, J., et al. (2003). Both the Fas ligand and inducible nitric oxide synthase are needed for control of parasite replication within lesions in mice infected with Leishmania major whereas the contribution of tumor necrosis factor is minimal. Infect. Immun. 71, 5287–5295. doi: 10.1128/iai.71.9.5287-5295.2003
Chen, X., Baumel, M., Mannel, D. N., Howard, O. M. Z., and Oppenheim, J. J. (2007). Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 179, 154–161. doi: 10.4049/jimmunol.179.1.154
Deng, W., Thiel, B., Tannenbaum, C. S., Hamilton, T. A., and Stuehr, D. J. (1993). Synergistic cooperation between T cell lymphokines for induction of the nitric oxide synthase gene in murine peritoneal macrophages. J. Immunol. 151, 322–329.
Diefenbach, A., Schindler, H., Donhauser, N., Lorenz, E., Laskay, T., MacMicking, J., et al. (1998). Type 1 interferon (IFN alpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8, 77–87. doi: 10.1016/S1074-7613(00)80460-4
Ding, M., Zhang, M., Wong, J. L., Rogers, N. E., Ignarro, L. J., and Voskuhl, R. R. (1998). Cutting Edge: antisense knockdown of inducible nitric oxide synthase inhibits induction of experimental autoimmune encephalomyelitis in SJL/J mice. J. Immunol. 160, 2560–2564.
Drapier, J.-C., Wietzerbin, J., and Hibbs, J. B. Jr., (1988). Interferon-gamma and tumor necrosis factor induce the L-arginine-dependent cytotoxic effector mechanism in murine macrophages. Eur. J. Immunol. 18, 1587–1592. doi: 10.1002/eji.1830181018
Elso, C., Kumar, B., Smyth, G., Foote, S., and Handman, E. (2004a). Dissociation of disease susceptibility, inflammation and cytokine profile in lmr1/2 congenic mice infected with Leishmania major. Genes Immun. 5, 188–196. doi: 10.1038/sj.gene.6364056
Elso, C. M., Roberts, L. J., Smyth, G. K., Thomson, R. J., Baldwin, T. M., Foote, S. J., et al. (2004b). Leishmaniasis host response loci (lmr1-3) modify disease severity through a Th1/Th2-independent pathway. Genes Immun. 5, 93–100. doi: 10.1038/sj.gene.6364042
Farlik, M., Reutterer, B., Schindler, C., Greten, F., Vogl, C., Muller, M., et al. (2010). Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity 33, 25–34. doi: 10.1016/j.immuni.2010.07.001
Green, S. J., Crawford, R. M., Hockmeyer, J. T., Meltzer, M. S., and Nacy, C. A. (1990). Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J. Immunol. 145, 4290–4297.
Heinzel, F. P., Sadick, M. D., Holaday, B. J., Coffman, R. L., and Locksley, R. M. (1989). Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine Leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 169, 59–72. doi: 10.1084/jem.169.1.59
Körner, H., Cook, M., Riminton, D. S., Lemckert, F. A., Hoek, R. M., Ledermann, B., et al. (1997). Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 27, 2600–2609. doi: 10.1002/eji.1830271020
Körner, H., McMorran, B., Schluter, D., and Fromm, P. (2010). The role of TNF in parasitic diseases: still more questions than answers. Int. J. Parasitol. 40, 879–888. doi: 10.1016/j.ijpara.2010.03.011
Laouar, Y., Sutterwala, F. S., Gorelik, L., and Flavell, R. A. (2005). Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat. Immunol. 6, 600–607. doi: 10.1038/ni1197
Launois, P., Maillard, I., Pingel, S., Swihart, K. G., XÈnarios, I., Acha-Orbea, H., et al. (1997). IL-4 rapidly produced by V[beta]4 V[alpha]8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity 6, 541–549. doi: 10.1016/S1074-7613(00)80342-8
Launois, P., Pingel, S., Himmelrich, H., Locksley, R., and Louis, J. (2007). Different epitopes of the LACK protein are recognized by V beta 4 V alpha 8 CD4+ T cells in H-2b and H-2d mice susceptible to Leishmania major. Microbes Infect. 9, 1260–1266. doi: 10.1016/j.micinf.2007.05.017
Liese, J., Schleicher, U., and Bogdan, C. (2007). TLR9 signaling is essential for the innate NK cell response in murine cutaneous Leishmaniasis. Eur. J. Immunol. 37, 3424–3434. doi: 10.1002/eji.200737182
Liese, J., Schleicher, U., and Bogdan, C. (2008). The innate immune response against Leishmania parasites. Immunobiology 213, 377–387. doi: 10.1016/j.imbio.2007.12.005
Liew, F. Y., Li, Y., and Millott, S. (1990). Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 145, 4306–4310.
Lykens, J. E., Terrell, C. E., Zoller, E. E., Divanovic, S., Trompette, A., Karp, C. L., et al. (2010). Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J. Immunol. 184, 877–885. doi: 10.4049/jimmunol.0902346
Meissner, A., Zilles, O., Varona, R., Jozefowski, K., Ritter, U., Marquez, G., et al. (2003). CC chemokine ligand 20 partially controls adhesion of naive B cells to activated endothelial cells under shear stress. Blood 102, 2724–2727. doi: 10.1182/blood-2003-01-0007
Morris, L., Troutt, A. B., Handman, E., and Kelso, A. (1992). Changes in the precursor frequencies of IL-4 and IFN-gamma secreting CD4+ cells correlate with resolution of lesions in murine cutaneous Leishmaniasis. J. Immunol. 149, 2715–2721.
Mosmann, T. R., and Coffman, R. L. (1989). TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173. doi: 10.1146/annurev.iy.07.040189.001045
Nashleanas, M., Kanaly, S., and Scott, P. (1998). Control of Leishmania major infection in mice lacking TNF receptors. J. Immunol. 160, 5506–5513.
Ohmori, Y., Schreiber, R., and Hamilton, T. A. (1997). Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem. 272, 14899–14907. doi: 10.1074/jbc.272.23.14899
Peschon, J. J., Torrance, D. S., Stocking, K. L., Glaccum, M. B., Otten, C., Willis, C. R., et al. (1998). TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 160, 943–952.
Prajeeth, C. K., Haeberlein, S., Sebald, H., Schleicher, U., and Bogdan, C. (2011). Leishmania-infected macrophages are targets of NK cell-derived cytokines but not of NK cell cytotoxicity. Infect. Immun. 79, 2699–2708. doi: 10.1128/IAI.00079-11
Reithinger, R., Dujardin, J. C., Louzir, H., Pirmez, C., Alexander, B., and Brooker, S. (2007). Cutaneous leishmaniasis. Lancet Infect. Dis. 7, 581–596. doi: 10.1016/S1473-3099(07)70209-8
Ritter, U., Mattner, J., Rocha, J. S., Bogdan, C., and Körner, H. (2004). The control of Leishmania (Leishmania) major by TNF in vivo is dependent on the parasite strain. Microbes Infect. 6, 559–565. doi: 10.1016/j.micinf.2004.02.008
Roebrock, K., Sunderkotter, C., Munck, N. A., Wolf, M., Nippe, N., Barczyk, K., et al. (2014). Epidermal expression of I-TAC (Cxcl11) instructs adaptive Th2-type immunity. FASEB J. 28, 1724–1734. doi: 10.1096/fj.13-233593
Ruuls, S. R., Hoek, R. M., Ngo, V. N., McNeil, T., Lucian, L. A., Janatpour, M. J., et al. (2001). Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity 15, 533–543. doi: 10.1016/S1074-7613(01)00215-1
Sacks, D., and Noben-Trauth, N. (2002). The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2, 845–858. doi: 10.1038/nri933
Sacks, D. L., and Perkins, P. V. (1985). Development of infective stage Leishmania promastigotes within phlebotomine sand flies. Am. J. Trop. Med. Hyg. 34, 456–459.
Sadick, M. D., Heinzel, F. P., Holaday, B. J., Pu, R. T., Dawkins, R. S., and Locksley, R. M. (1990). Cure of murine Leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell-dependent, interferon gamma-independent mechanism. J. Exp. Med. 171, 115–127. doi: 10.1084/jem.171.1.115
Sakthianandeswaren, A., Curtis, J. M., Elso, C., Kumar, B., Baldwin, T. M., Lopaticki, S., et al. (2010). Fine mapping of Leishmania major susceptibility Locus lmr2 and evidence of a role for Fli1 in disease and wound healing. Infect. Immun. 78, 2734–2744. doi: 10.1128/IAI.00126-10
Saura, M., Zaragoza, C., Bao, C., McMillan, A., and Lowenstein, C. J. (1999). Interaction of interferon regulatory factor-1 and nuclear factor [kappa]B during activation of inducible nitric oxide synthase transcription. J. Mol. Biol. 289, 459–471. doi: 10.1006/jmbi.1999.2752
Scharton-Kersten, T., Afonso, L. C., Wysocka, M., Trinchieri, G., and Scott, P. (1995). IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental Leishmaniasis. J. Immunol. 154, 5320–5330.
Schonian, G., Mauricio, I., Gramiccia, M., Canavate, C., Boelaert, M., and Dujardin, J. C. (2008). Leishmaniases in the Mediterranean in the era of molecular epidemiology. Trends Parasitol. 24, 135–142. doi: 10.1016/j.pt.2007.12.006
Schroder, K., Hertzog, P. J., Ravasi, T., and Hume, D. A. (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189. doi: 10.1189/jlb.0603252
Scott, P. (1991). IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous Leishmaniasis. J. Immunol. 147, 3149–3155.
Shoemaker, J., Saraiva, M., and O’Garra, A. (2006). GATA-3 directly remodels the IL-10 locus independently of IL-4 in CD4+ T cells. J. Immunol. 176, 3470–3479. doi: 10.4049/jimmunol.176.6.3470
Solbach, W., Forberg, K., Kammerer, E., Bogdan, C., and Rollinghoff, M. (1986). Suppressive effect of cyclosporin A on the development of Leishmania tropica-induced lesions in genetically susceptible BALB/c mice. J. Immunol. 137, 702–707.
Stenger, S., Donhauser, N., Thuring, H., Rollinghoff, M., and Bogdan, C. (1996). Reactivation of latent Leishmaniasis by inhibition of inducible nitric oxide synthase. J. Exp. Med. 183, 1501–1514. doi: 10.1084/jem.183.4.1501
Stenger, S., Thuring, H., Rollinghoff, M., and Bogdan, C. (1994). Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. J. Exp. Med. 180, 783–793. doi: 10.1084/jem.180.3.783
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., and Glimcher, L. H. (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. doi: 10.1016/S0092-8674(00)80702-3
Szabo, S. J., Sullivan, B. M., Stemmann, C., Satoskar, A. R., Sleckman, B. P., and Glimcher, L. H. (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338–342. doi: 10.1126/science.1065543
Titus, R. G., Sherry, B., and Cerami, A. (1989). Tumor necrosis factor plays a protective role in experimental murine cutaneous Leishmaniasis. J. Exp. Med. 170, 2097–2104. doi: 10.1084/jem.170.6.2097
Tough, D. F., Borrow, P., and Sprent, J. (1996). Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 272, 1947–1950. doi: 10.1126/science.272.5270.1947
Vieira, L. Q., Goldschmidt, M., Nashleanas, M., Pfeffer, K., Mak, T., and Scott, P. (1996). Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J. Immunol. 157, 827–835.
Wilhelm, P., Ritter, U., Labbow, S., Donhauser, N., Röllinghoff, M., Bogdan, C., et al. (2001). Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 166, 4012–4019. doi: 10.4049/jimmunol.166.6.4012
Yang, X. O., Pappu, B. P., Nurieva, R., Akimzhanov, A., Kang, H. S., Chung, Y., et al. (2008). T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39. doi: 10.1016/j.immuni.2007.11.016
Keywords: cutaneous leishmaniasis, IFN-γ, tumor necrosis factor, T cell subtypes, mouse models
Citation: Fromm PD, Kling JC, Remke A, Bogdan C and Körner H (2016) Fatal Leishmaniasis in the Absence of TNF Despite a Strong Th1 Response. Front. Microbiol. 6:1520. doi: 10.3389/fmicb.2015.01520
Received: 20 October 2015; Accepted: 17 December 2015;
Published: 22 January 2016.
Edited by:
Amy Rasley, Lawrence Livermore National Laboratory, USAReviewed by:
Katie Louise Flanagan, Monash University, AustraliaPaul Fisch, University Medical Center Freiburg, Germany
Copyright © 2016 Fromm, Kling, Remke, Bogdan and Körner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heinrich Körner, heinrich.korner@utas.edu.au
†Present address: Phillip D. Fromm, ANZAC Research Institute, Concord Hospital, Sydney, NSW, Australia