A Temporally Dynamic Gut Microbiome in Atlantic Salmon During Freshwater Recirculating Aquaculture System (RAS) Production and Post-seawater Transfer
 Marlene Lorgen-Ritchie1
Marlene Lorgen-Ritchie1  Michael Clarkson2 Lynn Chalmers2 John F. Taylor2 Herve Migaud2
Michael Clarkson2 Lynn Chalmers2 John F. Taylor2 Herve Migaud2  Samuel A. M. Martin1*
Samuel A. M. Martin1*- 1School of Biological Sciences, University of Aberdeen, Aberdeen, United Kingdom
- 2Institute of Aquaculture, University of Stirling, Stirling, United Kingdom
Atlantic salmon aquaculture is undergoing an expansion of land-based recirculating aquaculture systems (RAS), especially for freshwater (FW) stages of production. Juvenile salmon undergo parr-smolt transformation, also known as smoltification and become pre-adapted to tolerate seawater (SW). One aspect requiring study is the development of microbial communities during this time, especially in RAS systems. Here we analyzed temporal changes in microbiome associated with the intestine in Atlantic salmon during smolt production in a commercial RAS production facility and followed the same cohort of fish post-seawater transfer (SWT), using 16S rRNA gene sequencing. Microbial diversity and richness showed an increase over time across FW production, but declined sharply and significantly 1-week post-SWT before re-establishing itself with a completely different community structure after 4 weeks. Core microbial taxa could be assigned to three distinct categories; (1) omnipresent, (2) salinity specific, or (3) transient. By including diet and water samples in the analyses, we classified true core taxa associated with the host, those associated with the diet, and transient cores associated with microbial communities in tank water. The rising trend observed in microbial richness in the water may be a consequence of a temporal increase in organic load while dominance of Vibrionaceae may be attributed to the higher temperatures maintained during RAS production and above average natural water temperatures post-SWT. Functional analysis suggests modulation of metabolic pathways post-SWT, but downstream impacts on fish growth and health in a commercial setting remain to be elucidated. A deeper understanding of the interplay between microbial composition and functionality can play a role in optimizing fish performance in tightly regulated RAS production.
Introduction
The continued growth of the global population coupled with declining wild fish stocks continues to drive expansion of the global aquaculture industry worldwide, with production growing at 7.5% per year since 1970, providing 52% of global fish produced for human consumption in 2018 and generating 250 billion USD first sale value (FAO, 2020). A key stage in the life history of the anadromous Atlantic salmon is the process of smoltification, where the juvenile changes from a FW-adapted parr to SW-adapted smolt and is characterized by a myriad of physiological, morphological and behavioral changes (Björnsson et al., 2011; McCormick, 2012). The parr-smolt transformation (PST) is critical for the long-term health and performance of the stocks, and freshwater environmental conditions (e.g., light, temperature, microbiota, and water quality) and manipulations (e.g., smoltification regimes and vaccination) can impact on the robustness of smolts.
The production of smolts in land-based recirculating aquaculture systems (RAS) is expanding rapidly and globally as a means to provide a controlled stable environment for optimal growth, reduced water usage, biosecurity and minimize the impact on ecosystems (d’Orbcastel et al., 2009; Attramadal et al., 2014). However, fish farmed in RAS experience very different conditions than open water systems including microbial populations which are regulated by water physiochemical factors as well as available nutrients and space (De Schryver and Vadstein, 2014; Fossmark et al., 2020). Microbial communities in RAS play a vital role in converting waste nutrients from uneaten feed and feces to maintain high water quality, which in turn is critical to fish health (Sullam et al., 2012; Blancheton et al., 2013). Established biofilters in the RAS loop are critical to operational success and contain communities of microbes including nitrifying and denitrifying bacteria which convert potentially toxic by-products of nutrient metabolism such as ammonia into non-toxic forms (Blancheton et al., 2013; Fossmark et al., 2021). In addition, fish mucosal surfaces including skin, gill and gut are always in contact with microbes living in the surrounding water and, in the case of the gut, with feed-associated taxa, presenting opportunities for colonization.
The intestinal microbial community of fish consists of both autochthonous species which are attached to the intestinal mucosa as well as allochthonous species which do not attach due to inability or out-competition (Navarrete et al., 2012; Llewellyn et al., 2014; Givens et al., 2015). Microbial communities show extensive plasticity in response to environmental change, but may also reflect environmental or physiological history where the sequence of arrival of microbes into a community is important in determining microbiome composition, even under identical conditions (Vellend, 2016; Sprockett et al., 2018). Gut microbes also play a key role in the priming, protection and development of the host immune system and provide the hosts with exogenous nutrients and extracellular fatty acids and vitamins (Dhanasiri et al., 2011).
Host physiology and external environment provide niche environments that are colonized by microbes and form specialized microbial communities which may change in composition over time, for example across host development, or during a change in host environment. The gut microbiome of Atlantic salmon is strongly influenced by environmental factors including rearing system (Minich et al., 2020), diet (Schmidt et al., 2016; Jaramillo-Torres et al., 2019), seasonality (Zarkasi et al., 2014, 2016), and also by physiological factors such as developmental stage (Lokesh et al., 2019; Heys et al., 2020). In particular, a shift in the microbiome of Atlantic salmon has been observed following transition from FW to SW, often with a set of core microbes displaying stability across this transition (Llewellyn et al., 2016; Dehler et al., 2017b; Rudi et al., 2018; Fossmark et al., 2021). Water and diet are likely key environmental sources of microbes, with this being controlled to an extent by the fish retaining or expelling specific bacteria, ensuring that gut microbial communities are not a passive reflection of seeding communities (Sullam et al., 2012; Heys et al., 2020). The transfer of smolts to the hypertonic SW means these fish require to increase their drinking rates, as well as overall intestinal fluid re-absorption rates (McCormick, 2012), which is likely to impact microbial dynamics in the intestine dependent upon the surrounding environment.
While land-based RAS facilities are becoming the norm for Atlantic salmon smolt production, our understanding of gut microbiome, temporal changes throughout smoltification, and the associated water microbial composition is limited. Incidence of bacterial diseases are associated with seawater transfer (SWT) of smolts (Johansson et al., 2016) and for anadromous fish, a change in salinity means exposure to substantially different microbial communities in the water to which fish must be able to adapt (Schmidt et al., 2015). To this end, adaptive shifts in host-associated microbiota may be hypothesized to accompany the well-characterized and extensive physiological, morphological and behavioral adaptations of salmon undergoing PST. In addition, the RAS environment itself may also directly impact the colonization and succession of the FW gut microbial community with potential consequences post-SWT. In this study, we investigated the microbiome of Atlantic salmon hindgut reared in a commercial FW RAS facility, and following transfer to open seawater cages. Deep sequencing of the V3-V4 hypervariable region of the microbial 16S rRNA gene was performed to analyze the temporal stability of the gut microbiome pre- (FW) and post- (SW) PST.
Materials and Methods
Fish Maintenance and Sampling Schedule
Mixed sex juvenile Atlantic salmon were followed from parr to smolt stage in a single stream of a commercial RAS in Scotland. Water in the RAS was maintained at an average temperature of 15.4 ± 0.7°C, pH 7.0 ± 0.16 and oxygen saturation of 99.5 ± 5.5%. Fish were then transferred to a sea cage site. Fish were fed to satiation using automatic feeding systems in both FW and SW units (standard FW RAS diet, Skretting; standard SW diet, Mowi).
The sampling program is described in Figure 1. The first FW sampling (parr stage, FW1 – 14.05.2019, pre-winter photoperiod) was carried out from two replicate tanks under constant (24-h) artificial light (LL). Fish were then vaccinated (ALPHA JECT micro® 6, Pharmaq) and graded. Medium grade fish from the two tanks were then mixed and three experimental tanks were established. Fish were then exposed to a “winter” photoperiod (12L:12D) for 6–7 weeks and sampled after 4 weeks of winter (FW2 – 18.06.2019) and just prior to the onset of the spring photoperiod (FW3 – 02.07.2019). A “spring” signal in the form of LL was then applied until transfer to SW and fish were sampled prior to SWT (smolt stage, FW4 – 18.07.2019 to 25.07.2019). Details of degree days (dd – cumulative temperature over a number of days) associated with sampling points are described in Figure 1. At each sampling point post-grading, six fish were sampled from triplicate tanks (n = 3 tanks, 18 fish in total). Individuals from different tanks were mixed at the time of SWT. Fish in the open SW cages were exposed to natural ambient temperature and photoperiod. Fish (n = 2 cages, 12 fish in total) were sampled approximately 1 week and 4 weeks post-SW transfer.
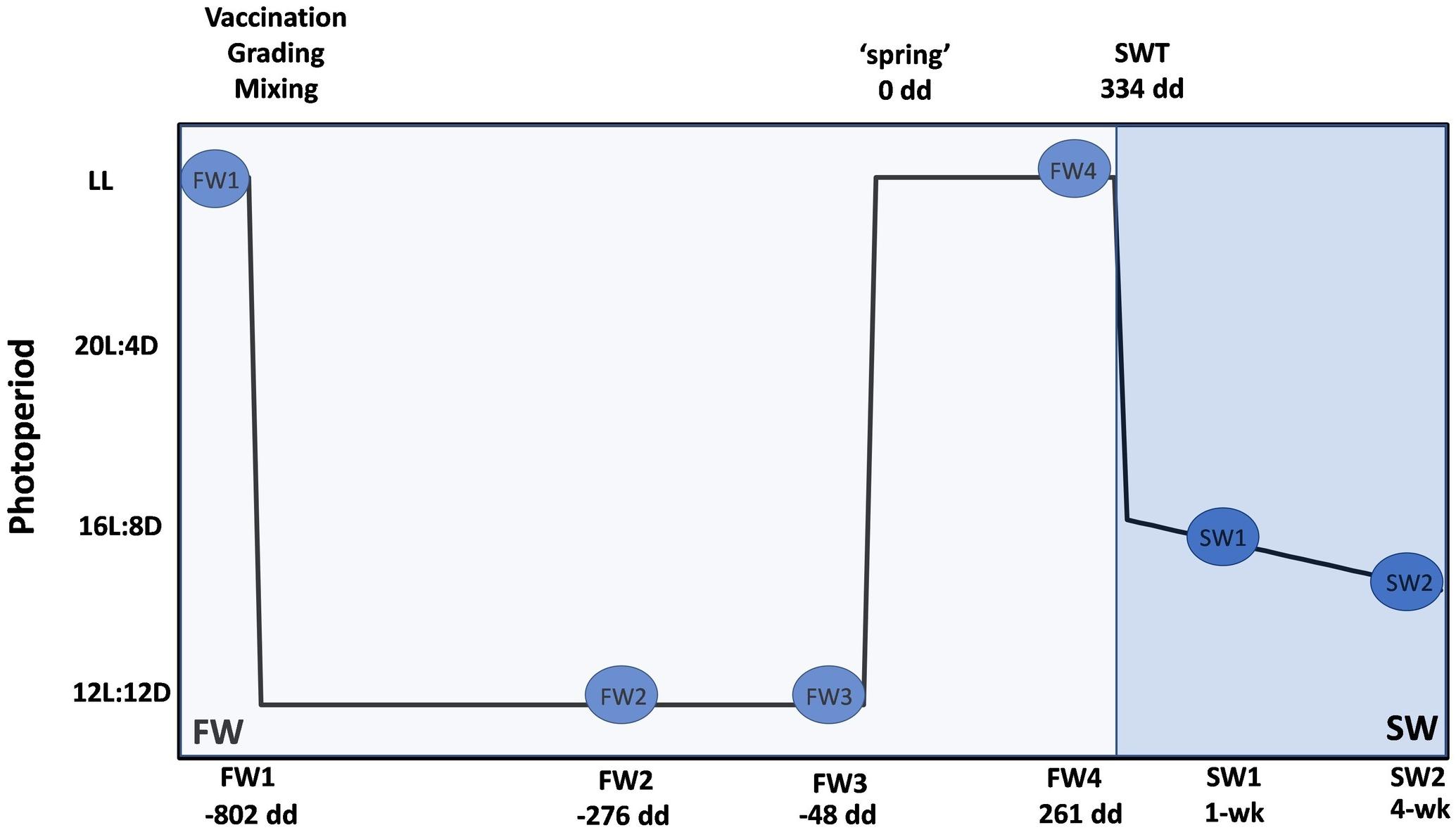
Figure 1. Experimental design for microbiome sampling at FW RAS and open SW cage site. Blue circles represent sampling points; four in FW and two in SW. Degree days is a measure used to determine smolt windows. All dd are calculated relative to application of spring photoperiod (0 dd) and represent mean dd across replicate tanks. At FW1 (–802 dd), unvaccinated fish were sampled from duplicate tanks (n = 2, 12 fish in total). Fish were then vaccinated (ALPHA HECT micro® 6, Pharmaq), graded and mixed, and three experimental tanks of medium grade fish were established. Post-grading, six fish from triplicate tanks were sampled at each FW point (n = 3, 18 fish in total). Fish were exposed to a “winter” photoperiod for a mean of 714 dd and sampled at a mean of –276 dd (FW2) and –48 dd (FW3). “Spring” photoperiod (24 h light – LL) was then applied until SWT and fish sampled at 261 dd (FW4) prior to SWT at a mean of 334 dd. Fish in duplicate open SW cages (n = 2, 12 fish in total) were sampled from duplicate open seawater cages at approximately 1 and 4 weeks post-SWT.
At each sampling point, fish were killed by anesthesia overdose (MS222) followed by a blow to the head. Individual weight (g) and fork length (cm) were recorded. Condition factor was calculated using the formula CF = 100 × [weight (g)/fork length (cm)–3]. Specific growth rates between sampling points were calculated using the formula SGR = 100∗((ln(final weight (g)) – ln(initial weight (g)))/days (Houde, 1981). Smolt index was recorded on all fish culled at each sampling point and from all fish at the time of transfer to sea using the following scale: 1, parr; 2, some silvering, parr marks visible; 3, fully silvered but parr marks visible; 4, smolt, no parr marks visible (Sigholt et al., 1995).
Gut samples were taken by aseptically dissecting out the hindgut with any digesta (100–150 mg of tissue) and adding to a 2 ml sample collection tube containing 1.5 ml RNAlaterTM (Ambion Inc., United States). Samples were stored at 4°C for 24 h followed by longer term storage at −80°C. For water analyses, 4 × 50 ml of tank or cage water were collected at each sample point. Water samples were transported at room temperature then stored at −20°C prior to filtration through 0.2 μM Whatman Cyclopore polycarbonate membrane filters (Sigma-Aldrich; WHA70634702) using a vacuum pump. Filters were stored at −80°C until extraction. Samples of each diet were also collected at each sampling point in FW. Diet samples were transported to the lab at room temperature and stored at −80°C prior to DNA extraction.
DNA Extraction
Hindgut samples in RNAlater containing digesta were thawed on ice, sliced open lengthwise and a scalpel used to scrape approximately 50 mg of digesta and mucosal layers from the interior of the gut to ensure collection of both adherent and allochthonous bacteria. Excess RNA later was removed by gently squeezing between tissue to remove residual salt from the storage solution before transferring to a 2 ml Eppendorf tube for extraction. The QIAamp Fast DNA Stool Mini Kit (Qiagen) was used for DNA extraction according to the manufacturers protocol with modifications described by Dehler et al. (2017b) and described briefly here. InhibitEx buffer was added to the sample tube along with two 3 mm tungsten carbide beads (Qiagen). The samples were then pre-treated with mechanical lysis using a TissueLyser for 4 min to avoid biases against tough-walled Gram-positive bacteria. Lysis temperature was 95°C to allow for cell-wall break-down of difficult to lyse bacteria. DNA was eluted in a final volume of 30 μl. Each batch of DNA extractions were randomized and ensured samples from all sampling timepoints were in each batch to mitigate against technical artifacts. A negative extraction control was carried out alongside each extraction batch. DNA was extracted from water filters and diets using this same protocol. Diet extractions were carried out in triplicate using 200 mg of feed pellets in each replicate. DNA quantity and purity were determined by NanoDrop spectrometry.
PCR Amplification and Sequencing
For primary PCR reactions, variable regions 3 and 4 of the 16S rRNA gene were targeted with the 341F/785R primer pair (Klindworth et al., 2013). Illumina adapter overhang sequences were added to the 5′ end of each primer. The forward primer (341F) had the sequence 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACG GGNGGGCWGCAG, and the reverse primer (785R) 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTA CHVGGGTATCTAATCC with the bold underlined sequence being the locus-specific V3–V4 primers. Triplicate PCR reactions were performed for each sample and pooled post-amplification to avoid PCR efficiency-related biases. PCR reactions were performed in a 10 μl reaction including 2 μl of each forward and reverse primer (1 μM stock, Sigma), 5 μl of 2x KAPA HiFi HotStart ReadyMix including high-fidelity polymerase (KAPA Biosystems Ltd., United Kingdom) and 1 μl of DNA. PCR conditions included an initial denaturation at 95°C for 3 min, followed by 26 cycles of 30 s at 98°C, 30 s at 57°C, and 30 s at 72°C after which a final extension of 72°C for 5 min was applied. Diet samples were diluted to 200 ngμl–1 prior to PCR amplification and only 22 cycles of initial amplification were utilized for water and diet samples. A subset of resulting PCR products was run on an Agilent 2200 TapeStation (Agilent Technologies, Italy) to verify amplification. Overall, DNA extracts from 90 fish hindgut samples were amplified for sequencing along with 12 water extracts, 12 diet extracts, 16 extraction negatives, three PCR negatives, and three positive controls consisting of a mock community (136 samples).
PCR products were cleaned with Agencourt AMPure XP beads on a BioMek 4000 Liquid handling machine (Beckman Coulter Genomics, Italy). The NextEra XT Index Kit (Illumina, San Diego, CA, United States) was used to attached dual indices and Illumina sequencing adapters (P5 and P7) by PCR to the amplicons to produce the final libraries. The index PCRs were carried out in 50 μl reactions containing 5 μl of DNA, 5 μl of NextEra XT Index Primer 1, 5 μl of NextEra XT Index Primer 2, 25 μl of 2x KAPA HiFi HotStart Ready Mix (Kapa Biosystems Ltd., United Kingdom), and 10 μl of nuclease-free water. The PCR conditions were as follows: 95°C for 3 min, 8 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and 72°C for 5 min. Prior to quantification, libraries were cleaned using AMPure XP Beads (Beckman Coulter Genomics, Italy) and the size of the amplicons were verified on an Agilent 4200 TapeStation (Agilent Technologies, Italy). The expected size of the final library was ∼630 bp. Libraries were quantified using a Quant-iT High-Sensitivity dsDNA Assay (Thermo Fisher Scientific, United States) using an Omega FLUOstar plate reader (BMG Labtech, United Kingdom). Final libraries were pooled equimolarly and quantification of pooled libraries confirmed by both qPCR using a KAPA Library Quantification Kit (Roche Sequencing Solutions, United States) and by fluorescence using Qubit dsDNA high-sensitivity (HS) assay (Invitrogen, United States). The final library was denatured and diluted to 1.2 nM prior to loading onto a MiSeq flow cell and sequencing on the Illumina MiSeq platform (Illumina, San Diego, CA, United States). 10% of PhiX Control library was spiked into the amplicon library. MiSeq reagent Kit v3 (600 cycles) (Illumina, San Diego, CA, United States) was used for library denaturing and for MiSeq sample loading. Sequencing was performed on an Illumina MiSeq platform using a 2 × 300 bp paired end protocol.
Sequencing Data Bioinformatics
Analysis of sequence data were carried out using DADA2 (Callahan et al., 2016) and phyloseq (McMurdie and Holmes, 2013) in RStudio v1.1.456 using R v3.6.1 (R Core Team, 2020). DADA2 infers an Illumina sequencing error profile to resolve true sequences from noise and quantifies the number of each actual sequence variant (ASV). Briefly, adapters and primers were removed using TrimGalore!1 and reads with an overall Phred quality score less than 30 were discarded. Forward reads were truncated to 250 bp and reverse reads to 200 bp. Remaining reads were denoised, merged, screened for chimeric sequences which were subsequently removed, and assigned as distinct actual sequence variants (ASVs) using DADA2. In total, 10,502,559 raw reads were obtained for both forward and reverse reads with a mean read depth of 77,225 ± 6512 (SE). After quality filtering, denoising and chimera removal in DADA2, 5,380,123 reads with a mean of 39,560 ± 3736 (SE) per sample were retained. Samples with less than 1000 reads were excluded from further analysis. Taxonomic classification of ASVs was carried out within phyloSeq using the Silva reference taxonomy v132 (Quast et al., 2013). Assignment of species was also conducted using the Silva species assignment v132, allowing for assignment of multiple species. Known contaminants including mitochondrial, eukaryotic, cyanobacteria and chloroplast sequences were removed along with singletons. Samples with less than 500 reads following removal of contaminants were excluded from further analysis. Two further samples were excluded as outliers. Of the 90 hindgut samples sequenced, 83 were retained for downstream analysis. Taxonomic composition of triplicate positive controls was in agreement with the mock community reference (Supplementary Figure 1).
Statistical Analysis
Statistical analysis was carried out in RStudio v1.1.456 using R v3.6.1 and the package phyloseq (McMurdie and Holmes, 2013). Growth parameters were analyzed by one-way ANOVA with Tukey’s HSD post hoc test. All samples were subsampled to an equal depth of 2,622 reads before calculation of alpha and beta diversity. Differences in alpha diversity across sampling points was determined by Kruskal–Wallis comparisons of Shannon (Shannon, 1948) and Chao1 measurements (Chao, 1984) followed by pairwise testing using the Wilcoxon rank sum test.
Community structure (beta diversity) determined by Bray-Curtis dissimilarity distance (Bray and Curtis, 1957) was visualized using non-metric multidimensional scaling (NMDS) ordination plots, implemented using the Vegan package (Oksanen et al., 2020) and plotted using ggplot2 (Wickham, 2016). Data ellipses based upon an assumed multivariate t-distribution were drawn at a level of 0.75 with stat-ellipse in ggplot2 to provide a visual summary. PERMANOVA (permutational multivariate statistical analysis of community separation) was carried out using the Adonis function in the Vegan package and pairwise comparisons computed using adonis.pair in the EcolUtils package (Salazar, 2020). Core microbiota were identified using the microbiome R package (Lahti and Shetty, 2017) with a prevalence cut-off of 80% and a lower relative abundance limit of 0.1%. Log2 relative abundances of core ASVs across samples were presented in heatmaps drawn with Pheatmap (Kolde, 2012) within R, using Euclidean distance clustering of ASVs.
In order to identify functional pathways based upon 16S communities, Piphillin was used to normalize the non-rarefied amplicon data by 16S rRNA gene copy number and to infer metagenomic contents (Iwai et al., 2016; Narayan et al., 2020). A sequence identity cut-off of 99% was implemented. The inferred metagenomic functions were assigned using the Kyoto Encyclopedia of Genes and Genomes database (KEGG; May 2020 Release) and KEGGREST (Tenenbaum, 2019) was utilized to obtain KEGG pathway names and BRITE hierarchies from pathway identifiers. STAMP v2.1.3 (Parks et al., 2014) was used to test for statistically significant differences in pathway contributions to parent terms using Welch’s t-test corrected for multiple-testing by Benjamini-Hochberg false discovery rate (FDR). Differences were considered significant at q < 0.05.
Results
Fish Growth and Smolt Indicators
Length and weight increased significantly throughout FW (p < 0.001), but no further significant increases were observed in the first 4 weeks post-SWT, considered to be due to initial loss of appetite upon transfer (Table 1). Condition factor showed a decline throughout the study period in line with smoltification (p < 0.001), and smolt index was 3.48 ± 0.5 at the time of SWT. Specific growth rate was positive throughout FW sampling points, but significantly declined upon initial SWT (p < 0.05). SGR across the entire study equated to 1.12% body weight per day.
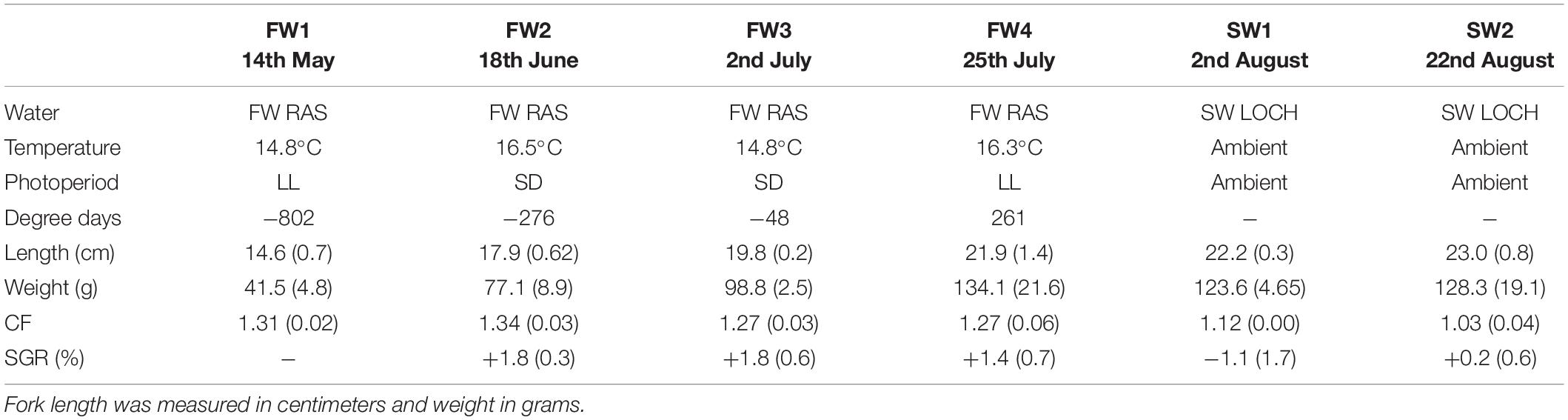
Table 1. Length, weight, condition, and specific growth rate in Atlantic salmon smolts during smolting (n = 2 or 3, 12–18 fish, ±SD).
Alpha Diversity
To maintain sample numbers across sub-groups, samples were limited to 2,622 sequences/samples prior to alpha and beta diversity comparisons, to account for different read depths between samples. Alpha diversity in the hindgut was generally higher in FW compared to SW and showed significant temporal changes (Shannon F = 35.341, p < 0.001), presented in Figure 2. Multiple comparisons revealed significant differences in Shannon diversity between FW3 and FW4 and both SW timepoints (p < 0.001 for all) although no significant differences were observed within FW or SW sampling points. Chao1 species richness was also significantly different between timepoints (Chao1 F = 22.252, p < 0.001), and multiple comparisons revealed that richness at SW1 was significantly lower than at FW3 (p < 0.001) and FW4 (p = 0.002). No significant differences in alpha diversity or richness were observed with sampling point in water or diet samples. Temporal trends in alpha diversity in hindgut samples did not mirror those found in water or diet samples, but a rising trend was observed in species richness in water samples from FW2 to FW4.
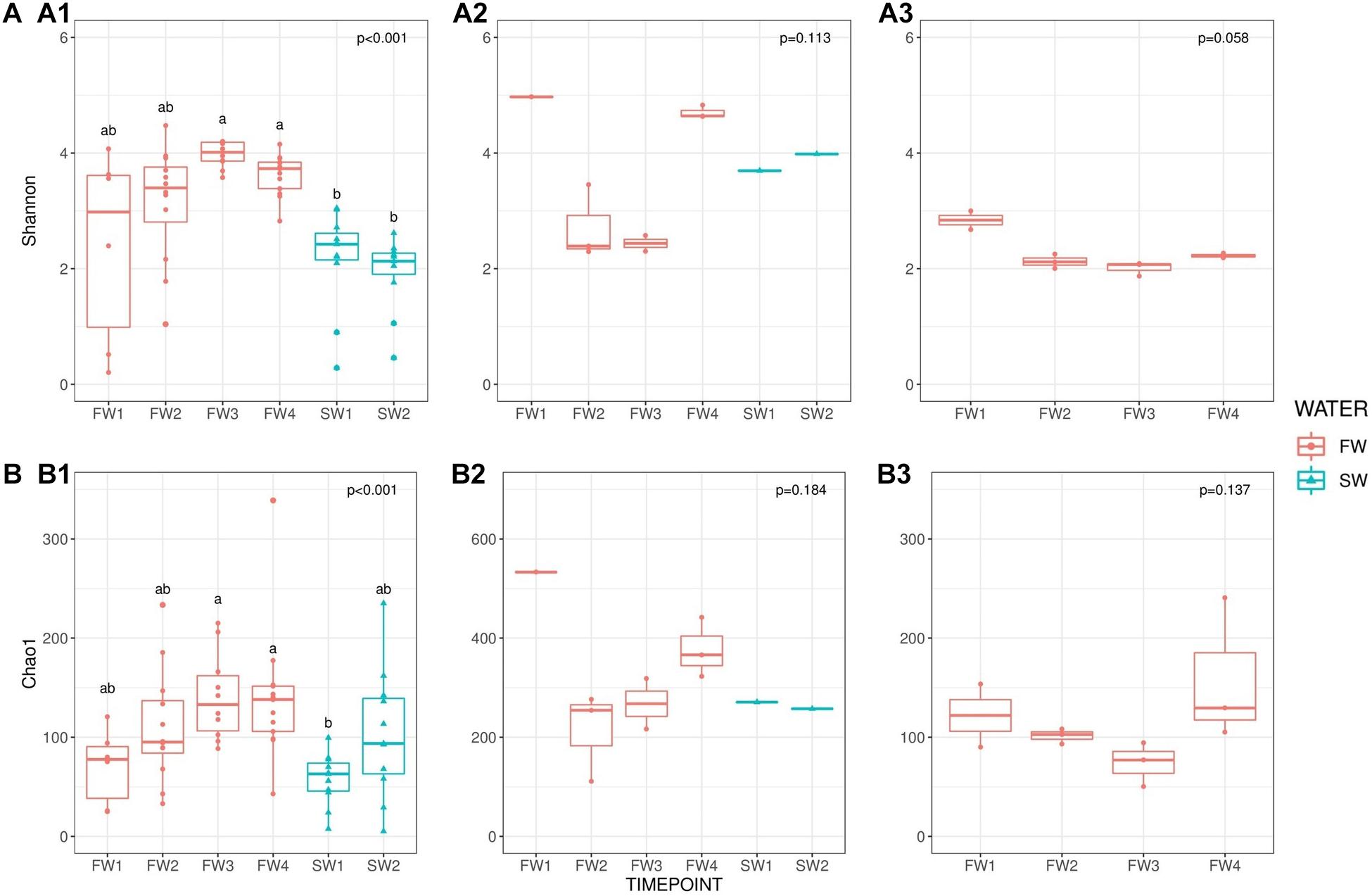
Figure 2. Alpha diversity [(A): Shannon] and richness [(B): Chao1] comparisons. (A1) Shannon: hindgut, (A2) Shannon: water and (A3) Shannon diet across sampling points. (B1) Chao1: hindgut, (B2) Chao1: water and (B3) Chao1: diet. Red circles indicate individual samples for FW RAS and blue triangles are individual samples from SW cages. Superscripts indicate significant differences between sampling points derived from pairwise testing.
Beta Diversity
PERMANOVA analysis revealed significant differences in the microbiome structure of the hindgut across sampling points (F5,57 = 4.13, R2 = 0.266, p < 0.001; Figure 3A). Pairwise comparisons identified significant differences in beta diversity between all sampling point contrasts (BH FDR < 0.01) with the exceptions of FW2 vs. FW3 (p = 0.070) and FW2 vs. FW4 (p = 0.070). Microbiome community structure in water (F5,4 = 10.6, R2 = 0.919, p < 0.001) and diet (F3,7 = 14.4, R2 = 0.851, p < 0.001) samples were also impacted by sampling point, but no significant pairwise differences were identified. Clear separation in communities was apparent between all sample types (F2,82 = 8.61, R2 = 0.174, p < 0.001; Figure 3B).
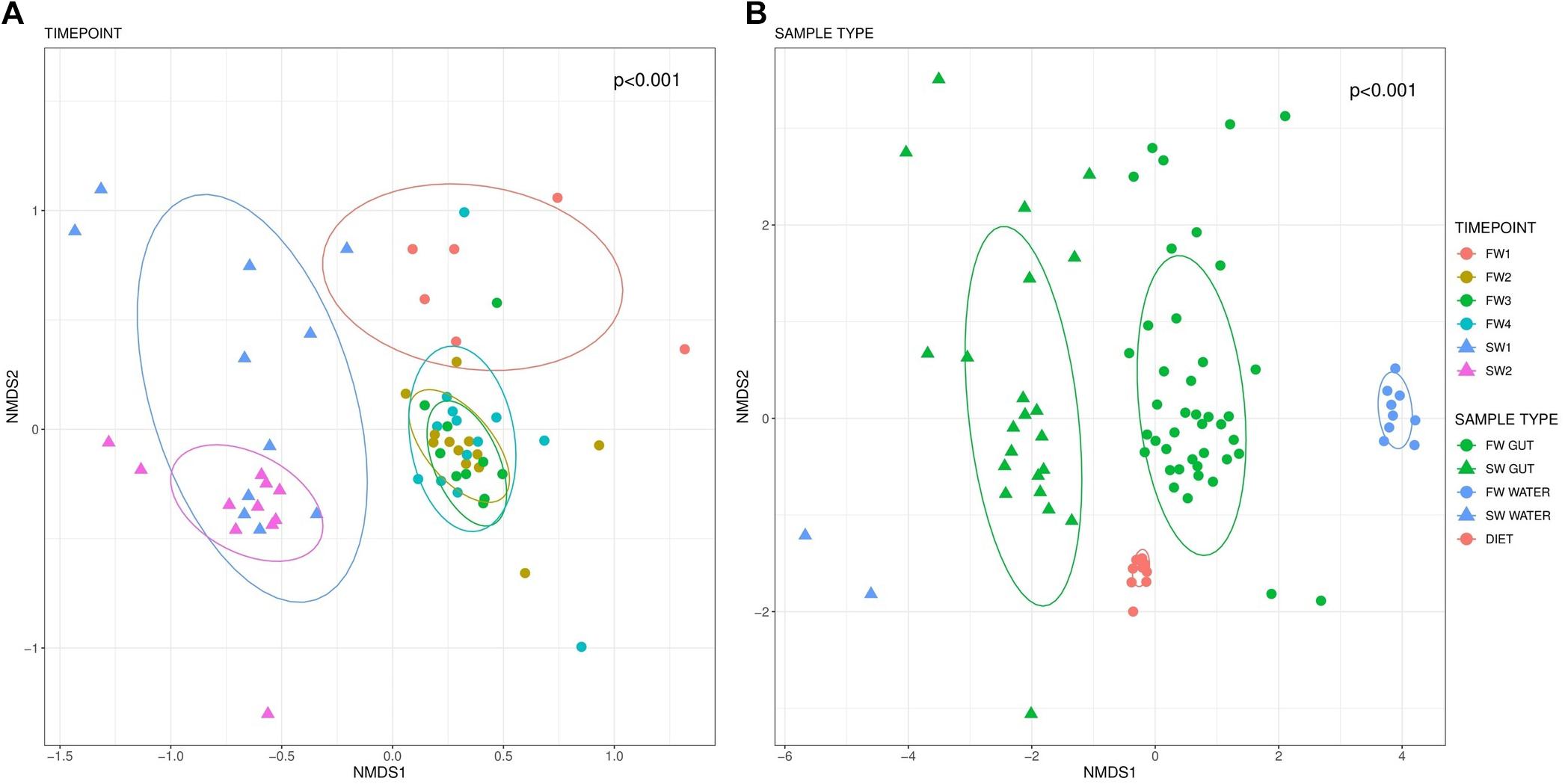
Figure 3. Beta-diversity based on Bray-Curtis distances visualized in an NMDS plot. Different colors indicate sampling points while circle markers indicate FW and triangles SW samples. Beta diversity within hindgut samples by sampling point is presented in (A). (B) Depicts beta diversity in different sample types. Data ellipses based upon an assumed multivariate t-distribution are drawn at a level of 0.75 to provide a visual summary.
Community Composition in Hindgut, Water, and Diet
Community composition was examined at the phylum (Figure 4A) and ASV level (Supplementary Table 1). Nineteen phyla were observed in hindgut samples. At all FW sampling points, communities were dominated by Proteobacteria and Firmicutes, supplemented by members of Bacteroidetes and Actinobacteria while at SW sampling points, Proteobacteria was the dominating genus, with Firmicutes still showing a strong presence. Relative abundances of the dominant taxa in hindgut at the ASV level are presented in Figure 4B. At FW1, ASV8, a Firmicutes from the family Ruminococcaceae, made up 17.9% and was also the most abundant ASV at FW2. ASV20 (Lactobacillus sp.) was the most abundant ASV at FW3 (5.9%) and FW4 (8.5%). At 1-week post-SWT (SW1), a single Proteobacteria of the genus Vibrio (ASV5) made up 19.4% of abundance, and after 4 weeks in SW (SW2), ASV7 (Vibrio sp.) constituted 24.9% of total relative abundance.
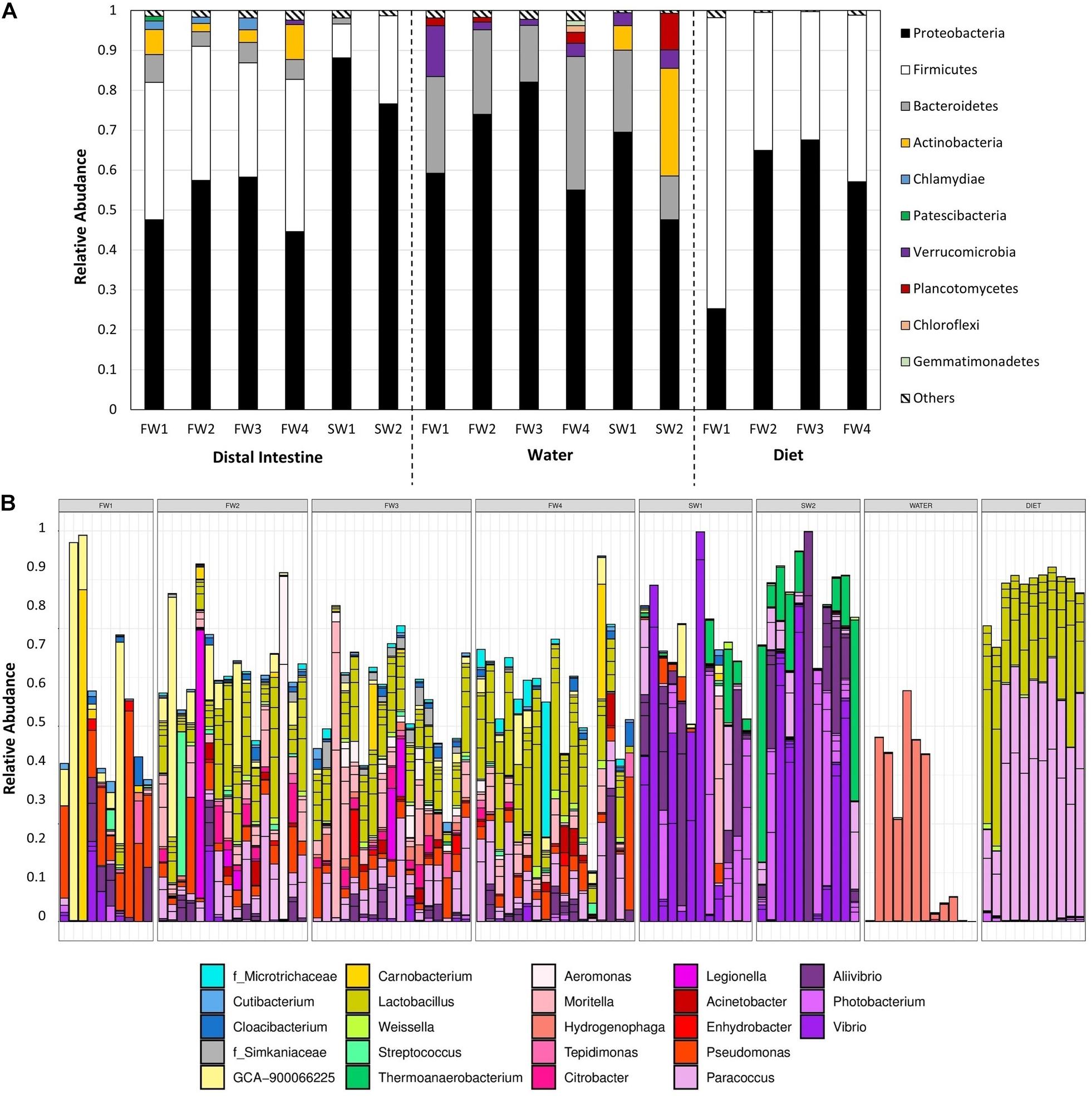
Figure 4. (A) Total relative abundance of each phyla across sampling points in hindgut, water, and diet samples. Only phyla constituting >1% of total relative abundance at each timepoint are presented individually, with those at lower abundances grouped together in “Others.” (B) Relative abundance of the 40 most abundant taxa in hindgut across all samples, colored by genus.
In water samples, 19 phyla were also present, 15 of which overlapped with those observed in the hindgut (Figure 4A). The microbial community in water in FW RAS tanks was dominated by Proteobacteria, Bacteroidetes and Verrucomicrobia while at both SW1 and SW2, Proteobacteria, Bacteroidetes, Actinobacteria and Verrucomicrobia dominated and Planctomycetes also became dominant at SW2 only. Relative abundances of the dominant taxa in water at the ASV level are presented in Supplementary Figure 2. At FW2, ASV13, and ASV24 (Hydrogenophaga) constituted 58.4% of relative abundance in total and by FW3, these two taxa made up 72.5% of total relative abundance. At SW1, Planktomarina (ASV65, Proteobacteria) was the most abundant at 17.6%, but at SW2, Candidatus_Actinomarina (ASV63, Actinobacteria) dominated.
In diet samples, 10 phyla were detected, all of which were also observed in the hindgut, with dominance of Proteobacteria and Firmicutes throughout (Figure 4A). Relative abundances of the dominant taxa in diet at the ASV level are presented in Supplementary Figure 3. At FW1, Firmicutes out-weighed Proteobacteria as the most dominant phylum, but this was reversed in all other sampling points. At FW1, ASV20 (Lactobacillus sp.) constituted 32.1% of total relative abundance. At all other FW sampling points, ASV6 (Paracoccus sp.) dominated constituting 40.2%, 42.1%, and 34.1%, respectively.
“Core” Microbiota Throughout Smoltification
A single “core ASV” was identified across all samples from the hindgut (ASV33; Pseudomonas sp., present at 0.1% or more, in ≥80% of all individuals). Considering FW samples alone, three additional core ASVs were observed: ASV6 (Paracoccus sp.), ASV23 (Lactobacillus sp.) and ASV41 (Lactobacillus sp.). Excluding FW1, a sampling point prior to vaccination, an additional 5 FW cores were observed: ASV16 (Paracoccus sp.), ASV20 (Lactobacillus sp.), ASV44 (Moritella sp.), ASV47 (Lactobacillus sp.) and ASV149 (Cutibacterium sp.). In SW individuals, no additional core taxa were identified at an 80% threshold, but ASV5 (Vibrio sp.) was present in 78% of SW samples. Considering timepoints individually, seven additional FW core ASVs were identified at FW2, six at FW3, six at FW4 and four additional SW cores were observed at SW2 (Supplementary Table 2). No additional cores were identified at FW1 or SW5, and the SW core ASV5 was not observed at SW1 alone. Patterns of abundance of all core taxa across sampling points are summarized in Figure 5. Core ASVs could be broadly separated into three groups; (1) omnipresent, (2) transient, and (3) salinity specific cores.
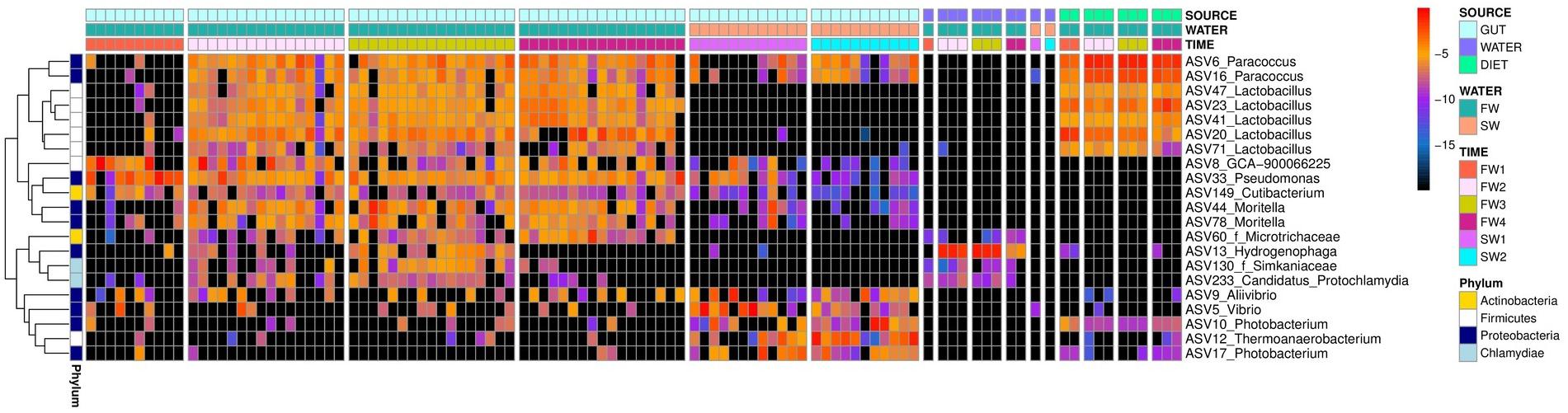
Figure 5. Heatmap depicting log2 relative abundance of core taxa identified in hindgut across timepoints in hindgut, water and diet samples. Colored blocks at the top of the figure depict different sample types (SOURCE), salinities (WATER) and timepoints (TIME) as detailed in the legend. Colored blocks at the left of the figure depict different phyla. ASV and genus names are presented for each row and where genus was undetermined, family is indicated with f prefix. Black squares indicate the absence of a taxon in that sample.
Functional Annotation
Piphillin inferred 374 KEGG pathways from 3730 ASVs present in the Atlantic salmon gut at an identity cut-off of 99%. A total of 970 ASVs had identity of 99% or more with a 16S sequence. Removing human diseases and top-level terms, 294 pathways remained and 156 (53.1%) of these pathways were related to metabolism (Figure 6A). Statistical analysis using STAMP revealed differing contributions of Metabolism level 2 categories pre- and post-seawater transfer (SWT). “Xenobiotics biodegradation and metabolism,” “Amino acid metabolism,” “Metabolism of other amino acids,” and “Metabolism of terpenoids and polyketides” had a higher contribution at FW4 while “Metabolism of cofactors and vitamins” and “Glycan biosynthesis and metabolism” a higher contribution at SW1 (Figure 6B).
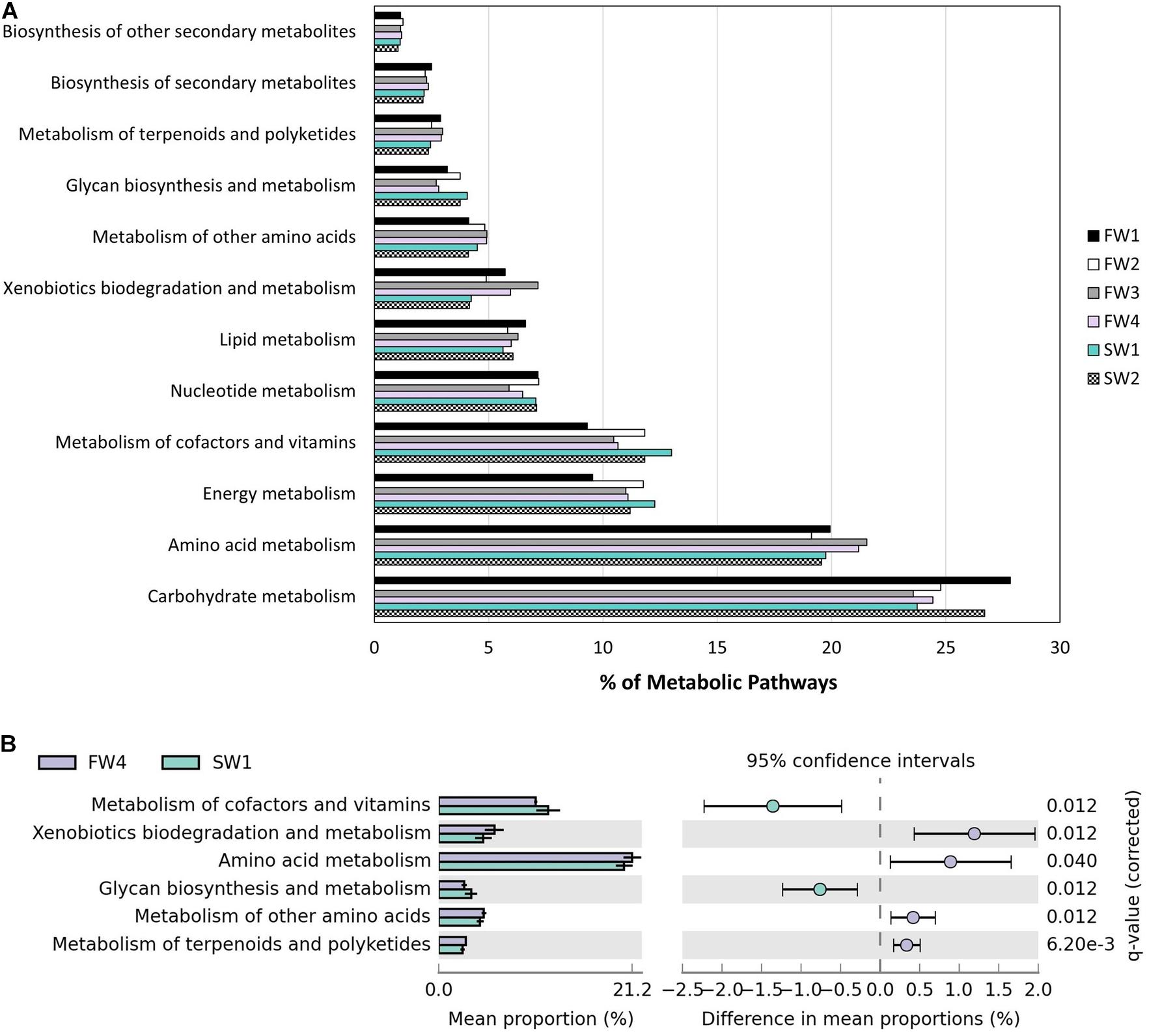
Figure 6. Functional annotation inferred by Piphillin. (A) Level 2 metabolic pathway abundance as a proportion of parent term “Metabolism” at all 6 at sampling points. (B) Significant differences in “Metabolism” level 2 terms as proportion of parent terms pre- and post-SWT (FW4 and SW1). Benjamini-Hochberg adjusted p-values are presented (q).
A total of 39 metabolic pathways showed significant differential contributions between pre- and post-SW transfer samples. Pathways with effect size >1% between pre- and post-SWT samples are shown in Table 2 (n = 24). These belonged to the level 1 metabolic categories which showed overall differential abundance (Figure 6B) as well as “Biosynthesis of other secondary metabolites,” “Lipid metabolism,” “Energy metabolism,” and “Carbohydrate metabolism.”
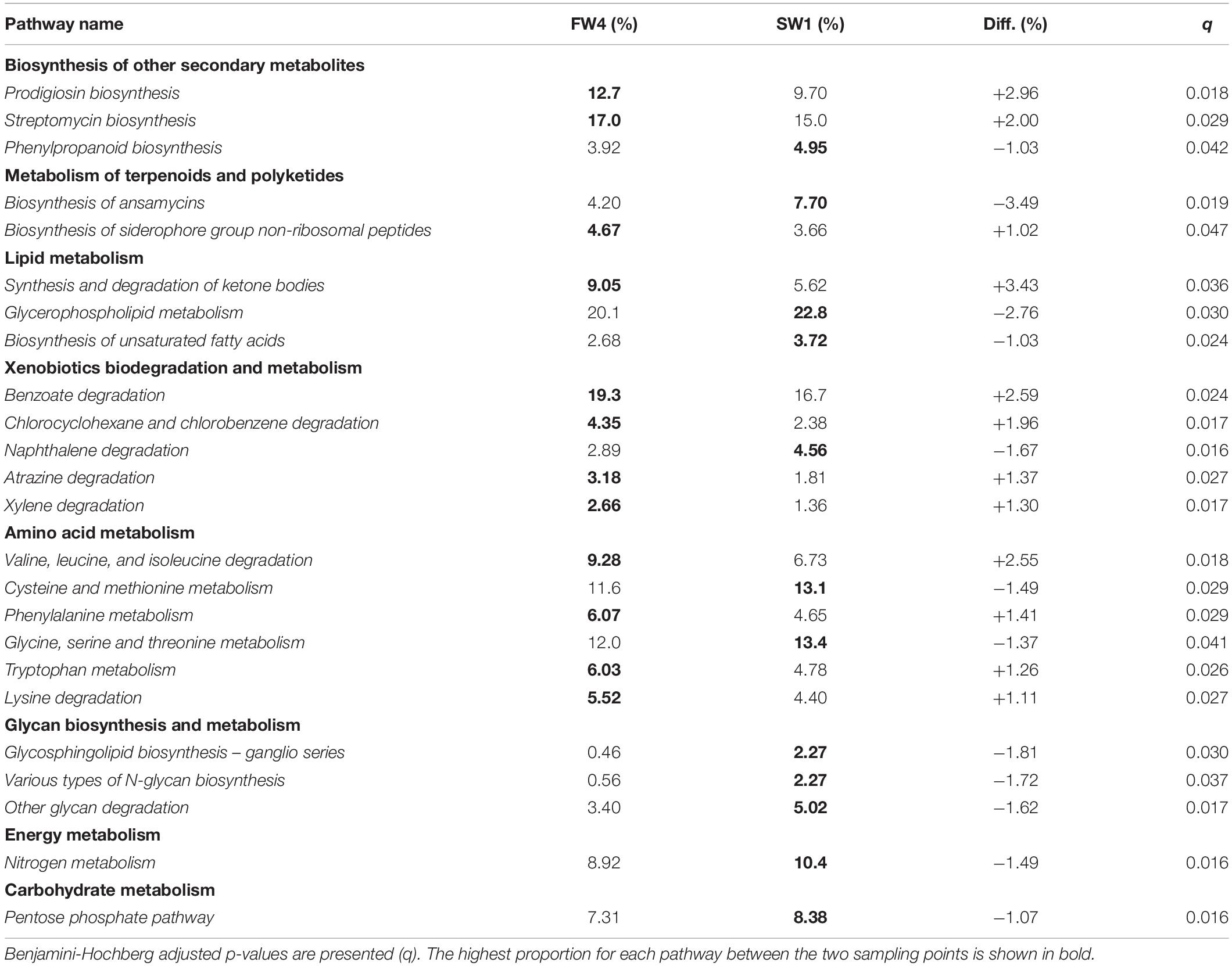
Table 2. Pathways showing significant differences in contribution to “Metabolism” level 3 pathways as proportion of level 2 parent terms pre- and post-SWT (FW4 and SW1).
Discussion
This study aimed to investigate the temporal stability of the gut microbiome in Atlantic salmon reared in FW RAS followed by transfer to open marine sea cages. A total of 6,999,913 quality filtered reads were classified into 3,730 ASVs from 16S sequencing of the Atlantic salmon hindgut. A rising trend in microbial richness was observed in the hindgut of Atlantic salmon parr undergoing smoltification in a FW RAS. Following transfer to SW, microbial diversity and richness declined and a distinct, less diverse, community structure was established, dominated by the Vibrionaceae family. Previous studies have reported a general decline in microbial diversity in the gut of Atlantic salmon as life history proceeds (Llewellyn et al., 2016; Lokesh et al., 2019; Heys et al., 2020). However, these previous studies were not conducted in RAS and covered more broadly a wide range of life history stages, while sampling intervals in the current study were designed to target the parr-smolt transformation window specifically, and the trend may be influenced by cycling of organic matter within RAS tanks.
Bacterial Composition and Core Taxa
Bacterial richness and diversity in the hindgut in the current study was similar to that observed in RAS-reared Chinook salmon (Steiner et al., 2020) and the most abundant bacterial phyla identified in digesta from all FW sampling points in RAS (i.e., Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria) were also in agreement with previous studies performed in FW RAS (Minich et al., 2020; Steiner et al., 2020). A single Pseudomonas sp. was identified as the only overall core taxon in the gut in the current study, as was also the case in Uren Webster et al. (2018), although additional cores were also observed when considering sampling points or FW and SW independently. Most core ASVs identified in the hindgut were also present in water and/or diet samples, with the exceptions of the omnipresent cores Pseudomonas ASV33, Ruminococcaceae ASV8, Moritella ASV44, Moritella ASV78, and Cutibacterium ASV149. Previous studies suggested little impact of the rearing water microbiota on the microbiome of gastrointestinal tract in fish (Gupta et al., 2019a). Core taxa identified transiently in the hindgut were generally also identified in tank water, highlighting a passing role, but suggest little to no persistent colonization.
The core taxa identified are in agreement with other studies covering FW juvenile stage, but these also reported more extensive core taxa from the phyla Firmicutes, Proteobacteria, Actinobacteria, and Tenericutes (Mycoplasma sp.) (Llewellyn et al., 2016; Dehler et al., 2017a, b; Jin et al., 2019). Mycoplasma sp. of the genus Tenericutes were notably absent in the current study, possibly as our studies focus on RAS in the FW stages, however, Tenericutes were also absent in flow-through FW systems investigated in a previous study (Jaramillo-Torres et al., 2019; Egerton et al., 2020), suggesting this observation is not specific to RAS-reared fish. Mycoplasma sp. colonization of the gut has been determined to be non-neutral, i.e., dependent upon the intra-host environment (Heys et al., 2020) and presence or absence may reflect exposure to Mycoplasma in early life. The low number of core taxa identified in the current study is likely a consequence of utilizing ASVs rather than OTUs, increasing stringency in taxon identification, but may be related also to a variety of additional factors including dietary regimes, sampling strategy, environmental factors or host genetic variation (Tarnecki et al., 2017). Distinct variability in the microbiota at the level of individual OTUs in a cohort of fish has been observed (Ciric et al., 2019). This suggests that a small number of core taxa is not surprising as the conservation of intestinal microbiota occurs primarily at the level of metabolic function, while the specific bacterial species fulfilling that function within an individual animal can vary significantly (Shafquat et al., 2014). Furthermore, fish with distinct microbiomes are often indistinguishable in terms of phenotype in a farm environment (Schmidt et al., 2016), which is interesting in the context of host-microbiome interactions, which are often considered species specific and of longstanding coevolutionary origin (Rosenberg and Zilber-Rosenberg, 2011; Franzenburg et al., 2013).
Taxa Associated With Nitrification Process in RAS
Temporal accumulation of organic matter and nitrogenous compounds are a consequence of the closed-nature of RAS, and environmental microbes play a key role in maintaining RAS water quality. Indeed, temporal increases in CO2 and nitrogenous compounds were observed in the current study (Supplementary Figure 4). When organic matter accumulates in a RAS, heterotrophic blooms can occur, outcompeting nitrifying microbes as heterotrophs obtain carbon and energy from organic matter (Leonard et al., 2000). Primary heterotrophic microbes associated with denitrification in RAS include Pseudomonas and Paracoccus, two genera identified as core in the current study, and primary autotrophs include Rhodobacter and Hydrogenophaga (Rurangwa and Verdegem, 2015). Dominance by two ASVs assigned to the facultative autotrophic denitrifiers Hydrogenophaga (Xing et al., 2018) in water samples from the RAS tank was observed at FW2 and FW3 sampling points and this was accompanied by a delayed significant increase in abundance in the hindgut. These results suggest that the water microbiome does have the potential to alter the microbial community in the gut, although it appears to have a lesser effect than diet (Uren Webster et al., 2018; Gupta et al., 2019a; Lokesh et al., 2019). Conversely, the gut microbiome itself may impact the microbial community of the water via excreted waste and it is challenging to determine the initial source of colonization and subsequent succession (Heys et al., 2020; Steiner et al., 2020). Primary hetero- and autotrophs characteristic of nitrogen cycling in RAS continued to be detected in the hindgut following SWT, albeit at lower relative abundance than in FW RAS, suggesting sustained colonization. Distinct microbial communities have been observed in biofilters, tank biofilms, tank water and mucosal samples (gut, skin and gill) but, as was also the case in this study, various biofilter-associated microbes were also detectable in mucus (Schmidt et al., 2016; Minich et al., 2020). The transient nature of such observations indicates the potential for temporary disturbances in water chemistry to impact upon the microbiomes of fish in the system via alterations to the biofilter and tank water communities.
Temporal Variation in Microbial Communities
A major question in this study was to determine if there were temporal changes of the microbiome during FW RAS followed by transfer to SW. We observed a rising trend in alpha diversity between sampling points in FW RAS as fish developed from parr to smolt, but no difference in overall community composition. Sampling point explained 27% of the variance in the hindgut microbiota suggesting that additional unidentified factors also play significant roles. In wild Atlantic salmon populations, microbiome signature within fresh and saltwater ecotypes (i.e., smolt vs. parr in FW) was not impacted by life-cycle stage (Llewellyn et al., 2016), in agreement with our findings. However, we also observed an increase in Chao1 richness between parr and smolt stages, and this temporal increase in FW was confirmed in a second RAS stream in the same facility despite differing smoltification regimes (unpublished data). The contrasting results in relation to richness during FW stages may arise from RAS vs. natural environment of the fish, as husbandry processes in RAS facilities such as disinfection may disrupt or steer microbial colonization and succession (Gupta et al., 2019b; Uren Webster et al., 2020). Furthermore, the closed-nature of RAS may support the accumulation of organic matter, providing additional substrate which may promote bacterial proliferation.
The gut microbiome of teleosts declines in diversity over time, becoming more stable and specialized, suggesting an increasingly important role for host-specific regulation, interaction between microbes and active dispersal (Burns et al., 2016; Stephens et al., 2016). Salinity is a restrictive environmental barrier for microbes (Logares et al., 2009, 2013) and a number of studies, including this one, have observed a significant decline in OTU richness and diversity, and re-structuring of the microbial community during the transition from FW to SW (Llewellyn et al., 2016; Schmidt et al., 2016; Dehler et al., 2017b). This was also the case in fish reared under a different smoltification regime in the same facility as those in the current study (Lorgen-Ritchie et al., in preparation). The final FW (FW4) and two post-SWT samples showed reciprocal patterns of change in abundance between Lactobacillus sp. (higher in FW4) and Clostridia sp. (higher in SW1). A number of lactic acid bacteria (LAB) genera previously showed differential distribution between FW and SW (Dehler et al., 2017b). Lactobacillus sp. can be added to commercial diets as a probiotic (Martínez Cruz et al., 2012) and have been observed to promote overdominance of the Lactobacillaceae family in the hindgut (Gupta et al., 2019a).
The intestinal microbial community in SW was dominated by the Vibrionaceae family. Dominance by Vibrionaceae has been observed in previous studies in salmonids in Tasmania and New Zealand where water temperatures exceeded 16°C for 4 months or spiked above average at the time of sampling (Zarkasi et al., 2014, 2016; Ciric et al., 2019). Water temperature was identified as a key factor in the prevalence and persistence of both Vibrio species in the hindgut of Atlantic salmon in Tasmania (Hatje et al., 2014). The salmon in this experiment were transferred to sea during the summer (August) and we may be echoing these previously reported findings as a spike in temperature compared to the 10-year average at a nearby climate monitoring site (15.2°C vs. 13.8°C at SW1) was observed, perhaps allowing Vibrionaceae taxa to out-compete other commonly observed SW taxa such as Mycoplasma sp. Temperature in the FW RAS itself was held at an average of 15.4°C, as the optimal temperature for growth, but higher than the 12°C generally observed in relevant studies which mostly examined fish from flow-through or aquarium systems (Dehler et al., 2017a; Lokesh et al., 2019). Furthermore, fluctuations saw water temperatures reach a maximum of 17.2°C in the RAS. The intestinal microbiome developed during FW RAS may act as a barrier to other species such as Mycoplasma sp. following SWT, however, this remains to be studied and confirmed. Interestingly, Vibrionaceae was still the dominant bacteria family at 4-weeks-post-SWT which suggests potential long-term consequences arising from the early post-transfer environment. Coincident with initial SWT and re-establishment of microbial communities, a reduction in food consumption can occur following SWT with as little as 10% of individuals feeding normally 1-week post-SWT (Stradmeyer, 1994) and we observed a decline in fish growth initially post-SWT. Additionally, the transfer of smolts to hypertonic SW results in increased drinking and overall intestinal fluid re-absorption rates (McCormick, 2012), which could impact microbial dynamics in the intestine dependent upon the surrounding environment, however, Vibrionaceae were present at very low relative abundance in water samples.
Role of the Hindgut Microbiome in Metabolic Function
Microbiome communities determined by 16S sequencing can be used to infer metagenomes and downstream functionality and the majority of identified pathways were related to metabolism. Pathways which showed the greatest magnitude of changes included biosynthesis of ansamycins and glycerophospholipid metabolism, which were more prevalent in SW compared to FW, while prodigiosin biosynthesis, synthesis and degradation of ketone bodies and “valine, leucine, and isoleucine” amino acid degradation were more prevalent in FW. Ansamycins are naturally occurring antimicrobial compounds which provide protection against a number of fish pathogens, and this pathway may be activated upon exposure to a new environment as protection against bacterial pathogens (Austin and Austin, 2012). In further support of a role in antimicrobial defense mechanisms, monobactams, which are beta-lactam antibiotics with the ability to inhibit peptidoglycan synthesis (Sykes et al., 1981; Allison and Nolan, 1994) were also more prevalent than in FW by SW2. Red-pigmented prodigiosins are also naturally occurring antibiotics (Darshan and Manonmani, 2015) which were more prominent at FW4. The branched amino acids valine, leucine and isoleucine are essential amino acids for fish (Halver et al., 1957) and play a role in energy metabolism (Roques et al., 2020) while ketone bodies produce an energy substrate which plays a role in maintaining energy homeostasis via the regulation of lipogenesis (Cabrera-Mulero et al., 2019). Increased contributions of glycerophospholipid, glycosphingolipid and glycan metabolism in SW are indicative of post-SWT structural modifications in gut mucosa.
Conclusion
Land-based salmon production has been increasing dramatically over recent years leading to new challenges in fish health, water chemistry and potential impacts on later life performance. The temporal dynamics of gut-associated microbial communities in RAS-reared fish will lead to a more comprehensive understanding of the dynamics of the RAS biological system and differences observed in performance and robustness of RAS versus loch-reared fish post-SWT. Microbial richness showed a rising temporal trend in FW RAS stages before declining and forming a distinct, less diverse, community structure post-SWT, dominated by the Vibrionaceae family. The identification of a temporally dynamic gut microbiome in RAS highlights the need to understand the impact of the RAS environment throughout a production cycle and comparative analyses in loch-reared fish are required to further understand the interplay between microbial dynamics in the FW rearing environment and performance of fish post-SWT.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA729215.
Ethics Statement
Ethical review and approval was not required for the animal study because the sampling of fish was carried out under established protocols for routine health assessments in accordance with RSPCA Assured Welfare Standards for farmed Atlantic salmon.
Author Contributions
SM, JT, and HM designed the experiment. MC, JT, and LC were responsible for management of fish parameters and smolt indicator analyses. ML-R, LC, and MC conducted the sample collection. ML-R performed the DNA extraction and preparation of samples for 16S sequencing and downstream bioinformatic data analyses, and wrote the initial manuscript draft which was reviewed and edited by SM, HM, and JT. JT and MC contributed smolt indicator analyses. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the UKRI project ROBUSTSMOLT (BBSRC BB/S004270/1 and BB/S004432/1). There was also co-funding from the Scottish Aquaculture Innovation Centre.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank John Richmond and staff at MOWI and the Centre for Genome Enabled Biology and Medicine (CGEBM) at the University of Aberdeen, particularly Dr. Ewan Campbell, for help with amplification protocols, conducting 16S library preparation and sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.711797/full#supplementary-material
Footnotes
References
Allison, D. G., and Nolan, R. D. (1994). Influence of growth rate and nutrient limitation on monobactam production and peptidoglycan synthesis in Pseudomonas aeruginosa. J. Basic Microbiol. 34, 217–224. doi: 10.1002/jobm.3620340403
Attramadal, K. J. K., Truong, T. M. H., Bakke, I., Skjermo, J., Olsen, Y., and Vadstein, O. (2014). RAS and microbial maturation as tools for K-selection of microbial communities improve survival in cod larvae. Aquaculture 432, 483–490. doi: 10.106/j.aquaculture.2014.05.052
Björnsson, B. T., Stefansson, S. O., and McCormick, S. D. (2011). Environmental endocrinology of salmon smoltification. Gen. Comp. Endocrinol. 170, 290–298. doi: 10.1016/j.ygcen.2010.07.003
Blancheton, J. P., Attramadal, K. J. K., Michaud, L., d’Orbcastel, E. R., and Vadstein, O. (2013). Insight into bacterial population in aquaculture systems and its implication. Aquacult. Eng. 53, 30–39. doi: 10.1016/j.aquaeng.2012.11.009
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Burns, A. R., Stephens, W. Z., Stagaman, K., Wong, S., Rawls, J. F., Guillemin, K., et al. (2016). Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 10, 655–664. doi: 10.1038/ismej.2015.142
Cabrera-Mulero, A., Tinahones, A., Bandera, B., Moreno-Indias, I., Macías-González, M., and Tinahones, F. J. (2019). Keto microbiota: a powerful contributor to host disease recovery. Rev. Endocr. Metab. Disord. 20, 415–425. doi: 10.1007/s11154-019-09518-8
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chao, A. (1984). Nonparametric estimation of the number of classes in a population. Scand. J. Statist. 11, 265–270.
Ciric, M., Waite, D., Draper, J., and Jones, J. B. (2019). Characterization of mid-intestinal microbiota of farmed Chinook salmon using 16S rRNA gene metabarcoding. Arch. Biol. Sci. 71, 577–587. doi: 10.2298/ABS190402040C
Darshan, N., and Manonmani, H. K. (2015). Prodigiosin and its potential applications. J. Food Sci. Technol. 52, 5393–5407. doi: 10.1007/s13197-015-1740-4
De Schryver, P., and Vadstein, O. (2014). Ecological theory as a foundation to control pathogenic invasion in aquaculture. ISME J. 8, 2360–2368. doi: 10.1038/ismej.2014.84
Dehler, C. E., Secombes, C. J., and Martin, S. A. M. (2017a). Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 467, 149–157. doi: 10.1016/j.aquaculture.2016.07.017
Dehler, C. E., Secombes, C. J., and Martin, S. A. M. (2017b). Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci. Rep. 7:13877. doi: 10.1038/s41598-017-13249-8
Dhanasiri, A. K. S., Brunvold, L., Brinchmann, M. F., Korsnes, K., and Bergh, Ø, and Kiron, V. (2011). Changes in the intestinal microbiota of wild Atlantic cod Gadus morhua L. upon captive rearing. Microb. Ecol. 61, 20–30. doi: 10.1007/s00248-010-9673-y
d’Orbcastel, E. R., Blancheton, J., and Aubin, J. (2009). Towards environmentally sustainable aquaculture: comparison between two trout farming systems using Life Cycle Assessment. Aquacult. Eng. 40, 113–119. doi: 10.1016/j.aquaeng.2008.12.002
Egerton, S., Wan, A., Murphy, K., Collins, F., Ahern, G., Sugrue, I., et al. (2020). Replacing fishmeal with plant protein in Atlantic salmon (Salmo salar) diets by supplementation with fish protein hydrolysate. Sci. Rep. 10:4194. doi: 10.1038/s41598-020-60325-7
Fossmark, R. O., Attramadal, K. J. K., Nordøy, K., Østerhus, S. W., and Vadstein, O. (2021). A comparison of two seawater adaptation strategies for Atlantic salmon post-smolt (Salmo salar) grown in recirculating aquaculture systems (RAS): nitrification, water and gut microbiota, and performance of fish. Aquaculture 532:735973. doi: 10.1016/j.aquaculture.2020.735973
Fossmark, R. O., Vadstein, O., Rosten, T. W., Bakke, I., Košeto, D., Bugten, A. V., et al. (2020). Effects of reduced organic matter loading through membrane filtration on the microbial community dynamics in recirculating aquaculture systems (RAS) with Atlantic salmon parr (Salmo salar). Aquaculture 524:735268. doi: 10.1016/j.aquaculture.2020.735268
Franzenburg, S., Walter, J., Künzel, S., Wang, J., Baines, J. F., Bosch, T. C. G., et al. (2013). Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl. Acad. Sci. U.S.A. 110, E3730–E3738. doi: 10.1073/pnas.1304960110
Givens, C. E., Ransom, B., Bano, N., and Hollibaugh, J. T. (2015). Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 518, 209–223. doi: 10.3354/meps11034
Gupta, S., Fečkaninová, A., Lokesh, J., Koščová, J., Sørensen, M., Fernandes, J., et al. (2019a). Lactobacillus dominate in the Intestine of Atlantic Salmon fed dietary probiotics. Front. Microbiol. 9:3247. doi: 10.3389/fmicb.2018.03247
Gupta, S., Fernandes, J., and Kiron, V. (2019b). Antibiotic-induced perturbations are manifested in the dominant intestinal bacterial Phyla of Atlantic salmon. Microorganisms 7:233. doi: 10.3390/microorganisms7080233
Halver, J. E., Delong, D. C., and Mertz, E. T. (1957). Nutrition of salmonoid fishes. V. classification of essential amino acids for Chinook salmon. J. Nutr. 63, 95–105. doi: 10.1093/jn/63.1.95
Hatje, E., Neuman, C., Stevenson, H., Bowman, J. P., and Katouli, M. (2014). Population dynamics of Vibrio and Pseudomonas species isolated from farmed Tasmanian Atlantic salmon (Salmo salar L.): a seasonal study. Microb. Ecol. 68, 679–687. doi: 10.1007/s00248-014-0462-x
Heys, C., Cheaib, B., Busetti, A., Kazlauskaite, R., Maier, L., Sloan, W. T., et al. (2020). Neutral processes dominate microbial community assembly in Atlantic salmon, Salmo salar. Appl. Environ. Microbiol. 86:e02283-19. doi: 10.1128/AEM.02283-19
Houde, E. D. (1981). Growth rates, rations and cohort consumption of marine fish larvae in relation to prey concentrations. Rapp. Reun. Cons. Int. Explor. Mer. 178, 441–453.
Iwai, S., Weinmaier, T., Schmidt, B. L., Albertson, D. G., Poloso, N. J., Dabbagh, K., et al. (2016). Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PLoS One 11:e0166104. doi: 10.137/journal.pone.0166104
Jaramillo-Torres, A., Rawling, M. D., Rodiles, A., Mikalsen, H. E., Johansen, L., Tinsley, J., et al. (2019). Influence of dietary supplementation of probiotic Pediococcus acidilactici MA18/5M during the transition from freshwater to seawater on intestinal health and microbiota of Atlantic Salmon (Salmo salar L.). Front. Microbiol. 10:2243. doi: 10.3389/fmicb.2019.02243
Jin, Y., Angell, I. L., Sandve, S. R., Snipen, L. G., Olsen, Y., and Rudi, K. (2019). Atlantic salmon raised with diets low in long-chain polyunsaturated n-3 fatty acids in freshwater have a Mycoplasma-dominated gut microbiota at sea. Aquacult. Environ. Interact. 11, 31–39. doi: 10.3354/aei00297
Johansson, L.-H., Timmerhaus, G., Afanasyev, S., Jørgensen, S. M., and Krasnov, A. (2016). Smoltification and seawater transfer of Atlantic salmon (Salmo salar L.) is associated with systemic repression of the immune transciprtome. Fish Shellf. Immunol. 58, 33–41. doi: 10.1016/j.fsi.2016.09.026
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kolde, R. (2012). Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. Available online at: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed April 6, 2021).
Lahti, L., and Shetty, S. A. (2017). Tools for Microbiome Analysis in R. Microbiome Package Version 1.7.21. Available online at: https://github.com/microbiome/microbiome (accessed April 6, 2021).
Leonard, N., Blancheton, J. P., and Guiraud, J. P. (2000). Populations of heterotrophic bacteria in an experimental recirculating aquaculture system. Aquacult. Eng. 22, 109–120. doi: 10.1016/S0144-8609(00)00035-2
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H., and Derome, N. (2014). Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5:207. doi: 10.3389/fmicb.2014.00207
Llewellyn, M. S., McGinnity, P., Dionne, M., Letourneau, J., Thonier, F., Carvalho, G. R., et al. (2016). The biogeography of the atlantic salmon (Salmo salar) gut microbiome. ISME J. 10, 1280–1284. doi: 10.1038/ismej.2015.189
Logares, R., Bråte, J., Bertilsson, S., Clasen, J. L., Shalchian-Tabrizi, K., and Rengefors, K. (2009). Infrequent marine–freshwater transitions in the microbial world. Trends Microbiol. 17, 414–422. doi: 10.1016/j.tim.2009.05.010
Logares, R., Lindström, E. S., Langenheder, S., Logue, J. B., Paterson, H., Laybourn-Parry, J., et al. (2013). Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J. 7, 937–948. doi: 10.1038/ismej.2012.168
Lokesh, J., Kiron, V., Sipkema, D., Fernandes, J. M. O., and Moum, T. (2019). Succession of embryonic and the intestinal bacterial communities of Atlantic salmon (Salmo salar) reveals stage-specific microbial signatures. Microbiologyopen 8:e00672. doi: 10.1002/mbo3.672
Martínez Cruz, P., Ibáñez, A. L., Monroy Hermosillo, O. A., and Ramírez Saad, H. C. (2012). Use of probiotics in aquaculture. ISRN Microbiol. 2012:916845. doi: 10.5402/2012/916845
McCormick, S. D. (2012). “5 - Smolt physiology and endocrinology,” in Fish Physiology, eds S. D. McCormick, A. P. Farrell, and C. J. Brauner (Cambridge, MA: Academic Press), 199–251. doi: 10.1016/b978-0-12-396951-4.00005-0
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Minich, J. J., Poore, G. D., Jantawongsri, K., Johnston, C., Bowie, K., Bowman, J., et al. (2020). Microbial ecology of Atlantic Salmon (Salmo salar) hatcheries: impacts of the built environment on fish mucosal microbiota. App. Environ. Microbiol. 86:e0411-20. doi: 10.1128/AEM.00411-20
Narayan, N. R., Weinmaier, T., Laserna-Mendieta, E., Claesson, M. J., Shanahan, F., Dabbagh, K., et al. (2020). Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genom. 21:56. doi: 10.1186/s12864-019-6427-1
Navarrete, P., Magne, F., Araneda, C., Fuentes, P., Barros, L., Opazo, R., et al. (2012). PCR-TTGE analysis of 16S rRNA from rainbow trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS One 7:e31335. doi: 10.1371/journal.pone.0031335
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2020). vegan: Community Ecology Package. R Package Version 2.5-7. Available online at: https://CRAN.R-project.org/package=vegan (accessed April 6, 2021)
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/mar/gks1219
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Roques, S., Deborde, C., Richard, N., Skiba-Cassy, S., Moing, A., and Fauconneau, B. (2020). Metabolomics and fish nutrition: a review in the context of sustainable feed development. Rev. Aquacult. 12, 261–282. doi: 10.111/raq.12316
Rosenberg, E., and Zilber-Rosenberg, I. (2011). “The hologenome concept,” in Beneficial Microorganisms in Multicellular Life Forms, eds E. Rosenberg and U. Gophna (Berlin: Springer), 323–340. doi: 10.1007/978-3-642-21680-0_24
Rudi, K., Angell, I. L., Pope, P. B., Vik, J. O., Sandve, S. R., and Snipen, L. (2018). Stable core gut microbiota across the freshwater-to-saltwater transition for farmed Atlantic Salmon. Appl. Environ. Microbiol. 84:e01974-17. doi: 10.1128/AEM.01974-17
Rurangwa, E., and Verdegem, M. C. J. (2015). Microorganisms in recirculating aquaculture systems and their management. Rev. Aquacult. 7, 117–130. doi: 10.1111/raq.12057
Salazar, G. (2020). EcolUtils: Utilities for Community Ecology Analysis. R Package Version 0.1. Available online at: https://github.com/GuillemSalazar/EcolUtils (accessed April 6, 2021).
Schmidt, V., Amaral-Zettler, L., Davidson, J., Summerfelt, S., and Good, C. (2016). Influence of fishmeal-free diets on microbial communities in Atlantic Salmon (Salmo salar) recirculation aquaculture systems. Appl. Environ. Microbiol. 82, 4470–4481. doi: 10.1128/AEM.00902-16
Schmidt, V. T., Smith, K. F., Melvin, D. W., and Amaral-Zettler, L. A. (2015). Community assembly of euryhaline fish microbiota during salinity acclimation. Mol. Ecol. 24, 2537–2550. doi: 10.1111/mec.13177
Shafquat, A., Joice, R., Simmons, S. L., and Huttenhower, C. (2014). Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol. 22, 261–266. doi: 10.1016/j.tim.2014.01.011
Sigholt, T., Staurnes, M., Jakobsen, H. J., and Åsgård, T. (1995). Effects of continuous light and short-day photoperiod on smolting, seawater survival and growth in Atlantic salmon (Salmo salar). Aquaculture 130, 373–388. doi: 10.1016/0044-8486(94)00349-S
Sprockett, D., Fukami, T., and Relman, D. A. (2018). Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 15, 197–205. doi: 10.1038/nrgastro.2017.173
Steiner, K., Heasman, K., Laroche, O., Pochon, X., Preece, M., Bowman, J. P., et al. (2020). The microbiome of Chinook salmon (Oncorhynchus tshawytscha) in a recirculation aquaculture system. Aquaculture 534:736227. doi: 10.1016/j.aquaculture.2020.736227
Stephens, W. Z., Burns, A. R., Stagaman, K., Wong, S., Rawls, J. F., Guillemin, K., et al. (2016). The composition of the zebrafish intestinal microbial community varies across development. ISME J. 10, 644–654. doi: 10.1038/ismej.2015.140
Stradmeyer, L. (1994). Survival, growth and feeding of Atlantic salmon, Salmo salar L., smolts after transfer to sea water in relation to the failed smolt syndrome. Aquacult. Res. 25, 103–112. doi: 10.1111/j.1365-2109.1994.tb00670.x
Sullam, K. E., Essinger, S. D., Lozupone, C. A., Lozupone, C. A., O’Connor, M. P., Rosen, G. L., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Sykes, R. B., Cimarusti, C. M., Bonner, D. P., Bush, K., Floyd, D. M., Georgopapadakou, N. H., et al. (1981). Monocyclic β-lactam antibiotics produced by bacteria. Nature 291, 489–491. doi: 10.1038/291489a0
Tarnecki, A. M., Burgos, F. A., Ray, C. L., and Arias, C. R. (2017). Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol. 123, 2–17. doi: 10.1111/jam.13415
Uren Webster, T. M., Consuegra, S., Hitchings, M., and Garcia de Leaniz, C. (2018). Interpopulation variation in the Atlantic salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol. 84:e0691-18. doi: 10.1128/AEM.00691-18
Uren Webster, T. M., Rodriguez-Barreto, D., Castaldo, G., Gough, P., Consuegra, S., and Garcia de Leaniz, C. (2020). Environmental plasticity and colonisation history in the Atlantic salmon microbiome: A translocation experiment. Mol. Ecol. 29, 886–898. doi: 10.1111/mec.15369
Vellend, M. (2016). The Theory of Ecological Communities (MPB-57). Princeton, NJ: Princeton University Press.
Xing, W., Li, J., Li, P., Wang, C., Cao, Y., Li, D., et al. (2018). Effects of residual organics in municipal wastewater on hydrogenotrophic denitrifying microbial communities. J. Environmental Sci. 65, 262–270. doi: 10.1016/j.jes.2017.03.001
Zarkasi, K. Z., Abell, G. C. J., Taylor, R. S., Neuman, C., Hatje, E., Tamplin, M. L., et al. (2014). Pyrosequencing-based characterization of gastrointestinal bacteria of Atlantic salmon (Salmo salar L.) within a commercial mariculture system. J. Appl. Microbiol. 117, 18–27. doi: 10.1111/jam.12514
Keywords: aquaculture, Atlantic salmon (Salmo salar), intestine, microbiome, recirculating aquaculture systems, temporal
Citation: Lorgen-Ritchie M, Clarkson M, Chalmers L, Taylor JF, Migaud H and Martin SAM (2021) A Temporally Dynamic Gut Microbiome in Atlantic Salmon During Freshwater Recirculating Aquaculture System (RAS) Production and Post-seawater Transfer. Front. Mar. Sci. 8:711797. doi: 10.3389/fmars.2021.711797
Received: 19 May 2021; Accepted: 15 June 2021;
Published: 06 July 2021.
Edited by:
Fotini Kokou, Wageningen University & Research, NetherlandsReviewed by:
Sanni Leea Aalto, Technical University of Denmark, DenmarkÜmit Acar, Çanakkale Onsekiz Mart University, Turkey
Copyright © 2021 Lorgen-Ritchie, Clarkson, Chalmers, Taylor, Migaud and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel A. M. Martin, sam.martin@abdn.ac.uk