 Marianna Nicoletta Rossi1,2†
Marianna Nicoletta Rossi1,2† Valentina Matteo1†
Valentina Matteo1† Francesca Diomedi-Camassei3
Francesca Diomedi-Camassei3 Ester De Leo4Olivier Devuyst5Mohamed Lamkanfi6
Ester De Leo4Olivier Devuyst5Mohamed Lamkanfi6 Ivan Caiello1
Ivan Caiello1 Elena Loricchio1
Elena Loricchio1 Francesco Bellomo4Anna Taranta4Francesco Emma4
Francesco Bellomo4Anna Taranta4Francesco Emma4 Fabrizio De Benedetti1
Fabrizio De Benedetti1 Giusi Prencipe1*
Giusi Prencipe1*- 1Laboratory of Immuno-Rheumatology, Bambino Gesù Children’s Hospital, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Roma, Italy
- 2Department of Science, University of Rome “Roma Tre”, Rome, Italy
- 3Department of Laboratories, Pathology Unit, Bambino Gesù Children’s Hospital, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Roma, Italy
- 4Division of Nephrology, Bambino Gesù Children’s Hospital, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Roma, Italy
- 5Mechanisms of Inherited Kidney Disorders Group, Institute of Physiology, University of Zurich, Zurich, Switzerland
- 6Laboratory of Medical Immunology, Department of Internal Medicine and Paediatrics, Ghent University, Ghent, Belgium
Cystinosis is a rare autosomal recessive disorder caused by mutations in the CTNS gene that encodes cystinosin, a ubiquitous lysosomal cystine/H+ antiporter. The hallmark of the disease is progressive accumulation of cystine and cystine crystals in virtually all tissues. At the kidney level, human cystinosis is characterized by the development of renal Fanconi syndrome and progressive glomerular and interstitial damage leading to end-stage kidney disease in the second or third decade of life. The exact molecular mechanisms involved in the pathogenesis of renal disease in cystinosis are incompletely elucidated. We have previously shown upregulation of NLRP2 in human cystinotic proximal tubular epithelial cells and its role in promoting inflammatory and profibrotic responses. Herein, we have investigated the role of NLRP2 in vivo using a mouse model of cystinosis in which we have confirmed upregulation of Nlrp2 in the renal parenchyma. Our studies show that double knock out Ctns-/- Nlrp2-/- animals exhibit delayed development of Fanconi syndrome and kidney tissue damage. Specifically, we observed at 4-6 months of age that animals had less glucosuria and calciuria and markedly preserved renal tissue, as assessed by significantly lower levels of inflammatory cell infiltration, tubular atrophy, and interstitial fibrosis. Also, the mRNA expression of some inflammatory mediators (Cxcl1 and Saa1) and the rate of apoptosis were significantly decreased in 4-6-month old kidneys harvested from Ctns-/- Nlrp2-/- mice compared to those obtained from Ctns-/-mice. At 12-14 months of age, renal histological was markedly altered in both genetic models, although double KO animals had lower degree of polyuria and low molecular weight proteinuria and decreased mRNA expression levels of Il6 and Mcp1. Altogether, these data indicate that Nlrp2 is a potential pharmacological target for delaying progression of kidney disease in cystinosis.
Introduction
Cystinosis is a rare multisystemic autosomal recessive disorder caused by mutations in the CTNS gene, coding for the lysosomal cystine transporter cystinosin (1). At the kidney level, cystinosis is characterized by accumulation of intracellular cystine, formation of cystine crystals, proximal tubular dysfunction causing renal Fanconi syndrome, and progressive glomerular damage leading to kidney failure (2). Cysteamine therapy reduces lysosomal cystine accumulation and delays but does not prevent progression of kidney failure (3). To date, the exact molecular mechanisms involved in the development and progression of the disease remain incompletely understood. Beyond lysosomal cystine accumulation, other pathogenic mechanisms have been shown to play an important role in the pathogenesis of cystinosis. These include increased oxidative stress (4–6), mitochondrial dysfunction (7–9), enhanced apoptosis (10), and abnormal autophagy (5, 11, 12). Consistent with the increasing evidence showing a major role of inflammation in the development and progression of chronic kidney diseases (13, 14), several studies have shown that inflammation is also involved in the pathogenesis of kidney disease in cystinosis (15–18).
Previous results obtained by our group have pointed to a possible causative association between Nod-like receptor protein (NLRP)-mediated inflammation and progressive kidney damage in cystinosis (15). Specifically, we have shown that cystine crystals activate the inflammasome in monocytes (15); and that NLRP2 is markedly overexpressed in human cystinotic proximal tubular epithelial cells (PTEC) in a kidney biopsy obtained from a patient with cystinosis, in primary cultured PTEC and in immortalized PTEC cell lines derived from cystinotic patients (19). In PTEC, overexpression of NLRP2 upregulated proinflammatory and profibrotic as well as anti-apoptotic responses through the modulation of NF-κB activity (19). Our data were consistent with evidence demonstrating that increased renal NLRP2 expression levels are associated with acute tubular damage in human as well as in mice (20, 21).
Based on these findings, we hypothesized that upregulation of NLRP2 in cystinotic kidneys plays a role in the development of the interstitial inflammation and fibrosis, contributing to the progression to the end-stage kidney disease. In order to test our hypothesis and explore the pathogenic role of NLRP2 in vivo, we analyzed Nlrp2 expression in the Ctns-/- mouse model, and generated a double knock out (KO) Ctns-/- Nlrp2-/- mouse model by crossing cystinotic mice with Nlrp2-/- mice.
Materials and methods
Generation of Ctns-/-Nlrp2-/- double-knockout mice, animal care and procedures
The C57BL/6 Ctns-/- mice were kindly provided by Prof. Corinne Antignac (22). C57BL/6 Nlrp2 -/- mice have been described (23). Animal care and experimental procedures were conducted in accordance with the European 2010/63/EU directive on the protection of animals used for scientific purposes and authorized by the Italian Ministry of Health (authorization number 898/2017-PR). Ctns-/- Nlrp2-/- double-knockout mice were generated first by crossing single knockout mice to produce compound heterozygotes and then by crossing compound heterozygous mice. Genotyping of mice was carried out as previously described (23, 24). Female Ctns-/- and Ctns-/- Nlrp2-/- mice (n= 7 animals per group) were followed longitudinally from birth; urine samples were collected every two months, starting from the fourth month of age. Mice were then sacrificed at 6, 12 and 14 months of age. At sacrifice, kidneys were rapidly dissected and snap-frozen or processed in paraffin for further analysis.
For urine collection, mice were acclimatized in metabolic cages for 24 hours before collecting urine in the following 24 hours, in the presence of 0.1% sodium azide and 1X protease inhibitors (Thermo Scientific). Mouse weight was recorded before entering the metabolic cages. Urine analyses were performed by the veterinary laboratory Appialab Srl (Rome, Italy). Urinary glucose (mg/dL), albumin (mg/L), calcium (mg/dL), and creatinine (mg/dL) were measured with the use of HITACHI Cobas C311 (Roche Diagnostics GmbH, Mannheim, Germany). Clara cell protein 16 (CC16) urinary levels were measured using the Immunosorbent Assay kit in accordance with manufacturer’s instructions (Biomatik, Wilmington, DE).
Isolation and primary cultures of proximal tubule cells was performed as previously described (25).
RNA isolation and quantitative real-time PCR
Total RNA was extracted from mouse kidney tissues using Trizol reagent (Ambion), and cDNA was obtained using the Superscript Vilo kit (Invitrogen). Real-time PCR assays were performed using TaqMan Universal PCR Master mix (Applied Biosystems) and the following gene expression assays: mouse Cxcl1, Cxcl5, Il6, Ccl2, Saa1. Gene expression data were normalized using mouse Hprt1 (Applied Biosystems) as endogenous controls. Data are expressed as arbitrary units (AU), determined using the 2−Δct method. Sybr green (Roche) was utilised to analyse Nlrp2 expression. Primer sequences are available on request.
Tissue histology
At sacrifice, kidneys were fixed overnight in 4% paraformaldehyde and then embedded in paraffin. Three-μm thick sections were obtained and stained with hematoxylin-eosin and Masson’s trichrome staining. Renal sections were scored blindly by Dr. Francesca Diomedi Camassei on a scale ranking 0 to 3+, to estimate the severity of tubulointerstitial changes. The following parameters were scored: immune cell infiltration, tubular atrophy, interstitial fibrosis, medullary tubular casts, cortical tubular casts, and glomerulosclerosis. A minimum of 10 fields for each kidney slide were examined. A global score was then calculated for each kidney by addition of all scores. For evaluating renal parenchyma damage, the scoring system was extrapolated from the Banff scoring system (26) and modified adding a category for very focal alterations. In our score, we considered 5 categories (instead of 4 in the Banff system): 0, no alterations (0%); 0.5, very focal alterations (1-10%); 1, mild alterations (11-25%); 2, moderate alterations (26-50%); 3, diffuse alterations (>50%).
Evaluation of apoptosis in kidney tissue
Immunohistochemistry was performed on 2 μm thick sections obtained from formalin-fixed tissue embedded in paraffin. After dewaxing and rehydrating, heat-induced epitope retrieval was performed by boiling the slides with sodium citrate (pH 6) (Dako, Glostrup, Denmark). Endogenous peroxidase was blocked with 3% hydrogen peroxide and then with 5% bovine serum albumin (BSA). Sections were incubated overnight at 4°C with rabbit monoclonal Cleaved Caspase-3 antibody (1:300) (9661S, Cell Signaling Technology, Danvers, MA, USA). Detection of the primary antibody was performed by using the appropriate secondary biotinylated antibody (K8024) (ready to use) (Dako, Carpinteria, CA, USA) and the peroxidase DAB kit (Dako, Carpinteria, CA, USA). Counterstaining was performed with hematoxylin solution Gill2.
The entire slides were scanned and captured by NanoZoomer S60 Digital scanner, 5 random 20X fields of the subcapsular cortex for each mouse were selected using the NDP.view2 Image viewing software. The mean intensity of caspase 3 staining was measured by Image J software.
Statistical analyses
Data are presented as mean ± SDs or as median and IQRs. Pairwise comparisons were evaluated by the Mann-Whitney U test. Group comparisons were performed by using 2-way Anova, followed by Tukey’s multiple comparison test.
All statistical analyses were performed using GraphPad Prism IX software. P-values lower than 0.05 were considered statistically significant.
Results
Cystinotic kidneys show increased inflammation markers
We previously demonstrated that the NOD like receptor protein 2 (NLRP2) is overexpressed in human cystinotic PTEC and plays a role in regulating proinflammatory and profibrotic responses. To extend our previous findings and investigate the in vivo role of NLRP2 in the pathogenesis of cystinosis, we first analysed the mRNA expression levels of Nlrp2 in whole kidney lysates obtained from Ctns-/- and wild type (WT) mice at different ages. We found that Nlrp2 expression levels were significantly higher in Ctns-/- kidneys compared to WT kidneys, both in the early (3-8 months of age) and in the late stage (10-16 months of age) of renal disease (Figure 1A). Accordingly, Nlrp2 mRNA expression levels were significantly increased in purified primary PTECs isolated from Ctns-/- mice at 6 months of age, compared to PTECs from WT mice (Figure 1B). In addition, we analysed the expression of chemokines/cytokines that we previously demonstrated to be modulated by NLRP2 in human cells, including C-X-C motif chemokine ligand-1 (Cxcl1), Cxcl5 and interleukin-6 (Il6). We observed significantly higher mRNA levels of Cxcl1 and Cxcl5 and a trend towards higher Il6 levels (p=0.056) in Ctns-/- compared to WT kidneys collected at 3-8 months of age, when renal damage begins to develop in Ctns-/- animals (Figure 1C). At 10-16 months of age, when overt kidney disease is established, Ctns-/- kidneys showed significant upregulation of all analysed inflammatory mediators, when compared to WT kidneys (Figures 1C–E). Collectively, these results confirmed that Nlrp2 expression is upregulated in vivo in total kidneys and in PTEC from cystinotic mice and that expression of some inflammatory chemokines/cytokines is increased in the kidney of cystinotic mice.
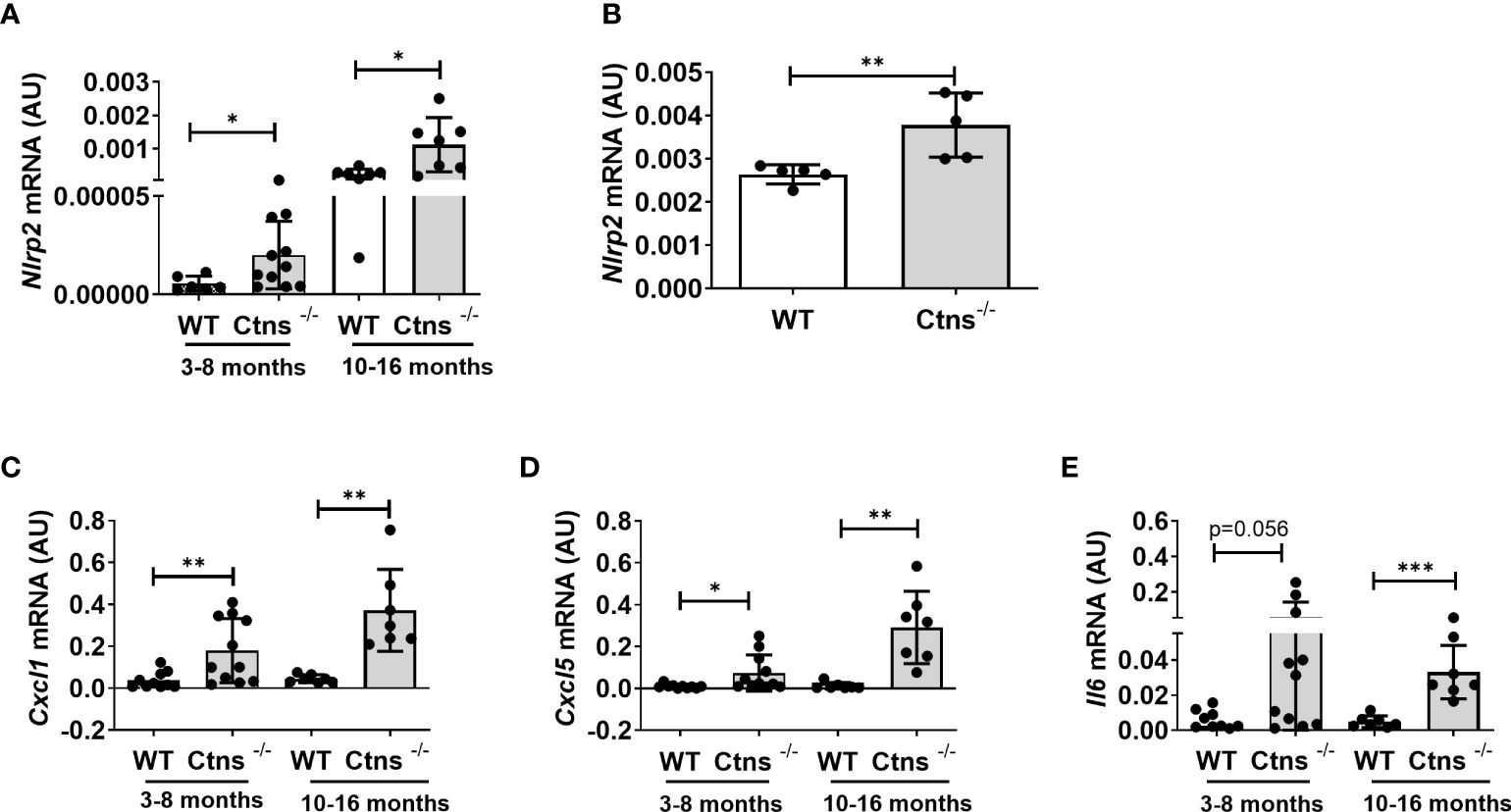
Figure 1 Nlrp2 and inflammation markers are increased in cystinotic kidneys. (A) Nlrp2 mRNA levels were evaluated by qPCR analysis in total kidneys from wild type (WT) and cystinotic (Ctns-/-) mice at 3-8 and 10-16 months of age and (B) in primary proximal tubular epithelial cells isolated from WT and Ctns-/- mice at 6 months of age. (C–E) Cxcl1, Cxcl5 and Il6 mRNA expression levels were evaluated by qPCR analysis in total kidneys from WT and Ctns-/) mice at 3-8 and 10-16 months of age. (A–E) Results were obtained after normalization with the housekeeping genes Hprt1 and are expressed as arbitrary units (AU). Differences between WT and Ctns-/- mice were compared using the Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001.
Genetic deletion of Nlrp2 partially ameliorates kidney functions in cystinotic mice
To investigate the in vivo role of Nlrp2 in the progression of cystinotic disease, we generated a double KO mouse model by crossing C57BL/6 Ctns-/- mice with C57BL/6 Nlrp2-/- mice. Double KO animals were vital and fertile and had normal growth and lifespan. Until 10 months of age double KO mice showed a significantly greater body weight compared to Ctns-/- mice (Figure 2). Unlike Ctns-/- mice that, as expected (22, 27), weighted significantly less than WT animals at 12-14 months of age, Ctns-/- Nlrp2-/- mice weighted similarly to WT mice at these same ages (Figure 2).
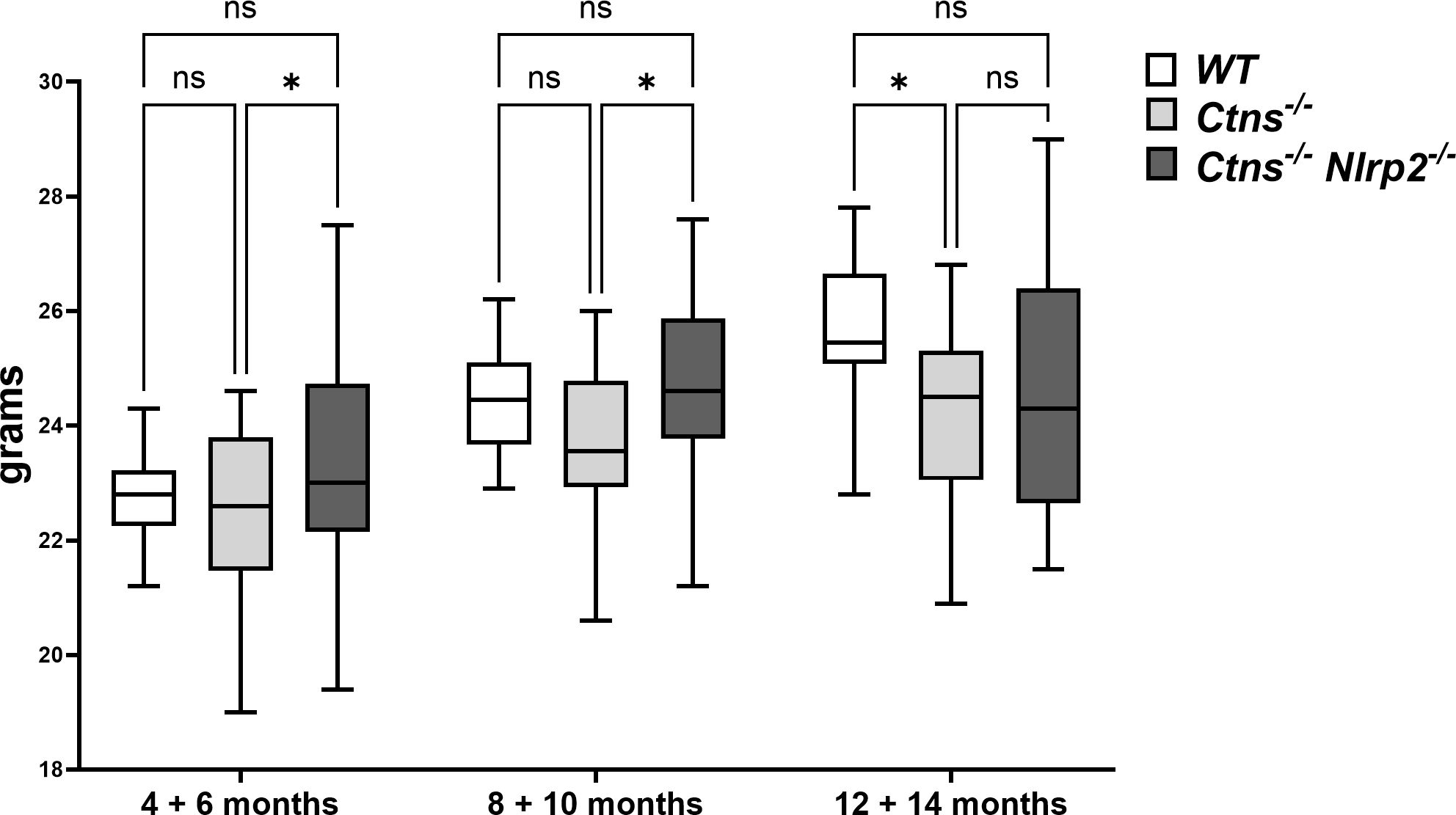
Figure 2 WT, Ctns-/- and Ctns-/- Nlrp2-/- mice body weight. WT, Ctns−/− and Ctns-/- Nlrp2-/- mice were weighed every two months from 4 to 14 months of age. Differences between groups were compared using 2-way Anova, followed by Tukey’s multiple comparison test. *p < 0.05; ns, not significant.
To investigate the development and progression of kidney disease, urine samples from Ctns-/- mice and Ctns-/- Nlrp2-/- double KO mice were collected every two months, starting from 4 and up to 14 months of age. We evaluated urinary levels of glucose, calcium, albumin, and the low molecular weight protein Clara cell protein 16 (CC16), a marker of proximal tubular renal proximal dysfunction (28), and measured urine volumes. Results are shown in Figure 3. As expected, Ctns-/- animals developed kidney disease around 4-6 months of age. In this experimental model of cystinosis, glucosuria peaks around 10 months of age and decreases thereafter; the reason for this biphasic evolution being unclear. When comparing Ctns-/- to Ctns-/- Nlrp2-/- mice, the onset of glucosuria and calciuria was delayed in the double KO animals (i.e. significant differences were observed at 4-6 months of age) (Figures 3A, B). Low molecular weight (LMW) proteinuria and polyuria were significantly lower in older animals (Figures 3D, E), whereas albuminuria was lower only at 8-10 months of age. Collectively, these results indicate that albeit the invalidation of the Nlrp2 gene cannot completely rescue the Fanconi syndrome, it does result in partial improvement.
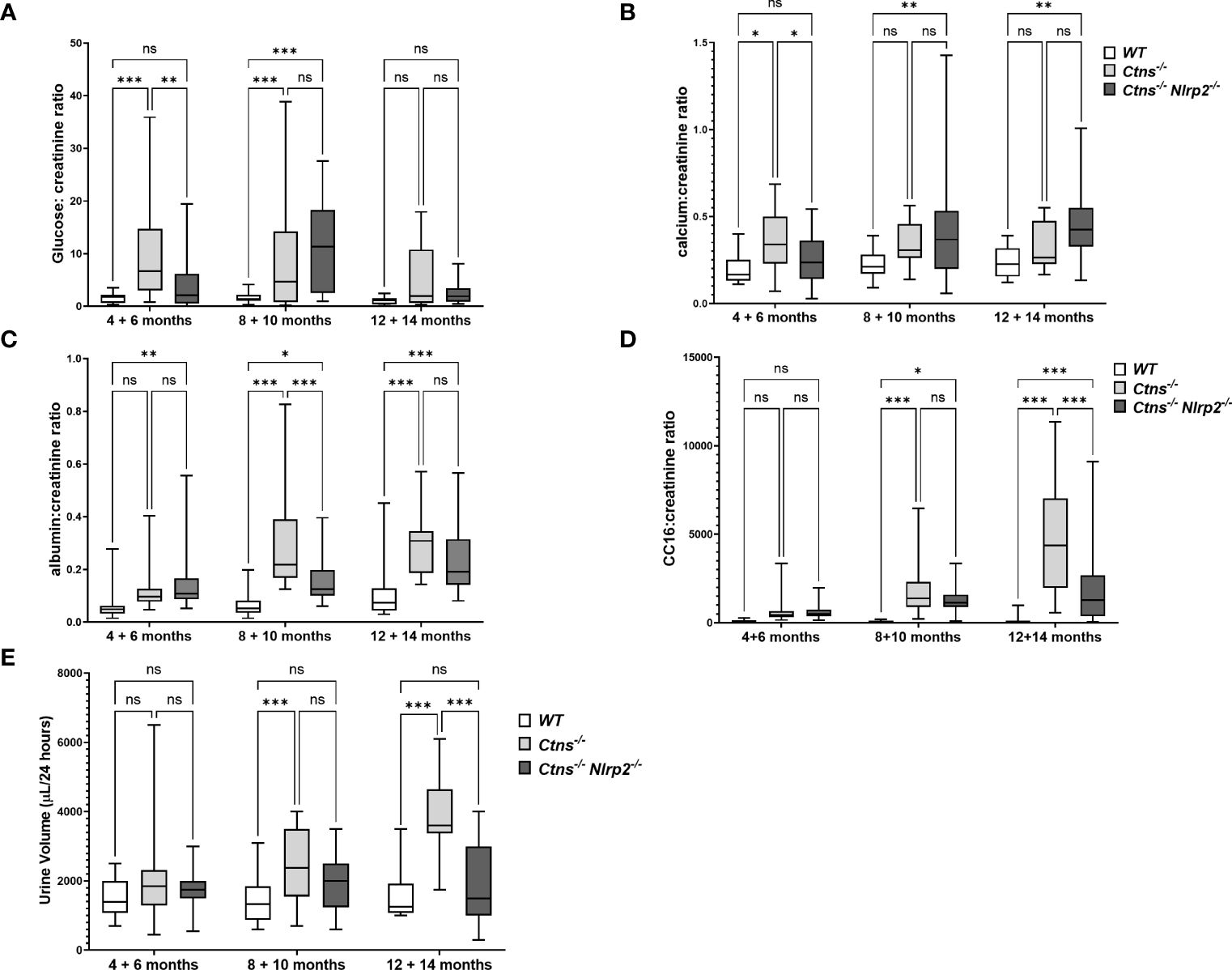
Figure 3 Evaluation of parameters of kidney function. (A–D) Glucose, calcium, albumin and Clara cell protein 16 (CC16) urinary levels were measured in WT, Ctns−/− and Ctns-/- Nlrp2-/- mice from 4 to 14 months of age and expressed as ratio against the corresponding creatinine levels. (E) 24h urine volume (expressed as µl/24 hours) were measured in WT, Ctns−/− and Ctns-/- Nlrp2-/- mice from 4 to 14 months of age. Differences between groups were compared using 2-way Anova, followed by Tukey’s multiple comparison test. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
Genetic deletion of Nlrp2 delays the appearance of histological markers of kidney damage
Kidney samples were examined at 6, 12 and 14 months of age for the presence of renal lesions, including immune cell infiltration, tubular atrophy, interstitial fibrosis, medullary tubular dysfunction casts, cortical tubular casts and glomerulosclerosis. Our results show a significant delay in the development of kidney lesions in double KO animals with highly significant differences observed in samples obtained from animals sacrificed at 6 months, whereas no significant differences being observed at later ages.
As expected, Ctns-/- mice showed evidence of kidney tissue damage at 6 months of age (Figures 4A–G). By contrast, double KO mice showed a marked attenuation of tubular atrophy (p=0.029) and interstitial fibrosis (p= 0.025), and significantly less cortical tubular casts (p=0.021), (Figures 4A–G). Inflammatory infiltrates were almost absent in the double KO animals (1/7 vs. 6/7 - p=0.029, Fisher’s exact test) (Figure 4G). The global kidney damage score confirms that genetic invalidation of Nlrp2 protected against the early development of kidney parenchymal lesions (p=0.009), (Figure 4H). The apoptosis rate was assessed by immunohistochemical staining for cleaved Caspase-3 and was also significantly decreased in double KO mice (p<0.001), (Figures 5A, B). By contrast, analyses of Ctns-/- and Ctns-/- Nlrp2-/- kidneys collected at 12 and 14 months of age showed severe renal lesions in all samples, without significant differences between the two genetic models (Figure 6A). The global kidney damage score was significantly increased compared to 6-month old animals and was not attenuated by Nlrp2 invalidation (Figure 6B).
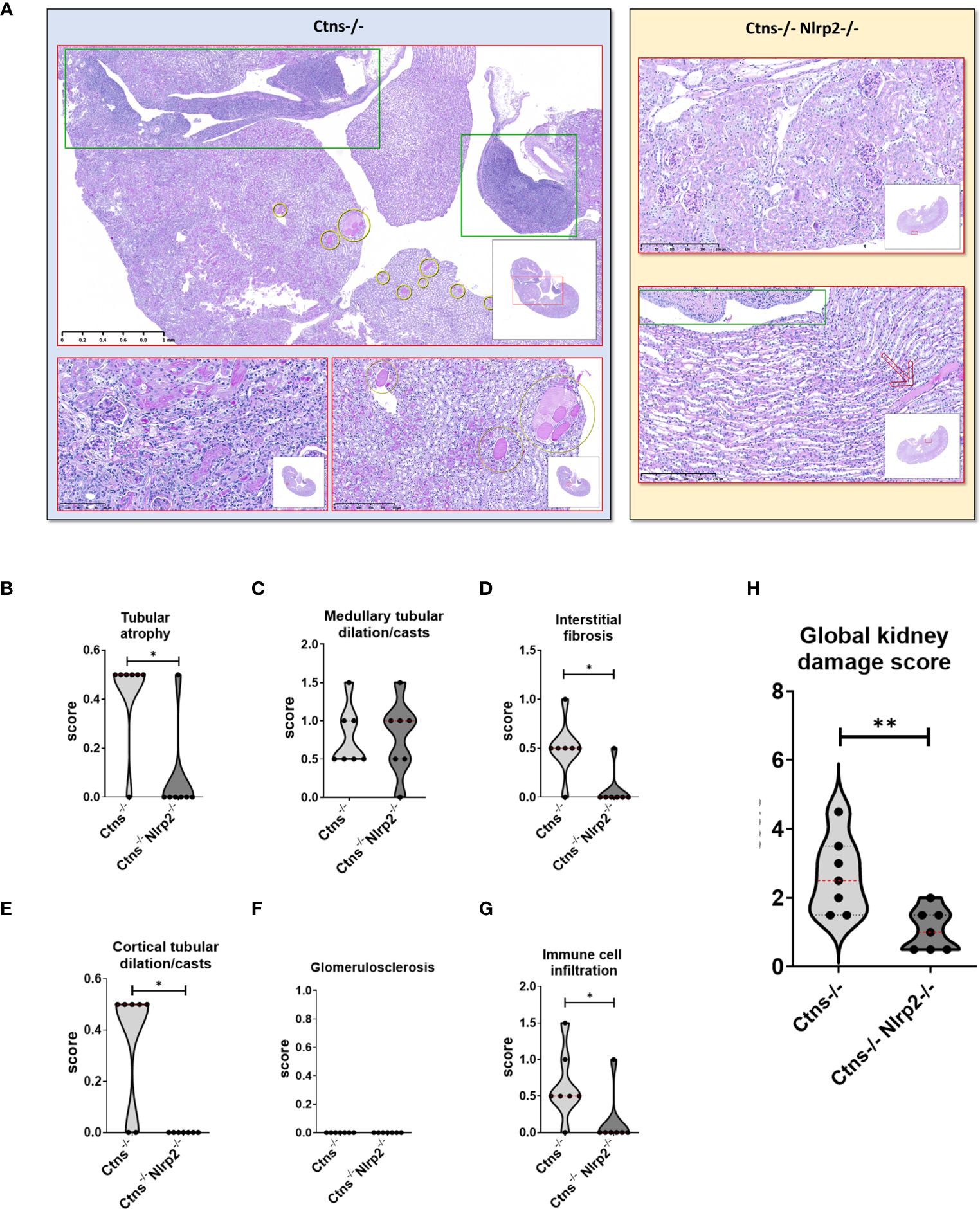
Figure 4 Renal histologic features of 6 months old Ctns-/- and Ctns-/- Nlrp2-/- mice. (A) PAS stain: Renal histologic features of 6 months old Ctns−/− (left panel) and Ctns-/- Nlrp2-/- (right panel) mice. In the left panel, the Ctns−/− mice renal parenchyma: marked interstitial inflammation, especially in the medulla and renal pelvis (green squares) was observed; numerous medullary casts were present (yellow circles). At higher magnification, interstitial inflammation extended in the cortex where mild fibrosis, tubular atrophy and small hyaline casts were also evident (lower left). Medullary casts resulted unevenly distributed and showed variable size (lower right). In the right panel, Ctns-/- Nlrp2-/- mice renal parenchyma: normal glomeruli were present the cortex and no inflammation, tubular atrophy or fibrosis were observed (up). Medulla showed preserved tubular structure and distribution. Renal pelvis was not inflamed (green square). Very occasional medullary casts were present (red arrow). Renal sections were scored using a scale ranging from 0 to 3 (0= absent damage - 3= very marked damage); tubular atrophy (B), medullary tubular dysfunction casts (C), interstitial fibrosis (D), cortical tubular casts (E), glomerulosclerosis (F) and immune cell infiltration (G) were evaluated. (H) The sum of all scores were used to calculate the global kidney damage score. Differences between Ctns-/- and Ctns-/- Nlrp2-/- mice were compared using the Mann-Whitney U test *p < 0.05; **p < 0.01.
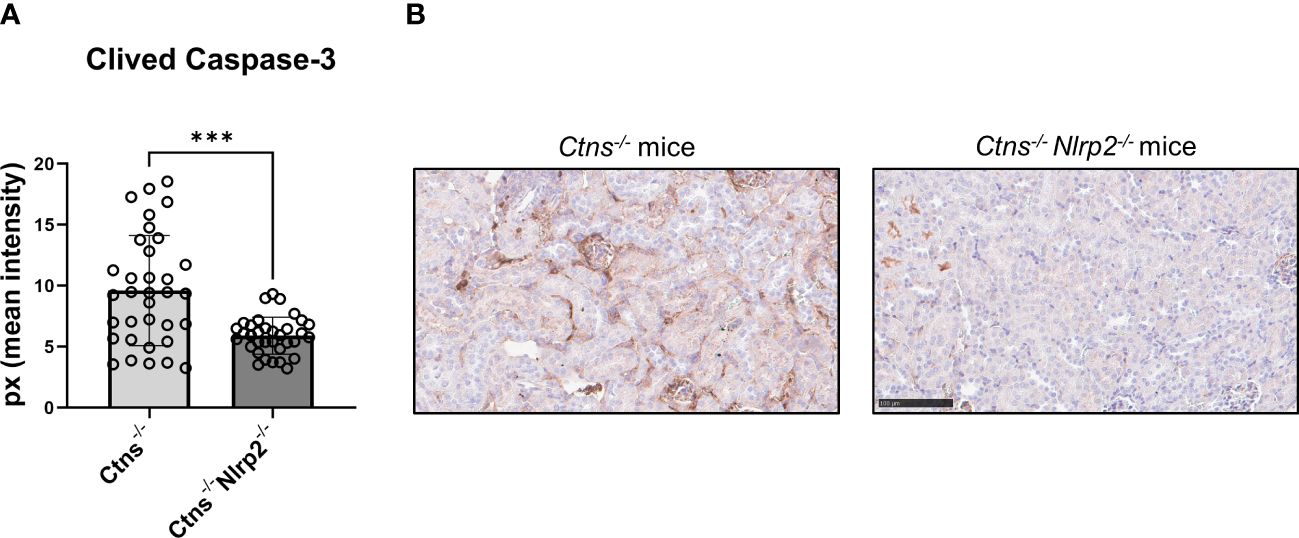
Figure 5 Apoptosis rate in kidney of Ctns-/- and Ctns-/- Nlrp2-/- mice. (A) Immunohistochemistry of cleaved Caspase-3 was performed in kidney sections from Ctns-/- (n= 7) and Ctns-/- Nlrp2-/- (n= 7) mice at 6 months of age. For each section, 5 fields were acquired. Total staining intensity were analysed with imageJ Software and reported as pixel mean intensity. (B) Representative image of kidney sections from Ctns-/- (left) and Ctns-/- Nlrp2-/- (right). Scale bar=100μm. Differences between WT and Ctns-/- mice were compared using the Mann-Whitney U test. ***p < 0.001.
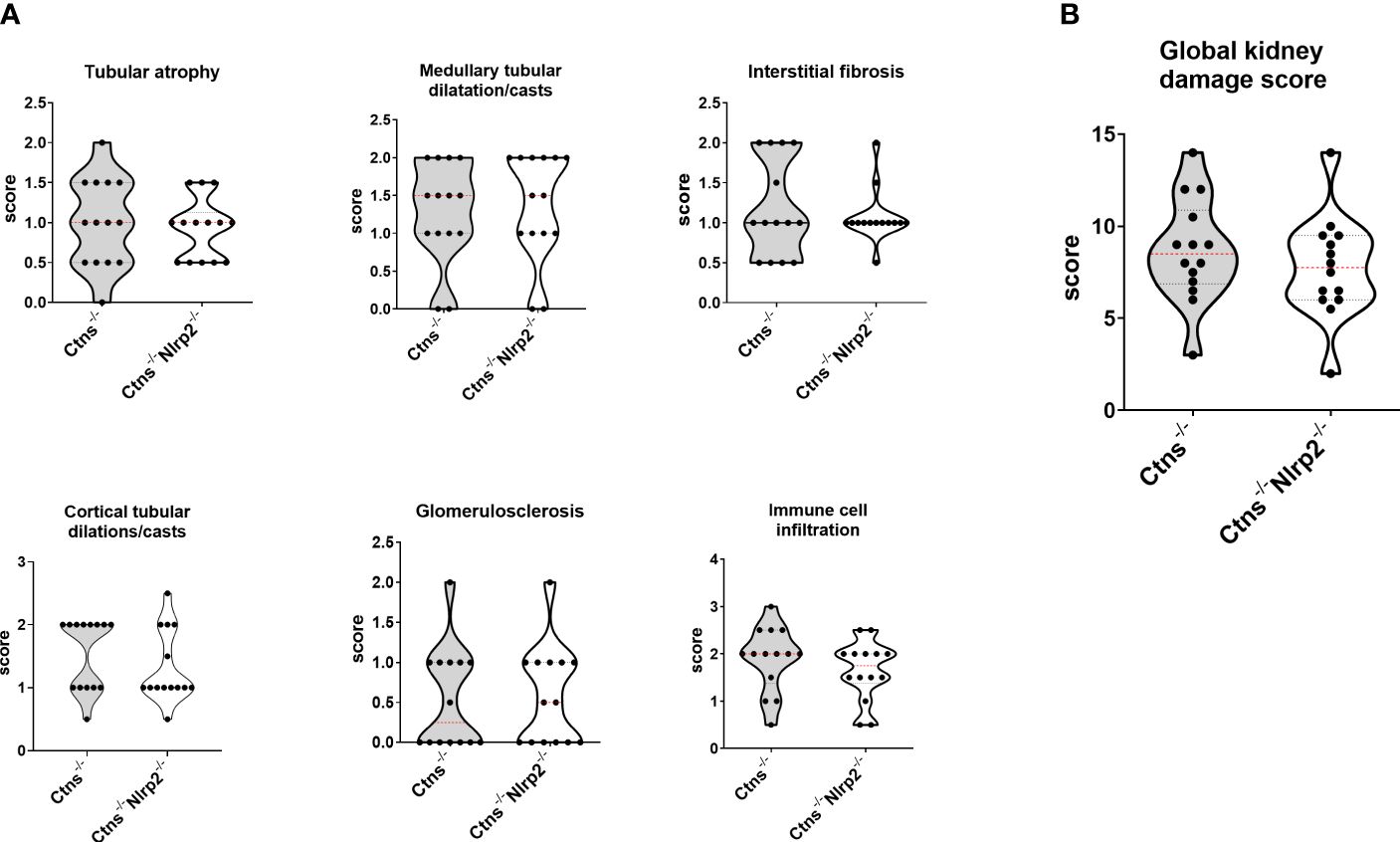
Figure 6 Renal histologic features of 12/14 old months Ctns-/- and Ctns-/- Nlrp2-/- mice. (A) Kidney sections from Ctns-/-and Ctns-/- Nlrp2-/- mice at 12/14 months of age were stained with haematoxylin-eosin and Masson’s trichrome staining. Renal sections were scored using a scale ranging from 0 to 3, (0= absent damage - 3= very marked damage), tubular atrophy, medullary tubular dysfunction casts, interstitial fibrosis, cortical tubular casts, glomerulosclerosis and immune cell infiltration were evaluated. (B) The sum of all scores were used to calculate the global kidney damage score. Differences between Ctns-/- and Ctns-/- Nlrp2-/- mice were compared using the Mann-Whitney U test.
Lastly, we evaluated in whole kidney lysates the mRNA expression levels of proinflammatory mediators, including Cxcl1, Il6, monocyte chemoattractant protein-1 (Mcp1), and serum amyloid A (Saa1) in 6- and 12/14-months old samples. Cxcl1 and Saa1 mRNA levels were significantly lower in Ctns-/- Nlrp2-/- mice at 6 months of age compared to Ctns-/- mice (Figures 7A, B), while comparable levels of Il6 and Mcp1 were observed (Figures 7C, D). At 12-14 months, the mRNA expression of Il6 and Mcp1 was attenuated in the double KO animals (Figures 7A, B), whereas expression of Cxcl1 and Saa1 increased to similar levels (Figures 7C, D).
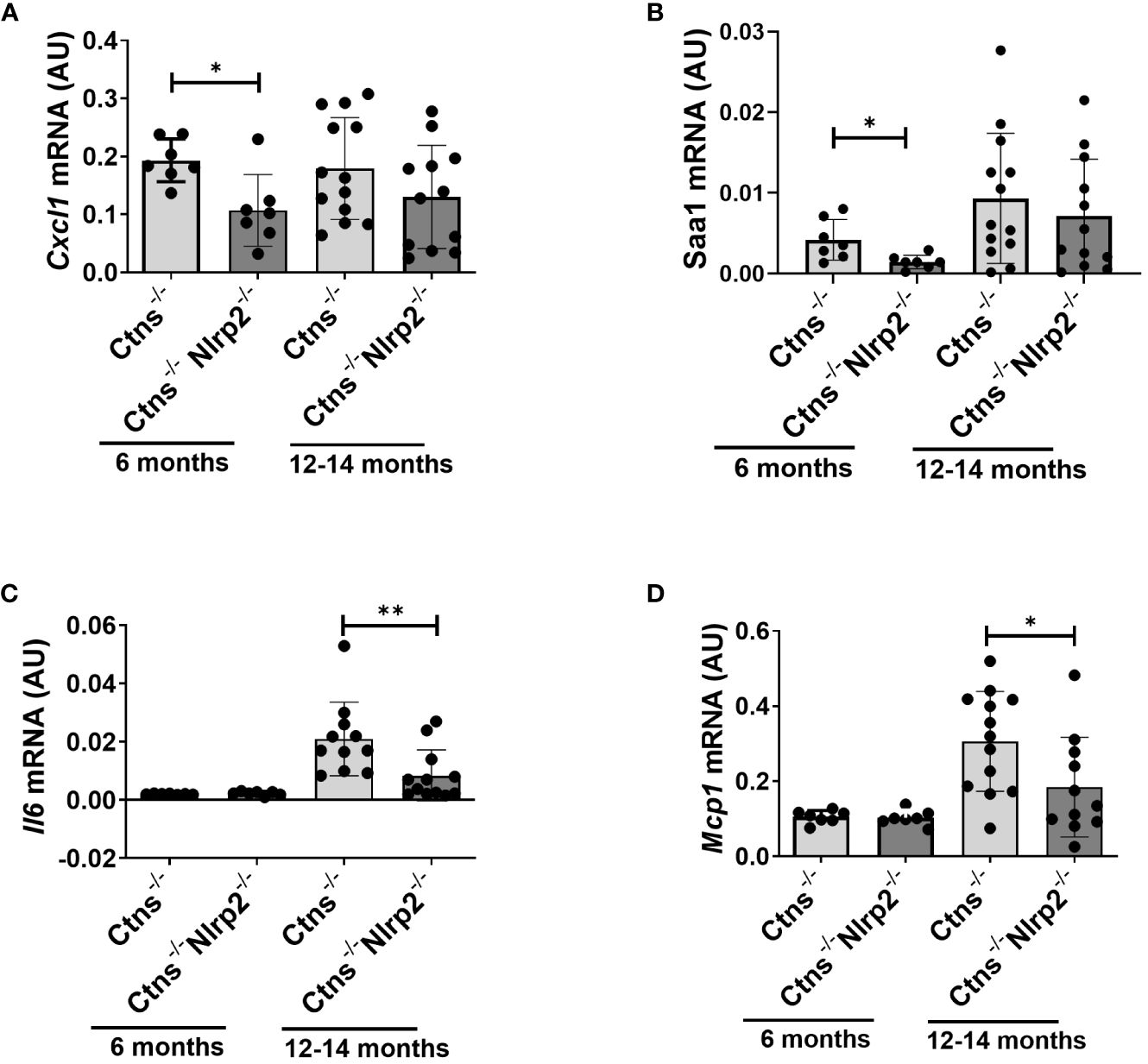
Figure 7 Inflammatory markers are decreased in kidney of Ctns-/- Nlrp2-/- mice. (A–D) Cxcl1, Saa1 Il6, and Mcp1, mRNA expression levels were evaluated by qPCR analysis in total kidneys from Ctns-/- and Ctns-/- Nlrp2-/- mice at 6 and 12-14 months of age. Results are obtained after normalization with the housekeeping gene Hprt1 and are expressed as arbitrary units (AU). Differences between Ctns-/- and Ctns-/- Nlrp2-/- mice were compared using the Mann-Whitney U test. *p < 0.05; **p < 0.01.
Taken together, results of this study indicate that the genetic ablation of the Nlrp2 gene delays the onset of kidney disease in Ctns-/- mice by reducing inflammatory infiltration, interstitial fibrosis, tubular lesions and apoptosis.
Discussion
We have previously reported that the NOD-like receptor protein NLRP2 is upregulated in a human cystinotic kidney and in PTECs, and that stable exogenous upregulation of NLRP2 in control PTEC induces proinflammatory and profibrotic responses (19). In the present study, we showed that Nlrp2 is upregulated in kidney samples and PTECs obtained from the C57BL/6 Ctns-/- mouse model. Consistent with previous findings (15, 17, 22), we have confirmed upregulation of several inflammatory mediators, including those modulated by NLRP2, namely Il6, Cxcl1 and Cxcl5 at early and late stages of the kidney disease.
Building on these findings, we next generated Ctns-/- Nlrp2-/- double KO mice to dissect the contribution of Nlrp2 in the development and progression of kidney disease in cystinosis. We observed that although genetic ablation of Nlrp2 did not abolish the development of Fanconi syndrome or renal parenchymal lesions, it did delay it. When comparing animals analysed at 4-6 months, double KO animals showed less glucosuria and calciuria, and later in life less polyuria and LMW proteinuria. The urinary biochemical analyses were supported by the histological data showing markedly preserved renal parenchymal tissue at 6 months of age, including a lower degree of inflammatory cell infiltration, tubular atrophy, cortical and tubular dilatations/casts and interstitial fibrosis. These findings were further supported by decreased expression of some inflammatory mediators, such as Cxcl1 and Saa1, and a lower apoptosis rate in kidney samples of Ctns-/- Nlrp2-/- double KO mice obtained at 6 months of age. However, when we analysed animals at 12/14 months of age, the severity of histological lesions increased to the same extent in all animals, despite double KO animals exhibiting less polyuria and LMW proteinuria and displaying lower mRNA expression of Il6 and Mcp1. Altogether, these results demonstrate a role for Nlrp2 in the progression of kidney disease in the Ctns-/- mouse model, and show that inhibition of this NOD-like receptor family member can partially delay disease onset.
The mode of action through which Nlrp2 deficiency induces these effects has not been investigated at the molecular level. However, the absence of significant inflammatory infiltrates and interstitial fibrosis in most 6-months old double KO animals strongly suggests that Nlrp2 is involved in modulating inflammatory and profibrotic responses in the kidneys of Ctns-/- mice at the early stages of the disease. These in vivo observations are in full agreement with our previous in vitro data, demonstrating in PTEC that NLRP2 overexpression upregulates the expression and production of many proinflammatory and profibrotic genes by modulating the activity of the transcription factor NF-κB (19). Indeed, it has been widely demonstrated that the local production of inflammatory mediators by hematopoietic and non-hematopoietic cells, including Cxcl1, Saa1, Il6 and Mcp1, which we found modulated in cystinotic kidney by Nlrp2 deletion, plays an important role in promoting immune and inflammatory responses which, in turn, may contribute to the exacerbation of kidney damage (29, 30). Contrary to results obtained in vitro showing a role for NLRP2 in promoting anti-apoptotic responses in PTECs, Nlrp2 deficiency in cystinotic mice induced a significant decrease in renal apoptotic rate, as assessed by cleaved Caspase-3 staining. In the literature, NLRP2 has been shown to either induce or decrease apoptosis, depending on cell or tissue type and on the model source (human or mouse) (21, 31–33). Therefore, the discrepancy that we observed could be related to a different behaviour of NLRP2 in mice compared to humans or in vitro compared to in vivo. However, recently Nlrp2 has been shown to have other functions, in addition to regulating NF-κB and apoptotic processes, including initiating inflammasome activation (34), modulating anti-viral responses by binding TBK-1 (35), and modulating DNA methylation at imprinted loci (36, 37). We cannot therefore exclude that Nlrp2 may regulate additional pathways involved in the progression of cystinosis; further studies are needed to better understand the precise role of Nlrp2 in renal pathophysiology and specifically in cystinosis.
In conclusion, our data add to the growing body of evidence demonstrating a role for inflammation in the development and progression of kidney disease in cystinosis (38). The activation of multiple inflammatory pathways and their interplay with other mechanisms involved in the pathogenesis of cystinosis, including increased oxidative stress, autophagy, mechanisms of cell death, and tissue fibrosis, have been reported (15–17, 19). Accordingly, treatments of cystinotic mice with drugs that have a primary or secondary anti-inflammatory effect, such as genistein, the IL-1 blocking agent anakinra or rapamycin, have been demonstrated to improve the cystinotic phenotype in experimental models (12, 18, 39, 40). Cysteamine per se, in addition to promoting cystine efflux from lysosomes, has been shown to exert anti-oxidative and anti-inflammatory effects (41–44). However, these therapeutic interventions cannot completely rescue the renal phenotype, but only ameliorate kidney disease, similarly to the genetic invalidation of Nlrp2. These results highlight the complexity of the physiopathology of cell dysfunction in cystinosis and the multiplicity of activities that the cystinosin protein plays in cells, beyond its function as a cystine/H+ symporter.
A limitation of our study was the use of a whole-body double KO model for Ctns and Nlrp2. Therefore, we cannot exclude that amelioration/delay in kidney damage we documented in Ctns-/- Nlrp2-/- mice are indirect as changes in other tissues, or cells other than PTEC, may have an effect on kidney disease progression. Generation of a PTEC-specific Nlrp2 knockout mouse would have allowed us to understand the exact role of Nlrp2 in PTEC and in the pathogenesis and progression of kidney disease in cystinosis.
In conclusion, our data show a partially protective effect of Nlrp2 inhibition in the cystinotic mouse model and provide additional rationale for testing anti-inflammatory strategies in addition to cysteamine to delay progression of kidney disease in patients with cystinosis.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal studies were approved by Animal care and experimental procedures were conducted in accordance with the European 2010/63/EU directive on the protection of animals used for scientific purposes and authorized by the Italian Ministry of Health (authorization number 898/2017-PR). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
MNR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. VM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. FD-C: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. EDL: Data curation, Investigation, Software, Writing – original draft, Writing – review & editing. OD: Data curation, Investigation, Writing – original draft, Writing – review & editing. ML: Investigation, Methodology, Writing – original draft, Writing – review & editing. IC: Data curation, Methodology, Writing – original draft, Writing – review & editing. EL: Investigation, Methodology, Writing – original draft, Writing – review & editing. FB: Data curation, Methodology, Writing – original draft, Writing – review & editing. AT: Data curation, Methodology, Writing – original draft, Writing – review & editing. FE: Investigation, Writing – original draft, Writing – review & editing, Data curation. FDB: Investigation, Methodology, Writing – original draft, Writing – review & editing. GP: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Cystinosis Research Foundation (Grant CRFF-2020-003 to GP) and by the University Research Priority Program (URPP) ITINERARE of the University of Zurich (to OD). This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Acknowledgments
We thank Prof. Corinne Antignac for kindly providing the Ctns−/− mouse model and Mrs. Huguette Debaix (UZH) for technical help.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. (2001) 20:5940–9. doi: 10.1093/emboj/20.21.5940
2. Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. (2017) 13:115–31. doi: 10.1038/nrneph.2016.182
3. Cherqui S. [Cystinosis: From the gene identification to the first gene therapy clinical trial]. Med sci: M/S. (2023) 39:253–61. doi: 10.1051/medsci/2023025
4. Sumayao R Jr., Newsholme P, McMorrow T. The role of cystinosin in the intermediary thiol metabolism and redox homeostasis in kidney proximal tubular cells. Antioxid (Basel Switzerland). (2018) 7:179. doi: 10.3390/antiox7120179
5. Festa BP, Chen Z, Berquez M, Debaix H, Tokonami N, Prange JA, et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat Commun. (2018) 9:161. doi: 10.1038/s41467-017-02536-7
6. Okamura DM, Bahrami NM, Ren S, Pasichnyk K, Williams JM, Gangoiti JA, et al. Cysteamine modulates oxidative stress and blocks myofibroblast activity in CKD. J Am Soc Nephrol. (2014) 25:43–54. doi: 10.1681/ASN.2012090962
7. Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ, et al. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol. (2010) 21:272–83. doi: 10.1681/ASN.2009040383
8. Bellomo F, Signorile A, Tamma G, Ranieri M, Emma F, De Rasmo D. Impact of atypical mitochondrial cyclic-AMP level in nephropathic cystinosis. Cell Mol Life sci: CMLS. (2018) 75:3411–22. doi: 10.1007/s00018-018-2800-5
9. De Rasmo D, Signorile A, De Leo E, Polishchuk EV, Ferretta A, Raso R, et al. Mitochondrial dynamics of proximal tubular epithelial cells in nephropathic cystinosis. Int J Mol Sci. (2019) 21:192. doi: 10.3390/ijms21010192
10. Ames EG, Thoene JG. Programmed cell death in cystinosis. Cells. (2022) 11:670. doi: 10.3390/cells11040670
11. Napolitano G, Johnson JL, He J, Rocca CJ, Monfregola J, Pestonjamasp K, et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med. (2015) 7:158–74. doi: 10.15252/emmm.201404223
12. Berquez M, Chen Z, Festa BP, Krohn P, Keller SA, Parolo S, et al. Lysosomal cystine export regulates mTORC1 signaling to guide kidney epithelial cell fate specialization. Nat Commun. (2023) 14:3994. doi: 10.1038/s41467-023-39261-3
13. Mulay SR. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. (2019) 96:58–66. doi: 10.1016/j.kint.2019.01.014
14. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. (2019) 15:144–58. doi: 10.1038/s41581-019-0110-2
15. Prencipe G, Caiello I, Cherqui S, Whisenant T, Petrini S, Emma F, et al. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol. (2014) 25:1163–9. doi: 10.1681/ASN.2013060653
16. Veys KRP, Elmonem MA, Van Dyck M, Janssen MC, Cornelissen EAM, Hohenfellner K, et al. Chitotriosidase as a novel biomarker for therapeutic monitoring of nephropathic cystinosis. J Am Soc Nephrol. (2020) 31:1092–106. doi: 10.1681/ASN.2019080774
17. Lobry T, Miller R, Nevo N, Rocca CJ, Zhang J, Catz SD, et al. Interaction between galectin-3 and cystinosin uncovers a pathogenic role of inflammation in kidney involvement of cystinosis. Kidney Int. (2019) 96:350–62. doi: 10.1016/j.kint.2019.01.029
18. Cheung WW, Hao S, Zheng R, Wang Z, Gonzalez A, Zhou P, et al. Targeting interleukin-1 for reversing fat browning and muscle wasting in infantile nephropathic cystinosis. J cachexia sarcopenia Muscle. (2021) 12:1296–311. doi: 10.1002/jcsm.12744
19. Rossi MN, Pascarella A, Licursi V, Caiello I, Taranta A, Rega LR, et al. NLRP2 regulates proinflammatory and antiapoptotic responses in proximal tubular epithelial cells. Front Cell Dev Biol. (2019) 7:252. doi: 10.3389/fcell.2019.00252
20. Perco P, Kainz A, Wilflingseder J, Soleiman A, Mayer B, Oberbauer R. Histogenomics: association of gene expression patterns with histological parameters in kidney biopsies. Transplantation. (2009) 87:290–5. doi: 10.1097/TP.0b013e318191b4c0
21. Yu X, Zhang X, Hu Z. NLRP2 is highly expressed and promotes apoptosis in a mouse model of kidney ischemia/reperfusion injury. Eur J Inflamm. (2019) 17:2058739219859805. doi: 10.1177/2058739219859805
22. Nevo N, Chol M, Bailleux A, Kalatzis V, Morisset L, Devuyst O, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. (2010) 25:1059–66. doi: 10.1093/ndt/gfp553
23. Kuchmiy AA, D'Hont J, Hochepied T, Lamkanfi M. NLRP2 controls age-associated maternal fertility. J Exp Med. (2016) 213:2851–60. doi: 10.1084/jem.20160900
24. Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. (2002) 22:7622–32. doi: 10.1128/MCB.22.21.7622-7632.2002
25. Raggi C, Luciani A, Nevo N, Antignac C, Terryn S, Devuyst O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum Mol Genet. (2014) 23:2266–78. doi: 10.1093/hmg/ddt617
26. Roufosse C, Simmonds N, Clahsen-van Groningen M, Haas M, Henriksen KJ, Horsfield C, et al. A 2018 reference guide to the banff classification of renal allograft pathology. Transplantation. (2018) 102:1795–814. doi: 10.1097/TP.0000000000002366
27. Cheung WW, Cherqui S, Ding W, Esparza M, Zhou P, Shao J, et al. Muscle wasting and adipose tissue browning in infantile nephropathic cystinosis. J cachexia sarcopenia Muscle. (2016) 7:152–64. doi: 10.1002/jcsm.12056
28. Martin-Granado A, Vazquez-Moncholi C, Luis-Yanes MI, Lopez-Mendez M, Garcia-Nieto V. Determination of Clara cell protein urinary elimination as a marker of tubular dysfunction. Pediatr Nephrol. (2009) 24:747–52. doi: 10.1007/s00467-008-1078-5
29. Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J Immunol Res. (2018) 2018:2180373. doi: 10.1155/2018/2180373
30. Kadatane SP, Satariano M, Massey M, Mongan K, Raina R. The role of inflammation in CKD. Cells. (2023) 12:1581. doi: 10.3390/cells12121581
31. Zhang X, Lu X, Yu L, Gu Y, Qu F. Downregulation of NLRP2 inhibits HUVEC viability by inhibiting the MAPK signaling pathway. Mol Med Rep. (2019) 19:85–92. doi: 10.3892/mmr
32. Jin L, Li T, Hong Y, Mao R, Li X, Zhu C, et al. Activation of NLRP2 in Triple-Negative Breast Cancer sensitizes chemotherapeutic therapy through facilitating hnRNPK function. Biochem Pharmacol. (2023) 215:115703. doi: 10.1016/j.bcp.2023.115703
33. Peng H, Chang B, Lu C, Su J, Wu Y, Lv P, et al. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PloS One. (2012) 7:e30344. doi: 10.1371/journal.pone.0030344
34. Matsuoka Y, Yamashita A, Matsuda M, Kawai K, Sawa T, Amaya F. NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain. (2019) 160:2149–60. doi: 10.1097/j.pain.0000000000001611
35. Yang Y, Lang X, Sun S, Gao C, Hu J, Ding S, et al. NLRP2 negatively regulates antiviral immunity by interacting with TBK1. Eur J Immunol. (2018) 48:1817–25. doi: 10.1002/eji.201847589
36. Mahadevan S, Sathappan V, Utama B, Lorenzo I, Kaskar K, Van den Veyver IB. Maternally expressed NLRP2 links the subcortical maternal complex (SCMC) to fertility, embryogenesis and epigenetic reprogramming. Sci Rep. (2017) 7:44667. doi: 10.1038/srep44667
37. Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, Yates JR, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PloS Genet. (2009) 5:e1000423. doi: 10.1371/journal.pgen.1000423
38. Elmonem MA, Veys KRP, Prencipe G. Nephropathic cystinosis: pathogenic roles of inflammation and potential for new therapies. Cells. (2022) 11:190. doi: 10.3390/cells11020190
39. De Leo E, Taranta A, Raso R, Polishchuk E, D'Oria V, Pezzullo M, et al. Genistein improves renal disease in a mouse model of nephropathic cystinosis: a comparison study with cysteamine. Hum Mol Genet. (2023) 32:1090–101. doi: 10.1093/hmg/ddac266
40. Hohenfellner K, Nießl C, Haffner D, Oh J, Okorn C, Palm K, et al. Beneficial effects of starting oral cysteamine treatment in the first 2 months of life on glomerular and tubular kidney function in infantile nephropathic cystinosis. Mol Genet Metab. (2022) 136:282–8. doi: 10.1016/j.ymgme.2022.06.009
41. Afolabi O, Alabi B, Omobowale T, Oluranti O, Iwalewa O. Cysteamine mitigates torsion/detorsion-induced reperfusion injury via inhibition of apoptosis, oxidative stress and inflammatory responses in experimental rat model. Andrologia. (2022) 54:e14243. doi: 10.1111/and.14243
42. Wang S, Bai M, Xu K, Shao Y, Yang Z, Xiong X, et al. Effects of coated cysteamine on oxidative stress and inflammation in weaned pigs. Anim an Open Access J MDPI. (2021) 11:2217. doi: 10.3390/ani11082217
43. Fraser-Pitt D, Mercer DK, Francis ML, Toledo-Aparicio D, Smith DW, O'Neil DA. Cysteamine-mediated blockade of the glycine cleavage system modulates epithelial cell inflammatory and innate immune responses to viral infection. Biochem Biophys Res Commun. (2023) 677:168–81. doi: 10.1016/j.bbrc.2023.08.021
Keywords: cystinosis, inflammation, fibrosis, NLRP2, chronic kidney disease
Citation: Rossi MN, Matteo V, Diomedi-Camassei F, De Leo E, Devuyst O, Lamkanfi M, Caiello I, Loricchio E, Bellomo F, Taranta A, Emma F, De Benedetti F and Prencipe G (2024) Nlrp2 deletion ameliorates kidney damage in a mouse model of cystinosis. Front. Immunol. 15:1373224. doi: 10.3389/fimmu.2024.1373224
Received: 19 January 2024; Accepted: 18 March 2024;
Published: 03 April 2024.
Edited by:
Lining Miao, Second Hospital of Jilin University, ChinaReviewed by:
Ruhao Yang, Renmin Hospital of Wuhan University, ChinaRaffaele Strippoli, Sapienza University of Rome, Italy
Copyright © 2024 Rossi, Matteo, Diomedi-Camassei, De Leo, Devuyst, Lamkanfi, Caiello, Loricchio, Bellomo, Taranta, Emma, De Benedetti and Prencipe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giusi Prencipe, giusi.prencipe@opbg.net
†These authors have contributed equally to this work