 Yikai Lu
Yikai Lu Mengmeng Zhao
Mengmeng Zhao Li Chen
Li Chen Yan Wang
Yan Wang Tianhao Liu
Tianhao Liu Haipeng Liu
Haipeng Liu- 1Central Laboratory, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai, China
- 2Research Center of Translational Medicine, Jinan Central Hospital Affiliated to Shandong First Medical University, Jinan, China
As a canonical cytoplasmic DNA sensor, cyclic GMP-AMP synthase (cGAS) plays a key role in innate immunity. In recent years, a growing number of studies have shown that cGAS can also be located in the nucleus and plays new functions such as regulating DNA damage repair, nuclear membrane repair, chromosome fusion, DNA replication, angiogenesis and other non-canonical functions. Meanwhile, the mechanisms underlying the nucleo-cytoplasmic transport and the regulation of cGAS activation have been revealed in recent years. Based on the current understanding of the structure, subcellular localization and canonical functions of cGAS, this review focuses on summarizing the mechanisms underlying nucleo-cytoplasmic transport, activity regulation and non-canonical functions of cGAS in the nucleus. We aim to provide insights into exploring the new functions of cGAS in the nucleus and advance its clinical translation.
1 Introduction
The innate immune system is the main contributor of the host defense against pathogen invasion and tissue damage (1). Pattern recognition receptors (PRRs) on innate immune cells recognize the pathogen-associated molecular patterns (PAMPs) carried by invading pathogens and the endogenous damage-associated molecular patterns (DAMPs) released by damaged cells, and then activates the innate immune response and prevents body damage (2, 3).
The discovery of cyclic GMP-AMP synthase (cGAS), the most interested PRRs in recent years, is a significant milestone in the field of DNA sensing, which plays a crucial role in infectious diseases (4), autoimmune diseases (5), cancers (6) and other diseases. cGAS recognizes both endogenous and exogenous DNA, such as pathogens DNA, mitochondrial DNA, micronuclei (MNi) DNA, genomic DNA, etc. (7, 8), then undergoes conformational changes and catalyzes the synthesis of the second messenger 2′3′-cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) using adenosine triphosphate (ATP) and guanosine triphosphate (GTP) (9). cGAMP binds with stimulator of interferon genes (STING) and activates STING on endoplasmic reticulum (ER). Activated STING transfers from ER to Golgi apparatus (GA) and recruits TANK binding kinase 1 (TBK1) and IκB kinase (IKK) complex to activate interferon regulatory factor 3 (IRF3) and nuclear factor kappa-B (NF-κB), respectively. Subsequently, IRF3 and NF-κB enter the nucleus and induce the expression of cytokines, including type I interferon (IFN), which activate the innate immune response (10–13).
Since Chen team first reported cGAS in 2013, the biological function of cGAS in the cytoplasm has been widely studied (9). However, in recent years, a growing number of studies have shown that cGAS was also located in the nucleus (14–17). In 2018, our team first elucidated the nuclear function of cGAS in inhibiting DNA repair, which is dispensable of its canonical function in inducing STING-mediated type I IFN response (7). Subsequently, more STING-independent nuclear functions of cGAS were discovered, such as DNA damage repair (15), DNA replication regulation (16), chromosome fusion (17), etc. However, the mechanisms underlying nuclear localization, nucleo-cytoplasmic transport, activity regulation and nuclear functions of cGAS are still uncovered. Therefore, this review will briefly summarize the structure, activation, canonical function and subcellular localization of cGAS. Importantly, we focus on the mechanisms underlying nucleo-cytoplasmic transport, activity regulation and non-canonical functions of cGAS in the nucleus, in order to provide a more comprehensive understanding of nuclear cGAS. Meanwhile, we hope that this review could provide researchers with perspectives on the functions of nuclear cGAS and shed lights on clinical translational study by targeting nuclear cGAS.
2 Brief introduction of cGAS and its canonical function
2.1 Structural characteristics of cGAS
Human cGAS (hcGAS), also known as C6ORF150 or MB21D1, is a protein composed of 522 amino acids with a molecular weight of about 60 kDa, and belongs to the cGAS/DncV-like nucleotidyltransferase (CD-NTase) superfamily (8, 18). The structure of cGAS in homo sapiens, sus scrofa and other species is relatively conserved, containing a disordered N-terminal domain and a C-terminal catalytic domain (19, 20). The N-terminal domain, consisting of amino acids 1-160, contains a high density of positively charged amino acid residues that are necessary for binding double-stranded DNA (dsDNA). cGAS probably contains two different DNA binding sites, which respectively plays a role in the conformational changes of cGAS and the cooperative binding of cGAS dimers and DNA (21–23). The C-terminal domain, composed of amino acids 161-522, contains an NTase core structural domain, a highly conserved male abnormal 21 (Mab21) domain and a site-C dsDNA-binding domain (9, 24, 25). The NTase core domain and Mab21 domain constitute the two-leaf structure of the catalytic domain. The N-terminal lobe consists of two helices and a highly twisted β-folded NTase fold, which contains all catalytic residues (Glu225, Asp227, Asp319). The C-terminal lobe is a tightly helical bundle and contains a conserved zinc-ion binding module that mediates the binding with DNA and its dimerization. The margin of the deep groove between the two lobes constitutes the substrate binding site of the enzyme (19, 20). The site-C dsDNA-binding domain is mainly composed of three marker fragments: α-region (261–286), KRKR-loop (299-302) and KKH-loop (427-432). This domain promotes multivalency-induced liquid-phase condensation and cGAMP production. In addition, the positively charged residues of the three fragments mediate the interaction of cGAS with nucleosomal DNA, which contributes to the formation of the 2:2 cGAS-nucleosome complex (24, 25).
Liquid-liquid phase separation (LLPS) of intracellular biomacromolecules ensures the independence, high efficiency and precision of intracellular reactions (26, 27). Interestingly, the positive charged and disordered N-terminal domain of cGAS promotes the cGAS-DNA phase separation under the induction of negatively charged DNA. The cGAS-DNA phase separation creates a relatively independent environment and thus avoids being inhibited by negative regulatory factors, such as nucleic acid exonucleases three prime repair exonuclease 1 (TREX1) and barrier to autointegration factor (BAF) (7, 23). LLPS depends not only on intrinsic disordered region, but also on factors such as polyvalent molecular interactions, concentration, and environmental conditions (19, 23, 27). Because the valence of long DNA is higher than short DNA, long DNA is more effective in promoting the LLPS and the enzyme activity of cGAS (23). In addition, the concentration of cGAS and DNA in the cytoplasm also appears to be crucial for LLPS, and the immune response is initiated only when their concentration reaches a certain threshold (19, 28). A recent study reported that a DNA agglutination molecule called spermine promoted the condensation of naked DNA (including viral DNA) but not nucleosomal DNA to enhance and stabilize the binding of cGAS and DNA and then activate downstream immune response, thus providing a self-nonself recognition mechanism (29). Notably, LLPS of the cGAS-DNA complex is inhibited by viral tegument proteins such as ORF52 and VP22 of the γ- and α-herpesvirinae, by which viruses escape the immune surveillance (30).
2.2 Molecular mechanism of cGAS activation
As the canonical PRR, cGAS is activated by endogenous and exogenous DNA, including invaded pathogens DNA, mitochondrial DNA, MNi DNA, genomic DNA, etc (7, 8). At the structural level, cGAS typically preferentially binds to dsDNA and incomplete nucleoid-like or bent DNA. Due to the two substitutions (K187 and L195) in the DNA-binding surface, which are sufficient to direct preferential detection of long DNA, the affinity of cGAS and DNA depends on the length of DNA (>45 bp) rather than the DNA sequence specificity (31–34). Although single-stranded DNA (ssDNA) also binds with cGAS and induces limited cGAMP production, its binding affinity (Kd ~1.5 μM) is significantly weaker than dsDNA (Kd ~87.6 nM) (35). Interestingly, cGAS is also activated by unpaired DNA nucleotides flanking short base-paired DNA stretches in a structural and sequence-dependent manner. An example is the unpaired guanosines within the stem-loop structure of ssDNA derived from human immunodeficiency virus (HIV) type 1 (36). In addition, RNA : DNA hybrid strands also activate cGAS (37).
The conformational change of cGAS is induced by DNA binding. cGAS binds to the two phospho-sugar main chains of DNA, while the DNA double strand binds to the platform between the spine-like α helix and zinc finger structure of cGAS, forming a 2:2 dimer or highly ordered complexes. The cGAS dimers then form a ladder network structure, positioned head to head next to the DNA, resulting in the activation of cGAS (20, 21). After activated, cGAS performs the enzymatic function, catalyzing the synthesis of cGAMP using ATP and GTP (22). As a second messenger, cGAMP binds and activates STING on the ER membrane (38, 39). In the inactive state, STING is anchored to ER in the form of a ‘V’ dimer, and its C-terminal domain (CTD) is exposed in the cytoplasm. The CTD of the activated STING dimers rotates 180° and unwraps around the connector between proteins (including a connector loop and a connector helix), forming a more closed lid-like structure covering the cGAMP binding site (40, 41). STING then activates transforming growth factor beta-activated kinase 1 (TAK1) prior to STING trafficking in a TAK1 binding protein 1 (TAB1)-dependent manner. The activated TAK1 directly mediates the phosphorylation of STING at S355, facilitating its interaction with STING ER exit protein (STEEP), thus promoting STING oligomerization and translocation to ER-Golgi intermediate compartments (ERGIC) for subsequent activation (42). Subsequently, STING is transferred from ER to GA via ERGIC, recruiting and activating downstream factors such as TBK1 and IKK complex (43–45). Activated TBK1 phosphorylates the CTD of STING, and the phosphorylated CTD recruits IRF3 through its conserved and positively charged phospho-binding domain. Recruited IRF3 is phosphorylated by TBK1 and forms a dimer. The activated IRF3 translocates into the nucleus and functions as a transcription factor, thereby inducing the expression of type I IFN such as IFN-β (11, 45, 46). The activated IKK complex phosphorylates NF-κB inhibitor IκBα, which promotes the translocation of NF-κB into the nucleus and induces the expression of pro-inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor (TNF) (11). IKK complex is composed of kinases IKKα and IKKβ, along with the regulatory subunit IKKγ (44, 47). IKKβ plays a key role in the activation of NF-κB mediated by cGAS-STING (44). Studies have shown that tripartite motif-containing protein (TRIM) 32 and TRIM56 is activated by cytoplasmic DNA, which add ubiquitin chains to NEMO and activate IKKβ, leading to the activation of NF-κB (44). Interestingly, the activated IKKβ is required for NF-κB and TBK1-IRF3 activation. Meanwhile, TBK1 is a reciprocal requirement for NF-κB activation, possibly by directly phosphorylating IKKβ. Thus, NF-κB/IRF3 activating cascade in which TBK1 and IKKβ forms a positive-feedback loop promotes the robust production of cytokines during cGAS-STING activation (44). Opposite to TRIM56 and TRIM32, TRIM29 has been reported to inhibit the innate immunity response. TRIM29 induces K48-linked ubiquitination of STING and promotes the degradation of STING, thus inhibiting the activation of innate immune response (Figure 1) (48).
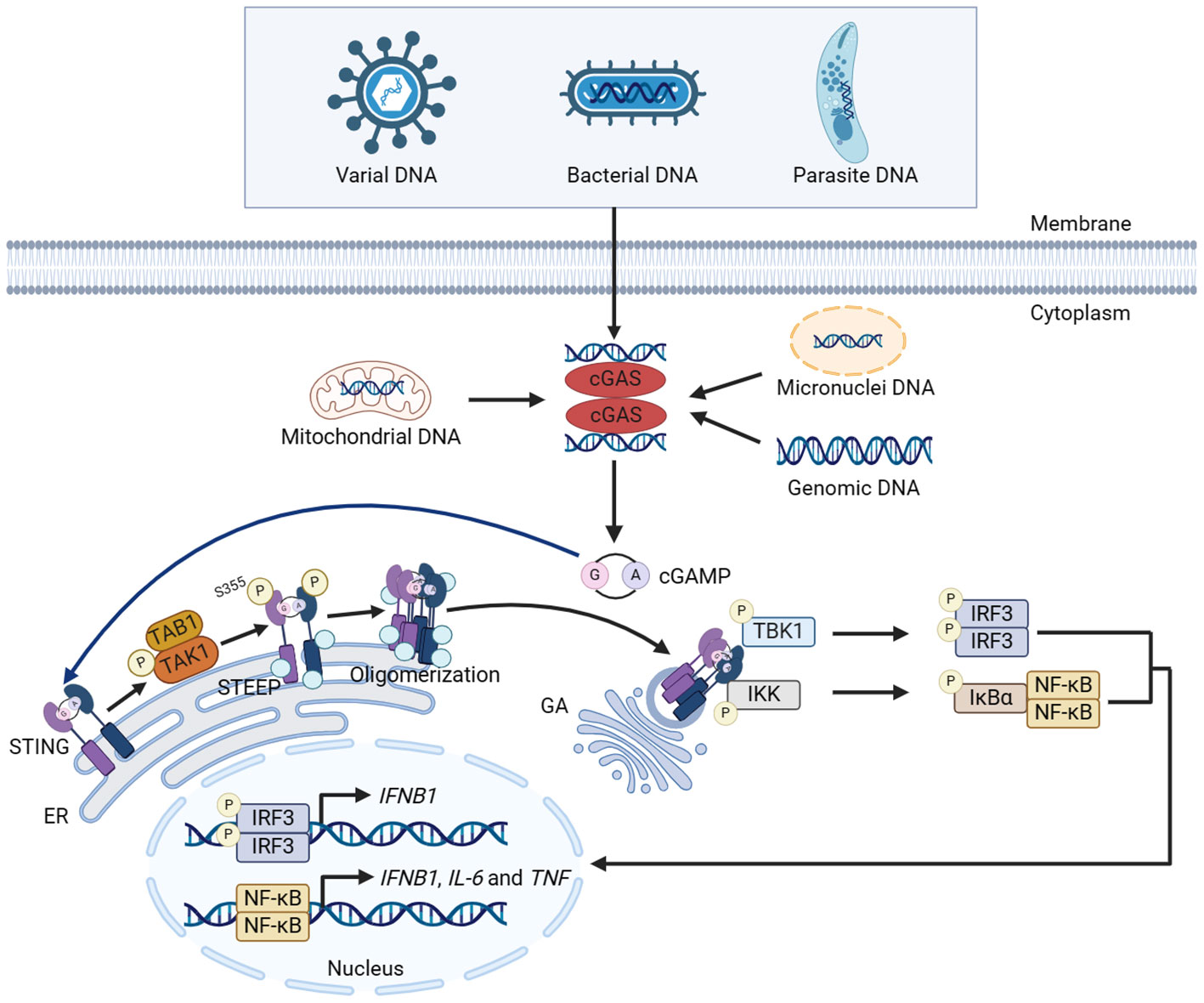
Figure 1 Canonical innate immune pathways induced by DNA. DNA from multiple sources induces the dimerization of cGAS and facilitates cGAS to catalyze the synthesis of cGAMP using ATP and GTP. cGAMP binds with STING and activates STING on the ER membrane. STING then activates TAK1, which directly mediates the phosphorylation of STING at S355 in a TAB1-dependent manner, facilitating the interaction between STING and STEEP. The interaction promotes STING oligomerization and translocation from ER to GA via ERGIC, recruiting and activating TBK1 and IKK complex. Activated TBK1 and IKK phosphorylate IRF3 and NF-κB, respectively, and promote the nuclear translocation of IRF3 and NF-κB, thereby inducing the expression of cytokines including type I IFN. The figures were created using scientific image and illustration software, BioRender (BioRender.com).
2.3 Canonical functions of cGAS
Abnormal distributed nucleic acid resulting from pathogen invasion or unstable genome induced by DNA damage caused by various stimuli is recognized by cGAS, and then initiates the canonical innate immune response. The role of cGAS on the innate immune regulation was first found in pathogen infection and autoimmune diseases (9). dsDNA from a variety of pathogenic microorganisms such as mycobacterium tuberculosis (49), adenovirus (50) and plasmodium falciparum (51) is recognized by cGAS, which then promotes the expression of type I IFN and proinflammatory cytokines to enhance the anti-infection immunity. However, not all the canonical functions of cGAS are favorable for defensing infection. It has been reported that the host usually inhibits cGAS-induced type I IFN expression through promoting the production of caspase-1, which avoids the excessive production of type I IFN and decreases the host’s resistance to mycobacterium bovis infection (52). Studies have shown that cGAS/STING-deficient mice showed stronger resistance to schistosoma mansoni (53) and lethal plasmodium yoelii YM (54), while showed no difference on the scavenging of adenovirus (55). Interestingly, cGAS-deficient mice show higher levels of IFN-α and IFN-β in vivo when infected with lethal plasmodium yoelii YM (54).
Hyperactivity of cGAS-STING pathway may lead to sustained immune response and result in autoimmune diseases, such as systemic lupus erythematosus (56), Aicardi-Goutières syndrome (29), STING-associated vasculopathy with onset in infancy (57), rheumatoid arthritis (58) and psoriasis (59). To maintain the immune balance, the body also evolves various negative regulatory mechanism to avoid the hyperactivation of innate immunity. As a 3’-5’ exonuclease anchored to ER, TREX1 degrades MNi DNA and cytoplasmic DNA in time, thereby inhibiting the hyperactivation of cGAS (60, 61). In addition, autophagy-related gene 9a (62), Unc-51-like autophagy activating kinase 1 (63), NOD-like receptor family CARD domain containing 3 (64) and other proteins inhibit the activation of STING, thus avoiding excessive activation of the cGAS-STING pathway. Therefore, the negative regulation of the cGAS-STING pathway is of great significance for immune homeostasis.
The cGAS-STING pathway also plays a vital role in tumorigenesis and tumor development. cGAS-STING pathway not only inhibits tumor progression, but also promotes tumor progression. On the one hand, cGAS-STING pathway plays an anti-tumor role in melanoma (65), small cell lung cancer (66), breast cancer (67, 68) and colon cancer (69) by promoting the activation of immune cells, cell apoptosis and tumor vascular remodeling. On the other hand, cGAS-STING pathway promotes tumor progression in melanoma (70), breast cancer (71, 72), lung cancer (73) and skin cancer (74) by promoting tumor immune escape and metastasis and maintaining chronic inflammation. Surprisingly, proteins of oncogenic DNA viruses such as E7 of human papilloma virus and E1A of adenovirus, could bind with STING and inhibit the activation of cGAS-STING pathway. Specifically, these proteins promote tumor progression via antagonizing DNA sensing and blocking tumor suppressor factors. The specific proteins encoded by these viruses contain the LXCXE motif (50), which provides some implications for the antagonists of STING and the treatment of viral tumors.
3 Cellular localization of cGAS
The precise localization of cGAS in cells is vital for regulating its function. However, cellular localization of cGAS remains controversial. With the further study of cGAS, a growing number of studies have shown that cGAS was not only located in the cytoplasm (9), but also located in the cytomembrane (75), MNi (76), stress granules (SG) (77), mitochondria (78), lysosome (79) and the highly anticipated nucleus (15). Therefore, it is vital to confirm the localization of cGAS under physiological and pathological conditions, which helps the prevention and treatment of pathogenic microbial infection, autoimmune diseases, tumors and other diseases.
3.1 Cytoplasm
Chen team first detected cGAS in the cytoplasmic extract of human monocytic THP-1 cells by western blot. In mouse fibroblast cell line L929 stimulated with interferon-stimulated DNA, cGAS co-locates with DNA in the cytoplasm observed by laser-scanning confocal microscopy (9). cGAS interacts with B-lymphoid tyrosine kinase (BLK) in the cytoplasm under physiological conditions, resulting in the phosphorylation of cGAS at Y215, which maintains the cytoplasmic localization of cGAS (15). Cytoplasmic localization of cGAS is conducive to the rapid identification of its own DNA in cells undergoing DNA damage or facing invading pathogens, and also prevents the recognition of its own DNA in the nucleus and organelles by cGAS (80, 81).
3.2 Nucleus
Actually, when cGAS was first found locating in the cytoplasm, it was also observed locating in the nuclear or perinuclear region by laser-scanning confocal microscopy (9). In recent years, an increasing number of studies have confirmed that cGAS can be located in the nucleus (14–17). Unexpectedly, the vast majority of cGAS is located in the nucleus, regardless of whether cells are rapidly dividing or post-mitosis (82). Under physiological conditions, endogenous cGAS is tethered tightly in the nucleus by a salt-resistant interaction and this tight tethering is comparable to that of histones in its strength (82). In chromatin tethering, nuclear cGAS is not activated by self-DNA due to the blocking of DNA binding sites by acidic patch (AP) formed by histones H2A and H2B (83). Moreover, the activity of nuclear cGAS is regulated both in nucleosome and non-nucleosome dependent way. The activity regulation and the functions of cGAS locating in the nucleus under physiological and pathological conditions will be described in part 4 and part 5.
3.3 Cytomembrane
In 2019, Kagan team made a groundbreaking discovery that cGAS, which was previously believed to be a cytoplasmic protein in the resting state, was located on the cytomembrane in human and mouse macrophages. Specifically, cGAS is located on the cytomembrane through the combination of its positively charged N-terminal domain with negatively charged phosphoinositide phosphate, thus preventing the recognition of self-DNA (75). However, the localization of cGAS on the cytomembrane via its N-terminal domain is debatable. Another study showed that the N-terminal domain 1-212 of cGAS encodes a cytoplasmic retention activity in interphase (84). This suggests that the cellular localization of cGAS through its N-terminal is influenced by cell type, cell cycle and other factors.
3.4 MNi
cGAS is also located to the MNi in the cytoplasm (76). MNi is formed due to the error of chromosome separation in the process of cell division, and consists of chromatin surrounded by micronuclear envelope (mNE). MNi is usually in a non-immunogenic state when it has an intact mNE, compact chromatin with intact nucleosome and supercoiled DNA. Once these conditions are damaged, the immunogenicity of MNi is enhanced, resulting in recognition of MNi DNA by cGAS. Such immunogenic transformation is manifested in various forms, such as the rupture of mNE, the loss of nucleosome integrity, and the loosening of the supercoiled structures of MNi DNA (72, 76, 80). Recognition of MNi DNA by cGAS leads to the cGAS-STING cytoplasmic DNA sensing pathway and activates innate immunity (72, 76). In addition, cGAS serves as a micronucleophagy receptor for the clearance of MNi, maintaining MNi homeostasis to avoid hyperactivation of innate immunity within the cells. To be specific, cGAS directly interacts with microtubule associated protein 1 light chain 3 beta (MAP1LC3B/LC3B) through its MAP1LC3-interacting region located at amino acid sites 355-360, by which cGAS promotes the micronucleophagy and degradation in lysosome (85).
3.5 SG
cGAS is also located in SG (77). SG is formed by untranslated messenger ribonucleoproteins resulting from mRNA stalled in translation initiation, and is a subcellular structure regulating the RNA localization, translation, and degradation (86). Studies have shown that Ras–guanosine triphosphatase activating protein Src homology 3 domain–binding protein 1 (G3BP1), an important protein of SG, interacted with cGAS and engaged cGAS in a primary liquid-phase condensation state. Under the stimulation of exogenous DNA, the pre-condensed cGAS forms LLPS more efficiently, which promotes the rapid binding of cGAS and DNA and induces the expression of IFNs (77, 86, 87). Intriguingly, it is reported that RNA regulates cGAS activity by modulating the formation of cGAS-containing condensates (88). Therefore, it is worth considering whether PKR dependent RNA plays a role in the process of cGAS pre-condensation induced by G3BP1. In conclusion, according to the existing studies, SG is considered as a hub of nucleic acid receptors that promotes the recognition of exogenous nucleic acids and the initiation of innate immunity response.
3.6 Mitochondria
cGAS has been shown to recognize mitochondrial DNA (mtDNA) released by damaged mitochondria in the cytoplasm (78). Interestingly, damaged mtDNA could synergizes with MNi to activate cGAS and induce a robust immune response (80). However, the localization of cGAS on mitochondria, as well as its mechanism of localization on other subcellular organelles, remains unclear. Fortunately, a recent study has shown that cGAS is located on the outer mitochondrial membrane in hepatocellular carcinoma cells. Specifically, cGAS is translocated to mitochondria via the mitochondrial targeting sequence with the assistance of outer mitochondrial membrane 70, and interacts with dynamin-related protein 1 to promote oligomerization. This interaction inhibits the accumulation of mitochondrial ROS and ferroptosis in cancer cells, which in turn promotes cancer progression (78).
3.7 Lysosome
In 2019, Chen team discovered that the primordial function of the cGAS-STING pathway autophagy induction via STING trafficking independent of the TBK1 activation and IFNs production (89). In 2020, a study on the role of cGAS in promoting inflammation and autophagy in huntington disease (HD) mentioned the localization of cGAS in lysosomes in HD-homo striatal cells (79). Therefore, it is worth considering whether cGAS is regulated in lysosomes in a STING-dependent but TBK1-independent way.
4 Distribution of cGAS in the nucleus and regulation of its activation
In the past, researchers observed that cGAS could not be activated by genomic DNA, and the widely accepted explanation for this was that cGAS was mainly distributed in the cytoplasm, where it did not have access to genomic DNA. However, in recent years, several studies have reported that endogenous cGAS is widely present in the nucleus in resting states (14, 82, 90, 91). It has been found that endogenous cGAS was tightly tethered in the nucleus by a force with significant resistance to salt extraction. Therefore, the presence of nuclear cGAS was omitted from the conventional cytoplasmic and nuclear extracts (89). These findings make the study of the function and mechanism of nuclear located cGAS become a research hotspot in this field.
4.1 Distribution of cGAS in the nucleus
Endogenous cGAS is tethered tightly in the nucleus and sometimes located to ‘chromatin bridges’ between adjacent cells (82). The formation of ‘chromatin bridges’ is thought to be associated with the fusions of chromosome and the incomplete segregation of DNA between daughter cells during mitosis (82, 92). It has been shown that ‘chromatin bridges’ occurred when telomere fusions were induced in various cell lines, and nuclear envelope rupture during interphase occurred more frequently in cells with ‘chromatin bridges’. It is worth noting that telomere crisis is closely related to cancer (93). So what role does ‘chromatin bridges’ play in cancer? Is the localization of cGAS in the ‘chromatin bridges’ related to the nucleo-cytoplasmic localization, and is it closely related to the occurrence and development of cancer? These are urgent issues remaining to be addressed.
4.2 Nucleo-cytoplasmic transport of cGAS
Nucleo-cytoplasmic transport of cGAS is precisely regulated which ensures that cGAS performs its function in the proper localization. cGAS is mainly located in the cytoplasm in nondividing cells and in the nucleus in mitotic proliferating cells (90). According to the existing reports, cytoplasmic retention and accumulation of cGAS may depend on the phosphorylation of Y215 and N-terminal domains mentioned above (Figure 2A) (15, 84). However, cGAS is translocated from the cytoplasm to the nucleus once nuclear damage occurs. When cells undergo DNA damage, the Y215 of cGAS is dephosphorylated, leading to its translocation from the cytoplasm to the nucleus through importin-α-dependent mechanisms. Nuclear cGAS is then recruited to double-strand breaks (DSBs) points in damaged DNA, where it inhibits the formation of the PARP1-Timeless complex through poly(ADP-ribose) interaction with PARP1 and impairs homologous-recombination-mediated DNA damage repair (Figure 2A) (15). Another study has shown that cGAS relied on lamin A/C to translocate into the nucleus and gather at the rupture site of the nuclear envelope (NE) when the NE broke (Figure 2A) (94).
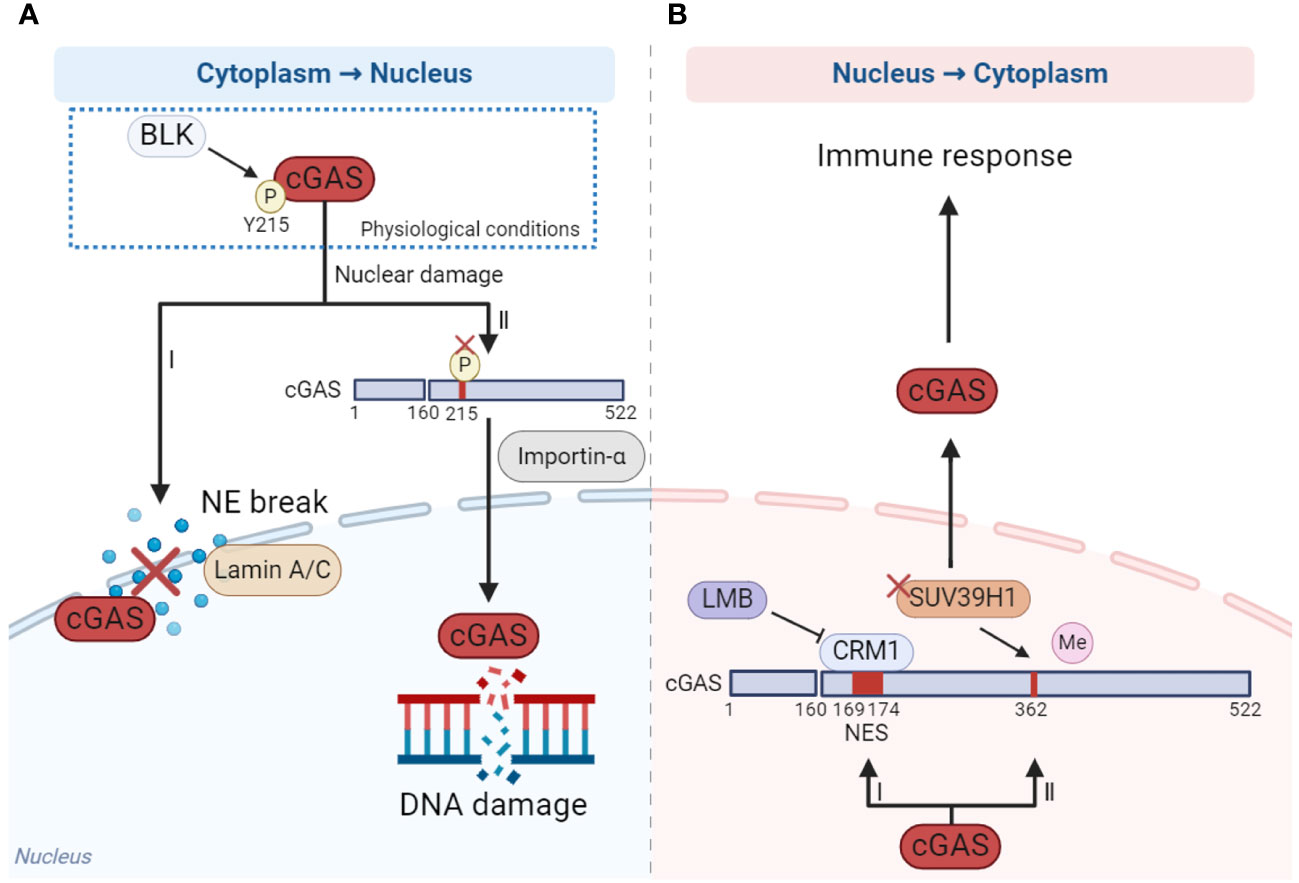
Figure 2 The mechanism of cGAS nucleo-cytoplasmic transport. (A) When DNA damage occurs, cGAS is dephosphorylated at Y215 and translocated from cytoplasm to the nucleus depending on importin-α (I), while the nuclear membrane is ruptured, the translocation of cGAS from cytoplasm to the nucleus is dependent on laminin A/C (II). (B) When cells are stimulated by cytoplasmic DNA, cGAS translocates from nucleus to cytoplasm in a CRM1-dependent manner under the effect of NES (I). In addition, reducing the methylation of cGAS by methyltransferase SUV39H1 promotes cGAS translocating to the cytoplasm (II). The figures were created using scientific image and illustration software, BioRender (BioRender.com).
How does the nuclear cGAS translocate to the cytoplasm? When stimulated by cytoplasmic DNA, cGAS translocates from the nucleus to the cytoplasm in a chromosome region maintenance 1 (CRM1)-dependent manner, facilitated by the nuclear export signal (NES) locating at amino acid sites 169-174 of cGAS. This translocation activates immune response. Once the NES is mutated or the CRM1 is inhibited by inhibitor leptomycin B (LMB), cGAS could not enter the cytoplasm from the nucleus and the activation of innate immune including IFN response is significantly inhibited (Figure 2B) (95). In addition, reducing the methylation of cGAS by methyltransferase SUV39H1 can promote cGAS translocation into the cytoplasm and thus promote the activation of the immune response (Figure 2B) (96).
4.3 Inactivation mechanism of nuclear cGAS
In recent years, some explanations have been proposed to elucidate why cGAS is not activated by self-DNA in the nucleus. Currently, the inhibition of cGAS activity in the nucleus is mainly explained from two perspectives: non-nucleosome regulation (97) and nucleosome regulation (14).
4.3.1 Non-nucleosome regulation
In 2017, Chen team found that cGAS entered the nucleus during mitosis in proliferating cells and was associated with chromatin DNA, suggesting that cGAS may regulate cell cycle and cellular senescence through an unrevealed mechanism (90). Further studies show that cGAS activity is selectively inhibited during mitosis in human cell lines and reveal two parallel mechanisms for this inhibition, both of which are dispensable of nucleosome. On the one hand, the N-terminal domain of cGAS is hyperphosphorylated by mitotic kinases, including Aurora kinase B, which leads to the blocking of chromatin sensing. On the other hand, oligomerization of chromatin-bound cGAS is prevented, leading to the inhibition of cGAS activation. These two mechanisms ensure that cGAS is inactive upon entry into the nucleus during mitosis, which may help prevent autoimmune responses (Figure 3A) (97). Also during mitosis, human cGAS is phosphorylated by mitotic cyclin-dependent kinase 1 (CDK1)-cyclin B complex at the highly conserved S305, and phosphorylation at this site inhibits cGAS activity and cGAMP synthesis (Figure 3A). However, the self-DNA sensing capability of cGAS is restored when cGAS is dephosphorylated by type 1 phosphatase PP1 at mitotic exit (98). In addition, BAF competitively binds self-DNA with cGAS during acute loss of nuclear membrane integrity, thereby limiting cGAS activation (Figure 3A) (99). Moreover, circular RNA antagonists cia-cGAS binds nuclear cGAS to avoid the detection of self-DNA and blocks its enzymatic activity, thus maintaining homeostasis of hematopoietic stem cells (Figure 3A) (100). Interestingly, spermine, one of the three polyamines synthesized in mammals, plays an important role in cGAS distinguishing between self and non-self DNA. Spermine effectively promotes the condensation of viral DNA rather than host nucleosome DNA, which is important for the immune system to protect against pathogens and maintain tolerance to the host and innocuous commensals during homeostasis (29).
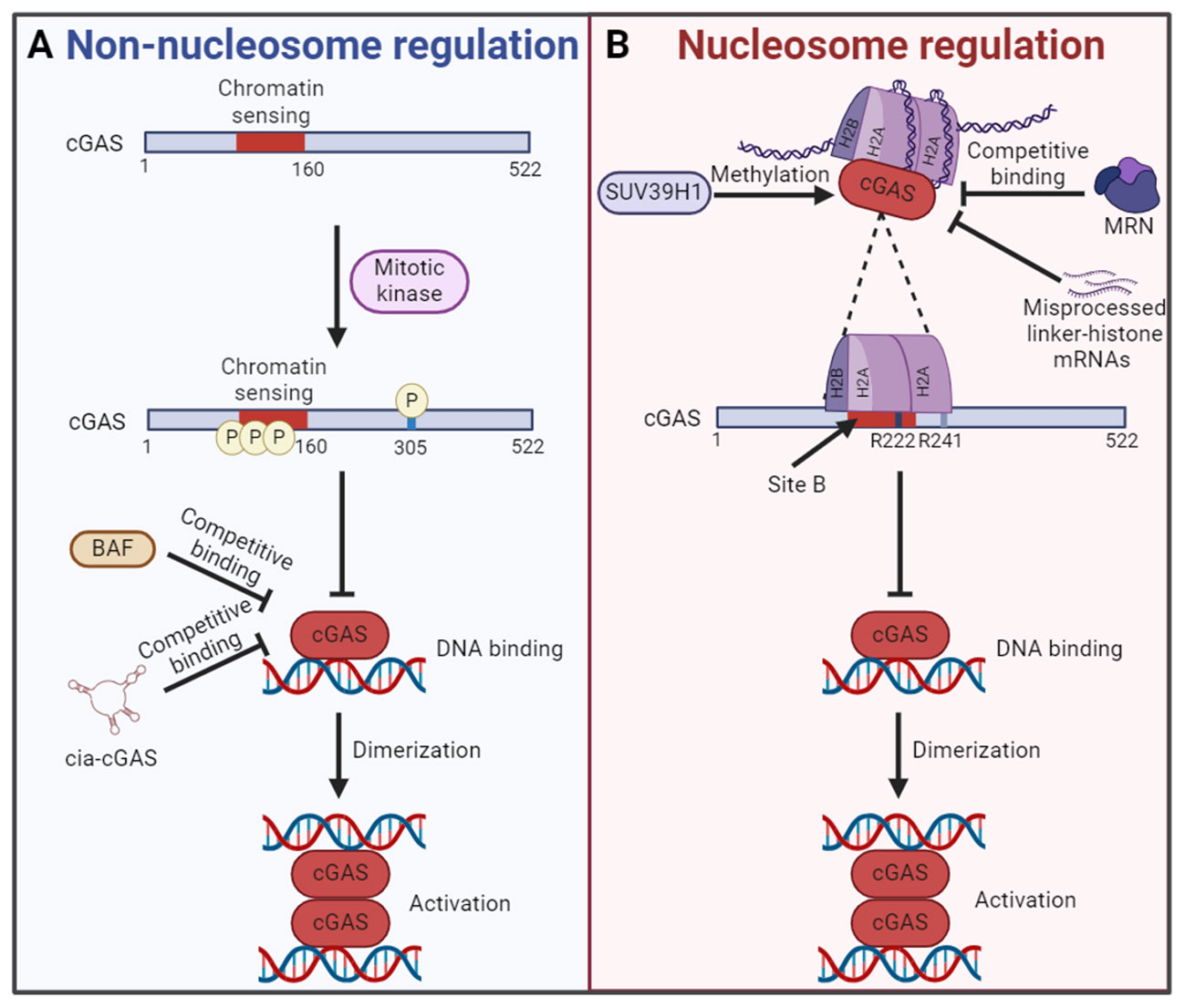
Figure 3 The mechanism of cGAS inactivation in the nucleus. (A) In nucleosome independent way, the N-terminal chromatin sensing region of cGAS or S305 is hyperphosphorylated by mitotic kinase. What’s more, the competitive binding of BAF or cia-cGAS to DNA also inhibits the cGAS activation. Both mechanisms lead to the failure of cGAS binding with self-DNA and the formation of cGAS dimers for activation. (B) In nucleosome dependent way, the DNA binding site B of cGAS is blocked by interacting with the AP. The AP is formed by histones H2A and H2B. Interact with the AP is cGAS conserved amino acid residues R222 and R241. In this regard, the chromatin tethering could be altered by the methylation of cGAS by SUV39H1, competitive binding of MRN and accumulation of misprocessed linker-histone mRNAs, etc. The figures were created using scientific image and illustration software, BioRender (BioRender.com).
4.3.2 Nucleosome regulation
Based on the discovery that cGAS was located in the nucleus and bound to chromatin (89), Li team found that the catalytic domain of cGAS promoted the binding of cGAS and nucleosome (14). The activity of cGAS in the nucleosome/cGAS complex is inhibited, which could not catalyze the synthesis of cGAMP. Interestingly, although oligonucleosome inhibits the activity of cGAS, a certain amount of cGAMP is synthesized when excessive cGAS is added to oligonucleosome/cGAS complex. The researchers suspect that the bare DNA at the junctions in the oligonucleosome may be responsible. The naked DNA activates cGAS that is not bound to the nucleosome (14). Further studies reveal that cGAS does not interact with nucleosome DNA. Actually, cGAS interacts with a negatively charged AP formed by histones H2A and H2B via its conserved amino acid residues R222 and R241 with nanomolar affinity. This interaction blocks the second DNA binding domain (site B) of cGAS and inhibits the formation of cGAS dimers, resulting in cGAS maintaining in an inactive conformation (14, 92). These findings confirm the critical role of nucleosome in inhibiting the activation of cGAS in the nucleus, which is a major breakthrough for the study of self and non-self discrimination of genomic DNA by cGAS (Figure 3B). Interestingly, it has been found that competitive uptake of methionine by tumor cells activated the methyltransferase SUV39H1 and promoted the methylation of cGAS, thus promoting the chromatin tethering of cGAS without activation (Figure 3B) (96). However, the chromatin tethering could be altered by molecular displacement. A recent study demonstrated that binding of the MRE11-RAD50-NBN (MRN) complex to nucleosome fragments was essential to displace cGAS from AP-mediated sequestration, which enabled its mobilization and activation by dsDNA (Figure 3B) (101). In addition, the chromatin tethering could also be disrupted by the accumulation of misprocessed linker-histone mRNAs and histone stoichiometry alteration, which are caused by mutations in the LSM11 (U7 snRNA-associated Sm-like protein) and RNU7-1 (RNA, U7 small nuclear 1) genes encoding core components of histone mRNA-preprocessing complex (Figure 3B) (102).
5 Functions of nuclear cGAS
5.1 DNA damage repair
There are many DNA damage factors that affect human health both in vivo and in vitro. For example, DSBs are potentially highly harmful. If not properly repaired, DSBs can lead to chromosomal deletion or translocation, which eventually result in diseases related to genomic instability, including tumorigenesis, accelerated aging, and other diseases (103). Powerful DNA damage repair mechanisms ensure the integrity of DNA. According to current reports, there are at least five major DNA damage repair pathways, including non-homologous end joining (NHEJ), base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR) and homologous recombination (HR) (104). Currently, most studies have proved that cGAS inhibited homologous recombination in response to DNA damage induced by different conditions and thus inhibited DNA damage repair (15, 105–107). Our study demonstrated that nuclear cGAS contributes to tumorigenesis by inhibiting HR-mediated DNA damage repair. Once the DNA damage occurred, cytoplasmic cGAS was dephosphorylated at Y215 and translocated into the nucleus. The C-terminal domain of nuclear cGAS is recruited to DNA damage sites after binding to phosphorylated H2AX and inhibits the formation of PAPR1/Timeless complex, thereby inhibiting HR and increasing genomic instability (15). Under ionizing radiation, demethylase ribosomal oxygenase 1 (RIOX1) removes monomethylation at K491 of cGAS, which promotes the separation of cGAS and methyl-lysine reader protein SAGA complex-associated factor 29 (SGF29). The released cGAS binds to PAPR1 to inhibit the formation of PAPR1/Timeless complex, thereby inhibiting HR (105). Moreover, nuclear translocated cGAS inhibits HR-mediated DNA damage repair and promotes mutagenesis in human hepatocytes after microcystiin-LR (MC-LR) treatment, while BLK inhibits nuclear translocation of cGAS and cell mutagenesis induced by MC-LR (106). Besides the important role of the cGAS-PARP1-Timeless axis in HR suppression, cGAS also inhibits HR through other mechanisms. Another study found that nuclear cGAS dimerization impaired HR efficiency by compacting dsDNA into a higher‐ordered state. This state hindered RAD51-coated ssDNA filaments invasion, thereby affecting D-loop formation during HR-mediated DNA damage repair. Eventually, it leads to genomic instability, the generation of MNi and cell death under genomic stress conditions (Figure 4A) (107). In addition, cGAS has also been reported to prevent excessive DNA damage by inhibiting long interspersed nuclear element (LINE, also called long retroposon)-1 (LINE-1) retrotransposition, thereby maintaining genomic stability (Figure 4A) (81, 108). It has been reported that cGAS was enriched in the LINE region in the nucleus (81, 109), and recognized the complementary DNA produced by reverse transcription of LINE-1 to induce pro-inflammatory cytokines production (110). However, whether nuclear cGAS directly participates in the regulation of LINE retrotransposition and the underlying mechanism have not been characterized. ORF2p, a protein encoded by the open reading frame in full-length LINE-1, is crucial for LINE-1 retrotransposition and causes DNA strand breaks which poses a threat to genome integrity via its endonuclease activity (111–113). Recently, Mao team revealed that upon the occurrence of DNA damage, the checkpoint kinase CHK2 interacted with and phosphorylated nuclear cGAS at S120 and S305 residues to facilitate the interaction of cGAS-TRIM41 and TRIM41-ORF2p, which promoted TRIM41-mediated ORF2p ubiquitination and degradation and restricted LINE-1 retrotransposition, thereby maintaining genomic stability (108).
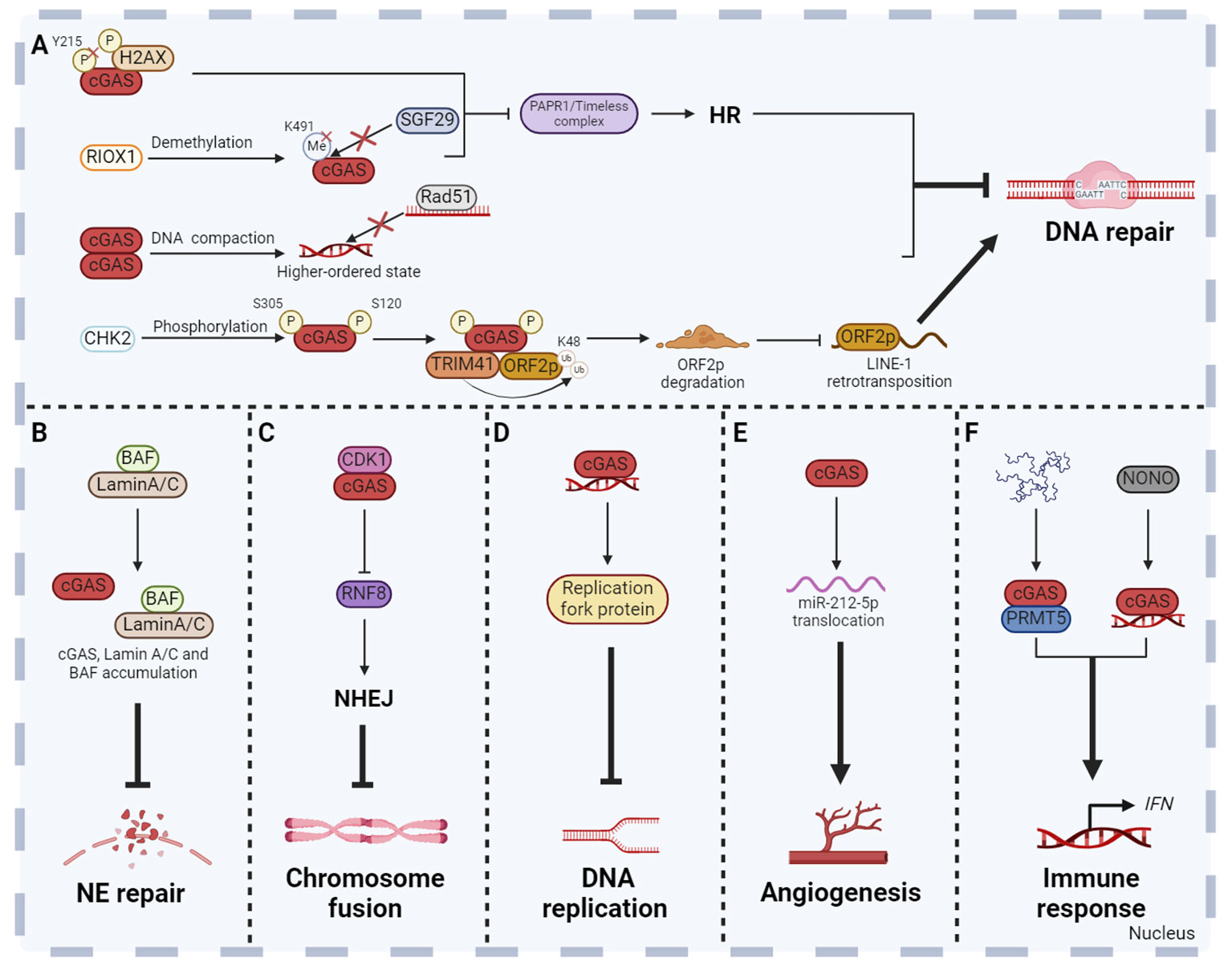
Figure 4 Non-canonical functions of cGAS in the nucleus. Besides the DNA sensor function, cGAS has been revealed many new functions in the nucleus. Nuclear cGAS plays a role in DNA repair (A), NE repair (B), chromosome fusion (C), DNA replication (D), angiogenesis (E) and non-cytoplasmic innate immune response (F). These functions collectively enhance the host defense, inflammatory response and anti-tumor immunity, so as to maintain human physiological health. The figures were created using scientific image and illustration software, BioRender (BioRender.com).
5.2 NE repair
Due to the weakening of the NE structural integrity, leakage of nuclear proteins containing a nuclear localization signal (NLS) into the cytoplasm has been observed under normal physiological and pathological conditions. However, the NE repair process is essential to prevent nuclear dysfunction due to accumulation of DNA damage and leakage of macromolecules into the cytoplasm (94, 114, 115). When NE is ruptured, laminin C rapidly accumulates at the site of rupture, which requires both the immunoglobulin-like fold domain that binds to BAF and the NLS. In addition, cGAS also co-aggregates with laminin C and BAF at this site, partly dependent on laminin A/C to facilitate rapid repair (Figure 4B) (94).
5.3 Chromosome fusion
Cellular senescence is a self-defense mechanism activated by intrinsic stimuli and/or exogenous stress, and is one of the central hallmarks of senescence (116). There are many factors inducing cellular senescence, including telomere attrition, loss of tumor suppressors, mitochondrial dysfunction, perturbed proteostasis, autophagy impairment, cytokines, oxidative stress, nutrient deprivation, epigenetic modifiers, genotoxic drugs and other internal and external factors. Senescent cells secrete a large number of factors, including IL, chemokines, growth factors, non-protein molecules, insoluble factors and so on, collectively termed the senescence messaging secretome (SMS) or senescent associated secretory phenotype (SASP) (117). It has been shown that the cellular senescence is inhibited in cGAS-deficient cell which accelerates spontaneous immortalization. Meanwhile, the SASP induced by DNA damage is also suppressed (90). cGAS activation and STING dimer formation were found in senescent cells, suggesting that the cGAS-STING pathway is involved in the regulation of cellular senescence (118). In addition, cGAS also regulates cellular senescence in a STING-independent manner through inhibiting chromosome fusion. Chromosome fusion is a manifestation of chromosomal instability, usually caused by the loss of telomeric repeat sequences or deficiencies in telomeric proteins (119). Recently, further studies revealed that cGAS could interact with CDK1 and localize them to chromosome ends (17). CDK1 then blocks RNF8 recruitment to inhibit NHEJ-mediated DSBs repair during mitosis. This process contributes to reducing the occurrence of end fusions on broken chromosomes and facilitating replicative senescence (17). However, the absence of cGAS can lead to problems in cellular senescence, showing chromosome end-to-end fusions, genomic instability, and long-term growth arrest (17). Cancer cells often have a high rate of spontaneous telomere loss, resulting in chromosome fusion and other chromosomal instability (119). The mechanism of cGAS inhibiting chromosome fusion and promoting replicative senescence may be a vital breakthrough in tumor therapy (Figure 4C).
5.4 DNA replication
Complete and accurate DNA replication is an important basis for cell proliferation and genome stability. Any errors of DNA replication will result in abnormal DNA replication, which can lead to diseases such as tumors. Compared to normal cells, the replication forks, replication origin, S-checkpoint and other replication stress are abnormal in tumor cells, which results in genomic instability (120). Lan team found that cGAS-deficient untransformed cancer cells exhibited uncontrolled DNA replication, leading to genomic instability and excessive proliferation (16). Further mechanism research found that cGAS promoted the genomic stability by acting as a ‘decelerator’ for DNA replication (16). cGAS interacts with replication fork proteins in a DNA binding-dependent manner in the nucleus to slow replication fork movement and reduce cell susceptibility to DNA damage, indicating that cGAS is an attractive target for exploiting genomic instability of cancer cells (16). It is deserved to be mentioned that STING is often inhibited in numerous cancers, which contributes to the resistance to tumor immunity (121). The new STING independent function of cGAS in replication dynamics is expected to be a potential therapeutic target for STING deficient tumors (Figure 4D).
5.5 Angiogenesis
Angiogenesis is the formation of new blood vessels from the existing vascular system, which is a dynamically regulated biological event (122). Excessive or insufficient angiogenesis has been linked to diseases such as cancer and diabetes (123). Interestingly, nuclear cGAS regulates vascular endothelial growth factor-A (VEGF-A)-mediated angiogenesis in an immune-independent manner (124). Mechanism studies showed that VEGF-A stimulation induced cGAS nuclear translocation through importin-β pathway. Subsequently, nuclear cGAS regulates the miR-212-5p-ARPC3 cascade to influence cytoskeletal dynamics and VEGF receptor 2 trafficking from the trans-Golgi network to the cytomembrane (124). This study suggests that cGAS may be a potential therapeutic target for pathologic angiogenesis related diseases (Figure 4E).
5.6 Non-cytoplasmic innate immune response
In fact, cGAS plays the role in innate immune response not only in the cytoplasm, but also in the nucleus. Nuclear cGAS enriched in centromeric satellite DNA and LINE synthesizes cGAMP and induces innate immune response in primary human monocyte derived dendritic cells (DCs). The level of cGAMP synthesized by nuclear cGAS is 200 times lower than that stimulated with exogenous DNA (84). This non-canonical innate immune response is usually caused by various nuclear pathogens, but the mechanism of cGAS activation is different. HIV activates nuclear cGAS through the interaction of nuclear cGAS and non-POU domain-containing octamer-binding protein in DCs and macrophages (125). Herpes simplex virus 1 infection results in the release of cGAS from chromatin into the nuclear soluble fraction. Released nuclear soluble cGAS senses viral DNA and produces cGAMP, which subsequently induces the expression of type I IFN and IFN-stimulated genes (126). Moreover, nuclear cGAS interacts with protein arginine methyltransferase 5 (PRMT5) to catalyze the symmetric dimethylation of the histone H3 arginine 2 at Ifnb and Ifna4 promoters, thereby promoting the entry of IRF3 and inducing IFN production (Figure 4F) (127).
6 Conclusion
Currently, our understanding of the role of cGAS as a DNA sensor has significantly advanced. Cytoplasmic cGAS is a crucial receptor for both endogenous and exogenous DNA which induces the activation of innate immune response and serves as a master regulator in inflammation, senescence, cancer and so on, making it an appealing target for the drug development (10, 128). Moreover, several important questions that have been puzzling researchers in this field have been more or less answered. Why is cGAS not activated by the DNA in the nucleus? What function it plays in the nucleus (14, 15, 97)? However, there are still a variety of interesting questions to be addressed. First, the localization of cGAS has long been controversial. cGAS is not only located in the cytoplasm as previously reported, but also located in the nucleus and cytomembrane (15, 75). It remains challenging to clarify the mechanisms underlying the tight regulation of the localization of cGAS. Second, cell cycle has been implicated to be correlated with the activity of cGAS (97), the exact underlying mechanism warrants further clarification. Third, it’s now well accepted that the enzymatic activity of cGAS can be inhibited by binding to chromatin nucleosome in the nucleus (14), however, whether cGAS participates in transcriptional regulation through modulating chromatin accessibility is unknown. LLPS has been previously reported to be important for regulating the activity of transcriptional factors such as IRF1 (129), and cGAS is capable of forming LLPS (23). Therefore, it’s interesting to question whether LLPS of cGAS serves as a mechanism underlying transcriptional regulation. It remains attractive to further characterize the novel function of cGAS in the nucleus. In a word, the study of the function of cGAS in the nucleus is still urgently needed, which may thereby provide the basis for drug development to precisely target nuclear cGAS.
Author contributions
YL: Writing – original draft, Writing – review & editing. MZ: Writing – original draft, Writing – review & editing. LC: Writing – review & editing. YW: Writing – original draft. TL: Writing – original draft. HL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (82271882 to HL, 82302068 to MZ, 82303553 to LC) and Beijing Xisike Clinical Oncology Research Foundation (Y-zai2021/ms-0160), and the Shanghai ShuGuang Program (20SG19), and the Shanghai Sailing Program (22YF1451400), and the China Postdoctoral Science Foundation (2021M701407 to MZ, 2022M712405 to LC), and Postdoctoral Innovation Project of Shandong Province (SDCX-ZG-202202005).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zheng W, Liu A, Xia N, Chen N, Meurens F, Zhu J. How the innate immune DNA sensing cGAS-STING pathway is involved in apoptosis. Int J Mol Sci. (2023) 24:3029. doi: 10.3390/ijms24033029
2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
3. Briard B, Place DE, Kanneganti TD. DNA sensing in the innate immune response. Physiol (Bethesda). (2020) 35:112–24. doi: 10.1158/2159-8290
4. Reinert LS, Rashidi AS, Tran DN, Katzilieris-Petras G, Hvidt AK, Gohr M, et al. Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J Clin Invest. (2021) 131:e136824. doi: 10.1172/JCI136824
5. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–49.e18. doi: 10.1016/j.cell.2020.09.020
6. de Mingo Pulido Á, Hänggi K, Celias DP, Gardner A, Li J, Batista-Bittencourt B, et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity. (2021) 54:1154–67.e7. doi: 10.1016/j.immuni.2021.04.019
7. Liu H, Wang F, Cao Y, Dang Y, Ge B. The multifaceted functions of cGAS. J Mol Cell Biol. (2022) 14:mjac031. doi: 10.1093/jmcb/mjac031
8. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signaling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
9. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
10. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science. (2019) 363:eaat8657. doi: 10.1126/science.aat8657
11. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
12. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
13. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. (2012) 5:ra20. doi: 10.1126/scisignal.2002521
14. Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature. (2020) 587:673–7. doi: 10.1038/s41586-020-2749-z
15. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
16. Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv. (2020) 6:eabb8941. doi: 10.1126/sciadv.abb8941
17. Li X, Li X, Xie C, Cai S, Li M, Jin H, et al. cGAS guards against chromosome end-to-end fusions during mitosis and facilitates replicative senescence. Protein Cell. (2022) 13:47–64. doi: 10.1007/s13238-021-00879-y
18. Kranzusch PJ. cGAS and CD-NTase enzymes: structure, mechanism, and evolution. Curr Opin Struct Biol. (2019) 59:178–87. doi: 10.1016/j.sbi.2019.08.003
19. Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. (2020) 53:43–53. doi: 10.1016/j.immuni.2020.05.013
20. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. (2013) 498:332–7. doi: 10.1038/nature12305
21. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
22. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
23. Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. (2018) 361:704–9. doi: 10.1126/science.aat1022
24. Cao D, Han X, Fan X, Xu RM, Zhang X. Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res. (2020) 30:1088–97. doi: 10.1038/s41422-020-00422-4
25. Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci U.S.A. (2019) 116:11946–55. doi: 10.1073/pnas.1905013116
26. Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, et al. Protein phase separation: A new phase in cell biology. Trends Cell Biol. (2018) 28:420–35. doi: 10.1016/j.tcb.2018.02.004
27. Uversky VN. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol. (2017) 44:18–30. doi: 10.1016/j.sbi.2016.10.015
28. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U.S.A. (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
29. Wang L, Li S, Wang K, Wang N, Liu Q, Sun Z, et al. Spermine enhances antiviral and anticancer responses by stabilizing DNA binding with the DNA sensor cGAS. Immunity. (2023) 56:272–88.e7. doi: 10.1016/j.immuni.2023.01.001
30. Xu G, Liu C, Zhou S, Li Q, Feng Y, Sun P, et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol Cell. (2021) 81:2823–37.e9. doi: 10.1016/j.molcel.2021.05.002
31. Tao J, Zhou X, Jiang Z. cGAS-cGAMP-STING: The three musketeers of cytosolic DNA sensing and signaling. IUBMB Life. (2016) 68:858–70. doi: 10.1002/iub.1566
32. Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. (2017) 549:394–8. doi: 10.1038/nature23890
33. Luecke S, Holleufer A, Christensen MH, Jønsson KL, Boni GA, Sørensen LK, et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. (2017) 18:1707–15. doi: 10.15252/embr.201744017
34. Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell. (2018) 174:300–11.e11. doi: 10.1016/j.cell.2018.06.026
35. Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. (2013) 3:1362–8. doi: 10.1016/j.celrep.2013.05.008
36. Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kübler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. (2015) 16:1025–33. doi: 10.1038/ni.3267
37. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. (2014) 33:2937–46. doi: 10.15252/embj.201488726
38. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. (2013) 339:826–30. doi: 10.1126/science.1229963
39. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
40. Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
41. Kato K, Omura H, Ishitani R, Nureki O. Cyclic GMP-AMP as an endogenous second messenger in innate immune signaling by cytosolic DNA. Annu Rev Biochem. (2017) 86:541–66. doi: 10.1146/annurev-biochem-061516-044813
42. Ma M, Dang Y, Chang B, Wang F, Xu J, Chen L, et al. TAK1 is an essential kinase for STING trafficking. Mol Cell. (2023) 83:3885–903.e5. doi: 10.1016/j.molcel.2023.09.009
43. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. (2015) 18:157–68. doi: 10.1016/j.chom.2015.07.001
44. Fang R, Wang C, Jiang Q, Lv M, Gao P, Yu X, et al. NEMO-IKKβ Are essential for IRF3 and NF-κB activation in the cGAS-STING pathway. J Immunol. (2017) 199:3222–33. doi: 10.4049/jimmunol.1700699
45. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
46. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ, et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. (2013) 341:1390–4. doi: 10.1126/science.1244040
47. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. (2004) 25:280–8. doi: 10.1016/j.it.2004.03.008
48. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
49. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe. (2015) 17:820–8. doi: 10.1016/j.chom.2015.05.005
50. Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. (2015) 350:568–71. doi: 10.1126/science.aab3291
51. Gallego-Marin C, Schrum JE, Andrade WA, Shaffer SA, Giraldo LF, Lasso AM, et al. Cyclic GMP-AMP synthase is the cytosolic sensor of plasmodium falciparum genomic DNA and activates type I IFN in malaria. J Immunol. (2018) 200:768–74. doi: 10.4049/jimmunol.1701048
52. Liao Y, Liu C, Wang J, Song Y, Sabir N, Hussain T, et al. Caspase-1 inhibits IFN-β production via cleavage of cGAS during M. bovis infection. Vet Microbiol. (2021) 258:109126. doi: 10.1016/j.vetmic.2021.109126
53. Souza C, Sanches RCO, Assis NRG, Marinho FV, Mambelli FS, Morais SB, et al. The role of the adaptor molecule STING during Schistosoma mansoni infection. Sci Rep. (2020) 10:7901. doi: 10.1038/s41598-020-64788-6
54. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity. (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
55. Anghelina D, Lam E, Falck-Pedersen E. Diminished innate antiviral response to adenovirus vectors in cGAS/STING-deficient mice minimally impacts adaptive immunity. J Virol. (2016) 90:5915–27. doi: 10.1128/JVI.00500-16
56. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
57. Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J Allergy Clin Immunol. (2019) 143:712–25.e715. doi: 10.1016/j.jaci.2018.04.034
58. Wang J, Li R, Lin H, Qiu Q, Lao M, Zeng S, et al. Accumulation of cytosolic dsDNA contributes to fibroblast-like synoviocytes-mediated rheumatoid arthritis synovial inflammation. Int Immunopharmacol. (2019) 76:105791. doi: 10.1016/j.intimp.2019.105791
59. Yu Y, Xue X, Tang W, Su L, Zhang L, Zhang Y. Cytosolic DNA‒Mediated STING-dependent inflammation contributes to the progression of psoriasis. J Invest Dermatol. (2022) 142:898–906.e894. doi: 10.1016/j.jid.2021.08.430
60. Atianand MK, Fitzgerald KA. Molecular basis of DNA recognition in the immune system. J Immunol. (2013) 190:1911–8. doi: 10.4049/jimmunol.1203162
61. Jiang H, Kong N, Liu Z, West SC, Chan YW. Human Endonuclease ANKLE1 Localizes at the Midbody and Processes Chromatin Bridges to Prevent DNA Damage and cGAS-STING Activation. Adv Sci (Weinh). (2023) 10:e2204388. doi: 10.1002/advs.202204388
62. Mitzel DN, Lowry V, Shirali AC, Liu Y, Stout-Delgado HW. Age-enhanced endoplasmic reticulum stress contributes to increased Atg9A inhibition of STING-mediated IFN-β production during Streptococcus pneumoniae infection. J Immunol. (2014) 192:4273–83. doi: 10.4049/jimmunol.1303090
63. Konno H, Chinn IK, Hong D, Orange JS, Lupski JR, Mendoza A, et al. Pro-inflammation associated with a gain-of-function mutation (R284S) in the innate immune sensor STING. Cell Rep. (2018) 23:1112–23. doi: 10.1016/j.celrep.2018.03.115
64. Zhao Y, Li R. Overview of the anti-inflammatory function of the innate immune sensor NLRC3. Mol Immunol. (2023) 153:36–41. doi: 10.1016/j.molimm.2022.11.014
65. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. (2018) 49:754–63.e754. doi: 10.1016/j.immuni.2018.09.016
66. Liu H, Su H, Wang F, Dang Y, Ren Y, Yin S, et al. Pharmacological boosting of cGAS activation sensitizes chemotherapy by enhancing antitumor immunity. Cell Rep. (2023) 42:112275. doi: 10.1016/j.celrep.2023.112275
67. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. (2017) 109:djw199. doi: 10.1093/jnci/djw199
68. Bhatelia K, Singh A, Tomar D, Singh K, Sripada L, Chagtoo M, et al. Antiviral signaling protein MITA acts as a tumor suppressor in breast cancer by regulating NF-κB induced cell death. Biochim Biophys Acta. (2014) 1842:144–53. doi: 10.1016/j.bbadis.2013.11.006
69. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. (2019) 129:4350–64. doi: 10.1172/JCI125413
70. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. (2016) 76:2076–81. doi: 10.1158/0008-5472
71. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–8. doi: 10.1038/nature18268
72. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
73. Zheng T, Liu H, Hong Y, Cao Y, Xia Q, Qin C, et al. Promotion of liquid-to-solid phase transition of cGAS by Baicalein suppresses lung tumorigenesis. Signal Transduct Target Ther. (2023) 8:133. doi: 10.1038/s41392-023-01326-6
74. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. (2014) 5:5166. doi: 10.1038/ncomms6166
75. Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, et al. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell. (2019) 176:1432–46.e1411. doi: 10.1016/j.cell.2019.01.049
76. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. (2017) 548:461–5. doi: 10.1038/nature23449
77. Hu S, Sun H, Yin L, Li J, Mei S, Xu F, et al. PKR-dependent cytosolic cGAS foci are necessary for intracellular DNA sensing. Sci Signal. (2019) 12:eaav7934. doi: 10.1126/scisignal.aav7934
78. Qiu S, Zhong X, Meng X, Li S, Qian X, Lu H, et al. Mitochondria-localized cGAS suppresses ferroptosis to promote cancer progression. Cell Res. (2023) 33:299–311. doi: 10.1038/s41422-023-00788-1
79. Sharma M, Rajendrarao S, Shahani N, Ramírez-Jarquín UN, Subramaniam S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc Natl Acad Sci U.S.A. (2020) 117:15989–99. doi: 10.1073/pnas.2002144117
80. Guo X, Hintzsche H, Xu W, Ni J, Xue J, Wang X. Interplay of cGAS with micronuclei: Regulation and diseases. Mutat Res Rev Mutat Res. (2022) 790:108440. doi: 10.1016/j.mrrev.2022.108440
81. MacDonald KM, Benguerfi S, Harding SM. Alerting the immune system to DNA damage: micronuclei as mediators. Essays Biochem. (2020) 64:753–64. doi: 10.1042/EBC20200016
82. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife. (2019) 8:e47491. doi: 10.7554/eLife.47491
83. Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, et al. Structural basis for the inhibition of cGAS by nucleosomes. Science. (2020) 370:455–8. doi: 10.1126/science.abd0237
84. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. (2019) 26:2377–93.e2313. doi: 10.1016/j.celrep.2019.01.105
85. Zhao M, Wang F, Wu J, Cheng Y, Cao Y, Wu X, et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy. (2021) 17:3976–91. doi: 10.1080/15548627.2021.1899440
86. Protter DSW, Parker R. Principles and properties of stress granules. Trends Cell Biol. (2016) 26:668–79. doi: 10.1016/j.tcb.2016.05.004
87. Zhao M, Xia T, Xing JQ, Yin LH, Li XW, Pan J, et al. The stress granule protein G3BP1 promotes pre-condensation of cGAS to allow rapid responses to DNA. EMBO Rep. (2022) 23:e53166. doi: 10.15252/embr.202153166
88. Chen S, Rong M, Lv Y, Zhu D, Xiang Y. Regulation of cGAS activity by RNA-modulated phase separation. EMBO Rep. (2023) 24:e51800. doi: 10.15252/embr.202051800
89. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
90. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U.S.A. (2017) 114:E4612–e4620. doi: 10.1073/pnas.1705499114
91. Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature. (2020) 587:668–72. doi: 10.1038/s41586-020-2750-6
92. Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature. (2020) 587:678–82. doi: 10.1038/s41586-020-2748-0
93. Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. (2015) 163:1641–54. doi: 10.1016/j.cell.2015.11.054
94. Kono Y, Adam SA, Sato Y, Reddy KL, Zheng Y, Medalia O, et al. Nucleoplasmic lamin C rapidly accumulates at sites of nuclear envelope rupture with BAF and cGAS. J Cell Biol. (2022) 221:e202201024. doi: 10.1083/jcb.202201024
95. Sun H, Huang Y, Mei S, Xu F, Liu X, Zhao F, et al. A nuclear export signal is required for cGAS to sense cytosolic DNA. Cell Rep. (2021) 34:108586. doi: 10.1016/j.celrep.2020.108586
96. Fang L, Hao Y, Yu H, Gu X, Peng Q, Zhuo H, et al. Methionine restriction promotes cGAS activation and chromatin untethering through demethylation to enhance antitumor immunity. Cancer Cell. (2023) 41:1118–33.e1112. doi: 10.1016/j.ccell.2023.05.005
97. Li T, Huang T, Du M, Chen X, Du F, Ren J, et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science. (2021) 371:eabc5386. doi: 10.1126/science.abc5386
98. Zhong L, Hu MM, Bian LJ, Liu Y, Chen Q, Shu HB. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discovery. (2020) 6:26. doi: 10.1038/s41421-020-0162-2
99. Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science. (2020) 369:823–8. doi: 10.1126/science.aaw6421
100. Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, et al. A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS-mediated exhaustion. Immunity. (2018) 48:688–701.e7. doi: 10.1016/j.immuni.2018.03.016
101. Cho MG, Kumar RJ, Lin CC, Boyer JA, Shahir JA, Fagan-Solis K, et al. MRE11 liberates cGAS from nucleosome sequestration during tumorigenesis. Nature. (2024) 625:585–92. doi: 10.1038/s41586-023-06889-6
102. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
103. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
104. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. (2017) 58:235–63. doi: 10.1002/em.22087
105. Xiao Y, Li J, Liao X, He Y, He T, Yang C, et al. RIOX1-demethylated cGAS regulates ionizing radiation-elicited DNA repair. Bone Res. (2022) 10:19. doi: 10.1038/s41413-022-00194-0
106. Wang X, Zhu Y, Lu W, Guo X, Chen L, Zhang N, et al. Microcystin-LR-induced nuclear translocation of cGAS promotes mutagenesis in human hepatocytes by impeding homologous recombination repair. Toxicol Lett. (2023) 373:94–104. doi: 10.1016/j.toxlet.2022.11.015
107. Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. (2019) 38:e102718. doi: 10.15252/embj.2019102718
108. Zhen Z, Chen Y, Wang H, Tang H, Zhang H, Liu H, et al. Nuclear cGAS restricts L1 retrotransposition by promoting TRIM41-mediated ORF2p ubiquitination and degradation. Nat Commun. (2023) 14:8217. doi: 10.1038/s41467-023-43001-y
109. Kramerov DA, Vassetzky NS. Short retroposons in eukaryotic genomes. Int Rev Cytol. (2005) 247:165–221. doi: 10.1016/S0074-7696(05)47004-7
110. Brégnard C, Guerra J, Déjardin S, Passalacqua F, Benkirane M, Laguette N. Upregulated LINE-1 activity in the fanconi anemia cancer susceptibility syndrome leads to spontaneous pro-inflammatory cytokine production. EBioMedicine. (2016) 8:184–94. doi: 10.1016/j.ebiom.2016.05.005
111. Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr. (2015) 3:Mdna3–0061-2014. doi: 10.1128/microbiolspec.MDNA3-0061-2014
112. Feng Q, Moran JV, Kazazian HH Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. (1996) 87:905–16. doi: 10.1016/s0092-8674(00)81997-2
113. Farkash EA, Kao GD, Horman SR, Prak ET. Gamma radiation increases endonuclease-dependent L1 retrotransposition in a cultured cell assay. Nucleic Acids Res. (2006) 34:1196–204. doi: 10.1093/nar/gkj522
114. Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. (2016) 352:353–8. doi: 10.1126/science.aad7297
115. Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. (2016) 352:359–62. doi: 10.1126/science.aad7611
116. Sun Y, Li Q, Kirkland JL. Targeting senescent cells for a healthier longevity: the roadmap for an era of global aging. Life Med. (2022) 1:103–19. doi: 10.1093/lifemedi/lnac030
117. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
118. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
119. Murnane JP. Telomere dysfunction and chromosome instability. Mutat Res. (2012) 730:28–36. doi: 10.1016/j.mrfmmm.2011.04.008
120. Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. (2015) 15:276–89. doi: 10.1038/nrc3916
121. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. (2016) 14:282–97. doi: 10.1016/j.celrep.2015.12.029
122. Linares PM, Chaparro M, Gisbert JP. Angiopoietins in inflammation and their implication in the development of inflammatory bowel disease. A review. J Crohns Colitis. (2014) 8:183–90. doi: 10.1016/j.crohns.2013.06.013
123. Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. (2012) 209:1611–27. doi: 10.1084/jem.20111580
124. Luo J, Lu C, Chen Y, Wu X, Zhu C, Cui W, et al. Nuclear translocation of cGAS orchestrates VEGF-A-mediated angiogenesis. Cell Rep. (2023) 42:112328. doi: 10.1016/j.celrep.2023.112328
125. Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell. (2018) 175:488–501.e22. doi: 10.1038/s42003-022-03400-1
126. Wu Y, Song K, Hao W, Li J, Wang L, Li S. Nuclear soluble cGAS senses double-stranded DNA virus infection. Commun Biol. (2022) 5:433. doi: 10.1038/s42003-022-03400-1
127. Cui S, Yu Q, Chu L, Cui Y, Ding M, Wang Q, et al. Nuclear cGAS Functions Non-canonically to Enhance Antiviral Immunity via Recruiting Methyltransferase Prmt5. Cell Rep. (2020) 33:108490. doi: 10.1016/j.celrep.2020.108490
128. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
Keywords: cyclic GMP-AMP synthase (cGAS), nucleus, structure, activation, localization, functions
Citation: Lu Y, Zhao M, Chen L, Wang Y, Liu T and Liu H (2024) cGAS: action in the nucleus. Front. Immunol. 15:1380517. doi: 10.3389/fimmu.2024.1380517
Received: 01 February 2024; Accepted: 23 February 2024;
Published: 07 March 2024.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesCopyright © 2024 Lu, Zhao, Chen, Wang, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haipeng Liu, haipengliu@tongji.edu.cn
†These authors have contributed equally to this work and share first authorship