The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms
 Teresa Salvatore1
Teresa Salvatore1  Pia Clara Pafundi2 Raffaele Galiero2 Gaetana Albanese2 Anna Di Martino2 Alfredo Caturano2 Erica Vetrano2
Pia Clara Pafundi2 Raffaele Galiero2 Gaetana Albanese2 Anna Di Martino2 Alfredo Caturano2 Erica Vetrano2  Luca Rinaldi2
Luca Rinaldi2  Ferdinando Carlo Sasso2*
Ferdinando Carlo Sasso2*- 1Department of Precision Medicine, University of Campania Luigi Vanvitelli, Naples, Italy
- 2Department of Advanced Medical and Surgical Sciences, University of Campania Luigi Vanvitelli, Naples, Italy
Individuals with diabetes mellitus (DM) disclose a higher incidence and a poorer prognosis of heart failure (HF) than non-diabetic people, even in the absence of other HF risk factors. The adverse impact of diabetes on HF likely reflects an underlying “diabetic cardiomyopathy” (DM–CMP), which may by exacerbated by left ventricular hypertrophy and coronary artery disease (CAD). The pathogenesis of DM-CMP has been a hot topic of research since its first description and is still under active investigation, as a complex interplay among multiple mechanisms may play a role at systemic, myocardial, and cellular/molecular levels. Among these, metabolic abnormalities such as lipotoxicity and glucotoxicity, mitochondrial damage and dysfunction, oxidative stress, abnormal calcium signaling, inflammation, epigenetic factors, and others. These disturbances predispose the diabetic heart to extracellular remodeling and hypertrophy, thus leading to left ventricular diastolic and systolic dysfunction. This Review aims to outline the major pathophysiological changes and the underlying mechanisms leading to myocardial remodeling and cardiac functional derangement in DM-CMP.
Introduction
Diabetic Cardiomyopathy (DM-CMP) is a form of heart disease associated with diabetes mellitus (DM), which causes significant structural and functional changes in the myocardium. The pathogenesis has been a hot topic of research since its first description (1), and it is still under active investigation, as a complex interaction among multiple factors play a role at systemic, myocardial, and cellular/molecular levels. The current pathogenic hypotheses mostly derive from translational models, with human evidence far less developed due to limited access to human tissue samples.
This review aims to outline the state of the art about the major pathophysiological changes and underlying mechanisms leading to myocardial remodeling and cardiac functional derangement in DM-CPM.
Diabetes Mellitus and Heart Failure: A Bidirectional Epidemiologic Association
The risk for heart failure (HF), as well as that for all components of cardiovascular disease (CVD), is higher in individuals with diabetes as compared to non-diabetic people.
The Framingham Heart Study, published in 1974, is among the first studies to demonstrate this association, reporting an incidence 2.4- and 5-fold higher, respectively, in men and women, after adjustment for common CVD risk factors (2).
A robust epidemiological evidence has confirmed that HF is among the most common complications of DM. A prevalence of ~20% (4–28%) has been found in clinical trials of glucose-lowering drugs in DM, consistent with a recent position paper of the Heart Failure Association of the European Society of Cardiology (ESC) (3).
DM patients without HF at baseline are more likely to develop this complication over time as compared to non-diabetic people (4), whereas subjects without diabetes at 45 years are more than 60% less likely to manifest HF (5). In the Kaiser Permanente system, out of more than 8,000 patients followed for up to 6 years, the risk of new-onset HF resulted 2.5-fold higher in patients with type 2 DM (T2DM) rather than their non-diabetic counterparts (6). In a large population-based study of 34,198 T2DM patients initially free from overt CVD, HF was even more common than myocardial infarction (MI) as first presentation of CVD (7). In T2DM subjects with newly-recognized HF, the incidence was almost 5-fold higher for HF with preserved ejection fraction (HFpEF) (about 23%) vs. HF with reduced EF (HFrEF) (about 5%) (8).
A low annual incidence of HF (0.2%) and myocardial dysfunction (−0.1%) is reported in type 1 DM (T1DM), likely dependent on the younger age of the studied population (9). Nevertheless, there is a well-documented prevalence of early subclinical cardiomyopathy in children and adolescents with T1DM (10). A meta-analysis of subjects included in clinical trials demonstrated that the presence vs. absence of DM in hypertensive individuals increased the risk of HF by more than 4-folds (11).
Subjects with impaired glucose tolerance (IGT) or insulin resistance (IR) have a 1.7-fold increased risk of HF (12). A community-based cohort study that followed patients for almost 30 years revealed that several biomarkers reflecting IR and dyslipidemia, predicted HF independently of ischemic CVD and other established CV risk factors (13).
HF is a frequent as serious complication of diabetes. Its prognosis is worse than in non-diabetic subjects, with a 75% higher risk of CV death or HF hospitalization (14), and a frequent progression to end-stage HF, which may require heart transplantation despite optimal medical therapy (15). In a prospective study from the mid-1990s, HF 1-year mortality was 30% in people with DM, about 1.5-fold higher than in those without (16). The HF mortality risk was 10-fold higher in a diabetic population older than 65 years (17). Currently, the clinical impact of DM–CMP and other chronic diseases in hospitalized elderly subjects is affected by both gender, and frailty (18–20).
In the CHARM (Candesartan in Heart Failure Assessment of MoRtality and Morbidity) study, DM was associated with a higher relative risk of HF hospitalizations or CV death in patients with HFpEF than HFrEF (21).
On the other hand, as HF is common in DM, so DM is highly prevalent in people with HF, hence one condition increases the incidence and worsens the prognosis of the respective other. Patients with HF have a 4-fold higher prevalence of T2DM (20%) than patients without (4–6%) (22). In a CHARM study group analysis, more than 25% of patients with HF has diabetes (23). When admitted with HF, one-third of patients without a previous diagnosis of diabetes results affected by DM or impaired glucose tolerance (IGT) (24). This prevalence rises to 40% in a large multicenter European study (25), as confirmed in the EVEREST analysis (26).
The mechanism responsible of the increased risk of T2DM in HF is the impaired insulin signaling induced by loss of skeletal muscle mass, sedentary lifestyle, and increased circulating cytokines, which trigger a vicious cycle in which IR and HF deteriorate each other (27). In patients with advanced HF, hemodynamic recovery after ventricular assist device placement is associated with improvements in both systemic and cardiac insulin sensitivity, glucose homeostasis, and toxic lipid products (28). Likewise, IR significantly affects HF prognosis (29).
The Diabetic Cardiomyopathy
Based on the observation that two-thirds of elderly patients with diabetes presented with a myocardial dysfunction, Lundbæk has firstly suggested in 1954 the concept of a specific DM-related cardiomyopathy (30). This term refers to the current definition proposed by the European Society of Cardiology (ESC), that is a “cardiomyopathy is defined as a heart muscle disease in which the myocardium is structurally and functionally abnormal in the absence of coronary artery disease (CAD) as well as hypertensive, valvular, or congenital heart disorders” (31). Almost 20 years later, Rubler et al. reported the post-mortem findings of four diabetic patients with glomerulosclerosis and advanced symptoms of HF unrelated to valvular, congenital or hypertensive heart disease, alcoholism or significant epicardial coronary artery atherosclerosis. These data thus provided evidence that a cardiomyopathy could directly result from DM, likely in dependence of myocardial microangiopathy or metabolic derangements (1).
Currently, DM-CMP is widely recognized as a specific form of cardiomyopathy which occurs independently of other cardiac risk factors and is promoted by the long-standing metabolic perturbations of diabetes, thus exerting a direct toxic effect on the myocardium (30). Although Rubler originally reported a dilated cardiomyopathy manifesting with the characteristic symptoms of HF, the restrictive LV remodeling with diastolic LV dysfunction is the more frequent picture in DM (32).
In the current clinical practice, DM-CMP diagnosis is still challenging, as it requires the identification of distinct functional and structural changes in the LV and the concomitant exclusion of other cardiac diseases and risk factors for CVD. Due to the very frequent confounding of other HF risk factors such as hypertension, CAD, and renal disease, the burden of a “pure” diabetic cardiomyopathy is conceivable not as high as the cardiomyopathy of heterogeneous etiology, with a calculated prevalence of 16.9% of diabetic patients in a small study (33).
Pathophysiology of Diabetic Cardiomyopathy
Several mechanisms determining molecular, cellular and interstitial changes, as well as activation of renin-angiotensin aldosterone axis and adrenergic systems, are involved in the development of DM-CPM. These include imbalance of myocardial energy substrates, gluco- and lipotoxicity, altered insulin signaling, mitochondrial defects, endoplasmic reticulum (ER) stress, deranged intracellular calcium handling, oxidative stress, endothelial dysfunction, deposition of advanced glycation end products (AGEs), maladaptive immune responses, and so on. Each of them contributes to the structural remodeling and functional defects in diabetic myocardium, including impairments in cardiac relaxation, compliance, and contractility (Figure 1).
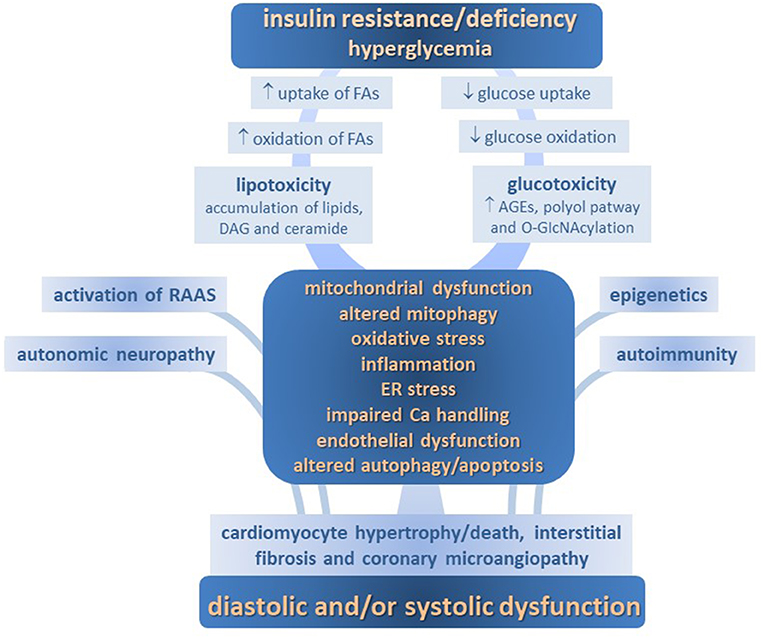
Figure 1. Pathophysiology of diabetic cardiomyopathy.
Metabolic Abnormalities in the Diabetic Heart
Changes to the metabolic milieu associated with DM, such as lipotoxicity, glucotoxicity and impaired insulin signaling, emerge as crucial pathogenic factors for DM-CMP. Together, they exert, both directly and indirectly, a detrimental increase in oxidative stress, endothelial dysfunction and inflammation, thus making a strong contribution to the myocardium structural and functional derangement.
Myocardial Energy Substrate Changes and Lipotoxicity
Energy Substrates in Healthy Myocardium
Due to the constant cardiac activity, the myocardium is the higher energy-demanding tissue in the body. To absolve this function, it is equipped with an efficient metabolic machinery, mainly represented by the mitochondrial oxidative phosphorylation (34). Under normal conditions, most of energy for myocardium (~60–90%) is supplied by fatty acid (FA) oxidation, whereas the remaining ~10–40% of ATP derives from the oxidation of pyruvate produced in equal amounts by glycolysis and lactate oxidation. Of note, the heart is a net consumer of lactate, both at baseline and upon increase in workload. Ketone bodies are not immediately available from food but produced in the liver by incomplete oxidation of FAs released from the adipose tissue in response either to fasting or energy depletion. They provide, mainly the D-beta-hydroxybutyrate, an alternative substrate for oxidative phosphorylation. Under physiological conditions, aminoacids represent a minor source of energy (21, 35).
The healthy heart is commonly defined a “metabolic omnivore,” due to its crucial capacity to shift between different substrates, according to their availability, in order to ensure a continuous energy supply (36). This metabolic flexibility is mainly determined by the “Randle cycle,” by which high circulating levels of glucose decrease the FA oxidation rates and vice-versa (37). Another metabolic regulator is 5′-AMP-activated protein kinase (AMPK), which acts as a cellular “fuel gauge” (38). In the long term, the nuclear peroxisome proliferator-activated receptor (PPAR)-α, abundantly expressed in the myocardium, play a pivotal role upregulating the transcription of genes related to FA uptake and oxidation (39). Overall, the relative substrate contribution to ATP production can vary mostly depending on energy demand, substrate availability and hormonal milieu. For instance, exercise induces a switch from FAs to glucose oxidation, whereas during prolonged fasting or poorly controlled diabetes, ketone bodies can represent the main energy supplier (34).
Changes of Energy Substrates in Failing Heart
In HF the mitochondrial oxidation of FAs is decreased, more likely due to the PPARα signaling suppression and the activation of hypoxia-inducible factor 1α (HIF1α)–PPARγ signaling axis, which impair FA transport into mitochondria and downregulate FA oxidative enzymes (34). Due to the stimulation of lipolysis by sympathetic activation in HF, an increased FA delivery to cardiac myocytes is responsible of this shift away from FA oxidation (40). This imbalance between FA uptake and oxidation leads to cytosolic overload of triglycerides and accumulation of metabolic intermediates generated by non-oxidative pathways such as ceramide and diacylglycerol (DAG), exerting toxic effects and maladaptive signaling, including IR (34, 41). These deposits promote inflammation, cell damage and, eventually, cell death, hence a condition of “lipotoxicity” which contributes to the HF progression (42, 43).
The glucose metabolism of the failing heart is characterized by an enhanced glucose uptake, not accompanied by a concomitant increase in glucose oxidation. Therefore, despite an increase in the relative contribution of glucose oxidation to ATP production, the absolute substrate flux through glucose oxidative pathways actually reduces (34). During the late stages of HF, the glucose availability for ATP production is further impaired by the association of a marked IR (44), even though some authors suggest that the cardiac IR may represent a beneficial mechanism protecting the heart from fuel overload (45).
Ketone bodies may represent a relevant energy source in the HF setting when metabolism of other energy substrates falls (46). Indeed, an upregulation of the enzymes involved in ketone body metabolism is reported in both murine models (47) and patients with advanced HF (48). Since the ATP production/oxygen consumption ratio of the β-hydroxybutyrate is higher (2.50) than that of FA palmitate (2.33), this ketone body has been proposed as a “super fuel” enhancing cardiac metabolic efficiency (34).
Metabolic Disturbances in Diabetic Heart
Exposure to hyperglycemia by itself decreases insulin signaling and glucose uptake in cardiomyocytes (49). Essentially, due to the impaired capacity to transport and metabolize glucose determined by insulin deficiency in T1DM and IR in T2DM, the diabetic heart shifts away from glucose as an energy source and gets in a “metabolically inflexible” and less efficient FA-dependent state. This is a crucial pathophysiological condition if considering that glucose is the unique cardiac substrate able to provide ATP during hypoxia or ischemia (50).
Consistent with a prevalent FA utilization, the diabetic heart shows an increased expression of the FA transporter CD36 on both sarcolemmal and endosomal membranes, with an enhanced subcellular vesicular recycling from endosomes to plasma membrane (51). Excess FAs activate PPAR-α, which increases expressions of genes involved in FA oxidation, but also suppresses glucose utilization (52). These typical derangements in the myocardial energy metabolism of diabetic heart are mimicked in mice with cardiac-restricted overexpression of PPAR-α (39). However, studies in diabetic patients either with or without HF argue against an activation of the PPAR-α signaling axis which drives the increase in FA uptake and oxidation (53, 54). Another proposed mechanism for enhanced FA oxidation may be the increased acetylation of mitochondrial β-oxidation enzymes observed in an obese animal model (55).
Excessive FA oxidation increases ATP expenditure for futile cycling of metabolic intermediates, inhibits ATP shuttling from mitochondria to the cytosol, and increases the expression of mitochondrial uncoupling protein (UCP) 3 through PPAR-α, thereby dissipating the mitochondrial proton gradient and deteriorating the ATP production efficiency (56, 57). Finally, these changes produce oxidative stress and mitochondrial dysfunction (58). Moreover, the dissipation of the mitochondrial membrane potential might interfere with excitation–contraction coupling and mitochondrial Ca2+ uptake, thus potentially underlying arrhythmias (59).
A deposition of lipids and their metabolites in the cytosol of cardiomyocytes has been documented in DM animal models (60). Human studies using Oil Red-O staining of explanted hearts at the time of heart transplantation have demonstrated cardiac steatosis (28, 41). This excess accumulation of lipids leads to myocardial IR and reduced bioavailability of nitric oxide (NO) (61). Increased levels of DAG in cardiomyocytes activates protein kinase C (PKC) isoforms, thus reducing insulin metabolic signaling and NO production. Similarly, ceramide directly activates atypical PKCs to phosphorylate and inhibit the insulin metabolic Akt signaling and disrupts endothelial NO synthase (NOS) signaling impairing NO bioavailability (62, 63). As well, ceramide may activate caspase 3 and stimulate cytochrome C release, thus inducing cellular apoptosis, and inhibit key pathways involved in defense against DNA damage, such as Poly ADP ribose polymerase (PARP) (64).
A relevant question is whether the altered substrate metabolism is cause or consequence of the failing heart in diabetes. A ventricular biopsy study has showed that even in the absence of contractile failure the diabetic heart exhibits a decreased mitochondrial capacity for β-oxidation, increased accumulation of intracellular lipids, ER stress, and a higher degree of apoptosis (65). Another very recent human bioptic study suggests the crucial role of the toxic metabolic milieu of DM in the early progression of DM-CMP (66). A lipid accumulation in cardiomyocyte was found after only 3 months in non-DM hearts transplanted to diabetic patients. Moreover, triacylglycerol and ceramide contents were both related with early dysfunctions in DM recipients after 12 months. Levels of myocardial insulin receptor were lower in healthy hearts transplanted in DM than non-DM recipients, and SREBP1c (sterol regulatory-element-binding protein-1c) and PPAR systems were highly expressed in cardiomyocytes of DM recipients.
Hyperglycemia and Glucotoxicity
Sustained exposure to high glucose levels is a major driver of cardiac pathology in DM (67–69). In an observational study on individuals with T1DM, the incidence of HF increased monotonically with the HbA1c, with a range of 1.42–5.20 per 1,000 patient-years between patients in the lowest (<6.5%) and highest (>10.5%) HbA1c categories (70). In a similar study on T2DM patients, each 1% increase in the HbA1c corresponded to an 8% increase in the HF risk (71). Conversely, in T2DM patients of UK Prospective Diabetes Study, each 1% reduction in HbA1c level corresponded to a 16% reduction in the risk of HF (72).
The detrimental effect of chronic hyperglycemia, referred to as “glucotoxicity,” is mainly mediated by oxidative stress, increased formation of AGEs and enhanced substrate flux through alternative metabolic pathways (50).
Oxidative Stress
Hyperglycemia contributes to oxidative stress in diabetic heart by excessive oxygen radical formation from the auto-oxidation of glucose, formation of glycated proteins, and impaired buffering capacity due to glycation of metfodant enzymes (73, 74).
The mitochondrial electron transport chain is among the first targets of high glucose levels, with a direct increase in superoxide anion formation. Moreover, high glucose activates protein kinase C (PKC), thus leading to up-regulation of NADPH oxidases (NOX), xanthine oxidase, uncoupling of NO synthase (NOS), microsomal P-450 enzymes, and arachidonic acid metabolism pathways (75). The consequent increased reactive oxygen species (ROS) impair cardiac structure and function by directly damage DNA, proteins and phospholipids, and promote myocytes apoptosis. Kuster et al. found that a short-period of exposure to H2O2 of in vitro rat ventricular myocytes determined a progressive decrease in cell shortening, followed by diastolic arrest. The possible mechanisms were the direct oxidative modification of sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA) and Na+/Ca2+ exchanger (NCX) (76). One harm of the superoxide generation stands in its interaction with NO to form peroxynitrite, a potent oxidant involved in enhanced apoptosis of both animal and human cardiomyocytes (77, 78).
Antioxidant response may be a determinant of the heart health in diabetes. Other findings reveal that the mitochondrial isoform of aldehyde dehydrogenase (ALDH2) may play a role in the development of DM-CMP, possibly through protection against oxidative stress and preservation of mitochondrial integrity (79). Evidence from literature indicates that diabetes upregulates the Ras-related small G protein RhoA, a factor that may impair cardiac function determining uncoupled eNOS, reduced NO bioavailability, and enhanced O. IGF-I is a crucial cardiac survival factor that downregulating RhoA produces beneficial effects also mimicked by the Rho kinase inhibitor Y27632 and BH4, a finding indicating that the selective IGF-I overexpression may represent a therapeutic potential for DM-CMP (80).
Enhancing cardiac endogenous antioxidant capacity is an attractive way to prevent DM-CMP. A pivotal target may be represented by Nrf2, an important regulator of cellular detoxification responses and redox status that can lead to antioxidant response elements (ARE)-mediated basal and inducible expression of more than 200 genes (81). Sulforaphane, a molecule within the isothiocyanate group of organosulfur compounds from cruciferous vegetables, such as broccoli, Brussel sprouts or cabbage, is a potent Nrf2 activator (82). A study on db/db mice fed with broccoli sprout extract or sulforaphane for 3 months showed significant prevention of diabetes-induced cardiac oxidative damage and inflammation by up-regulating Nrf2 transcriptional activity (83). A recent study on mice provided the direct evidence that the preventive effect of sulforaphane against DM-CMP depends on AMPK resulting from both improvement of AMPK-mediated lipid metabolism and potentiation of antioxidative pathway mediated by AMPK/AKT/GSK3β signaling (84).
Accumulation of Advanced Glycation End Products
Persistent hyperglycemia causes the non-enzymatic glycosylation of proteins and enzymes with production of toxic AGE adducts, irreversibly altering their structure and functions (85). As an example, AGEs formed on SERCA2a in diabetes impair the sarcoplasmic reticulum (SR) Ca2+ reuptake in cardiomyocytes and slow cardiac relaxation (86), whereas long-term treatment with an AGE crosslink breaker partially normalized SR Ca2+ signaling (87).
A significant increase in AGE compounds and their binding to cell surface specific receptors (RAGEs) trigger a cascade of pathophysiological responses responsible of severe cardiac damage. Among these, the activation of PKC and NOX lead to the fabrication of peroxide and, ultimately, of ROS, and to the maladaptive activation of mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NFκB) signaling, followed by the production of several inflammatory and/or profibrotic factors, as well as upregulation of apoptosis (via p53 and calcineurin signaling) and autophagy (88–92). All these mechanisms may cause functional and structural damage, till cardiomyocyte death and eccentric LV remodeling with systolic dysfunction.
Interestingly, metformin induces activation and phosphorylation of MAPK, which could mediate its several extraglicemic effects (93, 94).
As shown by light microscopic immune-histochemical visualization, AGEs also accumulate in the myocardial interstitium between cardiomyocytes (95). The non-structural compartment of extracellular matrix (ECM) is represented by a variety of proteins (including collagen IV, laminin, fibronectin, myelin, tubulin, plasminogen activator 1, and fibrinogen), vital for ECM plasticity and with glycosylation as a common denominator (96). Besides ECM disturbation by oxidative stress and inflammation, accumulation of AGEs in the interstitium stimulates the differentiation of fibroblasts into myofibroblasts (via Janus kinase-signal transducer and activator of transcription, JAK-STAT signaling), which produce excess matrix proteins, and the crosslink matrix metalloproteinases (MMPs), which indeed impair ECM degeneration. The increased resistance of connective tissue to enzymatic proteolysis and the enhanced collagen cross-linking lead to myocardial fibrosis and stiffness, thus resulting in impaired compliance and diastolic LV relaxation (97–99). This process is potentially mediated by the up-regulation of pro-fibrotic cytokines such as transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF) (100).
In DM-CMP, an abundant AGEs deposition even involves both endothelial and smooth muscle cells of myocardial microvasculature by triggering vascular inflammation and dampening endothelial NO production (101, 102).
As evidence for the role of AGEs in DM-CMP pathogenesis, the cleavage of preformed AGE crosslinks with ALT-711 attenuates the diabetes-associated cardiac abnormalities in rats (103), and the administration of a RAGE antagonist in a rat model of T1DM prevents AGEs/RAGE signaling-mediated increases in myocardial collagen, fibrosis, stiffness and diastolic dysfunction (104).
The soluble RAGE (sRAGE) is the circulant isoform of RAGE which, by competing with cellular RAGE, may inhibit the pro-inflammatory and pro-fibrotic activity of AGE (105). Unsurprisingly, lower levels of circulating soluble receptors for AGEs predict incident HF in patients with DM (106).
A recent study on experimental diabetes has demonstrated that the inhibition of AGE formation by aminoguanidine exerts a beneficial effect against cardiac remodeling and contractile dysfunction, likely through the regulation of autophagy and ER stress (107).
Activation of Polyol Pathway
In In a high-glucose state as diabetes, aldose reductase converts a part of glucose overload to sorbitol, which is oxidized to fructose by sorbitol dehydrogenase. The first reaction produces a depletion of NADPH, a molecule essential for the functioning of various endothelial enzymes, including cytochrome P450 and NO synthase, and a cofactor in the generation of the reduced glutathione. The second reaction increases the cytosolic NADH: NAD+ ratio, which can inhibit the glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and increase the concentrations of triose phosphate, with consequent formation of AGE and DAG (108).
The chronic elevation of DAG in diabetes (and in part the increased circulating levels of FAs) activates PKC, a central player in signal transduction and intracellular crosstalk, by phosphorylating a huge array of substrates on serine/threonine residues. PKCβ2 isoform is over-expressed in the myocardium of diabetic animal models and patients with HF (109, 110), and the activation of the PKC/DAG signaling pathway is associated with biochemical and structural changes typical of DM-CMP (e.g., reduced blood flow, increased vascular permeability, basal membrane thickening, ECM deposition, and cardiac hypertrophy) (111–113). On the contrary, PKC inhibition may reverse structural and functional derangements in the diabetic heart (114).
Maladaptive Hexosamine Biosynthesis
During chronic hyperglycemia, a small percentage of glucose is shuttled through the hexosamine biosynthesis pathway, thus generating the O-linked β-N-acetylglucosamine (O-GlcNAc). This metabolite may rapidly bind to a multitude of proteins altering their function via the O-GlcNAc transferase (115). The ones specifically involved in the progression of DM-CMP include Ca2+/calmodulin-dependent protein kinase II (CaMKII), phospholamban and myofilaments, with a negative impact on cardiac contractility and relaxation (116).
Several studies have suggested that O-Glc-N-Acylation of cardiomyocyte proteins might be associated with the development of cardiac hypertrophy (117, 118). This pathogenic mechanism of myocardial hypertrophy has been recently confirmed both in cultured cells and in vivo, as triggered by high carbohydrate diets (119). The reduction of the excess cellular O-Glc-N-Acylation, indeed, obtains beneficial effects on calcium handling and diabetic cardiac function (120).
Many mitochondrial proteins are highly susceptible to O-Glc-N-Acylation, which suggests another way for hexosamine pathway to induce cardiac dysfunction in diabetes (121).
Insulin Resistance
Increasing evidence points to IR as a primary etiologic factor in DM-CMP development.
IR impairs the myocardial glucose utilization and increases the expression of myocardial UCPs. The resulting decline in the efficiency of high-energy phosphate production prevents the myocardial adaptive response to injury, as observed in patients with HFpEF (122, 123).
IR impairs the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signal transduction pathway to elicit normal metabolic responses. The resultant reduction of glucose oxidation decreases the Ca2+ ATPase activity and moves Ca2+ back into the SR, thus increasing the intracellular content of ion (30, 124). Since PI3K/AKT can also activate endothelial NOS (125), the reduced NO production in IR states further increases the intracellular Ca2+ levels and Ca2+ sensitization in cardiomyocytes via the cGMP/PKG signaling pathway (30, 51, 126). On the other hand, through the PI3K/Akt pathway, the higher insulin levels associated to IR may induce the titin switching toward the stiff N2B isoform, thus impairing cardiomyocyte distensibility (127).
All these abnormalities may promote cardiac stiffness and diastolic dysfunction, being mainly relevant to restrictive/HFpEF phenotype of DM-CMP, especially in obese T2DM patients. Other contributing mechanisms of IR to myocardial injury are lipotoxicity, sympathetic up-regulation, inflammation, oxidative stress, and fibrosis (128).
The impact of IR on cardiac morphology and function has been extensively documented in clinical studies. In the Framingham Heart Study, LV mass and wall thickness increased with worsening glucose intolerance, and the relation between IR and LV mass observed only in women, was largely dependent on obesity (129). A recent longitudinal study with a 25-yrs follow-up period revealed that cumulative exposure to DM or higher IR adversely affects LV remodeling and function (130). A link between IR and concentric LV remodeling and hypertrophy is confirmed in studies using cardiac magnetic resonance imaging (131, 132).
Intriguingly, opioid system, which seems related to IR (133), play a role in HF (134).
Pathophysiological Mechanisms Promoting DM-CMP
A plethora of mechanisms mostly connected to the above-described metabolic alterations, act in unison to promote cardiomyocyte injury and cardiac dysfunction in DM.
Altered Calcium Homeostasis and Calcium/Calmodulin Dependent Protein Kinase II
Perturbations in the cytosolic calcium trafficking and ventricular excitation-contraction coupling at cardiomyocyte level are the mechanistic hallmark of cardiac dysfunction in diabetes (124). Physiologically, the excitation of the cardiomyocyte determines the actin-myosin interaction and contractile activity by inducing Ca2+ influx via L-type Ca2+ channels in the plasma lemma and subsequent Ca2+ transient, i.e., Ca2+ release from sarcoplasmic reticulum (SR) through ryanodine receptors. During cardiomyocyte relaxation, Ca2+ actively moves from cytoplasm into SR by SERCA, with the contribution of sarcolemma Ca2+ extrusion by NCX and Ca2+ATPase (135).
In diabetic cardiomyocytes, the activity of SERCA and NCX is impaired, likely by either reduction in protein levels or its post-translational modification because of non-enzymatic glycosylation (136). The slower Ca2+ transients and leaky Ca2+ release channel, result in an impaired calcium load of SR, which is the primary organelle for handling intracellular calcium. To support the correlation between Ca2+ handling and cardiac dysfunction, cardiac overexpression of SERCA2a significantly improves myocardial contractility in streptozotocin-induced diabetic rats (137). Being the calcium efflux from cytosol depressed, the cardiomyocyte relaxation impairs, and the action potential duration prolongs (138). These changes are likely associated with the clinical finding of diastolic dysfunction.
CaMKII is a multifunctional serine/threonine kinase physiologically activated in response to β-adrenergic receptor signaling, which targets a number of Ca2+ homeostatic proteins in the heart (139). During acute cardiomyocytes activation, CaMKII stimulates glucose uptake, energy production, sarcolemmal ion fluxes, SR Ca2+ release/reuptake and myocyte contraction/relaxation coupling, all mechanisms empowering the physiological cardiac adaptation. In diabetic myocardium, as a result of impaired Ca2+ handling and oxidative, nitrosative and hyperglycemic stresses, CaMKII is in a state of chronic maladaptive upregulation leading to inefficient substrate utilization, mitochondrial dysfunction, inflammation, fibrosis, ion channel remodeling, impaired intracellular Ca2+ handling, contractile dysfunction, and increased risk of arrhythmias (140, 141). In a recent study, the cardiac tissue from both T2DM patients and rats presents an elevated CaMKII activation as compared to non-diabetic controls. Moreover, the trabeculae from diabetic rats have reduced contraction and relaxation performance, which may be restored by the inhibition of this kinase (142).
Mitochondrial Dysfunction, ER Stress, and Altered Mitophagy
The increased β-oxidation exceeding the respiratory capacity of mitochondria in diabetic hearts induces accumulation of toxic lipid metabolites and generation of oxidative stress and inflammation, which further deteriorate mitochondrial function, possibly culminating in cardiomyocyte death (143). In addition, the signaling pathways by which AMPK activates the PPAR-γ coactivator-1α (PGC-1α), the master metabolic regulator of mitochondrial biogenesis and respiratory function, is impaired in advanced DM-CMP (51).
The hyperglycemia-stimulated ER stress may be the initiator, concomitantly with the FA overload of cardiomyocytes, of an adverse mitochondrial remodeling in human diabetic myocardium (144). ER stress is a condition of over-accumulation of misfolded proteins triggered by intracellular buildup of saturated FA and oxidative stress (145). If the activation of the “unfolded protein response” aiming to restore a normal ER function fails, the cardiomyocyte may go toward a profound mitochondrial dysfunction, including decreased ability to process FA up to self-destruction by apoptosis (146). Upregulation of GRP78 and induction of CHOP, two markers of ER stress response, has been recently described in LV myocardium from diabetic patients (65), consistent with previous findings in animal models of T2DM (147).
Mitophagy, a type of selective autophagy where the damaged or unnecessary mitochondria are sequestered by auto-phagosomes and degraded by lysosomes, is an essential step in maintaining mitochondrial homeostasis in the heart, together with mitochondrial fission, fusion, and biogenesis (148). Increasing lines of evidence suggest that mitophagy is significantly changed in diabetic cardiomyocytes, and some vital proteins involved in this process have been found altered in many diabetic tissues, including heart (149, 150). Even in the context of metabolic syndrome, cardiac mitophagy is altered (151).
Autophagy, Apoptosis, and Senescence of Myocytes
Adult cardiomyocytes rarely proliferate, thus their death may represent the primum movens for the cascade of hypertrophic and fibrotic LV remodeling leading to progressive heart dysfunction, till congestive HF. Higher rates of myocyte death, as determined by autophagy, apoptosis, and senescence, characterize DM-CMP (152).
Constitutive autophagy, a highly conserved process for bulk degradation and recycling of cytoplasmic components in lysosomes, is a homeostatic mechanism crucial to counter oxidative stress and AGE formation and to protect cardiomyocytes from aging-related and ischemia-induced cardiac hypertrophy (153, 154). Bellot et al. reported that ROS and autophagy mutually regulate and that elimination of ROS-damaged cells via autophagy is a protective mechanism (155). Indeed, if autophagy is suppressed and excessive ROS persists, the cardiomyocytes would eventually go toward apoptotic death (156). On the other hand, excessive induction of autophagy may indiscriminately destroy cytosol and organelles and determine hypertrophy and fibrosis, with an accelerated progression to ventricular dilatation and decline in systolic performance (157).
The concomitant release of autophagy-related factors, as observed under high-glucose conditions, may contribute to cell death and cardiac dysfunction (158).The activation of PI3K/Akt/mTOR signaling pathway, instead, an essential regulator of cardiac autophagy (159), ameliorates hyperglycemia–induced cardiac hypertrophy (160). A study convincingly supports insulin signaling as a significant regulator of myocardial autophagy, mediating in early life its physiological postnatal suppression, thereby linking nutrient sensing to postnatal cardiac development (161).
Whether the autophagic responses are adaptive or maladaptive remains controversial. Likewise, the role of autophagy in diabetic heart has been not fully understood yet. Several reports show an increased/decreased/unchanged autophagy in the hearts of either humans or animals with T2DM (162). In a study on animal models, autophagic adaptations in DM-CMP seem remarkably different between T1DM and T2DM, being overactivated in the first, but suppressed in the second (163), but even on this topic data are controversial (164). Likely, autophagy regulates both cell survival and cell death in diabetic heart through a strict cross-talk with apoptotic pathways (152), and apoptosis is involved in DM-CMP mainly as a consequence of autophagy dysregulation (165, 166).
A significant increase of apoptosis and cell necrosis characterizes both animal models and patients with DM. Endomyocardial biopsies in diabetic patients with dilated cardiomyopathy show a 4-fold increase of necrosis in cardiomyocytes, 9-fold in endothelial cells, and 6-fold in fibroblasts as compared to their non-diabetic counterparts (167). Hyperglycemia-induced ROS production speeds up apoptosis, some of which is elicited by angiotensin II and glycosylation (168). Many other factors (e.g., mitochondrion damage, oxidative stress, ER stress, inflammation, and even fibrotic signaling) can activate either pro-apoptotic or necrosis signaling pathways in the diabetic heart (169).
The phenomenon of senescence is typically attributed to telomere shortening after repeated cell division. Currently, we know that senescence is also inducible by a series of pathogenic stimuli involved in apoptosis, such genotoxic, mitochondrial and oxidative stresses, as well as inflammation. Moreover, the accumulation of senescent cells can itself cause persistent inflammation and oxidative stress via a so called “senescence-associated secretory phenotype” leading to organic dysfunction (169). It is also well-known that senescent cells contribute to the outcome of a variety of cardiac diseases, including age-related and -unrelated cardiac diseases like DM-CMP (170). In this context, DM may impair the in vitro proliferation and differentiation potential of adult cardiac stem/progenitor cells, further worsening their senescence phenotype, even when compared to non-diabetic ischemic patients (171).
Inflammation
Likewise to the known contribution of inflammation to other HF etiologies, both systemic and local maladaptive inflammation responses are strongly concerned with the progression of DM-CMP (172, 173).
Exposure of heart to glucose or FA excess activates NFκB, a protein complex which controls DNA transcription and induces the expression of proinflammatory cytokines (IL6, pro-IL18, pro-IL1β, and TNF-α) and the assembly of NLR family pyrin domain-containing 3 (NLRP3) inflammasome (30, 51). Similarly, AGE/RAGE signaling promotes NF-κB activation and mediates an inflammatory reaction by heterodimerizing with toll-like receptor-4, thus leading to the production of NLRP3, pro-IL1β, and pro-IL18 (104). Activated NLRP3 inflammasome plays a crucial role in the pathogenesis of HF in diabetes, resulting in amplification and infiltration of inflammatory cell, whereas a decrease in NLRP3 attenuates cardiomyopathy in a T2DM rat model (174–176).
Monocytes/macrophages are leading players in DM-CMP pathogenesis. Particularly, macrophage proinflammatory M1 polarization is increased and macrophage M2 anti-inflammatory response inhibited in diabetic heart (177). The recruitment of these cells to sites of inflammation is induced by the C-C chemokine receptor type 2 (CCR2) (178), and macrophages derived from CCR2+ monocytes are required for adverse left ventricle remodeling (179). A recent study on mice demonstrated that the heart expression of CCR2 associated to persistent hyperglycemia leads to DM-CMP development, whereas the inhibition of this chemokine could inhibit oxidative stress and M1 macrophage infiltration in diabetic hearts (180).
Apart from macrophages, an involvement of neutrophil and lymphocyte regulation in DM-CMP has emerged. Chronic systemic inflammation in diabetes leads to leukocyte activation and recruitment to various organs with further inflammatory tissue remodeling over time ultimately evolving in fibrosis. At heart level, this may result in reduced cardiac output that ultimately stimulates further cardiac inflammation and fibrosis leading to dilation and established heart failure (181). These pathways may be critical to the discovery of new targeted therapies for controlling DM-CMP progression. As an example, the T cell-specific deletion of sphingosine 1-phosphate receptor 1 (S1PR1), as well as the administration of the S1PR1 antagonist FTY720, are able to exert protection against cardiac fibrosis in a streptozotocin-induced diabetic model (182, 183).
Recently, the role of adipokines (e.g., adiponectin) on the cardiovascular outcome has been well-described (184). Moreover, several less investigated mechanisms might be involved in cardiovascular inflammation (185–187).
Endothelial Dysfunction
Regardless of the relevance for both accelerated atherogenesis and microvascular diabetic complications, the impaired endothelial function of coronary microvessels is a key feature of DM-CMP (188), especially contributing to diastolic dysfunction and HFpEF (189).
The hallmark of ED is the impaired endothelium-mediated arterial vasodilation as a consequence of depressed bioavailability of nitric oxide (NO), a short-living mediator generated from L-arginine by endothelial NOS (eNOS) (190). During the early stages of IR and DM-CPM, the impaired NO-induced vasodilatation may be balanced by the either preserved or even enhanced endothelium-derived hyperpolarizing factor (EDHF)-mediated vasodilatation. Later, even this mechanism degenerates, thereby promoting microvascular dysfunction (30, 191).
Exposure of endothelial cells to excessive and/or fluctuating blood glucose levels can stimulate the generation of ROS and AGEs, with the consequent downregulation of eNOS and production of NO and cGMP (192, 193). In addition, superoxide anion inactivates NO by forming the more powerful oxidant peroxynitrite, thus triggering nitrosative stress and premature endothelial senescence (188).
The low NO bioavailability to adjacent cardiomyocytes decreases cGMP production and protein kinase G (PKG) activity, with consequent increased ratio of titin isoform N2B:N2BA expression and of intracellular Ca2+ content and sensitization. These changes result in a slow relaxation, high diastolic stiffness, and impaired cardiomyocyte elastance (194). As support to the relevance of this mechanism, PKG administration to cardiomyocytes isolated from DM-CMP patients with this phenotype corrects their high resting tension (99). Similar alterations have been observed in cardiomyocytes isolated from patients suffering from both aortic stenosis and DM (195). In addition, ED is associated with microvascular inflammation due to an increased expression of adhesion molecules and local infiltration and accumulation of macrophages expressing TGF-β. As a consequence, myocardial fibroblasts transform into myofibroblasts responsible of interstitial fibrosis (188). The role of TNF-alpha on ED has also been observed (196).
Notably, an increased albuminuria, marker of renal ED, is strictly related to a poor CV outcome in diabetic patients (197–200).
Microvascular Rarefaction
Similar defects in endothelium-dependent/independent vasodilation involve coronary microcirculation in both T1DM and T2DM patients (201). In addition, structural microvascular alterations impairing the capacity of coronary vascular bed independently of coronary atherosclerosis, may also contribute to DM-CMP (202).
In the myocardium of a well-recognized murine model of diabetes, a significant decline in microvessel density, reduced expression of selected VEGF isoforms, and increase in oxidative stress have been described, all significantly associated with measures of LV performance (203). In a study on patients with end-stage HF, capillary rarefaction and pericyte loss, accompanied by decreased contractility and increased stiffness, characterize diabetic human myocardial explants as compared to non-diabetic samples (204). In the same study, in vitro experiments on murine endothelial cells have shown that hyperglycemia attenuates tube formation, migration, and pericyte attraction upon proangiogenic stimulation (204). Moreover, the relative microvascular rarefaction resulting from cardiomyocyte hypertrophy is itself sufficient to induce cardiac fibrosis and diastolic dysfunction (205).
Autoimmunity
Immune inflammation is involved in the pathogenesis of myocarditis and cardiomyopathy (206). An immune biopathology has also been suggested in the pathogenesis of DM-CMP, especially in autoimmune-prone T1DM patients.
MI has been reported to induce sustained proinflammatory CD4+ T-cell and auto-antibody responses against α-cardiac myosin heavy chain, a major autoantigen in myocarditis, both in mice models and in patients with T1DM, but not in control mice and T2DM subjects. Shared cardiac myosin autoantibody signatures between post-MI in T1DM patients and non-diabetic patients with myocarditis also suggests a post-infarction autoimmune syndrome in T1DM patients (207).
Some authors suggested that the cardiac insults of severe diabetic ketoacidosis might initiate the synthesis of antibodies directed to cardiac self-antigens involved in the early immunopathogenesis of cardiomyopathy in young patients with T1DM (208). By measuring prevalence and profiles of cardiac autoantibodies in longitudinal samples of T1DM patients from the Diabetes Control and Complications Trial, poor glycemic control has been demonstrated as associated with cardiac autoimmunity, as shown by the presence of multiple cardiac autoantibody types (209).
Epigenetics
Epigenetics, the inheritable changes in gene expression without change of DNA sequences, represents a significant link between environmental exposure, as hyperglycemia, inflammation, and oxidative stress, and alterations in gene activity (210).
MicroRNAs (miRNAs) are a group of small, single-strand RNA molecules belonging to the non-coding RNA family, which affect their target genes at a post-transcriptional level by either inhibiting mRNA or degrading protein production (211), whose dysregulated expression is highly implicated in the pathophysiology of DM-CMP.
Some miRNAs abundantly expressed in cardiomyocytes, such as miR-1 and miR-133a, are reduced in T2DM patients (212). In streptozotocin-induced diabetic rats, miR-133a overexpression is able to improve myocardial contractility through the upregulation of tyrosine aminotransferase, a known regulator of norepinephrine production and β-adrenergic receptors (213). Jeyabal et al. found a considerably decreased miR-9 expression in high glucose-cultivated cardiomyocytes and human DM myocardium (214). Downregulation of miR-30c mediates the pro-hypertrophic effects of hyperglycemia in diabetic cardiomyopathy by upregulating Cdc42 and Pak1 genes (215). Li et al. established that miR-30d leads to cardiomyocyte pyroptosis in DM-CMP by direct repression of Foxo3a expression (216). Cardiac-enriched miR-1 and miR-206 are responsive to hyperglycemia and favor the apoptosis of cardiomyocytes through the negative regulation of the heath shock protein 60 (217). Recent evidence demonstrates that miR-208 and miR-499, together with miR-1 and miR-133, might play a role in the differentiation of stem cells into cardiomyocytes (218). A proposed role for miR-208 in diabetic heart disease is the regulation of myosin heavy chain gene expression (219).
Some literature suggests an involvement of exosomes in DM-CMP, the extracellular vesicles containing a variety of biological components, including miRNAs, proteins and lipids, which mediate the intercellular communication (220). The stress induced by hypoxia, inflammation, and hyperglycemia has been reported to increase protein and mRNA content in endothelial cell-derived exosomes, and the exosomes released from diabetic cardiomyocytes could deliver detrimental components able to initiate endothelial cell dysfunction and impair angiogenesis (30). Of note, heat shock protein 20-engineered exosomes exert beneficial effects via the modulation of cardiomyocyte exosome secretion with restoration of normal cardiac function under hyperglycemic conditions (221).
Long non-coding RNAs (lncRNAs) are non-protein coding transcripts longer than 200 nucleotides with both nuclear and cytoplasmic location which regulate gene expression through a variety of molecular mechanisms, including the interaction or competition with other RNAs, DNA binding proteins, and specific regulatory DNA sequences (222). Recently, the lncRNA H19 has been found remarkably reduced in a murine model of DM-CMP as a consequence of hyperglycemia, and to regulate cardiomyocyte apoptosis by targeting VDAC1, a mitochondrial porin involved in ATP transport (223).
Histone acetylation is a rapid and dynamic process mainly regulated by histone acetyltransferases (promoting gene transcription) and histone deacetylases (preventing gene transcription), which represent a major epigenetic mechanism whose deregulation may induce the development of several diabetic complications (224). BRD4, a histone acetylated reader protein which regulates either the activation or repression of gene transcription, has been recently identified as a critical mediator of hyperglycemia-induced cardiomyocyte hypertrophy and cardiac fibrosis through the AKT pathway (225).
A study in streptozotocin-induced diabetic rats has recently found that DNA methyltransferase-1 enhances cardiac fibroblast autophagy in diabetic cardiac fibrosis through inhibiting androgen receptor axis (226).
Activation of the Renin-Angiotensin-Aldosterone System
In a context of IR and hyperglycemia, the inappropriate activation of RAAS despite a state of salt and volume excess, plays an important role in the development of DM-CMP (30), whereas the RAAS block protects against cardiac damage (227).
Beyond receptors AT1 and AT2, Ang-II interacts with NOX, resulting in an overload of oxidants and free radicals in the body, with the subsequent exacerbation of oxidative stress and inflammation (169). This effect is supported by studies showing the effectiveness of ramipril in preventing upregulation of p47phox, p22phox, and reducing NADPH driven oxide production (228). Blocking of Ang-II also reduces the expression of p22phox, NOX and hyperglycemia-induced p47phox (229).
Activation of RAAS may induce systemic and cardiac IR through the mTOR–S6K1 signal transduction pathway (230). Meanwhile, enhanced angiotensin II type 1 receptor and mineralocorticoid receptor signaling in the myocardium enhance the adaptive proinflammatory immune response and inflammation, including increases in leukocyte adhesion, cytokine expression and macrophage infiltration (231).
Cardiovascular Autonomic Neuropathy
Diabetes is often associated to both neurosensorial damage and neuropathy (232). In particular, diabetic Cardiac Autonomic Neuropathy (CAN), in the absence of cardiac disease, seems associated with LV systolic and mainly diastolic dysfunction, even though it is difficult to assess its independent role among the multitude of factors involved in DM-CMP (233).
Due to an initial predominant parasympathetic denervation, excessive sympathetic activation in the early stages of diabetic CAN may promote LV hypertrophy, thus affecting both sympathovagal balance and baroreflexes (234). Moreover, an abnormal norepinephrine signaling may induce myocardial injury and LV remodeling via the cytotoxic effects of the increased catecholamine heart content observed in diabetic rat ventricles (235), eventually mediated by oxidative stress, inflammation, and apoptosis (236–238).
On the other hand, the sympathetic denervation associated to long-lasting diabetic CAN may impair β-adrenergic signaling and reduce myocardial contractile strength, relaxation kinetics, and diastolic distensibility (63, 239).
By changes in myocardial neurotransmitters, CAN may also alter myocardial blood flow and directly deteriorate LV function. A diastolic dysfunction associated to abnormal cardiac sympathetic function appears early in the course of T1DM, as assessed by cardiac sympathetic imaging (240). Among subjects with T2DM or IGT referred for elective coronary angiography, those suffering from CAN have a higher prevalence and a more severe form of LV diastolic dysfunction (241). In a study based on cardiac magnetic resonance imaging in a large cohort of patients with T1DM, the presence of CAN is associated with an increased LV mass and concentric remodeling (242).
Structural Changes In Diabetic Cardiomyopathy
The above-described detrimental pathways elicited by diabetes at a systemic level and in the myocardium itself collectively promote myocardial hypertrophy and interstitial fibrosis, the two structural hallmarks identified in animal models and patients with both T1DM and T2DM (32, 243). Depending on the combination patterns of these two structural changes, the clinical phenotype of DM-CMP varies from a subclinical diastolic dysfunction to diastolic HFpEF and, eventually, to systolic dysfunction and HFrEF (30, 244). In the HFpEF phenotype, the LV is usually hypertrophied and stiff with normal LV volume. At the cellular level, cardiomyocytes appear hypertrophied with a normal structure of the sarcomere accompanied by increased collagen deposition in the interstitial space. HFrEF phenotype is usually associated with increased LV volume due to dilation, and cardiomyocytes appear damaged, with loss of sarcomeres, and at times replaced by fibrosis (63).
Cardiac Hypertrophy
Epidemiological data report diabetes and high-sugar diets as risk factors for cardiac hypertrophy and other complications (63, 243–245), a condition highly prevalent (up to 56%) in asymptomatic T2DM patients (246–248). Cardiac hypertrophy is strongly associated with the progression to HF, particularly if hypertension coexists (249), and with a higher incidence of other clinical events, including stroke and sudden death (250).
Cardiac myocytes are differentiated cells which have lost the propensity of proliferation after birth. When exposed to high glucose stress, they increase in size by enhanced protein synthesis and addition of sarcomeres, but not in number, with a resulting greater length (eccentric hypertrophy) or width (concentric hypertrophy) (251). A re-expression of fetal genes has been observed, such as myosin heavy chain (β-MHC) and GATA-1, and activation of early response genes (252).
The microvascular endothelial dysfunction may contribute to the cardiomyocyte enlargement through the parallel addition of sarcomeres due to the removal of a NO-dependent brake on pro-hypertrophic stimuli (188). The increased thickness of ventricular walls in hypertrophied diabetic hearts may partly depend on ECM enlargement. Accordingly, abnormally increased myocardial echodensity, more likely related to collagen deposition, has been detected in asymptomatic diabetic patients with normal ventricular mass (253).
Hypertrophy and fibrosis are two coexisting structural aspects of DM-CMP, likely generated by common pathophysiological mechanisms. As an example, the loss of cardiomyocytes typical of the diabetic heart stimulates the resident cardiomyocytes to compensatively work and become hypertrophic, but at the same time it evocates inflammation pathways generating fibrosis.
Extracellular Remodeling and Interstitial Fibrosis
Myocardial fibrosis is a main pathological feature of the diabetic heart which involves both left and right ventricular walls, and can lead to cardiac remodeling, dilation and dysfunction, as well as to arrhythmias and, eventually, congestive HF (101) (Figure 2).
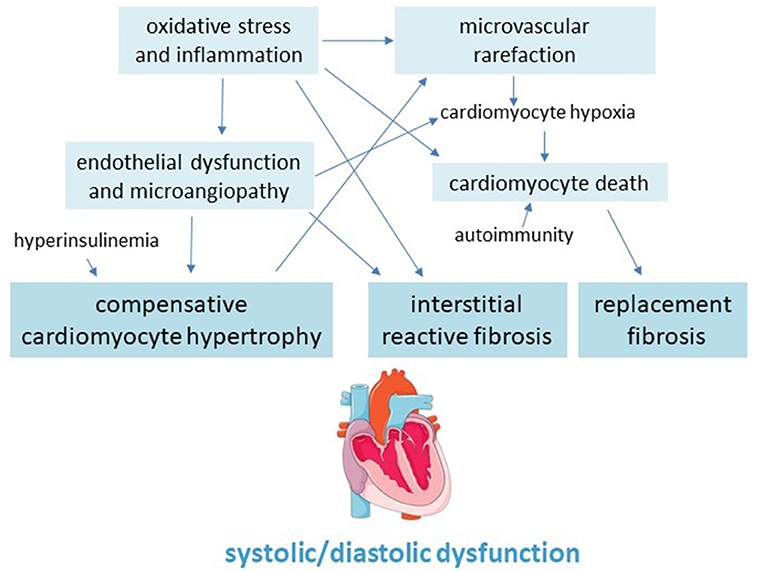
Figure 2. Pathogenic scheme of hypertrophy and fibrosis in the diabetic cardiomyopathy.
Cardiac fibroblasts, the primary matrix-producing cells in the myocardium, help maintaining ECM homeostasis in healthy hearts (254). The majority of resident cardiac fibroblasts responsible for fibrotic response arise from the embryonic epicardium. During development, these cells undergo epithelial-mesenchymal transition under the influence of several growth factors; subsequently, a portion of these mesenchymal cells invade the myocardium to become the resident cardiac fibroblasts. Studies have also revealed that cells of the endocardium, a specialized cardiac endothelial lining, and endothelial cells of the coronary vessel may migrate into the interstitium where they undergo endothelial-mesenchymal transition and respond to pro-fibrotic stimuli in a manner similar to resident fibroblasts. Other cells such as pericytes of cardiac vessels can differentiate into collagen-producing cells and may contribute to the fibroblast population following cardiac injury. Finally, the circulating fibrocytes are bone marrow-derived cells considered a potential source of fibroblasts in the fibrotic heart. They represent a unique fibroblast progenitor population that co-express fibroblast markers, along with typical hematopoietic markers (181).
Mechanical or bioactive pathological insults may induce the phenotypic transition of fibroblasts from a resting to an active state characterized by heightened proliferation, migration, contractility, and ECM production (255). A high activation of fibroblasts has been observed in hearts of db/db mice and atrial tissue derived from T2DM patients, resulting in a dynamic balance disorder of cardiac ECM synthesis and accumulation, along with an excessive collagen deposition (256–258).
A dysregulation of specific collagen degrading metalloproteinases (MMPs) and their tissue inhibitors (TIMPs), two crucial determinants of interstitial accumulation of secreted matrix proteins, also contributes to increased extracellular collagen content in the diabetic heart (259, 260). Recently, the enhanced expression of two isoforms of MMP-2 has been induced by high glucose in vitro and in a T1DM murine heart model (261).
The pathological processes referring to diabetes which mainly remodel ECM include hyperglycemia, AGE accumulation, inflammation, oxidative stress, and increased levels of neuro-hormones (258).
Excessive collagen deposition may derive from either hyper-expression of TGF-β or CTGF, two key modulators of collagen production. The former is either mediated by angiotensin II activation or induced by high glucose and leptin via increasing transcription, secretion, and activation (169, 262, 263). On the other hand, in a study on a murine model of obesity and IR given a diabetogenic diet for 11 weeks, cardiac fibroblasts acquired enhanced myofibroblastic/fibrotic gene expression but reduced responsiveness to TGF-β1 (264). Similarly, cardiac fibroblasts isolated from db/db mice exhibited elevated collagen synthesis but weakened TGF-β1 response (256).
Myocardium tissues of diabetic rats and cardiac fibroblasts treated with high glucose show a significant increased expression of the calcium sensing receptor (CaSR), a member of the C family of the G protein coupling receptor superfamily widely expressed in both prokaryotic and eukaryotic cells (265). A CaSR inhibitor may alleviate the myocardial fibrosis induced by high glucose (266).
The integrins, a family of transmembrane proteins able to integrate and transduce mechanical and biochemical signals, may have a key role in myocardial fibrosis by inducing myofibroblast differentiation (267). Collagen treated with methylglyoxal, a major cell-permeant precursor of AGEs, appears to initiate a forward-feedback loop where glycated ECM increases the expression of integrins. The stiffed myocardial matrix further activates integrins and up-regulates TGF-β, with worsened cardiac fibrosis. Indeed, the deletion of the α11 integrin in streptozotocin-treated diabetic animal models attenuates the cardiac fibrosis (64, 268).
In addition to fibroblasts, even fibrogenic actions by monocytes and macrophages, lymphocytes, endothelial cells and pericytes, mast cells, and cardiomyocytes may contribute to the diabetes-associated heart fibrosis (169).
Diabetes-Induced Left Ventricular Dysfunction
Even though some authors have postulated that HFpEF and HFrEF represent distinct phenotypes of DM-CMP (63), these clinical patterns are traditionally described as two stages occurring during diabetes progression. An early stage characterized by increased myocardial stiffness, enhanced atrial filling pressure and impaired diastolic function, may be followed, even though not so commonly, by a late stage of further impairment in diastolic function and appearance of a systolic dysfunction (269).
LV diastolic and systolic dysfunctions can be efficiently detected by echocardiography thanks to its large availability and low cost (243). Unfortunately, screening approaches including B-type natriuretic peptide, exercise stress testing, and more sensitive echocardiographic measurements, have not been fully validated yet to identify subclinical dysfunction in diabetic patients (270).
Diastolic Dysfunction
LV diastolic dysfunction displays from heart stiffening due to both myocardial fibrosis and hypertrophy (100) and represents the initial and most common functional deficit of diabetic heart, generally previous the appearance of systolic dysfunction (271, 272).
An impaired diastolic functioning of LV is detected in 40–75% of asymptomatic T1DM/T2DM patients by conventional echocardiography and tissue Doppler imaging, being characterized by a delayed and extended diastolic phase, with impaired early diastolic filling, prolongation of isovolumetric relaxation, increased atrial filling and increased myocardial stiffness, predominantly in late diastole (249).
Changes in diastolic function have also been widely reported in diabetic animals without evidence of heart disease by other factors (273). In a study of the 90s, diastolic dysfunction has been associated with aging, long duration of diabetes, increased blood pressure, interventricular septal thickness, dyslipidemia, and high HbA1c (274).
Systolic Dysfunction
As DM-CMP insidiously proceeds and eccentric cardiac remodeling develops, systolic dysfunction may appear, a condition associated with a poor prognosis with an annual mortality of 15–20% and a higher incidence of congestive HF and sudden death (249).
Defects in excitation-contraction coupling at the cardiomyocyte level, including impairment in cardiomyocyte contraction, relaxation, and cytosolic calcium trafficking, as well as epigenetic mechanisms and enhanced mitochondrial ROS generation, may all contribute to this progressive worsening (32, 124).
Even though systolic dysfunction usually follows diastolic dysfunction at a later stage of the DM-CMP course, some studies have detected systolic dysfunction in diabetic patients with normal diastolic function, suggesting that diastolic dysfunction may not necessarily be the first functional alteration (275). In a T2DM population with no documented cardiovascular disease and no signs of ischemia at stress test, asymptomatic LV dysfunction was detected in 262 patients. Among these, 27% had isolated systolic dysfunction and 16% isolated diastolic dysfunction (276).
Effects of Anti-hyperglycemic Drug Therapy on Heart Failure In Diabetes
Along with the classic outcome of major adverse CV events, recently published CV outcomes trials of anti-hyperglycemic drugs include analysis of HF data, especially the rate of hospitalization for this event.
The ancient drug metformin was absolutely contra-indicated in patients with HF until 2007 when FDA removed this limitation. The controversy regarding its safety and effectiveness in the setting of HF was resolved by the results of a later systematic review of observational studies including 34,000 patients favoring the metformin as the treatment of choice in patients with diabetes and HF (277).
In addition to raised concerns about increased MI, the use of the thiazolidinediones (TZDs) rosiglitazone and pioglitazone, was associated with fluid retention and increased risk of HF, as indicated by three randomized controlled trials, DREAM, ProACTIVE, and GSK211, reporting a respective relative risk of HF of 2.17 (95% CI 0.96–0.91), 1.49 (1.23–0.80), and 7.09 (1.60–0.96) (278). The main mechanisms accounting for TZD-related fluid retention is the PPAR-γ stimulation of EnaC-mediated renal salt absorption in the collecting duct, with the likely contribution of stimulation of sodium transporters in the proximal tubule. Concurrently, the reduction of systemic vascular resistance by TZD might expose the capillary networks to higher perfusion pressures thereby precipitating fluid extravasation. Additionally, TZDs increase the plasma concentration of the VEGF, a potent inducer of vascular permeability, further predisposing patients to oedema (279).
Three new classes of anti-hyperglycemic agents have been introduced in recent years.
The dipeptidyl peptidase-4 (DPP-4) inhibitors exhibited increased HF hospitalization in the SAVOR-TIMI 53 trial evaluating saxagliptin and in the secondary analysis of the EXAMINE trial for alogliptin. A recent pooled analysis illustrates that DPP-4 inhibitors do not increase the HF risk among T2DM patients with a previous history of HF, but they increased this risk among patients without history of HF (HR 1.21, 95% CI 1.04–1.41, p = 0.01), possibly because nearly all studied subjects had established CVD (280). Basic research suggests that the inhibition of DPP-4 may exert beneficial actions on heart, mainly by inhibiting the degradation of stromal cell-derive factor-1, a chemokine produced by stromal and endothelial cells that promotes regeneration and repair during organ damage, and that of GLP-1, thus restoring cardiac remodeling and apoptosis caused by the pathological decline in circulating GLP-1 in response to pressure overload (281, 282). On the other hand, since DPP-4 involves in the degradation of vasodilator factors and the NO-dependent mechanism, its inhibition can exert important systemic vasodilator effects that reduce heart load (283). Unfortunately, these beneficial results on animal studies were not replicated in humans.
The antagonists of the GLP-1 receptors (GLP-1RAs) represent the other incretin-based therapy potentiating endogenous GLP-1. Based on the evidence from RCTs, none of the six available GLP-1RAs has displayed benefits against HF, despite demonstration in animal models and humans of ameliorated endothelial dysfunction, improved myocardial function, and cardiomyocyte protection against glucolipotoxicity and ROS (280). A novel GLP-1RA, the oral hypoglycemic peptide 2 (OHP2), has demonstrated to protect against DM-CMP in high-fat diets and continuous streptozocin injection induced rat models. Both hyperlipidemia and myocardium lipid accumulation were decreased by OHP2 treatment. In addition, OHP2 reversed oxidative stress and mitochondrial dysfunction in diabetic hearts (284).
The inhibitors of the sodium glucose co-transporter 2 (SGLT2) are the first class of glucose-lowering agents that have demonstrated in large-scale studies an impressive reduction in the risk of serious new-onset HF events by ≈30% in T2DM patients with or without established CVD (285). Of note, in none of the trials this benefit is explained by the glycemic control. Several mechanisms have been postulated for such a striking cardioprotective effect. The primary action of SGLT2 inhibitors reducing sodium and glucose uptake in the nephron, leads to a decrease in preload and afterload through osmotic diuresis. Additional beneficial effects are improvement of the composition of proinflammatory and anti-inflammatory cytokines in the body, as well as reduction in cardiac fibrosis (286). Other potential cardioprotective mechanism includes the increase in hematocrit, determined by erythropoietin hyperproduction by renal fibroblasts when the stimuli of hyperglycemia and excess glucose reabsorption are removed, the increase in fasting levels of ketone bodies with enhanced utilization of this efficient metabolic fuel in the failing heart, and the inhibition of the sodium hydrogen exchanger-1 in the myocardium, whose overactivity may lead to increase in intracellular sodium and calcium (287).
Different Aspects of Cardiomyopathy In T1DM and T2DM
Various small and large animal models of T1DM and T2DM have been generated to investigate the impact of diabetes on the heart and a lot of clinical studies have been published in the last decades on DM-CMP. Nonetheless, the complex pathophysiology of this condition remains still less than fully clear. The topic is further complicated by the different etiology of T1DM and T2DM that make partially distinct the mechanisms involved in their cardiac dysfunction (288).
Although etiologically different, the two types of diabetes share common metabolic disturbances, including hyperglycemia, dyslipidemia and associated glucotoxicity, lipotoxicity, and oxidative stress that are the predominant pathological mechanisms driving the development of DM-CMP as determined by insulin deficiency in T1DM and insulin resistance in T2DM.
On the other side, the development of HF in T1DM appears more closely related to glycemic control than in T2DM as indicated by the reported different increase in HF risk, 30% in T1DM and 8% in T2DM patients, for each additional percentage point of HbA1c (270). Likely, a good metabolic control obtained by insulin therapy in patients with T1DM may normalizes the metabolic derangements induced by insulin deficiency and attenuate the detrimental effects of diabetes on the heart (288). Instead, the insulin resistance typical of T2DM leads to increase in circulating triacylglycerol levels and FA delivery to cardiomyocytes that result in impaired mitochondrial β-oxidation, with greater mitochondrial dysfunction and accumulation of toxic lipid metabolites in the heart of patients with T2DM than in patients with T1DM (30).
Differences in pathophysiology of heart damage between the two types of diabetes also result in different clinical pictures. In T2DM-associated DM-CMP, there is a prevalence of mechanisms mediating concentric LV remodeling and hypertrophy with increase in ventricular stiffness leading to diastolic dysfunction. The corresponding clinical features include reduced ventricular compliance with increased systemic and pulmonary venous pressures and congestion despite preserved systolic function. By contrast, T1DM-associated diabetic cardiomyopathy is characterized by cardiomyocyte loss, LV remodeling and increased myocardial collagen deposition, which increase LV end-diastolic volume and impair systolic function. As a consequence, symptoms of systolic dysfunction are more typical in patients with T1DM with earlier clinical manifestations of HFrEF (289).
A similar progression of DM-CMP has also emerged in preclinical studies in diabetic animal models. A study comparing cardiac performance in rat models of T1DM (streptozotocin induced) and T2DM (Zucker diabetic fatty rats) by a pressure-volume conductance catheter system, suggested that a decreased systolic performance and a delayed relaxation mainly characterize T1DM, whereas an increase in diastolic stiffness of the heart is more remarkably in T2DM (290). A recent study using speckle-tracking echocardiography with invasive hemodynamics for the detection of cardiac dysfunction in rat models of T1DM and T2DM confirmed these results (291). It was found that contractility and active relaxation were deteriorated to a greater extent in T1DM compared to T2DM. In contrast, diastolic stiffness was more pronounced in T2DM. Correspondingly, systolic function was markedly altered in T1DM but preserved in T2DM, a disease profile resembling that observed in T2DM patients with HFpEF.
Conclusion
Diabetic cardiomyopathy is a common complication of diabetes which deserves a special clinical attention due to its insidious subclinical progression that, in some cases, may culminate in a manifest and rapidly evolving HF burdened by a very poor outcome.
The main driving force of the pathological processes specific of DM-CMP is hyperglycemia, a factor centrally placed among multiple interwoven pathways involving complex cellular and molecular perturbations which affect both myocardial structure and function.
Despite the current large knowledge, the pathophysiology of DM-CMP development and progression is still far from being fully elucidated. Consequently, effective therapies targeting this diabetic complication are lacking.
In-depth knowledge of etiologic and pathogenic mechanisms is crucial for the development of target-specific treatments to reduce the risk of HF in diabetic patients. Since subclinical cardiac abnormalities could be reversible when early detected, prevention-oriented therapies can even hopefully be identified.
Author Contributions
TS and FS: conceptualization. TS, PP, RG, GA, AD, AC, EV, LR, and FS: investigation. TS: writing—original draft preparation. TS, FS, and PP: writing—review and editing. TS, FS, and RG: supervision. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AGE, advanced glycation and product; AMPK, 5′-AMP-activated protein kinase; CaSR, calcium sensing receptor; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CAN, cardiovascular autonomic neuropathy; CCR2, C-C chemokine receptor type 2; cGMP/PKG, cyclic guanosine 3′, 5′-monophosphate/ protein kinase G; CTGF, connective tissue growth factor; CV, cardiovascular; CVD, cardiovascular disease; DAG, diacylglycerol; DM, diabetes mellitus; DM-CMP, diabetic cardiomyopathy; ECM, extracellular matrix; ED, endothelial dysfunction; eNOS, endothelial NO synthase; ER, endoplasmic reticulum; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; HIF1α, hypoxia-inducible factor 1α; IGT, impaired glucose tolerance; FA, fatty acid; LV, left ventricle; FDA, Food and drug administration; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; lncRNAs, long non-coding RNAs; MI, myocardial infarction; MAPK, mitogen-activated protein kinase; miRNAs, MicroRNAs; MMPs, matrix metalloproteinases; NCX, Na+/Ca2+ exchanger; NFkB, nuclear factor kappa B; NO, nitric oxide; NOX, NADPH oxidase; NRLP3, NLR family pyrin domain-containing 3; O-GlcNAc, O-linked β-N-acetylglucosamine; PARP, poly ADP ribose polymerase; PGC-1α, PPAR-γ coactivator-1α; PI3K/Akt, phosphatidylinositol 3-kinase/protein kinase B; PKC, protein kinase C; PKG, protein kinase G; PPAR, peroxisome proliferator-activated receptor; RAAS, renin angiotensin aldosterone system; RAGEs, receptor for advanced glycation end products; ROS, reactive oxygen species; SERCA, sarcoplasmic/endoplasmic reticulum calcium-ATPase; SGLT2, sodium glucose co-transporter 2; sRAGE, soluble RAGE; S1PR1, sphingosine 1-phosphate receptor 1; SR, sarcoplasmic reticulum; SREBP-1c, sterol regulatory-element-binding protein-1c; T1DM, Type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TGF- β, transforming growth factor-β; TIMPs, tissue inhibitors MMPs; TZDs, thiazolidinediones; UCP, uncoupling protein; VEGF, vascular endothelial growth factor.
References
1. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. (1972) 30:595–602. doi: 10.1016/0002-9149(72)90595-4
2. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the framingham study. Am J Cardiol. (1974) 34:29–34. doi: 10.1016/0002-9149(74)90089-7
3. Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2018) 20:853–72. doi: 10.1002/ejhf.1170
4. He J, Ogden LG, Bazzano LA, Vupputuri S, Loria C, Whelton PK. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med. (2001) 161:996–1002. doi: 10.1001/archinte.161.7.996
5. Ahmad FS, Ning H, Rich JD, Yancy CW, Lloyd-Jones DM, Wilkins JT. Hypertension, obesity, diabetes, and heart failure-free survival: the cardiovascular disease lifetime risk pooling project. JACC Heart Fail. (2016) 4:911–9. doi: 10.1016/j.jchf.2016.08.001
6. Nichols GA, Gullion CM, Koro CE, Ephross SA, Brown JB. The incidence of congestive heart failure in type 2 diabetes: an update. Diabetes Care. (2004) 27:1879–84. doi: 10.2337/diacare.27.8.1879
7. Dinesh Shah A, Langenberg C, Rapsomaniki E, Denaxas S, Pujades-Rodriguez M, Gale CP, et al. Type 2 diabetes and incidence of a wide range of cardiovascular diseases: a cohort study in 1·9 million people. Lancet. (2015) 385(Suppl. 1):S86. doi: 10.1016/S0140-6736(15)60401-9
8. Boonman-de Winter LJ, Rutten FH, Cramer MJ, Landman MJ, Liem AH, Rutten GE, et al. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia. (2012) 55:2154–62. doi: 10.1007/s00125-012-2579-0
9. Konduracka E, Cieslik G, Galicka-Latala D, Rostoff P, Pietrucha A, Latacz P, et al. Myocardial dysfunction and chronic heart failure in patients with long-lasting type 1 diabetes: a 7-year prospective cohort study. Acta Diabetol. (2013) 50:597–606. doi: 10.1007/s00592-013-0455-0
10. Salem M, El Behery S, Adly A, Khalil D, El Hadidi E. Early predictors of myocardial disease in children and adolescents with type 1 diabetes mellitus. Pediatr Diabetes. (2009) 10:513–21. doi: 10.1111/j.1399-5448.2009.00517.x
11. Tocci G, Sciarretta S, Volpe M. Development of heart failure in recent hypertension trials. J Hypertens. (2008) 26:1477–86. doi: 10.1097/HJH.0b013e3282fe1d3d
12. Thrainsdottir IS Aspelund T Thorgeirsson G Gudnason V Hardarson T Malmberg K . The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. (2005) 28:612–6. doi: 10.2337/diacare.28.3.612
13. Ingelsson E, Arnlöv J, Sundström J, Zethelius B, Vessby B, Lind L. Novel metabolic risk factors for heart failure. J Am Coll Cardiol. (2005) 46:2054–60. doi: 10.1016/j.jacc.2005.07.059
14. Kristensen SL, Mogensen UM, Jhund PS, Petrie MC, Preiss D, Win S, et al. Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction: a report from the i-preserve trial (Irbesartan in heart failure with preserved ejection fraction). Circulation. (2017) 135:724–35. doi: 10.1161/CIRCULATIONAHA.116.024593
15. Cavender MA, Steg PG, Smith SC Jr, Eagle K, Ohman EM, Goto S, et al. REACH Registry Investigators. Impact of diabetes mellitus on hospitalization for heart failure, cardiovascular events, and death: outcomes at 4 years from the reduction of atherothrombosis for continued health (REACH) registry. Circulation. (2015) 132:923–31. doi: 10.1161/CIRCULATIONAHA.114.014796
16. Gustafsson I, Brendorp B, Seibaek M, Burchardt H, Hildebrandt P, Køber L, et al. Danish Investigatord of Arrhythmia and Mortality on Dofetilde Study Group. Influence of diabetes and diabetes-gender interaction on the risk of death in patients hospitalized with congestive heart failure. J Am Coll Cardiol. (2004) 43:771–7. doi: 10.1016/j.jacc.2003.11.024
17. Bertoni AG, Hundley WG, Massing MW, Bonds DE, Burke GL, Goff DC Jr. Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care. (2004) 27:699–703. doi: 10.2337/diacare.27.3.699
18. Corrao S, Santalucia P, Argano C, Djade CD, Barone E, Tettamanti M, et al. REPOSI Investigators. Gender-differences in disease distribution and outcome in hospitalized elderly: data from the REPOSI study. Eur J Intern Med. (2014) 25:617–23. doi: 10.1016/j.ejim.2014.06.027
19. Marcucci M, Franchi C, Nobili A, Mannucci PM, Ardoino I, REPOSI Investigators. Defining aging phenotypes and related outcomes: clues to recognize frailty in hospitalized older patients. J Gerontol A Biol Sci Med Sci. (2017) 72:395–402. doi: 10.1093/gerona/glw188
20. Lenti MV, Pasina L, Cococcia S, Cortesi L, Miceli E, Caccia Dominioni C, et al. Mortality rate and risk factors for gastrointestinal bleeding in elderly patients. Eur J Intern Med. (2019) 61:54–61. doi: 10.1016/j.ejim.2018.11.003
21. MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, et al. CHARM Investigators. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart failure: assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J. (2008) 29:1377–85. doi: 10.1093/eurheartj/ehn153
22. Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Heart J. (2018) 39:4243–54. doi: 10.1093/eurheartj/ehy596
23. Yusuf S, Ostergren JB, Gerstein HC, Pfeffer MA, Swedberg K, Granger CB, et al. Candesartan in Heart Failure-Assessment of Reduction in Mortality and Morbidity Program Investigators. Effects of candesartan on the development of a new diagnosis of diabetes mellitus in patients with heart failure. Circulation. (2005) 112:48–53. doi: 10.1161/CIRCULATIONAHA.104.528166
24. Matsue Y, Suzuki M, Nakamura R, Abe M, Ono M, Yoshida S, et al. Prevalence and prognostic implications of pre-diabetic state in patients with heart failure. Circ J. (2011) 75:2833–9. doi: 10.1253/circj.CJ-11-0754
25. Nieminen MS, Brutsaert D, Dickstein K, Drexler H, Follath F, Harjola VP, et al. EuroHeart Survey Investigators; Heart Failure Association, European Society of Cardiology. EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J. (2006) 27:2725–36. doi: 10.1093/eurheartj/ehl193
26. Sarma S, Mentz RJ, Kwasny MJ, Fought AJ, Huffman M, Subacius H, et al. EVEREST investigators. Association between diabetes mellitus and post-discharge outcomes in patients hospitalized with heart failure: findings from the EVEREST trial. Eur J Heart Fail. (2013) 15:194–202. doi: 10.1093/eurjhf/hfs153
27. Coats AJ, Anker SD. Insulin resistance in chronic heart failure. J Cardiovasc Pharmacol. (2000) 35(7 Suppl. 4):S9–14. doi: 10.1097/00005344-200000004-00002
28. Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. (2012) 125:2844–53. doi: 10.1161/CIRCULATIONAHA.111.060889
29. Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO, et al. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. (2005) 46:1019–26. doi: 10.1016/j.jacc.2005.02.093
30. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. (2018) 61:21–8. doi: 10.1007/s00125-017-4390-4
31. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
32. Marwick TH, Ritchie R, Shaw JE, Kaye D. Implications of underlying mechanisms for the recognition and management of diabetic cardiomyopathy. J Am Coll Cardiol. (2018) 71:339–51. doi: 10.1016/j.jacc.2017.11.019
33. Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ, Chen HH. The prevalence of diabetic cardiomyopathy: a population-based study in Olmsted County, Minnesota. J Card Fail. (2014) 20:304–9. doi: 10.1016/j.cardfail.2014.02.007
34. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0044-6
35. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.00006.2004
36. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. (2004) 1015:202–13. doi: 10.1196/annals.1302.017
37. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
38. Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem. (1997) 246:259–73. doi: 10.1111/j.1432-1033.1997.00259.x
39. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. (2002) 109:121–30. doi: 10.1172/JCI0214080
40. Opie LH, Knuuti J. The adrenergic-fatty acid load in heart failure. J Am Coll Cardiol. (2009) 54:1637–46. doi: 10.1016/j.jacc.2009.07.024
41. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. (2004) 18:1692–700. doi: 10.1096/fj.04-2263com
42. Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. (2010) 1801:311–9. doi: 10.1016/j.bbalip.2009.09.023
43. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. (2013) 10:109–21. doi: 10.1007/s11897-013-0133-0
44. Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, et al. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. (1997) 30:527–32. doi: 10.1016/S0735-1097(97)00185-X
45. Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol. (2013) 305:H1693–7. doi: 10.1152/ajpheart.00854.2012
46. Taegtmeyer H. Failing heart and starving brain: ketone bodies to the rescue. Circulation. (2016) 134:265–6. doi: 10.1161/CIRCULATIONAHA.116.022141
47. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
48. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
49. Joseph D, Kimar C, Symington B, Milne R, Essop MF. The detrimental effects of acute hyperglycemia on myocardial glucose uptake. Life Sci. (2014) 105:31–42. doi: 10.1016/j.lfs.2014.04.009
50. Battault S, Renguet E, Van Steenbergen A, Horman S, Beauloye C, Bertrand L. Myocardial glucotoxicity: mechanisms and potential therapeutic targets. Arch Cardiovasc Dis. (2020) 113:736–48. doi: 10.1016/j.acvd.2020.06.006
51. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
52. Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci. (2017) 24:5. doi: 10.1186/s12929-016-0309-5
53. Razeghi P, Young ME, Cockrill TC, Frazier OH, Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation. (2002) 106:407–11. doi: 10.1161/01.CIR.0000026392.80723.DC
54. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. (2009) 54:1891–8. doi: 10.1016/j.jacc.2009.07.031
55. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. (2014) 103:485–97. doi: 10.1093/cvr/cvu156
56. Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, et al. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J. (2001) 15:833–45. doi: 10.1096/fj.00-0351com
57. Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart–deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. (2013) 2:e000433. doi: 10.1161/JAHA.113.000433
58. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. (2008) 102:401–14. doi: 10.1161/CIRCRESAHA.107.165472
59. Turner JD, Gaspers LD, Wang G, Thomas AP. Uncoupling protein-2 modulates myocardial excitation-contraction coupling. Circ Res. (2010) 106:730–8. doi: 10.1161/CIRCRESAHA.109.206631
60. Harmancey R, Taegtmeyer H. The complexities of diabetic cardiomyopathy: lessons from patients and animal models. Curr Diab Rep. (2008) 8:243–8. doi: 10.1007/s11892-008-0042-x
61. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. (2012) 15:805–12. doi: 10.1016/j.cmet.2012.04.006
62. Atkinson LL, Kozak R, Kelly SE, Onay Besikci A, Russell JC, Lopaschuk GD. Potential mechanisms and consequences of cardiac triacylglycerol accumulation in insulin-resistant rats. Am J Physiol Endocrinol Metab. (2003) 284:E923–30. doi: 10.1152/ajpendo.00360.2002
63. Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. (2015) 36:1718–27. doi: 10.1093/eurheartj/ehv134
64. Meagher P, Adam M, Civitarese R, Bugyei-Twum A, Connelly KA. Heart failure with preserved ejection fraction in diabetes: mechanisms and management. Can J Cardiol. (2018) 34:632–43. doi: 10.1016/j.cjca.2018.02.026
65. Ljubkovic M, Gressette M, Bulat C, Cavar M, Bakovic D, Fabijanic D, et al. Disturbed fatty acid oxidation, endoplasmic reticulum stress, and apoptosis in left ventricle of patients with type 2 diabetes. Diabetes. (2019) 68:1924–33. doi: 10.2337/db19-0423
66. Marfella R, Amarelli C, Cacciatore F, Balestrieri ML, Mansueto G, D'Onofrio N, et al. Lipid accumulation in hearts transplanted from nondiabetic donors to diabetic recipients. J Am Coll Cardiol. (2020) 75:1249–62. doi: 10.1016/j.jacc.2020.01.018
67. Marfella R, Sasso FC, Cacciapuoti F, Portoghese M, Rizzo MR, Siniscalchi M, et al. Tight glycemic control may increase regenerative potential of myocardium during acute infarction. J Clin Endocrinol Metab. (2012) 97:933–42. doi: 10.1210/jc.2011-2037
68. Marfella R, Sasso FC, Siniscalchi M, Paolisso P, Rizzo MR, Ferraro F, et al. Peri-procedural tight glycemic control during early percutaneous coronary intervention is associated with a lower rate of in-stent restenosis in patients with acute ST-elevation myocardial infarction. J Clin Endocrinol Metab. (2012) 97:2862–71. doi: 10.1210/jc.2012-1364
69. Sasso FC, Rinaldi L, Lascar N, Marrone A, Pafundi PC, Adinolfi LE, et al. Role of tight glycemic control during acute coronary syndrome on CV outcome in type 2 diabetes. J Diabetes Res. (2018) 2018:3106056. doi: 10.1155/2018/3106056
70. Lind M, Bounias I, Olsson M, Gudbjörnsdottir S, Svensson AM, Rosengren A. Glycaemic control and incidence of heart failure in 20,985 patients with type 1 diabetes: an observational study. Lancet. (2011) 378:140–6. doi: 10.1016/S0140-6736(11)60471-6
71. Iribarren C, Karter AJ, Go AS, Ferrara A, Liu JY, Sidney S, et al. Glycemic control and heart failure among adult patients with diabetes. Circulation. (2001) 103:2668–73. doi: 10.1161/01.CIR.103.22.2668
72. Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microfibros of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. (2000) 321:405–12. doi: 10.1136/bmj.321.7258.405
73. Wolff SP. Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br Med Bull. (1993) 49:642–52. doi: 10.1093/oxfordjournals.bmb.a072637
74. Giugliano D, Ceriello A, Paolisso G. Diabetes mellitus, hypertension, and cardiovascular disease: which role for oxidative stress? Metabolism. (1995) 44:363–8. doi: 10.1016/0026-0495(95)90167-1
75. Evangelista I, Nuti R, Picchioni T, Dotta F, Palazzuoli A. Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. Int J Mol Sci. (2019) 20:3264. doi: 10.3390/ijms20133264
76. Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC, et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic Biol Med. (2010) 48:1182–7. doi: 10.1016/j.freeradbiomed.2010.01.038
77. Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem. (2003) 278:33972–7. doi: 10.1074/jbc.M303734200
78. Cai L, Wang J, Li Y, Sun X, Wang L, Zhou Z, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. (2005) 54:1829–37. doi: 10.2337/diabetes.54.6.1829
79. Zhang Y, Babcock SA, Hu N, Maris JR, Wang H, Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3β and mitochondrial function. BMC Med. (2012) 10:40. doi: 10.1186/1741-7015-10-40
80. Ren J, Duan J, Thomas DP, Yang X, Sreejayan N, Sowers JR, et al. IGF-I alleviates diabetes-induced RhoA activation, eNOS uncoupling, and myocardial dysfunction. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R793–802. doi: 10.1152/ajpregu.00713.2007
81. Sykiotis GP, Habeos IG, Samuelson AV, Bohmann D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr Opin Clin Nutr Metab Care. (2011) 14:41–8. doi: 10.1097/MCO.0b013e32834136f2
82. Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. (2007) 64:1105–27. doi: 10.1007/s00018-007-6484-5
83. Xu Z, Wang S, Ji H, Zhang Z, Chen J, Tan Y, et al. Broccoli sprout extract prevents diabetic cardiomyopathy via Nrf2 activation in db/db T2DM mice. Sci Rep. (2016) 6:30252. doi: 10.1038/srep30252
84. Sun Y, Zhou S, Guo H, Zhang J, Ma T, Zheng Y, et al. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism. (2020) 102:154002. doi: 10.1016/j.metabol.2019.154002
85. Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev. (2014) 19:49–63. doi: 10.1007/s10741-013-9374-y
86. Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, et al. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. (2004) 53:463–73. doi: 10.2337/diabetes.53.2.463
87. Kranstuber AL, Del Rio C, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, et al. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front Physiol. (2012) 3:292. doi: 10.3389/fphys.2012.00292
88. Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. (2013) 25:2185–97. doi: 10.1016/j.cellsig.2013.06.013
89. Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. (2006) 113:1235–43. doi: 10.1161/CIRCULATIONAHA.105.581397
90. Tikellis C, Thomas MC, Harcourt BE, Coughlan MT, Pete J, Bialkowski K, et al. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am J Physiol Endocrinol Metab. (2008) 295:E323–30. doi: 10.1152/ajpendo.00024.2008
91. Fukami K, Yamagishi S, Okuda S. Role of AGEs-RAGE system in cardiovascular disease. Curr Pharm Des. (2014) 20:2395–402. doi: 10.2174/13816128113199990475
92. Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. (2012) 29:613–8. doi: 10.3892/ijmm.2012.891
93. Della Corte CM, Ciaramella V, Di Mauro C, Castellone MD, Papaccio F, Fasano M, et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget. (2016) 7:4265–78. doi: 10.18632/oncotarget.6559
94. Morgillo F, Fasano M, Della Corte CM, Sasso FC, Papaccio F, Viscardi G, et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: a multicentre, open-label phase I-II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open. (2017) 2:e000132. doi: 10.1136/esmoopen-2016-000132
95. Donaldson C, Taatjes DJ, Zile M, Palmer B, VanBuren P, Spinale F, et al. Combined immunoelectron microscopic and computer-assisted image analyses to detect advanced glycation end-products in human myocardium. Histochem Cell Biol. (2010) 134:23–30. doi: 10.1007/s00418-010-0706-x
96. Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res. (2014) 114:872–88. doi: 10.1161/CIRCRESAHA.114.302533
97. Goh SY, Cooper ME. Clinical review: the role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. (2008) 93:1143–52. doi: 10.1210/jc.2007-1817
98. Berg TJ, Snorgaard O, Faber J, Torjesen PA, Hildebrandt P, Mehlsen J, et al. Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care. (1999) 22:1186–90. doi: 10.2337/diacare.22.7.1186
99. van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. (2012) 126:830–9. doi: 10.1161/CIRCULATIONAHA.111.076075
100. Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. (2000) 32:1805–19. doi: 10.1006/jmcc.2000.1215
101. van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. (2008) 117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
102. Zieman SJ, Melenovsky V, Clattenburg L, Corretti MC, Capriotti A, Gerstenblith G, et al. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens. (2007) 25:577–83. doi: 10.1097/HJH.0b013e328013e7dd
103. Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. (2003) 92:785–92. doi: 10.1161/01.RES.0000065620.39919.20
104. Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med. (2009) 13:1751–64. doi: 10.1111/j.1582-4934.2008.00547.x
105. Selvin E, Halushka MK, Rawlings AM, Hoogeveen RC, Ballantyne CM, Coresh J, et al. sRAGE and risk of diabetes, cardiovascular disease, and death. Diabetes. (2013) 62:2116–21. doi: 10.2337/db12-1528
106. Lazo M, Halushka MK, Shen L, Maruthur N, Rebholz CM, Rawlings AM, et al. Soluble receptor for advanced glycation end products and the risk for incident heart failure: the Atherosclerosis Risk in Communities Study. Am Heart J. (2015) 170:961–7. doi: 10.1016/j.ahj.2015.08.008
107. Pei Z, Deng Q, Babcock SA, He EY, Ren J, Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicol Lett. (2018) 284:10–20. doi: 10.1016/j.toxlet.2017.11.018
108. Gabbay KH. The sorbitol pathway and the complications of diabetes. N Engl J Med. (1973) 288:831–6. doi: 10.1056/NEJM197304192881609
109. Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA. (1992) 89:11059–63. doi: 10.1073/pnas.89.22.11059
110. Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. (1999) 99:384–91. doi: 10.1161/01.CIR.99.3.384
111. Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. (1998) 47:859–66. doi: 10.2337/diabetes.47.6.859
112. Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. (2006) 47:693–700. doi: 10.1016/j.jacc.2005.09.050
113. Way KJ, Isshiki K, Suzuma K, Yokota T, Zvagelsky D, Schoen FJ, et al. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes. (2002) 51:2709–18. doi: 10.2337/diabetes.51.9.2709
114. Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, et al. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail. (2009) 2:129–37. doi: 10.1161/CIRCHEARTFAILURE.108.765750
115. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. (2007) 446:1017–22. doi: 10.1038/nature05815
116. Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, et al. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacol Res. (2017) 116:45–56. doi: 10.1016/j.phrs.2016.12.016
117. Fülöp N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, et al. Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol. (2007) 292:C1370–8. doi: 10.1152/ajpcell.00422.2006
118. Marsh SA, Collins HE, Chatham JC. Protein O-GlcNAcylation and cardiovascular (patho)physiology. J Biol Chem. (2014) 289:34449–56. doi: 10.1074/jbc.R114.585984
119. Chen X, Zhang L, He H, Sun Y, Shen Q, Shi L. Increased O-GlcNAcylation induces myocardial hypertrophy. Vitro Cell Dev Biol Anim. (2020) 56:735–43. doi: 10.1007/s11626-020-00503-z
120. Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, et al. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. (2005) 96:1006–13. doi: 10.1161/01.RES.0000165478.06813.58
121. Ma J, Banerjee P, Whelan SA, Liu T, Wei AC, Ramirez-Correa G, et al. Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J Proteome Res. (2016) 15:2254–64. doi: 10.1021/acs.jproteome.6b00250
122. Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology. (2006) 21:250–8. doi: 10.1152/physiol.00008.2006
123. Phan TT, Abozguia K, Nallur Shivu G, Mahadevan G, Ahmed I, Williams L, et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol. (2009) 54:402–9. doi: 10.1016/j.jacc.2009.05.012
124. Lebeche D, Davidoff AJ, Hajjar RJ. Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat Clin Pract Cardiovasc Med. (2008) 5:715–24. doi: 10.1038/ncpcardio1347
125. Dhalla NS, Pierce GN, Innes IR, Beamish RE. Pathogenesis of cardiac dysfunction in diabetes mellitus. Can J Cardiol. (1985) 1:263–81.
126. Stühlinger MC, Abbasi F, Chu JW, Lamendola C, McLaughlin TL, Cooke JP, et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA. (2002) 287:1420–6. doi: 10.1001/jama.287.11.1420
127. Krüger M, Babicz K, von Frieling-Salewsky M, Linke WA. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J Mol Cell Cardiol. (2010) 48:910–6. doi: 10.1016/j.yjmcc.2010.02.012
128. Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J Am Coll Cardiol. (2008) 51:93–102. doi: 10.1016/j.jacc.2007.10.021
129. Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. (2003) 107:448–54. doi: 10.1161/01.CIR.0000045671.62860.98
130. Kishi S, Gidding SS, Reis JP, Colangelo LA, Venkatesh BA, Armstrong AC, et al. Association of insulin resistance and glycemic metabolic abnormalities with LV structure and function in middle age: the CARDIA study. JACC Cardiovasc Imaging. (2017) 10:105–14. doi: 10.1016/j.jcmg.2016.02.033
131. Velagaleti RS, Gona P, Chuang ML, Salton CJ, Fox CS, Blease SJ, et al. Relations of insulin resistance and glycemic abnormalities to cardiovascular magnetic resonance measures of cardiac structure and function: the Framingham Heart Study. Circ Cardiovasc Imaging. (2010) 3:257–63. doi: 10.1161/CIRCIMAGING.109.911438
132. Heckbert SR, Post W, Pearson GD, Arnett DK, Gomes AS, Jerosch-Herold M, et al. Traditional cardiovascular risk factors in relation to left ventricular mass, volume, and systolic function by cardiac magnetic resonance imaging: the Multiethnic Study of Atherosclerosis. J Am Coll Cardiol. (2006) 48:2285–92. doi: 10.1016/j.jacc.2006.03.072
133. Cozzolino D, Sessa G, Salvatore T, Sasso FC, Giugliano D, Lefebvre PJ, et al. The involvement of the opioid system in human obesity: a study in normal weight relatives of obese people. J Clin Endocrinol Metab. (1996) 81:713–8. doi: 10.1210/jcem.81.2.8636293
134. Cozzolino D, Sasso FC, Salvatore T, Torella M, Gentile S, Torella R, et al. Acute effects of beta-endorphin on cardiovascular function in patients with mild to moderate chronic heart failure. Am Heart J. (2004) 148:E13. doi: 10.1016/j.ahj.2004.01.029
135. Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. (2017) 121:181–95. doi: 10.1161/CIRCRESAHA.117.310230
136. Yilmaz S, Canpolat U, Aydogdu S, Abboud HE. Diabetic cardiomyopathy; summary of 41 years. Korean Circ J. (2015) 45:266–72. doi: 10.4070/kcj.2015.45.4.266
137. Trost SU, Belke DD, Bluhm WF, Meyer M, Swanson E, Dillmann WH. Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes. (2002) 51:1166–71. doi: 10.2337/diabetes.51.4.1166
138. Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. (2006) 55:608–15. doi: 10.2337/diabetes.55.03.06.db05-1284
139. Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. (2010) 48:322–30. doi: 10.1016/j.yjmcc.2009.10.016
140. Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. (2019) 127:246–59. doi: 10.1016/j.yjmcc.2019.01.001
141. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. (2013) 502:372–6. doi: 10.1038/nature12537
142. Daniels LJ, Wallace RS, Nicholson OM, Wilson GA, McDonald FJ, Jones PP, et al. Inhibition of calcium/calmodulin-dependent kinase II restores contraction and relaxation in isolated cardiac muscle from type 2 diabetic rats. Cardiovasc Diabetol. (2018) 17:89. doi: 10.1186/s12933-018-0732-x
143. Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta. (2011) 1813:1351–9. doi: 10.1016/j.bbamcr.2011.01.014
144. Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, et al. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. (2013) 45:438–47. doi: 10.1016/j.biocel.2012.09.017
145. Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate induces ER stress and autophagy in H9c2 cells: implications for apoptosis and adiponectin resistance. J Cell Physiol. (2015) 230:630–9. doi: 10.1002/jcp.24781
146. Prola A, Nichtova Z, Pires Da Silva J, Piquereau J, Monceaux K, Guilbert A, et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc Res. (2019) 115:328–42. doi: 10.1093/cvr/cvy197
147. Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, et al. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. (2009) 58:2863–72. doi: 10.2337/db09-0158
148. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The Role of Autophagy in the Heart. Annu Rev Physiol. (2018) 80:1–26. doi: 10.1146/annurev-physiol-021317-121427
149. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. (2015) 1852:252–61. doi: 10.1016/j.bbadis.2014.05.020
150. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. (2013) 288:18077–92. doi: 10.1074/jbc.M113.474650
151. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
152. Ouyang C, You J, Xie Z. The interplay between autophagy and apoptosis in the diabetic heart. J Mol Cell Cardiol. (2014) 71:71–80. doi: 10.1016/j.yjmcc.2013.10.014
153. Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. (2007) 13:482–91. doi: 10.1016/j.molmed.2007.08.004
154. Linton PJ, Gurney M, Sengstock D, Mentzer RM Jr, Gottlieb RA. This old heart: cardiac aging and autophagy. J Mol Cell Cardiol. (2015) 83:44–54. doi: 10.1016/j.yjmcc.2014.12.017
155. Bellot GL, Liu D, Pervaiz S. ROS, autophagy, mitochondria and cancer: Ras, the hidden master? Mitochondrion. (2013) 13:155–62. doi: 10.1016/j.mito.2012.06.007
156. Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. (2015) 1852:243–51. doi: 10.1016/j.bbadis.2014.05.005
157. Rifki OF Hill JA. Cardiac autophagy: good with the bad. J Cardiovasc Pharmacol. (2012) 60:248–52. doi: 10.1097/FJC.0b013e3182646cb1
158. Liu J, Tang Y, Feng Z, Hou C, Wang H, Yan J, et al. Acetylated FoxO1 mediates high-glucose induced autophagy in H9c2 cardiomyoblasts: regulation by a polyphenol -(-)-epigallocatechin-3-gallate. Metabolism. (2014) 63:1314–23. doi: 10.1016/j.metabol.2014.06.012
159. Pei H, Wang W, Zhao D, Su H, Su G, Zhao Z. G protein-coupled estrogen receptor 1 inhibits angiotensin II-induced cardiomyocyte hypertrophy via the regulation of PI3K-Akt-mTOR signalling and autophagy. Int J Biol Sci. (2019) 15:81–92. doi: 10.7150/ijbs.28304
160. Zhao LG, Li PL, Dai Y, Deng JL, Shan MY, Chen B, et al. Mibefradil alleviates high-glucose-induced cardiac hypertrophy by inhibiting PI3K/Akt/mTOR-mediated autophagy. J Cardiovasc Pharmacol. (2020) 76:246–54. doi: 10.1097/FJC.0000000000000844
161. Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest. (2013) 123:5319–33. doi: 10.1172/JCI71171
162. Delbridge LM, Mellor KM, Taylor DJ, Gottlieb RA. Myocardial autophagic energy stress responses–macroautophagy, mitophagy, and glycophagy. Am J Physiol Heart Circ Physiol. (2015) 308:H1194–204. doi: 10.1152/ajpheart.00002.2015
163. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. (2015) 11:1146–60. doi: 10.1080/15548627.2015.1051295
164. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. (2011) 50:1035–43. doi: 10.1016/j.yjmcc.2011.03.002
165. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. (2002) 51:1938–48. doi: 10.2337/diabetes.51.6.1938
166. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. (2013) 62:1270–81. doi: 10.2337/db12-0533
167. Kuethe F, Sigusch HH, Bornstein SR, Hilbig K, Kamvissi V, Figulla HR. Apoptosis in patients with dilated cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy? Horm Metab Res. (2007) 39:672–6. doi: 10.1055/s-2007-985823
168. Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal-Ginard B, et al. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes. (2001) 50:2363–75. doi: 10.2337/diabetes.50.10.2363
169. Hu X, Bai T, Xu Z, Liu Q, Zheng Y, Cai L. Pathophysiological fundamentals of diabetic cardiomyopathy. Compr Physiol. (2017) 7:693–711. doi: 10.1002/cphy.c160021
170. Cianflone E, Torella M, Biamonte F, De Angelis A, Urbanek K, Costanzo FS, et al. Targeting cardiac stem cell senescence to treat cardiac aging and disease. Cells. (2020) 9:1558. doi: 10.3390/cells9061558
171. Vecellio M, Spallotta F, Nanni S, Colussi C, Cencioni C, Derlet A, et al. The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in the human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes. (2014) 63:2132–47. doi: 10.2337/db13-0731
172. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. (2015) 131:1019–30. doi: 10.1161/CIRCULATIONAHA.114.008788
173. Frati G, Schirone L, Chimenti I, Yee D, Biondi-Zoccai G, Volpe M, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. (2017) 113:378–88. doi: 10.1093/cvr/cvx011
174. Li J, Ma W, Yue G, Tang Y, Kim IM, Weintraub NL, et al. Cardiac proteasome functional insufficiency plays a pathogenic role in diabetic cardiomyopathy. J Mol Cell Cardiol. (2017) 102:53–60. doi: 10.1016/j.yjmcc.2016.11.013
175. Parim B, Sathibabu Uddandrao VV, Saravanan G. Diabetic cardiomyopathy: molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart Fail Rev. (2019) 24:279–99. doi: 10.1007/s10741-018-9749-1
176. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE. (2014) 9:e104771. doi: 10.1371/journal.pone.0104771
177. Lee WS, Kim J. Diabetic cardiomyopathy: where we are and where we are going. Korean J Intern Med. (2017) 32:404–21. doi: 10.3904/kjim.2016.208
178. França CN, Izar MCO, Hortêncio MNS, do Amaral JB, Ferreira CES, Tuleta ID, et al. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci. (2017) 131:1215–24. doi: 10.1042/CS20170009
179. Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, et al. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci. (2018) 3:230–44. doi: 10.1016/j.jacbts.2017.12.006
180. Tan X, Hu L, Shu Z, Chen L, Li X, Du M, et al. Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes. (2019) 68:2063–73. doi: 10.2337/db18-1231
181. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
182. Abdullah CS, Li Z, Wang X, Jin ZQ. Depletion of T lymphocytes ameliorates cardiac fibrosis in streptozotocin-induced diabetic cardiomyopathy. Int Immunopharmacol. (2016) 39:251–64. doi: 10.1016/j.intimp.2016.07.027
183. Abdullah CS, Jin ZQ. Targeted deletion of T-cell S1P receptor 1 ameliorates cardiac fibrosis in streptozotocin-induced diabetic mice. FASEB J. (2018) 32:5426–35. doi: 10.1096/fj.201800231R
184. Sasso FC, Pafundi PC, Marfella R, Calabrò P, Piscione F, Furbatto F, et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: the prospective AIRE study. Cardiovasc Diabetol. (2019) 18:24. doi: 10.1186/s12933-019-0826-0
185. Marfella R, D' Amico M, Di Filippo C, Siniscalchi M, Sasso FC, Ferraraccio F, et al. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc Diabetol. (2007) 6:35. doi: 10.1186/1475-2840-6-35
186. Marfella R, Ferraraccio F, Rizzo MR, Portoghese M, Barbieri M, Basilio C, et al. Innate immune activity in plaque of patients with untreated and L-thyroxine-treated subclinical hypothyroidism. J Clin Endocrinol Metab. (2011) 96:1015–20. doi: 10.1210/jc.2010-1382
187. Torella D, Ellison GM, Torella M, Vicinanza C, Aquila I, Iaconetti C, et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J Am Heart Assoc. (2014) 3:e000434. doi: 10.1161/JAHA.113.000434
188. Gamrat A, Surdacki MA, Chyrchel B, Surdacki A. Endothelial dysfunction: a contributor to adverse cardiovascular remodeling and heart failure development in type 2 diabetes beyond accelerated atherogenesis. J Clin Med. (2020) 9:2090. doi: 10.3390/jcm9072090
189. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
190. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2007) 87:315–424. doi: 10.1152/physrev.00029.2006
191. Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S, et al. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes. (2004) 53:1418–23. doi: 10.2337/diabetes.53.6.1418
192. Verbeke P, Perichon M, Friguet B, Bakala H. Inhibition of nitric oxide synthase activity by early and advanced glycation end products in cultured rabbit proximal tubular epithelial cells. Biochim Biophys Acta. (2000) 1502:481–94. doi: 10.1016/S0925-4439(00)00071-5
193. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. (2011) 124:444–53. doi: 10.1161/CIRCULATIONAHA.110.014506
194. Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. (2013) 6:1239–49. doi: 10.1161/CIRCHEARTFAILURE.113.000539
195. Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. (2011) 124:1151–9. doi: 10.1161/CIRCULATIONAHA.111.025270
196. Esposito K, Ciotola M, Sasso FC, Cozzolino D, Saccomanno F, Assaloni R, et al. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: role of tumor necrosis factor-alpha. Nutr Metab Cardiovasc Dis. (2007) 17:274–9. doi: 10.1016/j.numecd.2005.11.014
197. Minutolo R, Gabbai FB, Provenzano M, Chiodini P, Borrelli S, Garofalo C, et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: pooled analysis of four cohort studies. Nephrol Dial Transplant. (2018) 33:1942–9. doi: 10.1093/ndt/gfy032
198. Minutolo R, Sasso FC, Chiodini P, Cianciaruso B, Carbonara O, Zamboli P, et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: a comparative analysis in nephrology, diabetology and primary care settings. J Hypertens. (2006) 24:1655–61. doi: 10.1097/01.hjh.0000239303.93872.31
199. Sasso FC, Chiodini P, Carbonara O, De Nicola L, Conte G, Salvatore T, et al. Nephropathy In Type 2 Diabetes Study Group. High cardiovascular risk in patients with Type 2 diabetic nephropathy: the predictive role of albuminuria and glomerular filtration rate. The NID-2 Prospective Cohort Study. Nephrol Dial Transplant. (2012) 27:2269–74. doi: 10.1093/ndt/gfr644
200. Giordano M, Ciarambino T, Castellino P, Malatino L, Cataliotti A, Rinaldi L, et al. Seasonal variations of hyponatremia in the emergency department: age-related changes. Am J Emerg Med. (2017) 35:749–52. doi: 10.1016/j.ajem.2017.01.018
201. Di Carli MF, Janisse J, Grunberger G, Ager J. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol. (2003) 41:1387–93. doi: 10.1016/S0735-1097(03)00166-9
202. Adameova A, Dhalla NS. Role of microangiopathy in diabetic cardiomyopathy. Heart Fail Rev. (2014) 19:25–33. doi: 10.1007/s10741-013-9378-7
203. Han B, Baliga R, Huang H, Giannone PJ, Bauer JA. Decreased cardiac expression of vascular endothelial growth factor and redox imbalance in murine diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2009) 297:H829–35. doi: 10.1152/ajpheart.00222.2009
204. Hinkel R, Howe A, Renner S, Ng J, Lee S, Klett K, et al. Diabetes mellitus-induced microvascular destabilization in the myocardium. J Am Coll Cardiol. (2017) 69:131–43. doi: 10.1016/j.jacc.2016.10.058
205. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. (2015) 131:550–9. doi: 10.1161/CIRCULATIONAHA.114.009625
206. Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. (2002) 4:411–7. doi: 10.1016/S1388-9842(02)00010-7
207. Gottumukkala RV, Lv H, Cornivelli L, Wagers AJ, Kwong RY, Bronson R, et al. Myocardial infarction triggers chronic cardiac autoimmunity in type 1 diabetes. Sci Transl Med. (2012) 4:138ra80. doi: 10.1126/scitranslmed.3003551
208. Hoffman WH, Sharma M, Cihakova D, Talor MV, Rose NR, Mohanakumar T, et al. Cardiac antibody production to self-antigens in children and adolescents during and following the correction of severe diabetic ketoacidosis. Autoimmunity. (2016) 49:188–96. doi: 10.3109/08916934.2015.1134509
209. Sousa GR, Pober D, Galderisi A, Lv H, Yu L, Pereira AC, et al. Glycemic control, cardiac autoimmunity, and long-term risk of cardiovascular disease in type 1 diabetes mellitus. Circulation. (2019) 139:730–43. doi: 10.1161/CIRCULATIONAHA.118.036068
210. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. (2004) 429:457–63. doi: 10.1038/nature02625
211. Bernardo BC, Charchar FJ, Lin RC, McMullen JR. A microRNA guide for clinicians and basic scientists: background and experimental techniques. Heart Lung Circ. (2012) 21:131–42. doi: 10.1016/j.hlc.2011.11.002
212. de Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JW, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. (2017) 7:47. doi: 10.1038/s41598-017-00070-6
213. Nandi SS, Zheng H, Sharma NM, Shahshahan HR, Patel KP, Mishra PK. Lack of miR-133a decreases contractility of diabetic hearts: a role for novel cross talk between tyrosine aminotransferase and tyrosine hydroxylase. Diabetes. (2016) 65:3075–90. doi: 10.2337/db16-0023
214. Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. (2016) 471:423–9. doi: 10.1016/j.bbrc.2016.02.065
215. Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, et al. miR-30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther. (2015) 33:89–97. doi: 10.1111/1755-5922.12113
216. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. (2014) 5:e1479. doi: 10.1038/cddis.2014.430
217. Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. (2003) 35:1135–43. doi: 10.1016/S0022-2828(03)00229-3
218. Rawal S, Manning P, Katare R. Cardiovascular microRNAs: as modulators and diagnostic biomarkers of diabetic heart disease. Cardiovasc Diabetol. (2014) 13:44. doi: 10.1186/1475-2840-13-44
219. Babiarz JE, Ravon M, Sridhar S, Ravindran P, Swanson B, Bitter H, et al. Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev. (2012) 21:1956–65. doi: 10.1089/scd.2011.0357
220. Westermeier F, Riquelme JA, Pavez M, Garrido V, Díaz A, Verdejo HE, et al. New molecular insights of insulin in diabetic cardiomyopathy. Front Physiol. (2016) 7:125. doi: 10.3389/fphys.2016.00125
221. Wang X, Gu H, Huang W, Peng J, Li Y, Yang L, et al. Hsp20-mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes. (2016) 65:3111–28. doi: 10.2337/db15-1563
222. De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C, Foti DP. Type 2 diabetes mellitus and cardiovascular disease: genetic and epigenetic links. Front Endocrinol. (2018) 9:2. doi: 10.3389/fendo.2018.00002
223. Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep. (2016) 6:36340. doi: 10.1038/srep36340
224. Bagchi RA, Weeks KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol. (2019) 130:151–9. doi: 10.1016/j.yjmcc.2019.04.003
225. Wang Q, Sun Y, Li T, Liu L, Zhao Y, Li L, et al. Function of BRD4 in the pathogenesis of high glucose-induced cardiac hypertrophy. Mol Med Rep. (2019) 19:499–507. doi: 10.3892/mmr.2018.9681
226. Tao H, Shi P, Xuan HY, Ding XS. DNA methyltransferase-1 inactivation of androgen receptor axis triggers homocysteine induced cardiac fibroblast autophagy in diabetic cardiac fibrosis. Arch Biochem Biophys. (2020) 692:108521. doi: 10.1016/j.abb.2020.108521
227. Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. (1999) 43:838–49. doi: 10.1016/S0008-6363(99)00145-5
228. Huynh K, Kiriazis H, Du XJ, Love JE, Gray SP, Jandeleit-Dahm KA, et al. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med. (2013) 60:307–17. doi: 10.1016/j.freeradbiomed.2013.02.021
229. Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, et al. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens. (2010) 28:340–52. doi: 10.1097/HJH.0b013e32833366cd
230. Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab. (2012) 302:E201–8. doi: 10.1152/ajpendo.00497.2011
231. Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, et al. Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension. (2015) 66:1159–67. doi: 10.1161/HYPERTENSIONAHA.115.06015
232. Sasso FC, Salvatore T, Tranchino G, Cozzolino D, Caruso AA, Persico M, et al. Cochlear dysfunction in type 2 diabetes: a complication independent of neuropathy and acute hyperglycemia. Metabolism. (1999) 48:1346–50. doi: 10.1016/S0026-0495(99)90141-5
233. Spallone V, Ziegler D, Freeman R, Bernardi L, Frontoni S, Pop-Busui R, et al. Toronto Consensus Panel on Diabetic Neuropathy. Cardiovascular autonomic neuropathy in diabetes: clinical impact, assessment, diagnosis, and management. Diabetes Metab Res Rev. (2011) 27:639–53. doi: 10.1002/dmrr.1239
234. La Rovere MT, Pinna GD, Maestri R, Robbi E, Caporotondi A, Guazzotti G, et al. Prognostic implications of baroreflex sensitivity in heart failure patients in the beta-blocking era. J Am Coll Cardiol. (2009) 53:193–9. doi: 10.1016/j.jacc.2008.09.034
235. Paulson DJ, Light KE. Elevation of serum and ventricular norepinephrine content in the diabetic rat. Res Commun Chem Pathol Pharmacol. (1981) 33:559–62.
236. Givertz MM, Sawyer DB, Colucci WS. Antioxidants and myocardial contractility: illuminating the “Dark Side” of beta-adrenergic receptor activation? Circulation. (2001) 103:782–3. doi: 10.1161/01.CIR.103.6.782
237. Kellogg AP, Converso K, Wiggin T, Stevens M, Pop-Busui R. Effects of cyclooxygenase-2 gene inactivation on cardiac autonomic and left ventricular function in experimental diabetes. Am J Physiol Heart Circ Physiol. (2009) 296:H453–61. doi: 10.1152/ajpheart.00678.2008
238. Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. (1998) 98:1329–34. doi: 10.1161/01.CIR.98.13.1329
239. Schnell O, Kirsch CM, Stemplinger J, Haslbeck M, Standl E. Scintigraphic evidence for cardiac sympathetic dysinnervation in long-term IDDM patients with and without ECG-based autonomic neuropathy. Diabetologia. (1995) 38:1345–52. doi: 10.1007/s001250050433
240. Pop-Busui R, Kirkwood I, Schmid H, Marinescu V, Schroeder J, Larkin D, et al. Sympathetic dysfunction in type 1 diabetes: association with impaired myocardial blood flow reserve and diastolic dysfunction. J Am Coll Cardiol. (2004) 44:2368–74. doi: 10.1016/j.jacc.2004.09.033
241. Dinh W, Füth R, Lankisch M, Bansemir L, Nickl W, Scheffold T, et al. Cardiovascular autonomic neuropathy contributes to left ventricular diastolic dysfunction in subjects with Type 2 diabetes and impaired glucose tolerance undergoing coronary angiography. Diabet Med. (2011) 28:311–8. doi: 10.1111/j.1464-5491.2010.03221.x
242. Pop-Busui R, Cleary PA, Braffett BH, Martin CL, Herman WH, Low PA, et al. DCCT/EDIC Research Group. Association between cardiovascular autonomic neuropathy and left ventricular dysfunction: DCCT/EDIC study (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications). J Am Coll Cardiol. (2013) 61:447–54. doi: 10.1016/j.jacc.2012.10.028
243. Tarquini R, Lazzeri C, Pala L, Rotella CM, Gensini GF. The diabetic cardiomyopathy. Acta Diabetol. (2011) 48:173–81. doi: 10.1007/s00592-010-0180-x
244. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. Authors/Task Force Members; Document Reviewers. 2016 ESC Guidelines for the diagnosis treatment of acute chronic heart failure: The Task Force for the diagnosis treatment of acute chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. (2016) 18:891–975. doi: 10.1002/ejhf.592
245. Sasso FC, Pafundi PC, Gelso A, Bono V, Costagliola C, Marfella R, et al. Telemedicine for screening diabetic retinopathy: the NO BLIND Italian multicenter study. Diabetes Metab Res Rev. (2019) 35:e3113. doi: 10.1002/dmrr.3113
246. Ortega-Loubon C, Fernández-Molina M, Singh G, Correa R. Obesity and its cardiovascular effects. Diabetes Metab Res Rev. (2019) 35:e3135. doi: 10.1002/dmrr.3135
247. Li T, Li G, Guo X, Li Z, Yang J, Sun Y. The influence of diabetes and prediabetes on left heart remodeling: a population-based study. J Diabetes Complications. (2021) 35:107771. doi: 10.1016/j.jdiacomp.2020.107771
248. Somaratne JB, Whalley GA, Poppe KK, ter Bals MM, Wadams G, Pearl A, et al. Screening for left ventricular hypertrophy in patients with type 2 diabetes mellitus in the community. Cardiovasc Diabetol. (2011) 10:29. doi: 10.1186/1475-2840-10-29
249. Felício JS, Koury CC, Carvalho CT, Abrahão Neto JF, Miléo KB, Arbage TP, et al. Present insights on cardiomyopathy in diabetic patients. Curr Diabetes Rev. (2016) 12:384–95. doi: 10.2174/1573399812666150914120529
250. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. (1990) 322:1561–6. doi: 10.1056/NEJM199005313222203
251. Bayes-Genis A. Hypertrophy and inflammation: too much for one heart. Eur Heart J. (2007) 28:661–3. doi: 10.1093/eurheartj/ehm008
252. Atale N, Yadav D, Rani V, Jin JO. Pathophysiology, clinical characteristics of diabetic cardiomyopathy: therapeutic potential of natural polyphenols. Front Nutr. (2020) 7:564352. doi: 10.3389/fnut.2020.564352
253. Di Bello V, Talarico L, Picano E, Di Muro C, Landini L, Paterni M, et al. Increased echodensity of myocardial wall in the diabetic heart: an ultrasound tissue characterization study. J Am Coll Cardiol. (1995) 25:1408–15. doi: 10.1016/0735-1097(95)00026-Z
254. Mishra PK, Givvimani S, Chavali V, Tyagi SC. Cardiac matrix: a clue for future therapy. Biochim Biophys Acta. (2013) 1832:2271–6. doi: 10.1016/j.bbadis.2013.09.004
255. Ivey MJ, Tallquist MD. Defining the cardiac fibroblast. Circ J. (2016) 80:2269–76. doi: 10.1253/circj.CJ-16-1003
256. Hutchinson KR, Lord CK, West TA, Stewart JA Jr. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS ONE. (2013) 8:e72080. doi: 10.1371/journal.pone.0072080
257. Sedgwick B, Riches K, Bageghni SA, O'Regan DJ, Porter KE, Turner NA. Investigating inherent functional differences between human cardiac fibroblasts cultured from nondiabetic and Type 2 diabetic donors. Cardiovasc Pathol. (2014) 23:204–10. doi: 10.1016/j.carpath.2014.03.004
258. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. (2016) 90:84–93. doi: 10.1016/j.yjmcc.2015.12.011
259. Westermann D, Rutschow S, Jäger S, Linderer A, Anker S, Riad A, et al. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes. (2007) 56:641–6. doi: 10.2337/db06-1163
260. Van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol. (2008) 103:319–27. doi: 10.1007/s00395-008-0715-2
261. Lee HW, Lee SJ, Lee MY, Park MW, Kim SS, Shin N, et al. Enhanced cardiac expression of two isoforms of matrix metalloproteinase-2 in experimental diabetes mellitus. PLoS ONE. (2019) 14:e0221798. doi: 10.1371/journal.pone.0221798
262. Toblli JE, Cao G, DeRosa G, Forcada P. Reduced cardiac expression of plasminogen activator inhibitor 1 and transforming growth factor beta1 in obese Zucker rats by perindopril. Heart. (2005) 91:80–6. doi: 10.1136/hrt.2003.022707
263. Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin Invest. (1994) 93:536–42. doi: 10.1172/JCI117004
264. Gorski DJ, Petz A, Reichert C, Twarock S, Grandoch M, Fischer JW. Cardiac fibroblast activation and hyaluronan synthesis in response to hyperglycemia and diet-induced insulin resistance. Sci Rep. (2019) 9:1827. doi: 10.1038/s41598-018-36140-6
265. Wang Y, Gao P, Wei C, Li H, Zhang L, Zhao Y, et al. Calcium sensing receptor protects high glucose-induced energy metabolism disorder via blocking gp78-ubiquitin proteasome pathway. Cell Death Dis. (2017) 8:e2799. doi: 10.1038/cddis.2017.193
266. Yuan H, Xu J, Xu X, Gao T, Wang Y, Fan Y, et al. Calhex231 alleviates high glucose-induced myocardial fibrosis via inhibiting itch-Ubiquitin proteasome pathway in vitro. Biol Pharm Bull. (2019) 42:1337–44. doi: 10.1248/bpb.b19-00090
267. Civitarese RA, Kapus A, McCulloch CA, Connelly KA. Role of integrins in mediating cardiac fibroblast-cardiomyocyte cross talk: a dynamic relationship in cardiac biology and pathophysiology. Basic Res Cardiol. (2017) 112:6. doi: 10.1007/s00395-016-0598-6
268. Talior-Volodarsky I, Connelly KA, Arora PD, Gullberg D, McCulloch CA. α11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc Res. (2012) 96:265–75. doi: 10.1093/cvr/cvs259
269. Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail Clin. (2019) 15:341–7. doi: 10.1016/j.hfc.2019.02.003
270. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
271. Ha JW, Lee HC, Kang ES, Ahn CM, Kim JM, Ahn JA, et al. Abnormal left ventricular longitudinal functional reserve in patients with diabetes mellitus: implication for detecting subclinical myocardial dysfunction using exercise tissue Doppler echocardiography. Heart. (2007) 93:1571–6. doi: 10.1136/hrt.2006.101667
272. Jensen MT, Sogaard P, Andersen HU, Bech J, Hansen TF, Galatius S, et al. Prevalence of systolic and diastolic ysfunction in patients with type 1 diabetes without known heart disease: the Thousand & 1 Study. Diabetologia. (2014) 57:672–80. doi: 10.1007/s00125-014-3164-5
273. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. (2002) 283:H976–82. doi: 10.1152/ajpheart.00088.2002
274. Celentano A, Vaccaro O, Tammaro P, Galderisi M, Crivaro M, Oliviero M, et al. Early abnormalities of cardiac function in non-insulin-dependent diabetes mellitus and impaired glucose tolerance. Am J Cardiol. (1995) 76:1173–6. doi: 10.1016/S0002-9149(99)80330-0
275. Ernande L, Bergerot C, Rietzschel ER, De Buyzere ML, Thibault H, Pignonblanc PG, et al. Diastolic dysfunction in patients with type 2 diabetes mellitus: is it really the first marker of diabetic cardiomyopathy? J Am Soc Echocardiogr. (2011) 24:1268–75.e1. doi: 10.1016/j.echo.2011.07.017
276. Faden G, Faganello G, De Feo S, Berlinghieri N, Tarantini L, Di Lenarda A, et al. The increasing detection of asymptomatic left ventricular dysfunction in patients with type 2 diabetes mellitus without overt cardiac disease: data from the SHORTWAVE study. Diabetes Res Clin Pract. (2013) 101:309–16. doi: 10.1016/j.diabres.2013.07.004
277. Eurich DT, Weir DL, Majumdar SR, Tsuyuki RT, Johnson JA, Tjosvold L, et al. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circ Heart Fail. (2013) 6:395–402. doi: 10.1161/CIRCHEARTFAILURE.112.000162
278. Singh S, Loke YK, Furberg CD. Thiazolidinediones and heart failure: a teleo-analysis. Diabetes Care. (2007) 30:2148–53. doi: 10.2337/dc07-0141
279. Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet. (2015) 385:2107–17. doi: 10.1016/S0140-6736(14)61402-1
280. Pan X, Xu S, Li J, Tong N. The effects of DPP-4 inhibitors, GLP-1RAs, and SGLT-2/1 inhibitors on heart failure outcomes in diabetic patients with and without heart failure history: insights from CVOTs and drug mechanism. Front Endocrinol. (2020) 11:599355. doi: 10.3389/fendo.2020.599355
281. Anderluh M, Kocic G, Tomovic K, Kocic R, Deljanin-Ilic M, Smelcerovic A. Cross-talk between the dipeptidyl peptidase-4 and stromal cell-derived factor-1 in stem cell homing and myocardial repair: potential impact of dipeptidyl peptidase-4 inhibitors. Pharmacol Ther. (2016) 167:100–7. doi: 10.1016/j.pharmthera.2016.07.009
282. Aoyama M, Kawase H, Bando YK, Monji A, Murohara T. Dipeptidyl peptidase 4 inhibition alleviates shortage of circulating glucagon-like peptide-1 in heart failure and mitigates myocardial remodeling and apoptosis via the exchange protein directly activated by cyclic AMP 1/Ras-related protein 1 axis. Circ Heart Fail. (2016) 9:e002081. doi: 10.1161/CIRCHEARTFAILURE.115.002081
283. Shah Z, Pineda C, Kampfrath T, Maiseyeu A, Ying Z, Racoma I, et al. Acute DPP-4 inhibition modulates vascular tone through GLP-1 independent pathways. Vascul Pharmacol. (2011) 55:2–9. doi: 10.1016/j.vph.2011.03.001
284. Qian P, Tian H, Wang Y, Lu W, Li Y, Ma T, et al. A novel oral glucagon-like peptide 1 receptor agonist protects against diabetic cardiomyopathy via alleviating cardiac lipotoxicity induced mitochondria dysfunction. Biochem Pharmacol. (2020) 182:114209. doi: 10.1016/j.bcp.2020.114209
285. Packer M, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, et al. Evaluation of the effect of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality of patients with chronic heart failure and a reduced ejection fraction: rationale for and design of the EMPEROR-reduced trial. Eur J Heart Fail. (2019) 21:1270–8. doi: 10.1002/ejhf.1536
286. Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia. (2018) 61:2108–17. doi: 10.1007/s00125-018-4670-7
287. Lan NSR, Fegan PG, Yeap BB, Dwivedi G. The effects of sodium-glucose cotransporter 2 inhibitors on left ventricular function: current evidence and future directions. ESC Heart Fail. (2019) 6:927–35. doi: 10.1002/ehf2.12505
288. Hölscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter?. Int J Mol Sci. (2016) 17:2136. doi: 10.3390/ijms17122136
289. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
290. Radovits T, Korkmaz S, Loganathan S, Barnucz E, BÖmicke T, Arif R, et al. Comparative investigation of the left ventricular pressure-volume relationship in rat models of type 1 and type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. (2009) 297:H125–33. doi: 10.1152/ajpheart.00165.2009
291. Mátyás C, Kovács A, Németh BT, Oláh A, Braun S, Tokodi M, et al. Comparison of speckle-tracking echocardiography with invasive hemodynamics for the detection of characteristic cardiac dysfunction in type-1 and type-2 diabetic rat models. Cardiovasc Diabetol. (2018) 17:13. doi: 10.1186/s12933-017-0645-0
Keywords: diabetes mellitus, cardiomyopathy, heart failure, pathophysiology, insulin resistance
Citation: Salvatore T, Pafundi PC, Galiero R, Albanese G, Di Martino A, Caturano A, Vetrano E, Rinaldi L and Sasso FC (2021) The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 8:695792. doi: 10.3389/fmed.2021.695792
Received: 15 April 2021; Accepted: 07 June 2021;
Published: 30 June 2021.
Edited by:
Qin Zhou, The First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Eliane Dias, Fluminense Federal University, BrazilJun Ren, University of Washington, United States
Copyright © 2021 Salvatore, Pafundi, Galiero, Albanese, Di Martino, Caturano, Vetrano, Rinaldi and Sasso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ferdinando Carlo Sasso, ferdinandocarlo.sasso@unicampania.it