 Margit Anthofer1
Margit Anthofer1 Markus Windisch1
Markus Windisch1 Rosa Haller1Sandra Ehmann1
Rosa Haller1Sandra Ehmann1 Sebastian Wrighton1†
Sebastian Wrighton1† Michael Miller1Lorenz Schernthanner1Iris Kufferath1Silvia Schauer1Barbara Jelušić1
Michael Miller1Lorenz Schernthanner1Iris Kufferath1Silvia Schauer1Barbara Jelušić1 Sabine Kienesberger2,3
Sabine Kienesberger2,3 Ellen L. Zechner2,3
Ellen L. Zechner2,3 Gernot Posselt4
Gernot Posselt4 Mar Vales-Gomez5
Mar Vales-Gomez5 Hugh T. Reyburn5
Hugh T. Reyburn5 Gregor Gorkiewicz1,3*
Gregor Gorkiewicz1,3*- 1Institute of Pathology, Medical University of Graz, Graz, Austria
- 2Institute of Molecular Biosciences, University of Graz, Graz, Austria
- 3Interuniversity Cooperation, BioTechMed-Graz, Graz, Austria
- 4Department of Biosciences and Medical Biology, Paris Lodron University of Salzburg, Salzburg, Austria
- 5Department of Immunology and Oncology, Spanish National Centre for Biotechnology, Madrid, Spain
Background: Helicobacter pylori (H. pylori) uses various strategies that attenuate mucosal immunity to ensure its persistence in the stomach. We recently found evidence that H. pylori might modulate the natural killer group 2, member 2 (NKG2D) system. The NKG2D receptor and its ligands are a major activation system of natural killer and cytotoxic T cells, which are important for mucosal immunity and tumor immunosurveillance. The NKG2D system allows recognition and elimination of infected and transformed cells, however viruses and cancers often subvert its activation. Here we aimed to identify a potential evasion of the NKG2D system in H. pylori infection.
Methods: We analyzed expression of NKG2D system genes in gastric tissues of H. pylori gastritis and gastric cancer patients, and performed cell-culture based infection experiments using H. pylori isogenic mutants and epithelial and NK cell lines.
Results: In biopsies of H. pylori gastritis patients, NKG2D receptor expression was reduced while NKG2D ligands accumulated in the lamina propria, suggesting NKG2D evasion. In vitro, H. pylori induced the transcription and proteolytic shedding of NKG2D ligands in stomach epithelial cells, and these effects were associated with specific H. pylori virulence factors. The H. pylori-driven release of soluble NKG2D ligands reduced the immunogenic visibility of infected cells and attenuated the cytotoxic activity of effector immune cells, specifically the anti-tumor activity of NK cells.
Conclusion: H. pylori manipulates the NKG2D system. This so far unrecognized strategy of immune evasion by H. pylori could potentially facilitate chronic bacterial persistence and might also promote stomach cancer development by allowing transformed cells to escape immune recognition and grow unimpeded to overt malignancy.
1 Introduction
Helicobacter pylori (H. pylori) is a major human pathogen causing chronic gastritis and gastroduodenal ulcer disease. It also drives the development of gastric adenocarcinoma and MALT lymphoma and is considered a class-I carcinogen (1). H. pylori infection typically occurs early in life (2), and persists life-long unless eradicated therapeutically (1). To establish chronic infection and mucosal persistence, H. pylori has evolved multiple strategies to overcome immunity (3). These strategies include avoiding recognition by TLRs, surviving phagocytosis by neutrophils and macrophages, modifying expression of immunomodulatory proteins (e.g. PD-L1) in stomach epithelial cells and modulating lymphocyte activation, proliferation and differentiation (1, 3, 4). Noteworthy, the induction of certain anti-inflammatory circuits in H. pylori infection, such as the promotion of interleukin 10 (IL-10)-producing regulatory T cells (Tregs), benefits the host by counteracting the development of atopic diseases like asthma (3, 5). However, weakened immunity also favors cancer development since transformed cells may escape immune surveillance and produce overt cancer.
The natural killer group 2, member D (NKG2D) system is a well characterized immune surveillance system, which is important for the elimination of infected and transformed cells (6). The NKG2D receptor is expressed on natural killer (NK) cells, cytotoxic T lymphocytes (CTLs) and gamma delta T cells (γδ T cells) (7, 8), all of which are part of mucosal immunity (9, 10). NKG2D recognizes the NKG2D ligands (NKG2D-Ls) MHC class I polypeptide-related sequence A (MICA), MHC class I polypeptide-related sequence B (MICB) and UL16 binding proteins 1-6 (ULBP 1-6) (6), which are mainly expressed intracellularly by epithelia and certain other cell types (11, 12). Cell surface expression of NKG2D-Ls is specifically increased under stress conditions such as infection (8, 13, 14), or oncogenic transformation (15). The receptor-ligand interaction, then, activates the cytotoxic immune cells to eliminate the ligand-expressing cells (7, 8, 13, 16). Notably, some viruses but also many cancers escape this mechanism, either by downregulating NKG2D-L expression or by releasing NKG2D-Ls as soluble proteins from the cell surface, which reduces the cells’ immunological visibility (17–19). Soluble NKG2D-Ls additionally attenuate the strength of the immune response by acting as suppressors of NKG2D-expressing effector cells. Their binding to the receptor leads to reduced NKG2D expression and thus diminished NKG2D-mediated immunity and an overall hypofunctional immune cell phenotype (20–23).
Several studies indicate an important role for the NKG2D system in mucosal homeostasis and the microbiota and certain microbial metabolites (e.g. short-chain fatty acids; SCFAs) are able to modulate NKG2D-L expression in the gut (14, 24–26). Interestingly, the system is dysregulated and contributes to inflammation in diseases such as celiac disease (27), and Crohn’s disease (28), conditions where H. pylori infection seems to be beneficial (5). We recently found indications that H. pylori might modulate the NKG2D system (26). Here we aimed to investigate this finding in more detail. By analyzing human stomach biopsies from H. pylori gastritis and gastric cancer cases and by employing cell-culture based infection models with epithelial and NK cells we demonstrate that H. pylori can actively subvert the NKG2D system. This so far unrecognized immune evasion strategy could potentially help the pathogen to persist in the stomach mucosa, and might also favor stomach cancer development.
2 Materials and methods
2.1 Human stomach biopsies
Formalin-fixed and paraffin-embedded (FFPE) tissue samples were derived from the archives of the Institute of Pathology at the Medical University of Graz (Supplementary Table 1). H. pylori presence was determined by Warthin–Starry staining (29), and IHC with an anti-H. pylori antibody (clone SP48; Ventana). Tissue use was approved by the institutional review board of the Medical University of Graz (EK-23-212ex10/11).
2.2 Histopathology and IHC
FFPE tissues were subjected to standard H&E staining. For immunohistochemistry, 2 μm thick sections were stained with antibodies against CD45 (Clones 2B11 + PD7/26, Cat# GA75161-2, Agilent Technologies, RRID: AB_2661839), CD8 (Clone C8/144B, Cat# GA62361-2, Agilent Technologies), CD56 (clone MRQ42; 760-4596, Ventana Medical Systems) and NKp46 (clone 195314, MAB1850, R and D systems, RRID: AB_2149153). For CD45 and CD8 detection, slides were stained on a Dako Omins machine using the EnVision FLEX staining system with Antigen-Retrieval using EnVision FLEX TRS, High pH for 30 min at 97°C for CD45 and using EnVision FLEX TRS, Low pH for 24 min at 95°C for CD8 detection. Blocking was achieved with the EnVision FLEX Peroxidase-Blocking-Reagent for 3 min, primary antibodies were incubated for 20 min at 36°C and for detection, the EnVision FLEX HRP and the EnVision FLEX Mouse Linker were used. For detection of CD56 and NKp46, slides were stained with the Ventana system using CC1 Standard (Ventana) at 95°C for 32 min for CD56 and at 95°C for 64 min for NKp46 detection. The CD56 antibody was ready-to-use and was incubated at 36°C for 16 min and the NKp46 antibody was diluted 1:100 and incubated at 36°C for 1 h. For detection, the ultraView Universal DAB Detection Kit (Ventana) was used. For MICA/B detection, antigen retrieval was performed in a microwave in sodium citrate buffer, pH 6.0, (Gatt-Koller) for 40 min. Endogenous peroxidase activity was inhibited with 3% H2O2 in methanol for 15 min. For blocking of nonspecific protein binding and for detection the UltraVision LP Detection System HRP Polymer (Ready-to-use) (TL-060-HL; Thermo Scientific) was used. Sections were incubated with primary MICA/B antibody (clone F-6; dilution 1:100; Santa Cruz Biotech, RRID: AB_2143751) for 1 h at RT. The sections were counterstained with Mayer’s hemalum solution (Merck) and Entellan (Merck) was used as mounting medium. A pathologist (GG) evaluated and scored all immunohistochemistry. CD45, CD8, CD56 and NKP46 were evaluated by quantifying positive cells per 1 mm2 of the tissue section. MICA/B staining was scored by assessing the intensity of staining in epithelial cells and in the lamina propria (0= absent, 0.5= very weak, 1= weak, 1.5= weak to medium, 2= medium, 2.5= medium to strong, 3= strong). In addition, individual snap-frozen (2-methylbutane) stomach biopsy sections were subjected to MICA/B staining.
2.3 qPCR
Total RNA from FFPE samples (7 sections, each 5 µm thick) was isolated with Deparaffinization Solution (Qiagen) and the RNeasy FFPE kit, which includes a DNase treatment step (Qiagen). Total RNA from cell cultures was isolated using the NucleoSpin® RNA extraction kit (Machery-Nagel) or TRIzol reagent (Invitrogen). RNA quality and quantity were determined spectrophotometrically with the NanoDrop™ 2000c (Thermo Fisher Scientific). For cDNA synthesis, the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) and the RNAase inhibitor (Applied Biosystems) were used. qPCR was performed with a CFX384 qPCR thermocycler (BioRad) using SYBR® Green PCR Master Mix (Applied Biosystems). A standard protocol for SYBR® Green was used with 10 min at 95°C, 39 cycles of 15 sec at 97°C and 1 min at 60°C, followed by 15 sec at 60°C and a final melting step until 95°C. The oligonucleotide primers used in this study are listed in Supplementary Table 2. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as reference gene. qPCR data are reported as –ΔCt in Figure 1C and as relative expression ratio in all other figures. The relative expression ratio was calculated according to Pfaffl’s method (equation 1) (30). As control group for the calculation of the relative expression ratio we used cells harvested at time point 0 h.
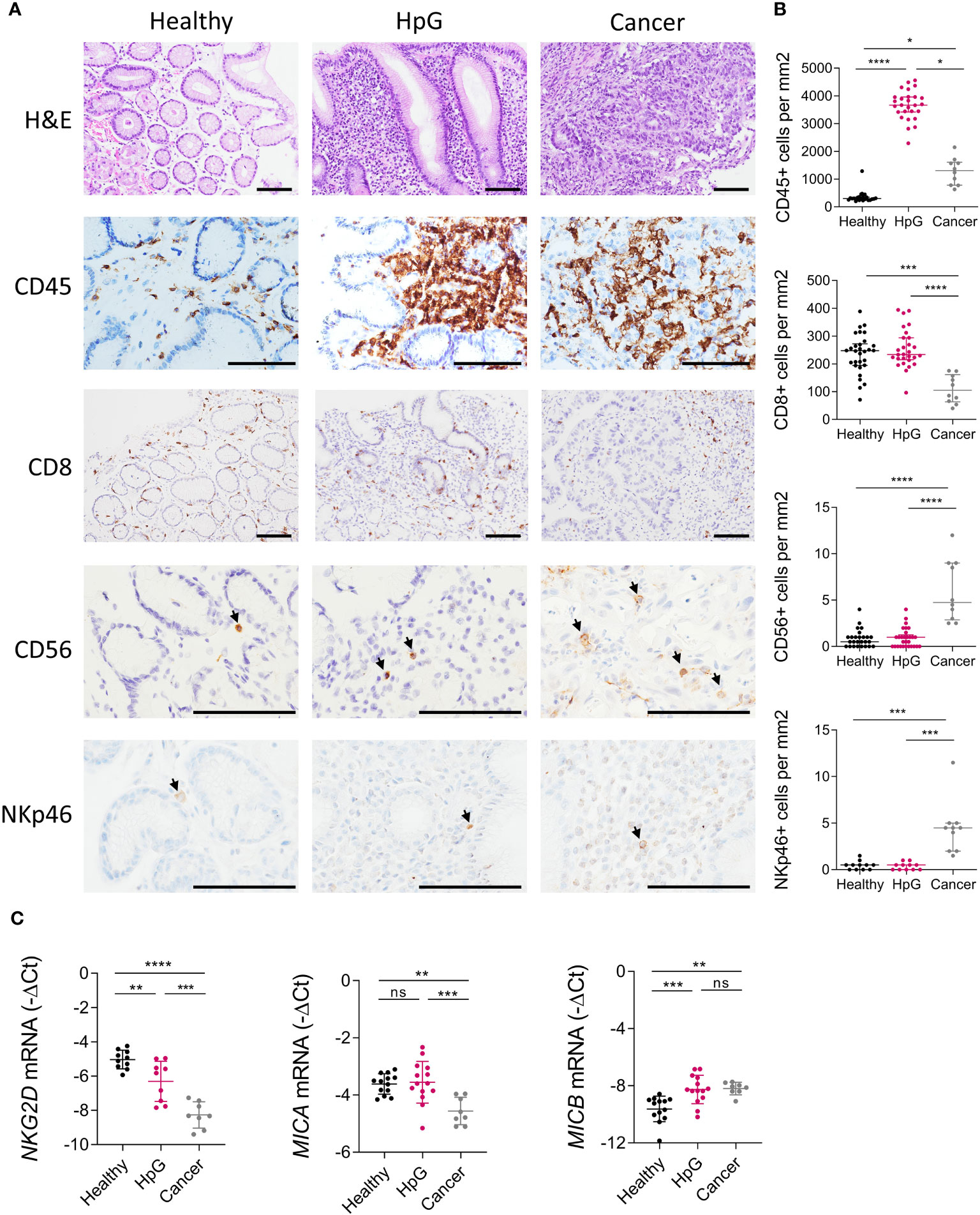
Figure 1 Inflammation and the NKG2D system in H. pylori infection and gastric cancer. Stomach biopsies from healthy controls (Healthy), H. pylori gastritis cases (HpG) and stomach adenocarcinoma cases (Cancer) were immune-phenotyped. (A, B) H&E and IHC staining of leucocytes (CD45), CTLs (CD8) and NK cells (CD56, NKp46). (A) Representative images, scale bars: 100 µm. (B) Quantification of positive cells per mm2 of tissue n=10-30 per group, the data do not follow a normal distribution, median ± interquartile range, Kruskal-Wallis test and Dunn’s test. (C) qPCR analysis of NKG2D, MICA and MICB, n=8-16 per group. The data passed Shapiro-Wilk normality test, mean ± SD, one-way ANOVA and Tukey’s test. *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. ns, not significant.
2.4 Cell lines
The human stomach epithelial cell line AGS (female, ATCC Number: CRL-1739, RRID: CVCL_0139) was obtained from Cell Lines Service (Eppelheim, Germany) and the human stomach epithelial cell line MKN28 (female, RRID : CVCL_1416) was kindly provided by Dr. Silja Wessler (Department of Biosciences and Medical Biology, Paris Lodron University of Salzburg, Austria) and originally obtained from the Japanese Collection of Research Bioresources (JCRB; http://cellbank.nibio.go.jp/). Adhesion of H. pylori to these cell lines was confirmed and cellular responses to the infection were characterized by Schneider et al. (31). AGS and MKN28 cells were cultivated in RPMI 1640 medium (Gibco), supplemented with 2 mmol/L L-glutamine (Gibco), 5 mmol/L HEPES (Gibco) and 10% fetal bovine serum (FBS) (Gibco). The human natural killer cell line NKL (male, RRID: CVCL_0466) was a kind gift of Dr. Francisco Borrego (Biocruces Bizkaia Health Research Institute, Barakaldo, Spain). The chronic myeloid leukemia cell line K562 (female, RRID: CVCL_0004) was obtained from ATCC (cat # CCL-243). NKL and K562 cells were cultured in RPMI 1640 medium, supplemented with 100 units/ml penicillin, 100 mg/ml streptomycin (Gibco), 1 mmol/L sodium pyruvate (Gibco), 0,1 mmol/L nonessential amino acids (Gibco) and 58 µmol/L 2-mercaptoethanol (Sigma Aldrich). K562 cells were additionally supplemented with 10% FBS (Gibco). NKL cells were additionally supplemented with 50 units/ml recombinant human IL-2 (Peprotech), 5% human normal serum (MP Biomedicals™) and 5% FBS (Gibco). All cells were incubated in a water-saturated atmosphere with 5% CO2 at 37°C.
2.5 Bacteria
H. pylori P12 wild type (expressing Western cagA EPIYA-ABCC, and with a vacA s1/m1 genotype) (32, 33), and isogenic mutant strains P12 ΔcagA, ΔcagL and ΔvacA were kindly provided by Dr. Silja Wessler (Department of Biosciences and Medical Biology, Paris Lodron University of Salzburg, Austria). H. pylori strains were cultured on agar plates containing 10% horse serum under microaerophilic conditions at 37°C. Source and cultivation of the C. acnes PA-2.2 strain were described recently (26).
2.6 Infection assay, butyrate treatment and metalloprotease inhibition
AGS, AGS-MICA and MKN28 cells were seeded in 6-well plates and grown for 48 h to 70% confluence. H. pylori strains were grown on agar plates for 48 h, then harvested in cell culture medium and used for infection. C. acnes was cultured on agar plates for 72 h, then harvested in cell culture medium to an optical density (OD600) of 0.1 and further grown for 24 h. Subsequently, cells were infected at multiplicities of infection (MOI) of 50 and incubated for 24 h and 48 h, according to other studies that performed infection experiments with H. pylori P12 and these stomach epithelial cell lines (34, 35). These infection conditions induce significant phenotypic changes in epithelia cell lines without causing excessive cell death (Supplementary Figure 1). Infection with the H. pylori WT strain induced the hummingbird phenotype [induced by CagA (36)] and vacuole formation [induced by VacA (37)] (Supplementary Figure 12). These phenotypic changes did not occur when cells were infected with the respective mutant strains (ΔcagA and ΔvacA). Infection with the ΔcagL strain did also not induce the hummingbird phenotype, consistent with the notion that CagL is essential for CagA transfer to host cells and the subsequent CagA-dependent cell modifications (38). As control treatment served 2 mmol/L butyrate (Sigma Aldrich), that was shown to induce MICA/B expression (26, 39). For metalloprotease inhibition, batimastat (BB94) (Sigma Aldrich) a broad-spectrum inhibitor of MMPs and of ADAM proteases was diluted in DMSO and added to AGS and MKN28 cells at 10 µmol/L (40), at the same time as H. pylori or butyrate. As solvent control, only DMSO was added to the cultures.
2.7 ELISA
Cell culture supernatants were passed through 0.22 µM sterile filters and were concentrated with Amicon® Ultra-2 mL Centrifugal Filters (MerckMillipore), according to the manufacturer’s instructions. We applied the same starting volume (2 ml) of all culture supernatants to the Amicon® Ultra-2 mL Centrifugal Filters and centrifuged until all samples were reduced to less than 500 µl. Because the procedure reduced all samples to slightly different volumes, we then added varying amounts of fresh reagent diluent to the samples, to adjust all samples to the equal volume of 500 µl. To take this 4-fold concentration into account, we divided the ELISA-readout values by 4 and reported these final values in Figures 2, 3. ELISA was performed using the Human MICA Duoset ELISA kit (R&D) and the Human MICB Duoset ELISA kit (R&D), according to the manufacturers protocol. Readouts were performed on a SPECTROstar Omega Plate reader.
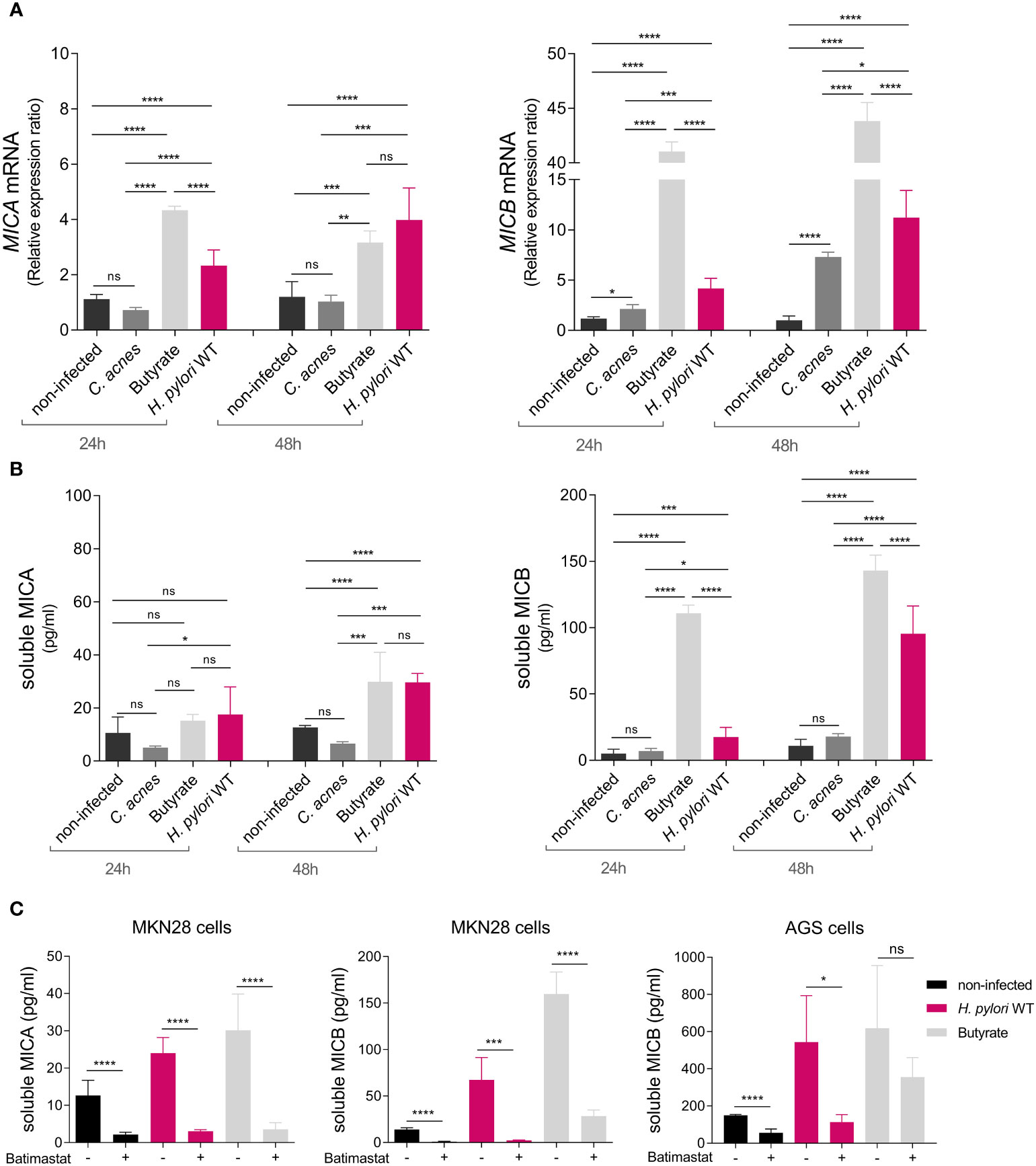
Figure 2 H. pylori-dependent modulation of NKG2D-L expression and soluble release via proteolytic shedding. MKN28 cells were challenged with C. acnes, butyrate, and H. pylori WT for 24 and 48 h (A) MICA and MICB mRNA levels were determined by qPCR. (B) Soluble MICA and MICB levels in cell culture supernatants were determined by ELISA. Experiments were performed three times. Mean ± SD, one-way ANOVA and Tukey’s test (ns, not significant, *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001). (C) MKN28 and AGS cells were challenged with H. pylori WT or 2 mM butyrate for 48 h Simultaneously, cells were treated with either 10 µmol/L batimastat dissolved in DMSO (= batimastat +) or with DMSO alone as solvent control (= batimastat –). Soluble MICA and MICB proteins in cell culture supernatants were determined by ELISA. Experiments were performed three times. Mean ± SD, t-test of batimastat – vs. batimastat +, for each treatment group (ns, not significant, *P <0.05; ***P <0.001; ****P <0.0001).
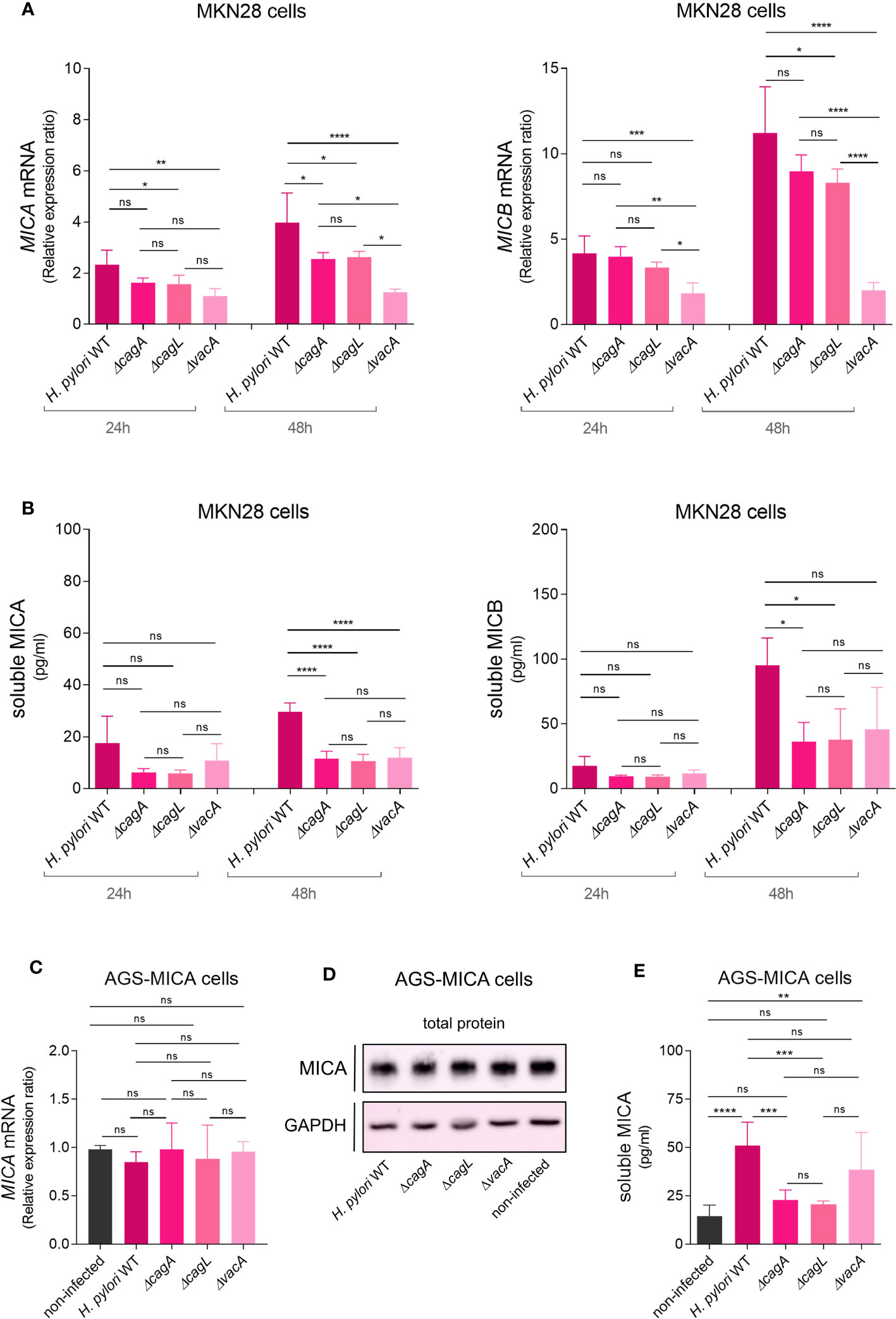
Figure 3 H. pylori virulence factor-dependent modulation of NKG2D-L expression and soluble release. MKN28 cells were challenged with H. pylori WT and isogenic mutants ΔcagA, ΔcagL and ΔvacA for 24 and 48 h (A) MICA and MICB mRNA levels were determined by qPCR. (B) Soluble MICA and MICB levels in cell culture supernatants were determined by ELISA. (C–E) AGS-MICA cells were challenged with H. pylori WT and isogenic mutants ΔcagA, ΔcagL and ΔvacA for 24 h (C) MICA mRNA levels were determined by qPCR. (D) MICA protein in cell lysates was determined by Western Blot. (E) Soluble MICA protein in cell culture supernatants was determined by ELISA. Experiments were performed three times. Mean ± SD, one-way ANOVA and Tukey’s test (ns, not significant, *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001).
2.8 Transfection of AGS cells
For the generation of AGS cells expressing the MICA allele *019 (A5) (denoted AGS-MICA cells), we used the lentiviral vector pHRSIN (41), a gift of Prof. Paul Lehner (Cambridge Institute for Medical Research, Cambridge, United Kingdom), expressing MICA*019 under control of the spleen focus-forming virus (SFFV) promoter for constitutive, high-level gene expression. MICA*019 encodes a full-length MICA protein with all the features that are typical for these allelic variants (42). Lentiviruses were generated by means of transfection of 293T cells with pHRSIN, together with the plasmids pCMVR8.91 and pMD2G. Two days after transfection, culture media containing the lentiviruses were harvested, filtered, and stored at −80°C. For each lentiviral transduction, 0.3 × 106 AGS cells were mixed with 0.75 mL of virus supernatant in the presence of 1 μmol/L of the TBK1 inhibitor BX795 (InvivoGen) and 8 μg/mL Polybrene (Sigma-Aldrich) and seeded into one well of a 6-well plate (BD Biosciences). The plates were centrifuged at 800 rpm at 33°C for 1 h. After centrifugation, without removing viral supernatants, the plates were incubated at 37°C in a 5% CO2 atmosphere for 4-6 h and then centrifuged again with the same conditions. Supernatants were removed from wells, and fresh growth medium was added. After two weeks of cultivation, AGS-MICA transfectants were stained with the Alexa Fluor® 647 anti-human MICA/MICB Antibody (BioLegend Cat# 320914, RRID: AB_2266419) and sorted by FACS.
2.9 Western blot
Protein lysates from cell cultures were prepared in RIPA buffer (Merck Millipore) containing 0.1 mmol/L Pefabloc, 1 mmol/L DTT, cOmplete Mini EDTA-free and PhosSTOP (Roche). Protein concentrations were determined using the BioRad Protein Assay Dye Reagent (BioRad Laboratories). Proteins were loaded onto 10% (v/v) (SDS)-polyacrylamide gels, subjected to electrophoresis and then blotted onto PVDF membranes (Immobilon-P, Merck Millipore). Blotting efficiency was determined by staining with Ponceau S solution (Sigma Aldrich). Non-specific binding was blocked for 1 h with 5% (w/v) non-fat dry milk (Bio-Rad Laboratories) in tris-buffered saline (TBS) with 0.1% (v/v) Tween 20 (Merck Millipore). Subsequently, the membranes were incubated with the primary antibody MICA biotinylated antibody (R&D Systems Cat# BAF1300, RRID: AB_355943, 1:2000) overnight at 4°C, followed by incubation with streptavidin-HRP (R&D Systems DY998, 1:5000) at RT for 1 h. For loading control, membranes were incubated with GAPDH antibody (Cell Signaling Technology #2118, 1:1000), overnight at 4°C, followed by incubation with rabbit IgG HRP linked F(ab′)2 (Merck, GENA9340-1ML, 1:5000) at RT for 1 h. Immunolabeling was detected using the ECL™ Select Western Blotting Detection Reagent (Merck, GERPN2235) and visualized with the ImageQuant™ LAS 500. Quantification of band intensities was performed using Image Lab Software (Bio-Rad).
2.10 Flow cytometry
Cells were harvested with Accutase® (Thermo Fisher Scientific) and stained with Alexa Fluor 647 anti-human MICA/B antibody (BioLegend, Cat# 320914, RRID: AB_2266419) at 4°C for 1 h. Flow cytometry was performed using a CytoFLEX S flow cytometer (Beckman Coulter) according to the manufacturer’s protocol. The software CytExpert (Beckman Coulter) was used for the analysis of flow cytometry data. Flow cytometry results are represented as median fluorescence intensity (MFI).
2.11 Immunofluorescence
Cells were grown on adhesive slides (QPath) with flexiPERM® (Sarstedt) and infection assays were performed as described above. Subsequently, cells were fixed in 4% formaldehyde (Thermo Scientific #28906) and permeabilized with 0,1% saponin before incubation with the Alexa Fluor® 647 anti-human MICA/MICB antibody (BioLegend Cat# 320914, RRID: AB_2266419, 1:100). 4’,6-Diamidin-2-phenylindol (DAPI) was used for nuclear counter-staining. Fluorescence staining was analyzed using the Zeiss LSM510 Meta Confocal Microscope. The Nikon software NIS-Elements General Analysis (GA3) was used for quantification of fluorescence intensity per cell.
2.12 MICA genotyping
Genotyping of the microsatellite repeat polymorphism in the transmembrane region of the MICA gene, was carried out as described (43). Fragment sizes were determined using the Peak Scanner v1.0 software (Applied Biosystems). The cell lines Jurkat (A5.1/A6), J82 (A5.1/A6), RT4 (A5.1) and RT112 (A4) were used as controls.
2.13 Analysis of NKG2D expression on NKL cells
AGS-MICA cells were either not infected or infected with H. pylori, as described above. After 24 h, cell culture supernatants were harvested, passed through 0.22 µM sterile filters and concentrated with centrifugal filters (Amicon Ultra-15, PLGC Ultracel-PL Membran, 10 kDa, Merck Millipore). Cell culture supernatants were concentrated 10-fold and then diluted with fresh RPMI medium by 2 (to generate 5-fold concentrated supernatants) and by 5 (to generate 2-fold concentrated supernatants). Next, we added 50 µl of these supernatants (10-fold, 5-fold and 2-fold concentrated) to 50 µl of NKL cells per well, resulting in a further 2-fold dilution and thus a final treatment of NKL cells with 5-fold, 2,5-fold and 1-fold concentrated supernatants. NKL cells were treated with these cell culture supernatants in the presence of 5 µg/ml of the human MICA/B antibody (R&D Systems Cat# MAB13001, RRID: AB_2143621) or the mouse IgG2A isotype control (R&D Systems Cat# MAB003, RRID: AB_357345). After 24 h, NKL cells were stained either with the FITC anti-human CD94 antibody (BioLegend Cat# 305504, RRID: AB_314534) or the human NKG2D/CD314 antibody (R&D Systems Cat# MAB139, RRID: AB_2133263), followed by staining with the goat F(ab’)2 anti-mouse IgG - (Fab)’2 (PE), pre-adsorbed (Abcam Cat# ab5889, RRID: AB_955482) at 4°C for 30 min. Flow cytometry readouts are represented as median fluorescence intensity (MFI).
2.14 Analysis of cytotoxic degranulation of NK cells
NKL cells were treated with cell culture supernatants from AGS-MICA cells, as described above. Subsequently, NKL cells were co-cultured with K562 cells at 37°C for 2 h. The cultures were then stained with the FITC anti-human CD94 antibody (BioLegend Cat# 305504, RRID: AB_314534) and the APC anti-human CD107a (LAMP-1) antibody (BioLegend Cat# 328620, RRID: AB_1279055) at 4°C for 30 min. Flow cytometry readouts are represented as % of the LAMP-1+ cells, out of the CD94+ cells.
2.15 Statistical analysis
For cell culture assays, data from three independent experiments were combined and parametric tests were performed to determine statistically significant differences between groups. One-way analysis of variance (ANOVA) was used to compare three or more groups with Dunnett’s multiple comparisons test to compare the mean of each group with the mean of one single control group or with Tukey’s multiple comparisons test to compare the mean of each group to the mean of every other group. An unpaired, two-tailed Student’s t-test was used to compare two independent groups and a one-sample t-test was used to compare the mean of one group to a theoretical mean of 1. Data from patient biopsies were tested for normality using the Shapiro-Wilk test. Subsequently, the data were subjected to statistical analysis by t-test or ANOVA for normally distributed sample sets, or to analysis by Kruskal Wallis test for nonparametric analyses. As post-hoc test, Dunn’s multiple comparisons test was applied to compare the mean rank of each group with the mean rank of every other group. A value of P<0.05 was considered significant (*P <0.05; **P <0.01; ***P <0.001; ****P <0.0001). Statistical analyses were performed with GraphPad Prism 9.
3 Results
3.1 The NKG2D system is altered in H. pylori infection and gastric cancer
H. pylori elicits a strong inflammation of the stomach mucosa, but the contribution of effector cells harboring the NKG2D receptor, like CTLs and NK cells, within the immune infiltrate is poorly described (44). We immuno-phenotyped biopsies from healthy controls (Healthy), H. pylori gastritis cases (HpG) and gastric adenocarcinoma cases (Cancer) by immunohistochemistry (IHC) (Figures 1A, B). HpG demonstrated substantially higher numbers of leukocytes (pan-leucocyte marker CD45) in the mucosa compared to healthy controls. Leucocytes were also significantly higher in cancer compared to heathy controls (Figures 1A, B). However, the numbers of CTLs (CD8) and NK cells (CD56, NKp46) were unchanged in HpG compared to healthy controls. In cancer, CTLs (CD8) were significantly lower while NK cells (CD56, NKp46) were significantly higher compared to healthy controls (Figures 1A, B). Overall, only sparse NK cell signals were detectable in HpG and controls with the NK cell markers CD56 and NKp46. Gene expression analysis of additional NK cell marker genes confirmed the observed lack of NK cell induction in HpG (Supplementary Figure 3). Next, we assessed expression of NKG2D system genes (NKG2D, MICA and MICB) via qPCR (Figure 1C). NKG2D receptor expression was significantly lower in HpG compared to healthy controls and even more so in cancer (Figure 1C). Expression of the ligand MICA was not changed in HpG but was lower in cancer compared to healthy controls. Expression of MICB was significantly higher in both conditions compared to healthy controls (Figure 1C). There were no significant differences in expression of lymphocyte markers and NKG2D system genes in relation to gender (data not shown). In summary, despite strong overall immune activation in H. pylori-associated pathologies, the numbers of CTLs and NK cells do not appear to be considerably changed in gastritis compared to the healthy state. Importantly, expression of the NKG2D receptor is significantly diminished and the NKG2D-L MICB is induced in both conditions. As most cancer samples were H. pylori-negative, the dysregulation of NKG2D and MICB was likely due to cancer-induced immune evasion. However, in HpG, the observed modulations of these genes suggest that H. pylori manipulates the NKG2D system.
3.2 H. pylori-dependent modulation of NKG2D-L expression and soluble release via proteolytic shedding
NKG2D evasion typically occurs either through repression of NKG2D-L expression or by the release of soluble NKG2D-Ls from the cell surface (17, 18). To determine a potential role of H. pylori in the modulation of MICA and MICB gene expression, we infected gastric epithelial cell line MKN28 with the H. pylori P12 wild-type (WT) strain. Treatment with Cutibacterium acnes (C. acnes) and the SCFA butyrate, both previously shown to induce NKG2D-L expression, served as controls (26, 39). After 24 and 48 h of treatment, MICA and MICB mRNA expression was determined by qPCR (Figure 2A). Butyrate induced both genes compared to non-infected cells, while C. acnes substantially affected only MICB (Figure 2A), consistent with previous findings (26, 39). Importantly, H. pylori WT infection resulted in significantly increased MICA and MICB mRNA levels compared to non-infected cells, and induction increased over time (Figure 2A). MICB was more induced than MICA and the degree of inducibility seemed to correlate negatively with the level of baseline mRNA expression of MICA and MICB in these cell lines (Supplementary Figure 4). H. pylori infection also upregulated gene expression of the NKG2D activator interleukin 15 (IL-15), the NKG2D ligands ULPB1 and ULBP2 and the NKG2D inhibitor transforming growth factor beta (TGF-β) compared to non-infected cells, whereas the NKG2D inhibitor macrophage migration inhibitory factor (MIF) was not induced (Supplementary Figures 5, 6) (45, 46).
We next asked whether H. pylori infection affects the release of soluble NKG2D-Ls. To determine this, we analyzed levels of soluble MICA (sMICA) and soluble MICB (sMICB) in cell culture supernatants of infected MKN28 cells via ELISA (Figure 2B). Butyrate induced sMICA and sMICB levels compared to non-infected cells, especially at 48 h, while C. acnes did not (Figure 2B), consistent with the notion that this bacterium activates the NKG2D system rather than evading it (26). Importantly, H. pylori WT infection significantly increased sMICA and sMICB compared to non-infected cells, especially at 48 h (Figure 2B). sMICB was also significantly increased in gastric epithelial cell line AGS after H. pylori infection, despite only minor induction of MICB gene expression in this cell line (Supplementary Figure 7). No sMICA was detected in the supernatants of AGS cells, since this cell line is MICA protein-deficient due to a single amino acid substitution (46) that interferes with protein folding (47).
We then aimed to elucidate the mode of soluble release of sMICA and sMICB. NKG2D-Ls can be released from the cell surface, either by proteolytic shedding or via extracellular vesicles (EVs), depending on the cell type, allele and treatment conditions (Supplementary Figure 8) (48, 49) Genotyping of our cell lines showed them to be homozygous for MICA alleles A5 (AGS) and A6 (MKN28), both of which amenable to proteolytic shedding. Regarding MICB, no alleles associated with extracellular vesicles release are known. Extracellular vesicle-enriched preparations from supernatants of H. pylori-infected cells showed only negligible amounts of MICA/B protein compared to extracellular vesicle-free supernatants, suggesting that there is no relevant extracellular vesicle release of MICA/B in these cell lines (Supplementary Figure 9). Next, we treated MKN28 and AGS cells with H. pylori WT or butyrate, to induce soluble release of MICA/B, and simultaneously added the broad-spectrum metalloprotease inhibitor batimastat dissolved in DMSO (40). As solvent control we only added DMSO. After 48 h of treatment, we assessed sMICA and sMICB by ELISA (Figure 2C). Batimastat significantly impaired the release of sMICA and sMICB from MKN28 cells and sMICB from AGS cells (Figure 2C). This indicates that H. pylori- and butyrate-induced release of sMICA/B, as well as its constitutive release is mediated by metalloproteases that can be specifically blocked (Figure 2C). Silencing of ADAM17 and MMP9, two proteases previously associated with proteolytic shedding of MICA/B (40, 50–53), had no significant impact on shedding frequencies (Supplementary results and Figure 10). Thus, the specific metalloproteases responsible for MICA/B shedding in H. pylori infection remain to be determined. Taken together, H. pylori WT infection induces gene expression and proteolytic shedding of NKG2D-Ls in stomach epithelial cells, suggesting evasion of the NKG2D system.
3.3 H. pylori virulence factor-dependent modulation of NKG2D-L expression and soluble release
To determine a potential role of the major H. pylori virulence factors in the modulation of NKG2D-Ls, we infected MKN28 cells with isogenic mutants of cytotoxin-associated gene A (ΔcagA), cytotoxin-associated gene L (ΔcagL) and vacuolating cytotoxin A (ΔvacA) (3). Infection with the ΔcagA and ΔcagL mutants resulted in only slightly lower levels of MICA and MICB mRNA expression, whereas infection with the ΔvacA mutant resulted in greatly lower levels of MICA and MICB mRNA expression, compared to infection with the WT (Figure 3A). Thus, vacA appears to be particularly important for inducing NKG2D-L mRNA expression. Regarding soluble NKG2D-Ls, infection with all three mutants (ΔcagA, ΔcagL and ΔvacA) resulted in lower sMICA levels and infection with the ΔcagA and ΔcagL mutants resulted in significantly lower sMICB levels compared to infection with the WT at 48 h (Figure 3B). We then speculated that cagA and cagL might be important for inducing the soluble release of NKG2D-Ls. Notably, cells infected with the ΔvacA mutant showed increasing sMICB levels from 24 h to 48 h without any increase in MICB mRNA levels. They also showed similar sMICB levels compared to cells infected with the ΔcagA and ΔcagL mutants, despite significantly lower MICB mRNA levels. Based on these observations, we hypothesized, that H. pylori might induce the release of soluble NKG2D-Ls independent of its effect on NKG2D-L gene expression.To specifically study the effect of H. pylori on the soluble release of NKG2D-Ls independent of its effect on NKG2D-L gene expression, we aimed to use a cell line where NKG2D-L gene and protein expression is unaffected by H. pylori. For this purpose, we used the cell line AGS where MICA mRNA expression was unaffected by H. pylori infection (Supplementary Figure 7A) and which is MICA protein-deficient (Supplementary Figure 7B) (47). To achieve constitutive MICA protein expression that is unlikely to be affected by H. pylori, we transfected AGS cells with a MICA-overexpression construct driven by the spleen focus forming virus (SFFV) promoter (the transfectants were termed AGS-MICA). As expected, infection of AGS-MICA transfectants with H. pylori WT or the ΔcagA, ΔcagL and ΔvacA mutants had no significant impact on total MICA mRNA expression (Figure 3C) and total MICA protein levels in whole cell lysates (Figure 3D; Supplementary Figure 11). Importantly, sMICA levels in cell culture supernatants of AGS-MICA cells significantly increased after challenge with H. pylori WT compared to non-infected cells (Figure 3E), indicating that H. pylori induces the release of soluble MICA, even when MICA gene and protein expression is unaffected. An increase in sMICA was also seen after infection with the ΔvacA strain but not with the ΔcagA and ΔcagL strains compared to non-infected cells (Figure 3E). Taken together, induction of MICA/B gene expression seems to be a vacA-dependent process, while soluble release of MICA appears to be cagA/L-dependent. Combined, these virulence factor-dependent effects could result in the release of large amounts of soluble NKG2D-Ls, while epithelial cells might have reduced levels of NKG2D-Ls at the cell surface, protecting them from immunosurveillance.
3.4 Translocation of NKG2D-Ls upon H. pylori infection
To determine the effect of H. pylori on the cell surface expression of NKG2D-Ls, we measured cell surface MICA/B proteins by flow cytometry (Figure 4A). In non-infected AGS-MICA cells, cell-surface MICA/B staining increased with time, likely due to continuous MICA/B production in these cells (Figure 4A). Cells infected with H. pylori showed significantly lower MICA/B surface staining compared to non-infected cells after 72 h, possibly due to a continuous release of soluble MICA/B from the cell surface (Figure 4A). Visualization with immunofluorescence confirmed these findings, wherein H. pylori-infected cells showed significantly less MICA/B staining compared to non-infected cells after 72 h (Figures 4B, C).
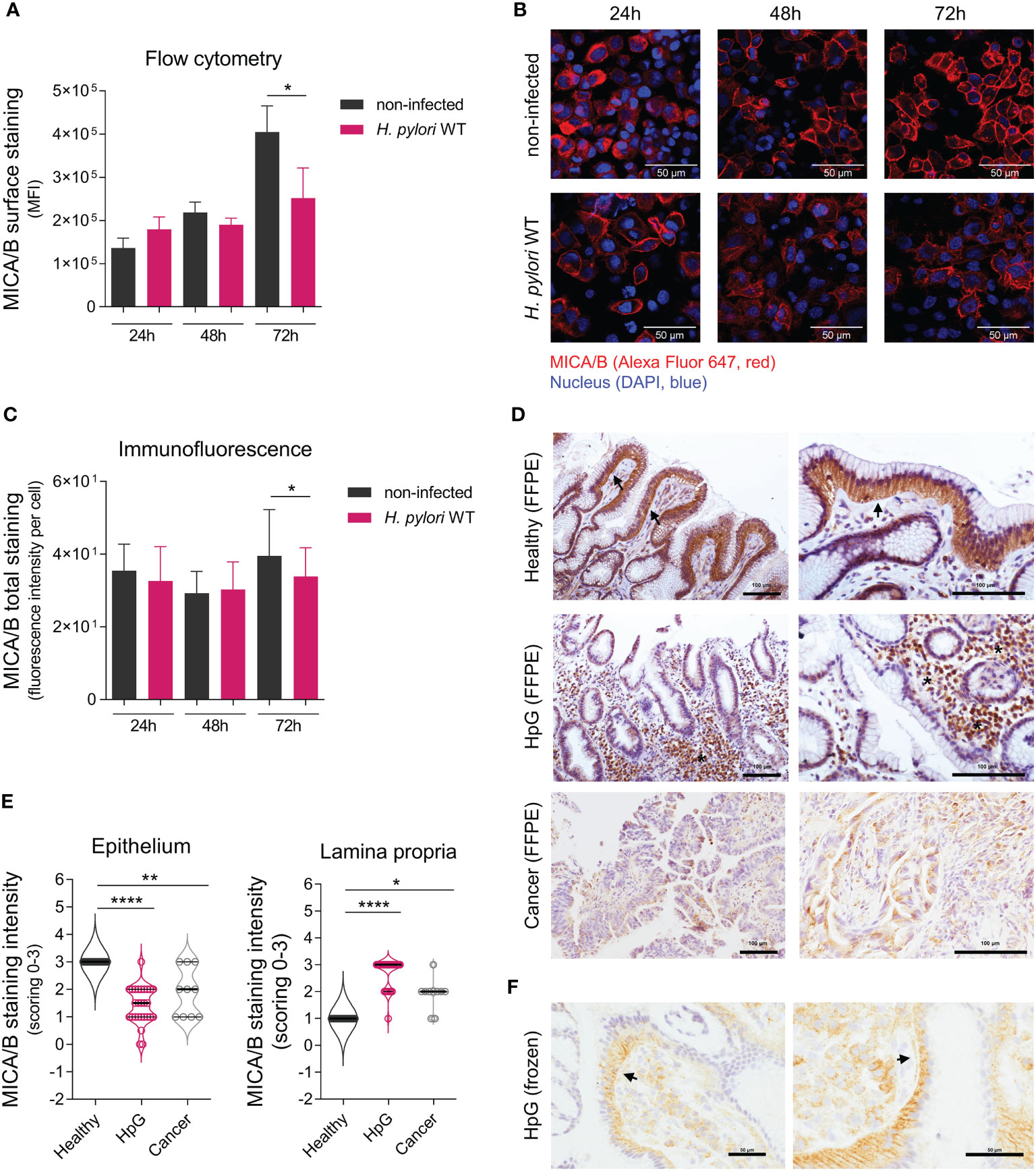
Figure 4 Translocation of NKG2D-Ls upon H. pylori infection. AGS-MICA cells were infected with H. pylori WT for 24, 48 and 72 h (A) MICA/B cell surface expression was quantified by flow cytometry. Experiments were performed three times. (B, C) Total cellular MICA/B protein in permeabilized cells was visualized by immunofluorescence (IF) staining with an anti-MICA/B antibody (red) and DAPI (blue). (B) Representative images, scale bars: 50µm. (C) Quantification of MICA/B staining-intensity per cell in IF micrographs. Experiments were performed six times. The data shown in a and c are represented as mean ± SD, unpaired, two-tailed Student’s t-test of infected versus non-infected cells at each timepoint (*P <0.05). (D, E) IHC staining of MICA/B in stomach biopsies (FFPE) from healthy controls (Healthy), H. pylori gastritis cases (HpG) and stomach adenocarcinoma cases (Cancer). (D) Representative images with arrows indicating basolateral epithelial staining and stars indicating lamina propria staining, scale bars: 100µm. (E) Quantification of MICA/B in biopsies by scoring of epithelial and lamina propria staining intensities from 0 (= no staining) to 3 (= strong staining), n=10-27 per group, the data do not follow normal distribution, violin plots (median, quartiles and all points), Kruskal-Wallis test and Dunn’s multiple comparisons test (*P <0.05; **P <0.01; ****P <0.0001). (F) Staining of frozen HpG biopsies, showing a distinct subepithelial MICA/B deposition (arrows), scale bars: 50µm.
We next assessed MICA/B protein localization in human stomach biopsies by IHC (Figures 4D–F). In healthy controls, strong intracellular MICA/B staining was observed in epithelial cells (Figures 4D, E), consistent with the presumed intracellular MICA/B protein localization (11, 12), and likely indicating the cells’ constitutive readiness to respond to stress. Only sparse MICA/B staining was detectable in the lamina propria in healthy controls (Figures 4D, E). In HpG, epithelial MICA/B staining was significantly lower compared to healthy controls, or completely absent (Figures 4D, E). However, abundant MICA/B staining was visible in the lamina propria of HpG cases, which was either associated with the immune infiltrate or free without any cell association (Figures 4D, E). MICA/B staining in HpG was also visible as a rim below the epithelial cells (Figure 4F), suggesting that epithelial MICA/B was translocated from the basolateral site to the lamina propria. In stomach cancer, epithelial MICA/B staining was also lower and lamina propria staining was also higher compared to healthy controls, but not as high as in HpG (Figures 4D, E). There were no significant differences in MICA/B expressions in relation to gender (data not shown). Altogether, these data suggest that in HpG and gastric adenocarcinoma, a translocation of NKG2D-Ls from the epithelia to the lamina propria likely happens. Consequently, soluble NKG2D-Ls in the lamina propria could encounter NKG2D-expressing effector cells and attenuate their cytotoxicity.
3.5 Soluble NKG2D-Ls from H. pylori-infected epithelia reduce surface expression of the NKG2D receptor and cytotoxic degranulation of NK cells
Soluble NKG2D-Ls act as strong repressors of NKG2D-mediated immunity by reducing NKG2D expression of effector cells and thereby diminishing NKG2D-mediated effector cell cytotoxicity (20–22). To determine the effect of H. pylori-induced soluble MICA/B on the expression of NKG2D on effector cells, we challenged NK cells (NKL cell line) with filter-sterilized cell culture supernatants from H. pylori-infected or non-infected AGS-MICA cells (termed ‘H. pylori-infected supernatant’ and ‘non-infected supernatant’). Supernatants were filter-concentrated and applied at three concentrations (1x, 2.5x and 5x concentrated). To neutralize the effects of sMICA/B, an anti-MICA/B neutralizing antibody (termed ‘α-MICA/B’), was added to the cultures. Alternatively, an isotype control antibody (termed ‘isotype’) was used. After 24 h of challenge, cell surface expressions of NK cell proteins were analyzed by flow cytometry (Figures 5A–C). Control measurements of the NK cell surface marker CD94 showed no changes after any treatment, ruling out non-specific effects of the supernatants on the expression of NK cell surface proteins (Figure 5B). NKG2D expression was reduced by treatment with H. pylori-infected supernatant + isotype compared to untreated NK cells, and the reduction was concentration-dependent, with the greatest reduction being induced by 5x concentrated supernatant (Figures 5B, C). Treatment with non-infected supernatant + isotype also reduced NKG2D surface expression compared to untreated NK cells, although to a lesser extent compared to H. pylori-infected supernatant + isotype (Figures 5B, C), consistent with the constitutive shedding of NKG2D-Ls from AGS-MICA cells (Figure 3E). Importantly, addition of the anti-MICA/B antibody significantly attenuated NKG2D downregulation (Figures 5B, C), indicating that sMICA/B proteins in the supernatants cause the reduction of NKG2D expression. The effect of H. pylori-infected supernatant was not completely abrogated by addition of the anti-MICA/B antibody (Figures 5B, C), suggesting that additional factors in these supernatants also contribute to NKG2D downregulation.
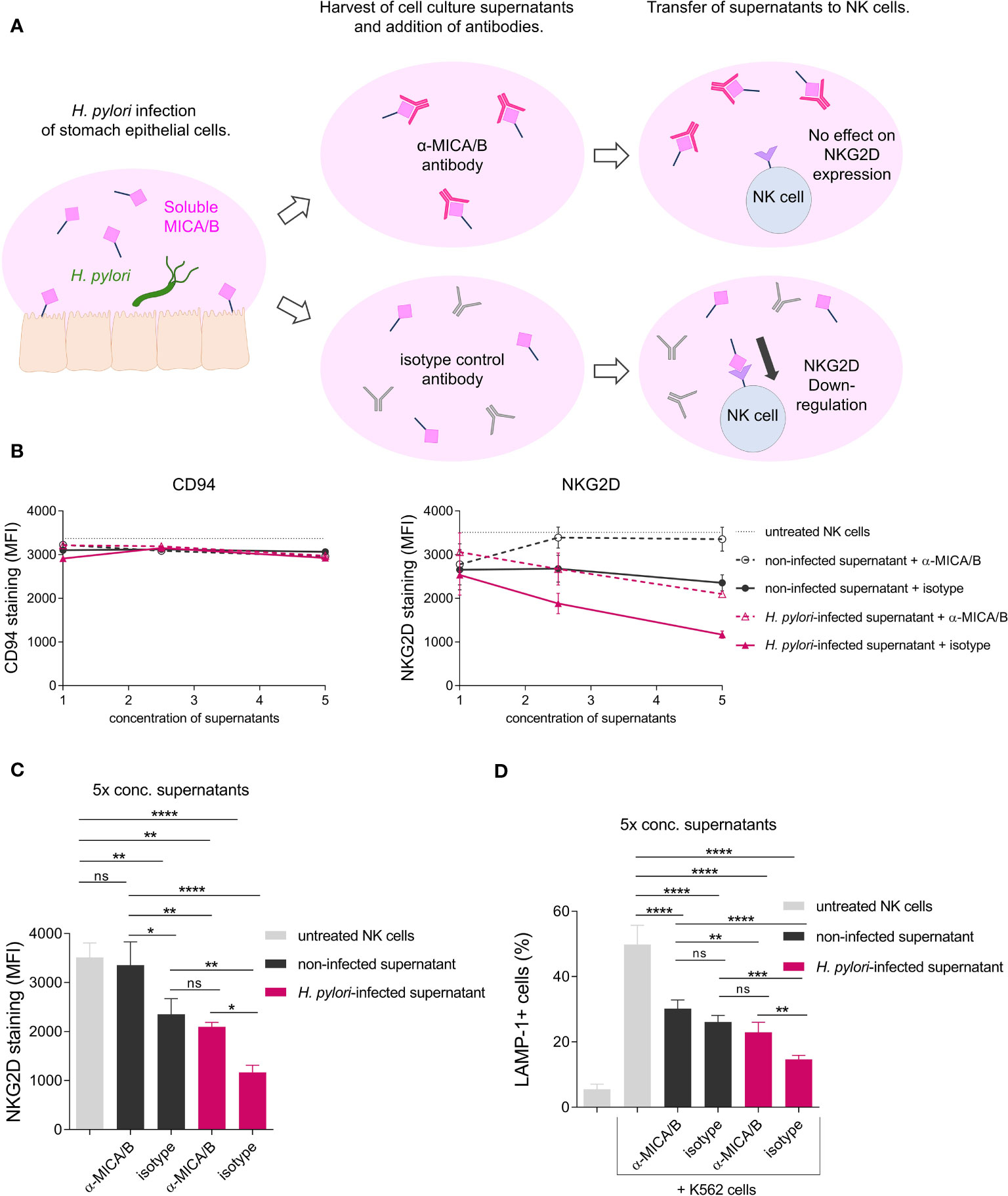
Figure 5 Soluble NKG2D-Ls from H. pylori-infected epithelia reduce surface expression of the NKG2D receptor and cytotoxic degranulation of NK cells. (A) Scheme of the experimental setup. (B, C) The NK cell line NKL was challenged with filter-sterilized cell culture supernatants from non-infected AGS-MICA cells (‘non-infected supernatant’) or from H. pylori-infected AGS-MICA cells (‘H. pylori-infected supernatant’). Supernatants were filter-concentrated and applied at three concentrations (1x, 2.5x, 5x) and either a neutralizing anti-MICA/B antibody (‘α-MICA/B’) or an isotype control antibody (‘isotype’) were added to the cultures. After 24 h, cell surface expressions of CD94 and NKG2D and were analyzed by flow cytometry. NKG2D expression after treatment with 5x concentrated supernatants is individually shown in (C, D) NKL cells were treated for 24 h with 5x concentrated supernatants as described above and then co-cultivated with K562 cells for 2 h Percentages of LAMP-1+ cells (from CD94+ cells) were determined by flow cytometry. Experiments were performed three times, mean ± SD. One-way ANOVA and Tukey’s test (*P <0.05; **P <0.01; ***P <0.001, ns, not significant).
To characterize the functional consequence of reduced NKG2D expression, we co-cultured NKL cells with the tumor cell line K562 for 2 h and then measured cell surface expression of lysosomal-associated membrane protein 1 (LAMP-1) on NKL cells as a marker for cytotoxic degranulation, indicating the cytotoxic reactivity of NK cells toward tumor cells (54). Co-cultivation with K562 cells induced a marked increase in LAMP-1 expression on untreated NKL cells (Figure 5D), confirming activation of NK cell cytotoxic degranulation upon exposure to tumor cells. Next, we pre-treated NKL cells with cell culture supernatants from H. pylori-infected or non-infected AGS-MICA cells as described above, before co-cultivation with K562 cells. NKL cells pre-treated with H. pylori-infected supernatant + isotype showed significantly reduced LAMP-1 expression compared to untreated NKL cells (Figure 5D). This reduction was significantly reversed by addition of the anti-MICA/B antibody during the pre-treatment (Figure 5D), indicating that sMICA/B proteins in H. pylori-infected supernatants act to suppress NK cell cytotoxic degranulation. NKL cells pre-treated with non-infected supernatant + isotype also showed reduced LAMP-1 expression compared to untreated NKL cells, but to a lesser extent compared to NKL cells pre-treated with H. pylori-infected supernatant + isotype (Figure 5D). This reduction was not reversed by addition of the anti-MICA/B antibody (Figure 5D), thus, it was not driven by MICA/B but likely by other immunomodulatory factors secreted by these stomach epithelial cells. Notably, tumor cell killing was also diminished when NKL cells were pre-treated with supernatants from H. pylori-infected epithelial cells, compared to untreated NKL cells or NK cells pre-treated with supernatants from non-infected epithelial cells (Supplementary Figure 13). In summary, these data demonstrate that H. pylori infection induces the release of soluble NKG2D-Ls from stomach epithelial cells which leads to reduced NKG2D receptor expression and attenuated cytotoxic degranulation of NK cells. Thus, impairment of the NKG2D system by H. pylori may weaken immune surveillance which could facilitate the development of gastric cancer (Figure 6).
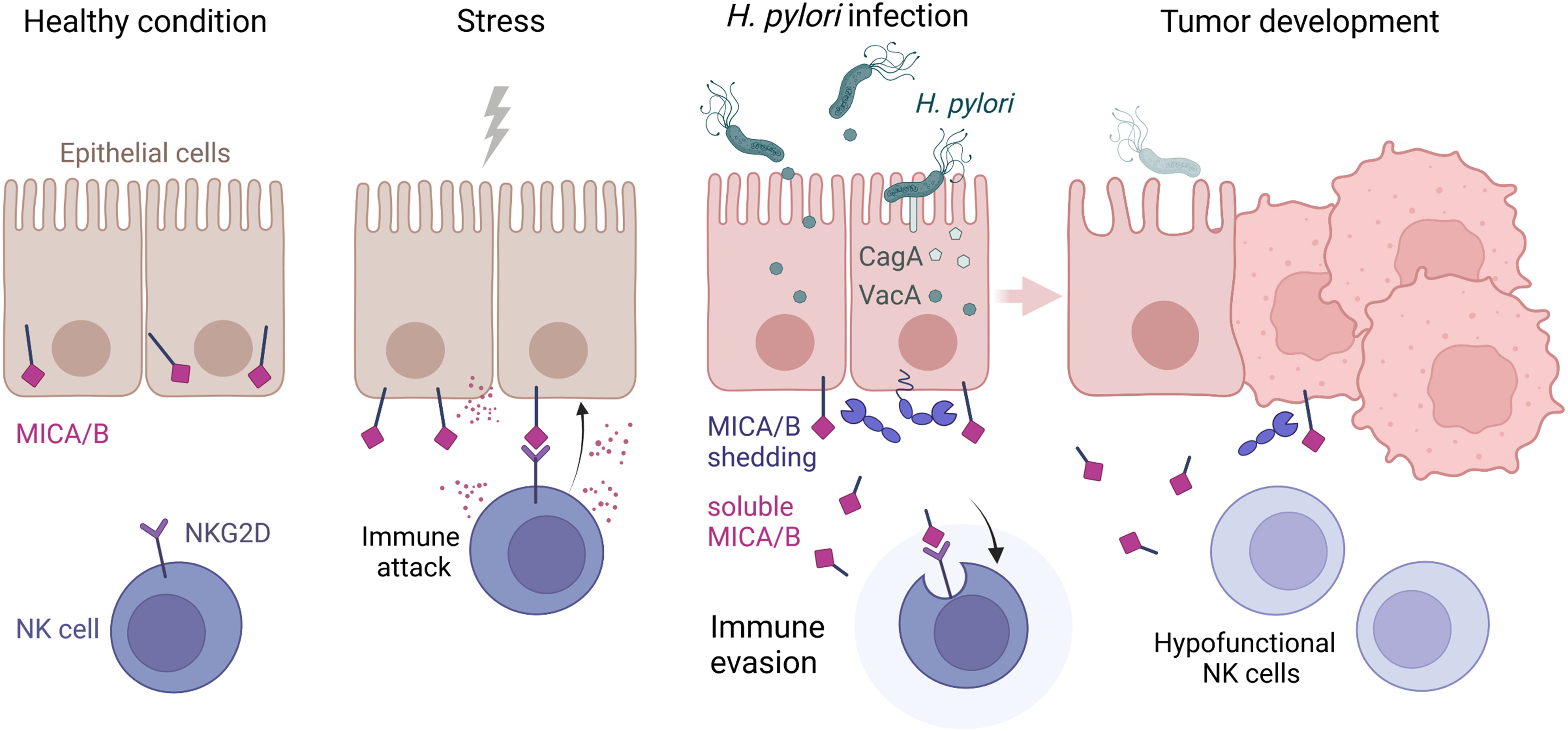
Figure 6 Scheme highlighting the NKG2D system modulation by H. pylori. Healthy epithelia store MICA and MICB proteins intracellularly. Upon stress, MICA/B are expressed at the cell surface and bound by the immunoreceptor NKG2D, which activates immune attack by NKG2D-harboring effector cells. During H. pylori infection, virulence factors CagA and VacA modify the expression and proteolytic shedding of MICA/B. Soluble MICA/B proteins lead to immune evasion by binding NKG2D, which results in internalization and downregulation of NKG2D and a suppression of lymphocyte cytotoxicity. Hypofunctional NK and cytotoxic T cells in the stomach lamina propria could allow transformed cells to escape immune surveillance and facilitate tumor development. Scheme drawn with BioRender (https://biorender.com).
4 Discussion
The immune activating receptor NKG2D and its ligands represent an immune recognition system important for the activation of NK cells, cytotoxic- and γδ T-cells in response to cellular stress such as infection or oncogenic transformation (6). The NKG2D system is involved in mucosal immunity (24), and altered in gastrointestinal pathologies such as gastritis, celiac disease and Crohn’s disease (26–28). It also represents a key component for tumor immune surveillance and tumors as well as viruses have acquired mechanisms to escape NKG2D-dependent immunity (17). Using human tissues and cell culture-based infection models we show that the NKG2D system can be influenced by H. pylori. Induction of NKG2D-L expression and the release of these ligands as soluble molecules were H. pylori virulence factor-dependent processes and mucosal NKG2D-L deposition was evident in H. pylori-associated pathologies. By modeling the effect of soluble NKG2D-Ls on effector cells, we verified H. pylori’s ability to attenuate NK cytotoxic activity, providing evidence for a so far unprecedented immune evasion mechanism by this prominent human pathogen.
Gastric H. pylori infection is characterized by pronounced inflammation of the mucosa, exhibiting abundant infiltration with plasma-, B- and T-cells with pro- (e.g., T helper cells Th1, Th17) but also anti- (e.g., Tregs) inflammatory properties. Individual compositions of these leucocytes influence disease courses (44, 55). The role of CTLs and NK cells in the stomach in H. pylori negative and positive individuals is less defined (44), and variable quantities of these cell types have been reported (56–59). We found no induction of NK cell and CTL numbers by IHC, despite a strong general immune infiltration in H. pylori gastritis. NKG2D is one of the main activating receptors of NK cells and a major co-stimulatory receptor of CTLs (6). We observed a significant downregulation of NKG2D gene expression by qPCR. In parallel, the NKG2D-L MICB was significantly upregulated in gastritis and adenocarcinoma cases, suggesting a dysregulation of the NKG2D system. Besides NKG2D-Ls (60) certain cytokines might also regulate NKG2D receptor expression, like IL-15 acting as an inducer or TGF-β and MIF acting as repressors (45, 46). We found that H. pylori infection induced IL-15 and TGF-β in AGS and MKN28 cells whereas MIF was not induced. Since these cytokines could also be produced by other cell types including leukocytes or stromal cells in addition to epithelial cells, various cell types might influence NKG2D system dysregulation in different gastric pathologies.
NKG2D-Ls are generally absent on cell surfaces in health (12). In the gut, their expression is also co-regulated by the microbiota (25). Stress conditions such as infections or neoplastic transformation, however, induce their de novo expression and surface display (8, 13–15). This induction of surface expression can be overcome by viruses and tumors, either by downregulating NKG2D-L expression or by releasing NKG2D-Ls as soluble proteins from the cell surface (17–19, 61, 62). Soluble NKG2D-Ls attenuate the strength of the immune response by acting as suppressors of NKG2D-expressing effector cells. Their binding to the receptor leads to NKG2D downregulation and thus diminished NKG2D-mediated immunity, as well as a general impairment of effector cell function (20–23). Using cell-culture based infection models, we found that H. pylori infection of stomach epithelial cells induced both gene expression and soluble release of the NKG2D-Ls MICA and MICB and reduced MICA/B cell surface expression. In vivo, MICA/B was reduced at epithelial cells but accumulated in the lamina propria in gastritis and adenocarcinoma cases. Since NK cells are essential for early detection of carcinogenesis (63), soluble NKG2D-Ls in the lamina propria in gastritis might impair the eradication of spontaneously transformed cells, thereby facilitating cancer development. In gastric adenocarcinoma, the elevated MICA/B levels in the lamina propria were likely a consequence of immune evasion by the cancer itself, since H. pylori is usually lost from the stomach with the development of advanced disease (64), and undetectable by the time of clinical cancer diagnosis. Release of soluble NKG2D-Ls is common in advanced cancer and high serum levels of soluble NKG2D-Ls correlate with a systemic reduction of NKG2D expression and with high cancer stage, metastatic disease and overall poor prognosis (20, 65–67). Therapeutic applications to overcome NKG2D system evasion in cancer could be a useful complement to other forms of tumor immunotherapy and are currently being explored (18). In our study we focused on NK cells, as they are the most extensively studied cell type in the context of the NKG2D-mediated immune response. Other NKG2D-expressing cell types like γδ T cells were also shown to play important roles in tumor immunity (15, 68). γδ T cells are highly abundant in the gut mucosa as intraepithelial lymphocytes (IELs) where they are important for immune surveillance and tissue homeostasis (69). Thus, inhibition of γδ T cells by H. pylori could potentially also contribute to bacterial persistence and the promotion of neoplastic processes.
H. pylori strains differ in their allelic compositions of the main virulence factors vacA and cagA. VacA is a pore-forming toxin and all strains harbor the vacA gene but different alleles exist wherein the alleles s1, m1 and i1 are associated with a higher risk for stomach cancer (1). CagA interferes with various host cell signaling processes and the cagA gene is considered an oncogene (70). CagA is injected into host cells by a type IV secretion system (T4SS), where the protein CagL is a crucial component for CagA translocation. Not all strains carry cagA and different cagA alleles vary in their virulence potential (71). Interestingly, cagA and vacA alleles show genetic associations. CagA positive strains typically contain the more pathogenic vacA alleles and the two toxins seem to counterbalance each other’s effects to reduce mucosal harm, allowing persistent colonization of H. pylori (1, 37). We identified another example for this synergism. While vacA seemed to induce NKG2D-L gene expression, which could activate the immune response against infected epithelial cells, cagA seemed to trigger NKG2D-L shedding, which can reduce the cells’ immunogenic visibility. The combined actions of both toxins might potentiate the amount of soluble NKG2D-Ls released, which could result in strong suppression of NKG2D-mediated immunity. The H. pylori strain P12 used in this study, which showed vacA-dependent induction of NKG2D-L gene expression, harbors the highly pathogenic vacA allele s1m1 (33). Notably, induction of NKG2D-L gene expression was absent in our previous study, where we used the H. pylori strains PMSS1 and SS1, which contain the less pathogenic vacA allele s2m2 (26, 72). Thus, induction of NKG2D-L gene expression is likely associated with specific H. pylori strains leading to varying disease outcomes (1). So far only a single report describes the effect of H. pylori on the NKG2D axis (73). Heat-killed H. pylori induced MICA gene expression but not MICB, driven by H. pylori-LPS stimulating TLR4 in gastric epithelial cell lines activating also the cytotoxicity of peripheral blood lymphocytes. An important mechanistic difference of our study is the use of live H. pylori, which does not stimulate TLR4, in contrast to H. pylori-LPS alone (74). Moreover, heat-killed H. pylori cannot produce the pathogenicity factors VacA and CagA, which we identified as the major drivers of NKG2D-L modulation.
Soluble release of NKG2D-Ls is mediated by proteolytic shedding via metalloproteases (40, 52, 75), or by release on EVs (42). Using the broad-spectrum metalloprotease inhibitor batimastat, we found that the H. pylori-induced release of soluble NKG2D-Ls was specifically mediated by metalloproteases in MKN28 and AGS cells. ADAM17 is the best studied metalloprotease in the context of NKG2D-L shedding. This host protease is embedded in the cell membrane and gets activated when dissociated from α5β1 integrin (76). Curiously, the CagL protein located on the tip of the T4SS apparatus was shown to bind α5β1 integrin and thereby dissociate and activate ADAM17 (34). In this study, deletion of either cagL or cagA, significantly abrogated induction of MICA shedding. Silencing of ADAM17 had no significant impact on shedding frequencies, nor did silencing of MMP9, an additional host protease that was induced in H. pylori infection in a cagA/cagL-dependent manner (77). Whether other proteases or a synergism of certain proteases is responsible for MICA/B cleavage remains to be determined (52, 53). Bacterial proteases might also take part in NKG2D-L release since they are key virulence factors and modulate host cells in various ways (78). The main H. pylori protease HtrA, however, could not account for the observed shedding since it is a serine-protease, not sensitive to batimastat inhibition (79).
The MICA and MICB genes are located in the highly polymorphic major histocompatibility complex (MHC)-class I locus and originate from a gene duplication event (80, 81). The two genes are highly homologous, but display numerous mutations and 531 MICA alleles and 244 MICB alleles are currently known (http://hla.alleles.org/alleles/classo.html, March 2023) (82). These genetic variations were shown to affect MICA/B expression and regulation (18, 51, 83–89). We detected lower baseline gene expression and greater inducibility of MICB compared to MICA in the cell lines AGS and MKN28 (Supplementary Figure 4). Regulatory differences in NKG2D-L expression might result from evolutionary selective pressure, in response to the evasion strategies of pathogens and tumors. In addition, these variations might allow for a diversified reaction of the system to variable stressors (6). Interestingly, specific MICA/B alleles are associated with certain diseases such as coeliac disease (90), ulcerative colitis (91), but also cancer, with gastric adenocarcinoma being associated with the MICA alleles A9 and *009/049 (80, 92). MICA A9 might show increased sensitivity to protease shedding due to a long transmembrane domain near the proteolytic cleavage site (52, 92, 93). In contrast, the frequent allele MICA*008 (A5.1), that is found in 20-55% of humans with varying frequencies in different populations (42, 82, 94–96), is less susceptible to proteolytic-shedding because of a truncated transmembrane domain (Supplementary Figure 8) (42). Thus, specific MICA/B alleles might differ in their susceptibility to H. pylori-induced shedding and this could influence the risk of developing H. pylori-induced pathologies, including gastric cancer (92).
In conclusion, our study shows that H. pylori actively manipulates the NKG2D system. This could help to protect the epithelial site of bacterial colonization from immune attack and thus shield H. pylori from eradication and contribute to its persistent colonization in the gastric mucosa. Furthermore, this mechanism might facilitate the development of stomach cancer by reducing the tumor surveillance represented by NK cells and CTLs (97). Our study provides a novel example of active immunoreceptor and/or -ligand modulation by the microbiota, similar to programmed cell death 1 ligand 1 (PD-L1) modulation by H. pylori (98), or proteolytic cleavage of Toll-like receptors (TLRs) by microbes (99). Our data provide new insights into the molecular principles governing microbe-induced proteolytic ectodomain shedding and enhance our knowledge regarding infection strategies employed by pathogens. Furthermore, these findings support a better characterization of tumor immune escape mechanisms, which could fuel the development of immunotherapy approaches.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Tissue use was approved by the institutional review board of the Medical University of Graz (EK-23-212ex10/11).
Author contributions
MA: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. MW: Formal analysis, Investigation, Writing – review & editing. RH: Formal analysis, Investigation, Writing – review & editing. SE: Formal analysis, Methodology, Writing – review & editing. SW: Formal analysis, Investigation, Writing – review & editing. MM: Formal analysis, Investigation, Writing – review & editing. LS: Formal analysis, Investigation, Writing – review & editing. IK: Methodology, Writing – review & editing. SS: Methodology, Writing – review & editing. BJ: Methodology, Writing – review & editing. SK: Methodology, Supervision, Writing – review & editing. EZ: Methodology, Supervision, Writing – review & editing. GP: Methodology, Resources, Writing – review & editing. MV-G: Conceptualization, Methodology, Resources, Supervision, Writing – review & editing. HR: Methodology, Resources, Supervision, Writing – review & editing. GG: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported by the Austrian Science Fund (FWF, DK-MOLIN W1241 and the “Cluster of Excellence: Microbiomes Drive Planetary Health”) and by the Spanish Ministry of Science and Innovation under Grants (PID2021-123795OB-I00, PID2020-115506RB-I00) [Ministerio de Ciencia, Innovación y Universidades (MCIU)/Agencia Estatal de Investigación (AEI)/European Regional Development Fund (FEDER, EU)].
Acknowledgments
We are grateful to Silja Wessler for providing gastric epithelial cell lines and H. pylori strains. For technical support and helpful discussions we thank Ana Montalban-Arques, Philipp Wurm, Nandhitha Madhusudhan, Marija Durdevic, Onur Özbeöz, Leon Gorkiewicz, Matthew Madsen, Ane Calvo, Carmen Campos-Silva, Gloria Esteso, Maren Kramer, Yaiza Cáceres-Martell, Christina Skofler, Florian Kleinegger, Anton Ibovnik, Peter Abuja, Paul Vessely, Alejandro Majali-Martinez, Nassim Ghaffari-Tabrizi-Wizsy, Agnes Mooslechner, Esther Föderl-Höbenreich, Antonio Kouros, Sonja Rittchen, Stefan Schild, Franz Zingl, Herbert Strobl, Karin Wagner, Birgit Galle, Hannes Angerer, Dominic Sudy, Markus Absenger, Jennifer Ober and Heimo Strohmeier.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1282680/full#supplementary-material.
References
1. Robinson K, Atherton JC. The spectrum of helicobacter-mediated diseases. Annu Rev Pathol Mech Dis (2021) 16:123–44. doi: 10.1146/annurev-pathol-032520-024949
2. Pérez-Pérez GI, Sack RB, Reid R, Santosham M, Croll J, Blaser MJ. Transient and persistent Helicobacter pylori colonization in native American children. J Clin Microbiol (2003) 41:2401–7. doi: 10.1128/JCM.41.6.2401-2407.2003
3. Salama NR, Hartung ML, Müller A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol (2013) 11:385–99. doi: 10.1038/nrmicro3016
4. Reyes VE, Peniche AG. Helicobacter pylori deregulates T and B cell signaling to trigger immune evasion. Curr Top Microbiol Immunol (2019) 421:229–65. doi: 10.1007/978-3-030-15138-6_10
5. Borbet TC, Zhang X, Müller A, Blaser MJ. The role of the changing human microbiome in the asthma pandemic. J Allergy Clin Immunol (2019) 144:1457–66. doi: 10.1016/j.jaci.2019.10.022
6. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
7. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress- inducible MICA. Science (1999) 285:727–9. doi: 10.1126/science.285.5428.727
8. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8αβ T cell by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol (2001) 2:255–60. doi: 10.1038/85321
9. Poggi A, Benelli R, Venè R, Costa D, Ferrari N, Tosetti F, et al. Human gut-associated natural killer cells in health and disease. Front Immunol (2019) 10:961. doi: 10.3389/fimmu.2019.00961
10. Ma H, Tao W, Zhu S. T lymphocytes in the intestinal mucosa: defense and tolerance. Cell Mol Immunol (2019) 16:216–24. doi: 10.1038/s41423-019-0208-2
11. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA (1996) 93:12445–50. doi: 10.1073/pnas.93.22.12445
12. Ghadially H, Brown L, Lloyd C, Lewis L, Lewis A, Dillon J, et al. MHC class i chain-related protein A and B (MICA and MICB) are predominantly expressed intracellularly in tumour and normal tissue. Br J Cancer (2017) 116:1208–17. doi: 10.1038/bjc.2017.79
13. Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, et al. MICA engagement by human Vγ2Vδ2 T cells enhances their antigen-dependent effector function. Immunity (2001) 15:83–93. doi: 10.1016/S1074-7613(01)00168-6
14. Tieng V, Le Bouguénec C, Du Merle L, Bertheau P, Desreumaux P, Janin A, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci USA (2002) 99:2977–82. doi: 10.1073/pnas.032668099
15. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived γδ T cells of MICA and MICB. Proc Natl Acad Sci USA (1999) 96:6879–84. doi: 10.1073/pnas.96.12.6879
16. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. Cutting edge: NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol (2001) 167:5527–30. doi: 10.4049/jimmunol.167.10.5527
17. Baugh R, Khalique H, Seymour LW. Convergent evolution by cancer and viruses in evading the nkg2d immune response. Cancers (Basel) (2020) 12:1–27. doi: 10.3390/cancers12123827
18. Schmiedel D, Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front Immunol (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
19. Reyburn H, Esteso G, Ashiru O, Vales-Gomez M. Viral strategies to modulate NKG2D-ligand expression in Human Cytomegalovirus infection. New Horizons Transl Med (2015) 2:159–66. doi: 10.1016/j.nhtm.2015.11.002
20. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419:734–8. doi: 10.1038/nature01112
21. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol (2003) 171:6891–9. doi: 10.4049/jimmunol.171.12.6891
22. De Andrade LF, En Tay R, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science (2018) 359:1537–42. doi: 10.1126/science.aao0505
23. Hanaoka N, Jabri B, Dai Z, Ciszewski C, Stevens AM, Yee C, et al. NKG2D initiates caspase-mediated CD3ζ Degradation and lymphocyte receptor impairments associated with human cancer and autoimmune disease. J Immunol (2010) 185:5732–42. doi: 10.4049/jimmunol.1002092
24. Antonangeli F, Soriani A, Cerboni C, Sciumè G, Santoni A. How mucosal epithelia deal with stress: Role of NKG2D/NKG2D ligands during inflammation. Front Immunol (2017) 8:1583. doi: 10.3389/fimmu.2017.01583
25. Hansen CHF, Holm TL, Krych L, Andresen L, Nielsen DS, Rune I, et al. Gut microbiota regulates NKG2D ligand expression on intestinal epithelial cells. Eur J Immunol (2013) 43:447–57. doi: 10.1002/eji.201242462
26. Montalban-Arques A, Wurm P, Trajanoski S, Schauer S, Kienesberger S, Halwachs B, et al. Propionibacterium acnes overabundance and natural killer group 2 member D system activation in corpus-dominant lymphocytic gastritis. J Pathol (2016) 240:425–36. doi: 10.1002/path.4782
27. Hüe S, Mention JJ, Monteiro RC, Zhang SL, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity (2004) 21:367–77. doi: 10.1016/j.immuni.2004.06.018
28. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132:2346–58. doi: 10.1053/j.gastro.2007.03.025
29. Cutler AF, Havstad S, Ma CK, Blaser MJ, Perez-Perez GI, Schubert TT. Accuracy of invasive and noninvasive tests to diagnose Helicobacter pylori infection. Gastroenterology (1995) 109:136–41. doi: 10.1016/0016-5085(95)90278-3
30. Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res (2001) 29:e45. doi: 10.1093/nar/29.9.e45
31. Schneider S, Carra G, Sahin U, Hoy B, Rieder G, Wessler S. Complex cellular responses of Helicobacter pylori-colonized gastric adenocarcinoma cells. Infect Immun (2011) 79:2362–71. doi: 10.1128/IAI.01350-10
32. Poppe M, Feller SM, Römer G, Wessler S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene (2007) 26:3462–72. doi: 10.1038/sj.onc.1210139
33. Schmitt W, Haas R. Genetic analysis of the Helicobacter pylori vacuoiating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol Microbiol (1994) 12:307–19. doi: 10.1111/j.1365-2958.1994.tb01019.x
34. Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology (2010) 139:239–48. doi: 10.1053/j.gastro.2010.03.036
35. Hoy B, Löwer M, Weydig C, Carra G, Tegtmeyer N, Geppert T, et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep (2010) 11:798–804. doi: 10.1038/embor.2010.114
36. Tegtmeyer N, Wessler S, Backert S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J (2011) 278:1190–202. doi: 10.1111/j.1742-4658.2011.08035.x
37. Palframan SL, Kwok T, Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol (2012) 2:92. doi: 10.3389/fcimb.2012.00092
38. Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature (2007) 449:862–6. doi: 10.1038/nature06187
39. Zhang C, Wang Y, Zhou Z, Zhang J, Tian Z. Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer Immunol Immunother (2009) 58:1275–85. doi: 10.1007/s00262-008-0645-8
40. Boutet P, Agüera-González S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, et al. Cutting edge: the metalloproteinase ADAM17/TNF-α-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol (2009) 182:49–53. doi: 10.4049/jimmunol.182.1.49
41. Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, et al. High-level transduction and gene expression in hematopoietic repopulating cells using a human imunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther (2002) 13:803–13. doi: 10.1089/10430340252898984
42. Ashiru O, López-Cobo S, Fernández-Messina L, Pontes-Quero S, Pandolfi R, Reyburn HT, et al. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem J (2013) 454:295–302. doi: 10.1042/BJ20130194
43. Gonzalez S, Martinez-Borra J, Torre-Alonso JC, Gonzalez-Roces S, Sanchez Del Río J, Rodriguez Pérez A, et al. The MICA-A9 triplet repeat polymorphism in the transmembrane region confers additional susceptibility to the development of psoriatic arthritis and is independent of the association of Cw*0602 in psoriasis. Arthritis Rheum (1999) 42:1010–6. doi: 10.1002/1529-0131(199905)42:5<1010::AID-ANR21>3.0.CO;2-H
44. White JR, Winter JA, Robinson K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J Inflammation Res (2015) 8:137–47. doi: 10.2147/JIR.S64888
45. Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γc cytokines and TGF-β1. Blood (2011) 118:3019–27. doi: 10.1182/blood-2011-04-346825
46. Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Hönig A, Häusler S, et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J Immunol (2008) 180:7338–48. doi: 10.4049/jimmunol.180.11.7338
47. Li Z, Groh V, Strong RK, Spies T. A single amino acid substitution causes loss of expression of a MICA allele. Immunogenetics (2000) 51:246–8. doi: 10.1007/s002510050039
48. Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol (2013) 78:120–9. doi: 10.1111/sji.12072
49. Fernández-Messina L, Reyburn HT, Valés-Gómez M. Human NKG2D-ligands: Cell biology strategies to ensure immune recognition. Front Immunol (2012) 3:299. doi: 10.3389/fimmu.2012.00299
50. Yamanegi K, Yamane J, Kobayashi K, Ohyama H, Nakasho K, Yamada N, et al. Downregulation of matrix metalloproteinase-9 mRNA by valproic acid plays a role in inhibiting the shedding of MHC class I-related molecules A and B on the surface of human osteosarcoma cells. Oncol Rep (2012) 28:1585–90. doi: 10.3892/or.2012.1981
51. Shiraishi K, Mimura K, Kua LF, Koh V, Siang LK, Nakajima S, et al. Inhibition of MMP activity can restore NKG2D ligand expression in gastric cancer, leading to improved NK cell susceptibility. J Gastroenterol (2016) 51:1101–11. doi: 10.1007/s00535-016-1197-x
52. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
53. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Fürst D, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer (2013) 133:1557–66. doi: 10.1002/ijc.28174
54. López-Cobo S, Pieper N, Campos-Silva C, García-Cuesta EM, Reyburn HT, Paschen A, et al. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology (2018) 7:1–13. doi: 10.1080/2162402X.2017.1392426
55. Sundquist M, Quiding-Järbrink M. Helicobacter pylori and its effect on innate and adaptive immunity: New insights and vaccination strategies. Expert Rev Gastroenterol Hepatol (2010) 4:733–44. doi: 10.1586/egh.10.71
56. Agnihotri N, Bhasin DK, Vohra H, Ray P, Singh K, Ganguly NK. Characterization of lymphocytic subsets and cytokine production in gastric biopsy samples from Helicobacter pylori patients. Scand J Gastroenterol (1998) 33:704–9. doi: 10.1080/00365529850171639
57. Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology (1998) 114:482–92. doi: 10.1016/S0016-5085(98)70531-1
58. Yun CH, Lundgren A, Azem J, Sjöling Å, Holmgren J, Svennerholm AM, et al. Natural killer cells and Helicobacter pylori infection: Bacterial antigens and interleukin-12 act synergistically to induce gamma interferon production. Infect Immun (2005) 73:1482–90. doi: 10.1128/IAI.73.3.1482-1490.2005
59. Lundgren A, Trollmo C, Edebo A, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ T cells home to and accumulate in the human Helicobacter pylori-infected gastric mucosa. Infect Immun (2005) 73:5612–9. doi: 10.1128/IAI.73.9.5612-5619.2005
60. Molfetta R, Quatrini L, Zitti B, Capuano C, Galandrini R, Santoni A, et al. Regulation of NKG2D expression and signaling by endocytosis. Trends Immunol (2016) 37:790–802. doi: 10.1016/j.it.2016.08.015
61. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous Malignancy. Immunity (2008) 28:723. doi: 10.1016/j.immuni.2008.04.001
62. McGilvray RW, Eagle RA, Watson NFS, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res (2009) 15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991
63. Huntington ND, Cursons J, Rautela J. The cancer–natural killer cell immunity cycle. Nat Rev Cancer (2020) 20:437–54. doi: 10.1038/s41568-020-0272-z
64. Karnes WE, Samloff IM, Siurala M, Kekki M, Sipponen P, Kim SWR, et al. Positive serum antibody and negative tissue staining for Helicobacter pylori in subjects with atrophic body gastritis. Gastroenterology (1991) 101:167–74. doi: 10.1016/0016-5085(91)90474-Y
65. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in Malignant diseases. Int J Cancer (2006) 118:684–7. doi: 10.1002/ijc.21382
66. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in Malignant diseases: Analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother (2006) 55:1584–9. doi: 10.1007/s00262-006-0167-1
67. Kumar V, Yi Lo PH, Sawai H, Kato N, Takahashi A, Deng Z, et al. Soluble MICA and a MICA variation as possible prognostic biomarkers for HBV-induced hepatocellular carcinoma. PloS One (2012) 7:5–10. doi: 10.1371/journal.pone.0044743
68. Wrobel P, Shojaei H, Schittek B, Gieselerà F, Wollenberg B, Kalthoff- H, et al. Lysis of a Broad Range of Epithelial Tumour Cells by Human cd T Cells: Involvement of NKG2D ligands and T-cell Receptor-versus NKG2D-dependent Recognition. Scand J Immunol (2007) 66:320–8. doi: 10.1111/j.1365-3083.2007.01963.x
69. Ribot JC, Lopes N, Silva-Santos B. γδ T cells in tissue physiology and surveillance. Nat Rev Immunol (2021) 21:221–32. doi: 10.1038/s41577-020-00452-4
70. Ohnishi N, Yuasa H, Tanaka S. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Chemtracts (2008) 21:121–3. doi: 10.1073/pnas.0711183105
71. Noto JM, Peek RM. The Helicobacter pylori cag Pathogenicity Island. Methods Mol Biol (2012) 921:41–50. doi: 10.1007/978-1-62703-005-2_7
72. Dey TK, Karmakar BC, Sarkar A, Paul S, Mukhopadhyay AK. A mouse model of helicobacter pylori infection. In: Methods in molecular biology. (Clifton, N.J.: Humana Press Inc) (2021) 2283:131–51. doi: 10.1007/978-1-0716-1302-3_14
73. Hernández C, Toledo-Stuardo K, García-González P, Garrido-Tapia M, Kramm K, Rodríguez-Siza JA, et al. Heat-killed Helicobacter pylori upregulates NKG2D ligands expression on gastric adenocarcinoma cells via Toll-like receptor 4. Helicobacter (2021) 26:1–14. doi: 10.1111/hel.12812
74. Mandell L, Moran AP, Cocchiarella A, Houghton JM, Taylor N, Fox JG, et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun (2004) 72:6446–54. doi: 10.1128/IAI.72.11.6446-6454.2004
75. Salih HR, Rammensee H-G, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
76. Grötzinger J, Lorenzen I, Düsterhöft S. Molecular insights into the multilayered regulation of ADAM17: The role of the extracellular region. Biochim Biophys Acta - Mol Cell Res (2017) 1864:2088–95. doi: 10.1016/j.bbamcr.2017.05.024
77. Mori N, Sato H, Hayashibara T, Senba M, Geleziunas R, Wada A, et al. Helicobacter pylori induces matrix metalloproteinase-9 through activation of nuclear factor κB. Gastroenterology (2003) 124:983–92. doi: 10.1053/gast.2003.50152
78. Posselt G, Crabtree JE, Wessler S. Proteolysis in helicobacter pylori-induced gastric cancer. Toxins (Basel) (2017) 9:134. doi: 10.3390/toxins9040134
79. Bernegger S, Vidmar R, Fonovic M, Posselt G, Turk B, Wessler S. Identification of Desmoglein-2 as a novel target of Helicobacter pylori HtrA in epithelial cells. Cell Commun Signal (2021) 19:108. doi: 10.1186/s12964-021-00788-x
80. Chen D, Gyllensten U. MICA polymorphism: biology and importance in cancer. Carcinogenesis (2014) 35:2633–42. doi: 10.1093/carcin/bgu215
81. Fukami-Kobayashi K, Shiina T, Anzai T, Sano K, Yamazaki M, Inoko H, et al. Genomic evolution of MHC class I region in primates. Proc Natl Acad Sci USA (2005) 102:9230–4. doi: 10.1073/pnas.0500770102
82. Klussmeier A, Massalski C, Putke K, Schäfer G, Sauter J, Schefzyk D, et al. High-throughput MICA/B genotyping of over two million samples: workflow and allele frequencies. Front Immunol (2020) 11:314. doi: 10.3389/fimmu.2020.00314
83. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol (2007) 178:961–9. doi: 10.4049/jimmunol.178.2.961
84. Toledano T, Vitenshtein A, Stern-Ginossar N, Seidel E, Mandelboim O. Decay of the stress-induced ligand MICA is controlled by the expression of an alternative 3′ Untranslated region. J Immunol (2018) 200:2819–25. doi: 10.4049/jimmunol.1700968
85. Rodríguez-Rodero S, González S, Rodrigo L, Fernández-Morera JL, Martínez-Borra J, López-Vázquez A, et al. Transcriptional regulation of MICA and MICB: A novel polymorphism in MICB promoter alters transcriptional regulation by Sp1. Eur J Immunol (2007) 37:1938–53. doi: 10.1002/eji.200737031
86. Nachmani D, Gutschner T, Reches A, Diederichs S, Mandelboim O. RNA-binding proteins regulate the expression of the immune activating ligand MICB. Nat Commun (2014) 5:1–13. doi: 10.1038/ncomms5186
87. Tsukerman P, Stern-Ginossar N, Gur C, Glasner A, Nachmani D, Bauman Y, et al. MiR-10b downregulates the stress-induced cell surface molecule MICB, a critical ligand for cancer cell recognition by natural killer cells. Cancer Res (2012) 72:5463–72. doi: 10.1158/0008-5472.CAN-11-2671
88. Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, et al. Host immune system gene targeting by a viral miRNA. Science (2007) 317:376–81. doi: 10.1126/science.1140956
89. del Toro-Arreola S, Arreygue-Garcia N, Aguilar-Lemarroy A, Cid-Arregui A, Jimenez-Perez M, Haramati J, et al. MHC class I-related chain A and B ligands are differentially expressed in human cervical cancer cell lines. Cancer Cell Int (2011) 11. doi: 10.1186/1475-2867-11-15
90. Lopez-Vazquez A, Rodrigo L, Fuentes D, Riestra S, Bousoño C, Garcia-Fernandez S, et al. MHC class I chain related gene A (MICA) modulates the development of coeliac disease in patients with the high risk heterodimer DQA1 *0501/DQB1 *0201. Gut (2002) 50:336–40. doi: 10.1136/gut.50.3.336
91. Fdez-Morera JL, Rodrigo L, López-Vázquez A, Rodero SR, Martínez-Borra J, Niño P, et al. MHC class I chain-related gene a transmembrane polymorphism modulates the extension of ulcerative colitis. Hum Immunol (2003) 64:816–22. doi: 10.1016/S0198-8859(03)00121-6
92. Toledo-Stuardo K, Ribeiro CH, Canals A, Morales M, Gárate V, Rodríguez-Siza J, et al. Major histocompatibility complex class I-related chain A (MICA) allelic variants associate with susceptibility and prognosis of gastric cancer. Front Immunol (2021) 12:645528. doi: 10.3389/fimmu.2021.645528
93. Mizuki N, Ota M, Kimura M, Ohno S, Ando H, Katsuyama Y, et al. Triplet repeat polymorphism in the transmembrane region of the MICA gene: A strong association of six GCT repetitions with Behçet disease. Proc Natl Acad Sci USA (1997) 94:1298–303. doi: 10.1073/pnas.94.4.1298
94. Petersdorf EW, Shuler KB, Longton GM, Spies T, Hansen JA. Population study of allelic diversity in the human MHC class I-related MIC-A gene. Immunogenetics (1999) 49:605–12. doi: 10.1007/s002510050655
95. Komatsu-Wakui M, Tokunaga K, Ishikawa Y, Kashiwase K, Moriyama S, Tsuchiya N, et al. MIC-A polymorphism in Japanese and a MIC-A-MIC-B null haplotype. Immunogenetics (1999) 49:620–8. doi: 10.1007/s002510050658
96. Pyo CW, Hur SS, Kim YK, Choi HB, Kim TY, Kim TG. Distribution of MICA alleles and haplotypes associated with HLA in the Korean population. Hum Immunol (2003) 64:378–84. doi: 10.1016/S0198-8859(02)00826-1
97. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev (2018) 32:1267–84. doi: 10.1101/gad.314617.118
98. Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased programmed death-ligand 1 is an early epithelial cell response to helicobacter pylori infection. PloS Pathog (2019) 15:1–30. doi: 10.1371/journal.ppat.1007468
Keywords: H. pylori, immune evasion, NK cells, cytotoxic T cells, stomach cancer, tumor immunity, stomach microbiota, NKG2D (natural killer group 2 member D)
Citation: Anthofer M, Windisch M, Haller R, Ehmann S, Wrighton S, Miller M, Schernthanner L, Kufferath I, Schauer S, Jelušić B, Kienesberger S, Zechner EL, Posselt G, Vales-Gomez M, Reyburn HT and Gorkiewicz G (2024) Immune evasion by proteolytic shedding of natural killer group 2, member D ligands in Helicobacter pylori infection. Front. Immunol. 15:1282680. doi: 10.3389/fimmu.2024.1282680
Received: 24 August 2023; Accepted: 02 January 2024;
Published: 22 January 2024.
Edited by:
Martin James Holland, University of London, United KingdomReviewed by:
Soraya Mezouar, Aix-Marseille University, FranceFarokh Jal Dotiwala, Ocugen, Inc., United States
Copyright © 2024 Anthofer, Windisch, Haller, Ehmann, Wrighton, Miller, Schernthanner, Kufferath, Schauer, Jelušić, Kienesberger, Zechner, Posselt, Vales-Gomez, Reyburn and Gorkiewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregor Gorkiewicz, gregor.gorkiewicz@medunigraz.at
†Present address: Sebastian Wrighton, Department of Clinical Sciences, Lund University, Lund, Sweden