Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment
 Amarpreet Kaur
Amarpreet Kaur Susan K. Mathai
Susan K. Mathai David A. Schwartz
David A. Schwartz- 1Department of Medicine, University of Colorado Denver School of Medicine, Aurora, CO, United States
- 2Department of Medicine, Division of Pulmonary Sciences and Critical Care Medicine, University of Colorado Denver School of Medicine, Aurora, CO, United States
Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia (IIP), is characterized by irreversible scarring of the lung parenchyma and progressive decline in lung function leading to eventual respiratory failure. The prognosis of IPF is poor with a median survival of 3–5 years after diagnosis and no curative medical therapies. Although the pathogenesis of IPF is not well understood, there is a growing body of evidence that genetic factors contribute to disease risk. Recent studies have identified common and rare genetic variants associated with both sporadic and familial forms of pulmonary fibrosis, with at least one-third of the risk for developing fibrotic IIP explained by common genetic variants. The IPF-associated genetic loci discovered to date are implicated in diverse biological processes, including alveolar stability, host defense, cell–cell barrier function, and cell senescence. In addition, some common variants have also been associated with distinct clinical phenotypes. Better understanding of how genetic variation plays a role in disease risk and phenotype could identify potential therapeutic targets and inform clinical decision-making. In addition, clinical studies should be designed controlling for the genetic backgrounds of subjects, since clinical outcomes and therapeutic responses may differ by genotype. Further understanding of these differences will allow the development of personalized approaches to the IPF management.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common of the idiopathic interstitial pneumonias (IIPs). IPF is characterized by progressive scarring of the lung parenchyma, which leads to dyspnea and declining pulmonary function and eventually to respiratory failure. The median survival after diagnosis of IPF is 3–5 years (1). In 2011, the American Thoracic Society/European Respiratory Society issued a new classification scheme in which they defined IPF as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown etiology, occurring mainly in older adults and associated with radiological and/or histopathological pattern of usual interstitial pneumonia (UIP) (2). The prognosis of IPF remains poor despite recently approved medical therapies (3, 4).
Numerous epidemiologic and genetic studies illustrate that genetic and environmental factors contribute to the risk of IPF (5, 6). The most convincing early evidence to support a genetic basis for IPF came from twin studies and studies focusing on familial clustering of the disease, a syndrome termed familial interstitial pneumonia (FIP) (7–9). Recent studies have identified several specific genetic variants that confer risk for development of IPF (10, 11). Discovery of disease-associated genetic variants has improved our understanding of the ways inherited risk factors influence disease risk. However, fundamental questions persist regarding the ways in which complex genetic risk factors interact with environmental exposures to influence disease pathogenesis.
In this review, we briefly discuss the current literature regarding the role of common and rare variants in disease pathogenesis and prognosis and how this may influence clinical management in the future. Genetic variants and loci associated with IPF involve abnormalities in alveolar stability, host defense, cell–cell barrier function, and cell senescence, all of which are all thought to contribute to the pathogenesis of IPF. We conclude by discussing how treatment decisions might be affected by these findings and how better understanding of genetic variation and disease could allow for a more personalized approach to the treatment of IPF.
Rare and Common Variants Associated with IPF
Genetic variants, both rare and common, are associated with sporadic and familial forms of pulmonary fibrosis. Numerous rare variants (those with minor allele frequency of <0.1%) play a role in FIP (≥2 members of the same family with interstitial pneumonia; FIP) (Table 1). Familial studies have identified FIP-associated variants related to alveolar stability [SFTPC (12, 13), SFTPA1 (14), SFTPA2 (15), ATP-binding cassette-type 3 (ABCA3) (16), and NAF1 (17)] as well as five genes linked to telomere biology [TERT (18), TERC (18), DKC1 (19), TINF2 (7, 20), RTEL1 (21–23), and PARN] (24).
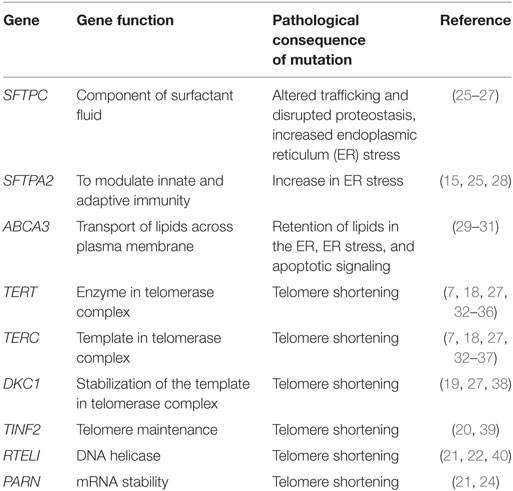
Table 1. Rare variants in idiopathic pulmonary fibrosis.
Common variants (defined as minor allele frequency of >5%) also appear to play a role in FIP risk (1). The most widely replicated risk variant (rs35705950), located in the promoter region of MUC5B, was initially identified in a combined linkage and association study (41) and has been strongly associated with IPF and FIP. Two large GWAS of IPF subjects (both familial and sporadic) with controls have been conducted in pulmonary fibrosis (10, 11). In addition to confirming the role of TERT at 5p15, MUC5B at 11p15, and the 3q26 region near TERC, the GWAS identified seven newly associated loci, including FAM13A (4q22), DSP (6q24), OBFC1 (10q24), ATP11A (13q34), DPP9 (19q13), and chromosomal regions 7q22 and 15q14-15 among others that have been nominally associated (Table 2).

Table 2. Common variants in idiopathic pulmonary fibrosis (IPF).
Rare variants are thought to be highly penetrant and to have a greater effect size, but given their low frequency, they account for a smaller proportion of overall disease risk in the general population (51). Alternatively, in general, common variants have a smaller effect size but are present at higher frequency and, in aggregate, may contribute to a larger proportion of disease risk (Figure 1). However, the MUC5B promoter variant rs35705950 is a common variant with a large effect size and therefore accounts for a substantial risk in IPF. In fact, it has been estimated the MUC5B promoter variant accounts for 30% of the risk of developing IPF (41, 51).
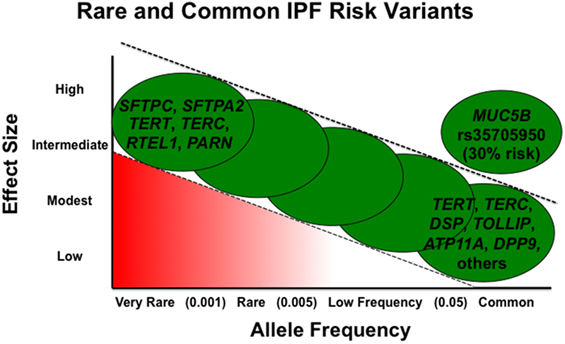
Figure 1. Adapted from figure published previously in BMC Medicine with permission (52).
Alveolar Stability
Surfactant proteins are synthesized in the endoplasmic reticulum (ER) of alveolar type II cells (AECII) and transported to and stored in the lamellar bodies until secretion into the alveolar space (25, 26). Rare variants identified in the genes encoding surfactant protein C and A (SFTPC, SFTPA1, and SFTPA2) have been associated with pulmonary fibrosis (53). SP-C is a small hydrophobic protein produced by AECIIs that requires the C-terminus for initial folding steps in the ER before secretion into the alveolar space (26). SFTPC rare variants are mutations that lie in the BRICHOS domain within the C-terminus of SP-C. The BRICHOS domain is critical for proper folding and trafficking (5, 26). Coding mutations in this region lead to accumulation of misfolded protein resulting in increased ER stress and activation of the unfolded protein response (26, 54). Mutations in the gene that encodes surfactant protein A (SFTPA2) have also been linked to FIP (15) and have been associated with increased ER stress as well (28, 55). Rare variants have also been identified in another gene involved with surfactant processing, ABCA3, in FIP families (16, 56). ABCA3 is a transporter protein mainly expressed in AECIIs and is involved in the transport of lipids across plasma membranes (29, 57). In AECIIs, ABCA3 mutations cause abnormal processing, trafficking, and functionality of the ABCA protein, leading to retention of lipids in the ER, ER stress, and apoptotic signaling (30). These mutations are expressed in a recessive manner, where as mutations in SFTPA2 and SFTPC are dominantly expressed (56).
In 2011, Lawson et al. (58) demonstrated that fibrotic remodeling in response to low-dose bleomycin was more severe in mice in which ER stress was induced, either through mutant SFTPC in AECIIs or by administration of tunicamycin, a chemical known to induce ER stress. In addition to effects on apoptosis, ER stress may induce biological pathways involved in cell differentiation (59, 60) through which epithelial cells acquire phenotypic characteristics of mesenchymal cells, a process known as epithelial-to-mesenchymal transition (EMT), in IPF lungs (61). EMT is hypothesized to increase the number of cells responsible for collagen production and matrix deposition thereby leading to fibrosis (13, 59, 60). To date, published data suggest that ER stress predisposes to AECII apoptosis and subsequent lung fibrosis. Surfactant proteins have been recognized as crucial in maintaining lung alveolar structure and function. However, the precise role of alveolar stability, ER stress, and EMT in IPF pathogenesis remains an area of active investigation.
Cell Senescence
Telomeres are repetitive nucleotide sequences at the ends of chromosomes that protect them from progressive shortening during the normal cell replication process (62). Telomerases restore telomere length and consist of two major components: telomerase reverse transcriptase (encoded by TERT) and telomerase RNA (encoded by TERC) (18, 37). Mutations in telomerase components were initially identified in the setting of dyskeratosis congenita (DKC), a rare inherited syndrome of telomere shortening characterized by oral leukoplakia, abnormal skin hyperpigmentation, and dystrophic nails, with pulmonary fibrosis present in about 20% of patients; bone marrow failure can also be a complication of DKC (32). More recent studies have found an association between numerous genes in the telomerase maintenance pathway and FIP, including those related to catalytic activity (TERT and TERC) (7, 32) and telomere stabilization (DKC1, PARN, and RTELI) (19, 21). These pathogenic variants cause dysfunction of telomerase activity leading to accelerated telomere shortening (32, 63) in peripheral blood and the lung (32–34). Thus far, TERT variants are the most frequently identified rare variants associated with pulmonary fibrosis; they are found in ~15% of FIP (7, 32) and in 1–3% of sporadic cases (34). A recent whole-exome sequencing study identified TERT, RTEL1, and PARN variants previously associated with FIP to be associated with sporadic IPF, further supporting the role of telomere dysfunction in IPF pathogenesis and highlighting the genetic commonalities between FIP and sporadic IPF (64).
Telomere dysfunction has further been implicated in IPF as evidence has suggested that short telomeres are not exclusively related to telomerase rare variant mutations. One study found that 25% of sporadic IPF subjects and 24% of familial IPF subjects, without identified mutations for TERT or TERC, had short telomeres. In addition, all subjects within this specific study who had a mutation in TERT or TERC and pulmonary fibrosis also had short telomeres (33).
The mechanisms by which telomere defects provoke lung disease are not fully understood. Defects in telomere maintenance have been linked to epithelial cell senescence and an impaired response to epithelial injury (65). During successive cycles of cell division, telomere shortening occurs and eventually leads to activation of the DNA-damage pathways, which result in apoptosis or senescence (32). In certain situations, cellular senescence is appropriate, but premature senescence can impair lung epithelial homeostasis and lead to stimulation of a lung remodeling response (66), resulting in fibrotic lesions (63). One study demonstrated increased epithelial cell senescence in IPF lung tissue by measuring B-galactosidase staining (a marker of senescence) and found that B-galactosidase staining was positive in all IPF cases but was not present in normal lung (67) supporting a role for senescent epithelial cells in IPF pathogenesis. Future studies are necessary to clarify the precise role of cellular senescence in lung injury response and fibrotic remodeling in IPF.
Host Defense
In 2011, genome-wide linkage analysis and targeted genetic sequencing identified a single nucleotide polymorphism (SNP) on chromosome 11 that was associated with both FIP and IPF (41). The SNP, rs35705950, was found to be a gain-of-function variant associated with increased expression of MUC5B. Heterozygous (GT) and homozygous (TT) individuals had an odds ratio for developing disease of 6.8 and 20.8 for FIP, and 9.0 and 21.8 for IPF, respectively, supporting the strength of the SNP’s association with development of both IPF and FIP (41). MUC5B encodes Mucin 5B, which is a major gel-forming mucin in mucus and expressed by airway epithelial cells (68, 69). The association of the MUC5B promoter polymorphism with IPF has been replicated and confirmed in nine independent cohorts (10, 11, 49, 50, 70–74), including in a 2013 GWAS (OR for T minor allele = 4.51; 95% CI = 3.91–5.21; P = 7.21 × 10−95) (10). Additional genotyping studies have noted that the MUC5B variant is associated with disease in a Mexican cohort of IPF patients, but not in Asian cohorts (75). Most recently, a study of select loci in various European cohorts, including Czech and Greek IPF patients, also confirmed the association between rs35705950 and IPF (76).
MUC5B expression in IPF is localized in the distal airway, respiratory bronchiole, honeycomb cyst (77), and the bronchiolar epithelium (78). Overexpression of MUC5B in these areas of the lung and especially in the honeycomb cysts, which are a histopathological finding in IPF (77), further supports the notion that MUC5B is important in the pathogenesis of IPF. Evans and colleagues (51) hypothesize that IPF is caused by recurrent injury/repair/regeneration at the bronchoalveolar junction secondary to overexpression of MUC5B, mucociliary dysfunction, retention of particles, ER stress, and disruption of normal reparative and regenerative mechanisms in the distal lung (51).
Interestingly, the MUC5B promoter polymorphism may be specific to IIP, since studies have illustrated that rs35705950 is not associated with increased risk of sarcoidosis and scleroderma-related ILD, two other fibrotic lung diseases (10, 50, 70). However, recent data have shown that MUC5B rs357057950 variant is associated with radiographic evidence of interstitial lung abnormalities (ILA) studied in the Framingham cohort (79–81). Increasing age and number of copies of MUC5B promoter polymorphism were associated with ILA progression, which has been linked to increased mortality (80, 81). In addition, there are some data to suggest that rs357057950 genotype may be associated with higher likelihood of radiographic UIP pattern in the setting of fibrotic IIP (82).
The mechanism by which variants in MUC5B confers risk of lung fibrosis is an active area of investigation. Given that mucins play a role in innate immunity (68, 83), immune dysregulation could be a possible mechanism by which increased mucin expression contributes to the pathophysiology of IPF (84). Alternatively, IPF may be a disease of mucociliary clearance in which overexpression of MUC5B leads to impaired ciliary function, thereby allowing retention of particles and, subsequently, recurrent lung injury (51).
Several studies have also implicated the human leukocyte antigen (HLA) region in IPF (85–89). The HLA region is located on chromosome 6p21.31 (90), and its main function is regulation of immune response. The DRB1*15:01 allele has been shown to be more prevalent among IPF patients and associated with greater impairment of gas exchange (89). Recently, a genome-wide imputation-based association analysis identified two risk alleles, DRB1*15:01 and DQB1*06:02, found to be associated with fibrotic idiopathic interstitial pneumonias (48). Although not definitive, HLA association with IPF may suggest that autoimmunity may play a role in pulmonary fibrosis; further characterizing the pathophysiologic connection between this genetic variation and disease this remains an area of active investigation.
Epithelial Integrity
The 2013 GWAS by Fingerlin et al. (10) identified multiple susceptibility loci for fibrotic IIP, including two cell–cell adhesion molecules, DSP and DPP9. DSP gene expression was increased in lung tissue of individuals with IIP and varied by genotype for a variant in intron 5 (10, 91). DSP encodes for desmoplakin, a critical component of desmosome structure important in cell–cell adhesion. Desmosomes mechanically link cells and stabilize tissue architecture. In addition, they are involved in the regulation of cell proliferation, differentiation, migration, and apoptosis (92). The association between DSP variants and IPF, as well as the relationship between DSP variants and lung expression of this gene, was confirmed more recently by Mathai et al. (91) IPF lung has higher gene expression of DSP. However, IPF subjects with the rs2076295 variant were found to have lower DSP expression, suggesting that differential DSP expression may play a role in a subset or sub-phenotype of IPF (91). This association further implicates the airway epithelium in the pathogenesis of IPF, as DSP appears to be localized primarily to the airway epithelia and not to alveolar epithelial cells. The role of DSP in IPF pathogenesis remains an area of active investigation.
Prognosis
Genetic variants, both rare (telomere related) and common (MUC5B and TOLLIP), may play a role in predicting disease outcomes and have prognostic implications. Short telomeres (<10th percentile adjusted for age) have been identified in a considerable portion of IPF patients, regardless of genetic mutations (33, 34). Patients with shorter telomeres have worse transplant-free survival in multiple independent cohorts (46, 93). Furthermore, a small observational study suggested that increased rates of bone marrow suppression and medication-related complications following lung transplantation are more common in IPF patients with telomerase mutations and/or short telomeres (94). Telomere length testing has been suggested as a component of pretransplant workup in IPF patients, although further prospective study is required before these observations can be utilized routinely in patient care (95).
Common polymorphisms, MUC5B and TOLLIP, have also shown promise as prognostic indicators (11, 50). A retrospective study of two separate IPF cohorts demonstrated improved survival in patients with the rs35705950 variant (49). In addition, carriers of at least 1 T allele of MUC5B polymorphism were found to have at least 50% improved survival and better lung function compared to those with the GG genotype (49). These findings were consistent with previous studies, which demonstrated an association between MUC5B variant and less severe pathological changes (96) and slower decline in FVC (50). Similarly, a TOLLIP variant was also associated with differential survival. The minor allele at rs5743890 (G) in TOLLIP is protective and associated with reduced susceptibility to IPF. However, those who developed IPF despite having the protective allele had increased mortality (11). At this time, there are no clinical guidelines suggesting genetic testing in the routine care and counseling of IPF patients (95), and further research is needed to identify the clinical implications of these preliminary findings.
Treatment
Approaches to therapy in IPF have been limited by the poorly understood pathophysiology of this progressive disease. In addition, the unpredictable clinical course of IPF, lack of validated biomarkers, and low clinical predictive value to animal models (97) have been barriers to identifying therapies. Despite these challenges, recent advances in understanding the pathophysiology of IPF have allowed for identification of novel treatment targets. Currently, two available medications, pirfenidone (4) and ninetedanib (3), have been shown to reduce the rate of lung function decline among IPF patients. However, neither approved drug is curative.
With survival-associated variants (e.g., MUC5B and TOLLIP) (74), it is possible that genotypes will define subtypes with differential responses to therapy. Identifying distinct sub-phenotypes in IPF may enable the application of targeted therapy on a pathway-specific basis. For example, it may be possible to use telomere length or TERT genotype to identify a group of patients who would benefit from telomere-directed therapy (95). Oldham and colleagues (98) found that some carriers with TOLLIP polymorphism may benefit from treatment with oral N-acetylcysteine (NAC). More specifically, of those that received NAC, subjects with TT genotype for rs3750920 (TOLLIP) had decreased risk for the trial’s composite end point of death, hospitalization, or 10% decrement in forced vital capacity. In contrast, subjects with the CC genotype for rs3750920 had increased risk for the composite endpoints of the NAC intervention study (98). While NAC has not been shown to be effective in IPF in aggregate (99), it is possible that patients have differential response to this therapy (or other therapies) based on TOLLIP genotype (100). More prospectively designed studies are needed before genetic variation can be utilized routinely when choosing therapies for individual patients.
Conclusion
This review focuses on the relationship between genetic variants and IPF. In addition to sequence variation, epigenetic changes (such as DNA methylation) (101–104) and gene expression changes are associated with disease risk and phenotype (103, 105, 106). Further studies are necessary to better understand the relationships between genetic variation and epigenetic and gene expression variation in terms of disease risk and phenotype.
Given the growing body of evidence that genetic variants influence disease risk as well as disease progression and clinically meaningful patient outcomes, it will be critical to account for genetic variation in future clinical trials. Such prospective studies and analyses that focus on the relationship between genotype and therapeutic response will be crucial in personalizing and improving IPF therapy.
Author Contributions
AK and SM researched and wrote first draft of manuscript; DS edited and revised document. All the authors read and approved final version of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This manuscript was supported by the National Institutes of Health NIH-NHLBI R01-HL097163.
References
1. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med (2007) 176(3):277–84. doi:10.1164/rccm.200701-044OC
2. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med (2011) 183(6):788–824. doi:10.1164/rccm.2009-040GL
3. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med (2014) 370(22):2071–82. doi:10.1056/NEJMoa1402584
4. King T Jr, Bradford W, Castro-Bernardini S, Fagan E, Glaspole I, Glassberg M, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med (2014) 370(22):2083–92. doi:10.1056/NEJMoa1402582
5. Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech (2013) 6(1):9–17. doi:10.1242/dmm.010736
6. Daccord C, Maher TM. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Res (2016) 5:F1000 Faculty Rev–1046. doi:10.12688/f1000research.8209.1
7. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A (2007) 104(18):7552–7. doi:10.1073/pnas.0701009104
8. Bonanni PP, Frymoyer JW, Jacox RF. A family study of idiopathic pulmonary fibrosis: a possible dysproteinemic and genetically determined disease. Am J Med (1965) 39(3):411–21. doi:10.1016/0002-9343(65)90208-1
9. Javaheri S, Lederer DH, Pella JA, Mark GJ, Levine BW. Idiopathic pulmonary fibrosis in monozygotic twins. The importance of genetic predisposition. Chest (1980) 78(4):591–4. doi:10.1378/chest.78.4.591
10. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet (2013) 45(6):613–20. doi:10.1038/ng.2609
11. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med (2013) 1(4):309–17. doi:10.1016/S2213-2600(13)70045-6
12. Nogee L, Dunbar AE, Wert S, Askin F. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med (2001) 344(8):573–9. doi:10.1056/NEJM200102223440805
13. Tanjore H, Cheng D-S, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem (2011) 286(35):30972–80. doi:10.1074/jbc.M110.181164
14. Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet (2016) 25(8):1457–67. doi:10.1093/hmg/ddw014
15. Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet (2009) 84(1):52–9. doi:10.1016/j.ajhg.2008.11.010
16. Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE, et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest (2010) 137(4):969–73. doi:10.1378/chest.09-0790
17. Stanley SE, Gable DL, Wagner CL, Carlile TM, Hanumanthu VS, Podlevsky JD, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med (2016) 8(351):ra107–351. doi:10.1126/scitranslmed.aaf7837
18. Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res (2012) 730(1–2):52–8. doi:10.1016/j.mrfmmm.2011.10.013
19. Kropski JA, Mitchell DB, Markin C, Polosukhin VV, Choi L, Johnson JE, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest (2014) 146(1):e1–7. doi:10.1378/chest.13-2224
20. Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest (2015) 147(5):1361–8. doi:10.1378/chest.14-1947
21. Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet (2015) 47(5):512–7. doi:10.1038/ng.3278
22. Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med (2015) 191(6):646–55. doi:10.1164/rccm.201408-1510OC
23. Kannengiesser C, Borie R, Ménard C, Réocreux M, Nitschké P, Gazal S, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J (2015) 46(2):474–85. doi:10.1183/09031936.00040115
24. Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest (2015) 125(5):2151–60. doi:10.1172/JCI78963
25. Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med (2010) 61(1):105–19. doi:10.1146/annurev.med.60.041807.123500
26. Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol (2005) 32(6):521–30. doi:10.1165/rcmb.2005-0009OC
27. van Moorsel CHM, Hoffman TW, van Batenburg AA, Klay D, van der Vis JJ, Grutters JC. Understanding idiopathic interstitial pneumonia: a gene-based review of stressed lungs. Biomed Res Int (2015) 2015:304186. doi:10.1155/2015/304186
28. Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem (2010) 285(29):22103–13. doi:10.1074/jbc.M110.121467
29. Yamano G, Funahashi H, Kawanami O, Zhao L-X, Ban N, Uchida Y, et al. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett (2001) 508(2):221–5. doi:10.1016/S0014-5793(01)03056-3
30. Weichert N, Kaltenborn E, Hector A, Woischnik M, Schams A, Holzinger A, et al. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res (2011) 12:4. doi:10.1186/1465-9921-12-4
31. Cheong N, Madesh M, Gonzales LW, Zhao M, Yu K, Ballard PL, et al. Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J Biol Chem (2006) 281(14):9791–800. doi:10.1074/jbc.M507515200
32. Armanios M, Chen JJ-L, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med (2007) 356(13):1317–26. doi:10.1056/NEJMoa066157
33. Cronkhite J, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med (2008) 178(7):729–37. doi:10.1164/rccm.200804-550OC
34. Alder JK, Chen JJ-L, Lancaster L, Danoff S, Su S, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A (2008) 105(35):13051–6. doi:10.1073/pnas.0804280105
35. Driscoll B, Buckley S, Bui KC, Anderson KD, Warburton D. Telomerase in alveolar epithelial development and repair. Am J Physiol Lung Cell Mol Physiol (2000) 279(6):L1191–8.
36. Liu T, Ullenbruch M, Choi YY, Yu H, Ding L, Xaubet A, et al. Telomerase and telomere length in pulmonary fibrosis. Am J Respir Cell Mol Biol (2013) 49(2):260–8. doi:10.1165/rcmb.2012-0514OC
37. Chen JL, Greider CW. Telomerase RNA structure and function: implications for dyskeratosis congenita. Trends Biochem Sci (2004) 29(4):183–92. doi:10.1016/j.tibs.2004.02.003
38. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet (1998) 19(1):32–8. doi:10.1038/ng0598-32
39. Kim SH, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet (1999) 23(4):405–12. doi:10.1038/13854
40. Vannier J-B, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol (2014) 24:416–25. doi:10.1016/j.tcb.2014.01.004
41. Seibold MA, Wise AL, Speer MC, Steel MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med (2012) 364(16):1503–12. doi:10.1056/NEJMoa1013660
42. Korthagen NM, van Moorsel CHM, Kazemier KM, Ruven HJT, Grutters JC. IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics (2012) 64(5):371–7. doi:10.1007/s00251-012-0604-6
43. Ahn M-H, Park B-L, Lee S-H, Park S-W, Park J-S, Kim D-J, et al. A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir Res (2011) 12(1):73. doi:10.1186/1465-9921-12-73
44. Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2012) 185(1):67–76. doi:10.1164/rccm.201101-0058OC
45. O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, et al. The toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2013) 188(12):1442–50. doi:10.1164/rccm.201304-0760OC
46. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med (2014) 2(7):557–65. doi:10.1016/S2213-2600(14)70124-9
47. Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J (2015) 45:1717–27. doi:10.1183/09031936.00163814
48. Fingerlin TE, Zhang W, Yang IV, Ainsworth HC, Russell PH, Blumhagen RZ, et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet (2016) 17(1):74. doi:10.1186/s12863-016-0377-2
49. Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA (2013) 309(21):2232–9. doi:10.1001/jama.2013.5827
50. Stock CJ, Sato H, Fonseca C, Banya WAS, Molyneaux PL, Adamali H, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax (2013) 68(5):436–41. doi:10.1136/thoraxjnl-2012-201786
51. Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, et al. Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol Rev (2016) 96(4):1567–91. doi:10.1152/physrev.00004.2016
52. Mathai SK, Yang IV, Schwarz MI, Schwartz DA. Incorporating genetics into the identification and treatment of idiopathic pulmonary fibrosis. BMC Med (2015) 13:191. doi:10.1186/s12916-015-0434-0
53. Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax (2004) 59(11):977–80. doi:10.1136/thx.2004.026336
54. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2012) 302(8):L721–9. doi:10.1152/ajplung.00410.2011
55. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng D-S, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol (2008) 294(6):L1119–26. doi:10.1152/ajplung.00382.2007
56. Campo I, Zorzetto M, Mariani F, Kadija Z, Morbini P, Dore R, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res (2014) 15(43):15. doi:10.1186/1465-9921-15-43
57. Zhou W, Wang Y. Candidate genes of idiopathic pulmonary fibrosis: current evidence and research. Appl Clin Genet (2016) 9:5–13. doi:10.2147/TACG.S61999
58. Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A (2011) 108(26):10562–7. doi:10.1073/pnas.1107559108
59. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A (2006) 103(35):13180–5. doi:10.1073/pnas.0605669103
60. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med (2009) 180(7):657–65. doi:10.1164/rccm.200903-0322OC
61. Marmai C, Sutherland RE, Kim KK, Dolganov GM, Fang X, Kim SS, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2011) 301(1):L71–8. doi:10.1152/ajplung.00212.2010
62. Mathai SK, Schwartz DA, Warg LA. Genetic susceptibility and pulmonary fibrosis. Curr Opin Pulm Med (2014) 20(5):429–35. doi:10.1097/MCP.0000000000000074
63. Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet (2009) 85(6):823–32. doi:10.1016/j.ajhg.2009.10.028
64. Petrovski S, Todd JL, Durheim MT, Wang Q, Chien JW, Kelly FL, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med (2017) 196(1):82–93. doi:10.1164/rccm.201610-2088OC
65. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet (2012) 13(10):693–704. doi:10.1038/nrg3246
66. Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med (2011) 184(8):904–12. doi:10.1164/rccm.201103-0520OC
67. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol (2011) 300(3):L391–401. doi:10.1152/ajplung.00097.2010
68. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature (2014) 505(7483):412–6. doi:10.1038/nature12807
69. Young HWJ, Williams OW, Chandra D, Bellinghausen LK, Pérez G, Suárez A, et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5’ elements. Am J Respir Cell Mol Biol (2007) 37(3):273–90. doi:10.1165/rcmb.2005-0460OC
70. Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One (2013) 8(8):e70621. doi:10.1371/journal.pone.0070621
71. Horimasu Y, Ohshimo S, Bonella F, Tanaka S, Ishikawa N, Hattori N, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology (2015) 20(3):439–44. doi:10.1111/resp.12466
72. Wang C, Zhuang Y, Guo W, Cao L, Zhang H, Xu L, et al. Mucin 5B promoter polymorphism is associated with susceptibility to interstitial lung diseases in Chinese males. PLoS One (2014) 9(8):e104919. doi:10.1371/journal.pone.0104919
73. van der Vis JJ, Snetselaar R, Kazemier KM, ten Klooster L, Grutters JC, van Moorsel CHM. Effect of Muc5b promoter polymorphism on disease predisposition and survival in idiopathic interstitial pneumonias. Respirology (2016) 21(4):712–7. doi:10.1111/resp.12728
74. Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res (2014) 163(5):494–502. doi:10.1016/j.trsl.2013.12.006
75. Peljto AL, Selman M, Kim DS, Murphy E, Tucker L, Pardo A, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest (2015) 147(2):460–4. doi:10.1378/chest.14-0867
76. Kishore A, Žižková V, Kocourková L, Petrkova J, Bouros E, Nunes H, et al. Association study for 26 candidate loci in idiopathic pulmonary fibrosis patients from four European populations. Front Immunol (2016) 7:274. doi:10.3389/fimmu.2016.00274
77. Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One (2013) 8(3):e58658. doi:10.1371/journal.pone.0058658
78. Nakano Y, Yang IV, Walts AD, Watson AM, Helling BA, Fletcher AA, et al. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2016) 193(4):464–6. doi:10.1164/rccm.201509-1872LE
79. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med (2013) 368(23):2192–200. doi:10.1056/NEJMoa1216076
80. Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, et al. Development and progression of interstitial lung abnormalities in the Framingham heart study. Am J Respir Crit Care Med (2016) 194(12):1514–22. doi:10.1164/rccm.201512-2523OC
81. Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Association between interstitial lung abnormalities and all-cause mortality. JAMA (2016) 315(7):672–81. doi:10.1001/jama.2016.0518
82. Chung JH, Peljto AL, Chawla A, Talbert JL, McKean DF, Rho B-H, et al. CT imaging phenotypes of pulmonary fibrosis in the MUC5B promoter site polymorphism. Chest (2016) 149(5):1215–22. doi:10.1016/j.chest.2015.11.009
83. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol (2011) 45:189–201. doi:10.1165/rcmb.2011-0011RT
84. Zhou Y, Wang X, Liu M, Hu Q, Song L, Ye L, et al. A critical function of toll-like receptor-3 in the induction of anti-human immunodeficiency virus activities in macrophages. Immunology (2010) 131(1):40–9. doi:10.1111/j.1365-2567.2010.03270.x
85. Falfán-Valencia R, Camarena A, Juárez A, Becerril C, Montaño M, Cisneros J, et al. Major histocompatibility complex and alveolar epithelial apoptosis in idiopathic pulmonary fibrosis. Hum Genet (2005) 118(2):235–44. doi:10.1007/s00439-005-0035-7
86. Aquino-Galvez A, Pérez-Rodríguez M, Camarena Á, Falfan-Valencia R, Ruiz V, Montaño M, et al. MICA polymorphisms and decreased expression of the MICA receptor NKG2D contribute to idiopathic pulmonary fibrosis susceptibility. Hum Genet (2009) 125(5–6):639–48. doi:10.1007/s00439-009-0666-1
87. Xue J, Gochuico BR, Alawad AS, Feghali-Bostwick CA, Noth I, Nathan SD, et al. The HLA class II Allele DRB1*1501 is over-represented in patients with idiopathic pulmonary fibrosis. PLoS One (2011) 6(2):e14715. doi:10.1371/journal.pone.0014715
88. Zhang J, Xu D, Xu K, Wu B, Zheng M, Chen J, et al. HLA-A and HLA-B gene polymorphism and idiopathic pulmonary fibrosis in a Han Chinese population. Respir Med (2012) 106(10):1456–62. doi:10.1016/j.rmed.2012.06.015
89. Zhang H-P, Zou J, Xie P, Gao F, Mu H-J. Association of HLA and cytokine gene polymorphisms with idiopathic pulmonary fibrosis. Kaohsiung J Med Sci (2015) 31(12):613–20. doi:10.1016/j.kjms.2015.10.007
90. Erlich HA, Opelz G, Hansen J. HLA DNA typing and transplantation. Immunity (2001) 14:347–56. doi:10.1016/S1074-7613(01)00115-7
91. Mathai SK, Pedersen BS, Smith K, Russell P, Schwarz MI, Brown KK, et al. Desmoplakin (DSP) variants are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2015) 193(10):1151–60. doi:10.1164/rccm.201509-1863OC
92. Huber O, Petersen I. 150th anniversary series: desmosomes and the hallmarks of cancer. Cell Commun Adhes (2015) 22(1):15–28. doi:10.3109/15419061.2015.1039642
93. Dai J, Cai H, Li H, Zhuang Y, Min H, Wen Y, et al. Association between telomere length and survival in patients with idiopathic pulmonary fibrosis. Respirology (2015) 20(6):947–52. doi:10.1111/resp.12566
94. Silhan LL, Shah PD, Chambers DC, Snyder LD, Riise GC, Wagner CL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J (2014) 44(1):178–87. doi:10.1183/09031936.00060014
95. Kropski JA, Young LR, Cogan JD, Mitchell DB, Lancaster LH, Worrell JA, et al. Genetic evaluation and testing of patients and families with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2016) 195(11):1423–8. doi:10.1164/rccm.201609-1820PP
96. Cosgrove GP, Groshong SD, Peljto AL, Talbert J, McKean D, Markin C, et al. The MUC5B promoter polymorphism is associated with a less severe pathological form of familial interstitial pneumonia (FIP). Am J Respir Crit Care Med (2012) 185:A6865. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A6865
97. O’Riordan TG, Smith V, Raghu G. Development of novel agents for idiopathic pulmonary fibrosis. Chest (2015) 148(4):1083–92. doi:10.1378/chest.14-3218
98. Oldham JM, Ma S-F, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA, et al. TOLLIP, MUC5B, and the response to N -acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2015) 192(12):1475–82. doi:10.1164/rccm.201505-1010OC
99. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med (2012) 366(21):1968–77. doi:10.1056/NEJMoa1113354
100. Mathai SK, Newton CA, Schwartz DA, Garcia CK. Pulmonary fibrosis in the era of stratified medicine. Thorax (2016) 71(12):1154–60. doi:10.1136/thoraxjnl-2016-209172
101. Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med (2015) 21(5):454–62. doi:10.1097/MCP.0000000000000191
102. Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol (2017) 57(1):91–9. doi:10.1165/rcmb.2017-0046OC
103. Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, Davidson EJ, Murphy E, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2014) 190(11):1263–72. doi:10.1164/rccm.201408-1452OC
104. Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2012) 186(6):525–35. doi:10.1164/rccm.201201-0077OC
105. Yang IV, Burch LH, Steele MP, Savov JD, Hollingsworth JW, McElvania-Tekippe E, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med (2007) 175(1):45–54. doi:10.1164/rccm.200601-062OC
Keywords: idiopathic pulmonary fibrosis, MUC5B, pulmonary fibrosis, interstitial lung disease, telomeres
Citation: Kaur A, Mathai SK and Schwartz DA (2017) Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 4:154. doi: 10.3389/fmed.2017.00154
Received: 07 June 2017; Accepted: 07 September 2017;
Published: 25 September 2017
Edited by:
Argyrios Tzouvelekis, Alexander Fleming Biomedical Sciences Research Center, GreeceReviewed by:
Brett Ley, University of California, San Francisco, United StatesMartin Petrek, Palacký University, Czechia
Copyright: © 2017 Kaur, Mathai and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan K. Mathai, susan.mathai@ucdenver.edu