The molecular mechanism of ferroptosis and its role in COPD
 Dandan Meng
Dandan Meng Chengfeng Zhu1
Chengfeng Zhu1 - 1Department of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Department of Second Department of Haematology, Jinan Haematology Hospital, Jinan, China
- 3Department of Basic Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
Ferroptosis, a new type of cell death, is mainly characterized by intracellular iron accumulation and lipid peroxidation. The complex regulatory network of iron metabolism, lipid metabolism, amino acid metabolism, p53-related signaling, and Nrf2-related signaling factors is involved in the entire process of ferroptosis. It has been reported that ferroptosis is involved in the pathogenesis of neurological diseases, cancer, and ischemia–reperfusion injury. Recent studies found that ferroptosis is closely related to the pathogenesis of COPD, which, to some extent, indicates that ferroptosis is a potential therapeutic target for COPD. This article mainly discusses the related mechanisms of ferroptosis, including metabolic regulation and signaling pathway regulation, with special attention to its role in the pathogenesis of COPD, aiming to provide safe and effective therapeutic targets for chronic airway inflammatory diseases.
In 2012, Dixon (1) first proposed a new type of iron-dependent programmed cell death named ferroptosis. As a new type of cell death, ferroptosis is mainly characterized by intracellular iron accumulation and lipid peroxidation. Ferroptosis differs from other forms of cell death such as apoptosis and necrosis in terms of morphology, biochemical characteristics, and genetics (2, 3). The morphological aspects of ferroptosis are mainly characterized by mitochondrial contraction and increased density of mitochondrial membranes with a decrease or disappearance of mitochondrial cristae and disintegration of the outer membrane (1). The biochemical features of ferroptosis are as follows: accumulation of ROS and iron ions, decreased cysteine uptake and GSH synthesis, activation of the mitogen-activated protein kinase system, and release of arachidonic acid (4). Iron metabolism, lipid metabolism, amino acid metabolism, p53-related signaling factors, and Nrf2-related signaling factors are involved in the entire process of regulating ferroptosis (5). Ferroptosis, a new programmed cell death mode, has been confirmed to be closely related to tumors, central nervous system diseases, arteriosclerosis, acute kidney injury, diabetes, and ischemia–reperfusion injury (6). Recent studies found that ferroptosis is also associated with the onset of chronic obstructive pulmonary disease (COPD) and has the potential to become a new therapeutic target. COPD is a common condition that can be prevented and treated, and it is characterized by persistent respiratory symptoms and airflow restriction; in addition, COPD is caused by airway and alveolar abnormalities due to heavy exposure to harmful particles or gases, and it is affected by host factors such as abnormal pulmonary development (7). COPD has developed into a major chronic respiratory disease that seriously threatens human health. Related studies showed that the disruption of iron homeostasis is related to the pathogenesis of COPD (8). With further study, the role of ferroptosis in the pathogenesis of COPD has gradually been revealed, and appropriate intervention in ferroptosis can delay the COPD process.
1. Mechanism of ferroptosis
The mechanism of ferroptosis is shown in Figure 1.
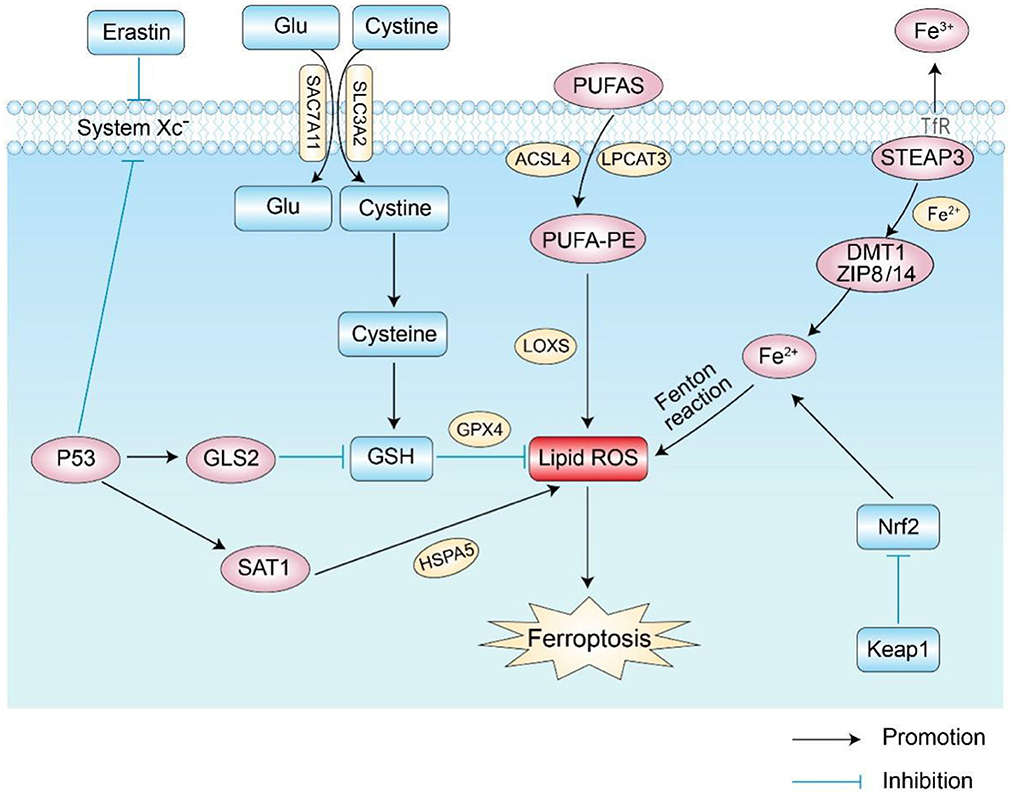
Figure 1. Main regulatory pathways of ferroptosis. There are two main ways of regulating ferroptosis shown in the figure: the first is the pathway of abnormal iron, amino acid, and lipid metabolism; the second involves the related signaling pathways that regulate ferroptosis, such as the P53 and Nrf2 pathways. Glu, glutamic acid; GSH, glutathione; GPX4, glutathione peroxidase 4; Nrf2, Nuclear factor erythroid 2-related factor 2; GLS2, glutaminase 2; LOXs, lipoxygenases; ROS, reactive oxygen species; DMT1, divalent metal ion transporter-1; SAT1, spermidine N1-acetyltransferase 1; TfR, transferrin receptor.
1.1. Abnormal iron metabolism
Iron is an element necessary for lipid peroxide accumulation and ferroptosis. Excessive iron load promotes the production of ROS through the Fenton reaction and iron-binding proteins, thereby promoting the occurrence of ferroptosis (4, 9, 10). Iron intake, transport, and storage affect ferroptosis (5). Under physiological conditions, intracellular iron absorption and metabolism should always be in a dynamic and stable state. Iron uptake and export proteins play important roles in the process of iron metabolism. On the one hand, transferrin receptor 1 (TFR1) and divalent metal transporter-1 (DMT1) take up extracellular iron in cells. On the other hand, ferroportin (FPN) transfers excess intracellular iron to the outside of the cell, a process that maintains the “on” and “off” state of intracellular iron homeostasis (4, 11). Iron in the daily diet is usually absorbed by intestinal epithelial cells in the form of Fe3+ and then enters cells through the transferrin receptor (TFR) on the cell membrane after binding to transferrin. Afterward, Fe3+ in cells is reduced to Fe2+ by six-transmembrane epithelial antigens of prostate 3 (STEAP3). Then, Fe2+ is released into the cytoplasmic iron pool by divalent metal transporter 1 (DMT1) or zinc–iron regulatory protein family 8/14 (ZIP8/14) to meet its own metabolic needs. Abnormal expression or dysfunction of related proteins increases the concentration of intracellular iron ions and leads to excess iron. Excess intracellular iron can generate lipid reactive oxygen species (ROS) through the Fenton and Haber–Weiss reactions, which further accumulate and initiate lipid peroxidation (LPO) to induce ferroptosis (12).
1.2. Abnormal lipid metabolism
Lipid metabolism is critical for regulating cellular susceptibility to ferroptosis. Because plasma membrane damage caused by iron-dependent excess accumulation is an important feature during ferroptosis (13), reducing oxidative damage from lipid peroxidation is a fundamental process to inhibit ferroptosis (14–16). ROS interacts with polyunsaturated fatty acids (PUFAs) on lipid membranes to form lipid ROS, and excessive ROS accumulation in cells induces ferroptosis (17). When PUFAs exist in large amounts, they lead to more lipid peroxides and aggravate the degree of cell ferroptosis (18). Studies found that lipoxygenases (LOXs), non-heme iron-containing proteins, are involved in the formation of iron-dependent lipid ROS. When cells contain a large amount of iron ions, they catalyze PUFAs to form lipid hydroperoxides and toxic lipid-free radicals, which cause cell damage and promote ferroptosis (19). It was later found that antioxidants significantly inhibit LOXs and further inhibit the formation of lipid hydroperoxides to prevent cell ferroptosis (19). Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are involved in helping PUFAs in cell membrane synthesis and esterification to generate PUFA-PEs, which accelerate the process of ferroptosis (20, 21). Therefore, PUFA-related biosynthetic enzymes may be potential targets for regulating ferroptosis.
1.3. Abnormal amino acid metabolism
Ferroptosis caused by abnormal amino acid metabolism is mainly related to the abnormal metabolism of GSH. GSH is a key substance in amino acid metabolism in ferroptosis and is mainly synthesized from cysteine, glutamate, and glycine (5). Extracellular cysteine and intracellular cysteine are essential for GSH biosynthesis. Cystine generates GSH through a series of enzymatic actions, and GSH is the basic substrate for the degradation of phospholipid hydrogen peroxide (PLOOH) by glutathione peroxidase 4 (GPX4). Decreased GPX4 activity leads to the accumulation of intracellular lipid peroxides, thereby inducing ferroptosis (22, 23).
Cystine/glutamate anti-transport system Xc- (System Xc-) on the cell membrane transports extracellular cystine and intracellular glutamate in a 1:1 ratio. Cells mainly acquire cystine from the extracellular space through System Xc-, where cystine is reduced to cysteine to participate in the synthesis of GSH, and erastin acts on System Xc- to inhibit the uptake of cystine by the cell membrane, thereby reducing the synthesis of GSH and further contributing to the accumulation of ROS (24). Because glutamate is a regulator of ferroptosis and is exchanged with cystine in a 1:1 ratio by System Xc-, the concentration level of glutamate affects the function of System Xc-. A previous study reported that a high concentration of extracellular glutamate further prevents the uptake of cystine by inhibiting the biological activity of System Xc-, thereby inducing ferroptosis (11, 25). Therefore, abnormal amino acid metabolism is another important mechanism in cell ferroptosis.
1.4. Pathways related to ferroptosis
The process of ferroptosis is affected by different signaling pathways. As a tumor suppressor gene for cell cycle inhibition, apoptosis, and senescence, p53 is also one of the main signaling pathways of ferroptosis in cells (26, 27). Related studies showed that p53 is involved in the ferroptosis process and is a key regulator of both the canonical and non-standard ferroptosis pathways (28–30). SLC7A11 is an important part of System Xc-, and p53 acts as a transcriptional repressor of SLC7A11, participates in the process of ferroptosis, and inhibits the acquisition of cysteine by downregulating the expression of SLC7A11 and reducing GPX activity and GSH synthesis ability; this allows ROS accumulation and induces ferroptosis (28). p53 also sensitizes ferroptosis by enhancing the expression of glutaminase 2 (GLS2) and spermidine/spermine N1-acetyl-transferase 1 (SAT1) (31, 32). In addition, p53 inhibits ferroptosis by directly inhibiting the activity of dipeptidyl peptidase 4 (DPP4) or by promoting the expression of cyclin-dependent kinase inhibitor 1A (CDKN1A/p21) (33, 34).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator of cellular antioxidant activity, and its targets play important roles in iron and lipid metabolism (35, 36). Studies found that Nrf2 further inhibits ferroptosis by increasing the expression of target genes related to iron and ROS metabolism (37). Under physiological conditions, Nrf2 expression is low, and its activity is tightly regulated by Keap1 (38). When oxidative stress occurs, Nrf2 dissociates from the Keap1 cytoplasmic inhibitor and activates the Nrf2 transcriptional gene and thus plays an antioxidant role in protecting cells from oxidative stress. Activation of Nrf2 greatly reduces iron absorption and inhibits the production of reactive oxygen species, thereby enhancing cellular antioxidant capacity (15, 39–41). Also, Nrf2 stimulates the expression of GPX4, thereby inhibiting the occurrence of ferroptosis under certain circumstances (42, 43). Thus, the regulation of the Nrf2 pathway inhibits ferroptosis.
2. Pathogenesis of COPD
2.1. Pathological mechanism of COPD
The pathogenesis of COPD is based on the response of the body to inhalation of harmful particles and gases, and it is complex and has not been fully elucidated. Smoking is the main cause of COPD, and the pathogenesis of COPD is mostly related to inflammatory mediators, inherent immunity, oxidative stress, and protease/antiprotease imbalance (7, 44). Among many theories regarding the pathogenesis of COPD, ferroptosis is likely to be a major internal manifestation (8), and environmental interaction that induces respiratory inflammation is the main factor leading to the pathogenesis (45). Chronic airway inflammation and defects in epithelial repair remain core problems in COPD (46).
Cigarette smoke (CS) and harmful particles induce the body to produce highly reactive molecules such as ROS and reactive nitrogen species (RNS), thus inducing oxidative stress. The accumulation of neutrophils and macrophages in airways and pulmonary blood vessels causes the release of a large number of inflammatory mediators, which induces an inflammatory response and leads to lung tissue damage and protease/antiprotease imbalance, ultimately accelerating the progression of COPD (47). Oxidative stress causes the release of IL-1β and tumor necrosis factor-α (TNF-α) by regulating redox transcription factors such as nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1), thereby enhancing the inflammatory response of lungs. It is worth noting that the increase in inflammatory cells and pro-inflammatory cytokines not only maintains the chronic inflammatory response in this population but also causes systemic damage (48).
2.2. Iron-dependent oxidative stress
Oxidative stress is an important pathogenic factor in COPD, and the presence of a large amount of ROS in the inflammatory response inactivates antiproteases and leads to lung tissue damage. Excessive secretion and accumulation of neutrophils lead to the production of a large amount of reactive oxygen species (ROS). The accumulation of ROS reduces the activity of histone deacetylases (HDACs) and increases the activity of histone acetyltransferases, leading to further accumulation of neutrophils, which exacerbates oxidative stress (49).
Iron homeostasis may be disrupted in inflammatory diseases, resulting in the production of excess reactive oxygen species with deleterious effects on cells and tissues. Epithelial cells and macrophages in lung tissues produce iron metabolism-related proteins, which regulate iron homeostasis and prevent the occurrence of oxidative stress (50). Disruption of pulmonary iron homeostasis is closely related to the development of COPD (51), and oxidative stress occurs due to excess iron in the lungs caused by endogenous or exogenous factors. Studies in rats showed high levels of pulmonary oxidative stress following intravenous injection of iron-containing compounds (iron dextran and iron carboxymaltose), which are mainly manifested by increased levels of nitrotyrosine and protein carbonyl modifications (52). Lung administration of Fe2O3 nanoparticles by inhalation induces ROS generation in rat lungs (53). Chio et al. found that cigarette smoke (CS) has an important effect on iron homeostasis in the lungs. Exposure of mouse and human bronchial epithelial cells to CS increases the concentrations of iron, Ft, serum ferritin, and non-heme iron in lung cells (54).
2.3. Lipid peroxidation
Lipid peroxidation is the loss of hydrogen atoms of intracellular lipids under the action of peroxidase or free radicals, resulting in oxidation, fragmentation, and shortening of carbon chains as well as lipid-free radicals, malondialdehyde (MDA), and 4-hydroxy-2-nonenal (4-HNE) peroxidation products, which eventually oxidatively degrade lipids and damage the lipid bilayer structure of cell membranes. (55) Currently, MDA, HNE, F2-isoprostanes (F2-isoP), and 8-iso-prostaglandin F2α (8-iso-PGF2α) are the main biomarkers for evaluating lipid peroxides.
Smoking increases the content of lipid peroxidation in the body, and lipid peroxidation is closely related to the pathological progression of COPD (56). A survey of community residents in Germany showed that the level of cotinine in plasma and the amount of smoking in community residents are proportional to the level of 8-iso-PGF2α in urine, and the survey also reported that the level of 8-iso-PGF2α in smokers is significantly higher than that in non-smokers. The study further showed that the smoking index constructed based on 71 smoking-related methylation sites has a positive dose–response relationship with the log value of 8-iso-PGF2α (57). In addition, air pollution is also a major risk factor for COPD, and air pollutants lead to elevated levels of lipid peroxides in the body. In a cohort study of 97 elderly people in the United States, Zhang et al. found that the levels of carbon monoxide, nitrogen oxides, and other related pollutants and ultrafine particulate matter (PM0.18) are closely related to the elevation of the MDA oxidative stress marker, and they also reported that the effect of the component with a smaller particle size is stronger. (58) A survey of adults commuting on the highway for 3 h during the morning rush hour in Atlanta reported that exhaled nitric oxide, C-reactive protein, and MDA levels of patients with asthma and without asthma are significantly higher than baseline levels, indicating that the inhalation of harmful gases causes pulmonary inflammation and oxidative stress (59).
3. Role of ferroptosis in COPD
The role of ferroptosis in COPD is shown in Figure 2.
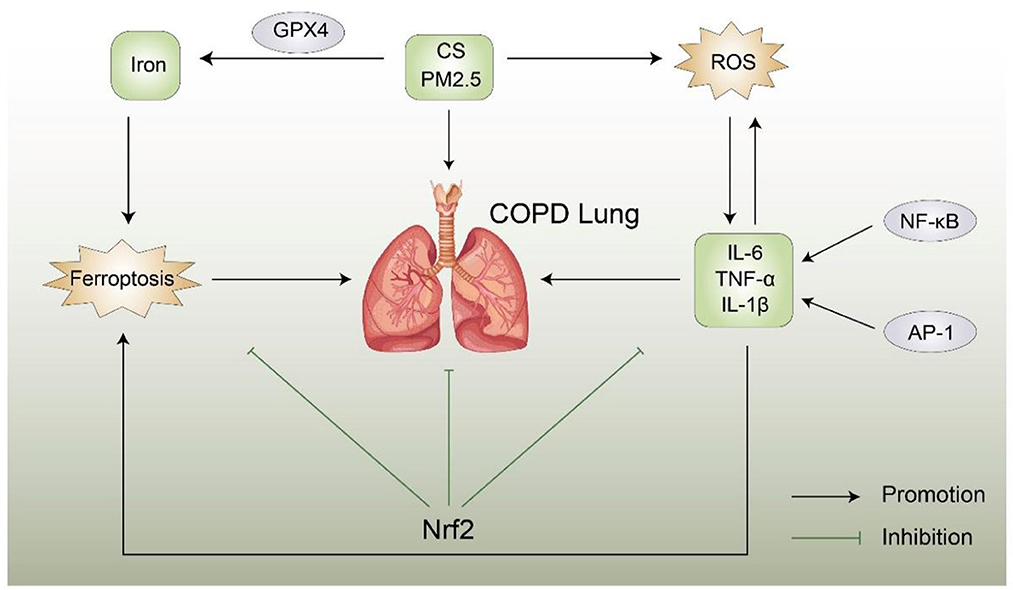
Figure 2. Possible relationship between ferroptosis and COPD, ROS, CS, and PM2.5 trigger COPD, resulting in an inflammatory reaction and abnormal levels of inflammatory factors. Conversely, abnormal inflammatory factors can aggravate COPD, Nrf2 can inhibit the production of reactive oxygen species and iron and the occurrence of COPD and ferroptosis. A direct link between ferroptosis and COPD remains unclear. Nrf2, Nuclear factor erythroid 2-related factor 2; CS, cigarette smoke; ROS, reactive oxygen species; AP-1, activator protein 1; NF-κB, nuclear factor of kappa B.
Ferroptosis is involved in the pathogenesis of COPD in a COPD mouse model. Murine lung epithelial cells exposed to CS exhibit unstable iron accumulation and increased lipid peroxidation accompanied by a non-apoptotic mode of cell death negatively regulated by GPX4. In addition, the mouse model further confirmed that the treatment of lung epithelial cells with deferoxamine and Fer-1 effectively reduces the lipid peroxidation induced by CSE, and inhibition of GPX4 also has the same effect (8). Therefore, related inhibitors such as deferoxamine and Fer-1 are potential approaches for the prevention of ferroptosis and COPD treatment. PM2.5 is one of the pathogenic factors of COPD. Studies found that, after inhalation of PM2.5 particles, the iron content and ROS concentration in human endothelial cells significantly increased, whereas the expression of GSH and NADPH decreased; in addition, the changes in the expression of TfR and Ft lead to an imbalance in cellular iron homeostasis, thereby inducing ferroptosis. The use of Fer-1 and deferoxamine improves GSH and nicotinamide adenine dinucleotide phosphate (NADPH) levels (60). Tang et al. found that cigarette smoke extract (CSE) aggravates the damage and death of BEAS-2B cells as well as increases the levels of IL-6 and TNF-α inflammatory factors, resulting in iron property changes. In vivo studies showed that CS causes lung injury in COPD rats, increases inflammatory cell infiltration, increases inflammatory cytokine secretion, and induces ferroptosis in lung tissue cells of COPD rats. In vitro and in vivo studies showed that CSE/CS increases the MDA content, increases the iron content, downregulates GPX4, decreases ferritin heavy chain levels, and upregulates transferrin receptor levels and also reported that curcumin reverses CS-induced lung inflammatory damage and epithelial cell ferroptosis (61).
Morphological aspects of ferroptosis mainly manifest as mitochondrial shrinkage, reduction or disappearance of mitochondrial cristae, and increased mitochondrial membrane density. Liu et al. found that dihydroquercetin (DHQ) inhibits CS-induced ferroptosis in the pathogenesis of COPD by activating the Nrf2-mediated pathway and attenuating CSE-induced mitochondrial morphological changes (62). In vitro and in vivo studies reported that the mRNA and protein expression levels of SLC7A11 and GPX4 are increased after DHQ treatment, and they also demonstrated that CSE-induced lipid peroxidation in HBE cells is significantly reduced after DHQ treatment. In addition, DHQ reverses CSE-induced excess MDA and ROS production, and it has also been reported that the DHQ-induced increase in SLC7A11 and GPX4 mRNA and protein levels is reversed by the use of an Nrf2-specific inhibitor (ML38.5). These findings suggest new options for the treatment of patients with COPD.
Nrf2 is a key factor in maintaining the oxidative/antioxidative balance, which can prevent the occurrence of COPD by resisting oxidative stress and lung inflammation. Recent studies reported that CpG hypermethylation in the promoter causes downregulation of Nrf2 expression in the lung tissue of patients with COPD (65). Zhang et al. found that, in HBE cells treated with CSE, the expression levels of reactive oxygen species (ROS), lipid peroxides, and MDA are increased, and they also reported that the levels of IL-1β and IL-8 are increased but that the expression levels of GPX4 and SOD are decreased, indicating that CSE induces ferroptosis in HBE cells and increases the release of inflammatory factors (63). However, increasing the expression of Nrf2 enhances the expression of GPX4 and SOD but inhibits the expression of ferroptosis and related inflammatory factors in the supernatant. Further studies found that CS-/CSE-induced hypermethylation may lead to abnormal expression of Nrf2, and targeting methylation and ferroptosis may prevent the progression of COPD, which may be a new strategy for the treatment of COPD in the future.
Macrophages are innate immune cells that play a key role in alleviating inflammation and defending against invasion by external pathogens (66, 67). Studies found that macrophage activation is closely related to the occurrence of COPD, and the total number of macrophages is proportional to the severity of smoking and COPD (68). Using both in vitro and in vivo studies, Liu et al. found increased levels of M2 macrophages, MMP9 expression, and MMP12 expression in patients with COPD, CS-exposed mice, and THP-M cells cocultured with CSE-treated human bronchial epithelial (HBE) cells, suggesting that NCOA4 and ferroptosis are involved in the pathogenesis of COPD (64). This trend is further reversed using NCOA4 siRNA and the ferrostatin-1 ferroptosis inhibitor. Therefore, blocking NCOA4 may be a promising therapeutic strategy for COPD, which provides a new direction for COPD diagnosis and treatment research. The Mechanism of ferroptosis in COPD is shown in Table 1.

Table 1. Mechanism of ferroptosis in COPD.
4. Ferroptosis as a potential therapeutic strategy in COPD
The imbalance of iron absorption and metabolism is an important factor affecting the progression of COPD. Therefore, correcting the local metabolism of iron is another key method for the treatment of COPD. Studies showed that iron chelators, antioxidants, iron supplementation, and dietary restriction are effective ways to treat COPD.
4.1. Iron chelators
Common iron chelators include deferoxamine (DFO), deferiprone, and deferasirox. DFO is one of the drugs that have been approved by the FDA for the treatment of iron overdose (69). In addition, a previous study found that DFO reduces the levels of inflammatory factors and reactive oxygen species in vitro and that it exerts an anti-inflammatory effect and reduces the inflammatory response (70). However, genetic factors are one of the important factors in the complex pathogenesis of COPD (71). Another study showed that iron regulatory protein 2 (IRP2, also known as IREB2) is increased in the lung tissue of patients with COPD, and IRP2 has been identified as a COPD susceptibility gene (72). Through COPD model mouse experiments, Cloonan et al. found that mice lacking the IRP2 gene are protected from CS-induced COPD, and they identified IRP2 as a regulator of mouse lung mitochondrial function. They further found that mice treated with a mitochondrial iron chelator (deferiprone) or fed a low-iron diet are protected from CS-induced COPD; in addition, CS-induced mucociliary clearance (MCC) impairment, lung inflammation, and lung injury in mice are attenuated (73). Therefore, these findings suggest that mitochondrial iron chelators may be a new potential approach for treating COPD.
4.2. Antioxidants
Oxidative stress is an important cause of airway inflammation, lung parenchyma destruction, and lung function decline in COPD, mainly induced by inhalation of air pollutants such as CS/CSE and dust. The acute exacerbation of COPD is closely related to oxidative stress and oxidant/antioxidant imbalance in the blood (74). Studies showed that H2S-activated Nrf2 pathway-mediated antioxidant effects play an important role in the progression of COPD (75). Wang et al. found that, after PM2.5 treatment of airway epithelial cells, the mitochondrial membrane density increases and characteristic changes in ferroptosis occur. Furthermore, COX2 expression is increased in patients with COPD. In the PM2.5-mediated mouse model and cell injury model, the levels of LIP ROS, total ROS, and MDA increased, and the levels of GSH, GSH Px, and GPX4 antioxidants decreased, indicating that PM2.5-mediated ferroptosis is involved in the pathogenesis of COPD (76). Further studies found that H2S inhibits lipid peroxidation-mediated ferroptosis by restoring the redox balance and regulating the Nrf2–PPAR ferritin in the phagocytosis pathway, thereby reducing PM2.5-induced emphysema and airway inflammation. These results suggest that inhibition of ferroptosis may be a potential therapeutic target for diseases with oxidative stress as the core pathogenesis, and H2S may be a potential antioxidant that blocks PM2.5 and causes COPD pathogenesis.
5. Conclusion and outlook
Ferroptosis, a new type of cell death, is involved in the pathogenesis of various diseases. Ferroptosis is a process involving abnormal metabolism of iron, amino acids, and lipids, which are involved in cell proliferation and differentiation. The metabolic process of ferroptosis is complex, and its mechanism has been preliminarily studied. Both in vitro and in vivo studies showed that ferroptosis is closely related to a variety of disease processes, and appropriate intervention in ferroptosis can treat the disease and delay the progression of related diseases. Although some progress has been made in the role of ferroptosis in lung cancer, ALI, and pulmonary fibrosis, the role of ferroptosis in COPD is not fully understood, and further clinical and experimental studies are needed. Although the existing ferroptosis inducers are effective, the therapeutic modalities for ferroptosis are still insufficient and only include iron inhibitors, iron chelators, and antioxidants. Identifying effective diagnostic markers of COPD may aid in the treatment of ferroptosis in COPD. For example, the known important regulator of iron metabolism, hepcidin, and its receptor, Fpn1, have been demonstrated to be effective diagnostic markers for COPD, and they may be promising targets for future drug development. In conclusion, with increasing studies on ferroptosis, new related regulators and their functions are constantly being explored. Considering ferroptosis as an entry point to develop COPD treatment regimens and targeted drugs has important clinical value.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
DM and CZ wrote the manuscript. RJ and ZL provided language assistance. WW proofread the manuscript. SS was involved in the design and review of this study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Shandong Province Postgraduate Education Quality Curriculum Construction Project (SDYKC20054) and the Qilu Medical School Traditional Chinese Medicine Inheritance Project (Lu Weihan [2022] No. 93).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
2. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
3. Gao M, Jiang X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr Opin Cell Biol. (2018) 51:58–64. doi: 10.1016/j.ceb.2017.11.001
4. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. (2014) 10:9–17. doi: 10.1038/nchembio.1416
5. Stockwell BR, Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
6. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
7. Global Initiative for Chronic Obstructive Lung Disease,. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease 2022 Report [EB/OL]. http://www.goldcopd.org.
8. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. (2019) 10:3145. doi: 10.1038/s41467-019-10991-7
9. Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology. (2000) 149:43–50. doi: 10.1016/S0300-483X(00)00231-6
10. Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci. (2017) 38:592–607. doi: 10.1016/j.tips.2017.04.005
11. Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. (1973) 7:253–66. doi: 10.1002/tera.1420070306
12. Fischbacher A, von Sonntag C, Schmidt TC. Hydroxyl radical yields in the Fenton process under various pH, ligand concentrations and hydrogen peroxide/Fe(II) ratios. Chemosphere. (2017) 182:738–44. doi: 10.1016/j.chemosphere.2017.05.039
13. Ye Z, Liu W, Zhuo Q, Hu Q, Liu M, Sun Q, et al. Ferroptosis: final destination for cancer? Cell Prolif. (2020) 53:e12761. doi: 10.1111/cpr.12761
14. Zilka O, Shah R, Li B, Angeli JPF, Griesser M, Conrad M, et al. On the Mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3:232–43. doi: 10.1021/acscentsci.7b00028
15. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
16. Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. (2015) 290:28097–106. doi: 10.1074/jbc.M115.680090
17. Florean C, Song S, Dicato M, Diederich M. Redox biology of regulated cell death in cancer: a focus on necroptosis and ferroptosis. Free Radic Biol Med. (2019) 134:177–89. doi: 10.1016/j.freeradbiomed.2019.01.008
18. D'Herde K, Krysko DV. Ferroptosis: oxidized pes trigger death. Nat Chem Biol. (2017) 13:4–5. doi: 10.1038/nchembio.2261
19. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
20. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
21. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
22. Kim S, Kang SW, Joo J, Han SH, Shin H, Nam BY, et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. (2021) 12:160. doi: 10.1038/s41419-021-03452-x
23. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22. doi: 10.1016/j.cell.2017.11.048
24. Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. (2019) 31:e1904197. doi: 10.1002/adma.201904197
25. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
26. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. (2009) 458:1127–30. doi: 10.1038/nature07986
27. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. (2014) 13:217–36. doi: 10.1038/nrd4236
28. Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
29. Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. (2021). doi: 10.1016/j.semcancer.2021.03.010
30. Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. (2022) 29:895–910. doi: 10.1038/s41418-022-00943-y
31. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
32. Ou Y, Wang S-J, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. (2016) 113:E6806–12. doi: 10.1073/pnas.1607152113
33. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
34. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. (2018) 22:569–75. doi: 10.1016/j.celrep.2017.12.077
35. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
36. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. (2018) 29:1756–73. doi: 10.1089/ars.2017.7176
37. Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol. (2019) 10:139. doi: 10.3389/fphys.2019.00139
38. Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. (2020) 14:267. doi: 10.3389/fnins.2020.00267
39. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. (2016) 64:488–500. doi: 10.1002/hep.28574
40. Chen G, Wei J, Lyu X. Research progress of nuclear factor-erythroid 2 related factor 2 in acute lung injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. (2018) 30:270–4. doi: 10.3760/cma.j.issn.2095-4352.2018.03.016
41. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
42. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. (2017) 68:224–32. doi: 10.1016/j.molcel.2017.09.009
43. Telorack M, Meyer M, Ingold I, et al. A glutathione-Nrf2-thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair. PLoS Genet. (2016) 12:e1005800. doi: 10.1371/journal.pgen.1005800
44. Cho WK, Lee CG, Kim LK. COPD as a disease of immunosenescence. Yonsei Med J. (2019) 60:407–13. doi: 10.3349/ymj.2019.60.5.407
45. Chen X, Que C, Yao Y, Han Y, Zhang H, Li X, et al. Susceptibility of individuals with lung dysfunction to systemic inflammation associated with ambient fine particle exposure: a panel study in Beijing. Total Environ. (2021) 788:147760. doi: 10.1016/j.scitotenv.2021.147760
46. Bernhagen J. A new cytokine target for chronic obstructive pulmonary disease? EBioMedicine. (2021) 69:103479. doi: 10.1016/j.ebiom.2021.103479
47. Szulakowski P, Crowther AJL, Jiménez LA, Donaldson K, Mayer R, Leonard TB, et al. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2006) 174:41–50. doi: 10.1164/rccm.200505-725OC
48. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2018) 18:454–66. doi: 10.1038/s41577-018-0006-6
49. Sahakian E, Chen J, Powers JJ, Chen X, Maharaj K, Deng SL, et al. Essential role for histone deacetylase 11 (HDAC11) in neutrophil biology. J Leukoc Biol. (2017) 102:475–86. doi: 10.1189/jlb.1A0415-176RRR
50. Ali MK, Kim RY, Karim R, Mayall JR, Martin KL, Shahandeh A, et al. Role of iron in the pathogenesis of respiratory disease. Int J Biochem Cell Biol. (2017) 88:181–95. doi: 10.1016/j.biocel.2017.05.003
51. Philippot Q, Deslée G, Adair-Kirk TL, Woods JC, Byers D, Conradi S, et al. Increased iron sequestration in alveolar macrophages in chronic obstructive pulmonary disease. PLoS ONE. (2014) 9:e96285. doi: 10.1371/journal.pone.0096285
52. Bailie GR, Schuler C, Leggett RE Li H, Li H-D, Patadia H, et al. Oxidative effect of several intravenous iron complexes in the rat. Biometals. (2013) 26:473–8. doi: 10.1007/s10534-013-9632-4
53. Sadeghi L, Yousefi BV, Espanani HR. Toxic effects of the Fe2O3 nanoparticles on the liver and lung tissue. Bratisl Lek Listy. (2015) 116:373–8. doi: 10.4149/BLL_2015_071
54. Ghio AJ, Hilborn ED, Stonehuerner JG, Dailey LA, Carter JD, Richards JH, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med. (2008) 178:1130–8. doi: 10.1164/rccm.200802-334OC
55. Ayala A, Muñoz MF, Argüelles S. A Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. (2014) 2014:360438. doi: 10.1155/2014/360438
56. Basu S, Helmersson J, Jarosinska D, Sällsten G, Mazzolai B, Barregård L. Regulatory factors of basal F(2)-isoprostane formation: population, age, gender and smoking habits in humans. Free Radic Res. (2009) 43:85–91. doi: 10.1080/10715760802610851
57. Gao X, Gào X, Zhang Y, Breitling LP, Schöttker B, Brenner H. Assoiations of self-reported smoking, cotinine levels and epigenetic smoking indicators with oxidative stress among older adults: a population-based study. Eur J Epidemiol. (2017) 32:443–56. doi: 10.1007/s10654-017-0248-9
58. Zhang X, Staimer N, Gillen DL, Tjoa T, Schauer JJ, Shafer MM, et al. Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ Res. (2016) 150:306–19. doi: 10.1016/j.envres.2016.06.019
59. Sarnat JA, Golan R, Greenwald R, Raysoni AU, Kewada P, Winquist A, et al. Exposure to traffic pollution, acute inflammation and autonomic response in a panel of car commuters. Environ Res. (2014) 133:66–76. doi: 10.1016/j.envres.2014.05.004
60. Wang Y, Tang M. PM25 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ Pollut. (2019) 254:112937. doi: 10.1016/j.envpol.2019.07.105
61. Tang X, Li Z, Yu Z, Li J, Zhang J, Wan N, et al. Effect of curcumin on lung epithelial injury and ferroptosis induced by cigarette smoke. Hum Exp Toxicol. (2021) 40:S753–62. doi: 10.1177/09603271211059497
62. Liu X, Ma Y, Luo L, Zong D, Li H, Zeng Z, et al. Dihydroquercetin suppresses cigarette smoke induced ferroptosis in the pathogenesis of chronic obstructive pulmonary disease by activating Nrf2-mediated pathway. Phytomedicine. (2022) 96:153894. doi: 10.1016/j.phymed.2021.153894
63. Zhang Z, Fu C, Liu J, Sai X, Qin C, Di T, et al. Hypermethylation of the Nrf2 Promoter Induces Ferroptosis by Inhibiting the Nrf2-GPX4 Axis in COPD. Int J Chron Obstruct Pulmon Dis. (2021) 16:3347–62. doi: 10.2147/COPD.S340113
64. Liu J, Zhang Z, Yang Y, Di T, Wu Y, Bian T. NCOA4-mediated ferroptosis in bronchial epithelial cells promotes macrophage M2 polarization in COPD emphysema. Int J Chron Obstruct Pulmon Dis. (2022) 17:667–81. doi: 10.2147/COPD.S354896
65. Vucic EA, Chari R, Thu KL, Wilson IM, Cotton AM, Kennett JY, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. (2014) 50:912–22. doi: 10.1165/rcmb.2013-0304OC
66. Gimenes JJ, Srivastava V, ReddyVari H, et al. Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis. Clin Sci. (2019) 133:983–96. doi: 10.1042/CS20181088
67. Wang L, Zhang Y, Zhang N, Xia J, Zhan Q, Wang C. Potential role of M2 macrophage polarization in ventilator-induced lung fibrosis. Int Immunopharmacol. (2019) 75:105795. doi: 10.1016/j.intimp.2019.105795
68. Feng H, Yin Y, Ren Y, Li M, Zhang D, Xu M, et al. Effect of CSE on M1/M2 polarization in alveolar and peritoneal macrophages at different concentrations and exposure in vitro. In Vitro Cell Dev Biol Anim. (2020) 56:154–64. doi: 10.1007/s11626-019-00426-4
69. Crisponi G, Nurchi VM, Lachowicz JI. Iron chelation for iron overload in thalassemia. Met Ions Life Sci. (2019) 19:9783110527872–009. doi: 10.1515/9783110527872-003
70. Ramezanpour M, Smith JLP, Ooi ML, Gouzos M, Psaltis AJ, Wormald PJ, et al. Deferiprone has anti-inflammatory properties and reduces fibroblast migration in vitro. Sci Rep. (2019) 9:2378. doi: 10.1038/s41598-019-38902-2
71. Siedlinski M, Tingley D, Lipman PJ, Cho MH, Litonjua AA, Sparrow D, et al. Dissecting direct and indirect genetic effects on chronic obstructive pulmonary disease (COPD) susceptibility. Hum Genet. (2013) 132:431–41. doi: 10.1007/s00439-012-1262-3
72. DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. (2009) 85:493–502. doi: 10.1016/j.ajhg.2009.09.004
73. Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. (2016) 22:163–74. doi: 10.1038/nm.4021
74. Babaoglu E, Kilic H, Hezer H, Dag O, Parlak E, Senturk A, et al. Comparison of thiol/disulphide homeostasis parameters in patients with COPD, asthma and ACOS. Eur Rev Med Pharmacol Sci. (2016) 20:1537–43. Available online at: https://pm.yuntsg.com/searchList.html
75. Han W, Dong Z, Dimitropoulou C, Su Y. Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid Redox Signal. (2011) 15:2121–34. doi: 10.1089/ars.2010.3821
Keywords: ferroptosis, COPD, inflammation, iron, lipid peroxidation
Citation: Meng D, Zhu C, Jia R, Li Z, Wang W and Song S (2023) The molecular mechanism of ferroptosis and its role in COPD. Front. Med. 9:1052540. doi: 10.3389/fmed.2022.1052540
Received: 24 September 2022; Accepted: 08 December 2022;
Published: 06 January 2023.
Edited by:
Bruno Guedes Baldi, University of São Paulo, BrazilReviewed by:
Jorge Boczkowski, Université Paris-Est Créteil Val de Marne, FranceChunbin Zou, University of Pittsburgh, United States
Copyright © 2023 Meng, Zhu, Jia, Li, Wang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wantao Wang,  wangwt0906@shutcm.edu.cn; Suhua Song, sdjnssh@163.com
wangwt0906@shutcm.edu.cn; Suhua Song, sdjnssh@163.com