Pharmacological Approach for Neuroprotection After Cardiac Arrest—A Narrative Review of Current Therapies and Future Neuroprotective Cocktail
 Rishabh C. Choudhary1,2†
Rishabh C. Choudhary1,2†  Muhammad Shoaib1,3†
Muhammad Shoaib1,3†  Samantha Sohnen4†
Samantha Sohnen4†  Daniel M. Rolston2,3,5 Daniel Jafari2,3,5
Daniel M. Rolston2,3,5 Daniel Jafari2,3,5  Santiago J. Miyara1,6
Santiago J. Miyara1,6  Kei Hayashida1,2 Ernesto P. Molmenti7 Junhwan Kim1,2,3 Lance B. Becker1,2,3*
Kei Hayashida1,2 Ernesto P. Molmenti7 Junhwan Kim1,2,3 Lance B. Becker1,2,3*- 1Laboratory for Critical Care Physiology, The Feinstein Institutes for Medical Research, Northwell Health, Manhasset, NY, United States
- 2Department of Emergency Medicine, Northshore University Hospital, Northwell Health, Manhasset, NY, United States
- 3Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, Hempstead, NY, United States
- 4Department of Anesthesiology, Dartmouth-Hitchcock Medical Center, Lebanon, NH, United States
- 5Department of Surgery, North Shore University Hospital, Northwell Health, Manhasset, NY, United States
- 6Elmezzi Graduate School of Molecular Medicine, Manhasset, NY, United States
- 7Department of Surgery, Northwell Health, Manhasset, NY, United States
Cardiac arrest (CA) results in global ischemia-reperfusion injury damaging tissues in the whole body. The landscape of therapeutic interventions in resuscitation medicine has evolved from focusing solely on achieving return of circulation to now exploring options to mitigate brain injury and preserve brain function after CA. CA pathology includes mitochondrial damage and endoplasmic reticulum stress response, increased generation of reactive oxygen species, neuroinflammation, and neuronal excitotoxic death. Current non-pharmacologic therapies, such as therapeutic hypothermia and extracorporeal cardiopulmonary resuscitation, have shown benefits in protecting against ischemic brain injury and improving neurological outcomes post-CA, yet their application is difficult to institute ubiquitously. The current preclinical pharmacopeia to address CA and the resulting brain injury utilizes drugs that often target singular pathways and have been difficult to translate from the bench to the clinic. Furthermore, the limited combination therapies that have been attempted have shown mixed effects in conferring neuroprotection and improving survival post-CA. The global scale of CA damage and its resultant brain injury necessitates the future of CA interventions to simultaneously target multiple pathways and alleviate the hemodynamic, mitochondrial, metabolic, oxidative, and inflammatory processes in the brain. This narrative review seeks to highlight the current field of post-CA neuroprotective pharmaceutical therapies, both singular and combination, and discuss the use of an extensive multi-drug cocktail therapy as a novel approach to treat CA-mediated dysregulation of multiple pathways, enhancing survival, and neuroprotection.
Introduction
Annually, there are over 356,000 out-of-hospital cardiac arrests (OHCA) and 209,000 in-hospital cardiac arrests (IHCA) in the United States with the survival to hospital discharge only 12% and 25%, respectively. Furthermore, only 8.4% of OHCA patients have favorable neurologic function, which can be determined using various scales, such as modified Rankin Scale (mRS), Cerebral Performance Categories (CPC), and Extended Glasgow Outcome Scale (1–3). Despite the heterogeneous etiologies of cardiac arrest (CA), the commonality is the decreased perfusion to vital organs, such as the brain (4). Hence, the best chances for increasing survival and clinical outcomes requires a combination of early recognition, early initiation of high-quality cardiopulmonary resuscitation (CPR) and Advanced Cardiovascular Life Support (ACLS), and appropriate post-resuscitation care that focus on protecting the brain (5). Despite proven benefits of these interventions on survival, the vast majority of survivors still have significant neurologic deficits; therefore, novel neuroprotective therapies that may be achieved through combination of evidence-based, pathway-specific, pharmacological agents can potentially improve outcomes for patients.
Ischemia-reperfusion injury (IRI) begins with decreased perfusion to tissues, depletion of energetic substrates, and subsequent upregulation of the anaerobic metabolism (6). Under physiological conditions, the brain consumes 20% of the basal metabolic rate despite representing only 2% of the total body weight. This high energetic demand, the virtual absence of glycogen reserves, and the dependance on oxidative phosphorylation makes the brain highly vulnerable (7, 8). Resuscitation with return of spontaneous circulation (ROSC) increases the chance of overall survival, however the subsequent reperfusion injury has been associated with further neurological damage (3).
Besides ACLS, there are limited evidence-based treatments that successfully improve survival and protect the brain after CA. The two major therapies used for treating CA that can aid in neuroprotection are extracorporeal cardiopulmonary resuscitation (E-CPR) and targeted temperature management (TTM); the former uses an external machine to oxygenate and circulate blood within the body (9), while the latter decreases and maintains the core temperature between 32 and 36°C to decrease cellular metabolism, decrease oxidative damage and stress signals, and decrease cellular death (6, 10–13). Although both E-CPR and TTM have therapeutic efficacy in CA patients, each has their limitations and can influence both protective and deleterious pathways (14). E-CPR is time, personnel, and resource intensive. Major complications of TTM include hemodynamic instability, arrhythmias, AV blocks, hydro-electrolyte disorders due to cold diuresis, endocrine and coagulation abnormalities, increased risk of infection, among others (15, 16).
Although improvements in technology may increase the general application of TTM and E-CPR, the major challenge resides in tackling the various molecular mechanisms that result in brain injury post-CA (14). Currently, preclinical and a few clinical studies have used individual pharmacologic agents to alleviate damage post-CA, yet these interventions are unable to provide sufficient protection to aid in survival with favorable neurological outcomes. As such, the focus of this review is to highlight therapies that have attempted to confer neuroprotection post-CA in preclinical and clinical studies, with their proposed targeted pathway(s). We also seek to propose the application of a multi-drug cocktail that is comprised of extensive pharmaceutical agents rather than a few in order to concurrently target the distinct, multiple metabolic, oxidative, and inflammatory alterations in the brain post-CA to potentially shift the current paradigm of neuroprotection to a multidimensional therapeutic approach.
Overview of the Various Cardiac Arrest-Mediated Pathway Alterations
CA and the ensuing global ischemia generate a variety of alterations, such as mitochondrial dysfunction, increased reactive oxygen species (ROS) generation (17, 18), metabolic disruption including protein and lipid dysfunction (19–23), increased neuroinflammation, intracellular calcium overload, and endoplasmic reticulum (ER) stress (24–26). Ultimately, CA pathologic alterations result in cellular dysfunction and brain injury of which substantial mitigation necessitates a change in the current paradigm of CA treatment from targeting singular altered pathways to using a multifunctional, multi-drug cocktail method (14). Figure 1A summarizes the various mechanisms of brain damage post-CA.
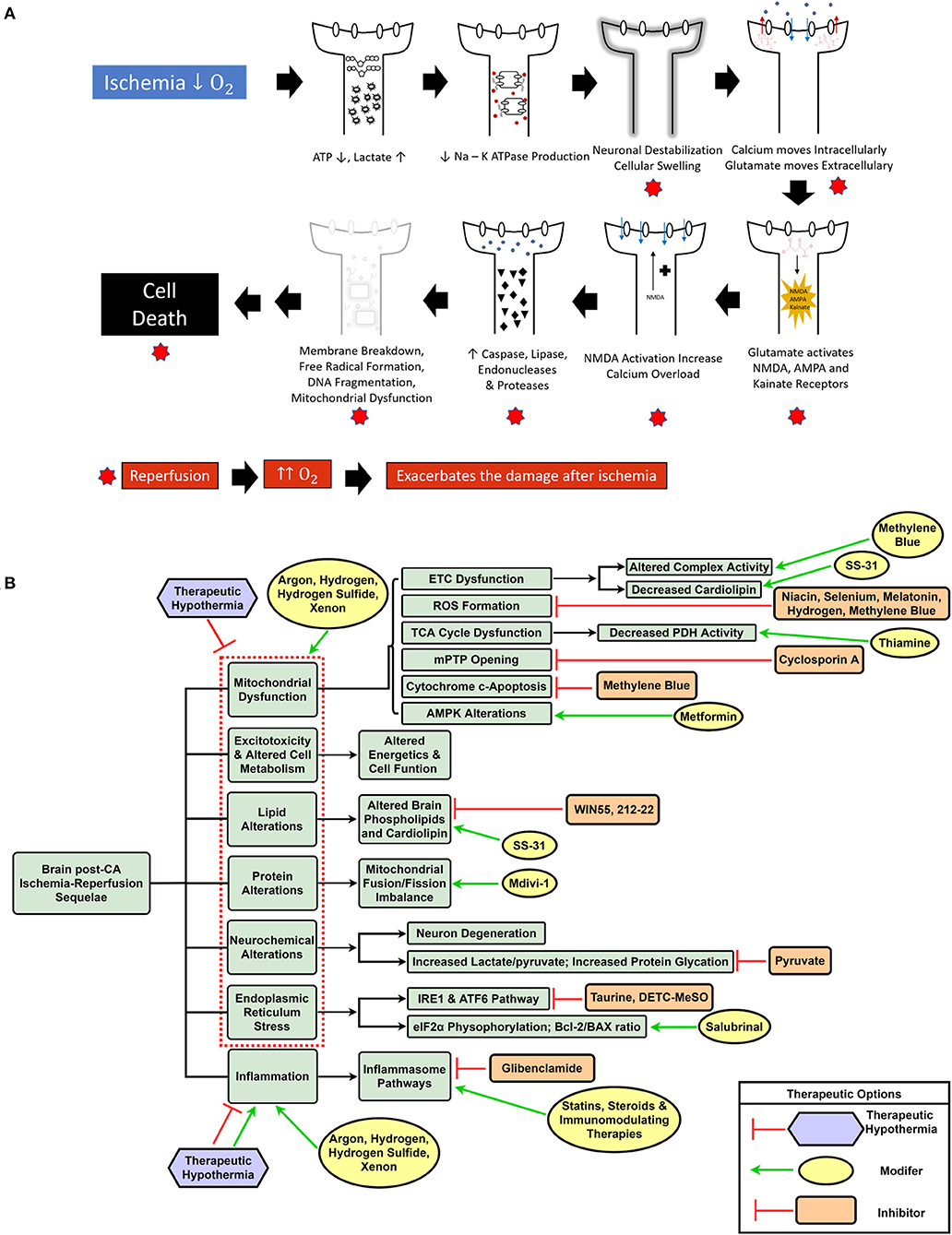
Figure 1. Schematic representation of ischemia-reperfusion injury after cardiac arrest and resuscitation resulting in brain injury (A). Overview of dysregulated pathways after cardiac arrest with a selection of inhibitory and modulatory interventions that can be combined in a cocktail therapy to confer neuroprotection (B).
Cardiac Arrest-Mediated Mitochondrial Dysfunction and ROS Generation With the Respective Therapeutic Interventions
Mitochondrial dysfunction has been correlated with poor neurologic outcomes secondary to impaired aerobic metabolism as well as increased oxidative stress, garnering attention as a target for neuroprotection after CA (17, 27, 28). Along with the general inefficiency in mitochondrial energy generation (19), CA patients exhibit low activity of pyruvate dehydrogenase (PDH) complex, the rate-limiting step in the Tricarboxylic acid (TCA) cycle (27, 29). Therefore, administering thiamine, a cofactor of the PDH complex, to mice after CA resulted in improved PDH activity and neurological outcomes (27). Decreased ATP production and impairment of the Na+/K+ ATPase (6) after CA results in intracellular calcium overload triggering the voltage-dependent anion channels (VDAC) and subsequent opening of the mitochondrial permeability transition pore (mPTP) (30, 31). Formation of the mPTP further activates the caspase cascade and endonuclease-mediated DNA damage resulting in mitophagy and cellular death (30). Cyclosporin A has been studied to provide neuroprotection by preventing mPTP opening both alone and in combination with hypothermia in rats, but human trials have not supported the therapeutic benefits observed in the preclinical setting (32–34).
The disruption of mitochondrial fusion and fission equilibrium has been documented in rats after CA, with temporary mitochondrial fusion shortly following CA, which shifts toward fission within 24 h, resulting in deficient cellular and mitochondrial function and eventual apoptosis (35). Inhibiting mitochondrial fission has been shown to prevent cardiomyocyte apoptosis and improve cardiac function in vitro, ex vivo, and in vivo (36, 37). Inhibition of the mitochondrial fission protein, Dynamin-related protein 1 (Drp1), using the well-known fission inhibitor, mitochondrial division inhibitor 1 (Mdivi-1), has shown to improve neurological dysfunction and survival in mice after CA (38, 39), prevent N-Methyl-d-aspartate (NMDA)-mediated excitotoxicity, calcium overload, cell death in rat neurons (40), and has cardioprotective properties in a rat model of cardiac IRI (41). Taken together, CA results in major metabolic alterations, including mitochondrial protein damage, that require targeting multiple pathways simultaneously for improved survival and neuroprotection.
Post-CA mitochondrial dysfunction generates ROS due to enhanced electron leak at complexes I and III in the electron transport chain, deglutathionylated complex II, and phosphorylated complex IV, among other mechanisms (42). ROS-mediated release of cytochrome c from the mitochondrial membrane upregulates pro-apoptotic genes causing apoptosis (30). General application of antioxidant therapies, such as vitamin C, α-tocopherol, and edaravone, have shown attenuation of ROS in preclinical and some clinical settings (43–45). Although there are multiple ongoing clinical trials evaluating the beneficial effects of antioxidant therapies in human CA patients, the mixed beneficial evidence from the preclinical setting does not support the translation of these singular antioxidant therapies (46–48). Site-specific inhibitors of ROS are more promising candidates because (1) they directly control the release of ROS at the source and (2) ROS generation at different sites can play either a beneficial, or a detrimental role (49). N-acetylcysteine, metformin, melatonin, and suppressor of site IQ electron leak (S1QEL) are a few examples of site-specific inhibitors that can decrease ROS generation after IRI and can help to potentiate survival and neurological functions (50–53). Inhaled hydrogen gas with its suppression of oxidative stress has also been observed to improve survival and neurologic outcomes in a rat CA model with an additive effect when compared to TTM alone (54). Similarly, other inert gases, such as Argon and Xenon seem to potentiate beneficial effects from standard treatments post-CA via a variety of proposed mechanisms (55–59). Mitochondrial dysfunction after IRI results from alterations in multiple pathways and further induces substantial metabolic and cellular disruption; therefore, utilizing a multi-drug cocktail that incorporates these therapies can target numerous mitochondrial pathways and may improve survival and brain function.
Cardiac Arrest-Mediated Endoplasmic Reticulum Stress With the Respective Therapeutic Interventions
Endoplasmic reticulum stress after intracellular calcium disruption or accumulation of unfolded/misfolded proteins in the ER lumen leading to the unfolded protein response (UPR) can be secondary to oxygen and glucose deprivation (30, 60). This, in turn, can expedite neurodegeneration through suppression of protein synthesis, protein degradation, and apoptosis (60). In a rat model of cerebral ischemia, the combination of taurine and S-Methyl-N, N-diethylthiocarbamate sulfoxide (DETC-MeSO), a partial NMDA antagonist, resulted in decreased expression of multiple ER stress pathways, which was insufficient when only using the individual therapies (61). Pretreatment using Salubrinal 30 min before CA improved neurological outcomes and cerebral mitochondrial morphology 24 h after CA by preservation of the mitochondrial membrane potential, increasing an antiapoptotic protein, Bcl-2, stabilizing the HIF-1α pathway, and inhibiting ER stress through the induction of eIF2α phosphorylation (62). Overall, these studies suggest that interventions targeting ER stress are important for neuroprotection post-CA and should be part of a larger cocktail therapy.
Cardiac Arrest-Mediated Metabolic Alterations and Therapeutic Interventions
Many studies have demonstrated the substantial and global metabolic changes that occur after CA and resuscitation. In a model of long duration CA and cardiopulmonary bypass (CPB) resuscitation, the severe metabolome dysregulation in the kidney and brain worsened post-resuscitation, as seen through alterations of fatty acids, amino acids, and TCA cycle metabolites (19). Kynurenine pathway alterations have been observed in rat, pig, and human plasma after CA, while in rats and pigs, higher levels of downstream metabolites were associated with worse survival and neurological outcomes observed through hippocampal lesions (20, 63). A study on plasma metabolomic profiling in pigs undergoing asphyxial cardiac arrest (ACA) or ventricular fibrillation cardiac arrest (VFCA) further supported major alterations in the plasma metabolites related with TCA and urea cycles (64). In a rat model of CA, pre-conditioning with metformin demonstrated early and sustained 5′-adenosine monophosphate-activated protein kinase (AMPK) activation in hippocampal brain tissue, reduced neuronal death, and improvement in overall survival with favorable neurologic outcomes (65). However, the role of AMPK after IRI has been controversial as a focal brain ischemia model suggested that downregulating AMPK may show better outcomes (66). AMPK is a mediator of metabolism and is involved in many cellular mechanisms (67) suggesting its role after different types of IRI pathologies and varying degrees of injury may either facilitate neuroprotection or participate in injury. This is an active area of exploration.
Along with individual metabolic alterations post-CA, specific changes in lipids and proteins are observed that implicate CA-induced mitochondrial injury. Lipids comprise a substantial category of biological materials that have unique and crucial functions in the body, such as cellular membrane composition, energy substrates and metabolites, transport mediators, as well as intracellular and hormonal signaling (68, 69). Free radical-mediated lipid peroxidation and decomposition of membrane phospholipids occurs after IRI (70, 71). Dogs undergoing 10 min CA had significantly increased levels of lipid oxidation in the frontal cortex, which continued to increase until 24 h post-resuscitation when ventilated with 100% oxygen; normoxic ventilation significantly lowered lipid oxidation and improved neurologic outcomes (21, 72). CA induces phospholipid changes in the brain, heart, kidney, and liver of rats post-CA with the brain having increased concentrations of lysophosphatidylethanolamine (LPE), lysophosphatidylcholine (LPC), and lysophosphatidylinositol (LPI), whereas only LPI was increased in the other organs; the brain's inability to regulate phospholipids could be related to decreased mitochondrial function (73–75). Decreased levels of brain cardiolipin (CL) post-CPB resuscitation in rats adversely affected mitochondrial integrity and decreased normal function resulting in decreased complex I and III activity in the brain (76). Plasma accumulation of brain-specific cardiolipin species after CA may represent the degree of neuronal injury after human and rat CA (77), while changes in mitochondrial lipids, phospholipids, and free fatty acids were noted after lethal ventricular tachyarrhythmia-mediated ischemia in rats (78). Administration of SS-31, a CL-targeting drug, in a severe CA rat model resulted in lower lactate levels and improved survival time, suggesting that improved support of CL can improve mitochondrial function (79). Furthermore, treatment with a cannabinoid receptor agonist, WIN55, 212-2, in combination with TTM was able to support lipid metabolism and improve neurologic outcomes after rodent CA (80). The use of probucol, a lipid-lowering agent used for hypercholesterolemia (81), increased survival time and decreased oxidative stress in post-CA rats potentially through antioxidant mechanisms (82). Collectively, these studies suggest that utilization of drugs that are lipid-based and/or target lipid pathways are vital components of a multi-drug cocktail for the treatment of CA.
Cardiac Arrest-Induced Neurochemical and Neuroinflammatory Alterations With Their Respective Therapeutic Interventions
Following cerebral ischemia, the acute efflux of dopamine and norepinephrine propagate brain injury (83), with the potential to significantly damage cerebral nerve terminals (84). Additionally, dopamine is considered a prerequisite for ischemic injury, since dopamine depletion protects the striatum from ischemic injury in a rat model of global brain ischemia by four-vessel occlusion (85). It is postulated that metabolic degradation of dopamine produces 2,4-dihydroxyphenylacetic acid (DOPAC), hydrogen peroxide, superoxide, and hydroxyl radicals, which have varying degrees of neurotoxic effects (86). In a small cohort of CA human patients undergoing TTM, lactate/pyruvate ratio measured using cerebral microdialysis progressively increased in the group of patients with an unfavorable neurologic outcome (87). In a swine model of CA, administration of intravenous pyruvate that can function as an antioxidant helped to minimize CA-induced protein glycation in the brain (88). Thus, the resultant neurochemical changes after IRI provide specific targets for therapeutic interventions.
Cerebral ischemia results in increased release of proinflammatory cytokines, such as IL-1 and TNF-α, microglial activation, disruption of the blood-brain barrier (BBB) and subsequent cerebral edema, all facilitating leukocyte migration into the brain (6, 83). Directly targeting inflammatory cytokine receptors is neuroprotective, as seen with reduced cortical infarct size after mild hypoxia/ischemia in knockout mice for IL-1-receptor (89) and with diminished brain injury in rats after blocking TNF-α via a neutralizing antibody (90). A majority of the neuroinflammation post-ischemia is microglia- and peripheral leukocyte-mediated (91). IRI stimulates IL-1β, IL-18, and NF-κB, activating the NLRP3 inflammasome, caspase-1, and other downstream mediators resulting in cell death and brain damage (92). Glibenclamide (GBC), a sulfonylurea drug used for diabetes, has been shown to inhibit the NLRP3 inflammasome pathway, suppress microglia and astrocyte activation, and support favorable neurologic outcomes following rodent CA (93). Furthermore, the immunomodulatory and antioxidative properties of statins have also shown efficacy in ischemic diseases (94). Thus, targeting the various pathways involved in neuroinflammation post-CA is a valuable therapeutic option, especially when used in a cocktail therapy.
Current Combination Therapies Applied for Conferring Neuroprotection After Cardiac Arrest
A detailed compilation of various pharmaceutical interventions attempted after CA in the preclinical and clinical settings along with their proposed mechanisms of actions and potential disadvantages is discussed in Table 1. Previous studies have examined some neuroprotective drugs in cerebral ischemia and CA (57, 58, 83, 107). While these studies have occasionally examined drug combinations, the drugs mentioned often target a singular pathway among the background of a global disease, potentially explaining the lower effectiveness of these therapies.
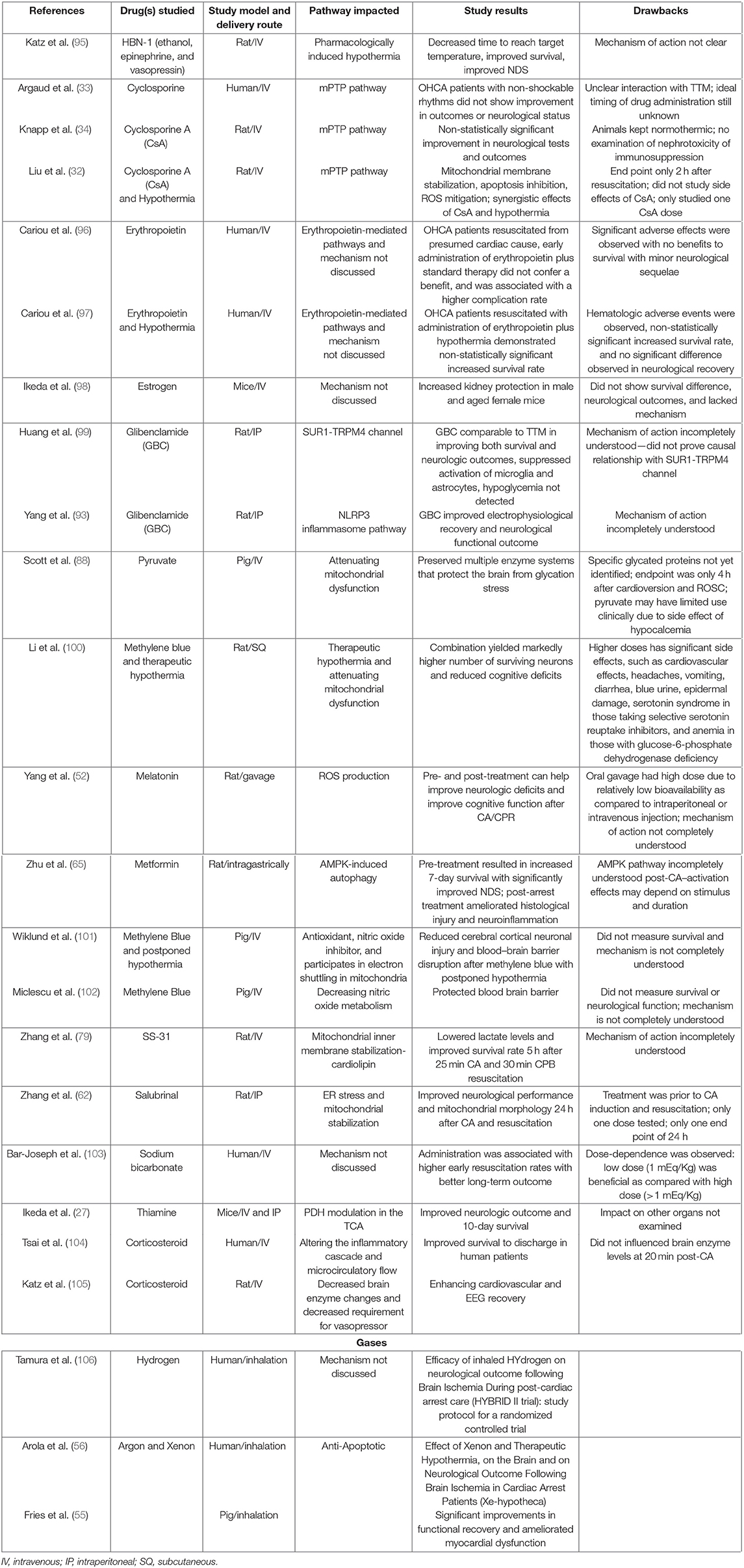
Table 1. Pharmacological interventions with their study results, mechanisms of action, and potential drawbacks for conferring survival and neuroprotection after cardiac arrest.
The use of combined therapy of adenosine triphosphate-magnesium chloride (ATP-MgCl2), norepinephrine, and vanadate improved protein synthesis and conferred neuroprotection after CA and resuscitation in rats as compared to individual drug treatment (108). Norepinephrine counteracts the vasodilatory effects of ATP and maintains blood pressure resulting in a selective vasodilatory effect in the brain as a result of this combination therapy. In ventricular fibrillation cardiac arrest (VFCA) rats, a combination therapy of niacin and selenium reduced ROS generation by enhancing glutathione (GSH) reductase activity and improved the GSH/GSSG ratio, attenuated brain injury, and improved the 7-day neurological outcomes by suppression of mechanisms that would normally increase caspase-mediated cell death (109). Another major limitation in these combination therapies is the use of very mild CA injury in the animals that does not reflect the injury of many human CA patients. A different combination of sevoflurane, Poloxamer-188, and TTM improved cardiac and neurologic functions after 17 min VF in a swine model as compared with controls that only received epinephrine and TTM (110). It is suggested that sevoflurane promotes endothelial protection by reducing leukocyte activation, affecting vascular tone (111), preventing apoptosis, and reducing cytokine production post-CA (112). Poloxamer-188 fills ischemia induced pores in the plasma membrane (113), prevents unregulated exchange of ions between cellular compartments, prevents cellular injury and apoptosis (114), preserves the BBB (115), and protects neurons against IRI (116). A major limitation with this combination is the lack of mechanistic insight of the pharmacologic compounds which is further complicated by TTM and the lack of dose optimization.
The combination therapy of epinephrine and vasopressin has shown potential beneficial effects in improving ROSC, and/or neurological and cerebral histopathological outcomes in animal models; however, this combination has only mixed results in the clinical setting (117–120). As potent vasoactive compounds, the potential adverse hemodynamic effects especially after a severe injury like CA are important considerations when combining these drugs, which may be one reason for the lack of translatability. A randomized control trial in humans with the combination of epinephrine, vasopressin, and methylprednisolone post-CA compared to epinephrine and saline demonstrated improved survival to hospital discharge with favorable neurological status (121–123). In fact, the singular administration of steroids during the post-arrest period has been associated with diminished brain enzyme changes, decreased requirement for vasopressor, and improved EEG and cardiovascular recovery in rodent CA (105) and improved survival to discharge in human patients (104, 124). The multisystemic effects of steroids present difficulty in isolating individual therapeutic actions, which can include hemodynamic, metabolic, and inflammatory modulation. The combination of vasopressin, epinephrine, and nitroglycerin has been shown to improve vital organ blood flow in pigs (125). Combination of epinephrine and naloxone significantly improved the survival and brain function post-CA in rats (126, 127). These studies emphasize that pharmaceutically managing hemodynamics during the resuscitation and post-resuscitation phases is critical for survival and protecting the brain.
Along with hemodynamic stabilization after arrest to maintain brain perfusion, managing the metabolic dysfunction is another critical requirement for post-CA cocktail therapy. The current, most effective method used is TTM, that is hypothesized to decrease the global metabolic demand, which is not without adverse effects (128). A new approach to achieve dampened metabolic stress post-arrest is to pharmacologically induce hypothermia (129). A combination of ethanol, vasopressin, lidocaine, known as HBN-1, after rodent CA was able to pharmacologically induce hypothermia by increasing heat loss without producing shivering and improve survival and neurological outcomes (95, 130). Similarly, HBN-1 with external hypothermia was shown to significantly decrease serum and cerebrospinal fluid levels of neuron specific enolase (NSE), a biomarker associated with poor neurologic outcomes (131). As is evident from previous preclinical and clinical post-CA trials, combination drugs have many benefits by simultaneously targeting different altered pathways; however, due to the severity of injury experienced by the whole body, a multi-drug cocktail should be similarly extensive in its therapeutic agents to appropriately manage the disease process.
Discussion
The global ischemia after CA results in a myriad of systemic insults, such as mitochondrial dysfunction (17), increased reactive oxygen species (ROS) generation (17, 18), metabolic alterations including lipid and protein dysfunction (19–21), along with other pathological sequelae. Although various single drug and some combination therapies have shown neuroprotection, to date, the overall outcomes have not been substantially improved. One potential reason is the lack of translatability of many of these agents to human patients; it is known that animal models are helpful in modeling disease, however, there are many challenges and limitations, especially when attempting to model a condition with a complex pathophysiology (19). Most preclinical studies use milder injury CA models that are unable to effectively represent the magnitude of human disease (132). Another explanation is that using singular drugs may effectively target the intended pathway, but are unable to alleviate the plethora of altered mechanisms by which brain damage occurs post-CA. A new shift in CA treatment can entail the incorporation of a multi-drug cocktail comprising a variety of drugs that can individually and synergistically confer neuroprotection based on their effects on the aforementioned diverse pathophysiological sequelae of CA. Furthermore, with the protective effects of TTM and E-CPR in eligible CA patients (9), it is conceivable that a cocktail therapy may provide additive benefits when combined with conventional interventions (14). Figure 1B highlights the various dysregulated pathways in the brain post-CA, and the respective interventions required for appropriate management that can be included in a cocktail therapy.
The important components for developing a cocktail therapy includes the types of pharmaceutical agents, formulations, dosages, modes of delivery and pharmacodynamics, as well as the potential translatability to human patients. One study of prolonged VFCA in pigs found that direct administration of the combination of epinephrine, vasopressin, amiodarone, sodium bicarbonate, and metoprolol worsened short-term outcomes as compared to serial administration (133). The treatment of CA involves a complex pathology at the arrest, ROSC, and the post-ROSC phases. Therefore, ideally, a cocktail therapy should be phase-based as well as incorporating the general parameters, such as types of drugs, dosages, and methods of administration. The phase-based approach of cocktail development implies that the cocktail comprises of drugs that during ROSC and post-ROSC phases can (1) stabilize hemodynamics, (2) maintain mitochondrial membrane and function integrity, (3) decrease ROS generation, (4) provide metabolic supplementation, (5) reduce neuronal excitotoxicity, and (6) modulate neuroinflammation and protect blood-brain barrier integrity.
The concept of cocktail therapy has been applied in both preclinical and clinical settings for stroke to target various altered ischemic cascades with and without TTM (134–139). Although the focal IRI in stroke contrasts the global brain ischemia and damage observed in CA, the general neuronal protection observed in stroke combination therapies may be directly utilized, supplemented with other agents, and repurposed in a cocktail treatment of brain injury post-CA. Targeting multiple pathways allows for maximal neuroprotection, as targeting solitary pathways does not address the other dysregulated pathways that cause organ damage. Ultimately, while a combination of drugs is needed to mitigate the damage post-CA, the drugs chosen should be mechanistically multi-functional in order to use the fewest number of drugs possible to create the most efficacious multi-drug cocktail.
Conclusion
This review has summarized the current landscape of CA interventions using single drug therapy or a few drug combination therapies to target one or a few pathways to potentiate neuroprotection in both the preclinical and clinical environments. Furthermore, the implicated pathways of CA pathophysiology are the targets of various interventions and provide a foundation for the development of an extensive multi-drug cocktail; the use of a cocktail, involving several pharmaceutical agents, can simultaneously target the multitude of altered pathways and may synergistically confer neuroprotection. This cocktail may be further combined with more advanced resuscitation procedures, such as TTM and E-CPR. A cocktail therapy that combines various studied agents that can stabilize the hemodynamic, mitochondrial, metabolic, oxidative, and inflammatory processes may aid in reducing brain injury after cardiac arrest and improve survival with favorable neurologic outcomes.
Author Contributions
RC, MS, and LB contributed to the conception of the review and edited and revised the manuscript critically for important intellectual content. MS, SS, and RC contributed to the writing of the paper and interpreting relevant literature. DR, DJ, SM, KH, EM, and JK edited and revised the manuscript critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
The authors received funding in the form of an institutional research grant to the Feinstein Institute from United Therapeutics to support some of the content of this manuscript. The funder was not involved in the study design, experimental design, methods, data or tissue collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
LB has a grant/research support from Philips Healthcare, the NIH, Nihon Kohden Corp., Zoll Medical Corp, PCORI, BrainCool, and United Therapeutics and owns several issued and pending patents involving the use of medical slurries as human coolant, devices to create slurries, reperfusion cocktails, and measurement of respiratory quotient.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to acknowledge Ms. Sayeen Nagarajan Mohanapriya for her assistance in literature search.
References
1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. (2019) 139:e56–528. doi: 10.1161/CIR.0000000000000659
2. Sandroni C, D'Arrigo S, Nolan JP. Prognostication after cardiac arrest. Crit Care. (2018) 22:150. doi: 10.1186/s13054-018-2060-7
3. Geocadin RG, Callaway CW, Fink EL, Golan E, Greer DM, Ko NU, et al. Standards for studies of neurological prognostication in comatose survivors of cardiac arrest: a scientific statement from the American Heart Association. Circulation. (2019) 140:e517–42. doi: 10.1161/CIR.0000000000000702
4. van den Brule JMD, van der Hoeven JG, Hoedemaekers CWE. Cerebral perfusion and cerebral autoregulation after cardiac arrest. Biomed Res Int. (2018) 2018:4143636. doi: 10.1155/2018/4143636
5. Myat A, Song KJ, Rea T. Out-of-hospital cardiac arrest: current concepts. Lancet. (2018) 391:970–9. doi: 10.1016/S0140-6736(18)30472-0
6. Tahsili-Fahadan P, Farrokh S, Geocadin RG. Hypothermia and brain inflammation after cardiac arrest. Brain Circ. (2018) 4:1–13. doi: 10.4103/bc.BC_4_18
7. Watts ME, Pocock R, Claudianos C. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mol Neurosci. (2018) 11:216. doi: 10.3389/fnmol.2018.00216
8. Raichle ME, Gusnard DA. Appraising the brain's energy budget. Proc Natl Acad Sci USA. (2002) 99:10237–9. doi: 10.1073/pnas.172399499
9. Dennis M, Lal S, Forrest P, Nichol A, Lamhaut L, Totaro RJ, et al. In-depth extracorporeal cardiopulmonary resuscitation in adult out-of-hospital cardiac arrest. J Am Heart Assoc. (2020) 9:e016521. doi: 10.1161/JAHA.120.016521
10. Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, et al. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. (2003) 108:118–21. doi: 10.1161/01.CIR.0000079019.02601.90
11. Donnino MW, Andersen LW, Berg KM, Reynolds JC, Nolan JP, Morley PT, et al. Temperature management after cardiac arrest: an advisory statement by the advanced life support Task Force of the International Liaison Committee on Resuscitation and the American Heart Association Emergency Cardiovascular Care Committee and the Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Resuscitation. (2016) 98:97–104. doi: 10.1016/j.resuscitation.2015.09.396
12. Song SS, Lyden PD. Overview of therapeutic hypothermia. Curr Treat Options Neurol. (2012) 14:541–8. doi: 10.1007/s11940-012-0201-x
13. Gonzalez-Ibarra FP, Varon J, Lopez-Meza EG. Therapeutic hypothermia: critical review of the molecular mechanisms of action. Front Neurol. (2011) 2:4. doi: 10.3389/fneur.2011.00004
14. Shoaib M, Becker LB. A walk through the progression of resuscitation medicine. Ann NY Acad Sci. (2020). doi: 10.1111/nyas.14507. [Epub ahead of print].
15. Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. (2009) 37(7 Suppl.):S186–202. doi: 10.1097/CCM.0b013e3181aa5241
16. Soleimanpour H, Rahmani F, Golzari SE, Safari S. Main complications of mild induced hypothermia after cardiac arrest: a review article. J Cardiovasc Thorac Res. (2014) 6:1–8. doi: 10.5681/jcvtr.2014.001
17. Han F, Da T, Riobo NA, Becker LB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Crit Care Med. (2008) 36(11 Suppl.):S447–53. doi: 10.1097/CCM.0b013e31818a8a51
18. Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. (2004) 61:461–70. doi: 10.1016/j.cardiores.2003.10.025
19. Choi J, Shoaib M, Yin T, Nayyar G, Shinozaki K, Stevens JF, et al. Tissue-specific metabolic profiles after prolonged cardiac arrest reveal brain metabolome dysfunction predominantly after resuscitation. J Am Heart Assoc. (2019) 8:e012809. doi: 10.1161/JAHA.119.012809
20. Ristagno G, Fries M, Brunelli L, Fumagalli F, Bagnati R, Russo I, et al. Early kynurenine pathway activation following cardiac arrest in rats, pigs, and humans. Resuscitation. (2013) 84:1604–10. doi: 10.1016/j.resuscitation.2013.06.002
21. Lei B, Tan X, Cai H, Xu Q, Guo Q. Effect of moderate hypothermia on lipid peroxidation in canine brain tissue after cardiac arrest and resuscitation. Stroke. (1994) 25:147–52. doi: 10.1161/01.STR.25.1.147
22. Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, Fiskum G, et al. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. (2000) 20:543–9. doi: 10.1097/00004647-200003000-00013
23. Xu M, Zhou GM, Wang LH, Zhu L, Liu JM, Wang XD, et al. Inhibiting high-mobility group box 1 (HMGB1) attenuates inflammatory cytokine expression and neurological deficit in ischemic brain injury following cardiac arrest in rats. Inflammation. (2016) 39:1594–602. doi: 10.1007/s10753-016-0395-2
24. Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. (2010) 47:122–9. doi: 10.1016/j.ceca.2010.01.003
25. Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. (2010) 17:189–202. doi: 10.1007/s12640-009-9110-5
26. Yin XL, Zhang W, Yang Y, Shen H. Increasing expression of (CCAAT enhancer binding protein) homologous protein induced by endoplasmic reticulum stress in myocardium after cardiac arrest and resuscitation in rat. Resuscitation. (2012) 83:378–85. doi: 10.1016/j.resuscitation.2011.08.008
27. Ikeda K, Liu X, Kida K, Marutani E, Hirai S, Sakaguchi M, et al. Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation. (2016) 105:138–44. doi: 10.1016/j.resuscitation.2016.04.024
28. Jiang J, Fang X, Fu Y, Xu W, Jiang L, Huang Z. Impaired cerebral mitochondrial oxidative phosphorylation function in a rat model of ventricular fibrillation and cardiopulmonary resuscitation. Biomed Res Int. (2014) 2014:192769. doi: 10.1155/2014/192769
29. Piao L, Fang YH, Kubler MM, Donnino MW, Sharp WW. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE. (2017) 12:e0185046. doi: 10.1371/journal.pone.0185046
30. Bakthavachalam P, Shanmugam PST. Mitochondrial dysfunction - silent killer in cerebral ischemia. J Neurol Sci. (2017) 375:417–23. doi: 10.1016/j.jns.2017.02.043
31. Briston T, Roberts M, Lewis S, Powney B, J MS, Szabadkai G, et al. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep. (2017) 7:10492. doi: 10.1038/s41598-017-10673-8
32. Liu J, Wang Y, Zhuang Q, Chen M, Wang Y, Hou L, et al. Protective effects of cyclosporine A and hypothermia on neuronal mitochondria in a rat asphyxial cardiac arrest model. Am J Emerg Med. (2016) 34:1080–5. doi: 10.1016/j.ajem.2016.02.066
33. Argaud L, Cour M, Dubien PY, Giraud F, Jossan C, Riche B, et al. Effect of cyclosporine in nonshockable out-of-hospital cardiac arrest: the CYRUS randomized clinical trial. JAMA Cardiol. (2016) 1:557–65. doi: 10.1001/jamacardio.2016.1701
34. Knapp J, Roewer J, Bruckner T, Bottiger BW, Popp E. Evaluation of cyclosporine a as a cardio- and neuroprotective agent after cardiopulmonary resuscitation in a rat model. Shock. (2015) 43:576–81. doi: 10.1097/SHK.0000000000000357
35. Li Y, Tang Q, Wang P, Qin J, Wu H, Lin J, et al. Dynamic changes of mitochondrial fusion and fission in brain injury after cardiac arrest in rats. Biomed Res Int. (2017) 2017:1948070. doi: 10.1155/2017/1948070
36. Maneechote C, Palee S, Chattipakorn SC, Chattipakorn N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J Cell Mol Med. (2017) 21:2643–53. doi: 10.1111/jcmm.13330
37. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. (2014) 28:316–26. doi: 10.1096/fj.12-226225
38. Sharp WW. Dynamin-related protein 1 as a therapeutic target in cardiac arrest. J Mol Med. (2015) 93:243–52. doi: 10.1007/s00109-015-1257-3
39. Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, et al. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit Care Med. (2015) 43:e38–47. doi: 10.1097/CCM.0000000000000817
40. Ruiz A, Alberdi E, Matute C. Mitochondrial division inhibitor 1 (mdivi-1) protects neurons against excitotoxicity through the modulation of mitochondrial function and intracellular Ca(2+) signaling. Front Mol Neurosci. (2018) 11:3. doi: 10.3389/fnmol.2018.00003
41. Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci. (2018) 132:1669–83. doi: 10.1042/CS20180510
42. Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. (2014) 2:702–14. doi: 10.1016/j.redox.2014.05.006
43. Tsai MS, Huang CH, Tsai CY, Chen HW, Cheng HJ, Hsu CY, et al. Combination of intravenous ascorbic acid administration and hypothermia after resuscitation improves myocardial function and survival in a ventricular fibrillation cardiac arrest model in the rat. Acad Emerg Med. (2014) 21:257–65. doi: 10.1111/acem.12335
44. Wallert M, Ziegler M, Wang X, Maluenda A, Xu X, Yap ML, et al. alpha-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. (2019) 26:101292. doi: 10.1016/j.redox.2019.101292
45. Qin T, Lei LY, Li N, Shi FR, Chen MH, Xie L. Edaravone improves survival and neurological outcomes after CPR in a ventricular fibrillation model of rats. Am J Emerg Med. (2016) 34:1944–9. doi: 10.1016/j.ajem.2016.06.084
46. Silbergleit R, Haywood Y, Fiskum G, Rosenthal RE. Lack of a neuroprotective effect from N-acetylcysteine after cardiac arrest and resuscitation in a canine model. Resuscitation. (1999) 40:181–6. doi: 10.1016/S0300-9572(99)00027-1
47. Motl J, Radhakrishnan J, Ayoub IM, Grmec S, Gazmuri RJ. Vitamin C compromises cardiac resuscitability in a rat model of ventricular fibrillation. Am J Ther. (2014) 21:352–7. doi: 10.1097/MJT.0b013e31824e2b9f
48. Cheng C, Li H, Liang L, Jin T, Zhang G, Bradley JL, et al. Effects of omega-3 PUFA and ascorbic acid combination on post-resuscitation myocardial function. Biomed Pharmacother. (2021) 133:110970. doi: 10.1016/j.biopha.2020.110970
49. Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis. (2016) 6:93–108.
50. He F, Zheng G, Hou J, Hu Q, Ling Q, Wu G, et al. N-acetylcysteine alleviates post-resuscitation myocardial dysfunction and improves survival outcomes via partly inhibiting NLRP3 inflammasome induced-pyroptosis. J Inflamm. (2020) 17:25. doi: 10.1186/s12950-020-00255-3
51. Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, et al. Mitochondrial complex i inhibition by metformin limits reperfusion injury. J Pharmacol Exp Ther. (2019) 369:282–90. doi: 10.1124/jpet.118.254300
52. Yang L, Wang J, Deng Y, Gong C, Li Q, Chen Q, et al. Melatonin improves neurological outcomes and preserves hippocampal mitochondrial function in a rat model of cardiac arrest. PLoS ONE. (2018) 13:e0207098. doi: 10.1371/journal.pone.0207098
53. Piao L, Fang YH, Hamanaka RB, Mutlu GM, Dezfulian C, Archer SL, et al. Suppression of superoxide-hydrogen peroxide production at site IQ of mitochondrial complex I attenuates myocardial stunning and improves postcardiac arrest outcomes. Crit Care Med. (2020) 48:e133–40. doi: 10.1097/CCM.0000000000004095
54. Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Ohta S, et al. Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest independently of targeted temperature management. Circulation. (2014) 130:2173–80. doi: 10.1161/CIRCULATIONAHA.114.011848
55. Fries M, Brucken A, Cizen A, Westerkamp M, Lower C, Deike-Glindemann J, et al. Combining xenon and mild therapeutic hypothermia preserves neurological function after prolonged cardiac arrest in pigs. Crit Care Med. (2012) 40:1297–303. doi: 10.1097/CCM.0b013e31823c8ce7
56. Arola OJ, Laitio RM, Roine RO, Gronlund J, Saraste A, Pietila M, et al. Feasibility and cardiac safety of inhaled xenon in combination with therapeutic hypothermia following out-of-hospital cardiac arrest. Crit Care Med. (2013) 41:2116–24. doi: 10.1097/CCM.0b013e31828a4337
57. Mangus DB, Huang L, Applegate PM, Gatling JW, Zhang J, Applegate RL 2nd. A systematic review of neuroprotective strategies after cardiac arrest: from bench to bedside (Part I - Protection via specific pathways). Med Gas Res. (2014). 4:9. doi: 10.1186/2045-9912-4-9
58. Huang L, Applegate PM, Gatling JW, Mangus DB, Zhang J, Applegate RL 2nd. A systematic review of neuroprotective strategies after cardiac arrest: from bench to bedside (part II-comprehensive protection). Med Gas Res. (2014). 4:10. doi: 10.1186/2045-9912-4-10
59. Zhuang L, Yang T, Zhao H, Fidalgo AR, Vizcaychipi MP, Sanders RD, et al. The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit Care Med. (2012) 40:1724–30. doi: 10.1097/CCM.0b013e3182452164
60. Nomura J, Hosoi T, Kaneko M, Ozawa K, Nishi A, Nomura Y. Neuroprotection by endoplasmic reticulum stress-induced HRD1 and chaperones: possible therapeutic targets for Alzheimer's and Parkinson's disease. Med Sci. (2016) 4:14. doi: 10.3390/medsci4030014
61. Prentice H, Gharibani PM, Ma Z, Alexandrescu A, Genova R, Chen PC, et al. Neuroprotective functions through inhibition of ER stress by taurine or taurine combination treatments in a rat stroke model. Adv Exp Med Biol. (2017) 975(Pt 1):193–205. doi: 10.1007/978-94-024-1079-2_17
62. Zhang J, Wang Y, Ju M, Song J, Zheng Y, Lin S, et al. Neuroprotective effect of the inhibitor salubrinal after cardiac arrest in a rodent model. Oxid Med Cell Longev. (2020) 2020:7468738. doi: 10.1155/2020/7468738
63. Ristagno G, Latini R, Vaahersalo J, Masson S, Kurola J, Varpula T, et al. Early activation of the kynurenine pathway predicts early death and long-term outcome in patients resuscitated from out-of-hospital cardiac arrest. J Am Heart Assoc. (2014) 3:e001094. doi: 10.1161/JAHA.114.001094
64. Varvarousis D, Xanthos T, Ferino G, Noto A, Iacovidou N, Mura M, et al. Metabolomics profiling reveals different patterns in an animal model of asphyxial and dysrhythmic cardiac arrest. Sci Rep. (2017) 7:16575. doi: 10.1038/s41598-017-16857-6
65. Zhu J, Liu K, Huang K, Gu Y, Hu Y, Pan S, et al. Metformin improves neurologic outcome via AMP-activated protein kinase-mediated autophagy activation in a rat model of cardiac arrest and resuscitation. J Am Heart Assoc. (2018) 7:e008389. doi: 10.1161/JAHA.117.008389
66. Li J, Benashski S, McCullough LD. Post-stroke hypothermia provides neuroprotection through inhibition of AMP-activated protein kinase. J Neurotrauma. (2011) 28:1281–8. doi: 10.1089/neu.2011.1751
67. Lim CT, Kola B, Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol. (2010) 44:87–97. doi: 10.1677/JME-09-0063
68. Glatz JF. Challenges in fatty acid and lipid physiology. Front Physiol. (2011) 2:45. doi: 10.3389/fphys.2011.00045
69. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. (2018) 19:281–96. doi: 10.1038/nrm.2017.138
70. Dohi K, Miyamoto K, Fukuda K, Nakamura S, Hayashi M, Ohtaki H, et al. Status of systemic oxidative stress during therapeutic hypothermia in patients with post-cardiac arrest syndrome. Oxid Med Cell Longev. (2013) 2013:562429. doi: 10.1155/2013/562429
71. Rink C, Khanna S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid Redox Signal. (2011) 14:1889–903. doi: 10.1089/ars.2010.3474
72. Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. (1998) 29:1679–86. doi: 10.1161/01.STR.29.8.1679
73. Kuschner CE, Yagi T, Kim J. Chapter 2 - thinking in polyunsaturated fatty acids, phospholipids, and the brain. In: Watson RR, Preedy VR, editors. Omega Fatty Acids in Brain and Neurological Health. 2nd ed. Cambridge, MA: Academic Press (2019). p. 21–32.
74. Kim J, Lampe JW, Yin T, Shinozaki K, Becker LB. Phospholipid alterations in the brain and heart in a rat model of asphyxia-induced cardiac arrest and cardiopulmonary bypass resuscitation. Mol Cell Biochem. (2015) 408:273–81. doi: 10.1007/s11010-015-2505-0
75. Kim J, Villarroel JP, Zhang W, Yin T, Shinozaki K, Hong A, et al. The responses of tissues from the brain, heart, kidney, and liver to resuscitation following prolonged cardiac arrest by examining mitochondrial respiration in rats. Oxid Med Cell Longev. (2016) 2016:7463407. doi: 10.1155/2016/7463407
76. Tam J, Hong A, Naranjo PM, Yin T, Shinozaki K, Lampe JW, et al. The role of decreased cardiolipin and impaired electron transport chain in brain damage due to cardiac arrest. Neurochem Int. (2018) 120:200–5. doi: 10.1016/j.neuint.2018.08.013
77. Anthonymuthu TS, Kenny EM, Lamade AM, Gidwani H, Krehel NM, Misse A, et al. Lipidomics detection of brain cardiolipins in plasma is associated with outcome after cardiac arrest. Crit Care Med. (2019) 47:e292–300. doi: 10.1097/CCM.0000000000003636
78. Wu J, Wu Q, Wang D, Kong J, Dai W, Wang X, et al. Common lipid features of lethal ventricular tarchyarrhythmias (LVTAs) induced by myocardial infarction and myocardial ion channel diseases. Sci Rep. (2017) 7:4220. doi: 10.1038/s41598-017-04620-w
79. Zhang W, Tam J, Shinozaki K, Yin T, Lampe JW, Becker LB, et al. Increased survival time with SS-31 after prolonged cardiac arrest in rats. Heart Lung Circ. (2019) 28:505–8. doi: 10.1016/j.hlc.2018.01.008
80. Xiao Y, Contaifer D, Huang W, Yang J, Hu Z, Guo Q, et al. Cannabinoid receptor agonist WIN55, 212-2 adjusts lipid metabolism in a rat model of cardiac arrest. Ther Hypothermia Temp Manag. (2020) 10:192–203. doi: 10.1089/ther.2019.0038
81. Buckley MM, Goa KL, Price AH, Brogden RN. Probucol. A reappraisal of its pharmacological properties and therapeutic use in hypercholesterolaemia. Drugs. (1989) 37:761–800. doi: 10.2165/00003495-198937060-00002
82. Xiao X, Hou H, Lin V, Ho D, Tran K, Che B, et al. Probucol protects rats from cardiac dysfunction induced by oxidative stress following cardiopulmonary resuscitation. Oxid Med Cell Longev. (2017) 2017:1284804. doi: 10.1155/2017/1284804
83. Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. (2006) 24:1–21. doi: 10.1016/j.ncl.2005.10.004
84. Weinberger J, Nieves-Rosa J, Cohen G. Nerve terminal damage in cerebral ischemia: protective effect of alpha-methyl-para-tyrosine. Stroke. (1985) 16:864–70. doi: 10.1161/01.STR.16.5.864
85. Globus MY, Ginsberg MD, Dietrich WD, Busto R, Scheinberg P. Substantia nigra lesion protects against ischemic damage in the striatum. Neurosci Lett. (1987) 80:251–6. doi: 10.1016/0304-3940(87)90463-0
86. Kahn RA, Weinberger J, Brannan T, Prikhojan A, Reich DL. Nitric oxide modulates dopamine release during global temporary cerebral ischemia. Anesth Analg. (1995) 80:1116–21. doi: 10.1213/00000539-199506000-00008
87. Hifumi T, Kawakita K, Yoda T, Okazaki T, Kuroda Y. Association of brain metabolites with blood lactate and glucose levels with respect to neurological outcomes after out-of-hospital cardiac arrest: a preliminary microdialysis study. Resuscitation. (2017) 110:26–31. doi: 10.1016/j.resuscitation.2016.10.013
88. Scott GF, Nguyen AQ, Cherry BH, Hollrah RA, Salinas I, Williams AG, et al. Featured article: pyruvate preserves antiglycation defenses in porcine brain after cardiac arrest. Exp Biol Med. (2017) 242:1095–103. doi: 10.1177/1535370217703353
89. Lazovic J, Basu A, Lin HW, Rothstein RP, Krady JK, Smith MB, et al. Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin-1 type 1 receptor-deficient mice conferring neuroprotection. Stroke. (2005) 36:2226–31. doi: 10.1161/01.STR.0000182255.08162.6a
90. Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. (1997) 28:1233–44. doi: 10.1161/01.STR.28.6.1233
91. Famakin B, Mou Y, Spatz M, Lawal M, Hallenbeck J. Downstream Toll-like receptor signaling mediates adaptor-specific cytokine expression following focal cerebral ischemia. J Neuroinflammation. (2012) 9:174. doi: 10.1186/1742-2094-9-174
92. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. (2016) 2016:2183026. doi: 10.1155/2016/2183026
93. Yang X, Wang Z, Jia X. Neuroprotection of glibenclamide against brain injury after cardiac arrest via modulation of NLRP3 inflammasome. Conf Proc IEEE Eng Med Biol Soc. (2019) 2019:4209–12. doi: 10.1109/EMBC.2019.8857285
94. Kamarainen A, Virkkunen I, Silfvast T, Tenhunen J. Statins for post resuscitation syndrome. Med Hypotheses. (2009) 73:97–9. doi: 10.1016/j.mehy.2009.01.021
95. Katz LM, Frank JE, Glickman LT, McGwin G Jr, Lambert BH, et al Effect of a pharmacologically induced decrease in core temperature in rats resuscitated from cardiac arrest. Resuscitation. (2015) 92:26–31. doi: 10.1016/j.resuscitation.2015.04.009
96. Cariou A, Deye N, Vivien B, Richard O, Pichon N, Bourg A, et al. Early high-dose erythropoietin therapy after out-of-hospital cardiac arrest: a multicenter, randomized controlled trial. J Am Coll Cardiol. (2016) 68:40–9. doi: 10.1016/j.jacc.2016.04.040
97. Cariou A, Claessens YE, Pene F, Marx JS, Spaulding C, Hababou C, et al. Early high-dose erythropoietin therapy and hypothermia after out-of-hospital cardiac arrest: a matched control study. Resuscitation. (2008) 76:397–404. doi: 10.1016/j.resuscitation.2007.10.003
98. Ikeda M, Swide T, Vayl A, Lahm T, Anderson S, Hutchens MP. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit Care. (2015) 19:332. doi: 10.1186/s13054-015-1049-8
99. Huang K, Wang Z, Gu Y, Hu Y, Ji Z, Wang S, et al. Glibenclamide is comparable to target temperature management in improving survival and neurological outcome after asphyxial cardiac arrest in rats. J Am Heart Assoc. (2016) 5:e003465. doi: 10.1161/JAHA.116.003465
100. Li L, Yang R, Li P, Lu H, Hao J, Li L, et al. Combination treatment with methylene blue and hypothermia in global cerebral ischemia. Mol Neurobiol. (2018) 55:2042–55. doi: 10.1007/s12035-017-0470-1
101. Wiklund L, Zoerner F, Semenas E, Miclescu A, Basu S, Sharma HS. Improved neuroprotective effect of methylene blue with hypothermia after porcine cardiac arrest. Acta Anaesthesiol Scand. (2013) 57:1073–82. doi: 10.1111/aas.12106
102. Miclescu A, Sharma HS, Martijn C, Wiklund L. Methylene blue protects the cortical blood-brain barrier against ischemia/reperfusion-induced disruptions. Crit Care Med. (2010) 38:2199–206. doi: 10.1097/CCM.0b013e3181f26b0c
103. Bar-Joseph G, Abramson NS, Kelsey SF, Mashiach T, Craig MT, Safar P, et al. Improved resuscitation outcome in emergency medical systems with increased usage of sodium bicarbonate during cardiopulmonary resuscitation. Acta Anaesthesiol Scand. (2005) 49:6–15. doi: 10.1111/j.1399-6576.2005.00572.x
104. Tsai MS, Chuang PY, Huang CH, Tang CH, Yu PH, Chang WT, et al. Postarrest steroid use may improve outcomes of cardiac arrest survivors. Crit Care Med. (2019) 47:167–75. doi: 10.1097/CCM.0000000000003468
105. Katz L, Vaagenes P, Safar P, Diven W. Brain enzyme changes as markers of brain damage in rat cardiac arrest model. Effects of corticosteroid therapy. Resuscitation. (1989) 17:39–53. doi: 10.1016/0300-9572(89)90078-6
106. Tamura T, Hayashida K, Sano M, Onuki S, Suzuki M. Efficacy of inhaled hydrogen on neurological outcome following brain ischemia during post-cardiac arrest care (HYBRID II trial): study protocol for a randomized controlled trial. Trials. (2017) 18:488. doi: 10.1186/s13063-017-2246-3
107. Lundin A, Djarv T, Engdahl J, Hollenberg J, Nordberg P, Ravn-Fischer A, et al. Drug therapy in cardiac arrest: a review of the literature. Eur Heart J Cardiovasc Pharmacother. (2016) 2:54–75. doi: 10.1093/ehjcvp/pvv047
108. Paskitti M, Reid KH. Use of an adenosine triphosphate-based 'cocktail' early in reperfusion substantially improves brain protein synthesis after global ischemia in rats. Neurosci Lett. (2002) 331:147–50. doi: 10.1016/S0304-3940(02)00588-8
109. Kwon WY, Suh GJ, Kim KS, Jung YS, Kim SH, Lee AR, et al. Niacin and selenium attenuate brain injury after cardiac arrest in rats by up-regulating DJ-1-Akt signaling. Crit Care Med. (2018) 46:e788–96. doi: 10.1097/CCM.0000000000003198
110. Bartos JA, Matsuura TR, Sarraf M, Youngquist ST, McKnite SH, Rees JN, et al. Bundled postconditioning therapies improve hemodynamics and neurologic recovery after 17 min of untreated cardiac arrest. Resuscitation. (2015) 87:7–13. doi: 10.1016/j.resuscitation.2014.10.019
111. Lucchinetti E, Ambrosio S, Aguirre J, Herrmann P, Harter L, Keel M, et al. Sevoflurane inhalation at sedative concentrations provides endothelial protection against ischemia-reperfusion injury in humans. Anesthesiology. (2007) 106:262–8. doi: 10.1097/00000542-200702000-00013
112. Meybohm P, Gruenewald M, Albrecht M, Muller C, Zitta K, Foesel N, et al. Pharmacological postconditioning with sevoflurane after cardiopulmonary resuscitation reduces myocardial dysfunction. Crit Care. (2011) 15:R241. doi: 10.1186/cc10496
113. Maskarinec SA, Hannig J, Lee RC, Lee KY. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys J. (2002) 82:1453–9. doi: 10.1016/S0006-3495(02)75499-4
114. Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, et al. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. (2010) 120:1140–50. doi: 10.1172/JCI41329
115. Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, et al. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res. (2012) 37:2856–67. doi: 10.1007/s11064-012-0880-4
116. Gu JH, Ge JB, Li M, Xu HD, Wu F, Qin ZH. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE. (2013) 8:e61641. doi: 10.1371/journal.pone.0061641
117. Guyette FX, Guimond GE, Hostler D, Callaway CW. Vasopressin administered with epinephrine is associated with a return of a pulse in out-of-hospital cardiac arrest. Resuscitation. (2004) 63:277–82. doi: 10.1016/j.resuscitation.2004.05.007
118. Callaway CW, Hostler D, Doshi AA, Pinchalk M, Roth RN, Lubin J, et al. Usefulness of vasopressin administered with epinephrine during out-of-hospital cardiac arrest. Am J Cardiol. (2006) 98:1316–21. doi: 10.1016/j.amjcard.2006.06.022
119. Varvarousi G, Johnson EO, Goulas S, Agrogiannis G, Valsamakis N, Perrea D, et al. Combination pharmacotherapy improves neurological outcome after asphyxial cardiac arrest. Resuscitation. (2012) 83:527–32. doi: 10.1016/j.resuscitation.2011.09.015
120. Ghafourian N, Maniae NH, Taherikalani M, Mehrazi M, Hossieni M, Valian F, et al. Combination of vasopressin -epinephrine as a novel candidate in patients with cardiac arrest. Recent Adv Cardiovasc Drug Discov. (2015) 10:65–9. doi: 10.2174/157489011001160111163856
121. Mentzelopoulos SD, Malachias S, Chamos C, Konstantopoulos D, Ntaidou T, Papastylianou A, et al. Vasopressin, steroids, and epinephrine and neurologically favorable survival after in-hospital cardiac arrest: a randomized clinical trial. JAMA. (2013) 310:270–9. doi: 10.1001/jama.2013.7832
122. Buddineni JP, Callaway C, Huang DT. Epinephrine, vasopressin and steroids for in-hospital cardiac arrest: the right cocktail therapy? Crit Care. (2014) 18:308. doi: 10.1186/cc13903
123. Mentzelopoulos SD, Zakynthinos SG, Tzoufi M, Katsios N, Papastylianou A, Gkisioti S, et al. Vasopressin, epinephrine, and corticosteroids for in-hospital cardiac arrest. Arch Intern Med. (2009) 169:15–24. doi: 10.1001/archinternmed.2008.509
124. Liu B, Zhang Q, Li C. Steroid use after cardiac arrest is associated with favourable outcomes: a systematic review and meta-analysis. J Int Med Res. (2020) 48:300060520921670. doi: 10.1177/0300060520921670
125. Lurie KG, Voelckel WG, Iskos DN, McKnite SH, Zielinski TM, Sugiyama A, et al. Combination drug therapy with vasopressin, adrenaline (epinephrine) and nitroglycerin improves vital organ blood flow in a porcine model of ventricular fibrillation. Resuscitation. (2002) 54:187–94. doi: 10.1016/S0300-9572(02)00096-5
126. Wang Y, Gao L, Meng L. Naloxone combined with epinephrine decreases cerebral injury in cardiopulmonary resuscitation. J Emerg Med. (2010) 39:296–300. doi: 10.1016/j.jemermed.2008.10.014
127. Lewanowitsch T, Irvine RJ. Naloxone methiodide reverses opioid-induced respiratory depression and analgesia without withdrawal. Eur J Pharmacol. (2002) 445:61–7. doi: 10.1016/S0014-2999(02)01715-6
128. Polderman KH. Application of therapeutic hypothermia in the intensive care unit. Opportunities and pitfalls of a promising treatment modality–Part 2: Practical aspects and side effects. Intensive Care Med. (2004) 30:757–69. doi: 10.1007/s00134-003-2151-y
129. Choudhary RC, Jia X. Hypothalamic or extrahypothalamic modulation and targeted temperature management after brain injury. Ther Hypothermia Temp Manag. (2017) 7:125–33. doi: 10.1089/ther.2017.0003
130. Katz LM, Frank JE, McGwin G Jr, Finch A, Gordon CJ. Induction of a prolonged hypothermic state by drug-induced reduction in the thermoregulatory set-point. Ther Hypothermia Temp Manag. (2012) 2:61–6. doi: 10.1089/ther.2012.0011
131. Katz LM, McGwin G Jr, Gordon CJ. Drug-induced therapeutic hypothermia after asphyxial cardiac arrest in swine. Ther Hypothermia Temp Manag. (2012) 2:176–82. doi: 10.1089/ther.2012.0017
132. Shoaib M, Choudhary RC, Choi J, Kim N, Hayashida K, Yagi T, et al. Plasma metabolomics supports the use of long-duration cardiac arrest rodent model to study human disease by demonstrating similar metabolic alterations. Sci Rep. (2020) 10:19707. doi: 10.1038/s41598-020-76401-x
133. Mader TJ, Coute RA, Kellogg AR, Nathanson BH. Blinded evaluation of combination drug therapy for prolonged ventricular fibrillation using a swine model of sudden cardiac arrest. Prehosp Emerg Care. (2016) 20:390–8. doi: 10.3109/10903127.2015.1086848
134. Xiong XY, Liu L, Yang QW. Refocusing neuroprotection in cerebral reperfusion era: new challenges and strategies. Front Neurol. (2018) 9:249. doi: 10.3389/fneur.2018.00249
135. Schmid-Elsaesser R, Hungerhuber E, Zausinger S, Baethmann A, Reulen HJ. Combination drug therapy and mild hypothermia: a promising treatment strategy for reversible, focal cerebral ischemia. Stroke. (1999) 30:1891–9. doi: 10.1161/01.STR.30.9.1891
136. Schmid-Elsaesser R, Zausinger S, Hungerhuber E, Baethmann A, Reulen HJ. Neuroprotective effects of combination therapy with tirilazad and magnesium in rats subjected to reversible focal cerebral ischemia. Neurosurgery. (1999) 44:163–71; discussion: 71–2. doi: 10.1097/00006123-199901000-00100
137. Zausinger S, Scholler K, Plesnila N, Schmid-Elsaesser R. Combination drug therapy and mild hypothermia after transient focal cerebral ischemia in rats. Stroke. (2003) 34:2246–51. doi: 10.1161/01.STR.0000083622.65684.21
138. Culmsee C, Junker V, Kremers W, Thal S, Plesnila N, Krieglstein J. Combination therapy in ischemic stroke: synergi stic neuroprotective effects of memantine and clenbuterol. Stroke. (2004) 35:1197–202. doi: 10.1161/01.STR.0000125855.17686.6d
139. Ozyener F, Cetinkaya M, Alkan T, Goren B, Kafa IM, Kurt MA, et al. Neuroprotective effects of melatonin administered alone or in combination with topiramate in neonatal hypoxic-ischemic rat model. Restor Neurol Neurosci. (2012) 30:435–44. doi: 10.3233/RNN-2012-120217
Glossary
ACA: asphyxial cardiac arrest
ACLS: advanced cardiovascular life support
AMPK: 5′-adenosine monophosphate-activated protein kinase
ATP-MgCl2: adenosine triphosphate-magnesium chloride
CA: cardiac arrest
CL: cardiolipin
CPB: cardiopulmonary bypass
CPR: cardiopulmonary resuscitation
DETC-MeSO: S-Methyl-N, N-diethylthiocarbamate sulfoxide
DRP1: dynamin-related protein 1
E-CPR: extracorporeal cardiopulmonary resuscitation
eIF2α: eukaryotic Initiation Factor 2 alpha
ER: endoplasmic reticulum
GBC: glibenclamide
HIF-1α: hypoxia-inducible factor 1-alpha
IHCA: in-hospital cardiac arrest
IRI: Ischemia- Reperfusion Injury
IL1-α: interleukin-1 alpha
IL-1β: interleukin-1 beta
IL-18: interleukin-18
LPC: lysophosphatidylcholine
LPE: lysophosphatidylethanolamine
LPI: lysophosphatidylinositol
MCAO: middle cerebral artery occlusion
Mdivi-1: mitochondrial division inhibitor 1
MLCL: monolysocardiolipin
mPTP: mitochondrial permeability transition pore
NDS: neurodeficit score
NF-κB: nuclear factor-kappa B
NLRP3: nucleotide-binding oligomerization domain, leucine rich, repeat and, pyrin domain-containing 3
NMDA: N-Methyl- d-aspartate
NSE: neuron specific enolase
OHCA: out-of-hospital cardiac arrest
PDH: pyruvate dehydrogenase
ROS: reactive oxygen species
ROSC: return of spontaneous circulation
S1QEL: suppressor of site IQ electron leak
SS-31: Szeto-Schiller peptide-31
TCA: tricarboxylic acid
TH: therapeutic hypothermia
TNF-α: tumor necrosis factor alpha
TTM: targeted temperature management
UPR: unfolded protein response
VFCA: ventricular fibrillation cardiac arrest
Keywords: cardiopulmonary arrest, ischemia and reperfusion injury, resuscitation, neuroprotection, cerebral ischemia, pharmacological intervention, cocktail therapy
Citation: Choudhary RC, Shoaib M, Sohnen S, Rolston DM, Jafari D, Miyara SJ, Hayashida K, Molmenti EP, Kim J and Becker LB (2021) Pharmacological Approach for Neuroprotection After Cardiac Arrest—A Narrative Review of Current Therapies and Future Neuroprotective Cocktail. Front. Med. 8:636651. doi: 10.3389/fmed.2021.636651
Received: 09 December 2020; Accepted: 12 April 2021;
Published: 18 May 2021.
Edited by:
Vidula Vachharajani, Case Western Reserve University, United StatesReviewed by:
Adriana Miclescu, Uppsala University, SwedenWillard Sharp, University of Chicago, United States
Copyright © 2021 Choudhary, Shoaib, Sohnen, Rolston, Jafari, Miyara, Hayashida, Molmenti, Kim and Becker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lance B. Becker, lance.becker@northwell.edu
†These authors have contributed equally to this work