Mechanisms of Tumor Necrosis Factor-Alpha Inhibitor-Induced Systemic Lupus Erythematosus
 Chung-Yang Yen1,2
Chung-Yang Yen1,2  Sheng-Jie Yu3
Sheng-Jie Yu3  Yi-Ming Chen4
Yi-Ming Chen4  Kuo-Lung Lai4
Kuo-Lung Lai4  Yi-Da Wu4
Yi-Da Wu4  En-Chih Liao5†
En-Chih Liao5†  Ching-Liang Hsieh6,7*†
Ching-Liang Hsieh6,7*†- 1Department of Dermatology, Taichung Veterans General Hospital, Taichung City, Taiwan
- 2School of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan
- 3Department of Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung City, Taiwan
- 4Division of Allergy, Immunology and Rheumatology, Department of Internal Medicine, Taichung Veterans General Hospital, Taichung City, Taiwan
- 5Department of Medicine, MacKay Medical College, New Taipei City, Taiwan
- 6Chinese Medicine Research Center, China Medical University, Taichung City, Taiwan
- 7Department of Chinese Medicine, China Medical University Hospital, Taichung City, Taiwan
Systemic lupus erythematosus induced by biologics mainly results from tumor necrosis factor-alpha remains unclear. The objectives of the study were to investigate the mechanisms of tumor necrosis factor-alpha inhibitor-induced systemic lupus erythematosus. Peripheral blood mononuclear cells obtained from thirteen psoriasis patients were cultured and treated with the following: untreated control, Streptococcus pyogenes with or without different biologics. The supernatants were collected for cytokines assay. Analysis of cytokine expression revealed that IL-2 and IL-10 levels decreased only in the TNF-α inhibitor-treated groups but not in the groups treated with biologics involving IL-17, IL-12/IL-23 or IL-23 inhibitor mechanisms (p < 0.001, p < 0.05). The IFN-γ/IL-13 ratio increased significantly in patients with SLE inducing biologics to S. pyogenes induction only compared with non-SLE inducing biologics to S. pyogenes induction only (p = 0.001). IL-2 and IL-10 depletion and a shift to the Th-1 pathway in the innate response are the correlated mechanism for tumor necrosis factor-alpha inhibitor-induced systemic lupus erythematosus.
Introduction
Biologics are widely used in treating moderate to severe psoriasis, psoriatic arthritis, rheumatoid arthritis, and hidradenitis suppurativa (1–4). Systemic lupus erythematosus (SLE) induced by biologics mostly occurs during the treatment of psoriasis with tumor necrosis factor-alpha (TNF-α) inhibitors, especially in patients with psoriatic and rheumatoid arthritis. TNF-α inhibitors were calculated with an odds ratio of 3.64 for SLE development (5). The onset of symptoms ranges from <1 month to more than 4 years. High antinuclear antibody (ANA) levels, double stranded DNA (dsDNA) autoantibody positivity, thrombocytopenia, leukopenia, hypocomplementemia, skin rash, and arthritis are relatively common symptoms in TNF-α inhibitor-induced SLE (TAILS) (6). Further pathological findings characterizing classical drug-induced lupus include an increased frequency of significant anti-dsDNA antibody titers and a decreased incidence of anti-histone antibodies (7). The incidence of TAILS in patients with ulcerative colitis/Crohn's, etc., is 0.5–1% (8). However, how anti-TNF-α agents induce autoantibody expression and lead to the development of SLE remain incompletely understood. Streptococcus pyogenes (S. pyogenes) can trigger the immune responses to activate psoriasis outbreaks (9, 10). Furthermore, the innate immune system has been shown to be activated by S. pyogenes in both guttate and chronic plaque psoriasis (11). The specific IgA response against to S. pyogenes was correlated with a cutaneous lymphocyte-associated antigen+ T-cell-depend IL-17F response (12). In this study, peripheral blood mononuclear cells (PBMCs) from psoriasis patients and S. pyogenes was used to challenge the PBMCs to simulate a real clinical psoriasis outbreak. We describe the cases of three patients with psoriasis who developed TAILS and investigated the underlying mechanisms. Two of these patients developed TAILS after treatment with adalimumab and one after treatment with etanercept. All three patients met the 1997 American College of Rheumatology (ACR) criteria for SLE.
Materials and Methods
Participants
Thirteen psoriasis patients with or without psoriatic arthritis and three healthy controls were enrolled in this study. All participants were selected from the clinic of the Dermatology or Rheumatology Departments of Taichung Veterans General Hospital. All participants provided written informed consent. The protocols and all research involving human participants were approved by the Institutional Review Board of Taichung Veterans General Hospital (TCVGH-CE16265B; TCVGH-CE20043B).
Materials
Streptococcus pyogenes group A was identified and provided by the Department of Pathology and Laboratory Medicine of Taichung Veterans General Hospital. After heat inactivation, S. pyogenes group A was placed on blood agar plates for 1 week.
Cell Culture
For PBMC culture, 16 mL of blood was collected from each patient in sodium citrate tubes (Vacutainer® CPT™, BD, USA), and PBMCs were purified through centrifugation over a density gradient. The cells were washed with PBS and subsequently cultured in RPMI-1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2. S. pyogenes group A was prepared at a concentration of 2 × 107 CFU/mL under the similar cell viability to normal control and proper immune induction response on PBMC (data not shown).
A total of 6 × 105 cells per milliliter were then cultured in a 12-well plate and treated for 24 h with the following: control, S. pyogenes only, S. pyogenes + golimumab (0.5 μg per milliliter), S. pyogenes + ixekizumab (3.5 μg per milliliter), S. pyogenes + ustekinumab (0.25 μg per milliliter), S. pyogenes + adalimumab (4 μg per milliliter), S. pyogenes + secukinumab (16.7 or 34 μg per milliliter), S. pyogenes + guselkumab (1.2 μg per milliliter), and S. pyogenes + etanercept (1.9 μg per milliliter). Supernatants were collected for the subsequent measurement of cytokine levels. The concentrations of the biological agents we tested are the trough serum concentrations at a steady-state indicated in the pharmacokinetic section of the reference list.
Cell Viability Test
The separated peripheral blood mononuclear cells (PBMCs) were cultured using different concentrations of S. pyogenes for 24 h, and then, 0.5 mg per milliliter of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added. After reacting for 1 h, the mixtures were centrifuged and the supernatants were removed. Then, 200 μL of dimethyl sulfoxide was added to lyse the cells and dissolve purple crystals, and cell viability was analyzed using an enzyme-linked immunosorbent assay reader at a wavelength of 570 nm.
Multiplex Assay for Cytokine Levels
To measuring cytokine levels, culture supernatants were collected, and the concentrations of IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-17A, IFN-γ, and TNF-α were determined using a protein multiplex immunoassay system (Bio-Plex Cytokine Array System, Bio-Rad Laboratories, Hercules, CA, USA). The IFN-γ/IL-13 ratio was calculated in different patients undergoing different biologics in vitro. If the patient uses this biological agent and eventually causes SLE, it can be classified into the SLE group. Conversely, if the patient uses another biological agent lead to eventually doesn't get SLE, it can be classified as a non-SLE group.
Statistical Analyses
All statistical analyses were performed using SPSS version 22 (IBM, Armonk, NY, U.S.A.). Analysis of cytokine expression was with the use of the Mann-Whitney U test. The ratio of IFN-γ to IL-13 between TAILS-inducing biologics and non-TAILS-inducing biologics was analyzed with t-test. Data were presented as the mean ± standard deviation (SD). Two-sided P-values of 0.05 or less were considered to indicate statistical significance.
Results
Patients
Thirteen patients were enrolled in our study including, three of which developed TAILS that met the 1997 ACR criteria for SLE. Patients 1 and 2 received adalimumab for 6 and 47 months, respectively, whereas Patient 3 received etanercept for 24 months (Table 1). In addition, we assessed the patients' skin condition by using the Psoriasis Area and Severity Index (PASI). Patient 1's psoriasis worsened after adalimumab treatment from absolute PASI:15 to PASI:16; Patient 2's psoriasis was largely stable, going from PASI:3.8 to PASI:2; and Patient 3's psoriasis improved, going from PASI:26 to PASI:13 (Table 2). Of the three cases, Patient 1 experienced an outbreak of psoriasis and progressed to SLE most quickly. TNF-α inhibitor administration was subsequently discontinued for all three patients.
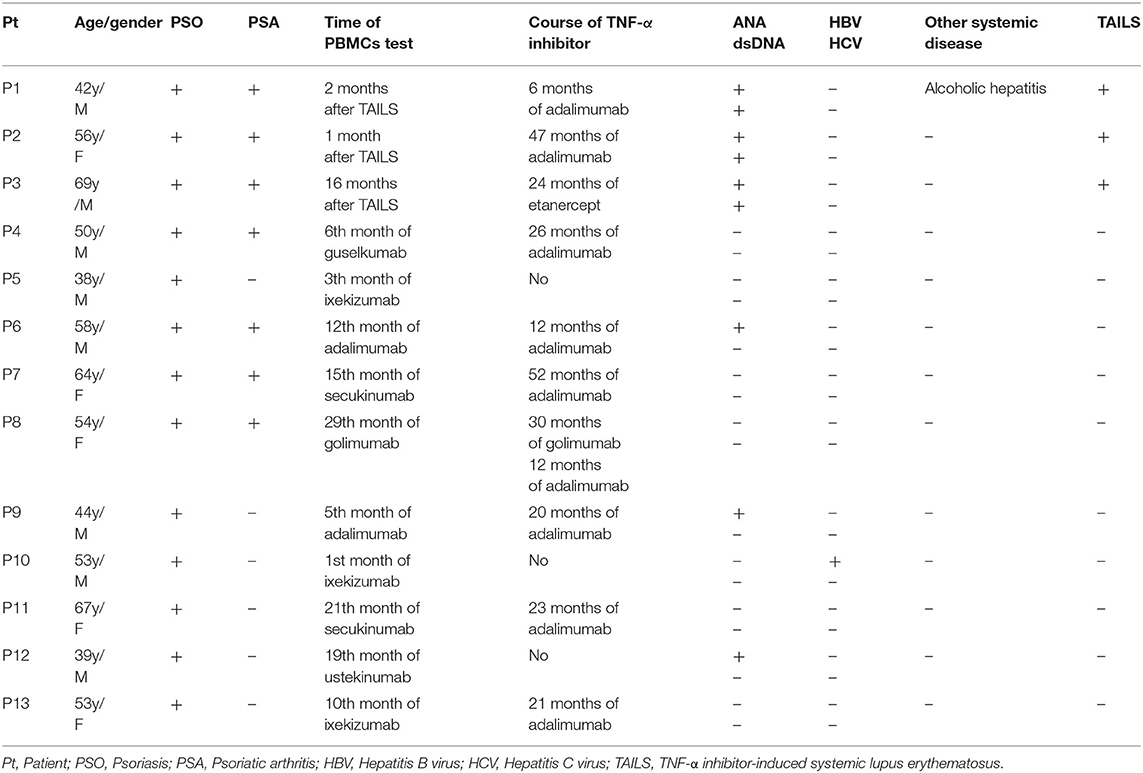
Table 1. Demographic and clinical characteristics of psoriasis patients (n = 13).
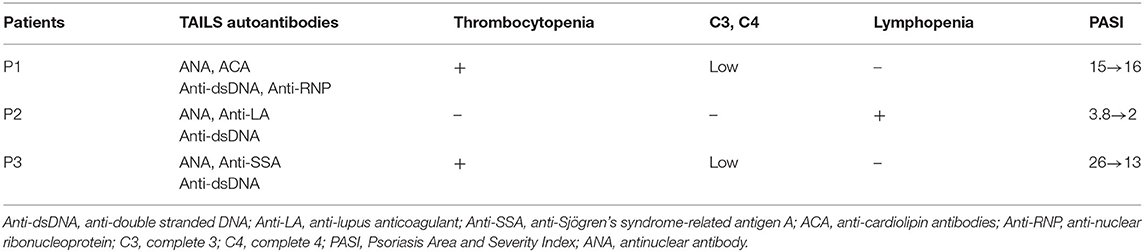
Table 2. Clinical characteristics of patients with TNF-α inhibitor-induced systemic lupus erythematosus.
Response to Treatment
Patient 1 refused oral prednisolone and received ustekinumab for psoriasis and subsequently recovered in skin condition, going from PASI:16 to PASI:0.2. Patient 2 received secukinumab (150 mg per week), following which skin absolute PASI declined to 0. Patient 3 received Methotrexate (7.5 mg per week) for controlling psoriasis and thrombocytopenia, and skin absolute PASI was maintained to 18.2. ANA remained positive in all three cases. In addition, dsDNA autoantibody titer levels remained high in Patient 1 after 6 months of ustekinumab treatment. The level of the dsDNA autoantibody titer declined to normal in Patient 2 after only 2 months and after 1.5 years in Patient 3.
Cytokine Expression Analysis
The analysis of cytokine levels on psoriasis patients and healthy controls revealed no obvious difference (Supplementary Table S1). The analysis of cytokine expression treated with experienced or naïve biological agents revealed that IL-2 and IL-10 levels decreased in only the TNF-α inhibitor-treated groups but not in the groups treated with biologics involving inhibitor mechanisms for IL-17, IL-12/IL-23 or IL-23 (p < 0.001, p < 0.05, respectively; Figure 1). IL-2 and IL-10 levels also decreased in only the TNF-α inhibitor-treated groups but not in the groups treated with other biologics in healthy controls (Supplementary Table S1). The data between the concentration of IL-2 and TNF-α in vitro is a proportional correlation (Figure 2). We hypothesize that IL-2 secreting requires not only pathogens but also TNF-α, explaining IL-2 depletion in the presence of TNF-α inhibitors. The hypothesis needs more evidence to prove. The anti-IL-17 inhibitors, including ixekizumab and secukinumab, apparently did not attenuate IL-17, which could result from the action of different epitopes used for verifying the concentration in the multiplex assay and their blocking by biologics. In our thirteen patients, most of them experienced two or more biologics. Twenty-eight clinical treatment courses of biologics in total were as follows: adalimumab, 9; etanercept, 1; golimumab, 1; ustekinumab, 8; ixekizumab, 3; secukinumab, 5; and guselkumab, 1. The ratio of IFN-γ to IL-13 was significantly higher (p = 0.001) between after induction with S. pyogenes plus SLE-inducing biologics and after induction with S. pyogenes only than after induction with S. pyogenes plus non-SLE-inducing biologics and after induction with S. pyogenes only (Table 3; Figure 3). Because three patients with SLE were PSO + PSA, only four patients with PSO+PSA without SLE (P4, P6, P7, P8) were selected and the ratio of IFN-γ/IL13 was also significantly increased in the SLE group (p = 0.02; Supplementary Figure S1). This ratio could be used in the future for identifying the risk of TAILS. Taken together, low levels of TNF-α was correlated to low levels of IL-2. IL-2 and IL-10 depletion could create an environment for developing SLE. The ratio of IFN-γ/IL13 in SLE-inducing biologics was significantly higher than in non-SLE-inducing biologics.
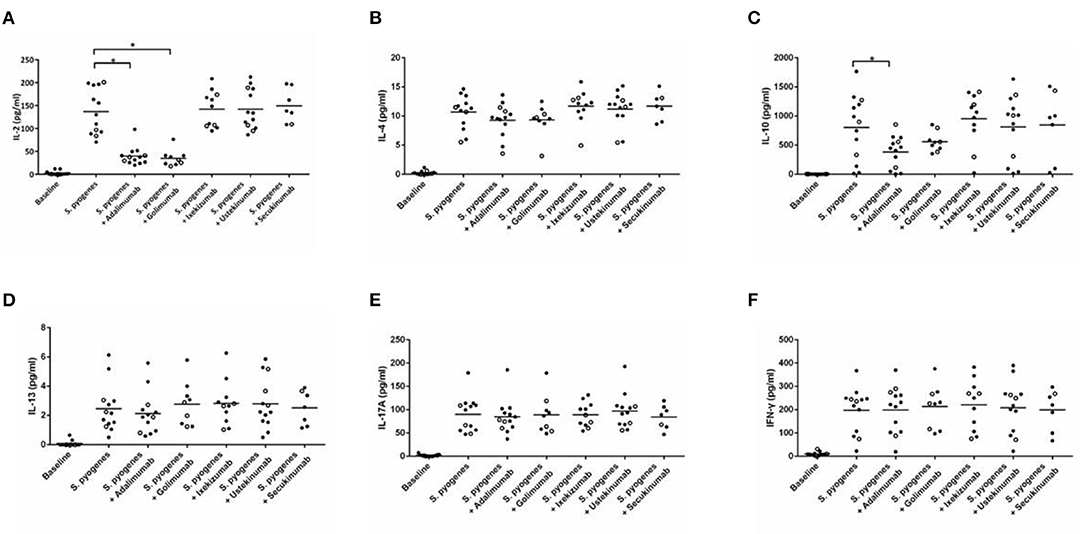
Figure 1. Cytokine expression in peripheral blood mononuclear cells after induction with S. pyogenes and treated with different biologics. (A–F) depict the levels of cytokines (shown on the y-axis) in response to the application of listed biologics (shown on the x-axis) after induction by S. pyogenes. Each circle represents cell cultures from one patient. In all panels, the three TNF-α inhibitor-induced SLE patients are marked with hollow circles. Black bars in each column indicate the mean cytokine concentration. TNF-α inhibitors significantly inhibited S. pyogenes-induced IL-2 (A) (p < 0.001 in adalimumab, p < 0.001 in golimumab, respectively) and IL-10 (C) expression (p = 0.039 in adalimumab).
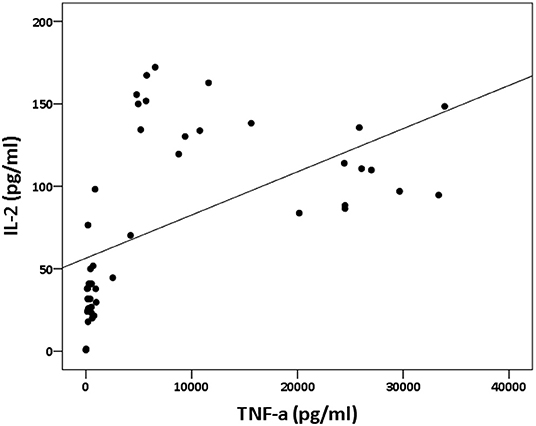
Figure 2. Relation between IL-2 and TNF-α concentration in vitro. The panel depicts the levels of IL-2 (shown on the y-axis) and the levels of TNF-α (shown on the x-axis). The data between the concentration of IL-2 and TNF-α in vitro is a proportional correlation (p < 0.000; rs = 0.74).
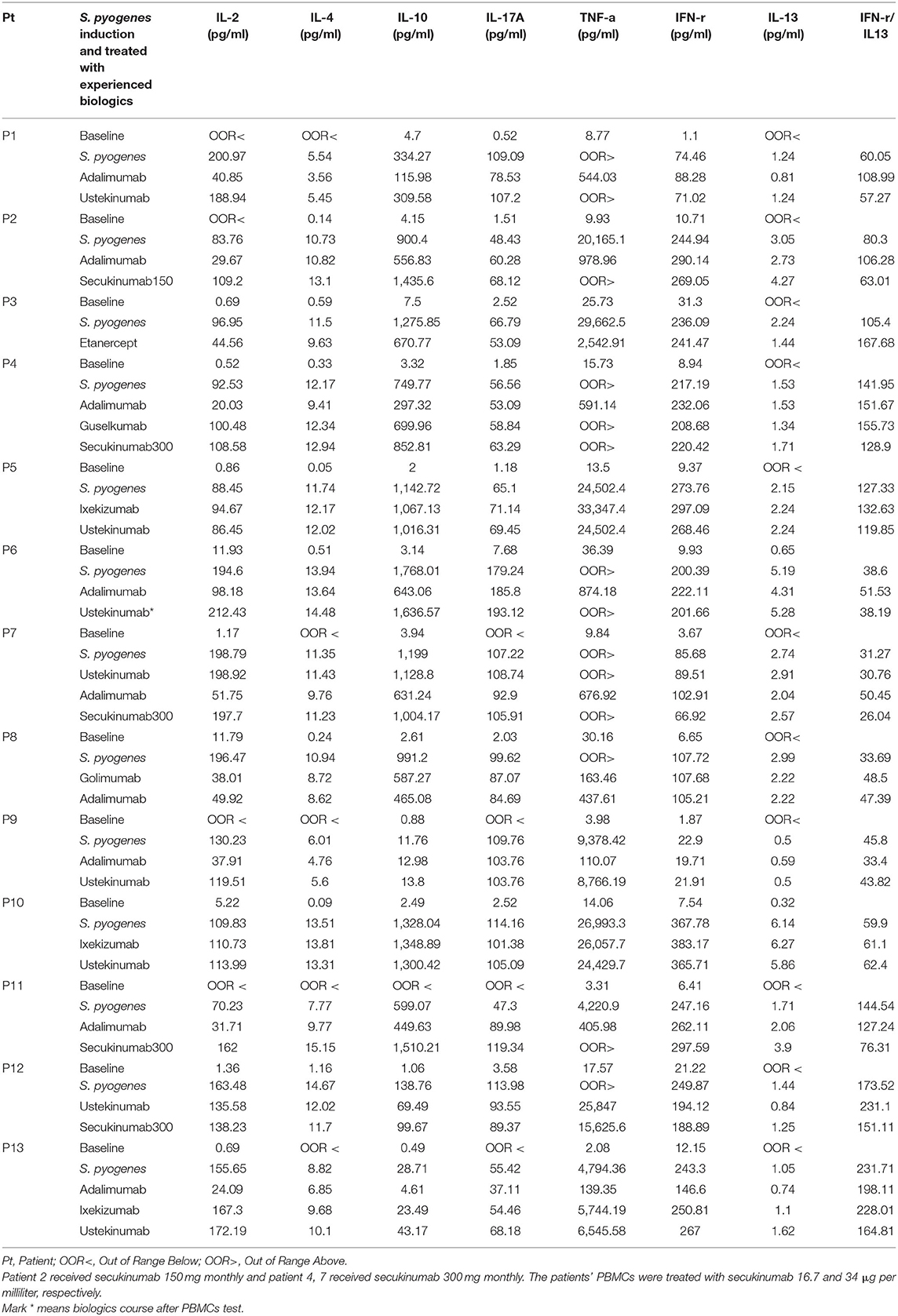
Table 3. Laboratory profile of psoriasis patients with administered biologics (n = 13).
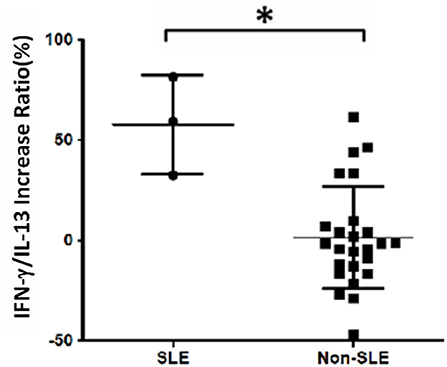
Figure 3. Increased IFN-γ/IL-13 ratio in patients with systemic lupus erythematosus (SLE). Cell samples were obtained from 13 patients, and 28 treatment courses of biologics were as follows: adalimumab, 9; etanercept, 1; golimumab, 1; ustekinumab, 8; ixekizumab, 3; secukinumab, 5; and guselkumab, 1. The increase in the ratio of IFN-γ to IL-13 levels was calculated as the increase in percentage following treatment with biologics compared with S. pyogenes induction only: (IFNγ/IL13 w/S. pyogenes & biologics) − (IFNγ/IL13 w/S. pyogenes only)/(IFNγ/IL13 w/S. pyogenes only). We found that the ratio was significantly increased in the SLE group (p = 0.001).
Discussion
Decreased expression of IL-2 and IL-10 is associated with SLE development. A lack of IL-2 leads to the inhibition of activation-induced cell death and enhances the longevity of autoreactive T cells (13). Vaccinia recombinant viruses expressing the human IL-2 gene showed prolonged survival, decreased autoantibody in SLE mice (14). Limited IL-10 expression can increase the secretion of proinflammatory cytokines in pathogenic T cells and upregulate the presentation of antigens by dendritic cells to naïve T cells, thus promoting differentiation into pathogenic cells (15). Yin et al. (16) discussed the protective role of IL-10 in the development of lupus. They found that IL-10 depletion was closely related to severe and early-onset lupus and was also associated with IFN-γ production and an increased serum concentration of anti-dsDNA autoantibodies, similar to our observations in patients with TAILS.
As a chronic autoimmune disease, SLE is characterized by the presence of nuclear antigen autoantibodies. Recent studies have implicated innate immunity as a key switch for coordinating B cells, T cells, and macrophages in the pathogenesis of SLE (17). Innate immunity and the Th1 reaction are major initiation responses against infections including streptococcus, staphylococcus, and various viruses. In a second aspect, cytokines involved in Th2 immunity, including IL-4 and IL-13, counteract the Th1 response. Macrophage polarization is a key step that drives the immune response toward either the M1 or M2 pathway. In the early stages of lupus, the M1 pathway is mediated by cytokines including IFN-γ, whereas the M2 pathway is mediated by IL-4 and IL-13. IFN-γ induction favors the production of M1 macrophages that are involved in inflammation and tissue damage. An increased concentration of IFN-γ can alter the ratio of Th1 to Th2 with predominance by Th1 cells (18). In lupus immunopathology, IFN-γ is elevated in the serum of lupus patients, particularly those in the active stage (19). Th1/Th2 balance shift and elimination of IFN-gamma by IFN-γ targeting gene therapy was developed in treating SLE (20). However, the role of Th2 immune responses in autoimmune development remains controversial, although it is known that IL-4 plays a crucial role in the Treg-mediated suppressive immune response. Plasma IL-4 concentrations were found to be significantly lower in SLE patients than in healthy controls (21). Furthermore, IL-4 can downregulate Th1-mediated IgG subclasses of autoantibodies to prevent the development of lupus-like autoimmune disease (22). Although IL-13 is a strong anti-inflammatory cytokine that modulates macrophages, monocytes, and lymphocytes (23), a previous study found that plasma IL-13 levels were significantly higher in SLE patients than in controls (24). We hypothesize that different stages and timing are key factors governing the IL-13 concentration. Moreover, a disproportionate increase in IFN-γ or reduced IL-13 could be the mechanism in SLE. These differences could be due to possible feedback mechanism in vivo (24), which needs to be investigated by further or future studies. Alternatively, this could be due to different evolution—primary SLE and drug-induced SLE (TAILS).
In the present study, the IL-13 concentration increased within 24 h following the onset of inflammation caused by streptococcus infection. However, we are uncertain whether IL-13 promotes inflammation, rather than acting as a feedback cytokine that reduces inflammation. In our study, different biologics appeared to react differently to IL-13, but the detailed mechanisms remain unknown. In the three TAILS patients, we observed the lowest IL-13 concentrations for pathogenic drugs compared with other biologics including anti-IL-17, anti-IL-23, and other anti-TNF-α. IL-17 is apparently an important cytokine in the pathogenesis of SLE because it can amplify the immune response by increasing autoantibodies through B-cell stimulation (25). However, our data indicated no obvious differences in the IL-17 concentration between SLE and non-SLE groups. Anti-IL-17 inhibitors, including ixekizumab and secukinumab, apparently did not attenuate IL-17, which could result from the action of different epitopes used for verifying the concentration in the multiplex assay vs. blocking by biologics.
Adaptive transfer to M2 macrophages reduces the severity of SLE, whereas IL-4 and IL-13 drive the production of M2 macrophages (26). A similar condition was observed in innate lymphoid cells (ILCs). Group 2 ILCs produce IL-4, IL-9, and IL-13. Hou et al. found significantly reduced numbers of Group 2 ILCs in SLE patients (27). Restoring Group 2 ILCs with IL-33 reduced immune cell infiltration and improved survival in a mouse model (28). During outbreaks of psoriasis and SLE, the ratio of M1 to M2 macrophages is increased (26, 29). Other recent studies have highlighted M2 promotion and immunomodulation as another avenue for SLE treatment (30, 31). Hence, maintaining balance of M1/M2 and Th1/Th2 ratios is an ideal way to prevent SLE. Otherwise, IL-13 induction will favor the production of M2 macrophages that are involved in tissue repair.
In the present study, the data revealed that the three TAILS cases had high ratios of IFN-γ to IL-13 compared with those of biologics targeting other mechanisms in the same patients. Taken together with previous findings, our results suggest that the depletion of IL-2 and IL-10, along with the deterioration of the Th1/Th2 ratio, are potentially a major underlying mechanism for TAILS. To verify this, we analyzed the symptom severity of the three TAILS patients. Of the three cases, Patient 1's symptoms were the most severe, whereas Patient 2's was the least severe. Patients 1 and 3 were revealed to have low levels of IL-4, whereas Patient 2's IL-4 level was normal when compared with other biologics. Less deviation in the ratio of IFN-γ to IL-13 and a normal level of IL-4 could explain why Patient 2's symptoms were the least severe of the three patients. Hence, the degrees of deviation in the IFN-γ/IL-13 ratio and IL-4 impairment could be regarded as indicators of disease severity.
In the present study, we tested two types of anti-TNF-α (golimumab and adalimumab) in all three patients and an additional anti-TNF-α (etanercept) in Patient 3. The IFN-γ/IL-13 ratio in adalimumab and golimumab was 108.99 and 22.59, respectively, in Patient 1. The IFN-γ/IL-13 ratio in adalimumab and golimumab was 106.28 and 92.69, respectively, in Patient 2. The IFN-γ/IL-13 ratio in adalimumab, golimumab, and etanercept were 145.37, 114.5, and 167.68, respectively, in patient 3. These findings support the previous hypothesis that drug-induced SLE can be drug-specific rather than class-specific (6). Therefore, it is crucial to identify which patients will develop drug-induced SLE and what types of drugs will induce it. We believe the findings presented herein will provide a clear understanding for TAILS. We also believe the incidence of TAILS is underestimated.
Limitation
This study has some limitations. First, the results of the study need to be validated in a larger group to assess the variability and validity of our findings. Second, in vitro experiments on different cells line about cytokine expression, Th1/Th2 related cells subpopulation, and how the mechanisms of different biologics are affected by the IFN-γ or IL-13 concentration requires further investigation.
Conclusions
In conclusion, our results suggest that the underlying expression of TNF-α inhibitor-induced SLE correlates with depletion of IL-2 and IL-10 and Th1/Th2 related cytokine imbalance. Our findings provide a clearer understanding of SLE and suggest more appropriate treatments such as low-dose courses of IL-2 and IL-10. Although the sample sized analyzed was small, the observations in this study highlight possible mechanisms of etiology. However, more sample size researches are needed in the future.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Taichung Veterans General Hospital (TCVGH-CE16265B; TCVGH-CE20043B). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
C-YY performed the experiments, protocol design, and wrote the manuscript. S-JY and E-CL performed the experiments. Y-MC, K-LL, and Y-DW participated in protocol design and discussion. C-LH participated in protocol design and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded in part by the Chinese Medicine Research Center, China Medical University from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (CMRC-CENTER-0). And also from Ministry of Science and Technology in Taiwan (MOST 109-2320-B-39-044). The funders had no role in study design, data collection and analysis, interpretation of findings, manuscript writing, and target journal selection.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the participants who volunteered for this study, the Biostatistics Task Force of Taichung Veterans General Hospital for analyses and the Department of Pathology and Laboratory Medicine of Taichung Veterans General Hospital for S. pyogenes group A identification.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.870724/full#supplementary-material
Abbreviations
ACR, American College of Rheumatology; ANA, antinuclear antibody; dsDNA, double stranded DNA; IFN-γ, interferon-gamma; IL, interleukin; PASI, Psoriasis Area Severity Index; PBMC, peripheral blood mononuclear cell; SLE, Systemic lupus erythematosus; TAILS, TNF-α inhibitor-induced SLE; Th1, T helper type 1; Th2, T helper type 2; S. pyogenes, Streptococcus pyogenes.
References
1. Mourad AI, Gniadecki R. Biologic drug survival in psoriasis: a systematic review & comparative meta-analysis. Front Med. (2020) 7:625755. doi: 10.1016/j.jaad.2020.06.551
2. Chen HH, Lin CH, Wang CY, Chao WC. Association of hospitalised infection with socioeconomic status in patients with rheumatoid arthritis receiving biologics or tofacitinib: a population-based cohort study. Front Med. (2021) 8:696167. doi: 10.3389/fmed.2021.696167
3. Chia AYT, Ang GWX, Chan ASY, Chan W, Chong TKY, Leung YY. Managing psoriatic arthritis with inflammatory bowel disease and/or uveitis. Front Med. (2021) 8:737256. doi: 10.3389/fmed.2021.737256
4. Frew JW, Singh N, Jiang CS, Vaughan R, Krueger JG. The impact of body mass index upon the efficacy of adalimumab in hidradenitis suppurativa. Front Med. (2021) 8:603281. doi: 10.3389/fmed.2021.603281
5. Haugaard JH, Kofoed K, Gislason G, Dreyer L, Egeberg A. Association between drug use and subsequent diagnosis of lupus erythematosus. JAMA Dermatol. (2020) 156:1199–207. doi: 10.1001/jamadermatol.2020.2786
6. Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. (2011) 50:619–25. doi: 10.1111/j.1365-4632.2011.04871.x
7. Katz U, Zandman-Goddard G. Drug-induced lupus: an update. Autoimmun Rev. (2010) 10:46–50. doi: 10.1016/j.autrev.2010.07.005
8. Lupu A, Tieranu C, Constantinescu CL, Diculescu M. TNFalpha inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Curr Health Sci J. (2014) 40:285–8. doi: 10.12865/CHSJ.40.04.11
9. Muto M, Fujikura Y, Hamamoto Y, Ichimiya M, Ohmura A, Sasazuki T, et al. Immune response to Streptococcus pyogenes and the susceptibility to psoriasis. Australas J Dermatol. (1996) 37 (Suppl. 1):S54–55. doi: 10.1111/j.1440-0960.1996.tb01087.x
10. Groot J, Blegvad C, Nybo Andersen AM, Zachariae C, Jarlov JO, Skov L. Presence of streptococci and frequent tonsillitis among adolescents with psoriasis. Br J Dermatol. (2021) 184:758–9. doi: 10.1111/bjd.19672
11. Fry L, Baker BS, Powles AV, Fahlen A, Engstrand L. Is chronic plaque psoriasis triggered by microbiota in the skin? Br J Dermatol. (2013) 169:47–52. doi: 10.1111/bjd.12322
12. De Jesus-Gil C, Sans-De San Nicolas L, Ruiz-Romeu E, Ferran M, Soria-Martinez L, Chiriac A, et al. Specific IgA and CLA(+) T-Cell IL-17 response to Streptococcus pyogenes in psoriasis. J Invest Dermatol. (2020) 140:1364–70.e1361. doi: 10.1016/j.jid.2019.12.022
13. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
14. Gutierrez-Ramos JC, Andreu JL, Revilla Y, Vinuela E, Martinez C. Recovery from autoimmunity of MRL/lpr mice after infection with an interleukin-2/vaccinia recombinant virus. Nature. (1990) 346:271–4. doi: 10.1038/346271a0
15. Uchida M, Ooka S, Goto Y, Suzuki K, Fujimoto H, Ishimori K, et al. Anti-IL-10 antibody in systemic lupus erythematosus. Open Access Rheumatol. (2019) 11:61–5. doi: 10.2147/OARRR.S191953
16. Yin Z, Bahtiyar G, Zhang N, Liu L, Zhu P, Robert ME, et al. IL-10 regulates murine lupus. J Immunol. (2002) 169:2148–55. doi: 10.4049/jimmunol.169.4.2148
17. Herrada AA, Escobedo N, Iruretagoyena M, Valenzuela RA, Burgos PI, Cuitino L, et al. Innate immune cells' contribution to systemic lupus erythematosus. Front Immunol. (2019) 10:772. doi: 10.3389/fimmu.2019.00772
18. Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. (1999) 42:1644–8. doi: 10.1002/1529-0131(199908)42:8<1644::AID-ANR12>3.0.CO;2-L
19. Al-Janadi M, Al-Balla S, Al-Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol. (1993) 13:58–67. doi: 10.1007/BF00920636
20. Hayashi T. Therapeutic strategies for SLE involving cytokines: mechanism-oriented therapies especially IFN-gamma targeting gene therapy. J Biomed Biotechnol. (2010) 2010:461641. doi: 10.1155/2010/461641
21. Yu HH, Liu PH, Lin YC, Chen WJ, Lee JH, Wang LC, et al. Interleukin 4 and STAT6 gene polymorphisms are associated with systemic lupus erythematosus in Chinese patients. Lupus. (2010) 19:1219–28. doi: 10.1177/0961203310371152
22. Santiago ML, Fossati L, Jacquet C, Muller W, Izui S, Reininger L. Interleukin-4 protects against a genetically linked lupus-like autoimmune syndrome. J Exp Med. (1997) 185:65–70. doi: 10.1084/jem.185.1.65
23. Hamzei Taj S, Le Blon D, Hoornaert C, Daans J, Quarta A, Praet J, et al. Targeted intracerebral delivery of the anti-inflammatory cytokine IL13 promotes alternative activation of both microglia and macrophages after stroke. J Neuroinflammation. (2018) 15:174. doi: 10.1186/s12974-018-1212-7
24. Wang R, Lu YL, Huang HT, Qin HM, Lan Y, Wang JL, et al. Association of interleukin 13 gene polymorphisms and plasma IL 13 level with risk of systemic lupus erythematosus. Cytokine. (2018) 104:92–7. doi: 10.1016/j.cyto.2017.09.034
25. Li D, Guo B, Wu H, Tan L, Chang C, Lu Q. Interleukin-17 in systemic lupus erythematosus: a comprehensive review. Autoimmunity. (2015) 48:353–61. doi: 10.3109/08916934.2015.1037441
26. Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage polarization modulates development of systemic lupus erythematosus. Cell Physiol Biochem. (2015) 37:1279–88. doi: 10.1159/000430251
27. Hou M, Liu S. Innate lymphoid cells are increased in systemic lupus erythematosus. Clin Exp Rheumatol. (2019) 37:676–9.
28. Duster M, Becker M, Gnirck AC, Wunderlich M, Panzer U, Turner JE. T cell-derived IFN-gamma downregulates protective group 2 innate lymphoid cells in murine lupus erythematosus. Eur J Immunol. (2018) 48:1364–75. doi: 10.1002/eji.201747303
29. Lu CH, Lai CY, Yeh DW, Liu YL, Su YW, Hsu LC, et al. Involvement of M1 macrophage polarization in endosomal toll-like receptors activated psoriatic inflammation. Mediators Inflamm. (2018) 2018:3523642. doi: 10.1155/2018/3523642
30. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. (2017) 26:1318–27. doi: 10.1177/0961203317701842
Keywords: systemic lupus erythematosus, tumor necrosis factor-alpha inhibitors, IL-2, IL-10, IFN-γ/IL-13 ratio
Citation: Yen C-Y, Yu S-J, Chen Y-M, Lai K-L, Wu Y-D, Liao E-C and Hsieh C-L (2022) Mechanisms of Tumor Necrosis Factor-Alpha Inhibitor-Induced Systemic Lupus Erythematosus. Front. Med. 9:870724. doi: 10.3389/fmed.2022.870724
Received: 07 February 2022; Accepted: 16 May 2022;
Published: 06 June 2022.
Edited by:
Devinder Mohan Thappa, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), IndiaReviewed by:
Irina Khamaganova, Pirogov Russian National Research Medical University, RussiaMark Christopher Arokiaraj, Pondicherry Institute of Medical Sciences, India
Copyright © 2022 Yen, Yu, Chen, Lai, Wu, Liao and Hsieh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ching-Liang Hsieh, clhsieh@mail.cmuh.org.tw
†These authors have contributed equally to this work