The Microbiome of the Worldwide Invasive Ascidian Didemnum vexillum
 Maria Casso
Maria Casso Marta Turon
Marta Turon Núria Marco
Núria Marco Marta Pascual
Marta Pascual Xavier Turon
Xavier Turon- 1Department of Marine Ecology, Center for Advanced Studies of Blanes, Consejo Superior de Investigaciones Científicas, (CEAB, CSIC), Blanes, Spain
- 2Department of Genetics, Microbiology and Statistics, and Institut de Recerca de la Biodiversitat (IRBio), University of Barcelona, Barcelona, Spain
All multicellular organisms, including ascidians, host diverse microbial communities that are essential for their evolution. The global invader Didemnum vexillum is a colonial species native to Japan with two main genetic clades, A (the only invasive) and B, which provides a unique opportunity to assess if the microbiome remains stable in the colonization process or shifts according to local environment. We have analyzed, using 16S amplicon sequencing, the microbiome of 65 D. vexillum colonies from 13 populations worldwide including the two clades in the native area, plus samples from a congeneric species and seawater from one of the localities. We found 3,525 zero-radius operational taxonomic units (ZOTUs) in D. vexillum, belonging to 36 bacterial and 3 archaeal phyla. The microbiome of this species had a markedly different composition from surrounding seawater and from the congeneric species. For the globally invasive clade A, we found 3,154 ZOTUs, and 8 of them were present in all colonies, constituting a core microbiome with high abundance (69.57% of the total reads) but low diversity (0.25% of the total number of ZOTUs). The variable component was quantitatively much less important but comprised a highly diverse assemblage. In a multiple regression model, global microbiome structure correlated with differences in temperature range across localities and also with geographic distances, pointing to horizontal acquisition of the symbionts. However, the ascidian may have a strong capacity to select and enrich its microbiome, as we found that the most abundant ZOTUs from tunic samples had low abundance in seawater samples from the same locality. The microbiome structure also correlated with the genetic distances between colonies obtained in a previous genome-wide analysis, suggesting some potential for vertical transmission. In geographically restricted comparisons, temperature and genetic makeup, but not geography, explained microbiome structure. The combination of a quantitatively dominant core component and a highly diverse variable fraction in the microbiome of D. vexillum can contribute to the success of this global invader in different environments.
Introduction
All multicellular organisms can be considered holobionts hosting diverse microbial communities that are essential for the evolution of the organism (Uriz et al., 2012; Rosenberg and Zilberg-Rosenberg, 2016, 2018; Leite et al., 2018; Pollock et al., 2018; Cleary et al., 2019). The ubiquity and importance of bacteria in the living world have been recently showcased by the application of new genomic technologies (McFall-Ngai et al., 2013). Holobionts rely on their microbiome for vital processes (Mandrioli and Manicardi, 2013), which led to the concept of the hologenome (the collective genomes of the holobiont) as a unit of selection (Zilber-Rosenberg and Rosenberg, 2008; Rosenberg and Zilberg-Rosenberg, 2018). Numerous studies used different methods to determine how the microbiome provides mutualistic benefits to the host. Some of these benefits are nutrient fixation or improved metabolism (Newton et al., 2008; Barott et al., 2011; Kellogg, 2019), protection against predation or other competitors (Barott et al., 2011; Kwan et al., 2012), and adaptation to diverse environments (Rosenberg et al., 2010; Mortzfeld et al., 2016; Ziegler et al., 2017; Morrissey et al., 2019; Wilkins et al., 2019).
Invertebrates are crucial to the analysis of animal–microbe interactions (Petersen and Osvatic, 2018). Among marine invertebrates, most research to date has been performed on corals and sponges (e.g., Webster and Taylor, 2012; Blackall et al., 2015; Hill and Sacristán-Soriano, 2017; Pita et al., 2018; Kellogg, 2019; van Oppen and Blackall, 2019). Comparatively, ascidians have attracted less attention, despite harboring a highly diverse assemblage of symbionts in their tunics that can fulfill diverse functions (Erwin et al., 2014; Cahill et al., 2016; Dou and Dong, 2019). These symbionts have been investigated mostly from the point of view of their potential role in the production of ascidian secondary metabolites (Schmidt, 2014; Tianero et al., 2015; Chen et al., 2018; Dou and Dong, 2019). However, other roles have also been pointed out, such as photosymbiosis (Donia et al., 2011; López-Legentil et al., 2011; Hirose, 2015), nitrification (Martínez-García et al., 2008; Erwin et al., 2014), or vanadium accumulation (Ueki et al., 2019).
Ascidians are a group featuring many notorious introduced species around the world (Lambert, 2007; Shenkar and Swalla, 2011; Zhan et al., 2015). They have several characteristics of successful colonizers, such as a rapid growth rate, high fecundity, and short time to maturity (e.g., Rius et al., 2009; Pineda et al., 2013; Casso et al., 2018). They are also known to adapt to a range of temperatures, salinities, and pollution levels (Naranjo et al., 1996; Pineda et al., 2012; Nagar and Shenkar, 2016; Rocha et al., 2017). Colonial ascidians can also reproduce asexually by fragmentation and fuse to form chimeras with zooids of different genotypes, which may enhance the colony’s adaptive capacity (Ben-Shlomo, 2017; Casso et al., 2019a). For introduced marine species in general, it has been pointed out that the microbial symbiont community can contribute to the survival and adaptation of the holobiont during the invasion process. Several mechanisms have been proposed, such as heavy metal detoxification (Aires et al., 2016), production of toxic chemicals that can increase competitive ability on a new habitat (Amsellem et al., 2017), or enhanced nutrient fixation (Arnaud-Haond et al., 2017). Likewise, broadly introduced species face diverse temperature regimes, and microbial symbionts have been shown to play a role in adaptation of invertebrates to temperature changes (e.g., in corals: Ziegler et al., 2017; Osman et al., 2020). In this context, the tunic ascidian microbiome has been suggested to enhance to the adaptive capacity and invasion success of this group (Evans et al., 2017; Novak et al., 2017; Dror et al., 2019).
Didemnum vexillum Kott (2002) is an invasive colonial ascidian, native from the NW Pacific, with two main mitochondrial clades, named clade A and clade B, of which only clade A has spread in temperate seas worldwide (New Zealand, East and West coasts of North America, Atlantic and Mediterranean coasts of Europe, and South East of Russia: Lambert, 2009; Stefaniak et al., 2012; Ordóñez et al., 2015; Zvyagintsev et al., 2016). This clade shows a strong population structure, with three main invasive genetic groups and a high genetic differentiation between populations (Casso et al., 2019b). Environmental conditions are different between the main regions of distribution of the species and even between localities in the same region (e.g., Mediterranean and Atlantic localities in Europe). Therefore, the ascidian faced different environmental stressors during the introduction events; thus, quick responses such as those mediated by their microbiota could enhance adaptation and invasive potential. In the present study, we (i) describe and compare the microbiome of D. vexillum at different levels (species, mitochondrial clades, localities, and invasive genetic groups); (ii) identify the core microbiome of the invasive clade; and (iii) evaluate the relationship between microbiome composition and several environmental and genetic variables. We had the unique opportunity to assess individually the relationship between host genetics and microbiome as the same colonies analyzed in the present study were previously genotyped at a panel of genome-wide loci (Casso et al., 2019b).
Materials and Methods
Sampling
A total of 65 colonies of D. vexillum from 12 locations spanning the native (NW Pacific) and the known introduced distribution of the species were sampled during 2015 and 2016 in marinas and artificial substrates (Table 1 and Figure 1). Sequences of the specimens showed that five colonies were D. vexillum clade B from Sugashima (Japan), whereas the remaining 60 colonies were D. vexillum clade A from different temperate regions of the world (Casso et al., 2019b). Thus, only in one of the two populations (Sugashima and Aomori) corresponding to the native range of the species did we find the two described mitochondrial clades in sympatry. Moreover, five colonies of a different species (Didemnum sp.), likely a new species (authors’ unpublished research), were sampled in a marina in Roscoff (France) and included in the present study for comparative purposes (Table 1). All 70 colonies were collected in marinas and other artificial substrates, at least 2 m apart from each other to avoid pseudoreplication (Smith et al., 2012), and preserved at −20°C in 96% ethanol. Samples were coded by geographic region as defined in previous studies (Stefaniak et al., 2012; Casso et al., 2019b). Clade A introduced samples were assigned to the previously defined genetic groups (Casso et al., 2019b): Europe (EUR), West North America (WNA), and New Zealand plus East North America (NZ-ENA).
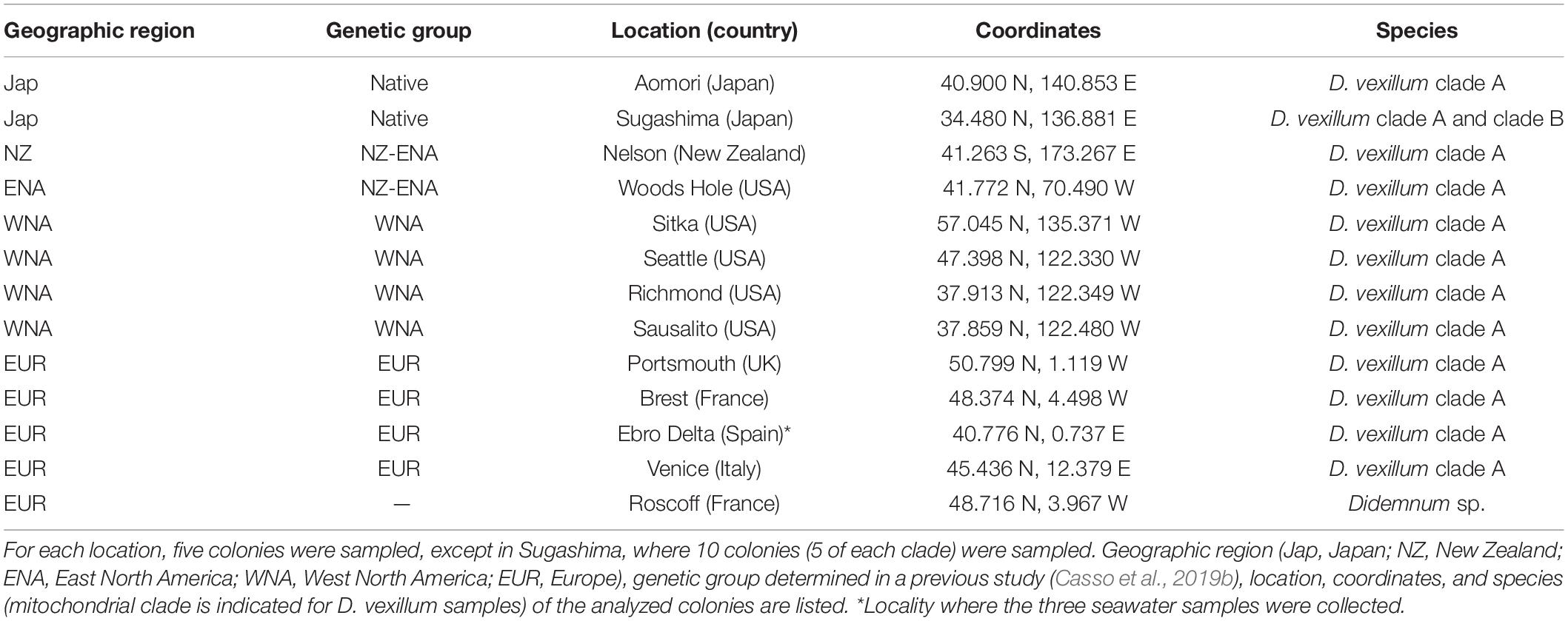
Table 1. Origin of samples for microbiome analyses.
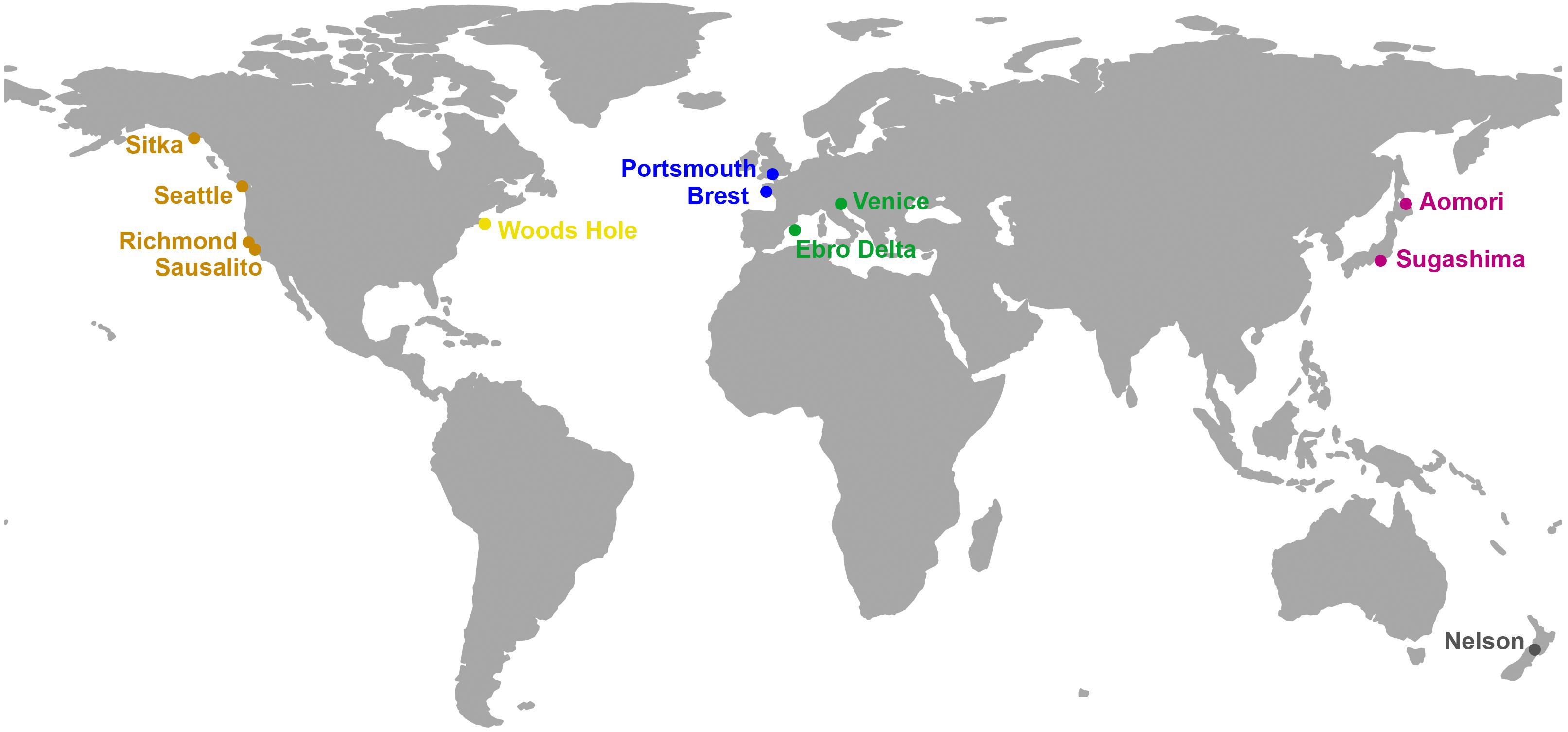
Figure 1. Map of the localities sampled in the present study. Colors follow the palette used in the nmMDS plots. The map has been drawn with the R package “rworldmap (https://cran.rproject.org/web/packages/rworldmap/index.html).”
Additionally, three seawater samples of 1 L were collected adjacent to the sampling site in the Ebro Delta location (Table 1). They were separately pre-filtered using 47-mm-diameter polycarbonate 5 μm pore-size filters to eliminate large particles and then filtered using a total of four 47 mm diameter polycarbonate 0.2 μm pore-size filters per sample (250 mL of seawater per filter). All 0.2 μm pore-size filters were preserved in 500 μL of lysis buffer (Tris 50 mM pH 8.3, EDTA 40 mM, sucrose 0.75 M) at −20°C.
DNA Extraction and Sequencing
From each colony, a fragment of ca. 4–16 mm2 of distal layer of the tunic (up to 0.5 mm thick) was cleaned of zooids and used for DNA extraction. We avoided using basal surfaces in contact with the substrate to prevent epibiont contamination (see section Discussion). We used the QIAamp mini kit (Qiagen, Venlo, The Netherlands) and followed the manufacturer’s recommendations, with a final elution in 80 μL of AE buffer. DNA extractions of the seawater filters were performed using both the filter and the buffer. As described in Turon et al. (2018), membranes were enzymatically digested with lysozyme, proteinase K, and sodium dodecyl sulfate. DNA was extracted with phenol:chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) and chloroform:isoamyl alcohol (24:1, vol/vol). Purification and concentration of the DNA was carried out with Amicon Ultra 4 Centrifugal Filter Units–100,000 NMWL (Merck Millipore, Burlington, Massachusetts, United States). The average concentration of DNA in samples was of 13.17 ng/μL (SE ± 1.08) in a volume of 20 μL of AE buffer. A fragment of the V4 region of the bacterial 16S rRNA gene was amplified using the F515/R806 primers (Caporaso et al., 2011) and paired-end sequenced (2 × 250 bp fragments) in a MiSeq Illumina platform, at the Research Technology Support Facility of the Michigan State University (USA). All sequences obtained were deposited in the Short Read Archive of the NCBI under Bioproject PRJNA608680.
Sequence Data Processing
Raw reads were processed using USEARCH v.10.0.240 (Edgar, 2010) and VSEARCH v.1.4.1 (Rognes et al., 2016). First, paired-ends were merged (using USEARCH fastq_mergepairs command: fastq_maxdiffs = 20, setting the maximum number of mismatches in the alignment to 20 bp), and then they were truncated to 250 bp (VSEARCH fastq_filter: fastq_trunclen = 250, fastq_maxee = 0.25). Only the remaining quality-filtered sequences were used to find the set of unique sequences (USEARCH fastx_uniques). Unique sequences were then denoised using the UNOISE algorithm (Edgar, 2016), which performs error correction and chimera removal on amplicon reads (USEARCH unoise3: unoise_alpha = 2 and minsize = 8 as per default settings), to obtain a list of zero-radius operational taxonomic units (ZOTUs) (100% identity; Edgar and Flyvbjerg, 2015; Edgar, 2016). Using an identity of 100% has been shown to be optimal for the V4 16S rDNA sequences (Edgar, 2018). Finally, we constructed the final table of ZOTU abundances by mapping the reads to the list of ZOTUs using the USEARCH otutab command with the default identity threshold of 0.97. All subsequent analyses were based on this ZOTU table.
Taxonomic assignment of each ZOTU was performed using the SILVA_132_SSURef_NR99 database with SINA v.1.2.11 (Pruesse et al., 2007) (SINA: search-max-result = 7, search-min-sim = 0.7). Sequences with an alignment quality score below 90% and those identified as mitochondria or chloroplasts were removed from the ZOTU table. Reads from the four filters of each seawater sample were averaged. Finally, to standardize sequencing depths among the samples, the ZOTU table was read into R (R Core Team, 2018), rarefied 100 times to the lowest read count (n = 31,206) using the R package1 “vegan” (Oksanen et al., 2019) and averaged to get the working ZOTU table.
Microbial Community Analysis
Microbial composition was assessed by relative read abundance and by relative number of ZOTUs of the major groups of symbionts in each of the 73 samples (i.e., 70 tunic and 3 seawater samples). Core microbial communities, defined as ZOTUs present in all samples, were identified without applying a minimum abundance threshold. Unique ZOTUs of a given locality were defined as those present only in that locality, and in at least two replicates, thus excluding singletons (i.e., ZOTUs found in only one sample). Shannon’s diversity and Pielou’s evenness indices were calculated for each sample using the R package “vegan” and averaged for each locality. Diversity and evenness measures were further compared between localities with a Kruskal–Wallis (K-W) and a Dunn test using the “dunn.test” R package (Dinno, 2017). We used the Benjamini–Hochberg (White et al., 2019) false discovery rate method to correct p-values for multiple comparisons. Rarefaction curves of ZOTU richness at increasing number of reads were obtained with rarecurve function in “vegan.” Using the same package, the Bray–Curtis dissimilarity matrix was calculated on rarefied read abundance values with square-root transformation (Legendre and Gallagher, 2001) and used to perform non-metric multidimensional scaling (nmMDS) analyses using the metaMDS function. PERMANOVA (permutational analysis of variance; Anderson, 2001) tests were done with function adonis of the “vegan” package to compare different groups of samples. When this test was significant, we performed permutational pairwise tests with package “pairwiseAdonis” (Martinez-Arbizu, 2019) to assess differences between pairs of levels of the significant factor, using again the Benjamini–Hochberg method to correct p-values. For the worldwide distributed mitochondrial clade (clade A), the dissimilarity of microbiome samples was compared within localities, between localities for each genetic group (Casso et al., 2019b) and between genetic groups using density plots and analyzed with a K-W and a Dunn test with the “dunn.test” R package as described above.
Comparison With Other Variables
For the clade A, we tested the correlation between the ordination scores of the nmMDS configuration and the temperature variables in each locality (minimum, maximum, mean, and range of temperatures), obtained from World Sea Temperatures2, using the envfit test in “vegan.”
We further explored in the clade A dataset the role of the hosts’ genotypes and environmental parameters (thermal and geographic distances between localities) on the microbiome composition. Prevosti genetic distances between colonies were obtained from a previous study (Casso et al., 2019b), in which exactly the same colonies were analyzed using multiple loci recovered with a genotyping-by-sequencing procedure. Thermal distances were calculated as differences in the temperature variable that best explained the ordination found in the previous analysis (envfit). Geographic distances were calculated as the shortest distance by sea (excluding routes through the Panama Canal) obtained from SEA-DISTANCES.ORG3. We performed a stepwise multiple regression analysis to model microbiome structure (Bray–Curtis distance) as a function of genetic, thermal, and geographic distances. The analysis was run with the “MASS” R package (Venables and Ripley, 2002) in forward mode (adding variables sequentially) using the Akaike Information Criterion (AIC) to evaluate the fitness gain as variables are added to the model. A global analysis was performed for the complete dataset and two restricted analyses were done for EUR (Europe) and WNA (West North America) as these areas were colonized by a single ascidian genetic group (Casso et al., 2019b), allowing comparisons within geographically restricted and genetically similar populations.
Results
Microbial Community Composition and Diversity
On average, 100,326 reads per sample were retained in the ZOTU table, with a minimum of 31,206 (subsample size for rarefaction) and a maximum of 226,123 reads. A total of 4,323 ZOTUs were identified among the 73 samples, with an average of 266.4 ZOTUs per sample (SE ± 30.78). Supplementary Table S1 lists the ZOTUs retained, with their taxonomic affiliation, abundance per sample, and sequence. Except for seawater samples, rarefaction curves reached clear asymptotes, indicating that sequencing depth was sufficient to capture microbiome diversity per sample (Supplementary Figure S1). Most of the ZOTUs (4,236) corresponded to 36 different bacteria phyla and the remaining ones (87) to 3 Archaea phyla, which represented less than 0.1% of the reads. Among bacteria, the most abundant phylum in number of reads was Proteobacteria (79.08% of the total reads of bacteria), with Alphaproteobacteria representing 41.79% of bacterial reads, and Gammaproteobacteria, 36.77% (Figure 2). Other dominant phyla were Spirochaetes (11.25% of bacterial reads), Bacteroidetes (5.24%), Epsilonbacteraeota (1.08%), and Cyanobacteria (0.89%). The remaining phyla combined accounted for ca. 2.5% of the reads (Figure 2). One sample from Sugashima clade A showed a visibly different composition (marked with an asterisk in Figure 2), which may indicate a contamination or some artifact during processing. In terms of number of ZOTUs (Supplementary Figure S2), Proteobacteria was again the most diverse (i.e., ZOTU-rich) phylum, with Gammaproteobacteria accounting for 1,183 ZOTUs (27.89%) and Alphaproteobacteria totaling 683 ZOTUs (16.12%). The second most diverse phylum was Bacteroidetes (846 ZOTUs, 19.97%). Spirochaetes, despite their abundance in some samples, had a low diversity (21 ZOTUs, 0.5%).
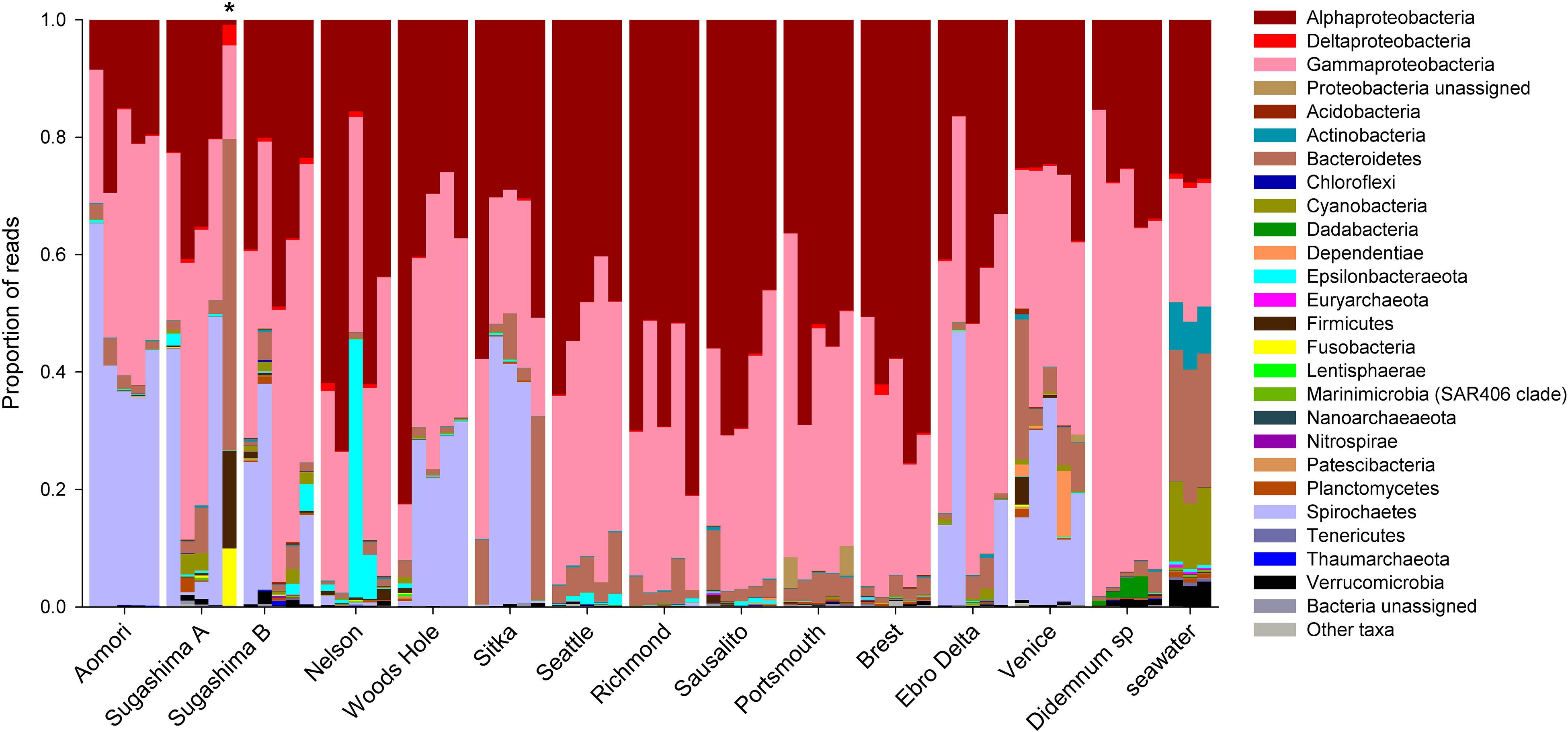
Figure 2. Taxonomic composition of the microbial community for each of the 73 samples. The proportions of number of reads at phylum level are shown, except for Proteobacteria, which are divided into their major classes. *Sample with a visibly different microbial taxonomic composition.
When analyzing the 64 samples from D. vexillum (excluding the sample from Sugashima clade A with a different composition), we found 3,525 ZOTUs (mean = 210.40; SE ± 14.91), and the taxonomic composition was similar to that of the global dataset with the 73 samples. The ZOTUs corresponded to the same 36 bacteria phyla (3,491 ZOTUs) and 3 Archaea phyla (34 ZOTUs) as in the global dataset. Archaea phyla also represented less than 0.1% of the reads, and the same most abundant bacteria phyla were identified using both datasets (Supplementary Table S1).
Seawater samples had the highest number of ZOTUs (1,738), unique ZOTUs (31.19%), and diversity values (H = 4.810; J = 0.664) (Table 2). Among D. vexillum samples, the maximum numbers of ZOTUs and unique ZOTUs were found in Sugashima clade B samples (n = 1,381 and 1.74% unique) and Brest (n = 1,028, of which 4.47% were unique) (Table 2). The highest values of Shannon and Pielou Indices were found in Venice (H = 2.946; J = 0.578) (Table 2). In comparison to D. vexillum, Didemnum sp. samples from Roscoff showed a high number of ZOTUs (n = 1,025, of which 1.85% were unique) and the lowest Shannon and Pielou Indices (H = 1.884; J = 0.329). The K-W analyses of diversity (Shannon) and evenness (Pielou) showed significant differences between sample groups (K-W χ2, 38.942 and 44.190, respectively, df = 14, p < 0.001 for both variables). The multiple comparisons performed with the Dunn test are summarized in Supplementary Table S2. Most the significant comparisons for the Shannon index involved seawater (significantly higher values in five comparisons) and Woods Hole (significantly lower in four comparisons). For the Pielou Index, most significant comparisons involved Didemnum sp. (significantly lower evenness in seven comparisons), followed by seawater (significantly higher in six comparisons).
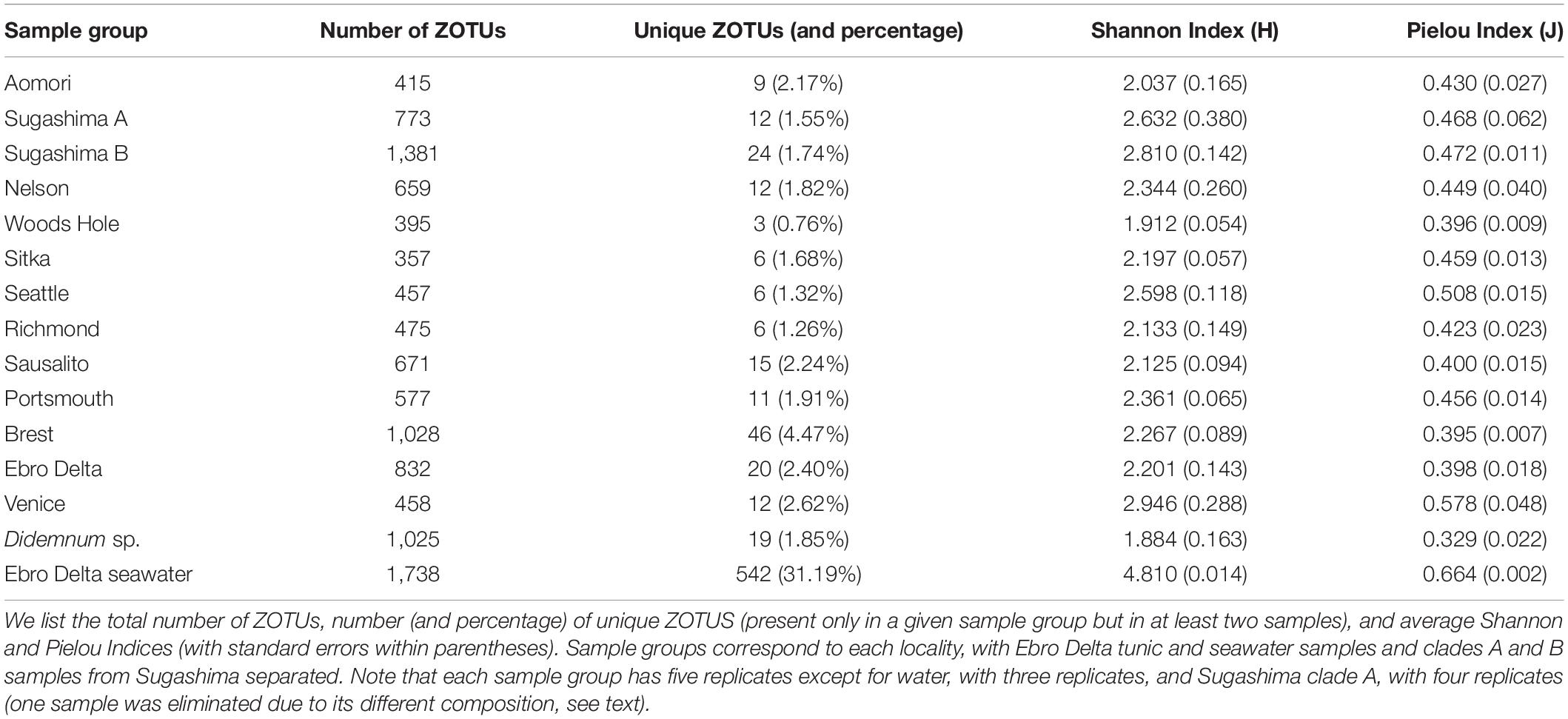
Table 2. Microbial diversity per sample group.
Microbial Community Structure
The global nmMDS (Figure 3) showed differences between D. vexillum, Didemnum sp., and seawater samples. The sample from Sugashima clade A with a different composition (marked with an asterisk in Figure 2) also appeared as an outlier in the ordination results (Figure 3) and was therefore removed from further analyses. Samples from clade B showed differentiation from clade A samples (albeit with some overlap), and some substructure was detected within clade A (Figure 3). The PERMANOVA analyses comparing Sugashima clade B, seawater and clade A samples showed a highly significant effect (Supplementary Table S3), and all pairwise comparisons proved significant.
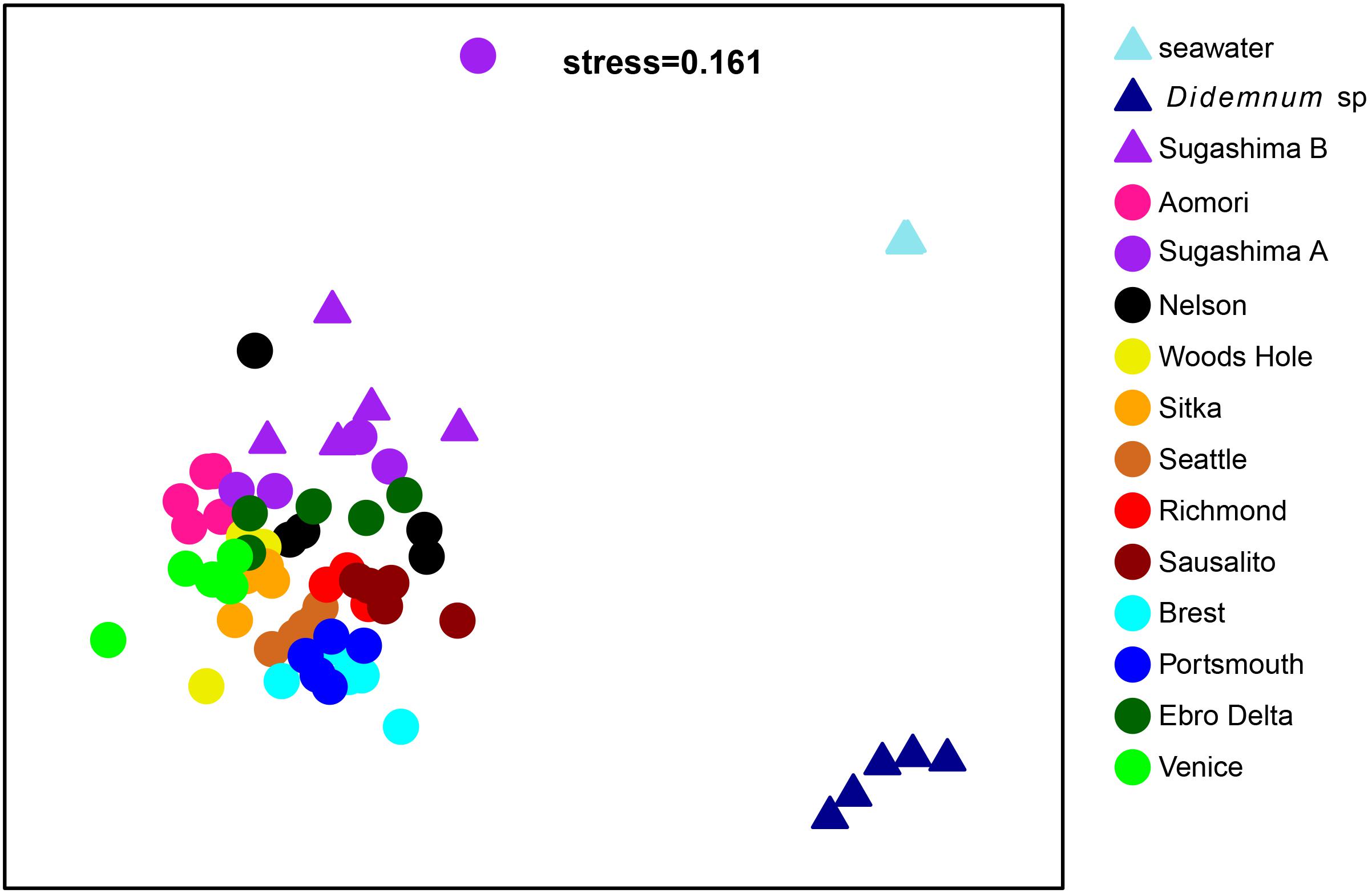
Figure 3. Non-metric MDS with all samples. Each color represents a locality. Dots correspond to clade A samples, and triangles to samples from clade B, Didemnum sp. and seawater from Ebro Delta. The Sugashima A sample with a different composition appears as an outlier in the positive values of the second axis. Note that the three seawater samples were almost completely superimposed and appear as a single point.
Seawater samples (n = 3) were compared with tunic samples (n = 5) from the same locality (Ebro Delta). Among the 832 ZOTUs present in tunic samples, 526 were also found in seawater samples. The 10 most abundant ZOTUs from tunic samples represented the 90.79% of the total reads of the tunic samples and only 1.39% of the total reads of the seawater samples. Conversely, the 10 most abundant ZOTUs from seawater samples represented the 37.51% of the total reads of the seawater samples and only 1.22% of the total reads of the tunic samples. All this pointed to clearly differentiated microbiome structure between ascidians and surrounding seawater.
To further analyze the differences between D. vexillum clades and within clade A, we decided to analyze separately the samples from Japan (the area where both clades were present). Among the 14 samples from Japan, 15 ZOTUs were shared by all samples, which represented 77.3% of the reads from these samples. The same 15 ZOTUs were shared by the nine Aomori and Sugashima clade A individuals (making up for 83.57% of the reads of these individuals), 17 ZOTUs (77.02% of the reads) were shared by Aomori and Sugashima clade B, and 25 ZOTUs were shared by Sugashima clades A and B (comprising 84.70% of the reads of these samples). The Japan nmMDS (Figure 4) clearly differentiated samples from Aomori, Sugashima clade A and Sugashima clade B. The PERMANOVA analysis (Supplementary Table S3) showed overall significant differences among the three groups, and all pairwise comparisons were also significant.
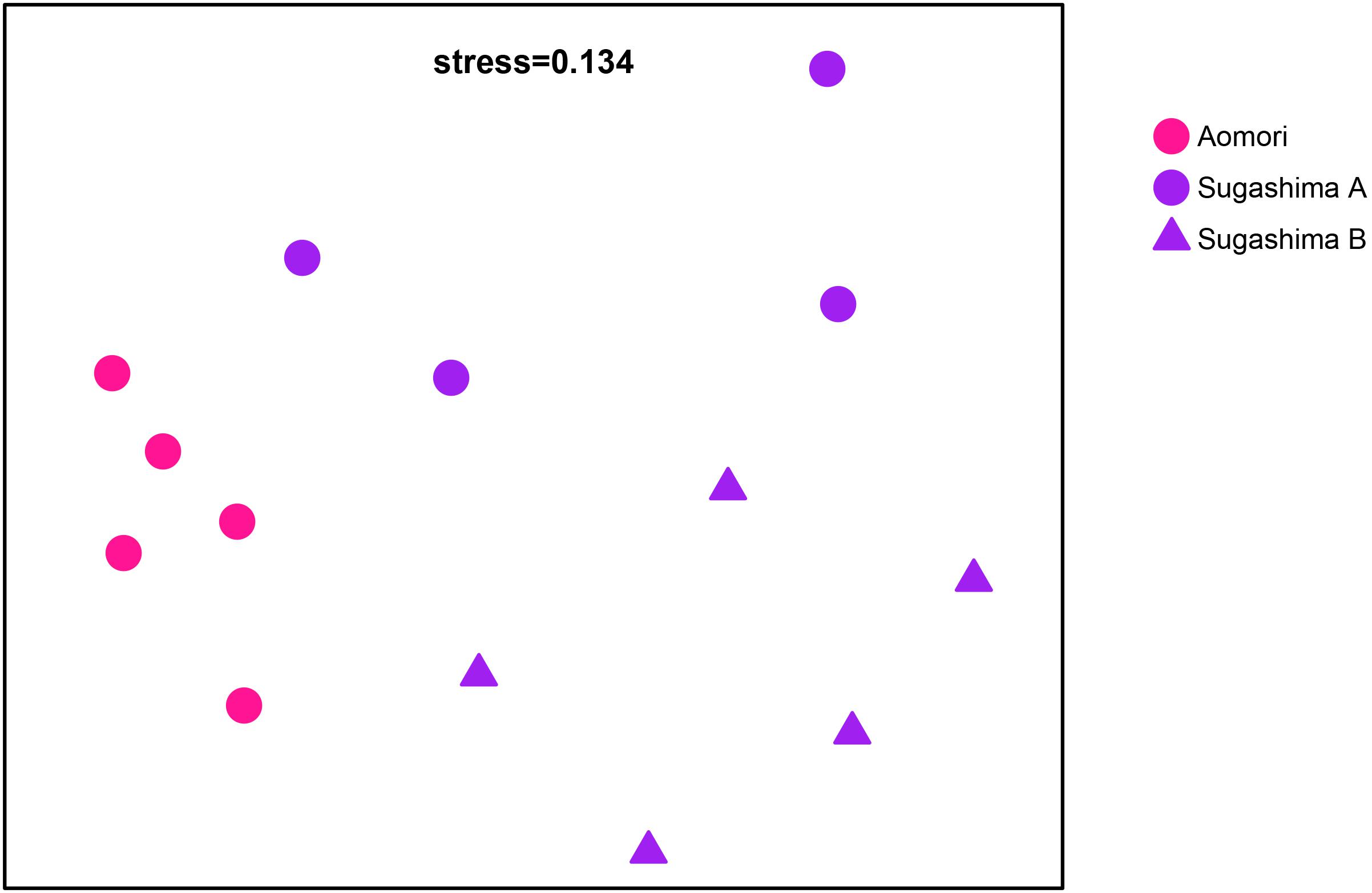
Figure 4. Non-metric MDS with the 14 samples from Japan (five from Aomori, four from Sugashima clade A, and five from Sugashima clade B). Each color represents a locality. Dots correspond to clade A samples and triangles to clade B samples.
Among the 59 microbiome samples belonging to clade A, we found a total of 3,154 ZOTUs. The core community was constituted by eight ZOTUs shared by all samples, which represented 69.57% of the reads, but only 0.25% of the total number of ZOTUs from these samples. The eight core ZOTUs were all bacteria: four Alphaproteobacteria, three Gammaproteobacteria, and one Spirochaetes (Table 3). These ZOTUs were also present in all clade B and Didemnum sp. specimens, but with decreasing abundance as genetic distance increased. Thus, these ZOTUs accounted for 50.89% of the reads of clade B specimens and only 20.10% of the reads in Didemnum sp. samples (Table 3). Finally, the eight core ZOTUs were also present in the seawater samples, where they represented only 1.39% of the reads, implying they were enriched ca. 50-fold in the clade A ascidian tunics (Table 3).
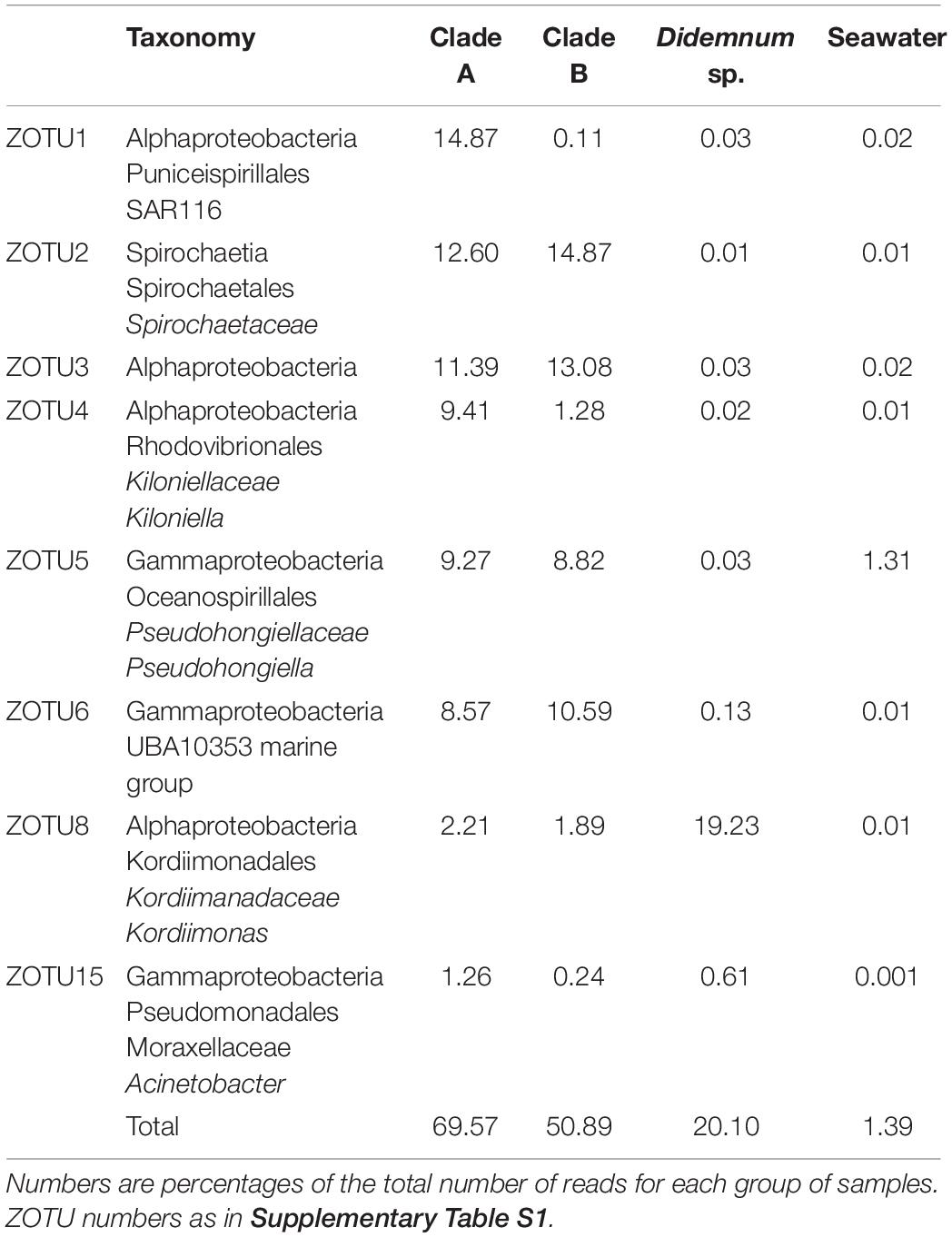
Table 3. Taxonomy assigned and read frequency of the eight clade A core ZOTUs in clade A, clade B, Didemnum sp., and seawater samples.
Using the clade A dataset, pairwise Bray–Curtis distances between samples from the same locality averaged 0.512 (SE ± 0.009), between samples from different localities in the same genetic group (i.e., within Japan, WNA, NZ-ENA or EUR, as defined in Casso et al. (2019b) note that these groups coincide with geographic regions except in the case of NZ-ENA), they averaged 0.650 (SE ± 0.004), and between samples from different genetic groups, the mean distance was 0.696 (SE ± 0.002) (Figure 5). These mean distances were significantly different from each other (K-W χ2 = 291.81, df = 2, p < 0.001, followed by pairwise Dunn tests, all p < 0.001).
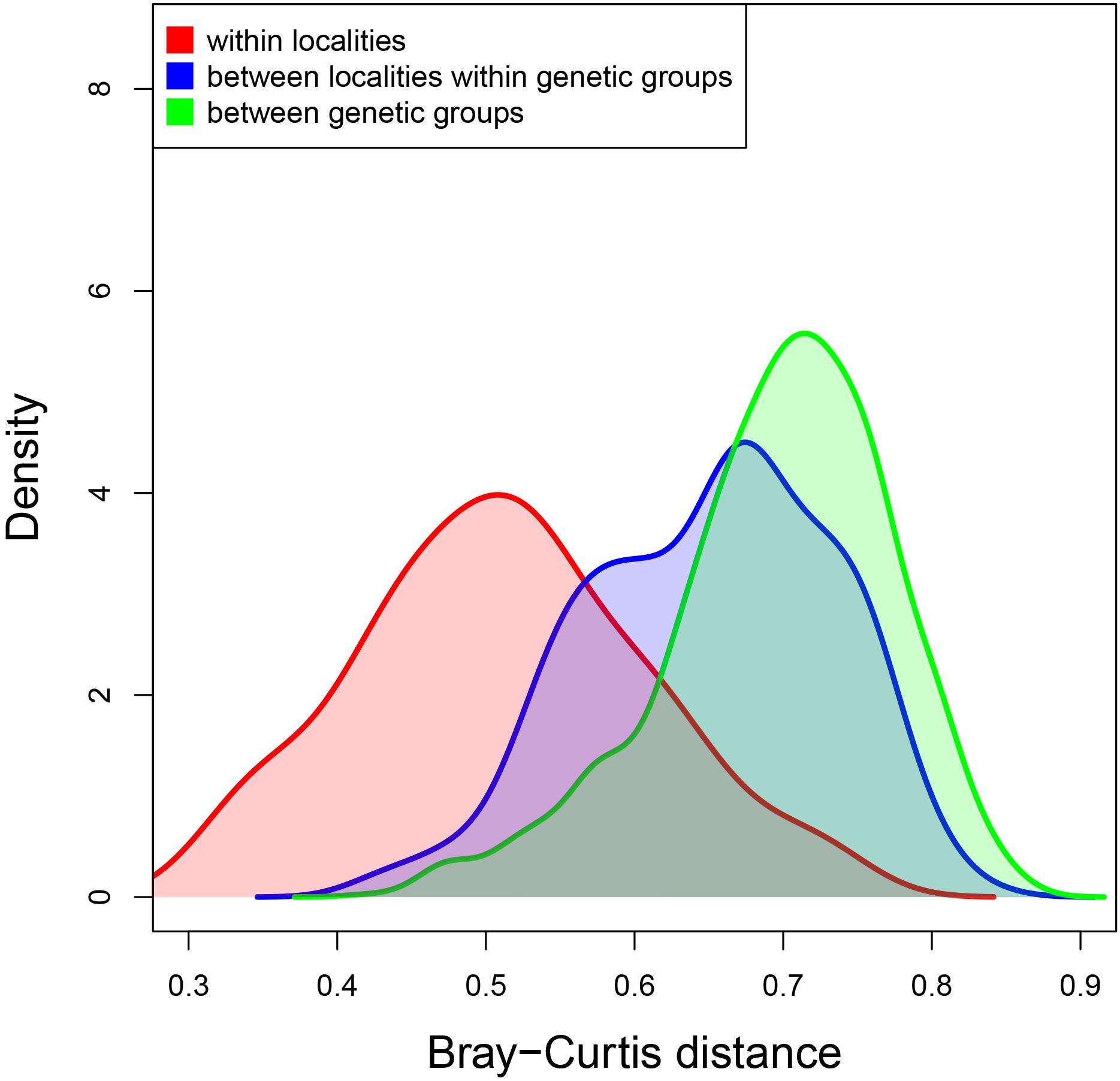
Figure 5. Density plot of the Bray–Curtis distances between samples from the same locality (in red), from different localities of the same genetic group (in blue) and from different genetic groups (in green). Genetic groups of introduced populations are those identified in Casso et al. (2019b).
The clade A microbiome nmMDS (Figure 6) showed a general pattern of sample grouping by locality, although the degree of dispersion is variable. Atlantic samples (Portsmouth and Brest) grouped together; San Francisco Bay (Richmond and Sausalito) and Seattle samples also grouped together. According to the main axis, Japanese samples (Sugashima and Aomori) grouped together. Although with more dispersion, the main axis also grouped together both Mediterranean samples (Delta and Venice), Woods Hole and Alaska (Sitka). New Zealand samples (from Nelson) appear more dispersed. The PERMANOVA analysis (Supplementary Table S3) demonstrated significant differentiation, and the pairwise tests were all significant after FDR correction except for the comparison between Richmond and Sausalito.
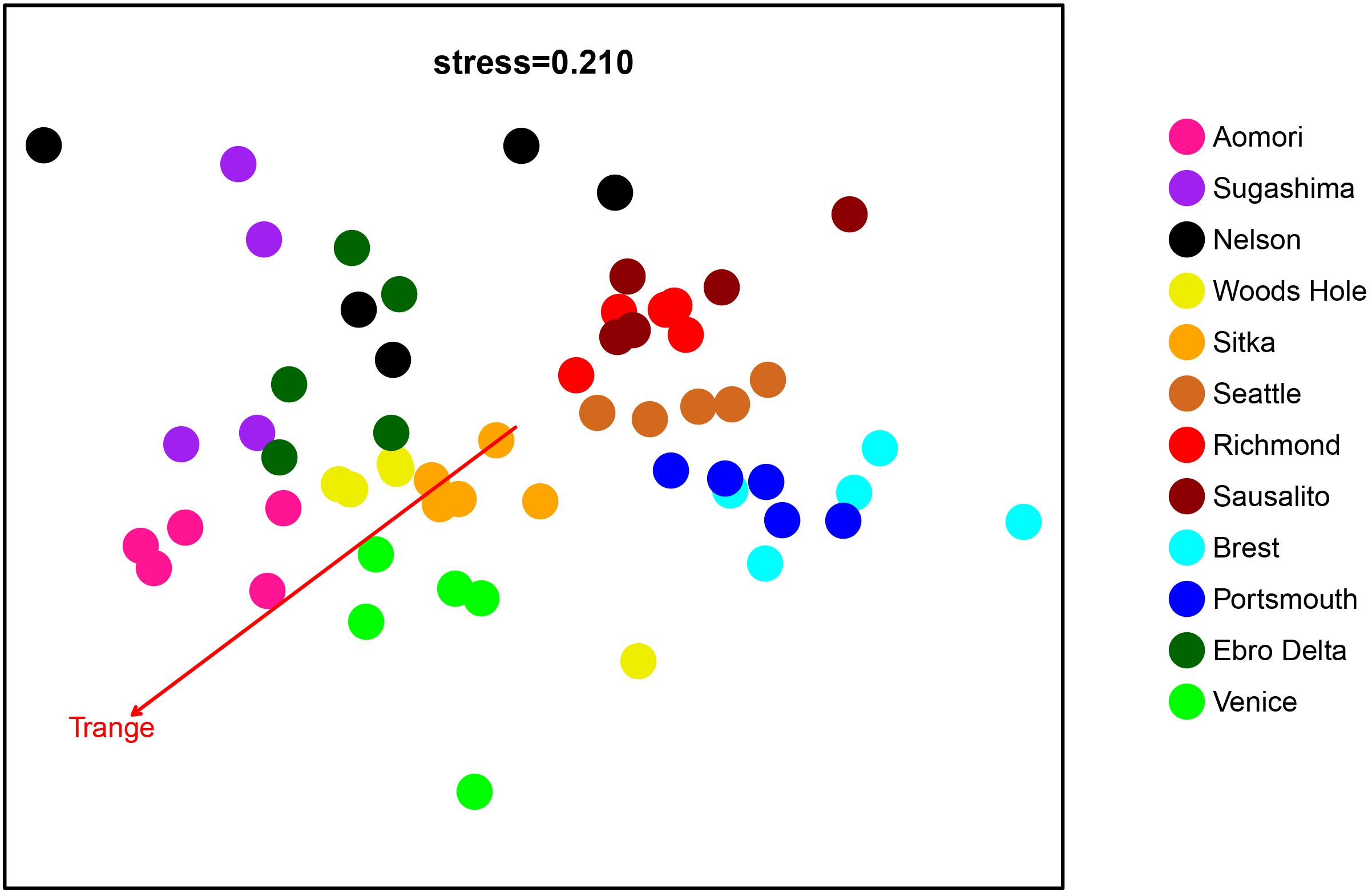
Figure 6. Non-metric MDS with the 59 microbiome samples from clade A localities. Each color represents a locality. The vector corresponding to the best-fitting variable in the envfit analysis is represented.
For the restricted datasets analyzed, in the case of Europe (Supplementary Figure S3), the nmMDS showed a clear separation of the Atlantic and Mediterranean localities, and also between localities in each sea. Accordingly, the PERMANOVA results (Supplementary Table S3) showed a significant effect of locality, and all pairwise comparisons were significant. For the North West Atlantic (NWA) dataset (Supplementary Figure S4), the nmMDS configuration showed a sharp differentiation between the two San Francisco Bay localities and those further North (Seattle and Sitka). This is confirmed in the PERMANOVA analysis, which detected overall significant differentiation. The pairwise tests showed that only the comparison of Sausalito and Richmond was not significant.
Comparison With Other Variables
Using the clade A samples (Figure 6), the environmental temperature variables (minimum, maximum, mean, and range; Supplementary Table S1) were fitted to the nmMDS ordination scores. All the relationships were significant, but the better fit was with temperature range (Tmin r2 = 0.372; Tmax r2 = 0.490; Tmean r2 = 0.219; Trange r2 = 0.731; all p < 0.01). The corresponding vector is plotted in Figure 6. Temperature range differences were therefore chosen to calculate thermal distances used in the following analysis.
According to the forward stepwise regression procedure, temperature range was the best variable explaining the microbiome structure (as ascertained by the AIC minimization), the second variable added was the genetic distance, and in the third place, geographic distance. The three variables were retained in the final model, and the fit was highly significant (adjusted r2 = 0.280, p < 0.001, Supplementary Table S4).
As for the restricted datasets, in the case of Europe (Supplementary Figure S3), the envfit analysis showed that the maximal temperature provided the best fit to the ordination scores (Tmax, r2 = 0.934, vector depicted in Supplementary Figure S3). The multiple regression analysis showed a best fit model including first temperature (as Tmax) and, next, genetic distances, but excluding geographic distances (final model, adjusted r2 = 0.634, p < 0.001, Supplementary Table S4).
In the NWA localities (Supplementary Figure S4), the temperature variable best correlated with the ordination scores was the mean temperature (Tmean, r2 = 0.910, vector depicted in Supplementary Figure S4). The multiple regression analysis revealed again that the best final model included temperature (using Tmean), then genetic distance as the second explanatory variable, and geographic distance was excluded from the final model (adjusted r2 = 0.679, p < 0.001, Supplementary Table S4). Thus, contrary to what we found in the global analysis, in the spatially restricted datasets, geographic distance effects did not explain the microbiome composition once the other variables have been accounted for.
Discussion
In this study, we provide the first characterization of the tunic microbiome of the invasive colonial ascidian D. vexillum. We were able to analyze the microbiome of both mitochondrial clades of this species, from exactly the same samples genetically genotyped in a previous study that investigated the population genomics of the host (Casso et al., 2019b). These samples were collected from representative localities of the whole range of distribution of D. vexillum, providing an exceptional opportunity to study the differences in the microbiota composition of the different populations worldwide and their relationship with the host genetic relatedness. We also analyzed the microbiome of a population from a closely related species (Didemnum sp.) plus seawater samples from one of the localities, for comparison.
We found that the number of ZOTUs found among D. vexillum samples (3,525 ZOTUs) represents a highly diverse microbiota comparable to that found in other ascidians and marine invertebrates (Erwin et al., 2014; Cleary et al., 2019). The large difference between abundance and diversity of Archaea and bacteria (the former representing ca. 2% of ZOTUs and 0.1% of reads) in our results is likely to be confounded by primer amplification biases downplaying the representation of Archaea (Klindworth et al., 2013; Turon and Uriz, 2020).
Given the encrusting nature of the colonies and the existence of common colonial cavities in contact with the external environment in D. vexillum, as in other colonial ascidians, it is not possible to analyze only internal tunic. In our samples, we avoided basal surfaces in contact with the substrate, but had to include distal surfaces with cuticle where seawater bacteria could be found (López-Legentil et al., 2016). However, as the fixative for the sampling of the colonies was changed several times, and colonies were rinsed during manipulation, we are confident that any remaining non-internal bacteria were truly attached to the ascidian surface. Little difference was found between the microbiome of the inner tunic of a solitary ascidian and that contained in the outer tunic including the cuticle surfaces (Blasiak et al., 2014). Likewise, similar bacterial composition was detected between tunic and tunic with cuticle in several ascidian species (Cahill et al., 2016), indicating that few prokaryotic groups occur exclusively in the exterior of the tunic.
Seawater samples showed the most diverse bacterial communities, with 1,738 ZOTUs, which is 2.5 times greater than the average number of ZOTUs in the ascidian tunics (679, Table 2). This ratio may be even higher because we had only three seawater replicates, and the sequencing depth was insufficient to capture adequately the microbiome diversity in these samples, as the rarefaction curve did not reach a clear asymptote (Supplementary Figure S1). The difference in diversity we found between seawater and tunic samples is consistent with other studies comparing seawater and microbiomes from other ascidians (Erwin et al., 2014). There was also a marked difference in composition between seawater and tunic samples, as shown in the global nmMDS analysis (Figure 3). A similar degree of differentiation was also observed between D. vexillum and the congeneric Didemnum sp. samples. This indicates that the microbiome composition is clearly different between species, even if they are closely related and living in the same kind of environment, a result found in previous studies in ascidians (Erwin et al., 2014; Cahill et al., 2016) and other marine invertebrates such as sponges, chitons, corals, or sea cucumbers (Turon et al., 2018; Cleary et al., 2019). The reduction in abundance of the clade A core microbial community when increasing the host genetic distance (Table 3) also points to the evolution of species specific microbial communities. Samples from clade B individuals showed some differences in composition with respect to clade A specimens from the same locality (Sugashima) (Figures 3, 4). These differences may be explained by the possible speciation process occurring between D. vexillum clades A and B (Casso et al., 2019b).
The core community of D. vexillum clade A samples was highly abundant (69.57% of reads) but with low diversity (eight ZOTUs). On the contrary, the variable component has low abundance and high diversity. The microbiome is related to changes in the environment (such as temperature variables and geographic distance between localities in the global analysis), which points to horizontal acquisition of the symbionts from the seawater. However, there seems to be a selective enrichment in the host of particular bacteria. For instance, the 10 most abundant ZOTUs from tunic samples in the Ebro Delta represented the 90.79% of the total reads of the tunic samples and only 1.39% of the total reads of the seawater samples, implying that there is a considerable difference in abundance in the ZOTUs present in both tunic and seawater samples. This further suggests the role of the environment on the horizontal transfer, but with an enrichment promoted by the host. A small but abundant core and a highly diverse variable microbiome component, with marked selective enrichment from the pool of water prokaryotes, has also been found in sponges (Turon et al., 2018) and may contribute to the host resilience to environmental changes (Leite et al., 2018; Turon et al., 2019).
We found a relationship between microbiome structure and the genetic makeup of the colonies, both in the global analysis and in the restricted analyses of particular genetic groups (Europe and North Western America, where genetic distances, and not geographic distances, explained microbiome composition). These results indicate either a vertical transmission of some of the symbionts, or the heritability of traits related to the selection of symbionts from the seawater. We cannot, at present, resolve these factors unambiguously, but vertical transmission seems the most likely one. The presence of prokaryotes in the embryos and larvae of colonial ascidians has been noted (Moss et al., 2003; Martínez-García et al., 2007; López-Legentil et al., 2011), and transmission of symbionts via larvae has been well documented in photosymbiotic relationships between ascidians and the cyanobacterial Prochloron species (Hirose, 2015; Hirose and Nozawa, 2020). Moreover, the larvae of other species of Didemnum are known to harbor bacteria (López-Legentil et al., 2015), making vertical transmission a highly plausible trait in our case. Vertical transmission is not necessarily fixed and consistent (Björk et al., 2019), but can contribute to propagate particular symbionts. We suggest that a combination of horizontal and vertical transmission shapes the microbiome of D. vexillum, as suggested for other invertebrates such as sponges (Sipkema et al., 2015).
It is noteworthy that all temperature variables and, particularly, the temperature range in the global analysis correlate with the microbiome structure of D. vexillum. In the restricted analyses of Europe and North Western America, temperature range differences were not so large, but nevertheless maximal and mean temperature, respectively, were closely correlated with the ordination of the samples, again confirming the role of temperature in shaping the microbial community. Didemnum vexillum is defined as a temperate species, however, it can live from subtropical seas such as the Mediterranean to cold regions such as Alaska. Hibernation and estivation processes may help the species to cope with unfavorable temperatures (Bullard et al., 2007; Ordóñez et al., 2015). Our results indicate that temperature adaptation seems to be a driving force in the colonization process of D. vexillum, and the microbiome can play a role in the thermal adaptation of the species, as has been suggested in fish and corals (Kokou et al., 2018; Osman et al., 2020).
In the marine realm, many instances of successful introduction of species outside their native ranges have occurred (Carlton and Geller, 1993), which implies fast adaptation to new conditions (Rius et al., 2015). It has been pointed out that the microbial symbiont community can contribute to the survival and adaptation of the holobiont during the invasion process (Aires et al., 2016; Amsellem et al., 2017; Arnaud-Haond et al., 2017). Among other mechanisms, thermal adaptation and the production of active substances may be key for the introduction success and can be mediated by the microbiome (Dou and Dong, 2019; Osman et al., 2020). Didemnum vexillum must be able to adapt to a wide range of environments in its worldwide introduction range. Chimerism (Casso et al., 2019a) and epigenetic changes (Hawes et al., 2019) have been suggested as potential mechanisms enhancing its adaptive capacity, and the microbiome is likely to play a significant role as well. We found that the microbiome composition of the introduced clade of D. vexillum is significantly influenced by temperature, genetics of the host, and geographic distance. The combination of a quantitatively dominant core microbiome with a highly diverse variable component, likely acquired by a combination of horizontal and vertical transmission, can contribute to the success of this global invader in different environments.
Data Availability Statement
The datasets generated for this study can be found in the Short Read Archive (SRA) of the NCBI under Bioproject PRJNA608680.
Author Contributions
XT and MP designed the research. MC, MP, and XT collected samples. MC performed laboratory work and wrote the first draft of the manuscript. MC, MT, and NM ran the bioinformatics pipeline, analyzed results, and prepared tables and figures. All authors discussed and interpreted results, and revised the manuscript.
Funding
This research was funded by the projects CHALLENGEN and PopCOmics (CTM2013-48163 and CTM2017-88080, MCIU/AEI/FEDER/UE) from the Spanish Government. MC was funded by a predoctoral FPI contract of the Spanish Government. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). This is a contribution from the Consolidated Research Group Benthic Biology and Ecology SGR2017-1120 (Catalan Government).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to all people that provided samples for this study: Gretchen Lambert, Ian Davidson, Mike Page, Judith Pederson, and Frédérique Viard. We also thank Margarita Fernández, Carles Bori, Davide Tagliapietra, Marco Sigovini, and Irene Guarneri for their help during sampling in Ebro Delta and Venice; Marc Rius for help while sampling in United Kingdom, and Gaku Kumano for assistance with the Aomori sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00201/full#supplementary-material
Footnotes
References
Aires, T., Serrão, E. A., and Engelen, A. H. (2016). Host and environmental specificity in bacterial communities associated to two highly invasive marine species (genus Asparagopsis). Front. Microbiol. 7:559. doi: 10.3389/fmicb.2016.00559
Amsellem, L., Brouat, C., Duron, O., Porter, S. S., Vilcinskas, A., and Facon, B. (2017). “Importance of microorganisms to macroorganisms invasions: is the essential invisible to the eye?,” in The Little Prince, 1st Edn, ed. A. de Saint-Exupéry (Amsterdem: Elsevier Ltd), doi: 10.1016/bs.aecr.2016.10.005
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46.
Arnaud-Haond, S., Aires, T., Candeias, R., Teixeira, S. J. L., Duarte, C. M., Valero, M., et al. (2017). Entangled fates of holobiont genomes during invasion: nested bacterial and host diversities in Caulerpa taxifolia. Mol. Ecol. 26, 2379–2391. doi: 10.1111/mec.14030
Barott, K. L., Rodriguez-Brito, B., Janouškovec, J., Marhaver, K. L., Smith, J. E., Keeling, P., et al. (2011). Microbial diversity associated with four functional groups of benthic reef algae and the reefbuilding coral Montastraea annularis. Environ. Microbiol. 13, 1192–1204. doi: 10.1111/j.1462-2920.2010.02419.x
Ben-Shlomo, R. (2017). Invasiveness, chimerism and genetic diversity. Mol. Ecol. 26, 6502–6509. doi: 10.1111/mec.14364
Björk, J. R., Díez-Vives, C., Astudillo-García, C., Archie, E. A., and Montoya, J. M. (2019). Vertical transmission of sponge microbiota is inconsistent and unfaithful. Nat. Ecol. Evol. 3, 1172–1183. doi: 10.1038/s41559-019-0935-x
Blackall, L. L., Wilson, B., and Van Oppen, M. J. H. (2015). Coral-the world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347. doi: 10.1111/mec.13400
Blasiak, L. C., Zinder, S. H., Buckley, D. H., and Hill, R. T. (2014). Bacterial diversity associated with the tunic of the model chordate Ciona intestinalis. ISME J. 8, 309–320. doi: 10.1038/ismej.2013.156
Bullard, S. G., Lambert, G., Carman, M. R., Byrnes, J., Whitlatch, R. B., Ruiz, G., et al. (2007). The colonial ascidian Didemnum sp. A: current distribution, basic biology and potential threat to marine communities of the northeast and west coasts of North America. J. Exp. Mar. Biol. Ecol. 342, 99–108. doi: 10.1016/j.jembe.2006.10.020
Cahill, P. L., Fidler, A. E., Hopkins, G. A., and Wood, S. A. (2016). Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep. 8, 470–478. doi: 10.1111/1758-2229.12391
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carlton, J. T., and Geller, J. B. (1993). Ecological Roulette: the global transport of nonindigenous marine organisms. Science 261, 78–82.
Casso, M., Navarro, M., Ordóñez, V., Fernández-Tejedor, M., Pascual, M., and Turon, X. (2018). Seasonal patterns of settlement and growth of introduced and native ascidians in bivalve cultures in the Ebro Delta (NE Iberian Peninsula). Reg. Stud. Mar. Sci. 23, 12–22. doi: 10.1016/j.rsma.2017.11.002
Casso, M., Tagliapietra, D., Turon, X., and Pascual, M. (2019a). High fusibility and chimera prevalence in an invasive colonial ascidian. Sci. Rep. 9:15673. doi: 10.1038/s41598-019-51950-y
Casso, M., Turon, X., and Pascual, M. (2019b). Single zooids, multiple loci: independent colonisations revealed by population genomics of a global invader. Biol. Invasions 21, 3575–3592. doi: 10.1007/s10530-019-02069-8
Chen, L., Hu, J. S., Xu, J. L., Shao, C. L., and Wang, G. Y. (2018). Biological and chemical diversity of ascidian associated microorganisms. Mar. Drugs 16, 1–33. doi: 10.3390/md16100362
Cleary, D. F. R., Swierts, T., Coelho, F. J. R. C., Polónia, A. R. M., Huang, Y. M., Ferreira, M. R. S., et al. (2019). The sponge microbiome within the greater coral reef microbial metacommunity. Nat. Commun. 10:11033. doi: 10.1038/s41467-019-09537-8
Dinno, A. (2017). Package ‘dunn.test’ v.1.3.5. Dunn’s test of multiple comparisons using rank sums. Available online at: https://CRAN.R-project.org/package=dunn.test.
Donia, M. S., Fricke, W. F., Partensky, F., Cox, J., Elshahawi, S. I., White, J. R., et al. (2011). Complex microbiome underlying secondary and primary metabolism in the tunicate-Prochloron symbiosis. Proc. Natl. Acad. Sci. U.S.A. 108, 1–10. doi: 10.1073/pnas.1111712108
Dou, X., and Dong, B. (2019). Origins and bioactivities of natural compounds derived from marine ascidians and their symbionts. Mar. Drugs 17:670. doi: 10.3390/md17120670
Dror, H., Novak, L., Evans, J. S., López-Legentil, S., and Shenkar, N. (2019). Core and dynamic microbial communities of two invasive ascidians: can host–symbiont dynamics plasticity affect invasion capacity? Microb. Ecol. 78, 170–184. doi: 10.1007/s00248-018-1276-z
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv [Preprint]. doi: 10.1101/081257
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. doi: 10.1093/bioinformatics/bty113
Edgar, R. C., and Flyvbjerg, H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482. doi: 10.1093/bioinformatics/btv401
Erwin, P. M., Pineda, M. C., Webster, N., Turon, X., and López-Legentil, S. (2014). Down under the tunic: bacterial biodiversity hotspots and widespread ammonia-oxidizing archaea in coral reef ascidians. ISME J. 8, 575–588. doi: 10.1038/ismej.2013.188
Evans, J. S., Erwin, P. M., Shenkar, N., and López-Legentil, S. (2017). Introduced ascidians harbor highly diverse and host-specific symbiotic microbial assemblages. Sci. Rep. 7:11033. doi: 10.1038/s41598-017-11441-4
Hawes, N. A., Amadorou, A., Tremblay, L. A., Pochon, X., Dunphy, B., Fidler, A. E., et al. (2019). Epigenetic patterns associated with an ascidian invasion: a comparison of closely related clades in their native and introduced ranges. Sci. Rep. 9:14275. doi: 10.1038/s41598-019-49813-7
Hill, M. S., and Sacristán-Soriano, O. (2017). “Molecular and Functional Ecology of Sponges and Their Microbial Symbionts,” in Climate Change, Ocean Acidification and Sponges, eds J. L. Carballo and J. J. Bell (Berlin: Springer International Publishing), 105–142. doi: 10.1007/978-3-319-59008-0
Hirose, E. (2015). Ascidian photosymbiosis: diversity of cyanobacterial transmission during embryogenesis. Genesis 53, 121–131. doi: 10.1002/dvg.22778
Hirose, E., and Nozawa, Y. (2020). Convergent evolution of the vertical transmission mode of the cyanobacterial obligate symbiont distributed in the tunic of colonial ascidians. J. Zool. Syst. Evol. Res. doi: 10.1111/jzs.12370
Kellogg, C. A. (2019). Microbiomes of stony and soft deep-sea corals share rare core bacteria. Microbiome 7:90. doi: 10.1186/s40168-019-0697-3
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Qast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nuclei Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kokou, F., Sasson, G., Nitzan, T., Doron-Faigenboim, A., Harpaz, S., Cnaani, A., et al. (2018). Host genetic selection for cold tolerance shapes microbiome composition and modulates its response to temperature. eLife 7:e36398. doi: 10.7554/eLife.36398
Kwan, J. C., Donia, M. S., Han, A. W., Hirose, E., Haygood, M. G., and Schmidt, E. W. (2012). Genome streamlining and chemical defense in a coral reef symbiosis. Proc. Natl. Acad. Sci. U.S.A. 109, 20655–20660. doi: 10.1073/pnas.1213820109
Lambert, G. (2007). Invasive sea squirts: a growing global problem. J. Exp. Mar. Biol. Ecol. 342, 3–4.
Lambert, G. (2009). Adventures of a sea squirt sleuth: unravelling the identity of Didemnum vexillum, a global ascidian invader. Aquat. Inv. 4, 5–28. doi: 10.3391/ai.2009.4.1.2
Legendre, P., and Gallagher, E. D. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280. doi: 10.1007/s004420100716
Leite, D. C. A., Salles, J. F., Calderon, E. N., Castro, C. B., Bianchini, A., Marques, J. A., et al. (2018). Coral bacterial-core abundance and network complexity as proxies for anthropogenic pollution. Front. Microbiol. 9:833. doi: 10.3389/fmicb.2018.00833
López-Legentil, S., Song, B., Bosch, M., Pawlik, J. R., and Turon, X. (2011). Cyanobacterial diversity and a new Acaryochloris-like symbiont from bahamian sea-squirts. PLoS One 6:e0023938. doi: 10.1371/journal.pone.0023938
López-Legentil, S., Turon, X., and Erwin, P. M. (2016). Feeding cessation alters host morphology and bacterial communities in the ascidian Pseudodistoma crucigaster. Front. Zool. 12:2. doi: 10.1186/s12983-016-0134-4
López-Legentil, S., Turon, X., Espluga, R., and Erwin, P. M. (2015). Temporal stability of bacterial symbionts in a temperate ascidian. Front. Microbiol. 6:1022. doi: 10.3389/fmicb.2015.01022
Mandrioli, M., and Manicardi, G. (2013). Evolving aphids: one genome-one organism insects or holobionts? Invertebr. Surviv. J. 10, 1–6.
Martinez-Arbizu, P. (2019). pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0.3.
Martínez-García, M., Díaz-Valdés, M., Wanner, G., Ramos-Esplá, A., and Antón, J. (2007). Microbial community associated with the colonial ascidian Cystodytes dellechiajei. Environ. Microbiol. 9, 521–534. doi: 10.1111/j.1462-2920.2006.01170.x
Martínez-García, M., Stief, P., Díaz-Valdés, M., Wanner, G., Ramos-Esplá, A., Dubilier, N., et al. (2008). Ammonia-oxidizing Crenarchaeota and nitrification inside the tissue of a colonial ascidian. Environ. Microbiol. 10, 2991–3001. doi: 10.1111/j.1462-2920.2008.01761.x
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Morrissey, K. L., Çavaş, L., Willems, A., and De Clerck, O. (2019). Disentangling the influence of environment, host specificity and thallus differentiation on bacterial communities in siphonous green seaweeds. Front. Microbiol. 10:717. doi: 10.3389/fmicb.2019.00717
Mortzfeld, B. M., Urbanski, S., Reitzel, A. M., Künzel, S., Technau, U., and Fraune, S. (2016). Response of bacterial colonization in Nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18, 1764–1781. doi: 10.1111/1462-2920.12926
Moss, C., Green, D. H., Pérez, B., Velasco, A., Henríquez, R., and McKenzie, J. D. (2003). Intracellular bacteria associated with the ascidian Ecteinascidia turbinata: phylogenetic and in situ hybridisation analysis. Mar. Biol. 143, 99–110. doi: 10.1007/s00227-003-1060-5
Nagar, L. R., and Shenkar, N. (2016). Temperature and salinity sensitivity of the invasive ascidian Microcosmus exasperatus Heller, 1878. Aquat. Invasions 11, 33–43. doi: 10.3391/ai.2016.11.1.04
Naranjo, S. A., Carballo, J. L., and García-Gómez, J. C. (1996). Effects of environmental stress on ascidian populations in Algeciras Bay (southern Spain). Possible marine bioindicators? Mar. Ecol. Prog. Ser. 144, 119–131. doi: 10.3354/meps144119
Newton, I. L., Girguis, P. R., and Cavanaugh, C. M. (2008). Comparative genomics of vesicomyid clam (Bivalvia: Mollusca) chemosynthetic symbionts. BMC Genomics 9:585. doi: 10.1186/1471-2164-9-585
Novak, L., López-Legentil, S., Sieradzki, E., and Shenkar, N. (2017). Rapid establishment of the nonindigenous ascidian Styela plicata and its associated bacteria in marinas and fishing harbors along the Mediterranean coast of Israel. Mediterr. Mar. Sci. 18, 324–331. doi: 10.12681/mms.2135
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). Package ‘vegan’ v. 2.5-6. Community Ecology Package. Available online at: https://github.com/vegandevs/vegan
Ordóñez, V., Pascual, M., Fernández-Tejedor, M., Pineda, M. C., Tagliapietra, D., and Turon, X. (2015). Ongoing expansion of the worldwide invader Didemnum vexillum (Ascidiacea) in the Mediterranean Sea: high plasticity of its biological cycle promotes establishment in warm waters. Biol. Invasions 17, 2075–2085. doi: 10.1007/s10530-015-0861-z
Osman, E. O., Suggett, D. J., Voolstra, C. R., Pettay, D. T., Clark, D. R., Pogoreutz, C., et al. (2020). Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome 8:8. doi: 10.1186/s40168-019-0776-5
Petersen, J. M., and Osvatic, J. (2018). Microbiomes In Natura: importance of invertebrates in understanding the natural variety of animal-microbe interactions. mSystems 3:e00179-17. doi: 10.1128/mSystems.00179-17
Pineda, M. C., López-Legentil, S., and Turon, X. (2013). Year-round reproduction in a seasonal sea: biological cycle of the introduced ascidian Styela plicata in the Western Mediterranean. Mar. Biol. 160, 221–230. doi: 10.1007/s00227-012-2082-7
Pineda, M. C., Turon, X., and López-Legentil, S. (2012). Stress levels over time in the introduced ascidian Styela plicata: the effects of temperature and salinity variations on hsp70 gene expression. Cell Stress Chaperones 17, 435–444. doi: 10.1007/s12192-012-0321-y
Pita, L., Rix, L., Slaby, B. M., Franke, A., and Hentschel, U. (2018). The sponge holobiont on a changing ocean: from microbes to ecosystems. Microbiome 6:46. doi: 10.1186/s40168-018-0428-1
Pollock, F. J., McMinds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921. doi: 10.1038/s41467-018-07275-x
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rius, M., Pineda, M. C., and Turon, X. (2009). Population dynamics and life cycle of the introduced ascidian Microcosmus squamiger in the Mediterranean Sea. Biol. Invasions 11, 2181–2194. doi: 10.1007/s10530-008-9375-2
Rius, M., Turon, X., Bernardi, G., Volckaert, F. A. M., and Viard, F. (2015). Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol. Invasions 17, 869–885. doi: 10.1007/s10530-014-0792-0
Rocha, R., Castellano, G., and Freire, C. (2017). Physiological tolerance as a tool to support invasion risk assessment of tropical ascidians. Mar. Ecol. Prog. Ser. 577, 105–119. doi: 10.3354/meps12225
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rosenberg, E., Sharon, G., Atad, I., and Zilber-Rosenberg, I. (2010). The evolution of animals and plants via symbiosis with microorganisms. Environ. Microbiol. Rep. 2, 500–506. doi: 10.1111/j.1758-2229.2010.00177.x
Rosenberg, E., and Zilberg-Rosenberg, I. (2018). The hologenome concept of evolution after 10 years. Microbiome 6:78. doi: 10.1186/s40168-018-0457-9
Rosenberg, E., and Zilber-Rosenberg, I. (2016). Microbes drive evolution of animals and plants: the hologenome Concept. mBio 7:e01395-15. doi: 10.1128/mBio.01395-15
Schmidt, E. W. (2014). The secret to a successful relationship: lasting chemistry between ascidians and their symbiotic bacteria. Invertebr. Biol. 134, 88–102. doi: 10.1111/ivb.12071
Shenkar, N., and Swalla, B. J. (2011). Global diversity of Ascidiacea. PLoS One 6:e20657. doi: 10.1371/journal.pone.0020657
Sipkema, D., De Caralt, S., Morillo, J. A., Al-Soud, W. A., Sorensen, S. J., Smidt, H., et al. (2015). Similar sponge-associated bacteria can be acquired via both vertical and horizontal transmission. Environ. Microbiol. 17, 3807–3821. doi: 10.1111/1462-2920.12827
Smith, K. F., Stefaniak, L., Saito, Y., Gemmill, C. E. C., Cary, S. C., and Fidler, A. E. (2012). Increased intercolony fusion rates are associated with reduced COI haplotype diversity in an invasive colonial ascidian Didemnum vexillum. PLoS One 7:e30473. doi: 10.1371/journal.pone.0030473
Stefaniak, L., Zhang, H., Gittenberger, A., Smith, K., Holsinger, K., Lin, S., et al. (2012). Determining the native region of the putatively invasive ascidian Didemnum vexillum Kott, 2002. J. Exp. Mar. Biol. Ecol. 42, 64–71. doi: 10.1016/j.jembe.2012.04.012
Tianero, M. D. B., Kwan, J. C., Wyche, T. P., Presson, A. P., Koch, M., Barrows, L. R., et al. (2015). Species specificity of symbiosis and secondary metabolism in ascidians. ISME J. 9, 615–628. doi: 10.1038/ismej.2014.152
Turon, M., Cáliz, J., Garate, L., Casamayor, E. O., and Uriz, M. J. (2018). Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 8:15201. doi: 10.1038/s41598-018-33545-1
Turon, M., Cáliz, J., Triadó-Margarit, X., Casamayor, E. O., and Uriz, M. J. (2019). Sponges and their microbiomes show similar community metrics across impacted and well-preserved reefs. Front. Microbiol. 10:1961. doi: 10.3389/fmicb.2019.01961
Turon, M., and Uriz, M. J. (2020). New insights into the Archaeal consortium of tropical sponges. Front. Mar. Sci. 6:789. doi: 10.3389/fmars.2019.00789
Ueki, T., Fujie, M., Romaidi, and Satoh, N. (2019). Symbiotic bacteria associated with ascidian vanadium accumulation identified by 16S rRNA amplicon sequencing. Mar. Genomics 43, 33–42. doi: 10.1016/j.margen.2018.10.006
Uriz, M. J., Agell, G., Blanquer, A., Turon, X., and Casamayor, E. O. (2012). Endosymbiotic calcifying bacteria: a new cue to the origin of calcification in Metazoa? Evolution 66, 2993–2999. doi: 10.5061/dryad.ff586c8k
van Oppen, M. J. H., and Blackall, L. L. (2019). Coral microbiome dynamics, functions and design in a changing world. Nat. Rev. Microbiol. 17, 557–567. doi: 10.1038/s41579-019-0223-4
Venables, W. N., and Ripley, B. D. (2002). Modern Applied Statistics with S, Fourth Edi. New York, NY: Springer.
Webster, N. S., and Taylor, M. W. (2012). Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. 14, 335–346. doi: 10.1111/j.1462-2920.2011.02460.x
White, T., van der Ende, J., and Nichols, T. E. (2019). Beyond Bonferroni revisited: concerns over inflated false positive research findings in the fields of conservation genetics, biology, and medicine. Cons. Genet. 20, 927–937. doi: 10.1007/s10592-019-01178-0
Wilkins, L. G. E., Leray, M., O’Dea, A., Yen, B., Peixoto, R. S., Pereira, T. J., et al. (2019). Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17:e3000533. doi: 10.1371/journal.pbio.3000533
Zhan, A., Briski, E., Bock, D. G., Ghabooli, S., and MacIsaac, H. J. (2015). Ascidians as models for studying invasion success. Mar. Biol. 162, 2449–2470. doi: 10.1007/s00227-015-2734-5
Ziegler, M., Seneca, F. O., Yum, L. K., Palumbi, S. R., and Voolstra, C. R. (2017). Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat. Commun. 8:14213. doi: 10.1038/ncomms14213
Zilber-Rosenberg, I., and Rosenberg, E. (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol. Rev. 32, 723–735. doi: 10.1111/j.1574-6976.2008.00123.x
Keywords: holobiont, symbiosis, vertical transmission, horizontal transmission, adaptation, introduced species
Citation: Casso M, Turon M, Marco N, Pascual M and Turon X (2020) The Microbiome of the Worldwide Invasive Ascidian Didemnum vexillum. Front. Mar. Sci. 7:201. doi: 10.3389/fmars.2020.00201
Received: 12 December 2019; Accepted: 13 March 2020;
Published: 22 April 2020.
Edited by:
Laura Steindler, University of Haifa, IsraelReviewed by:
Heidi M. Luter, Australian Institute of Marine Science (AIMS), AustraliaJames Evans, University of North Carolina at Wilmington, United States
Copyright © 2020 Casso, Turon, Marco, Pascual and Turon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xavier Turon, xturon@ceab.csic.es
†These authors have contributed equally to this work and share senior authorship