 Olivia G. Thomas
Olivia G. Thomas Tomas Olsson
Tomas Olsson- 1Therapeutic Immune Design, Centre for Molecular Medicine, Department of Clinical Neuroscience, Karolinska Institute, Stockholm, Sweden
- 2Neuroimmunology Unit, Department of Clinical Neuroscience, Centre for Molecular Medicine, Karolinska Institute, Stockholm, Sweden
T cells have an essential role in adaptive immunity against pathogens and cancer, but failure of thymic tolerance mechanisms can instead lead to escape of T cells with the ability to attack host tissues. Multiple sclerosis (MS) occurs when structures such as myelin and neurons in the central nervous system (CNS) are the target of autoreactive immune responses, resulting in lesions in the brain and spinal cord which cause varied and episodic neurological deficits. A role for autoreactive T cell and antibody responses in MS is likely, and mounting evidence implicates Epstein-Barr virus (EBV) in disease mechanisms. In this review we discuss antigen specificity of T cells involved in development and progression of MS. We examine the current evidence that these T cells can target multiple antigens such as those from pathogens including EBV and briefly describe other mechanisms through which viruses could affect disease. Unravelling the complexity of the autoantigen T cell repertoire is essential for understanding key events in the development and progression of MS, with wider implications for development of future therapies.
Introduction
MS is the second most common cause of neurological disability amongst young adults after trauma affecting approximately 2.8million people globally (1). MS is more common in females with onset generally between 20 to 40 years of age, and occurs due to the formation of focal inflammatory demyelinating lesions in the CNS. Initial disease has a predominantly inflammatory component and approximately 85% of cases present with a relapsing-remitting phenotype, but over time accumulated damage and neurodegenerative mechanisms can lead to permanent disability. Genetic aetiology is estimated to be around 20-30% for MS with the remaining risk lying with stochastic events and environmental factors such as obesity, smoking, low serum vitamin D and EBV (2).
The brain and spinal cord, once thought to be an immune privileged compartment (3), are surveyed by patrolling T cells which guard against infection and also have roles in brain development and behaviour (4–8). However, the CNS is sensitive to inflammation-mediated damage and therefore, under healthy conditions, a carefully controlled balance is maintained between protection from infections and prevention of injurious inflammation. Loss or impairment of such CNS T cell immunosurveillance leads to an increased risk of CNS infections or malignancies (9).
Focal CNS lesions in early MS disease show widespread inflammatory infiltrates which contain a variety of immune cells including CD4+ T cells, CD8+ T cells B cells and monocytes. Genome-wide association studies (GWAS), as well as clinical therapeutic observations, indicate that T cells and B cells alongside innate immune cells have a key roles in MS neuroinflammatory mechanisms (10–15). The majority of MS-associated genes have functions in antigen presentation, cytokine production, proliferation, T helper (TH) cell differentiation, co-stimulation, signal transduction and function. Associated polymorphisms have been identified in the human leukocyte antigen (HLA) locus as well as in IL-2Rα, IL-7Rα, CXCR5, CD40, CD86, STAT3 and many other genes (15–17). However, the strongest known genetic risk factor associated with MS is the HLA-DRB1*15:01 allele with an odds ratio (OR) of approximately 3 (15–18), and has also been shown to interact with multiple other environmental risk factors to increase risk further (19–24). The HLA locus also contains several other class II alleles which confer risk for developing MS and several identified HLA class I alleles which protect from disease such as HLA-A*02:01 (17, 25). CD4+ T cells recognise peptides presented in the context of HLA class II molecules, whereas CD8+ T cells recognise class I-presented peptides. Given that the function of HLA is to present peptides to T cells for recognition via T cell receptors (TCR), the association of HLA-DRB1*15:01 with MS development suggests a role for class II-presented peptides and autoreactive CD4+ T cell responses and has led to substantial investigation of the autoantigens responsible for priming of pathogenic responses.
Early research established in particular the role of CD4+ T cells which target these antigens due to several observations, such as the presence of CNS-infiltrating CD4+ T cells in MS brain lesions, genetic risk conferred by HLA-DR and HLA-DQ alleles, increased experimental autoimmune encephalomyelitis (EAE) susceptibility of transgenic mice expressing MS-associated HLA class II molecules, and it is likely that CD4+ T cells also contribute to MS pathogenesis via their influence on both adaptive and innate immune processes such as antibody production by B cells and CD8+ T cell maturation.
To prevent production of T cells which can target self-tissues and trigger autoimmunity, T cells undergo central tolerance mechanisms in the thymus during their maturation, where cells expressing T cell receptors (TCRs) that bind strongly to peptide:HLA complexes on thymic epithelial cells and dendritic cells are negatively selected, and those that do not bind at all die by neglect (26). Additional mechanisms in the periphery also act to remove self-reactive T cells which escape central tolerance (27). Due to these mechanisms, TCRs which pass thymic tolerance quality control and bind foreign antigens generally have high affinity for their cognate antigen, however some responses may also have the ability to bind other peptides presented on HLA at different affinity which could include those from self-antigens. The classical view of T cells is that a single TCR expressed at the cell surface allows them to bind one peptide:HLA complex but the reality is that a single TCR can likely bind multiple peptides, and possibly also different HLA molecules (28). This existence of cross-reactivity might be an evolutionary advantage of a limited genome-encoded TCR repertoire against the myriad of possible peptide combinations which can occur in nature, estimated to be around 1015 possible peptides (29, 30). This T cell degeneracy indicates a potential for TCRs which were originally selected during exposure to prior foreign antigens to also bind self-peptides presented by HLA, and therefore the pathogens which we encounter throughout life shape both the memory T cell repertoire as a whole and also its autoreactive potential.
As well as T cells, there is strong evidence supporting a role for B cells in MS development, in particular due to the dramatic therapeutic effect of anti-CD20 therapies (11, 12). The reasons for which include B cell antigen presentation to T cells, production of pro-inflammatory cytokines, elimination of Epstein-Barr virus (EBV), and finally production of pathogenic autoantibodies – not in prime focus for this review. This review will discuss the current knowledge surrounding T cell specificity in MS, evaluating the existing evidence that CNS autoreactive responses may originally have been generated in response to non-self antigens and briefly describing other mechanisms through which viruses could affect disease.
Selected evidence for the role of T cells in MS
For many years CD4+ T cells have been considered an important cell type involved in MS pathogenesis due to early observations in experimental autoimmune encephalomyelitis (EAE) – the animal model for MS – that CNS demyelinating disease can be transferred by adoptive transfer of myelin-reactive CD4+ T cells (31, 32), and further evidenced by the observation that EAE cannot be transferred by antibodies alone. The role of CD4+ T cells in MS has been further demonstrated by the strongest genetic susceptibility conferred by HLA class II alleles (15), susceptibility of HLA class II-carrying mice to demyelinating disease (33–36), presence of CD4+ T cells in inflammatory brain lesions (37), and the involvement of CD4+ T cells in several other arms of adaptive immunity such as antibody production and CD8+ T cell maturation.
TH1 and TH17 CD4+ T cells have been linked to MS disease with identification of these specific subsets in MS brain lesions and correlation of TH1 cytokine-producing cells in peripheral blood with MS relapses (38–41). Further studies have also demonstrated a role for a unique intermediate population of TH1-like TH17 CD4+ T cells in MS which are associated with relapse, predominant in the CSF of early disease pwMS and can be isolated from MS brain lesions (42, 43). High avidity CD4+ T cells with specificity for selected myelin antigens have also been shown to produce interferon-γ (IFNγ) and have a TH1 phenotype in persons with MS (pwMS) (44–47). A clinical trial of a myelin basic protein (MBP) altered peptide ligand (APL) showed exacerbations in some MS patients, and further investigation showed cross-recognition between the APL and MBP driven by CD4+ T cells which were skewed towards a TH1 phenotype (45). In addition to TH1, CD4+ T cells with a TH17 phenotype have also been detected in MS brain lesions and have been shown to be necessary for the development of EAE (48, 49). Follicular helper CD4+ T cells (TFH) cells provide help to B cells for their maturation, affinity maturation and antibody production, and germinal centre formation, and this essential link between humoral and cellular immunity makes them of key interest in MS pathology due to the involvement of B cells in disease. Activated TFH cells have been shown to be increased in peripheral blood, their frequency correlated with disability and are detectable in brain lesions in MS (50–52).
The role of CD8+ T cells is less clear though several observations suggest their involvement in MS such as high abundance in MS lesions, low or transient expression of HLA class I molecules on the surface of microglia, oligodendrocytes and neurons (53, 54), and observations that EAE does not develop in B2-microglobulin knockout mice (55). Several HLA class I associations with MS have also been identified, such as the strongest known protective alleles HLA-A*02:01 and HLA-B*38:01 (25) mentioned above, although untangling HLA associations with disease is notoriously difficult due to linkage disequilibrium and also other factors such as killer-immunoglobulin receptor (KIR) type, which can have a significant effect on immune activation in natural killer (NK) cell subsets (56). In addition, HLA-A*02:01 protection against MS may be related to actions in the type I interferon system rather than peptide binding and activation of CD8+ T cells (57). Myelin-reactive CD8+ T cells have also been characterised – although to a lesser extent than CD4+ T cells – and have been isolated from both pwMS and healthy individuals. Studies of brain-infiltrating CD8+ T cells in MS have shown their TCR repertoire to be oligoclonal, suggesting antigen-specific migration or expansion within the CNS (58, 59), and other studies have directly enumerated autoantigen-specific CD8+ T cells from peripheral blood of pwMS (60, 61).
Mechanisms of pathogenic cross-reactivity
A long array of infectious agents have been associated with MS however the strongest evidence lies with EBV and – to some extent – human herpesvirus 6A (HHV-6A) (2, 62–65). In addition to this, the long-list of MS-associated autoantigens has grown in recent years which has broadened the focus of research in this area and led to some debate on which antigens and pathogens are pathologically relevant in neuroimmunological demyelinating disease (66–72). The association of these viruses and other pathogens in the context of the molecular mimicry with CNS autoantigens in MS will be discussed in this review.
MS has long been associated with previous EBV infection (73) and, while the exact mechanism remains to be fully characterised, the different theories have been summarised previously (65) (Figure 1). Despite uncertainty surrounding the sequence of events which eventually lead to MS, it has been established that EBV infection almost always precedes disease development. There is in fact a delay between infection and onset of neurological symptoms and also a lack of neurological symptoms in individuals with acute symptomatic EBV infection, also known as infectious mononucleosis (IM) (73–76). This interval between infection and neurological disease onset could potentially reflect a time delay between initial priming of pathogenic immune responses and epitope spreading within antigens or to new ones, which over time leads to inflammatory demyelinating disease. In support of this view, altered adaptive immune responses to EBV antigens have been identified in MS (77–80) and some have been found to cross-react with human proteins, leading researchers to conclude that molecular mimicry may have a key role in MS development.
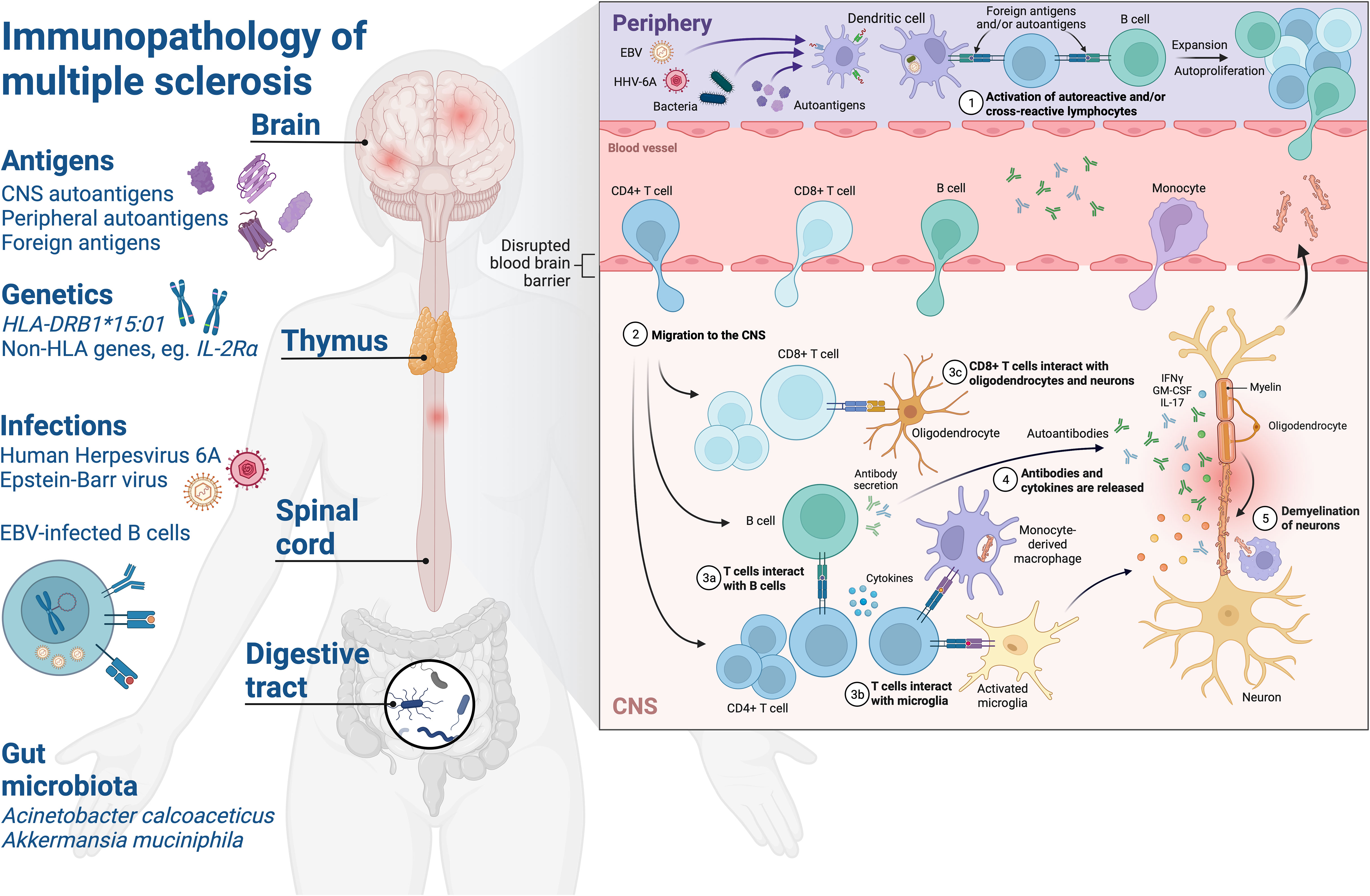
Figure 1 Schematic of the immunopathology of multiple sclerosis. Environmental and genetic factors affect development of neuroinflammatory mechanisms in MS. Exposure to foreign antigens primes humoral and cellular immune responses with cross-reactivity to self-antigens in the CNS. Human leukocyte antigen (HLA) – HLA-DRB1*15:01 – on the surface of APC present foreign or autoantigen peptides and shape the CD4+ T cell repertoire via central tolerance mechanisms in the thymus. Infections such as EBV and HHV-6A are associated with increased MS risk and drive elevated antibody responses to viral proteins which have been observed in pwMS, such as increased EBNA1-specific antibodies. The role of the EBV-infected memory B cell compartment in MS is not fully understood, but may be attributed to several factors which could predispose to autoimmunity such as: providing a source of molecular mimicry epitopes throughout life which can prime pathogenic and cross-reactive CD4+ T cell and antibody responses, processing and presentation of antigens to prime T cells, production of proinflammatory cytokines, rescuing of autoreactive B cells from apoptosis and/or modulation of innate and adaptive immune mechanisms via expression of viral immune evasion proteins. Gut microbiota associated with MS such as Acinetobacter calcoaceticus and Akkermansia muciniphila may influence CNS autoimmunity by providing molecular mimicry epitopes or driving a proinflammatory milieu which could lead to breakdown of immune tolerance via bystander activation of autoreactive T and B cells. (1) Priming of T cells and B cells with reactivity to foreign antigens and/or autoantigens in the periphery, autoproliferation is driven via interactions between T cells and B cells in pwMS and leads to clonal expansion. Elevation of antibodies with reactivity to viral and/or autoantigen in the periphery. (2) Blood-brain barrier alterations lead to migration of lymphocytes to the CNS where they mediate inflammation. (3) CD4+ T cells interact with B cells and activated microglia in the CNS, CD8+ T cells interact with neurons and oligodendrocytes via HLA class I molecules. (3) Antibodies and cytokines are released driving inflammation and lesion formation. (4) Breakdown of myelin sheaths leads to neuronal axonal damage and demyelination over time. CNS, central nervous system; HLA, human leucocyte antigen; EBV, Epstein-Barr virus; HHV-6A, Human herpesvirus 6A; IFNγ, interferon-γ; IL-2Rα, interleukin-2 receptor α; IL-17, interleukin-17; GM-CSF, granulocyte macrophage stimulating factor. Created with BioRender.com.
Despite many links between viruses and MS, direct evidence that viruses can trigger molecular mimicry leading to initial CNS autoimmune demyelinating disease is lacking in humans. However, proof of concept was shown in an EAE model using recombinant Theiler’s murine encephalomyelitis virus (TMEV) expressing a naturally occurring proteolipid protein (PLP) molecular mimic from H. influenzae. Early onset EAE could be induced in SJL/J mice by TMEV infection, but disease was not triggered when the same peptide was used to vaccinate in complete Freud’s adjuvant, suggesting that virus activation of antigen presenting cells (APC) is necessary for disease development in this model (81). An additional study by Ji et al. used a recombinant Vaccinia virus expressing myelin basic protein (MBP) to trigger autoimmunity in Rag2-/- mice expressing an MBP-specific CD8+ TCR (82). Disease was also triggered in mice after infection with the wild type Vaccinia vector which did not contain MBP but immunisation with peptide plus adjuvant only did not trigger disease, indicating that viral infection was necessary to break tolerance to CNS antigens in these models. A further study investigating the role of CD8+ T cells used a mouse model where oligodendrocytes expressed ovalbumin (OVA) and showed that even high numbers of high avidity OVA-specific CD8+ T cells could not induce EAE, and that these cells were in fact deleted from the immune repertoire to prevent autoimmunity under normal non-infected CNS conditions or during peripheral infection with OVA-expressing Listeria bacteria. In contrast, when mice were intracerebrally infected, OVA-specific CD8+ T cells destroyed oligodendrocytes and induced demyelination (83).
These examples from EAE indicate that autoreactive T cell responses to myelin antigens are not solely sufficient to initiate CNS autoimmunity and that additional triggers are required to break tolerance, such as virus-mediated activation of APC or a blood-brain barrier permeabilisation event. In support of this view is the observation that myelin-specific T cells can be detected in healthy individuals (84, 85), however it could be that activation of these autoreactive T cells in the periphery through molecular mimicry to foreign antigens skews them towards a pathogenic phenotype capable of migrating to and targeting CNS tissue (Figure 1).
Particular examples of cross-reactivity between CNS and foreign antigens in MS
A general problem with most descriptions of molecular mimicry T cell specificities is to know if these indeed have pathogenic roles or whether they represent innocuous epiphenomena, which is difficult to prove in humans. However, for some, there is epidemiological evidence for an association to disease and in the following paragraphs we discuss a set of autoantigen mimicry suspects. A full list of the foreign antigen cross-reactivity with CNS proteins discussed in this article is summarised in Table 1.
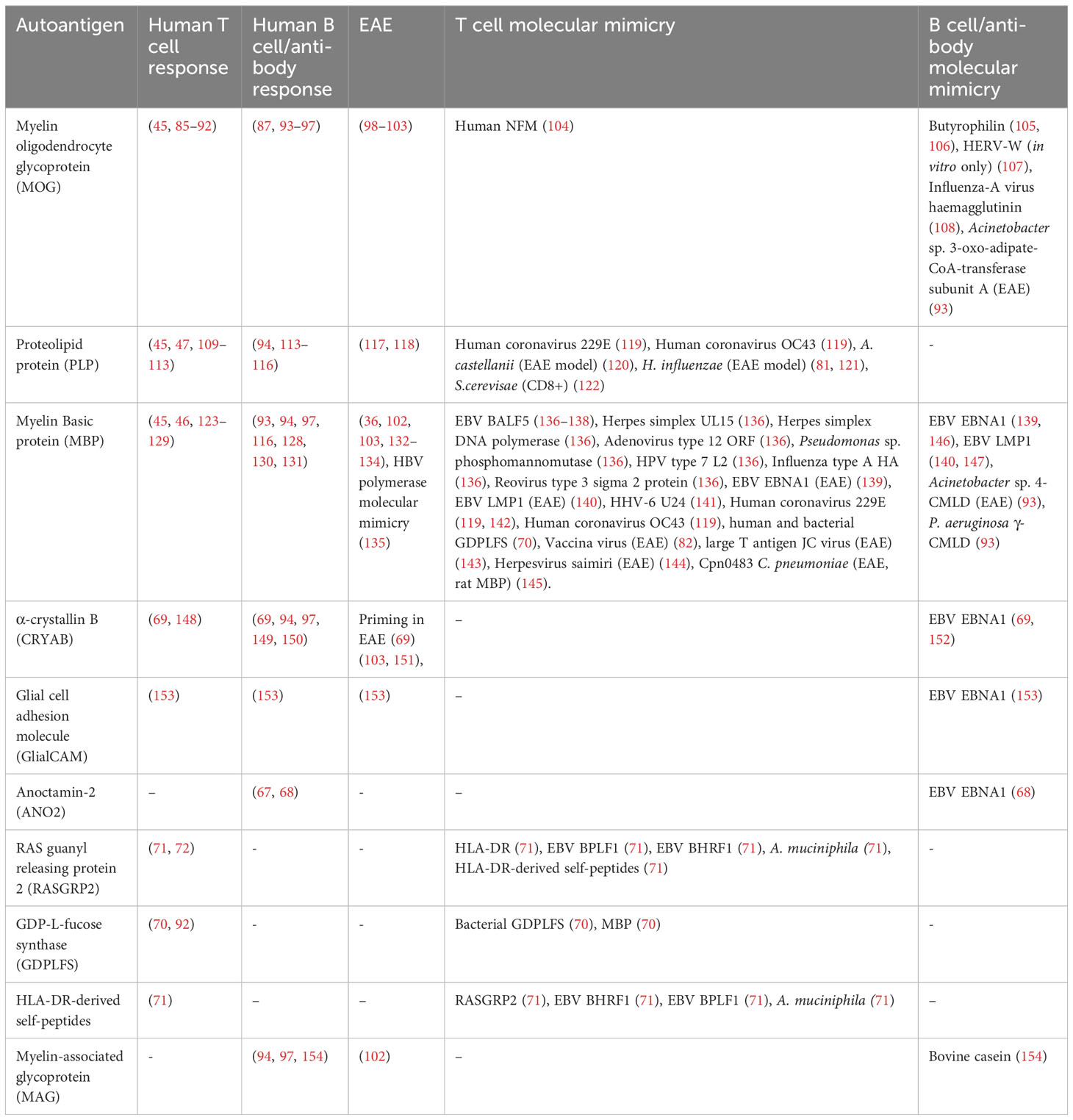
Table 1 Selected molecular mimicry between foreign and host CNS antigens in MS.
EBNA1 as a source of mimicry epitopes to Anoctamin-2, α-crystallin B and Glial cell adhesion molecule
Several studies have shown that elevated antibody responses to certain antigens from EBV are elevated in MS, in particular immunoglobulin G (IgG) responses to the EBNA1380-440 region have a MS odds ratio of approximately 8 and have also been shown to interact with HLA-DRB1*15:01 to increase disease risk further (19, 24, 64). In addition, elevated serum neurofilament levels have been shown to positively correlate with EBNA1 IgG in MS, indicating that there is a relationship between CNS injury and humoral responses to EBNA1 which occurs before disease onset (73). However it is not fully understood what is causing elevated EBNA1 antibody responses in MS and, as this elevation is specific to MS and interacts with HLA-DRB1*15:01, may suggest that molecular mimicry with CNS antigens are driving the EBNA1 antibody response. In support of this view, several epitopes within the MS-associated EBNA1 region have been identified with similar amino acid sequences to CNS autoantigens (68, 69, 139, 153). On the other hand, increased EBNA1 antibody responses in MS may be due to a frustrated EBV-specific immune response, where HLA-DRB1*15:01 is a poor class II allele in the context of EBV immune control. In one study humanised mice that were immune reconstituted from HLA-DRB1*15:01+ donors had increased steady state activation of CD4+ and CD8+ T cells and poor virus control evidenced by high EBV viral loads, compared to mice which were reconstituted with an allele not associated with increased MS risk (155). These findings suggests a synergistic interaction between EBV infection and HLA-DRB1*15:01 which primes a hyperactive adaptive immune compartment, leading to poor viral control and facilitating the generation of cross-reactive pathogenic T cell responses.
Α-crystallin B (CRYAB) is a small heat shock protein which is expressed in oligodendrocytes and has been shown to have paradoxical roles in MS: both in protection from harmful inflammatory innate immune mechanisms via chaperone activity and also conversely as the target of adaptive T cell responses in a proinflammatory environment (148–150, 156). This multifaceted role somewhat confounded the MS field and cast doubt on CRYAB’s role as an autoantigen in MS, however antibody responses to CRYAB were recently revisited in a large Swedish cohort and showed 27.6% of pwMS and 16.9% of controls to have IgG responses to CRYAB peptides with homology to EBNA1, and were associated with MS (OR=1.98) (69). Risk was further increased with an OR of 8.99 when combined with high EBNA1 IgG responses in individuals. Reciprocal blocking experiments showed that CRYAB IgG responses were blocked when the homologous EBNA1 epitope was spiked into sera, and the core homologous epitope between these antigens was mapped to a RRPFF motif at CRYAB11-15 and EBNA1402-406. Similarly, another group identified cross-reactive antibodies targeting the RRPFF motif in oligoclonal bands of pwMS (152), and interestingly the CRYAB sequence contains a PxxP motif similar to that found in MBP and several antigens from herpesviruses discussed later in this review. In addition, the high frequencies of EBNA1- and CRYAB-specific T cells observed in natalizumab-treated pwMS produced IFNγ and immunisation of mice with CRYAB or EBNA1 protein elicited T cell responses to the reciprocal antigen, also indicating cross-reactivity on the T cell level (69). The role of CRYAB as an autoantigen is complex and autoantibodies such as those that target intracellular antigens may not be directly pathogenic, however generation of high affinity antibodies depends on T cell help, and therefore they could be markers of a T cell response which is able to target intracellular antigens in autoimmune disease. In support of this view, B cells have been shown process and present epitopes from the antigen that they have Ig specificity with greater efficiency to T cells which have TCRs that respond to the same antigen (157). In addition, LMP1 expression has been shown to enhance antigen presentation and co-stimulation in EBV-transformed B cells via CD70, OX40 ligand and 4-1BB, which may have a role in priming of pathogenic autoreactive T cell responses (158).
The presence of CRYAB IgG in only a subset of pwMS suggests involvement of other autoantigens in the non-responders, and a recent study by Lanz et al. identified clonally expanded plasmablasts in the CNS of pwMS and identified their target epitope as a sequence shared between EBNA1 and glial cell adhesion molecule (GlialCAM) (153). These antibodies bound a core epitope within the MS-associated region of EBNA1 at residues 394-399 which is directly next to the CRYAB homologous epitope and cross-reacted with GlialCAM377-383, again both sequences contain PxxP motifs. In addition to this, the authors noted that affinity for the GlialCAM epitope was increased with the phosphorylation of the serine residue at position 376, and increased antibody reactivity to these core epitopes was also demonstrated in the plasma of pwMS compared to controls. SJL/J mice which were initially immunised with EBNA1386-405 peptide had a worse EAE disease course compared to a scrambled peptide control and the mice developed antibody responses to GlialCAM indicating generation of cross-reactive responses. Also similar to CRYAB, GlialCAM is expressed by oligodendrocytes and astrocytes in the CNS and is also present in chronic active lesions of MS (159, 160). Whilst no study so far has investigated both GlialCAM and CRYAB responses in individuals, it would be interesting to understand whether pwMS have responses to both antigens or whether these are restricted by different HLA. It is possible that the close proximity of these epitopes could lead them to be differentially processed and presented on surface HLA, or it could lead to their restriction by the same HLA.
Anoctamin-2 (ANO2) is a calcium-activated chloride channel with 8 membrane-spanning domains which is predominantly expressed in neurons and glial cells in the CNS, and also has high expression in the retina and in MS lesions (67). ANO2 was identified as a target of autoantibodies in MS in a large screening study and responses were found to be positively associated with HLA-DRB1*15:01, with an adjusted OR of 17.3 (67). Furthermore, combination of several risk factors including ANO2 IgG positivity, high EBNA1 IgG, presence of HLA-DRB1*15:01 and absence of HLA-A*02:01, produced a combined OR of over 26 (68). A later study went on to confirm the association of ANO2 IgG with MS in a larger cohort of almost 16,000 individuals, and identified cross-reactivity of ANO2140-149 antibody responses with EBNA1431-440 which is also in the MS-associated region (68). In addition to HLA-DRB1*15:01, 14 other HLA alleles were found to be associated with ANO2 IgG levels, indicating that epitopes from ANO2 can be presented by multiple different HLA and providing indirect evidence that there may also be ANO2 T cells in MS. Interestingly the strongest HLA effect on ANO2 IgG levels was a protective effect for HLA-DRB1*04:01, which the authors speculate could be due to increased elimination of high affinity ANO2-specific T cells in the thymus; the same effect was also observed for EBNA1 IgG levels, again indicating potential cross-reactivity on the T cell level.
Given that EBNA1-specific antibodies have now been reported in several studies to cross-react with multiple human proteins including MBP (discussed later in this review), CRYAB, GlialCAM and ANO2 (68, 69, 139, 146, 153), all of which were found to be elevated in pwMS and were associated with disease, what evidence is that EBV infection may have triggered these responses? Tengvall et al. showed that ANO2 IgG responses could be detected in a pre-MS cohort (68), indicating that responses appear before clinical onset of disease, an observation that has also been published for EBNA1 IgG (73). Additionally, it is very rare to detect antibody responses to ANO2, GlialCAM and CRYAB in individuals without evidence for a prior EBV infection, particularly EBNA1 IgG, indicating that EBV infection may be a prerequisite for the development of these autoantibodies and implying that molecular mimicry may be driving development of these responses. Presence of a CRYAB IgG response was also shown to be negatively correlated with ANO2 IgG in individuals, suggesting that these autoantibodies likely do not develop in the same individuals which could be due to factors such as HLA type (69) and that different autoantigens lead to MS disease in individuals.
However, the autoantibodies with cross-reactivity to EBNA1 described above were each only detected in a relatively small subset of pwMS, suggesting that these cross-reactive antibodies are not necessary for disease in all patients. It is likely that further undiscovered autoantigen cross-reactivity exists or it could be that the cross-reactive antibodies are not themselves pathogenic in the majority of MS cases, but are instead biomarkers for a T cell response which is responsible for mediating autoimmune damage, as has been observed for other autoimmune diseases such as Addison’s disease and diabetes mellitus (161, 162). Additionally, GlialCAM and ANO2 epitopes are both within intracellular domains and CRYAB is expressed intracellularly and are therefore not exposed, making it difficult to assume direct autoantibody-mediated damage to the CNS; although this cannot necessarily be excluded as several examples exist of pathogenic autoantibodies which bind intracellular targets such as GAD65, proinsulin and IA-2 in diabetes (163). Plasmapheresis is only therapeutically effective in a subset of MS patients (164) which suggests that it is not the antibodies themselves that are responsible for MS disease but the pathogenic T cell responses that they mark. Although positive responses to plasmapheresis in patients with histological lesion patterns type I and II, and particularly in individuals who showed signs of a humoral response, could indicate that autoantibodies drive disease in some individuals (165). In addition to this, evidence for T cell responses to EBNA1 mimics has been shown both in EAE immunisation models and in humans (69, 139, 153) and, although no direct evidence for cross-reactivity on the single T cell level has been so far shown, this is likely to be present, although the potential relevance of these responses to MS development and progression remains to be determined.
Other scant reports of further antibody cross-reactivity between EBV proteins and autoantigens have been described such as that of EBNA1 IgG with heterogeneousnuclear ribonucleoprotein L (HNRNPL) (166), although these were not found to be increased in the plasma of pwMS compared to controls. Antibody cross-reactivity has also been reported for BFRF3 with septin-9 and BRRF2 with the mitochondrial protein dihydrolipoyllysine-residue succinyltransferase (DLST), although again these responses were only in a subset of patients and need to be confirmed in larger cohorts (167). As with most of these reports, validation in large cohorts are required to identify the frequency of these responses in patients and their relevance to neuroinflammatory disease, however the mounting reports of cross-reactivity between EBV and self-antigens suggest that there are multiple disease relevant autoantigens in MS and that each individual may have specific profiles of reactivity.
Another potential consideration is the promotion of tolerance breakdown and molecular mimicry by EBV-mediated immune dysregulation. EBV encodes several viral mimics of human proteins with essential roles in immunity and have evolved over thousands of years to facilitate viral escape from the immune system, and one such example is the viral CD40 mimic latent membrane protein 1 (LMP1). CD40 is a co-stimulatory molecule expressed on APC and is particularly essential for B cell activation via its function as a co-receptor for the B cell receptor (BCR), interaction with CD40 ligand (CD40L) on T cells and amplification of innate mechanisms such as TLR signalling (168–172). LMP1 has been demonstrated to self-aggregate and facilitate downstream signalling in B cells with promote activation, germinal centre formation and production of cytokines and antibodies (173). One study of a transgenic mouse model which constitutively expressed the cytoplasmic tail of LMP1 showed that animals were prone to autoantibody production and immune dysregulation but had no signs of clinical disease (174). However, when these animals were immunised with EBNA1, they showed markedly increased inflammatory cellular and humoral responses compared to animals without LMP1 with T cells producing IFNγ and IL-17 cytokines. Additionally, expression of LMP1 was shown to drive molecular mimicry between EBNA1 and the systemic lupus erythematosus-associated autoantigen Sm (174). Incidentally, the Sm-homologous epitope is at EBNA1398-404 and overlaps almost identically with the region reported to contain homology to GlialCAM and CRYAB in MS (69, 153). Whilst the authors do not report increased neurological symptoms in these animals, it would be pertinent to also investigate whether transgenic expression of LMP1 also facilitates molecular mimicry with CNS autoantigens.
Other EBV-encoded mimics of host antigens include BCRF1 which contains homology to interleukin-10 (IL-10) and BHRF1 which is a mimic of Bcl-2, proteins which limit host immune responses to pathogens and promote survival of infected B cells respectively (175, 176). In addition to this, multiple EBV proteins have been shown to modulate antigen processing and presentation in infected B cells, suggesting even further ways in which the virus may shape adaptive immune responses to mimicry epitopes (176–179), and this is an avenue in which there has been very little research in the context of autoimmunity. Given these observations, one can easily imagine how high expression of EBV-encoded immune mimics and modulators such as LMP1 during acute infection or IM may facilitate the breakdown of tolerance, and indeed history of IM has been demonstrated to increase MS risk (19, 22, 23). Of further relevance to MS immunopathology is how EBV-mediated modulation of immunity differs throughout life from childhood to adolescence given the increased risk of developing MS with delayed EBV primary infection (62). For example, studies in mice have shown that a specific population of early-differentiated natural killer (NK) cells expand prior to CD8+ T cells and is involved in virus control during early infection (180, 181). Further study of the analogous population in humans showed that early differentiated NK cells diminished with age and may be involved in protection of children from EBV infection and IM (182). Furthermore, proliferative and cytokine production of CD56BRIGHT NK cell have been demonstrated to be diminished in pwMS (183) indicating that this population may also be impaired in its response against EBV, but further study of NK cell function in pwMS are needed to establish their relevance.
It is evident that cross-reactivity occurs between CNS and viral antigens, however the time and space of exposure to virus antigens may be one of the key determinants for developing MS. Indeed, does EBV’s unique life cycle and persistence in the B cell compartment throughout life mean that this virus is uniquely positioned to trigger CNS autoimmunity? Evidence for this could be derived from observations that the T cell and antibody response to EBNA1 only emerge 3-6 months post primary infection (184) and the delay also exists between seroconversion and development of MS in individuals (74).
Myelin basic protein
Proof of concept that virus peptides could induce CNS autoimmunity to was shown by Fujinami and Oldstone in 1985, where rabbits immunised with either MBP or a homologous peptide from Hepatitis B virus (HBV) polymerase developed EAE (135). Since then, multiple studies in both human and animal models have demonstrated the ability of T cells generated against MBP to target other antigens.
T cell molecular mimicry between myelin and EBV antigens in humans was first reported by Wucherpfenning and Strominger who isolated T cell clones which responded to MBP85-99 (136). Amongst the epitopes which activated the MBP85-99-specific T cell clones was a peptide from EBV DNA polymerase (BALF5627-641) in the context of MS-associated alleles HLA-DRB1*15:01 and HLA-DRB5*01:01 respectively, and these clones were subsequently tracked to the cerebrospinal fluid (CSF) of patients and the TCR:peptide-HLA structure solved (137, 138). This cross-recognition of BALF5 and MBP peptides in the context of different HLA molecules demonstrated that TCRs can even recognise different complexes as long as there is similar overall structure and charge of residues. Interestingly, the same study showed that MBP85-99-specific T cell clones were also activated by multiple other viral and bacterial epitopes – some of them with no clear amino acid homology to the original MBP peptide – including peptides from human papilloma virus (HPV), herpes simplex virus and influenza A virus amongst others (136).
The long-established association of elevated EBNA1 IgG with MS suggests that this response may have a role in disease pathology. Early research by Bray et al. discovered two homologous epitopes in EBNA1 and MBP and isolated antibodies with specificity for the MBP-homologous EBNA1 epitope from oligoclonal bands in the cerebrospinal fluid (CSF) of 85% pwMS in their cohort (146). This provided early evidence that, rather than simply biomarkers, elevated EBNA1-specific IgG responses may target myelin and have a role in MS disease mechanisms. A later study identified antibody responses to EBNA1411-426 which were specific to untreated MS-patients and were also able to bind MBP205-224 (139). Whilst the study cohort was small, IgG responses to EBNA1411-426 were higher in untreated pwMS but low/no responses could be detected in individuals undergoing interferon-β therapy. Furthermore, mice immunised with EBNA1411-426 amounted both T cell and antibody responses to MBP, despite low amino acid sequence homology between these two regions (139). Whilst this data from a mouse model is intriguing and suggests T cell cross-reactivity, there are currently no examples of dual-reactive human EBNA1-specific T cells which have been investigated on the single cell level. It is also important to note that oligoclonal bands in MS have been shown to contain specificities for multiple viruses in addition to EBV EBNA1, and therefore there is some debate around their role in CNS autoimmunity (185), however EBNA1 remains a top candidate for molecular mimicry.
Sequence similarity between MBP and the EBV latent antigen LMP1 was also identified by a small study which used phage display to recreate clones from B cells in peripheral blood of pwMS (147). Reverse engineering of sequences from MBP-specific antibody variable domains showed similarity to previously identified LMP1-specific antibody sequences, and were subsequently found to bind recombinant LMP1 by Western blot. Further in vivo analysis showed that MBP- or LMP1-immunised mice produced antibody responses to the reciprocal antigen, and comparison of responding B cell repertoire clonality was suggestive of greater epitope spreading in the LMP1-immunised animals (140). CD4+ T cells from LMP1-immunised mice also showed proliferation following in vitro MBP re-stimulation suggesting cross-priming of T cell responses in vivo (140). Despite these findings, no obvious amino acid homology exists between MBP and LMP1 antigens, however this does not necessarily exclude the presence of structurally similar epitope/s as has been previously shown (186). However, as for EBNA1 T cell responses, comprehensive analysis of LMP1-specific adaptive responses in MS cohorts is needed to determine the relevance of this cross-reactivity for disease pathogenesis.
Epidemiological evidence has also linked HHV-6A to increased risk for developing MS, and antibody responses to immediate early protein 1 (IE1) from HHV-6A in a pre-MS cohort showed an increased MS risk with an OR of 2.22 (63). Interestingly, this effect was only observed for IE1 IgG responses to HHV-6A and not for the HHV-6B strain which was instead negatively associated with disease (OR=0.74) (63). As the study used sequence variation in IE1 to distinguish between infection with HHV-6A and 6B, it is so far not known whether the risk associated with HHV-6A IE1 IgG responses is due to the IE1 response itself or the virus that it marks. However, a previous study by Tejada-Simon et al. identified dual-specific T cell responses with reactivity to MBP96-102 epitope and HHV-6 U244-10 which share 6 out of 6 identical amino acids across a PxxP motif (141). Approximately 50% of T cells which responded to MBP also responded to this HHV-6 U24 epitope and produced predominantly TH1 cytokines and patients with dual-specific responses also had increased antibody titres to both peptides, indicating a direct link between dual-specific cellular and humoral responses to the same epitopes (141). HHV-6A and B strains share over 90% sequence identity and the U24 PxxP amino acid motif which is relevant for cross-recognition with MBP is conserved between both A and B strains. However, different phosphorylation patterns in the MBP-homologous U24 region may affect immune recognition of this epitope or interaction with cellular proteins (187). Alternatively, the difference in MS risk between these strains could be due to increased susceptibility to HHV-6A infection of KIR2DL2-carrying MS patients or to U24-mediated disruption of MBP-Fyn interactions which stabilise myelin (188, 189), and further research is needed to elucidate the exact mechanisms of HHV-6A in MS.
Proof that a virus peptide with homology to a myelin antigen with a core of only 5 amino acids could induce disease in EAE was first demonstrated in by Gautam et al. in 1998, where a PxxP motif peptide from Herpesvirus Saimiri with amino acid homology to MBP1-11 was able to induce disease in EAE (144). PxxP amino acid motifs can be found in MBP and multiple other proteins from herpesviruses such as HHV-6 U24, Human Herpesvirus 7 (HHV-7) U24, human Cytomegalovirus (HCMV) UL25 and UL42, Varicella Zoster Virus (VZV) ORF0, Herpes Simplex Virus-1 (HSV-1) UL56 and EBV LMP2A – the latter of which contains four PxxP motifs (189). These viruses are all in the Herpesviridae family and persist in the human host throughout life, although they have vastly different cell tropisms and immune evasion mechanisms which may affect the availability of antigens to prime responses. As previously mentioned, several of these viruses are associated with MS risk – with EBV and HHV-6A showing increased odds ratios (62, 63, 190) whilst CMV is associated with protection (191, 192). So far, T cell molecular mimicry has only been identified between HHV-6 U24 and MBP in humans, and it is possible that sequence of infection with these viruses throughout childhood and early adulthood shapes the T cell repertoire, predisposing some individuals to CNS autoimmunity through cross-reactivity. Further research is needed to establish the relevance of cross-reactive MBP PxxP motifs with viral antigens, and in particular how this might develop throughout challenge with multiple homologous epitopes from viruses.
It seems evident that multiple virus infections have the potential to induce immune responses which cross-recognise MBP, however this may be in part due to pre-existing T cells in the periphery with low to moderate avidity for MBP which escape thymic tolerance mechanisms, despite some expression of MBP in the thymus (193, 194). The escape of MBP-specific T cells from negative selection during central tolerance may be due to the generally lower avidity with which MBP peptides bind to HLA-DRB1*15:01 rendering them unstable, and peptides from MBP have also been reported as promiscuous binders to multiple HLA class II alleles (123, 195, 196). One study demonstrated the ability of an MBP-specific TCR to bind peptide:HLA with a wide range of orientation angles which is possibly due to the scarcity of hydrogen bonds between at the TCR:peptide interface, and this low affinity interaction may explain TCR degeneracy and escape from thymic negative selection (197). MBP-specific T cells can also be detected in healthy individuals which suggests that they occur naturally but are less frequent, have a less pathogenic phenotype, do not gain access to the CNS or are prevented from causing disease by regulatory or other mechanisms under normal conditions (46, 124, 198–202). Differences in MBP peptide immunodominance have also been identified between pwMS and controls (40, 124) although other studies have found no changes in peptides targeted (109, 203, 204). However, it is plausible that environmental exposure to pathogens which contain molecular mimics to MBP – such as to Herpesviruses – could prime or skew responses to different epitopes with higher potential to cause CNS inflammation leading to MS development; so far there is only sero-epidemiological evidence supporting for a role for EBV in MS and to some extent HHV-6A.
More recently, microbiome studies in MS cohorts have led researchers to investigate elevated antibody responses to some bacterial species – such as Acinetobacter calcoaceticus, Akkermansia muciniphila and Pseudomonas aeruginosa – for potential molecular mimicry (205, 206). These studies showed that antibodies from pwMS with reactivity to MBP43-57 could bind to epitopes from both A. calcoaceticus and P. aeruginosa 4- and γ-carboxymuconolactone decarboxylase (CMLD) respectively (93). A similar homology between myelin oligodendrocyte glycoprotein (MOG) and 3-oxo-adipate-CoA-transferase subunit A from Acinetobacter species was identified (93), however only the MOG43-57-immunised animals showed any sign of disease activity in ABH mice and disease could not be induced by immunisation with the homologous bacterial peptides. On the other hand, molecular mimicry is not the only mechanism through which these bacteria have been suggested to play a role in MS, and studies have shown that A. calcoaceticus and A.muciniphila species are increased in the gut microbiota of pwMS. Mice which are mono-colonised with these bacterial species have a more severe EAE disease course and produce more proinflammatory adaptive immune responses with fewer IL-10-producing regulatory T cells (TREG) (207). In theory, induction of a proinflammatory environment by these bacteria in the MS host could help to skew pre-existing molecular mimicry responses to a pathogenic phenotype which could lead to or influence progression of CNS autoimmunity.
Proteolipid protein
PLP is the most abundant protein in myelin and has two main isoforms: the full-length version which is almost exclusively expressed in the CNS, and the slightly shorter DM20 variant which is missing a loop of 35 amino acids and is only expressed in the periphery, thymus and lymph nodes (208, 209). The sequence excluded from the thymus-expressed DM20 variant contains the immunodominant PLP139-151 epitope which is a strong encephalitogen in some EAE models such as SJL/J (117, 210). SJL/J is a mouse model which is strongly predisposed to develop EAE with epitope spreading during subsequent relapses to other PLP epitopes and to MBP (117, 210). PLP139-151 is a frequent target of high avidity T cells in MS (45) – most likely due to its exclusion from thymic tolerance mechanisms – but several other encephalitogenic PLP peptides have been identified in humans such as PLP104−117, PLP142−153, PLP184−199, and PLP190−209, all of which can be presented by the MS risk allele HLA-DRB1*15:01 (47, 109). PLP is also the target of antibody responses, with up to 58% of pwMS in some studies showing antibody responses which are sensitive to protein conformation (114, 115).
Although fewer examples of CNS cross-reactivity between PLP and non-self antigens have been reported than for MBP, homology between human coronaviruses (HCoV) and PLP led to the isolation of several T cell clones from pwMS with dual-specificity for PLP and HCoV 229E and OC43 strains. Clonality and TCRVβ chain usage off cross-reactive T cell clones was confirmed, although the study did not enumerate frequency of these cells in peripheral blood of pwMS (119). The relevance of HCoV T cell molecular mimicry with CNS antigens is not certain and so far no further studies have replicated these findings with no large-scale sero-epidemiological studies have been presented. However, several reports globally of new MS cases and disease exacerbations following severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection or vaccination could suggest that exposure to SARS-CoV-2 antigens may trigger autoimmune attack on the CNS in some individuals (211), although there is currently no published functional evidence to support this. However, a recent in silico study showed that the nucleocapsid protein from SARS-CoV-2 shares significant overlap with several MS-associated myelin proteins including PLP (212), although this has not yet been investigated in vitro. On the other hand, increased numbers of MS cases following SARS-CoV-2 exposure could be attributed to bystander activation of pre-existing myelin-reactive T cells, or simply chance occurrences due to the immense number of people who were infected or vaccinated during the global Covid-19 pandemic. Further investigation is warranted to determine if cross-reactivity between coronavirus and neuronal antigens occurs in vivo.
As for MBP, sequence similarity between PLP and bacterial antigens have been reported, and one study demonstrated that EAE could be induced by both immunisation with PLP139-151 peptide or with homologous epitopes from Haemophilus influenzae and Acanthamoeba castellanii (81, 120, 121). The homologous peptide from A. castellanii was able to induce EAE in SJL/J mice and interestingly adoptive transfer of A. castellanii-specific T cells from female mice could also induce disease, however the same T cells derived from males could not (120). Further investigation of the PLP-homologous epitope from H. influenzae showed that induction of EAE required delivery of the pathogenic epitope in a recombinant Theiler’s encephalomyelitis virus (TMEV) vector, suggesting that CNS autoimmunity requires virus-specific activation of innate immune mechanisms in APCs such as Toll-like receptors (TLR) to fully break immune tolerance and lead to disease (81). In addition, further study of this model showed that mice infected with the recombinant H. influenzae TMEV had a TH1 CD4+ response to the homologous PLP139-151 peptide but no epitope spreading to PLP178-191. This was in contrast to the PLP139-151 TMEV, where epitope spreading to PLP178-191 could be detected and marked initial disease relapse in the SJL/J model (213). Amino acid substitution in the primary contact residue of PLP139-151 removed the ability of the virus to induce early EAE and therefore indicated that this residue was necessary for induction of pathogenic CD4+ T cell responses which drive early disease (121). Together these data indicate the strong adjuvant effect of viruses on autoreactive responses which are able to induce pathogenic TH1 CD4+ responses with rapid onset disease when combined with molecular mimics to myelin antigens. However, in this model epitope spreading readily occurred between PLP epitopes and mediated disease relapse and/or progression but this was not achieved between the foreign peptide and PLP. This suggests that further factors may be needed to sustain chronic CNS autoinflammation and long-term disease in this setting.
Although current evidence suggests a more important role for CD4+ T cells in MS pathogenesis, the high abundance of CD8+ T cells in MS lesions and oligoclonal TCR repertoires suggest that these expand and may be antigen-specific (58, 59). However, other studies have also shown myelin-specific CD8+ T cells to be present in peripheral blood at the same frequency in pwMS as in healthy individuals (214). The generally higher avidity of CD8+ TCR interactions with peptide:HLA and lower degeneracy of CD8+ T cells make cross-reactivity in this compartment less likely (215), features which perhaps emerged via evolution in order to limit cross-reactive responses with the ability to bind peptide:HLA class I complexes that are expressed almost ubiquitously on nucleated cells. However, examples of CD8+ T cell cross-reactivity do occur and Honma et al. described a HLA-A*03:01-restricted CD8+ T cell clone with specificity for PLP45-53 that could cross-recognise a peptide from Saccharomyces cerevisae in the context of HLA-A*02:01 (122). Although infrequent reports of myelin-reactive CD8+ T cell degeneracy may be in part due to the focus on CD4+ T cells in the MS research field. However, evidence from TCR sequencing of blood, CSF and MS lesions all suggest a clonal expansion of the CD8+ compartment in MS which may indicate migration of antigen-specific CD8+ T cells to the CNS during disease (58, 59), although the targets remain to be characterised and these could equally have a regulatory or suppressive phenotype. Interestingly, acute EBV infection – also known as infectious mononucleosis (IM) – is also characterised by enormously expanded oligoclonal CD8+ T cell repertoires directed against EBV antigens which subside over several weeks to months (216–218), however how these compare to the CD8+ compartment of MS patients remains to be determined.
Even though few examples exist of direct mimicry between PLP and foreign antigens, PLP remains one of the strongest encephalitogens, and in vivo inter- and intramolecular epitope spreading from initial PLP epitope responses is well-characterised in some EAE models as is described above (117, 210, 219). Further studies have shown EAE to be dependent on B cell presentation of PLP and MOG antigens to CD4+ T cells (220, 221), and efficacy of B cell depletion therapy in MS is thought to be partially due to removal of the antigen-presenting function of B cells. In contrast, fewer examples of epitope spreading from an initial PLP response have been reported for MS, however there are reports of spreading from MBP epitopes to PLP, and it is likely that each patient has a unique sequence of responses which develop through disease depending on their HLA type and other factors (222). However, this does not discount the possibility that an immunodominant T cell response to PLP139-151 in humans could, under the right circumstances, lead to an inflammatory event causing breakdown of the blood-brain barrier and lesion formation, after which epitope spreading to other CNS autoantigens may occur. These events are extremely difficult to investigate in humans due to the long prodromal phase of MS and also the difficulty and ethical barriers to sampling the affected tissue, ie. the brain and spinal cord.
Myelin oligodendrocyte glycoprotein
MOG is a minor myelin component and is a transmembrane protein of the Ig superfamily. Overall, it constitutes less than 0.05% of myelin and is located in the outer membrane, and this location contributes to its relevance as an autoantigenic target as it is readily accessible by autoantibodies targeting its extracellular domain (223, 224). MOG was identified as a candidate autoantigen following observations that immunisation induces EAE and also the presence of MOG-specific autoantibodies and T cell responses in MS (45, 85–87, 94, 95, 98). In the EAE model, MOG-specific autoantibodies work synergistically with T cells to induce an inflammatory demyelinating disease which replicates observations from MS pathology (99, 225). An additional factor is MOG’s almost undetectable expression in the thymus (208), leading to escape of MOG-specific T cells from central tolerance mechanisms and which also partly accounts for the detection of MOG-specific T cells in some healthy controls as well as pwMS (85). MOG-reactive T cells retained in the immune repertoire may then become activated by environmental antigens with homology, which could lead to CNS inflammation and disease.
Mode and situation of cross-reactivity has been demonstrated to be important for priming of pathogenic cross-reactive responses, such as that demonstrated for cross-reactivity between MOG and the milk protein butyrophilin (BTN). BTN is a protein expressed in mammary tissue and is a major component of milk fat globules and has homology to MOG76-87 in its extracellular IgV-like domain. Immunisation of rats with MOG76-87 caused disseminated CNS inflammation characterised by infiltration of macrophages and CD4+ T cells, but interestingly this could be ameliorated by administration of the homologous BTN peptide either intravenously or intranasally with animals showing markedly decreased clinical scores (105). This amelioration is potentially due to induction of TREG cells by the homologous BTN peptide, and this protective effect could in theory also occur in humans who consume bovine milk products into adulthood. However, further studies have shown that mechanisms of oral tolerance are poorly developed in babies, and tolerance may only be maintained when oral exposure is continued past a certain age – as has been demonstrated for oral tolerance to MBP in EAE (226). On the other hand, consumption of dairy products has also been linked to MS (227) and, although this finding remains controversial, cross-reactivity of antibodies which could bind homologous epitopes from both BTN and MOG were detected in the blood and CSF of pwMS (106). In addition to BTN, animals immunised with bovine casein were demonstrated to develop severe spinal cord pathology and demyelination which was attributed to induction of antibodies which bind to casein and cross-react with myelin associated glycoprotein (MAG) (154). The authors also noted increased antibody responses in pwMS compared to other neurological disease controls, suggesting that loss of tolerance to casein and molecular mimicry with MAG could contribute to disease in a subset of patients. However, the contribution of dietary milk proteins to MS pathogenesis remains to be fully elucidated in large cohort studies.
In addition to dietary antigens, sequence similarity has been identified between MOG and the human endogenous retrovirus W (HERV-W) envelope protein. This led to the finding that MOG autoantibodies could bind HERV-W protein (107) and one study used a nanotechnology approach to show a proof of principle that antibodies raised against MOG could cross-react with HERV-W (107). HERV are remnant genetic material left in the human genome following infection with retroviruses and constitute around 7% of the human genome (228). Initial investigation of HERV-W in MS brains was driven by isolation of HERV-W protein from sera, CSF and brain samples of affected individuals (229). It has also been demonstrated that HERV can also lead to activation of innate immune mechanisms through activation of Toll-like receptor 4 (TLR4) in APC and contributing to a proinflammatory milieu by driving TH1 responses. However, under these conditions, it is also possible that autoreactive cells could become activated via bystander mechanisms, and therefore the biological relevance of this MOG cross-reactivity with HERV-W remains to be verified in an MS cohort. Further studies by Sutkowski et al. demonstrated that the env protein of HERV-K18 – a superantigen capable of activating T cells expressing TCRVB13 – can become transcriptionally activated by EBV (230, 231). This activation of TCRBV13+ T cells was HLA-dependent but not restricted and may be involved in autoproliferative mechanisms in MS.
Although several factors likely contribute to epitope spreading as was discussed above for EAE, T cells isolated in response to the encephalitogenic epitope MOG35-55 have also been characterised as polyreactive to a similar sequence in neurofilament medium protein (NFM15-35) in mice (104). Both epitopes share the same TCR contact residues and could therefore potentially contribute to epitope spreading in disease. However, observations from EAE showed that NFM peptide did not expand MOG-specific T cells to a sufficient threshold to induce disease and NFM knockout mice had identical EAE disease to wild type animals indicating that, although NFM is immunogenic at the polyclonal level, it fails to expand high affinity MOG-specific T cells necessary for EAE induction (104).
Guanosine diphosphate-L-fucose synthase, RAS guanyl releasing protein 2 and HLA-DR
Reactivity to guanosine diphosphate (GDP)-L-fucose synthase (GDPLFS) was first identified by systematically screening brain-infiltrating CD4+ T cell clones from MS patients for reactivity to a peptide library (70). T cell clones which responded to this antigen secreted TH2 cytokines and were identified in the CSF and brain of HLA-DRB3*02:02 positive individuals. Interestingly, GDPLFS clones were found to cross-recognise a number of bacterially-derived antigens as well as epitopes from other autoantigens such as MBP, PLP and MOG (70). T cell clones which reacted to GDPLFS could also respond to the dominant MBP83-99 epitope, with other clones showing reactivity to PLP139-154. Given the dual reactivity with bacterial epitopes, secretion of TH2 cytokines by the cross-reactive GDPLFS T cell clone is interesting as TH2 cells have a role in chronic inflammation and tissue repair (232). Given that the T cell clones respond to bacterial epitopes it is possible that they were initially primed in the gut. Also intriguing is that they were isolated from an individual with pattern II lesion pathology which is characterised by autoantibodies and complement deposition (233, 234), and an interesting avenue of future investigation would be to characterise whether the organ in which they were primed affects pathogenic T cell phenotype or subsequent lesion pathology in MS.
Auto-proliferation is defined as a spontaneous in vitro T cell proliferation without stimulus and is an observed feature of MS patient peripheral blood mononuclear cells (PBMC) in several studies (72, 235). Thorough examination of this phenomenon in pwMS was performed by Jelcic et al. who identified that there was a significant overlap of TCRVβ sequences from brain-infiltrating T cells in lesions and the auto-proliferating CD4+ T cell compartment in peripheral blood (72). T cell clones were isolated from the auto-proliferating blood compartment and their TCRVβ sequence was compared to those recovered from active lesions of the same HLA-DRB1*15:01 homozygous MS patient. The authors were then able to investigate specific clones which had infiltrated the brain and map their the antigen specificity using combinatorial peptide libraries. The cognate peptide from one T cell clone was mapped to a sequence from RAS guanyl-releasing protein 2 (RASGRP2), a previously unidentified MS autoantigen which is expressed in B cells, striatal neurons and cortical grey matter in the brain but is not a constituent of myelin, indicating that autoantigenic targets in MS do not necessarily need to be myelin proteins (72).
Identification of this situation in MS pathogenesis identifies a crucial link between peripheral activation of autoreactive CD4+ T cells by B cells and their subsequent infiltration into the CNS. In this scenario, B cells expressing high levels of HLA-DR present self-peptides from autoantigens such as RASGRP2 and stimulate auto-proliferative and auto-aggressive CD4+ T cells which subsequently enter the brain. After their infiltration into the CNS, these cells recognise RASGRP2 peptides presented in neuronal tissue and mediate inflammation. However, in this case the activating autoantigen in the periphery is the same as the target in the CNS, indicating that there should in theory be removal of autoreactive T cells via central tolerance mechanisms if the specified autoantigen is expressed in the thymus. RASGRP2’s expression in the thymus is not completely certain and it was not detected in thymic epithelial cells (71, 236). However, RASGRP2 expression was detected in some APCs in the thymus, indicating that RASGRP2-reactive T cells may be negatively selected from the repertoire via interaction with B cells and plasmacytoid dendritic cells (pDC) in the thymus (236).
Evidence that RASGRP2 autoreactivity could be stimulated by foreign antigens was demonstrated by Wang et al. who showed that peptides from MS associated pathogens EBV and A. muciniphila could also stimulate RASGRP2-specific CD4+ T cell clones in vitro (71). Interestingly, the cross-reactive EBV peptide sequences were derived from two lytic cycle antigens BHRF1 and BPLF1 which are expressed at high levels during acute infection where their functions are as a viral Bcl-2 homologue and a tegument deubiquitinase respectively (175, 237). No responses were observed to peptides from human HCMV or the bacterium Prevotella histicola, two pathogens have been negatively associated with MS (191, 206, 238). RASGRP2-specific T cell clones could also recognise HLA-derived self-peptides (HLA-SP) from HLA-DRB1 albeit with lower avidity, indicating that self-derived peptides are also partial agonists for CD4+ T cells targeting RASGRP2 and may help to maintain these T cells in the peripheral tissues via molecular mimicry (71). Some RASGRP2-specific T cell clones were shown to have an IFNγ+ TH1 phenotype, although the clone which was stimulated by EBV and A. muciniphila peptides had a TH2 phenotype and was isolated from a MS patient with type II lesion pathology (71, 72). Ex vivo analysis of T cell responses to peptides from RASGRP2, EBV and A. muciniphila showed these to be targets of responses in natalizumab-treated MS patients, indicating responses that had been primed in vivo and supporting their pathological relevance in this setting (71).
These data together suggest that HLA-DRB1*15:01-restricted RASGRP2-specific T cells can become activated in the periphery via both self-antigens and foreign peptides derived from EBV and A. muciniphila, and also that these CD4+ T cells specifically infiltrate the brain where they can be found in active lesions. This supports the hypothesis in MS that CNS autoimmunity is either initiated or maintained by pathogenic CD4+ T cell responses which are initially primed by exogenous antigens but can respond to autoantigens, leading to their recruitment to the brain where they mediate inflammatory tissue damage. These data are striking, and further investigation of RASGRP2 in large MS cohorts to establish the frequency of responses to this autoantigen are warranted.
Conclusion
It is clear that cross reactivity between a variety of microbial agents and host CNS autoantigens has been demonstrated, though in most cases of unclear relevance for MS pathogenesis due to experiments with low numbers of T cell clones or in artificial experimental animal models. However, in large sero-epidemiological studies, EBV – and to some extent HHV-6A – stand out. Especially for EBV, clear evidence for molecular mimicry epitopes has been demonstrated and antibody responses directed against these epitopes strikingly associate to an increased risk for MS. This strongly supports their role in MS pathogenesis, perhaps as markers for a concomitant T cell autoimmunity.
These observations make a case for therapeutic intervention, in particular if EBV drives the chronicity of the disease, perhaps with EBV-specific low molecular antiviral agents, or vaccination of individuals at risk for MS or at onset of disease. However, due to the multiple CNS autoantigen mimics now identified in EBNA1390-440, development of future EBV vaccines or adoptive T cell therapies which include this antigen should either be designed with extreme caution or avoided altogether lest MS disease be triggered or exacerbated. The MS research field has reached an exciting stage, however further extensive research is needed to fully elucidate the role of foreign antigens which mimic autoantigens in the development and progression of CNS demyelinating disease.
Author contributions
OT: Conceptualization, Writing – original draft, Writing – review & editing. TO: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study has received grant support from the Swedish Research Council, the Swedish Brain Foundation, Knut and Alice Wallenbergs Foundation and Margaretha af Ugglas Foundation.
Conflict of interest
TO has received unrestricted MS research grants from Biogen, Merck, Novartis and Sanofi, as well as lecture and/or advisory board honoraria from the same companies. None of which have had any relation to the content of this review.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler Houndmills Basingstoke Engl (2020) 26(14):1816–21. doi: 10.1177/1352458520970841
2. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol (2017) 13(1):25–36. doi: 10.1038/nrneurol.2016.187
3. Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol (1948) 29(1):58–69.
4. Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci (2006) 9(2):268–75. doi: 10.1038/nn1629
5. Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, et al. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol Baltim Md (1950) 182(7):3979–84. doi: 10.4049/jimmunol.0801218
6. Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A (2004) 101(21):8180–5. doi: 10.1073/pnas.0402268101
7. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523(7560):337–41. doi: 10.1038/nature14432
8. Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. eLife (2017) 6:e29738. doi: 10.7554/eLife.29738
9. Schmidt-Hieber M, Zweigner J, Uharek L, Blau IW, Thiel E. Central nervous system infections in immunocompromised patients: update on diagnostics and therapy. Leuk Lymphoma (2009) 50(1):24–36. doi: 10.1080/10428190802517740
10. Farh KKH, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518(7539):337–43. doi: 10.1038/nature13835
11. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med (2008) 358(7):676–88. doi: 10.1056/NEJMoa0706383
12. Salzer J, Svenningsson R, Alping P, Novakova L, Björck A, Fink K, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. (2016) 87(20):2074–81. doi: 10.1212/WNL.0000000000003331
13. Fox RJ, Cree BAC, De Sèze J, Gold R, Hartung HP, Jeffery D, et al. MS disease activity in RESTORE: a randomized 24-week natalizumab treatment interruption study. Neurology. (2014) 82(17):1491–8. doi: 10.1212/WNL.0000000000000355
14. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med (2006) 354(9):899–910. doi: 10.1056/NEJMoa044397
15. Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476(7359):214–9. doi: 10.1038/nature10251
16. Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet (2013) 45(11):1353–60. doi: 10.1038/ng.2770
17. Moutsianas L, Jostins L, Beecham AH, Dilthey AT, Xifara DK, Ban M, et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet (2015) 47(10):1107–13. doi: 10.1038/ng.3395
18. Brynedal B, Duvefelt K, Jonasdottir G, Roos IM, Akesson E, Palmgren J, et al. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PloS One (2007) 2(7):e664. doi: 10.1371/journal.pone.0000664
19. Hedström AK, Huang J, Michel A, Butt J, Brenner N, Hillert J, et al. High levels of epstein-barr virus nuclear antigen-1-specific antibodies and infectious mononucleosis act both independently and synergistically to increase multiple sclerosis risk. Front Neurol (2019) 10:1368. doi: 10.3389/fneur.2019.01368
20. De Jager PL, Simon KC, Munger KL, Rioux JD, Hafler DA, Ascherio A. Integrating risk factors: HLA-DRB1*1501 and Epstein-Barr virus in multiple sclerosis. Neurology. (2008) 70(13 Pt 2):1113–8. doi: 10.1212/01.wnl.0000294325.63006.f8
21. Hedström AK, Lima Bomfim I, Barcellos L, GianFrancesco M, Schaefer C, Kockum I, et al. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. (2014) 82(10):865–72. doi: 10.1212/WNL.0000000000000203
22. Disanto G, Hall C, Lucas R, Ponsonby AL, Berlanga-Taylor AJ, Giovannoni G, et al. Assessing interactions between HLA-DRB1*15 and infectious mononucleosis on the risk of multiple sclerosis. Mult Scler Houndmills Basingstoke Engl (2013) 19(10):1355–8. doi: 10.1177/1352458513477231
23. Nielsen TR, Rostgaard K, Askling J, Steffensen R, Oturai A, Jersild C, et al. Effects of infectious mononucleosis and HLA-DRB1*15 in multiple sclerosis. Mult Scler Houndmills Basingstoke Engl (2009) 15(4):431–6. doi: 10.1177/1352458508100037
24. Sundström P, Nyström M, Ruuth K, Lundgren E. Antibodies to specific EBNA-1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J Neuroimmunol (2009) 215(1–2):102–7. doi: 10.1016/j.jneuroim.2009.08.004
25. International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility.. Science (2019) 365(6460):eaav7188. doi: 10.1126/science.aav7188
26. Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol (2009) 9(12):833–44. doi: 10.1038/nri2669
27. Walker LSK, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol (2002) 2(1):11–9. doi: 10.1038/nri701
28. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287(2):1168–77. doi: 10.1074/jbc.M111.289488
29. Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol (2012) 12(9):669–77. doi: 10.1038/nri3279
30. Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today (1998) 19(9):395–404. doi: 10.1016/S0167-5699(98)01299-7
31. Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol Baltim Md 1950 (1981) 127(4):1420–3. doi: 10.4049/jimmunol.127.4.1420
32. Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol (1990) 8:579–621. doi: 10.1146/annurev.iy.08.040190.003051
33. Das P, Drescher KM, Geluk A, Bradley DS, Rodriguez M, David CS. Complementation between specific HLA-DR and HLA-DQ genes in transgenic mice determines susceptibility to experimental autoimmune encephalomyelitis. Hum Immunol (2000) 61(3):279–89. doi: 10.1016/S0198-8859(99)00135-4
34. Kawamura K, Yamamura T, Yokoyama K, Chui DH, Fukui Y, Sasazuki T, et al. Hla-DR2-restricted responses to proteolipid protein 95-116 peptide cause autoimmune encephalitis in transgenic mice. J Clin Invest (2000) 105(7):977–84. doi: 10.1172/JCI8407
35. Forsthuber TG, Shive CL, Wienhold W, de Graaf K, Spack EG, Sublett R, et al. T cell epitopes of human myelin oligodendrocyte glycoprotein identified in HLA-DR4 (DRB1*0401) transgenic mice are encephalitogenic and are presented by human B cells. J Immunol Baltim Md 1950 (2001) 167(12):7119–25. doi: 10.4049/jimmunol.167.12.7119
36. Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet (1999) 23(3):343–7. doi: 10.1038/15525
37. Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cell subsets within active chronic lesions. Science. (1983) 219(4582):308–10. doi: 10.1126/science.6217550
38. Nakajima H, Fukuda K, Doi Y, Sugino M, Kimura F, Hanafusa T, et al. Expression of TH1/TH2-related chemokine receptors on peripheral T cells and correlation with clinical disease activity in patients with multiple sclerosis. Eur Neurol (2004) 52(3):162–8. doi: 10.1159/000081856
39. Shimizu Y, Ota K, Kubo S, Kabasawa C, Kobayashi M, Ohashi T, et al. Association of Th1/Th2-related chemokine receptors in peripheral T cells with disease activity in patients with multiple sclerosis and neuromyelitis optica. Eur Neurol (2011) 66(2):91–7. doi: 10.1159/000329576
40. Tejada-Simon MV, Zang YC, Yang D, Hong J, Li S, Singh RA, et al. Aberrant T cell responses to myelin antigens during clinical exacerbation in patients with multiple sclerosis. Int Immunol (2000) 12(12):1641–50. doi: 10.1093/intimm/12.12.1641
41. Calabresi PA, Fields NS, Farnon EC, Frank JA, Bash CN, Kawanashi T, et al. ELI-spot of Th-1 cytokine secreting PBMC’s in multiple sclerosis: correlation with MRI lesions. J Neuroimmunol (1998) 85(2):212–9. doi: 10.1016/S0165-5728(98)00008-3
42. van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. (2018) 141(5):1334–49. doi: 10.1093/brain/awy069
43. Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-γ–expressing TH17 cells in multiple sclerosis. Ann Neurol (2009) 66(3):390–402. doi: 10.1002/ana.21748
44. Olsson T, Zhi WW, Höjeberg B, Kostulas V, Jiang YP, Anderson G, et al. Autoreactive T lymphocytes in multiple sclerosis determined by antigen-induced secretion of interferon-gamma. J Clin Invest (1990) 86(3):981–5. doi: 10.1172/JCI114800
45. Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol Baltim Md 1950 (2004) 172(6):3893–904. doi: 10.4049/jimmunol.172.6.3893
46. Olsson T, Sun J, Hillert J, Höjeberg B, Ekre HP, Andersson G, et al. Increased numbers of T cells recognizing multiple myelin basic protein epitopes in multiple sclerosis. Eur J Immunol (1992) 22(4):1083–7. doi: 10.1002/eji.1830220431
47. Correale J, McMillan M, McCarthy K, Le T, Weiner LP. Isolation and characterization of autoreactive proteolipid protein-peptide specific T-cell clones from multiple sclerosis patients. Neurology. (1995) 45(7):1370–8. doi: 10.1212/WNL.45.7.1370
48. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi: 10.1084/jem.20041257
49. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13(10):1173–5. doi: 10.1038/nm1651
50. Romme Christensen J, Börnsen L, Ratzer R, Piehl F, Khademi M, Olsson T, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PloS One (2013) 8(3):e57820. doi: 10.1371/journal.pone.0057820
51. Kaufmann M, Evans H, Schaupp AL, Engler JB, Kaur G, Willing A, et al. Identifying CNS-colonizing T cells as potential therapeutic targets to prevent progression of multiple sclerosis. Med. (2021) 2(3):296–312.e8. doi: 10.1016/j.medj.2021.01.006
52. Morille J, Mandon M, Rodriguez S, Roulois D, Leonard S, Garcia A, et al. Multiple sclerosis CSF is enriched with follicular T cells displaying a th1/eomes signature. Neurol Neuroimmunol Neuroinflammation (2022) 9(6):e200033. doi: 10.1212/NXI.0000000000200033
53. Höftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, et al. Expression of Major Histocompatibility Complex class l Molecules on the Different Cell Types in Multiple Sclerosis Lesions. Brain Pathol (2004) 14(1):43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x
54. Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J Immunol Baltim Md 1950 (1998) 160(6):3056–9. doi: 10.4049/jimmunol.160.6.3056
55. Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice1. J Immunol (2001) 166(12):7579–87. doi: 10.4049/jimmunol.166.12.7579
56. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J Autoimmun (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010
57. Lundtoft C, Pucholt P, Imgenberg-Kreuz J, Carlsson-Almlöf J, Eloranta ML, Syvänen AC, et al. Function of multiple sclerosis-protective HLA class I alleles revealed by genome-wide protein-quantitative trait loci mapping of interferon signalling. PloS Genet (2020) 16(10):e1009199. doi: 10.1371/journal.pgen.1009199
58. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of cd8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192(3):393–404. doi: 10.1084/jem.192.3.393
59. Salou M, Garcia A, Michel L, Gainche-Salmon A, Loussouarn D, Nicol B, et al. Expanded CD8 T-cell sharing between periphery and CNS in multiple sclerosis. Ann Clin Transl Neurol (2015) 2(6):609–22. doi: 10.1002/acn3.199
60. Sabatino JJ Jr, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti-CD20 therapy depletes activated myelin-specific CD8+ T cells in multiple sclerosis. Proc Natl Acad Sci U S A (2019) 116(51):25800–7. doi: 10.1073/pnas.1915309116
61. Liu PJ, Yang TT, Fan ZX, Yuan GB, Ma L, Wang ZY, et al. Characterization of antigen-specific CD8+ memory T cell subsets in peripheral blood of patients with multiple sclerosis. Front Immunol (2023) 14:1110672. doi: 10.3389/fimmu.2023.1110672
62. Biström M, Jons D, Engdahl E, Gustafsson R, Huang J, Brenner N, et al. Epstein-Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur J Neurol (2021) 28(2):579–86. doi: 10.1111/ene.14597
63. Engdahl E, Gustafsson R, Huang J, Biström M, Lima Bomfim I, Stridh P, et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol (2019) 10:2715. doi: 10.3389/fimmu.2019.02715
64. Sundqvist E, Sundström P, Lindén M, Hedström AK, Aloisi F, Hillert J, et al. Epstein-Barr virus and multiple sclerosis: interaction with HLA. Genes Immun (2012) 13(1):14–20. doi: 10.1038/gene.2011.42
65. Thomas OG, Rickinson A, Palendira U. Epstein-Barr virus and multiple sclerosis: moving from questions of association to questions of mechanism. Clin Transl Immunol (2023) 12(5):e1451. doi: 10.1002/cti2.1451
66. Bronge M, Högelin KA, Thomas OG, Ruhrmann S, Carvalho-Queiroz C, Nilsson OB, et al. Identification of four novel T cell autoantigens and personal autoreactive profiles in multiple sclerosis. Sci Adv (2022) 8(17):eabn1823. doi: 10.1126/sciadv.abn1823
67. Ayoglu B, Mitsios N, Kockum I, Khademi M, Zandian A, Sjöberg R, et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc Natl Acad Sci U S A (2016) 113(8):2188–93. doi: 10.1073/pnas.1518553113
68. Tengvall K, Huang J, Hellström C, Kammer P, Biström M, Ayoglu B, et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc Natl Acad Sci U S A (2019) 116(34):16955–60. doi: 10.1073/pnas.1902623116
69. Thomas OG, Bronge M, Tengvall K, Akpinar B, Nilsson OB, Holmgren E, et al. Cross-reactive EBNA1 immunity targets alpha-crystallin B and is associated with multiple sclerosis. Sci Adv (2023) 9(20):eadg3032. doi: 10.1126/sciadv.adg3032
70. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-l-fucose synthase is a CD4(+) T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci Transl Med (2018) 10(462):eaat4301. doi: 10.1126/scitranslmed.aat4301
71. Wang J, Jelcic I, Mühlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell. (2020) 183(5):1264–1281.e20. doi: 10.1016/j.cell.2020.09.054
72. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell. (2018) 175(1):85–100. doi: 10.1016/j.cell.2018.08.011
73. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
74. Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol (2010) 67(6):824–30. doi: 10.1002/ana.21978
75. Yea C, Tellier R, Chong P, Westmacott G, Marrie RA, Bar-Or A, et al. Epstein-Barr virus in oral shedding of children with multiple sclerosis. Neurology. (2013) 81(16):1392–9. doi: 10.1212/WNL.0b013e3182a841e4
76. Dunmire SK, Grimm JM, Schmeling DO, Balfour HHJ, Hogquist KA. The incubation period of primary epstein-barr virus infection: viral dynamics and immunologic events. PloS Pathog (2015) 11(12):e1005286. doi: 10.1371/journal.ppat.1005286
77. Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Büssow K, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest (2005) 115(5):1352–60. doi: 10.1172/JCI200523661
78. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain J Neurol (2008) 131(Pt 7):1712–21. doi: 10.1093/brain/awn108
79. Höllsberg P, Hansen HJ, Haahr S. Altered CD8+ T cell responses to selected Epstein-Barr virus immunodominant epitopes in patients with multiple sclerosis. Clin Exp Immunol (2003) 132(1):137–43. doi: 10.1046/j.1365-2249.2003.02114.x
80. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Transl Immunol (2017) 6(1):e126.
81. Croxford JL, Olson JK, Anger HA, Miller SD. Initiation and exacerbation of autoimmune demyelination of the central nervous system via virus-induced molecular mimicry: Implications for the pathogenesis of multiple sclerosis. J Virol (2005) 79(13):8581–90. doi: 10.1128/JVI.79.13.8581-8590.2005
82. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol (2010) 11(7):628–34. doi: 10.1038/ni.1888
83. Na SY, Hermann A, Sanchez-Ruiz M, Storch A, Deckert M, Hünig T. Oligodendrocytes enforce immune tolerance of the uninfected brain by purging the peripheral repertoire of autoreactive CD8+ T cells. Immunity. (2012) 37(1):134–46. doi: 10.1016/j.immuni.2012.04.009
84. Burns J, Rosenzweig A, Zweiman B, Lisak RP. Isolation of myelin basic protein-reactive T-cell lines from normal human blood. Cell Immunol (1983) 81(2):435–40. doi: 10.1016/0008-8749(83)90250-2
85. Bronge M, Ruhrmann S, Carvalho-Queiroz C, Nilsson OB, Kaiser A, Holmgren E, et al. Myelin oligodendrocyte glycoprotein revisited-sensitive detection of MOG-specific T-cells in multiple sclerosis. J Autoimmun (2019) 102:38–49. doi: 10.1016/j.jaut.2019.04.013
86. Wallström E, Khademi M, Andersson M, Weissert R, Linington C, Olsson T. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur J Immunol (1998) 28(10):3329–35. doi: 10.1002/(SICI)1521-4141(199810)28:10<3329::AID-IMMU3329>3.0.CO;2-B
87. Sun J, Link H, Olsson T, Xiao BG, Andersson G, Ekre HP, et al. T and B cell responses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J Immunol Baltim Md 1950 (1991) 146(5):1490–5. doi: 10.4049/jimmunol.146.5.1490
88. Koehler NKU, Genain CP, Giesser B, Hauser SL. The human T cell response to myelin oligodendrocyte glycoprotein: a multiple sclerosis family-based study. J Immunol Baltim Md 1950 (2002) 168(11):5920–7. doi: 10.4049/jimmunol.168.11.5920
89. Kerlero de Rosbo N, Hoffman M, Mendel I, Yust I, Kaye J, Bakimer R, et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol (1997) 27(11):3059–69. doi: 10.1002/eji.1830271144
90. Weissert R, Kuhle J, de Graaf KL, Wienhold W, Herrmann MM, Müller C, et al. High immunogenicity of intracellular myelin oligodendrocyte glycoprotein epitopes. J Immunol Baltim Md 1950 (2002) 169(1):548–56. doi: 10.4049/jimmunol.169.1.548
91. Lindert RB, Haase CG, Brehm U, Linington C, Wekerle H, Hohlfeld R. Multiple sclerosis: B- and T-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain J Neurol (1999) 122(Pt 11):2089–100. doi: 10.1093/brain/122.11.2089
92. Cruciani C, Puthenparampil M, Tomas-Ojer P, Jelcic I, Docampo MJ, Planas R, et al. T-cell specificity influences disease heterogeneity in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2021) 8(6):e1075. doi: 10.1212/NXI.0000000000001075
93. Hughes LE, Smith PA, Bonell S, Natt RS, Wilson C, Rashid T, et al. Cross-reactivity between related sequences found in Acinetobacter sp., Pseudomonas aeruginosa, myelin basic protein and myelin oligodendrocyte glycoprotein in multiple sclerosis. J Neuroimmunol (2003) 144(1–2):105–15. doi: 10.1016/s0165-5728(03)00274-1
94. Kuerten S, Lanz TV, Lingampalli N, Lahey LJ, Kleinschnitz C, Mäurer M, et al. Autoantibodies against central nervous system antigens in a subset of B cell-dominant multiple sclerosis patients. Proc Natl Acad Sci U S A (2020) 117(35):21512–8. doi: 10.1073/pnas.2011249117
95. Wang H, Munger KL, Reindl M, O’Reilly EJ, Levin LI, Berger T, et al. Myelin oligodendrocyte glycoprotein antibodies and multiple sclerosis in healthy young adults. Neurology. (2008) 71(15):1142–6. doi: 10.1212/01.wnl.0000316195.52001.e1
96. Pröbstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. (2011) 77(6):580–8. doi: 10.1212/WNL.0b013e318228c0b1
97. Hecker M, Fitzner B, Wendt M, Lorenz P, Flechtner K, Steinbeck F, et al. High-density peptide microarray analysis of igG autoantibody reactivities in serum and cerebrospinal fluid of multiple sclerosis patients. Mol Cell Proteomics MCP (2016) 15(4):1360–80. doi: 10.1074/mcp.M115.051664
98. Abdul-Majid KB, Jirholt J, Stadelmann C, Stefferl A, Kjellén P, Wallström E, et al. Screening of several H-2 congenic mouse strains identified H-2(q) mice as highly susceptible to MOG-induced EAE with minimal adjuvant requirement. J Neuroimmunol (2000) 111(1–2):23–33. doi: 10.1016/S0165-5728(00)00360-X
99. Weissert R, Wallström E, Storch MK, Stefferl A, Lorentzen J, Lassmann H, et al. MHC haplotype-dependent regulation of MOG-induced EAE in rats. J Clin Invest (1998) 102(6):1265–73. doi: 10.1172/JCI3022
100. Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol (1995) 25(7):1951–9. doi: 10.1002/eji.1830250723
101. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med (2003) 197(9):1073–81. doi: 10.1084/jem.20021603
102. Berger T, Weerth S, Kojima K, Linington C, Wekerle H, Lassmann H. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Investig J Tech Methods Pathol (1997) 76(3):355–64.
103. Ellmerich S, Mycko M, Takacs K, Waldner H, Wahid FN, Boyton RJ, et al. High incidence of spontaneous disease in an HLA-DR15 and TCR transgenic multiple sclerosis model. J Immunol Baltim Md 1950 (2005) 174(4):1938–46. doi: 10.4049/jimmunol.174.4.1938
104. Blanchfield L, Sabatino JJJ, Lawrence L, Evavold BD. NFM cross-reactivity to MOG does not expand a critical threshold level of high-affinity T cells necessary for onset of demyelinating disease. J Immunol Baltim Md 1950 (2017) 199(8):2680–91. doi: 10.4049/jimmunol.1700792
105. Stefferl A, Schubart A, Storch M, Amini A, Mather I, Lassmann H, et al. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J Immunol (2000) 165(5):2859–65. doi: 10.4049/jimmunol.165.5.2859
106. Guggenmos J, Schubart AS, Ogg S, Andersson M, Olsson T, Mather IH, et al. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol Baltim Md 1950 (2004) 172(1):661–8. doi: 10.4049/jimmunol.172.1.661
107. de Luca V, Martins Higa A, Malta Romano C, Pimenta Mambrini G, Peroni LA, Trivinho-Strixino F, et al. Cross-reactivity between myelin oligodendrocyte glycoprotein and human endogenous retrovirus W protein: nanotechnological evidence for the potential trigger of multiple sclerosis. Micron Oxf Engl 1993 (2019) 120:66–73. doi: 10.1016/j.micron.2019.02.005
108. Markovic-Plese S, Hemmer B, Zhao Y, Simon R, Pinilla C, Martin R. High level of cross-reactivity in influenza virus hemagglutinin-specific CD4+ T-cell response: implications for the initiation of autoimmune response in multiple sclerosis. J Neuroimmunol (2005) 169(1–2):31–8. doi: 10.1016/j.jneuroim.2005.07.014
109. Greer JM, Csurhes PA, Cameron KD, McCombe PA, Good MF, Pender MP. Increased immunoreactivity to two overlapping peptides of myelin proteolipid protein in multiple sclerosis. Brain J Neurol (1997) 120(Pt 8):1447–60. doi: 10.1093/brain/120.8.1447
110. Pelfrey CM, Trotter JL, Tranquill LR, McFarland HF. Identification of a novel T cell epitope of human proteolipid protein (residues 40-60) recognized by proliferative and cytolytic CD4+ T cells from multiple sclerosis patients. J Neuroimmunol (1993) 46(1–2):33–42. doi: 10.1016/0165-5728(93)90231-M
111. Pelfrey CM, Trotter JL, Tranquill LR, McFarland HF. Identification of a second T cell epitope of human proteolipid protein (residues 89-106) recognized by proliferative and cytolytic CD4+ T cells from multiple sclerosis patients. J Neuroimmunol (1994) 53(2):153–61. doi: 10.1016/0165-5728(94)90025-6
112. Kondo T, Yamamura T, Inobe J, Ohashi T, Takahashi K, Tabira T. TCR repertoire to proteolipid protein (PLP) in multiple sclerosis (MS): homologies between PLP-specific T cells and MS-associated T cells in TCR junctional sequences. Int Immunol (1996) 8(1):123–30. doi: 10.1093/intimm/8.1.123
113. Greer JM, Csurhes PA, Muller DM, Pender MP. Correlation of blood T cell and antibody reactivity to myelin proteins with HLA type and lesion localization in multiple sclerosis. J Immunol Baltim Md 1950 (2008) 180(9):6402–10. doi: 10.4049/jimmunol.180.9.6402
114. Owens GP, Fellin TJ, Matschulat A, Salas V, Schaller KL, Given KS, et al. Pathogenic myelin specific antibodies in multiple sclerosis target conformational proteolipid protein 1 anchored membrane domains. J Clin Invest (2023) 10:e162731. doi: 10.1172/JCI162731
115. Greer JM, Trifilieff E, Pender MP. Correlation between anti-myelin proteolipid protein (PLP) antibodies and disease severity in multiple sclerosis patients with PLP response-permissive HLA types. Front Immunol (2020) 11:1891. doi: 10.3389/fimmu.2020.01891
116. Quintana FJ, Farez MF, Izquierdo G, Lucas M, Cohen IR, Weiner HL. Antigen microarrays identify CNS-produced autoantibodies in RRMS. Neurology. (2012) 78(8):532–9. doi: 10.1212/WNL.0b013e318247f9f3
117. Kennedy MK, Tan LJ, Dal Canto MC, Tuohy VK, Lu ZJ, Trotter JL, et al. Inhibition of murine relapsing experimental autoimmune encephalomyelitis by immune tolerance to proteolipid protein and its encephalitogenic peptides. J Immunol Baltim Md 1950 (1990) 144(3):909–15. doi: 10.4049/jimmunol.144.3.909
118. Waldner H, Whitters MJ, Sobel RA, Collins M, Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc Natl Acad Sci U S A (2000) 97(7):3412–7. doi: 10.1073/pnas.97.7.3412
119. Boucher A, Desforges M, Duquette P, Talbot PJ. Long-term human coronavirus-myelin cross-reactive T-cell clones derived from multiple sclerosis patients. Clin Immunol Orlando Fla (2007) 123(3):258–67. doi: 10.1016/j.clim.2007.02.002
120. Massilamany C, Thulasingam S, Steffen D, Reddy J. Gender differences in CNS autoimmunity induced by mimicry epitope for PLP 139-151 in SJL mice. J Neuroimmunol (2011) 230(1–2):95–104. doi: 10.1016/j.jneuroim.2010.09.011
121. Olson JK, Croxford JL, Calenoff MA, Dal Canto MC, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest (2001) 108(2):311–8. doi: 10.1172/JCI200113032
122. Honma K, Parker KC, Becker KG, McFarland HF, Coligan JE, Biddison WE. Identification of an epitope derived from human proteolipid protein that can induce autoreactive CD8+ cytotoxic T lymphocytes restricted by HLA-A3: evidence for cross-reactivity with an environmental microorganism. J Neuroimmunol (1997) 73(1–2):7–14. doi: 10.1016/S0165-5728(96)00161-0
123. Valli A, Sette A, Kappos L, Oseroff C, Sidney J, Miescher G, et al. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest (1993) 91(2):616–28. doi: 10.1172/JCI116242
124. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. (1990) 346(6280):183–7. doi: 10.1038/346183a0
125. Martino G, Olsson T, Fredrikson S, Hojeberg B, Kostulas V, Grimaldi LM, et al. Cells producing antibodies specific for myelin basic protein region 70-89 are predominant in cerebrospinal fluid from patients with multiple sclerosis. Eur J Immunol (1991) 21(12):2971–6. doi: 10.1002/eji.1830211211
126. Chou YK, Vainiene M, Whitham R, Bourdette D, Chou CH, Hashim G, et al. Response of human T lymphocyte lines to myelin basic protein: association of dominant epitopes with HLA class II restriction molecules. J Neurosci Res (1989) 23(2):207–16. doi: 10.1002/jnr.490230211
127. Schmidt S, Linington C, Zipp F, Sotgiu S, de Waal Malefyt R, Wekerle H, et al. Multiple sclerosis: comparison of the human T-cell response to S100 beta and myelin basic protein reveals parallels to rat experimental autoimmune panencephalitis. Brain J Neurol (1997) 120(Pt 8):1437–45. doi: 10.1093/brain/120.8.1437
128. Wucherpfennig KW, Catz I, Hausmann S, Strominger JL, Steinman L, Warren KG. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA-DR2-restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B-cell and T-cell epitopes. J Clin Invest (1997) 100(5):1114–22. doi: 10.1172/JCI119622
129. Vergelli M, Kalbus M, Rojo SC, Hemmer B, Kalbacher H, Tranquill L, et al. T cell response to myelin basic protein in the context of the multiple sclerosis-associated HLA-DR15 haplotype: peptide binding, immunodominance and effector functions of T cells. J Neuroimmunol (1997) 77(2):195–203. doi: 10.1016/S0165-5728(97)00075-1
130. García-Merino A, Persson MA, Ernerudh J, Díaz-Gil JJ, Olsson T. Serum and cerebrospinal fluid antibodies against myelin basic protein and their IgG subclass distribution in multiple sclerosis. J Neurol Neurosurg Psychiatry (1986) 49(9):1066–70. doi: 10.1136/jnnp.49.9.1066
131. Olsson T, Baig S, Höjeberg B, Link H. Antimyelin basic protein and antimyelin antibody-producing cells in multiple sclerosis. Ann Neurol (1990) 27(2):132–6. doi: 10.1002/ana.410270207
132. Jansson L, Olsson T, Höjeberg B, Holmdahl R. Chronic experimental autoimmune encephalomyelitis induced by the 89-101 myelin basic protein peptide in B10RIII (H-2r) mice. Eur J Immunol (1991) 21(3):693–9. doi: 10.1002/eji.1830210323
133. Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. (1993) 72(4):551–60. doi: 10.1016/0092-8674(93)90074-Z
134. Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. (1994) 78(3):399–408. doi: 10.1016/0092-8674(94)90419-7
135. Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. (1985) 230(4729):1043–5. doi: 10.1126/science.2414848
136. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. (1995) 80(5):695–705. doi: 10.1016/0092-8674(95)90348-8
137. Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol (2002) 3(10):940–3. doi: 10.1038/ni835
138. Holmøy T, Kvale EØ, Vartdal F. Cerebrospinal fluid CD4+ T cells from a multiple sclerosis patient cross-recognize Epstein-Barr virus and myelin basic protein. J Neurovirol (2004) 10(5):278–83. doi: 10.1080/13550280490499524
139. Jog NR, McClain MT, Heinlen LD, Gross T, Towner R, Guthridge JM, et al. Epstein Barr virus nuclear antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J Autoimmun (2020) 106:102332. doi: 10.1016/j.jaut.2019.102332
140. Lomakin Y, Arapidi GP, Chernov A, Ziganshin R, Tcyganov E, Lyadova I, et al. Exposure to the epstein–barr viral antigen latent membrane protein 1 induces myelin-reactive antibodies in vivo. Front Immunol (2017) 8:777. doi: 10.3389/fimmu.2017.00777
141. Tejada-Simon MV, Zang YCQ, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol (2003) 53(2):189–97. doi: 10.1002/ana.10425
142. Talbot PJ, Paquette JS, Ciurli C, Antel JP, Ouellet F. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann Neurol (1996) 39(2):233–40. doi: 10.1002/ana.410390213
143. Mao YS, Lu CZ, Wang X, Xiao BG. Induction of experimental autoimmune encephalomyelitis in Lewis rats by a viral peptide with limited homology to myelin basic protein. Exp Neurol (2007) 206(2):231–9. doi: 10.1016/j.expneurol.2007.04.015
144. Gautam AM, Liblau R, Chelvanayagam G, Steinman L, Boston T. A viral peptide with limited homology to a self peptide can induce clinical signs of experimental autoimmune encephalomyelitis. J Immunol Baltim Md 1950 (1998) 161(1):60–4. doi: 10.4049/jimmunol.161.1.60
145. Lenz DC, Lu L, Conant SB, Wolf NA, Gérard HC, Whittum-Hudson JA, et al. A chlamydia pneumoniae-specific peptide induces experimental autoimmune encephalomyelitis in rats1. J Immunol (2001) 167(3):1803–8. doi: 10.4049/jimmunol.167.3.1803
146. Bray PF, Luka J, Bray PF, Culp KW, Schlight JP. Antibodies against Epstein-Barr nuclear antigen (EBNA) in multiple sclerosis CSF, and two pentapeptide sequence identities between EBNA and myelin basic protein. Neurology. (1992) 42(9):1798–804. doi: 10.1212/WNL.42.9.1798
147. Gabibov AG, Belogurov AAJ, Lomakin YA, Zakharova MY, Avakyan ME, Dubrovskaya VV, et al. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J Off Publ Fed Am Soc Exp Biol (2011) 25(12):4211–21. doi: 10.1096/fj.11-190769
148. van Noort JM, van Sechel AC, Bajramovic JJ, Ouagmiri M, Polman CH, Lassmann H, et al. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature. (1995) 375(6534):798–801. doi: 10.1038/375798a0
149. Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O’Connor KC, Hafler DA, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. (2007) 448(7152):474–9. doi: 10.1038/nature05935
150. van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, et al. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol (2010) 69(7):694–703. doi: 10.1097/NEN.0b013e3181e4939c
151. Thoua NM, van Noort JM, Baker D, Bose A, van Sechel AC, van Stipdonk MJ, et al. Encephalitogenic and immunogenic potential of the stress protein alphaB-crystallin in Biozzi ABH (H-2A(g7)) mice. J Neuroimmunol (2000) 104(1):47–57. doi: 10.1016/S0165-5728(99)00246-5
152. Rand KH, Houck H, Denslow ND, Heilman KM. Molecular approach to find target(s) for oligoclonal bands in multiple sclerosis. J Neurol Neurosurg Psychiatry (1998) 65(1):48–55. doi: 10.1136/jnnp.65.1.48
153. Lanz TV, Brewer RC, Ho PP, Moon JS, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. (2022) 603(7900):321–7. doi: 10.1038/s41586-022-04432-7
154. Chunder R, Weier A, Mäurer H, Luber N, Enders M, Luber G, et al. Antibody cross-reactivity between casein and myelin-associated glycoprotein results in central nervous system demyelination. Proc Natl Acad Sci U S A (2022) 119(10):e2117034119. doi: 10.1073/pnas.2117034119
155. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol (2021) 51(1):64–75. doi: 10.1002/eji.202048655
156. Bsibsi M, Peferoen LAN, Holtman IR, Nacken PJ, Gerritsen WH, Witte ME, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-γ and alpha B-crystallin. Acta Neuropathol (Berl) (2014) 128(2):215–29. doi: 10.1007/s00401-014-1317-8
157. Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature (1985) 314(6011):537–9. doi: 10.1038/314537a0
158. Choi IK, Wang Z, Ke Q, Hong M, Qian Y, Zhao X, et al. Signaling by the Epstein-Barr virus LMP1 protein induces potent cytotoxic CD4(+) and CD8(+) T cell responses. Proc Natl Acad Sci U S A (2018) 115(4):E686–95. doi: 10.1073/pnas.1713607115
159. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. (2015) 347(6220):1260419. doi: 10.1126/science.1260419
160. Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. (2008) 451(7182):1076—1081. doi: 10.1038/nature06559
161. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
162. Dawoodji A, Chen JL, Shepherd D, Dalin F, Tarlton A, Alimohammadi M, et al. High frequency of cytolytic 21-hydroxylase-specific CD8+ T cells in autoimmune Addison’s disease patients. J Immunol Baltim Md 1950 (2014) 193(5):2118–26. doi: 10.4049/jimmunol.1400056
163. Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes. (2005) 54 Suppl 2:S52–61. doi: 10.2337/diabetes.54.suppl_2.S52
164. Lipphardt M, Wallbach M, Koziolek MJ. Plasma exchange or immunoadsorption in demyelinating diseases: A meta-analysis. J Clin Med (2020) 9(5):1597. doi: 10.3390/jcm9051597
165. Stork L, Ellenberger D, Beißbarth T, Friede T, Lucchinetti CF, Brück W, et al. Differences in the reponses to apheresis therapy of patients with 3 histopathologically classified immunopathological patterns of multiple sclerosis. JAMA Neurol (2018) 75(4):428–35. doi: 10.1001/jamaneurol.2017.4842
166. Lindsey JW, deGannes SL, Pate KA, Zhao X. Antibodies specific for Epstein-Barr virus nuclear antigen-1 cross-react with human heterogeneous nuclear ribonucleoprotein L. Mol Immunol (2016) 69:7–12. doi: 10.1016/j.molimm.2015.11.007
167. Lindsey JW. Antibodies to the Epstein-Barr virus proteins BFRF3 and BRRF2 cross-react with human proteins. J Neuroimmunol (2017) 310:131–4. doi: 10.1016/j.jneuroim.2017.07.013
168. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol (2009) 21(5):293–300. doi: 10.1016/j.smim.2009.05.012
169. Haxhinasto SA, Bishop GA. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem (2004) 279(4):2575–82. doi: 10.1074/jbc.M310628200
170. Haxhinasto SA, Hostager BS, Bishop GA. Cutting edge: molecular mechanisms of synergy between CD40 and the B cell antigen receptor: role for TNF receptor-associated factor 2 in receptor interaction. J Immunol Baltim Md 1950 (2002) 169(3):1145–9. doi: 10.4049/jimmunol.169.3.1145
171. Buchta CM, Bishop GA. Toll-like receptors and B cells: functions and mechanisms. Immunol Res (2014) 59(1–3):12–22. doi: 10.1007/s12026-014-8523-2
172. Vanden Bush TJ, Bishop GA. TLR7 and CD40 cooperate in IL-6 production via enhanced JNK and AP-1 activation. Eur J Immunol (2008) 38(2):400–9. doi: 10.1002/eji.200737602
173. Stunz LL, Busch LK, Munroe ME, Sigmund CD, Tygrett LT, Waldschmidt TJ, et al. Expression of the cytoplasmic tail of LMP1 in mice induces hyperactivation of B lymphocytes and disordered lymphoid architecture. Immunity. (2004) 21(2):255–66. doi: 10.1016/j.immuni.2004.07.008
174. Munroe ME, Anderson JR, Gross TF, Stunz LL, Bishop GA, James JA. Epstein-barr functional mimicry: pathogenicity of oncogenic latent membrane protein-1 in systemic lupus erythematosus and autoimmunity. Front Immunol (2021) 11:606936. doi: 10.3389/fimmu.2020.606936
175. Fitzsimmons L, Cartlidge R, Chang C, Sejic N, Galbraith LCA, Suraweera CD, et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ (2020) 27(5):1554–68. doi: 10.1038/s41418-019-0435-1
176. Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PloS Pathog (2012) 8(5):1–11. doi: 10.1371/journal.ppat.1002704
177. Quinn LL, Zuo J, Abbott RJM, Shannon-Lowe C, Tierney RJ, Hislop AD, et al. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PloS Pathog (2014) 10(8):e1004322. doi: 10.1371/journal.ppat.1004322
178. Quinn LL, Williams LR, White C, Forrest C, Zuo J, Rowe M. The missing link in epstein-barr virus immune evasion: the BDLF3 gene induces ubiquitination and downregulation of major histocompatibility complex class I (MHC-I) and MHC-II. J Virol (2016) 90(1):356–67. doi: 10.1128/JVI.02183-15
179. Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ, et al. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J Virol (2011) 85(4):1604–14. doi: 10.1128/JVI.01608-10
180. Chijioke O, Müller A, Feederle R, Barros MHM, Krieg C, Emmel V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep (2013) 5(6):1489–98. doi: 10.1016/j.celrep.2013.11.041
181. Chijioke O, Landtwing V, Münz C. NK cell influence on the outcome of primary epstein-barr virus infection. Front Immunol (2016) 7:323. doi: 10.3389/fimmu.2016.00323
182. Azzi T, Lünemann A, Murer A, Ueda S, Béziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood. (2014) 124(16):2533–43. doi: 10.1182/blood-2014-01-553024
183. Lünemann A, Tackenberg B, DeAngelis T, da Silva RB, Messmer B, Vanoaica LD, et al. Impaired IFN-γ production and proliferation of NK cells in multiple sclerosis. Int Immunol (2011) 23(2):139–48. doi: 10.1093/intimm/dxq463
184. Long HM, Chagoury OL, Leese AM, Ryan GB, James E, Morton LT, et al. MHC II tetramers visualize human CD4+ T cell responses to Epstein-Barr virus infection and demonstrate atypical kinetics of the nuclear antigen EBNA1 response. J Exp Med (2013) 210(5):933–49. doi: 10.1084/jem.20121437
185. Sindic CJM, Monteyne PH, Laterre EC. The intrathecal synthesis of virus-specific oligoclonal IgG in multiple sclerosis. J Neuroimmunol (1994) 54(1):75–80. doi: 10.1016/0165-5728(94)90233-X
186. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. (2014) 157(5):1073–87. doi: 10.1016/j.cell.2014.03.047
187. Pi KS, Bortolotti D, Sang Y, Schiuma G, Beltrami S, Rizzo S, et al. Studying the interactions of U24 from HHV-6 in order to further elucidate its potential role in MS. Viruses (2022) 14(11):2384. doi: 10.3390/v14112384
188. Rizzo R, Gentili V, Casetta I, Caselli E, De Gennaro R, Granieri E, et al. Altered natural killer cells’ response to herpes virus infection in multiple sclerosis involves KIR2DL2 expression. J Neuroimmunol (2012) 251(1–2):55–64. doi: 10.1016/j.jneuroim.2012.07.004
189. Pi KS, Sang Y, Straus SK. Viral proteins with pxxP and PY motifs may play a role in multiple sclerosis. Viruses (2022) 14(2):281. doi: 10.3390/v14020281
190. Ascherio A, Munch M. Epstein-Barr virus and multiple sclerosis. Epidemiol Camb Mass (2000) 11(2):220–4. doi: 10.1097/00001648-200003000-00023
191. Sundqvist E, Bergström T, Daialhosein H, Nyström M, Sundström P, Hillert J, et al. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler Houndmills Basingstoke Engl (2014) 20(2):165–73. doi: 10.1177/1352458513494489
192. Grut V, Biström M, Salzer J, Stridh P, Jons D, Gustafsson R, et al. Cytomegalovirus seropositivity is associated with reduced risk of multiple sclerosis—a presymptomatic case–control study. Eur J Neurol (2021) 28(9):3072–9. doi: 10.1111/ene.14961
193. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med (2004) 199(2):155–66. doi: 10.1084/jem.20031677
194. Pribyl TM, Campagnoni C, Kampf K, Handley VW, Campagnoni AT. The major myelin protein genes are expressed in the human thymus. J Neurosci Res (1996) 45(6):812–9. doi: 10.1002/(SICI)1097-4547(19960915)45:6<812::AID-JNR18>3.0.CO;2-X
195. Wucherpfennig KW, Sette A, Southwood S, Oseroff C, Matsui M, Strominger JL, et al. Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J Exp Med (1994) 179(1):279–90. doi: 10.1084/jem.179.1.279
196. Vogt AB, Kropshofer H, Kalbacher H, Kalbus M, Rammensee HG, Coligan JE, et al. Ligand motifs of HLA-DRB5*0101 and DRB1*1501 molecules delineated from self-peptides. J Immunol Baltim Md 1950 (1994) 153(4):1665–73. doi: 10.4049/jimmunol.153.4.1665
197. Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J (2005) 24(17):2968–79. doi: 10.1038/sj.emboj.7600771
198. Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, et al. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol Baltim Md 1950 (1990) 145(2):540–8. doi: 10.4049/jimmunol.145.2.540
199. Tejada-Simon MV, Hong J, Rivera VM, Zhang JZ. Reactivity pattern and cytokine profile of T cells primed by myelin peptides in multiple sclerosis and healthy individuals. Eur J Immunol (2001) 31(3):907–17. doi: 10.1002/1521-4141(200103)31:3<907::AID-IMMU907>3.0.CO;2-1
200. Moldovan IR, Rudick RA, Cotleur AC, Born SE, Lee JC, Karafa MT, et al. Interferon gamma responses to myelin peptides in multiple sclerosis correlate with a new clinical measure of disease progression. J Neuroimmunol (2003) 141(1):132–40. doi: 10.1016/S0165-5728(03)00221-2
201. Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. (2008) 125(2):161–9. doi: 10.1111/j.1365-2567.2008.02837.x
202. Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med (1994) 179(3):973–84. doi: 10.1084/jem.179.3.973
203. Greer JM, Csurhes PA, Pender MP, McCombe PA. Effect of gender on T-cell proliferative responses to myelin proteolipid protein antigens in patients with multiple sclerosis and controls. J Autoimmun (2004) 22(4):345–52. doi: 10.1016/j.jaut.2004.03.004
204. Pender MP, Csurhes PA, Greer JM, Mowat PD, Henderson RD, Cameron KD, et al. Surges of increased T cell reactivity to an encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects1. J Immunol (2000) 165(9):5322–31. doi: 10.4049/jimmunol.165.9.5322
205. Hughes LE, Bonell S, Natt RS, Wilson C, Tiwana H, Ebringer A, et al. Antibody responses to Acinetobacter spp. and Pseudomonas aeruginosa in multiple sclerosis: prospects for diagnosis using the myelin-acinetobacter-neurofilament antibody index. Clin Diagn Lab Immunol (2001) 8(6):1181–8. doi: 10.1128/CDLI.8.6.1181-1188.2001
206. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun (2016) 7(1):12015. doi: 10.1038/ncomms12015
207. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci U S A (2017) 114(40):10713–8. doi: 10.1073/pnas.1711235114
208. Bruno R, Sabater L, Sospedra M, Ferrer-Francesch X, Escudero D, Martínez-Cáceres E, et al. Multiple sclerosis candidate autoantigens except myelin oligodendrocyte glycoprotein are transcribed in human thymus. Eur J Immunol (2002) 32(10):2737–47. doi: 10.1002/1521-4141(2002010)32:10<2737::AID-IMMU2737>3.0.CO;2-0
209. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A (1994) 91(23):10859–63. doi: 10.1073/pnas.91.23.10859
210. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol (2002) 2(2):85–95. doi: 10.1038/nri724
211. Tavazzi E, Pichiecchio A, Colombo E, Rigoni E, Asteggiano C, Vegezzi E, et al. The potential role of SARS-coV-2 infection and vaccines in multiple sclerosis onset and reactivation: A case series and literature review. Viruses (2023) 15(7):1569. doi: 10.3390/v15071569
212. Lake CM, Breen JJ. Sequence similarity between SARS-CoV-2 nucleocapsid and multiple sclerosis-associated proteins provides insight into viral neuropathogenesis following infection. Sci Rep (2023) 13(1):389. doi: 10.1038/s41598-022-27348-8
213. Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med (1997) 3(10):1133–6. doi: 10.1038/nm1097-1133
214. Berthelot L, Laplaud DA, Pettré S, Ballet C, Michel L, Hillion S, et al. Blood CD8+ T cell responses against myelin determinants in multiple sclerosis and healthy individuals. Eur J Immunol (2008) 38(7):1889–99. doi: 10.1002/eji.200838023
215. Wilson DB, Wilson DH, Schroder K, Pinilla C, Blondelle S, Houghten RA, et al. Specificity and degeneracy of T cells. Mol Immunol (2004) 40(14–15):1047–55. doi: 10.1016/j.molimm.2003.11.022
216. Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O’Callaghan CA, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med (1998) 187(9):1395–402. doi: 10.1084/jem.187.9.1395
217. Callan MF, Steven N, Krausa P, Wilson JD, Moss PA, Gillespie GM, et al. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med (1996) 2(8):906–11. doi: 10.1038/nm0896-906
218. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
219. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med (2005) 11(3):335–9. doi: 10.1038/nm1202
220. Wilhelm CR, Upadhye MA, Eschbacher KL, Karandikar NJ, Boyden AW. Proteolipid protein-induced mouse model of multiple sclerosis requires B cell-mediated antigen presentation. J Immunol Baltim Md 1950 (2023) 7:ji2200721. doi: 10.4049/jimmunol.2200721
221. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210(13):2921–37. doi: 10.1084/jem.20130699
222. Muraro PA, Wandinger K, Bielekova B, Gran B, Marques A, Utz U, et al. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain. (2002) 126(1):20–31. doi: 10.1093/brain/awg021
223. Macrini C, Gerhards R, Winklmeier S, Bergmann L, Mader S, Spadaro M, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain J Neurol (2021) 144(8):2375–89. doi: 10.1093/brain/awab105
224. Tea F, Lopez JA, Ramanathan S, Merheb V, Lee FXZ, Zou A, et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun (2019) 7(1):145. doi: 10.1186/s40478-019-0786-3
225. Storch MK, Stefferl A, Brehm U, Weissert R, Wallström E, Kerschensteiner M, et al. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol Zurich Switz (1998) 8(4):681–94. doi: 10.1111/j.1750-3639.1998.tb00194.x
226. Miller A, Lider O, Abramsky O, Weiner HL. Orally administered myelin basic protein in neonates primes for immune responses and enhances experimental autoimmune encephalomyelitis in adult animals. Eur J Immunol (1994) 24(5):1026–32. doi: 10.1002/eji.1830240503
227. Malosse D, Perron H, Sasco A, Seigneurin JM. Correlation between milk and dairy product consumption and multiple sclerosis prevalence: a worldwide study. Neuroepidemiology. (1992) 11(4–6):304–12. doi: 10.1159/000110946
228. Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev (1999) 9(6):657–63. doi: 10.1016/S0959-437X(99)00031-3
229. Ryan FP. Human endogenous retroviruses in multiple sclerosis: potential for novel neuro-pharmacological research. Curr Neuropharmacol (2011) 9(2):360–9. doi: 10.2174/157015911795596568
230. Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. Epstein-barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. (2001) 15(4):579–89. doi: 10.1016/S1074-7613(01)00210-2
231. Sutkowski N, Palkama T, Ciurli C, Sekaly RP, Thorley-Lawson DA, Huber BT. An Epstein-Barr virus-associated superantigen. J Exp Med (1996) 184(3):971–80. doi: 10.1084/jem.184.3.971
232. Walker JA, McKenzie ANJ. T(H)2 cell development and function. Nat Rev Immunol (2018) 18(2):121–33. doi: 10.1038/nri.2017.118
233. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol (2000) 47(6):707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
234. Planas R, Metz I, Ortiz Y, Vilarrasa N, Jelčić I, Salinas-Riester G, et al. Central role of Th2/Tc2 lymphocytes in pattern II multiple sclerosis lesions. Ann Clin Transl Neurol (2015) 2(9):875–93. doi: 10.1002/acn3.218
235. Mohme M, Hotz C, Stevanovic S, Binder T, Lee JH, Okoniewski M, et al. HLA-DR15-derived self-peptides are involved in increased autologous T cell proliferation in multiple sclerosis. Brain J Neurol (2013) 136(Pt 6):1783–98. doi: 10.1093/brain/awt108
236. Gabrielsen ISM, Helgeland H, Akselsen H, D. Aass HC, Sundaram AYM, Snowhite IV, et al. Transcriptomes of antigen presenting cells in human thymus. PloS One (2019) 14(7):1–18. doi: 10.1371/journal.pone.0218858
237. Lui WY, Bharti A, Wong NHM, Jangra S, Botelho MG, Yuen KS, et al. Suppression of cGAS- and RIG-I-mediated innate immune signaling by Epstein-Barr virus deubiquitinase BPLF1. PloS Pathog (2023) 19(2):1–33. doi: 10.1371/journal.ppat.1011186
Keywords: autoimmunity, Epstein-Barr virus, T cell, multiple sclerosis, cross-reactivity, antibody, central nervous system, autoreactive B and T cells
Citation: Thomas OG and Olsson T (2023) Mimicking the brain: Epstein-Barr virus and foreign agents as drivers of neuroimmune attack in multiple sclerosis. Front. Immunol. 14:1304281. doi: 10.3389/fimmu.2023.1304281
Received: 29 September 2023; Accepted: 23 October 2023;
Published: 03 November 2023.
Edited by:
Daniela Latorre, ETH Zürich, SwitzerlandReviewed by:
Christian Muenz, University of Zurich, SwitzerlandJens Geginat, University of Milan, Italy
Copyright © 2023 Thomas and Olsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivia G. Thomas, olivia.thomas@ki.se