 Hoyul Lee
Hoyul Lee Jae-Han Jeon
Jae-Han Jeon Eun Soo Kim3*
Eun Soo Kim3*- 1Research Institute of Aging and Metabolism, Kyungpook National University, Daegu, Republic of Korea
- 2Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Republic of Korea
- 3Division of Gastroenterology, Department of Internal Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, Republic of Korea
Mitochondria has emerged as a critical ruler of metabolic reprogramming in immune responses and inflammation. In the context of colitogenic T cells and IBD, there has been increasing research interest in the metabolic pathways of glycolysis, pyruvate oxidation, and glutaminolysis. These pathways have been shown to play a crucial role in the metabolic reprogramming of colitogenic T cells, leading to increased inflammatory cytokine production and tissue damage. In addition to metabolic reprogramming, mitochondrial dysfunction has also been implicated in the pathogenesis of IBD. Studies have shown that colitogenic T cells exhibit impaired mitochondrial respiration, elevated levels of mROS, alterations in calcium homeostasis, impaired mitochondrial biogenesis, and aberrant mitochondria-associated membrane formation. Here, we discuss our current knowledge of the metabolic reprogramming and mitochondrial dysfunctions in colitogenic T cells, as well as the potential therapeutic applications for treating IBD with evidence from animal experiments.
1 Introduction
Inflammatory bowel disease (IBD) is a multifactorial immune disorder, characterized by chronic relapsing inflammation of the gastrointestinal tract accompanied with impaired immune homeostasis resulting from inappropriate and persistent activation of the mucosal immune system. Crohn’s disease (CD) and ulcerative colitis (UC) are the two major forms of the condition. Despite the accumulating evidences that immune dysregulation, intestinal microbiota, environmental factors, and genetic susceptibility are implicated in the pathogenesis of IBD (1), the exact causes remain unclear. The intestinal tract is the largest immune organ in the body, composing of complex immune-cell populations with a persistent exposure to the luminal antigens and pathogens. Therefore, immune homeostasis is essential for tolerance to luminal antigens and protection against pathogens.
The aberrant mucosal infiltration by innate and adaptive immune cells has been considered as a key player in the pathogenesis of IBD. According to the GWAS (genome-wide association study) based on human studies, NOD2 (pattern recognition receptor), CARD9 (inflammation), IL23R (Th17 cell responses), ATG16L1 (autophagy), PTPN22 (T cell activation) and FUT2 (microbiome) are well known causative IBD genes (2, 3). This set of identified susceptibility genes for IBD simply implies vulnerability or hyper-activation of innate and adaptive immune systems (Figure 1A), although the opening inflammatory response is thought to remove foreign luminal antigens.
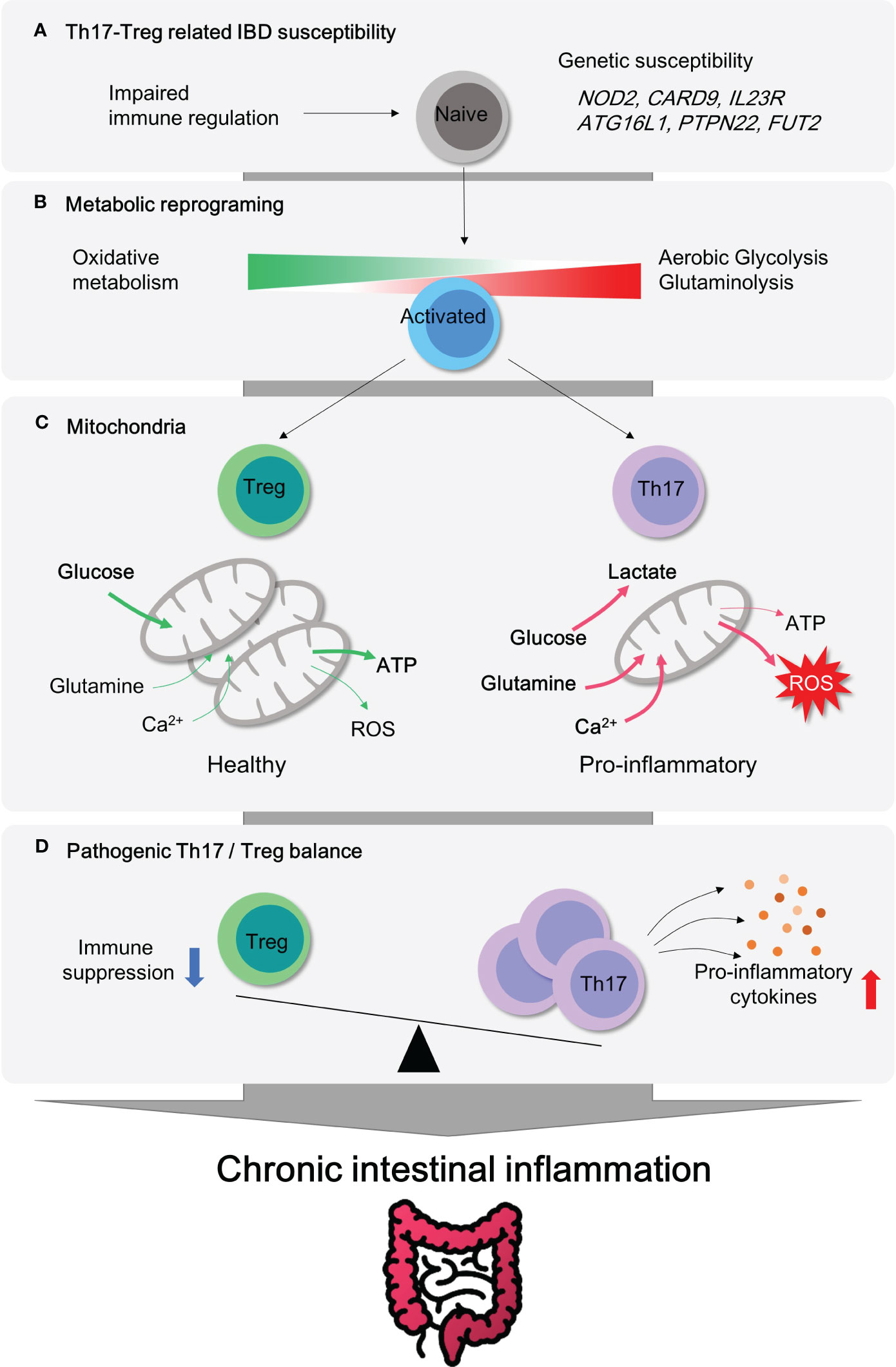
Figure 1 Metabolic Reprogramming and mitochondrial activity in CD4+ T subsets for pathogenesis of IBD. (A) Genetic susceptibility factors can trigger T cell-mediated immune dysregulation, leading to the onset of intestinal inflammation. (B) Pro-inflammatory CD4+ T cells become hyper-activated and demand metabolic reprogramming to meet their cellular needs for proliferation and effector functions. This metabolic shift involves the utilization of aerobic glycolysis, glutaminolysis, and mitochondrial oxidative respiration. (C) Regulatory T cells possess a higher number of healthy mitochondria that can efficiently utilize glucose oxidation to produce ATP without excess ROS generation. In contrast, pro-inflammatory Th17 cells rely on aerobic glycolysis and glutamine to fuel mitochondria and produce lactate. (D) Treg cells play a critical role in maintaining immune homeostasis in the gut mucosa by inhibiting immune responses. The imbalance between Treg and Th17 cells is a significant factor observed in patients with autoimmune diseases such as inflammatory bowel disease. Thus, targeting this imbalance could be an important strategy for the treatment of IBD.
Uncontrolled activation of effector CD4+ T cell responses (Th1, Th2 and Th17) and defects in immunosuppressive activity by regulatory T cell (Treg) in lamina propria (LP) were well characterized in IBD, including both CD (4) and UC (5). In line with this, recent studies using single cell RNA sequencing have revealed that distinct subsets of CD4+ T cells are considered to be an essential contributing factor in the immune landscape of IBD despite heterogeneity of T cells (6–8). Depletion of CD4+ T by chimeric monoclonal anti-CD4 antibody is affirmative in treating patients with Crohn’s disease (9). The ablation of tissue resident memory CD4+ T cells protects from experimental colitis in mice (10). Transcriptomic analysis reveals that human intestinal CD4+ T cells display exclusive gene signatures from circulating CD4+ T cells, such as differential chemokine and activation gene, Th17-related transcription factors, tumor necrosis factor (TNF) receptor signaling pathways (11). These clinical observations advocate a noteworthy role of CD4+ T cells in IBD. Moreover, colitogenic CD4+ T cells have been shown to undergo metabolic changes that support their pathogenicity. For instance, hypoxia-inducible factor 1 alpha (Hif1α) transcription is exclusively over-expressed in lamina propria CD4+ T cells, as compared to the intestinal epithelial CD4+ T and circulating CD4+ T cells (11). As gut tissue resident memory CD8+ T cells exhibit distinct metabolic signatures to the naive T cells (12), colitogenic tissue resident memory CD4+ T cells (10) are perhaps expected to experience similar metabolic rewiring (Figure 1, 2).
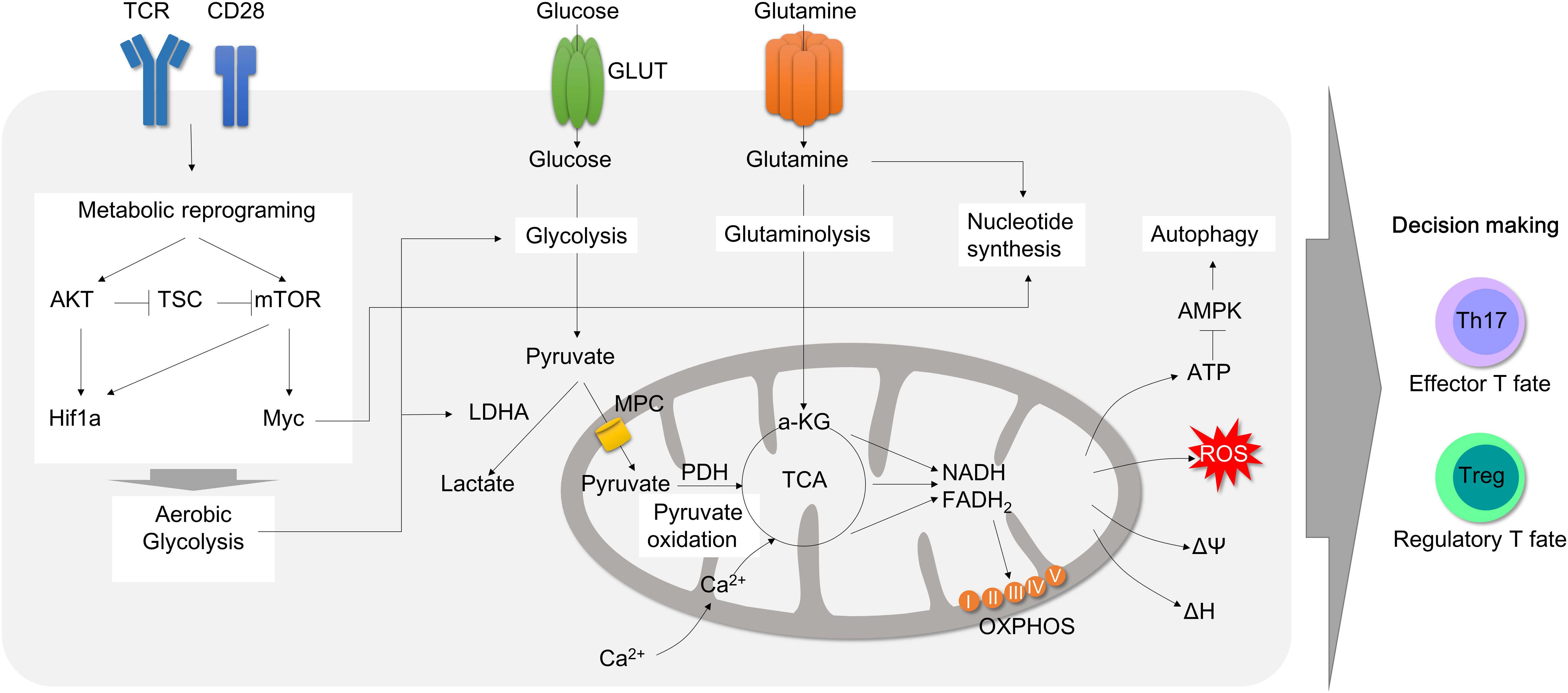
Figure 2 Mitochondrial involvement in T cell activation and metabolic reprogramming during inflammation. T cell activation by T cell receptor with CD28 promotes metabolic reprogramming. CD28 activation leads to AKT and mTOR activation. Consequently, Hif1α and Myc transcription factors are upregulated, which accelerates aerobic glycolysis by increasing glucose uptake and lactate secretion to rapidly generate cellular ATP and carbon building blocks. Upon activation, pyruvate oxidation is decreased and glutaminolysis rather fuels TCA cycles in mitochondria in order to produce ATP. Glutamine is also rapidly consumed to process de novo nucleotide synthesis. Metabolic adaptation during T cell activation consequently leads to mitochondrial instability followed by mitochondrial ROS production, which can promote effector T cell polarization. Therefore, mitochondria is a pivot to all T cell metabolic reprogramming upon activation and differentiation. TCR, T cell receptor; GLUT, glucose transporter; AKT, protein kinase B; mTOR, mammalian target of rapamycin; TSC, tuberous sclerosis complex; LDHA, lactate dehydrogenase A; MPC, mitochondria pyruvate carrier; TCA, tricarboxylic acid cycle; OXPHOS, oxidative phosphorylation; AMPK, AMP-activated protein kinase.
The last two decade, there have been numerous research papers published in terms of immunometabolism. T cells face extensive metabolic changes to sustain the energy generation to achieve activation, clonal expansion, differentiation, effector cytokine production, and survival in inflammatory environments. Naïve and resting T cells utilize mitochondrial oxidative respiration coupled to TCA (tricarboxylic acid) cycle, OXPHOS (oxidative phosphorylation) and adenosine triphosphate (ATP) biosynthesis for a stable energy generation using a limited nutrient source. Fatty acid oxidation is especially prominent in naïve, memory and regulatory T cells.
Once engaged with antigen recognition or T cell receptor (TCR) activation by CD3 and co-stimulatory receptor CD28, T cell rapidly changes in their metabolic program toward glycolysis. Although glycolysis is not so efficient for ATP production, only 2 ATP molecules per glucose molecule, compared to mitochondrial respiration, which produces 36 ATP molecules per glucose molecule, glycolysis can rapidly substitute the cellular building blocks for proliferation, differentiation and effector function. This metabolic change is known as ‘aerobic glycolysis’ or the ‘Warburg effects’, which was first described phenomenon in cancer pathogenesis. Later, mitochondrial dysfunction appears to be a prominent cause of aerobic glycolysis. Today, Warburg effects and mitochondrial dysfunction gained attention again and furthermore extended to immunology field (Figures 1B, C).
Mitochondria have a central role in energy metabolism and cellular homeostasis. Primarily, mitochondria provide the bioenergetics ATP as a power plant and regulate metabolic activity within the cell. Upon activation, T cells expand mitochondrial mass along with the mitochondrial ribosomal proteins, OXPHOS proteins, and one-carbon metabolism (13, 14). Besides, mitochondria contribute to cellular calcium homeostasis, apoptosis regulation, reactive oxygen species (ROS) generation, and inflammatory signaling pathways. Given the accumulated understanding of mitochondrial alterations in the process of T cell activation and Th17/Treg differentiation (Figure 1D), it is still a puzzle whether manipulating mitochondrial function and metabolism could modulate the inflammatory signaling of these cells and alleviate the symptoms of IBD. Recently, several reviews have been published describing how immune cell metabolism is associated with inflammatory responses in autoimmunity (15–19) and the role of mitochondria in intestinal epithelial defense regarding pathogenesis of IBD (20–22). In this review, we examine recent studies that have implicated aerobic glycolysis and mitochondrial dysfunctions in the pathogenesis of IBD (Figure 1). Our analysis focuses on CD4+ T cells, which have been identified as a key factor in the disease’s progression. We have provided experimental evidence that specifically highlights these findings (Tables 1–3).
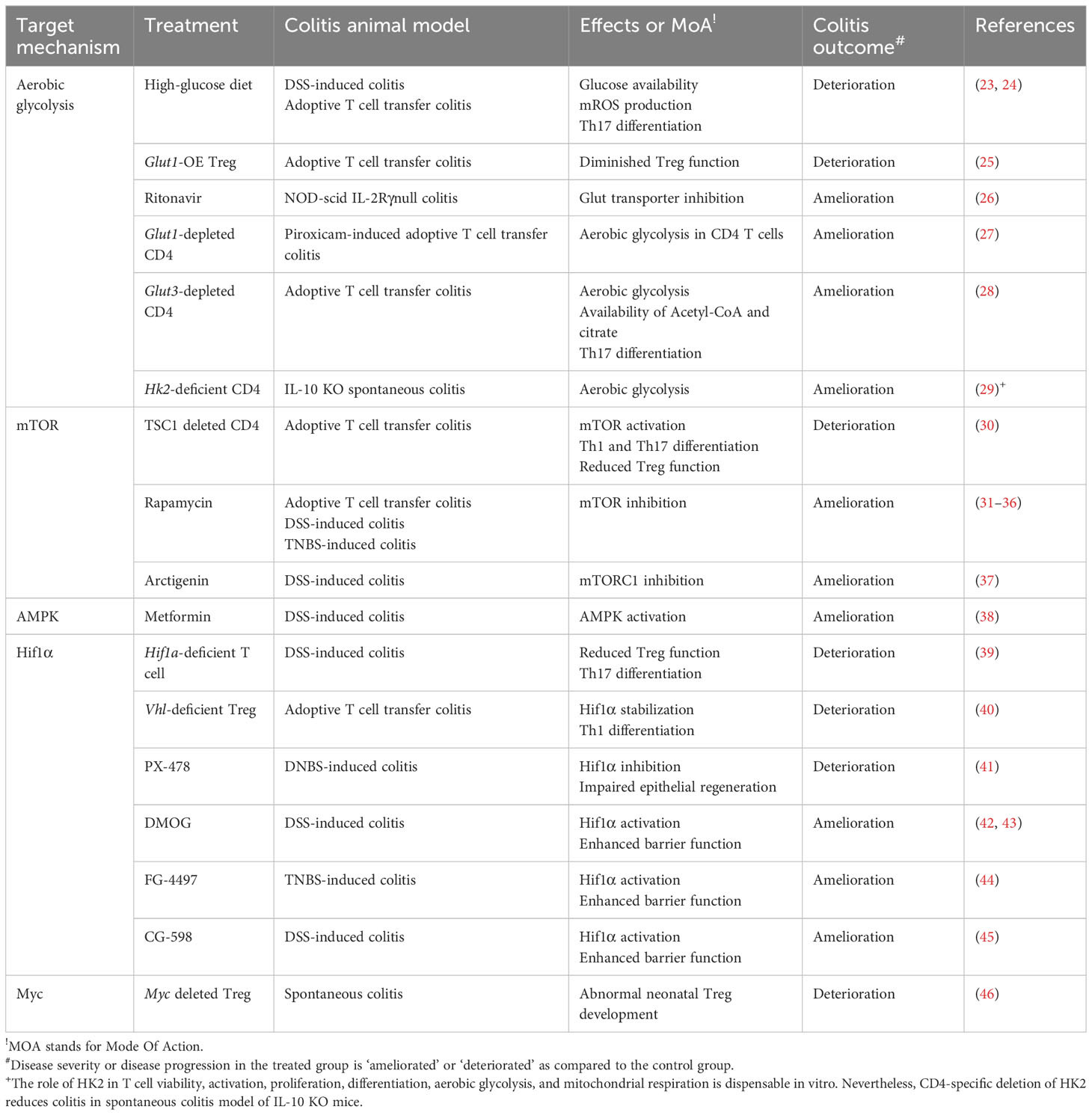
Table 1 Targeting CD4+ T cell glucose metabolism for treating IBD - evidence from an in vivo animal study.
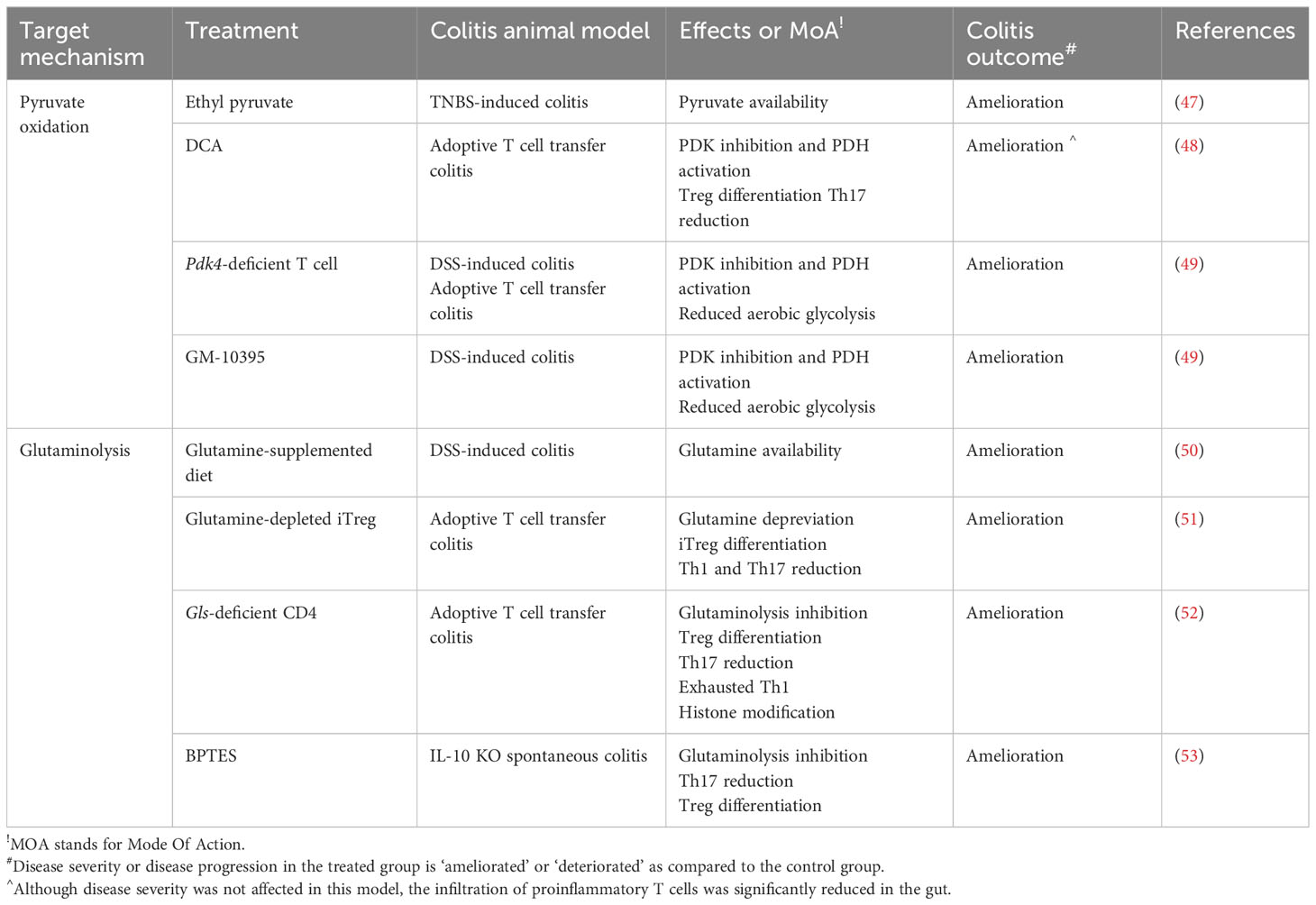
Table 2 Targeting CD4+ T pyruvate and glutamine metabolism for treating IBD - evidence from an in vivo animal study.
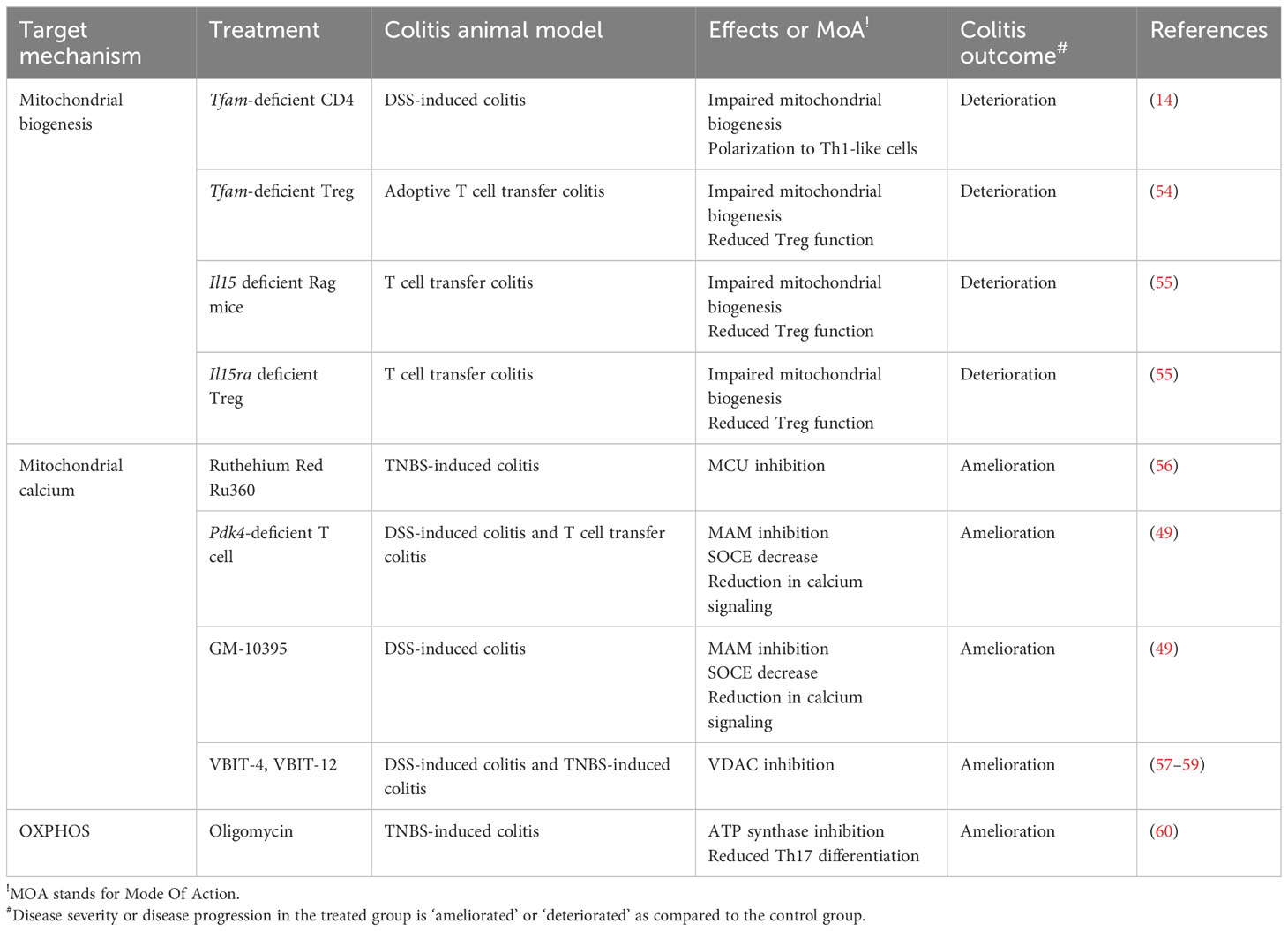
Table 3 Targeting CD4+ T mitochondrial fitness for treating IBD - evidence from an in vivo animal study.
2 Targeting T cell metabolism for treating colitis
2.1 Aerobic glycolysis in colitogenic T cells
CD3/CD28 co-stimulation in a naïve CD4+ T cells during TCR activation induces glycolysis (Figure 2) while producing lactate by increasing the expression of glucose transporter (61), hexokinase 2 (HK2) (29), and consumption of glucose (61), and prevents it from becoming anergy. Inhibition of glycolysis by pan-hexokinase blocker, 2-dehydroxy-D-glucose (2-DG), during T cell activation impedes cytokine secretion co-related with mTOR signaling pathway (62). Moreover, glucose availability is a key factor for effector T cell functions. Glucose availability significantly affects secretion of IL-2 and IFN-γ (63, 64), aerobic glycolysis (65), Ca2+/NFAT signaling pathway (64), proliferation, and viability (66) in T cells. Therefore, targeting elevated glycolysis in T cells could be fascinating approaches in treating inflammatory diseases like rheumatoid arthritis, multiple sclerosis (MS) and systemic lupus erythematosus (67).
2.1.1 Glucose availability
In patients with UC, there is a higher prevalence of hyperglycemia than in controls (68). Interestingly, patients who have both psoriasis and IBD have a significantly higher prevalence rate of diabetes than those who have sole psoriasis (69). Moreover, patients with IBD and diabetes mellitus exhibit a significantly higher levels of C-reactive protein, erythrocyte sedimentation rate, eosinophil counts, monocyte counts in blood (70). Furthermore, use of metformin, a medication for the treatment of type 2 diabetes, known as an 5’ adenosine monophosphate-activated protein kinase (AMPK) activator, significantly reduces the hazardous ratio of new-onset IBD by almost a half (71), and positively prevents dextran sulfate sodium (DSS)-induced colitis in mice (38). These clinical observations may suggest that systemic glucose metabolism and intolerance is notably associated with IBD.
Hyper-glycolysis in cells often leads to mitochondrial dysfunctions by a hyper-polarizing the mitochondrial membrane potential, and oversupply of mitochondrial ROS (mROS). A high glucose concentration induces more mROS, and drives Th17 cell generation. Accordingly, the treatment of the ROS scavenger, N-acetyl-L-cysteine (NAC), or the mitochondria-targeted anti-oxidant, mitoquinone (MitoQ), in T cells significantly inhibits Th17 cell differentiation even in the presence of high glucose (23).
In animal models of colitis, high glucose availability has been shown to exacerbate the development of inflammation. Colonic expression of glycolysis-associated proteins such as HK2, lactate dehydrogenase A (LDHA), phosphate fructose kinase, and c-MYC are evidently enriched in inflamed tissues of DSS-induced colitis mouse model (72). Likewise, a high-sugar diet clearly deteriorates lymphocyte infiltration, epithelial damage, and cytokine expressions in the gut after colitic DSS challenges in mice (24). High glucose consumption (10% in a drinking water) also exacerbates T cell transfer colitis model in mice by increasing proinflammatory Th17 populations in the gut (23). Conversely, overexpressing a transgenic glucose transporter (Glut) 1 receptor in regulatory T cells fails to achieve disease remission in an adoptive T cell transfer colitis model (25) (Figure 3A; Table 1).
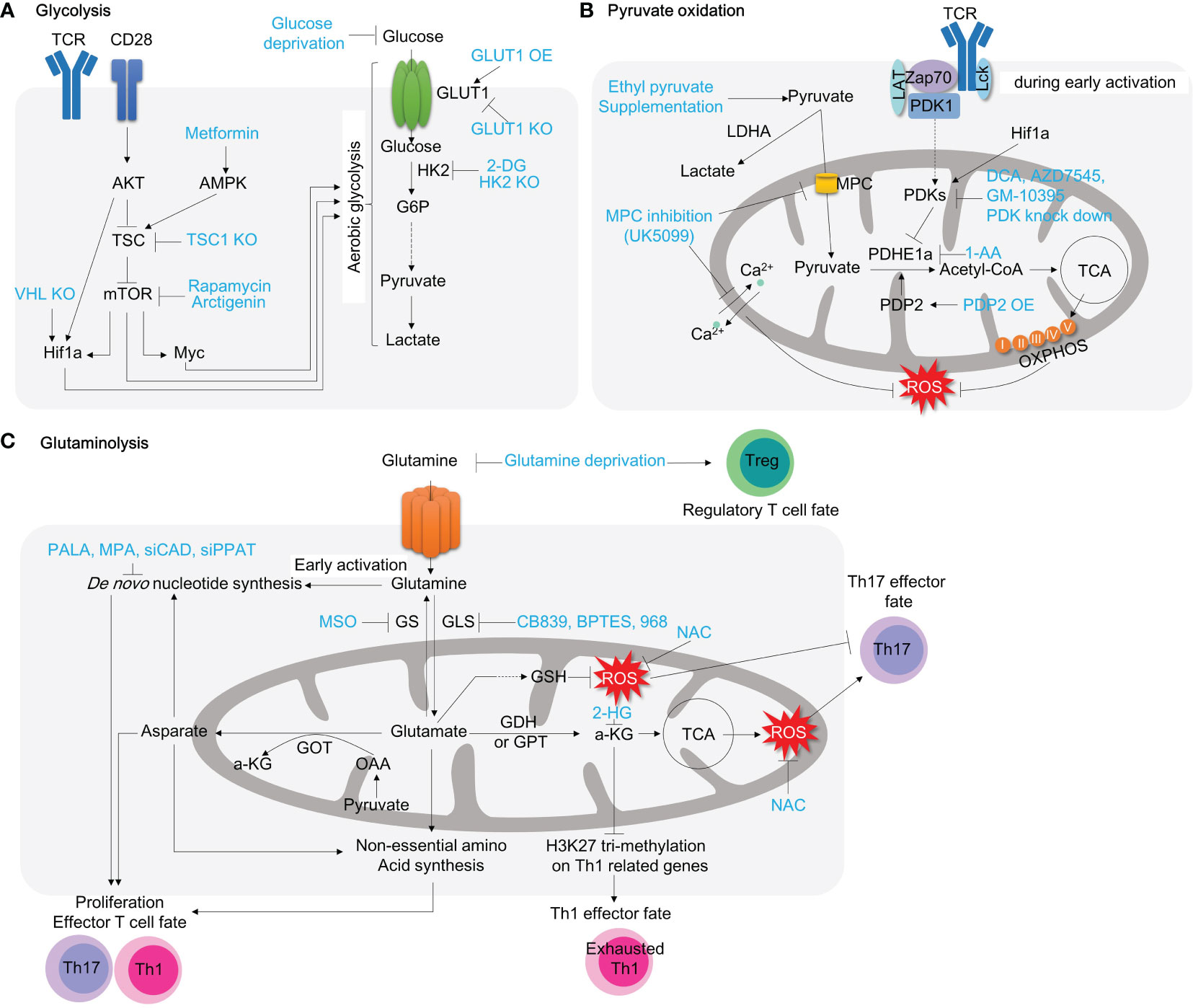
Figure 3 Targeting T cell metabolic reprogramming for inflammation treatment. (A) Targeting general aerobic glycolysis drivers, such as mTOR, Myc, and Hif1a, with inhibitors like rapamycin, arctigenin, and 10058-F4, has demonstrated anti-inflammatory effects in autoimmune CD4+ T cells. Inhibiting the glucose transporter GLUT1 on the plasma membrane also restricts glycolysis, which slows T cell activation. Additionally, inhibiting HK2, the first glycolytic enzyme converting glucose to G6P, with 2-DG significantly decreases T cell activation. (B) Pyruvate oxidation can be enhanced by pyruvate supplementation. Inhibiting the MPC with compounds like UK5099 not only induces aerobic glycolysis by increasing pyruvate levels in the cytosol but also lowers mitochondrial calcium concentration, resulting in significant mitochondrial dysfunction in CD4+ T cells. Inhibiting PDHE1α with 1-AA alters mitochondrial pyruvate oxidation and can impact T cell development. Conversely, promoting pyruvate oxidation by inhibiting PDK with compounds like DCA, AZD7545, and GM10395 may be a potential therapeutic strategy for intestinal inflammation. Glut3 depletion reduces acetyl-CoA availability and histone acetylation (C) Glutaminolysis is associated with mitochondrial function and T cell activation. Glutamine serves as a source for de novo nucleotide synthesis during T cell activation and effector T cell polarization. Inhibiting de novo nucleotide synthesis with compounds like PALA and MPA or withdrawing glutamine with compounds like MSO or glutamine deprivation suppresses effector T cell polarization. Glutaminase inhibition with compounds like CB839, BPTES, and 968 leads to alterations in Th1/Th17 polarization and shows promise as a highly effective approach for inflammation treatment. TCR, T cell receptor; AKT, protein kinase B; TSC, tuberous sclerosis complex; mTOR, mammalian target of rapamycin; Hif1a, hypoxia-inducible factor 1; HK2, hexokinase 2; G6P, glucose-6-phosphate; GLUT1, glucose transporter 1; 2-DG, 2-dehydroxy-D-glucose; LDHA, lactate dehydrogenase A; MPC, mitochondria pyruvate carrier; PDK, pyruvate dehydrogenase kinase; LAT, Linker for activation of T cells; TCA, tricarboxylic acid cycle; PDP2, pyruvate dehydrogenase phosphatase 2; DCA, dichloroacetate; GS, glutamine synthetase;GLS, glutaminase; GSH, glutathione; PALA, N-(phosphonacetyl)-L-aspartate; MPA, mycophenolic acid; MSO, methionine sulfoximine; NAC, N-acetylcysteine; 2-HG, 2-Hydroxyglutarate; a-KG, alpha ketoglutarate; GDH, glutamate dehydrogenase; GPT, glutamate pyruvate transaminase; GOT, glutamate OAA transaminase; OAA, oxaloacetate.
Glucose restriction achieves an effective prevention of colitis disease. Treatment of potential Glut blocker, Ritonavir, has been reported to ameliorate the disease severity in NOD-scid IL-2Rγnull colitis animal model (26). Glut1-depleted CD4+ T cells fail to trigger intestinal inflammation in nonsteroidal anti-inflammatory drug-induced T cell transfer colitis model in mice (27). Adoptive transfer of Glut3-deficient T cells fail to induce intestinal inflammation (28). T cell specific-HK2 deficiency partially recovers intestinal inflammation in a spontaneous colitis model of IL-10 knockout (KO) mice, although HK2-deficient CD4+ T cells appear to have normal proliferation, viability, activation and differentiation in vitro (29) (Figure 3A; Table 1).
2.1.2 Transcriptional control of glucose metabolism
This metabolic shifts from OXPHOS to glycolysis reliance often accompanies induction of the mammalian target of rapamycin (mTOR), c-Myc, and Hif1α pathways (73). The importance of these factors in regulating T cell metabolism and effector functions is well described in the previous literature and review (73, 74).
Tuberous sclerosis 1 (TSC1) is a negative regulator of mTORC1. Constitutive activation of mTORC1 by CD4+ specific deletion of TSC1 promotes Th1 and Th17 cell differentiation and suppresses immunosuppressive activity of Treg, resulting in an enhanced disease severity in an adoptive T cell transfer colitis model (30). Conversely, mTOR deficient T cells fail to differentiate into effector T cells, including Th1, Th2, and Th17 (75). The therapeutic efficacy of rapamycin, a classic pharmacological inhibitor of mTORC1, has been substantially demonstrated to ameliorate intestinal inflammations in experimental animal colitis models in a diverse perspective (31–36). Another pharmacological mTORC1 inhibitor, arctigenin, has been reported to decrease Th1 and Th17 cell differentiation, leading to amelioration of disease severity in DSS-induced colitis model (37) (Figure 3A).
The activation of mTOR signaling pathway further stimulates the activity of Hif1α, a master regulator of oxygen homeostasis. Although Hif1α-deficient T cells have better cellular growth and proliferation in response to TCR engagement with IL-3 (76), HIF1α–mediated metabolic reprogramming toward ‘aerobic glycolysis’ (76, 77) promotes Th17 cell differentiation and declines Treg cell differentiation in both in vitro experiment, and in vivo animal model of MS (77, 78) (Figure 3A). In patients with CD, and UC, Hif1α expression is highly augmented in the inflamed colonic mucosa (39), especially in Th17 cells (79). These experimental evidences may suggest that HIF-signaling pathway is hypothetically to be a fascinating therapeutic target. Despite this, the modulation of HIF levels for IBD has several limitations to induce clinical responses. First, the role of Hif1α in regulatory T cells seems to be debatable in an animal model of colitis (39, 40, 80, 81). That is presumably because Hif1α under hypoxic condition not only facilitates glycolysis but also paradoxically protects mitochondria from ROS by reducing complex I activity, pyruvate oxidation, autophagy, and mtDNA encoded mRNA levels (82). Certainly, T cell specific deletion of Hif1α significantly downregulates Foxp3 over IL-17 expression, leading to the uncontrolled immune responses in DSS-induced colitis model (39). In contrast, Von Hippel-Lindau (VHL)-deficient (i.e. Hif1α-stabilized) Treg cells favor Th1 differentiation over Treg differentiation, resulting in colitis development in adoptive T cell transfer model (40) (Figure 3A). Besides, HIF signaling pathways plays an important role in the maintenance of the epithelial barrier functions. In an animal colitis model, the systemic administration of Hif1α inhibitor, PX-478, attenuates the protective effects of exogenous H2S and epithelial regeneration in dinitrobenzene sulfonic acid-treated rat colitis model (41), whereas Hif1α activation by DMOG (42, 43), FG-4497 (44) or CG-598 (45) results in enhancement of epithelial barrier functions against animal colitis models. Collectively, Hif1α, a master transcription factor driving aerobic glycolysis, regulates T cell differentiation as well as intestinal barrier functions (Table 1).
Myc is a proto-oncogene that acts as a transcriptional factor downstream of the mTOR signaling pathway. It is strongly upregulated in both CD4+ and CD8+ T cells after TCR activation (83, 84). Myc regulates the expression of glycolytic enzymes at both mRNA (84) and protein (83) levels, and affects the expression of lactate transporter (Slc16a1), glycolytic flux (83), and PPP flux (84). Myc also supports the de novo pyrimidine/purines synthesis, which are essential for nucleotide production and proliferation in activated T cells (83, 84). Moreover, Myc enhances the expression of amino acid transporter and glutaminolysis in the early phase of TCR activation (4 to 8 hours). Thus, Myc modulates metabolic pathways in CD4+ T cells during activation.
Regulatory T cells have a different metabolic signature from conventional effector T cells, and Myc plays a different role in them. Foxp3, the key transcription factor for Treg cells, inhibits c-Myc expression by binding directly to the TATA box of the c-Myc gene (85). In addition, overexpression of Glut1 in Treg cells impairs their functions and exacerbates colitis in a colitis model with adoptive transfer (25) (Table 1). However, c-Myc expression is critical for effector Treg generation during early neonatal Treg development (46). Thus, deleting Myc specifically in Foxp3+ cells causes early-onset inflammation in multiple organs such as skin, pancreas, liver, lung, and colon in mice (46) (Figure 3A). Therefore, the use of Myc inhibitors against proinflammatory T cells should be carefully taken into account for optimal therapeutic effects.
2.2 Pyruvate oxidation
Pyruvate can be oxidized into acetyl-CoA, which is a substrate for citrate synthase to generate citrate in the mitochondria (Figure 2). Pyruvate oxidation is catalyzed by pyruvate dehydrogenase complex or PDC, which consists of three enzymes: PDHE1, PDHE2 and PDHE3. The activity of PDHE1α is controlled by three phosphorylation sites, Ser232, Ser293 and Ser300, which are tightly regulated by PDC phosphatases (PDPs) and kinases (PDKs). PDC kinase (PDK) is a serine/threonine kinase that inactivates PDC activity by reversible phosphorylation. Therefore, the activity of PDK plays an important role in controlling energy balance and metabolic fitness in cells. PDK has four isoforms (PDK1, PDK2, PDK3 and PDK4). Expression of PDK isoforms is highly enriched in metabolic diseases. Inhibition of PDKs has been suggested as a potential therapeutic target for obesity, diabetes, heart failure, hepatic steatosis and cancer (86, 87). Moreover, the role of PDKs has been investigated in the mitochondrial respiration, activation and polarization of myeloid (88) and lymphoid cells (88).
2.2.1 T cell development and maturation
Mitochondrial pyruvate oxidation is a metabolic checkpoint for cell cycle and cellular functions during T cells development. T cells progenitor originate from hematopoietic stem cells (HSC) in the bone marrow. Under physiological conditions in the bone marrow, long-term hematopoietic stem cells (LT-HSC) show a high glycolytic-dependent metabolic profile and impaired oxygen consumption. LT-HSC can preserve their stemness and quiescence by increasing Hif1α-dependent expression of PDKs and reducing mitochondrial mass (89), indicating that pyruvate oxidation is not essential for T cell development at BM. Consistently, defects in PDHA1, PDK2/PDK4 or mitochondrial pyruvate carrier 1 (MPC1) do not affect T cell development at BM (89–91). However, PDK2 or PDK4 knock-in can rescue Hif1α deletion-induced mROS and cell death. Interestingly, only LT-HSC can survive in the in vitro cell culture condition with PDH inhibitor, 1-aminoethylphosphinic acid (1-AA), while short-term HSC and multipotent progenitors comes to cell death (89). Thus, pyruvate oxidation is not a requirement for T cell progenitor development at BM (Figure 4).
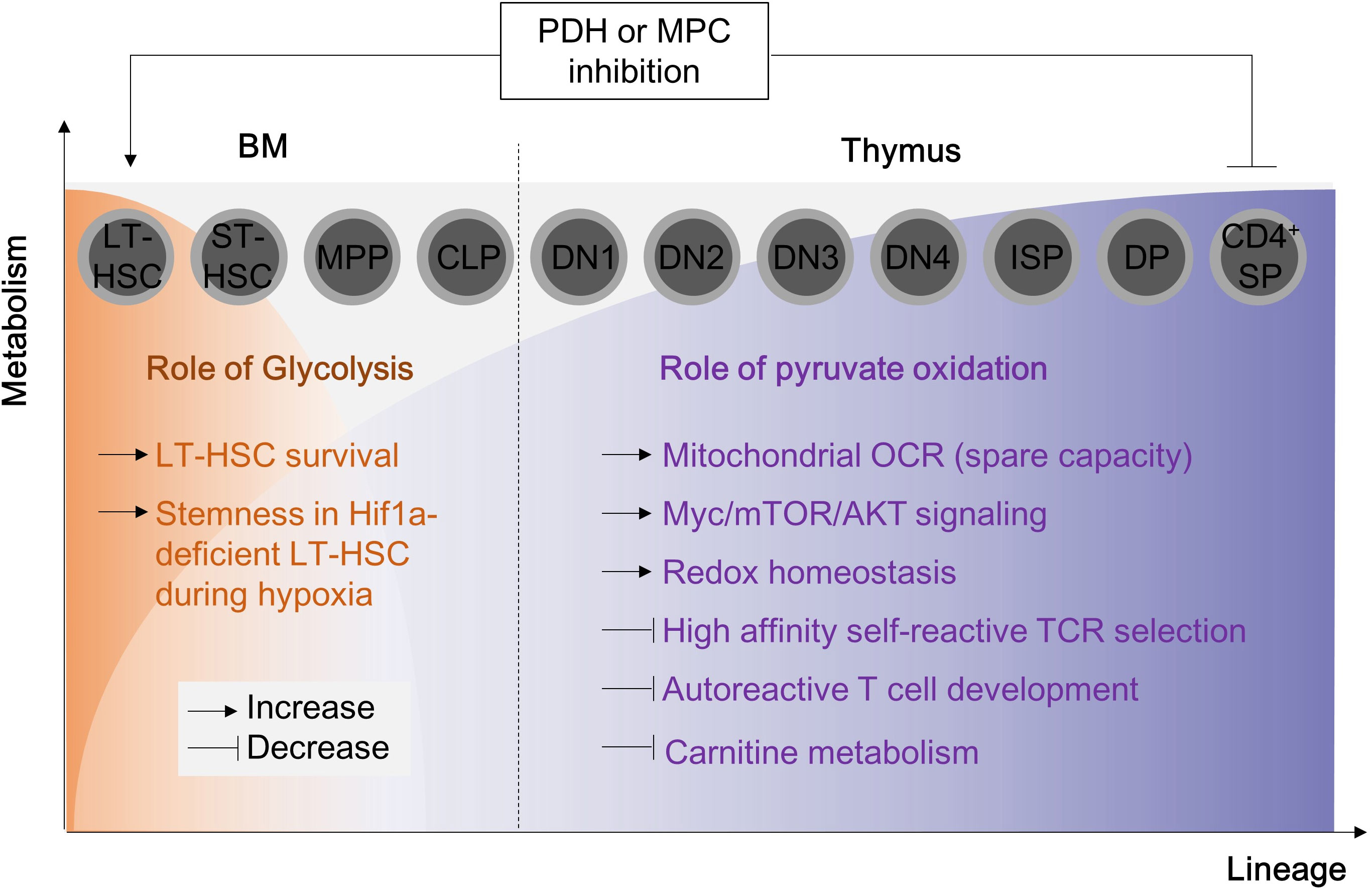
Figure 4 Pyruvate oxidation disruption in hematopoietic stem cells and T cell progenitors alters normal T cell development. The metabolic alterations in hematopoietic stem cells and T cell progenitors can disrupt the normal development of T cells. During early hematopoietic development, pyruvate oxidation is not essential. The double deletion of PDK2/4 has no visible impact on long-term hematopoietic stem cells, but PDH inhibition (for example, with 1-AA) inhibits ST-HSC and MPP cells. Inhibiting MPC1 or PDHE1a, which results in the complete termination of pyruvate oxidation, can lead to severe mitochondrial dysfunction, affecting thymic selection and leading to the development of autoreactive T cells. MPC, mitochondria pyruvate carrier; LT-HSC, long term-hematopoietic stem cell; ST-HSC, short term-hematopoietic stem cell; DN, double negative cells; ISP, immature single-positive cells; DP, double positive cells; SP, single positive cells.
In the later stage, thymocytes mature and differentiate into double negative (DN) T, double positive (DP) T and finally CD4+ or CD8+ single positive (SP) T cells. During this maturation, T cells metabolically need glucose-derived pyruvate oxidation (90, 91). Blocking pyruvate flux into mitochondria by MPC1 deletion reduces OXPHOS gene expression, cellular oxygen consumption rate, and thymic T cell development (90). PDHE1α deficiency in the thymocytes also affects redox balance and carnitine metabolism (91). Loss of PDHE1α remarkably compromise thymic DP T cell survival and PTEN deletion-induced malignant proliferation in thymocytes (91) (Figure 4).
Fully differentiated T cells during homeostasis are largely quiescent and demand a relatively low level of cellular activity. Therefore, they require somewhat higher mitochondrial glucose-derived pyruvate oxidation (65). Stimulation of TCR with CD28 drives expedited metabolic reprogramming through upregulating glycolysis, the pentose phosphate pathway, and glutaminolysis, presumably in the cytosol, yet downregulating pyruvate oxidation (65) in a mitochondrion (84). Certainly, the levels of p-PDHE1 and PDK4, which signify inhibition of pyruvate oxidation, were raised in T cells after early TCR/CD28 activation (13, 49), demonstrating that mitochondrial pyruvate oxidation is essential (Figure 3B).
2.2.2 Pyruvate oxidation in T cell responses
In addition to the T cell activation, cellular pyruvate availability also affects T cell differentiation. Ethyl pyruvate (EP) supplementation notably enhances Treg cell proliferation and differentiation in both in vivo and in vitro (92, 93), whereas Th1 and Th17 differentiation are not affected by EP supplementation in vitro (93). Furthermore, the role of pyruvate oxidation in activated CD4+ T cells is more important because in vivo activated CD8+ T cells favor carbon sources from PDH flux (pyruvate oxidation) over pyruvate carboxylase (PC) flux, while in vitro activated CD8+ T cells prefer PC over PDH flux (94). In addition, CD4-specific deletion of PDHE1α (95) or Glut3 (28) in CD4+ T cells compromises glucose-derived acetyl-CoA availability, leading to reduction in the histone acetylation and Th17 polarization (28, 95).
Defeats in mitochondrial pyruvate influx induced by MPC inhibition disrupts mitochondrial calcium homeostasis. This is evident by decreasing carbonyl cyanide 3-chlorophenylhydrazone-induced mitochondrial Ca2+ release as well as ATP-induced mitochondrial Ca2+ uptake (96). Consequently, nutrient stress by restricted pyruvate oxidation substantially compromises mitochondrial respiration (oxygen consumption rate, OCR) and ATP production and compensatorily enhances autophagy flux (96). This may suggest that mitochondrial pyruvate oxidation sustains mitochondrial health. MPC1 deletion leads to abnormal activation in splenic T cell and results in expansion of auto-reactive T cells in the response to CD3/CD28 stimulation (90) (Figure 3B).
In colonic mucosal biopsies from patients with IBD, phosphorylation of PDHE1α, a substrate of PDK4, has been strongly correlated with CD4+ T cells (Figure 3B). In line with this, the experimental DSS-induced colitis model also displays the significantly upregulated expressions of PDK4 and p-PDHE1α after DSS challenge. Interestingly, CD45+ hematopoietic cells including CD4+ T cells, neutrophils, macrophages and dendritic cells appears to be a central player in charge with PDK and PDHE1α expression (49) (Table 2). Collectively, the restoration of pyruvate oxidation via supplementation of pyruvate or inhibition of PDKs may be directly connected to therapeutic strategy for IBD.
2.2.3 PDH activation through PDK inhibition or PDP activation
Dichloroacetate (DCA) is a structural analogue of pyruvate that inhibits PDKs activity (PDK1 at most among PDKs). Previously, its ability to modulate mitochondrial respiration, ROS generation, and metabolic reprograming has been demonstrated in respect of metabolic syndrome (97, 98), osteoporosis (99) and cancer metabolism (100, 101) as well as immune response (65, 88, 102). In human alloreactive-human peripheral blood mononuclear cell (PBMC), DCA treatment significantly decrease glucose uptake, lactate production, and protein expressions of aerobic glycolysis related enzymes such as Glut1, HK2, LDHA, p-PDH and Myc (103, 104). Furthermore, DCA markedly increased regulatory T cell signature (IL-10 secretion and Foxp3 protein expression), and yet decreased effector T cell signature (such as T-bet, GATA3, and RORγT expression) in alloreactive human PBMC. These data suggests that PDK blocker, DCA, mitigate aerobic glycolysis and favor differentiation toward immunosuppressive regulatory T cell rather than proinflammatory effector T cells (103, 104) (Figure 3B). In line with this observation, activated CD4+ T cells (isolated from BALB/c normal mice) by CD3/CD28-mediated TCR stimulation had decreased lactate production and effector cytokine productions (IFN-γ, IL-5, and IL-17) in response to DCA for 48 hours (105) (Figure 3B).
Under hypoxia, both glycolysis and pyruvate oxidation are critical for the survival of effector memory (EM) T cells Glucose deprivation or 2-DG treatment significantly reduces mitochondrial membrane potentials and consequently induces apoptosis in EM T cells. However, the addition of sodium pyruvate or DCA dramatically restores 2DG-induced mitochondrial membrane potential declines and enhances survival under hypoxic stress (106). This finding suggests that glucose oxidation via pyruvate metabolism stabilizes mitochondrial membrane potentials and thereby increases survivability under hypoxic conditions.
PDK also modulates CD4+ T cell differentiation. First, its pharmacological inhibitor, DCA, has been clearly demonstrated to induce Treg differentiation and suppress Th17 differentiation (48, 107, 108) in part through ROS generation (Figure 3B). However, the current understanding of the mechanisms by which PDK inhibitors regulate T cell differentiation is contradictory. The predominant isoforms of PDKs in naïve, Th17 and regulatory T cells are PDK1 and PDK3 (48, 107). One paper shows that PDK1 deficiency, the strongest target isoform among PDKs that is possibly inhibited by DCA, compromises Th17 differentiation and accelerates Treg differentiation (48), which suggests that the effects of DCA are dependent on PDK –presumably PDK1- activity. On the other hand, it has been shown that the immunosuppressive effects of PDK inhibitors were ironically independent of PDKs because the effect of DCA on manipulating T cell differentiation is persistent even under the knockdown of PDK1 and PDK3 (107). This may suggest that the anti-inflammatory effects of DCA are likely followed through the inhibition of PDK2 and PDK4, or unidentified non-canonical pathways such as ROS production (107). Despite the differences in methods of isolation and differentiations of naïve CD4+ T cells that were used in those papers, PDK activity is undoubtedly pivotal in T cell differentiation. In addition, PDK1 (60) or PDK4 (49) knockdown significantly reduces IL-17 secretion in in vitro-polarized Th17 cells (Figure 3B).
The effects of PDK inhibition on Th1 and Th2 differentiation have not been thoroughly investigated. DCA treatment significantly enhances IFN-γ secretion and production in splenocytes (109) as well as Th1-polarized CD4+ T cells (108), indicating enhanced Th1 differentiation. In contrast, one report showed that DCA treatment fails to reduce Th1 differentiation and effector function (48).
PDK1 has been implicated in the early stages of TCR signaling pathways during T cell activation, in addition to its role in the mitochondrial matrix (Figure 3B). In PA-R CD8+ T cells, TCR activation by CD3 results in the downstream signaling complex, including ZAP70 and Lck, which binds and activates PDK1 within a few minutes (65). Consequently, due to the impediment of mitochondrial pyruvate influx, the cumulative cytosolic pyruvate allows an augmentation in lactate biosynthesis. In this way, the TCR complex rapidly utilizes aerobic glycolysis during the activation process (65, 110). In summary, these data suggest that inhibition of PDKs reduces anaerobic glycolysis, improves mitochondrial function, promotes ROS production, decreases Th17 differentiation, and increases immunosuppressive Treg function. Therefore, targeting pyruvate oxidation flux to manipulate T cell functions is a promising strategy in the treatment of inflammatory bowel disease (IBD).
Notably, EP has been shown to significantly increase the population of CD4+CD25+Foxp3+ regulatory T cells in both peritoneal cells, lamina propria, and the Peyer’s patches (92) (Figure 3B). Furthermore, EP treatment attenuates Th17 cell infiltration in the intestine and prevents trinitrobenzene sulphonic acid (TNBS)-induced colitis in mice (47). In vivo DCA treatment (2g/L, ad libitum) significantly reduces CD4+ T cell accumulation, especially Th17 cells, in the spleen and mesenteric lymph nodes in a naive T cell adoptive transfer colitis model (48). Unfortunately, DCA treatment fails to prevent intestinal inflammation due to the non-responsive Th1 cells to DCA. However, PDK inhibition by DCA successfully attenuates EAE progression, which is another Th17-mediated neuronal disease (48). Although DCA treatment is an efficient way to restrain the development of Th17-mediated inflammation, other approaches inhibiting PDKs may be warranted.
Recently, we have reported the pathological role of PDK4 in CD4+ T cell (49, 91). PDK4 is highly expressed during early T cell activation, and its deletion leads to the suppression of aerobic glycolysis. Certainly, PDK4 KO mice were found to be more resistant to DSS-induced colitis. In addition, CD4+ T cells deficient in PDK4 induce less intestinal inflammation in both DSS-induced colitis and naïve T cell adoptive transfer colitis. Furthermore, treatment with the pharmacological PDK4 inhibitor, GM-10395, compromises CD4+ T cell activation and attenuates DSS-induced colitis (49) (Table 2).
Pyruvate dehydrogenase phosphatase (PDP) is a mitochondrial matrix enzyme that sustains PDC activity. PDP1 is predominantly expressed in mitochondria from skeletal muscle, whereas the PDP catalytic subunit 2 (PDP2) is expressed in the liver and many other cells, including white blood cells. In agreement with the effects of DCA on T cell differentiation, PDP2 overexpression, which enhances PDC and pyruvate oxidation, inhibits glycolysis and Th17 differentiation (111) (Figure 3B).
2.3 Glutaminolysis
Glutaminolysis is a catabolic mechanism by which the amino acid glutamine is degraded to glutamate, α-ketoglutarate (α-KG), aspartate, and pyruvate (Figure 3C). Glutamine is initially catalyzed by mitochondrial glutaminase (GLS) to produce glutamate, which is an essential amino acid playing various roles in cellular physiology. Glutamate can be further oxidized by glutamate dehydrogenase (GDH), glutamic oxaloacetic transaminase (GOT), and glutamic pyruvic transaminase (GPT) to produce α-KG, which supports TCA cycle intermediate via an anaplerotic route for mitochondrial ATP generation and a substrate for histone/DNA methylation during epigenetic modifications (Figure 3C). Particularly, the glutamate-α-KG cycle plays the most important role in cancer cells or other proliferating cells to maintain nitrogen metabolism. α-KG gathers nitrogen atoms from excess amino acids, while glutamine may contribute nitrogen atoms to synthesize nucleotides or non-essential amino acids to support cellular proliferation, protein synthesis, and nucleotide synthesis (Figure 3C). Thus, a therapeutic strategy targeting glutaminolysis could be a superior resolution to make not only cancer cells but also inflammatory, proliferating CD4+ T cells vulnerable.
2.3.1 Glutamine availability
In activated T cells, glutaminolysis plays a vital role in their proliferation and effector functions. In mouse splenocytes or T cells, removal of glutamine completely blocks their effector functions (i.e., proliferation and secretion of IL-2 and IFN-γ (112)) in spite of comparable expression of activation cell surface marker (CD25, CD69, and CD98 (112)). This dependence of glutamine for proliferation of activated splenocytes or PBMC cannot be replaced by supplementation of glutamate, α-KG, asparagine, or proline, which are substrates that can substitute for glutamine (112, 113).
In addition, activated human CD4+ T cell for 24 hours do not utilize glucose or glutamine as a carbon source to fuel mitochondrial respiration, as the pharmacological treatment using MPC inhibitor (UK5099), GLS inhibitor (CB839 or BPTES), or a combination of both fail to decrease mitochondrial oxygen consumption (52, 114). However, glutamine deprivation reduces mitochondrial oxygen consumption and ATP production (51) (Figure 3C).
Moreover, glutamine availability is still critical in the differentiation of naïve CD4+ T cells. Glutamine deprivation leads to the differentiation of naïve CD4+ T cells into Foxp3+ regulatory T cells with robust in vivo proliferative potentials and immune suppressive effects (51, 113) and a reduction in effector cytokine production such as IFN-γ and IL-17A (52) (Figure 3C). Strikingly, the skewing of naïve CD4+ T cells to Foxp3+ Tregs with glutamine restriction cannot be reversed by glutamate or α-KG supplementation, indicating that glutamine is not required for carbon source (113).
Moreover, naïve CD4+ T cells grown with low glutamine availability, even under Th1 or Th17-polarizing conditions, favor the differentiation into Foxp3+ Tregs while blocking the differentiation into Th1 and Th17 cells (51, 52), respectively (Figure 3C). These observations suggest that glutamine itself, not those intermediates of glutaminolysis such as glutamate and α-KG, plays an essential role in modulating effector functions, mitochondrial ATP synthesis, and differentiation in CD4+ T cells.
De novo nucleotide synthesis requires glutamine-driven aspartate as a source of nitrogen atoms for the formation of the purine/pyridine ring (115). It is well established that aspartate biosynthesis from glutamate via GOT1/2 is required for proliferation in mammalian cells (116, 117). In CD4+ T cells, inhibition of de novo purine/pyrimidine synthesis by N-phosphonacetyl-l-aspartate (PALA) and mycophenolic acid (MPA) promotes the differentiation of Foxp3+ Tregs, which is consistent with the effects of glutamine deprivation (Figure 3C). In addition, the generation of Foxp3+ Tregs is abolished under low glutamine conditions when glutamine synthetase (GS), which synthesizes endogenous glutamine from glutamate, is inhibited by Methionine sulfoximine (MSO) (113) (Figure 3C). These findings suggest that the nitrogen atoms in the amide group of glutamine are primarily used for nucleotide synthesis, rather than the carbon backbone of glutamine, in proliferating CD4+ T cells.
Despite that glutamine supplementation attenuates intestinal inflammation in the DSS-induced colitis animal model (50) (Table 2), Foxp3+ T cells induced by glutamine restriction have shown superior immunosuppressive capacity in vivo to prevent IBD using the adoptive T cell transfer mouse model. Injection of either CD4+Foxp3+ natural Tregs or Foxp3+ T cells grown under glutamine-limited conditions fully protects against colonic infiltration of immune cells, weight loss, and activation of effector T cells. Moreover, glutamine withdrawal has been shown to enhance the proliferation of Foxp3+ T cell in vivo (51) (Figure 3C).
2.3.2 Glutaminase
GLS enzyme activity and mRNA expression are highly induced during early T cell activation (112, 118). However, GLS activity is considerably lower than the activity of GOT or GDH in T cells (112). This indicates that GLS, which is the first enzymes in the process of deamidation of glutamine, is likely to be a limiting factor in T cell activation and presumably T cell differentiation. Under Th0 activating condition by CD3 and CD28 stimulation, GLS inhibition by compound 968 or BPTES significantly impairs the expression of T cell activation marker (CD25 and CD226), secretion of effector cytokines such as IFN-γ, TNF-α, IL-2, and IL-17 and chemokine receptors (CCR6 and CXCR3) (118). Nevertheless, the mode of action by GLS inhibition is quite astonishingly different from that by glutamine deprivation on T cell differentiation. Treatment with GLS inhibitors (CB839 or BPTES) significantly decreases proliferation, IL-17 cytokine production (52, 119) and ATP-coupled OCR (120) in Th17 cells but increases proliferation and IFN-γ production in Th1 cells with an enhanced exhausted phenotype (i.e. PD-1, Lag3, and Tim3) (52) (Figure 3C). Moreover, the genetic deletion of GLS leads to the attenuation of RORγt expression in Th17 cells but the accumulation of t-bet expression in Th1 cells while not affecting Foxp3 expression in Treg (Figure 3C). In addition, the generation of α-KG decreases in CB839-treated Th1 but not Th17 cells, whereas 2-HG increased in both Th1 and Th17 (Figure 3C). Given that α-KG is an important cofactor for both histone and DNA demethylation, GLS deficiency may influence T cell differentiation mechanistically through not only TCA anaplerotic intermediate but also alteration of epigenetic modifications. CB839-treated Th1 cells display decreased global H3K27 trimethylation and more Th1 related genes such as Ifng with opened chromatin accessibility, and supplementation of a-KG reverses the decreased global methylation and opened chromatin status in CB839-treated Th1 cells (52) (Figure 3C). Of note, α-KG has been previously reported to regulate IL-2-sensitive effector gene expression in Th1 cells through the association of CCCTC-binding factor in part (121). Taken together, it is suggested that GLS deficiency has distinct mechanisms of differentiation of Th1 and Th17 cells. Patients with CD possess significantly higher numbers of GLS1-positive cells in the inflamed regions of the lamina propria than the control patients (53). Collectively, targeting GLS may impair the immune responses of the infiltrated T cell, particularly Th17 in vivo for treating IBD. Adoptive transfer of GLS-deficient naive CD4+ T cell to Rag1 KO mice indeed fails to induce weight loss and intestinal inflammation (52) (Figure 3C). Treatment of BPTES also effectively ameliorates spontaneous intestinal inflammation in IL-10 deficient mice by restoring Th17/Treg balance (53) (Figure 3C; Table 2).
Metabolic adaptations has emerged as a critical process in T cell activation, survival, and differentiation. Manipulating the metabolism of T cells can be promising strategies for treating IBD. However, metabolic modulators may have unwanted effects on not just only pathogenic T cells, but also intestinal epithelial cells, endocrine system, bile acid production, and intestinal microbial metabolism to some extent. Thus, further studies are required.
3 Targeting T cell mitochondria for treating colitis
3.1 Mitochondrial biogenesis
Mitochondrial biogenesis is a fundamental process for supplying sufficient energy demands during inflammation in CD4+ T cells. In quiescent circulating CD4+ T cells, the expression levels of glycolytic enzymes (such as HK2, Pyruvate kinase M2, LDHA) and the basal glycolysis rate determined by extracellular acidification rate are relatively higher than those in CD8+ T cells (122). At the same time, unstimulated CD4+ T cells possess a higher number of mitochondrial masses than CD8+ T cells despite maintaining mitochondrial respiration at the same level, suggesting that quiescent CD4+ T cells stock a decent amount of mitochondrial biogenesis for later use (122). Indeed, TCR stimulation rapidly drives mitochondrial reworking in CD4+ T cells. Upon activation by TCR ligation, splenic T cells increase mitochondrial membrane potentials after 12 hours and robust mitochondrial biogenesis (mtDNA and mitochondrial mass) after 48 hours (123) (Figure 5B). In accordance with splenic T cell activation, the mitochondria in activated CD4+ T cells turn into hyper-fused form after 9 hours and become highly energetic with re-fragmentation of mitochondrial morphology, enhanced mitochondrial biogenesis, and accumulated TCA intermediates after 24-hour exposure to CD3/CD28 antibodies (124) (Figure 5B). During this early activation, T cells accelerate the fatty acid biosynthesis and uptake process (125). Meanwhile, mitochondria rearrange one-carbon metabolism within mitochondria, which enhances redox homeostasis and promotes de novo purine biosynthesis (124). After prolonged activation (more than 96 hours), CD4+ T cells continuously increase mitochondrial biogenesis and respiration without mROS-induced mitochondrial dysfunction (126) (Figure 5B). In addition to activation, mitochondrial biogenesis and fitness also orchestrate immune-senescence in CD4+ T cells during aging (127). Therefore, controlling mitochondrial biogenesis could provide therapeutic opportunities in resolving CD4+ T cell-mediated inflammation.
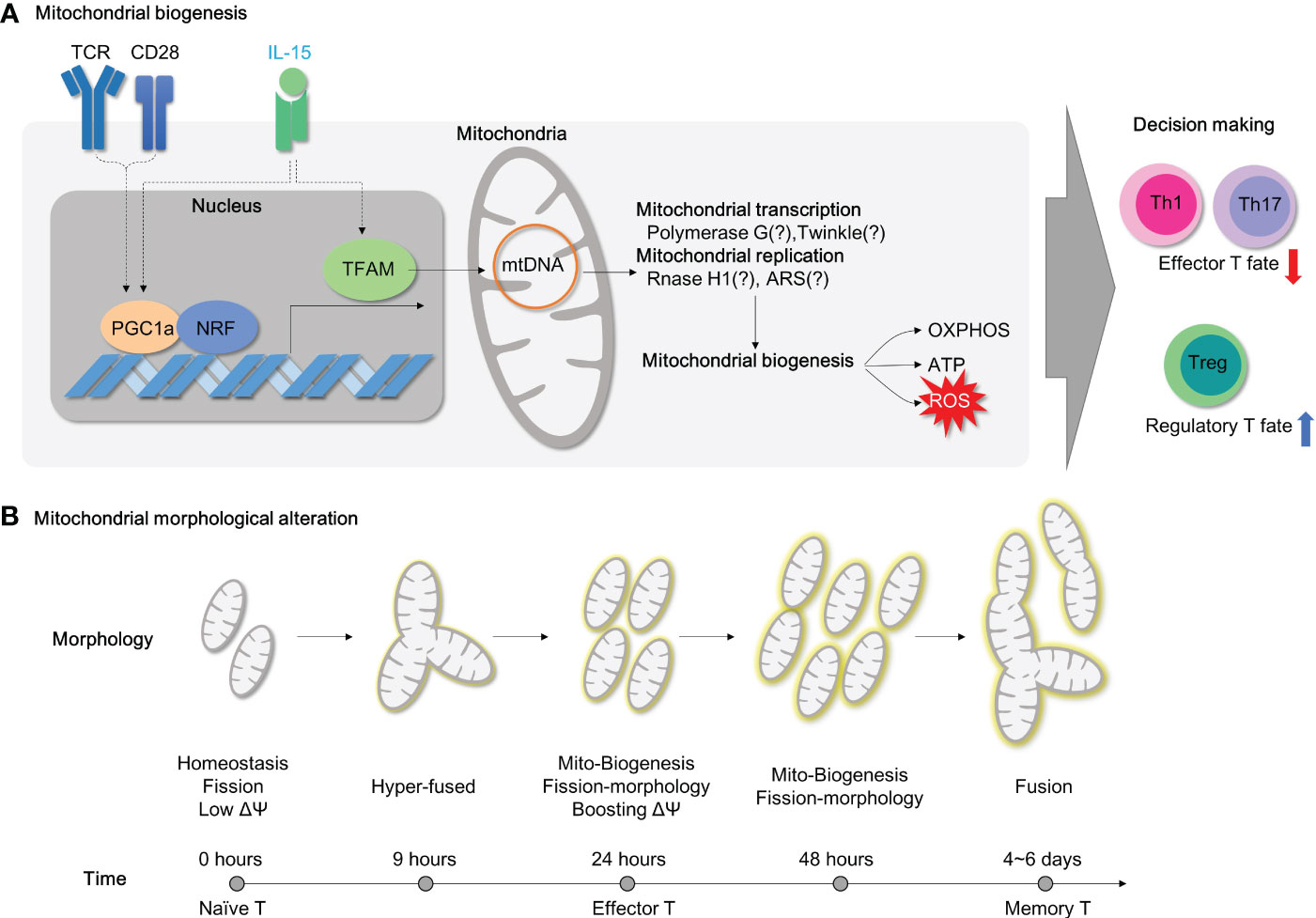
Figure 5 Mitochondrial biogenesis and morphology changes during T cell activation. (A) For T cell activation, it is necessary to undergo mitochondrial biogenesis to meet the energy requirements of the cell. When TFAM is inhibited, energy homeostasis is disrupted, and this results in the polarization of effector T cells. Additionally, PGC1a/TFAM activation is stimulated by IL-15, which leads to mitochondrial biogenesis. (B) The morphology of mitochondria in CD4+ T cells changes during TCR-mediated activation as time progresses. TCR, T cell receptor;PGC1a, peroxisome proliferator-activated receptor-gamma coactivator 1; NRF, Nuclear Respiratory Factor; TFAM, transcriptional factor A mitochondrial; OXPHOS, oxidative phosphorylation; ATP, Adenosine triphosphate; ROS, Reactive oxygen species; ARS, aminoacyl tRNA synthetase.
Mitochondrial biogenesis is primarily regulated by the nuclear transcription factor peroxisome proliferator-activated receptor γ coactivator‐1 (PGC‐1) family of transcription coactivators, which includes PGC1α, PGC1β, and PPRC1 (Figure 5A). PGC1α, together with NRF-1, initiates the transcription of various mitochondrial proteins, including mitochondrial transcription factor A (TFAM), which is then imported into mitochondria (128). Among mitochondrial nucleoids, TFAM is the most abundant DNA-binding protein, which functions in mitochondrial DNA stability and replication. Notably, Tfam mRNA or protein expressions are remarkably upregulated in CD4+ T cells after 4- or 24-hour exposure to CD3/CD28 stimulation, respectively (129) in line with mitochondrial biogenesis.
3.1.1 Mitochondrial transcription factor A
Deficiency of TFAM in CD4+ T cells leads to mitochondrial dysfunction characterized by reduced mtDNA copy number, decreased transcription of mitochondrial proteins, and impaired fatty acid oxidation, despite compensatory increases in mitochondrial mass. Strikingly, Tfam-deficient CD4+ T cells also have dysfunctions in transcription factor EB (TFEB)-induced endolysosomal biogenesis, impaired autophagic flux, and abnormal accumulation of lipids such as sphingomyelin and triglycerides (14). Furthermore, the mitochondrial dysfunctions induced by Tfam deficiency lead to effector T cell differentiation favoring Th1 cell differentiation under Th1 or Th2 polarizing conditions, as well as more pathogenic IFN-γ-secreting Th17 cells (14). Consistent with this, Tfam deletion in regulatory T cells induces mROS and switches metabolism towards glycolysis (54). Tregs in the absence of Tfam have significantly reduced Foxp3 expression, which is indispensable for sustaining immune-tolerance (54). Moreover, Tfam deficiency leads to the development of more proinflammatory IFN-γ (54), IL-13 or IL-17a secreting Tregs (54) (Figure 5A). Collectively, mitochondrial Tfam in CD4+ T cells governs mitochondrial fitness, the balance between effector and regulatory T cells, and autoimmunity. Therefore, it is suggested that Tfam deficiency in CD4+ T cells is likely to prompt the development of autoimmune diseases such as IBD. Undoubtedly, CD4-specific Tfam KO mice are more susceptible to chemical-induced experimental colitis model using 3% DSS (14) (Table 3). Of note, CD4-specific Tfam-deficient mice over 2 months of age exhibit premature aging syndromes with multiple organ dysfunctions associated with aging (130). Moreover, the infusions of Tfam-deficient Tregs functionally fail to resolve the accumulation of pathogenic CD4+ effector T cells in the adoptive T cell transfer colitis model and accordingly advance disease severity (54) (Figure 5A; Table 3).
In addition to TFAM, there are more gene sets related to mitochondrial biogenesis regarding mitochondrial DNA replication (e.g., Polymerase G and Twinkle) and mitochondrial transcription (e.g., RNase H1 and mitochondrial aminoacyl-tRNA synthetase) (131, 132). Yet, none of them have been thoroughly elucidated in mitochondrial functions and T cell responses. Recently, one paper shows that T cells with sti mutation (defects in alanyl-tRNA synthetase) have compromised TCR signal initiation machinery in T cells (133). Therefore, targeting mitochondrial biogenesis is still considered a viable option for IBD therapy and new developments in this field are expected to appear in the near future.
3.1.2 Interleukin-15
IL-15 is a cytokine known for T cell homeostasis, generation of memory T cells and prevention from cell death (134–136). Nevertheless, IL-15 has been recognized as a critical cytokine in mitochondrial biogenesis in T cells. IL-15-treated CD8+ T cells have elongated hyper-fused mitochondria with increased mitochondrial mass (137, 138) and TFAM expression (139) compared to unstimulated or IL-2 treated CD8+ T cells. In addition, IL-15 has also been shown to play a central role in CD4+ T cell proliferation (140), Th17 effector function (141), T helper cell differentiation (142), and neuronal-autoimmunity (141). Moreover, IL-15 remarkably restored augmented mitochondrial biogenesis (TFAM and PGC1α expression) (Figure 5A), diminished mitochondrial mass (mitotracker-green staining), and impaired proliferation in cycling Tregs of immune non-responder subjects after HIV infection (143). In addition, deprivation of the IL-15 cytokine presumably shifts immunosuppressive Foxp3-positive cells to proinflammatory RORγt-positive cells (55). Collectively, it is highly affirmative that IL-15 has a possible implication in controlling mitochondrial biogenesis in CD4+ T cells and its therapeutic applications in IBD. Although genetic deletion of IL-15 (i.e. using IL-15 whole-body KO mice) is not successful in treating DSS-induced colitis model in mice (144) (Table 3), adoptive transfer of B6 CD4+ T cells to IL-15-deficient Rag KO mice induces colonic inflammation compared to the control, and adoptive transfer of IL-15 receptor-deficient Treg fails to suppress severe colonic inflammations with extensive mucosa damage in CD4+ T cell transfer colitis model (55) (Table 3), suggesting a direct role of IL-15 in colitogenic CD4+ T cells.
3.2 Mitochondrial oxidative phosphorylation
Mitochondria produces the bioenergetics molecule ATP from NADH and FADH2 by oxidative phosphorylation via complex I, II, III, IV, and V. As expanding mitochondrial respiration is a critical progression during early T cell activation, as mentioned above, inhibition of mitochondrial respiratory complexes might be helpful in buffering T cell immune responses (145) and treating T cell–mediated inflammation such as IBD.
T cell metabolism is strongly influenced by the surrounding microenvironment, including nutrient (146) and oxygen availability (147, 148). As a result, the distinct in vivo conditions (characterized by relatively high lactate and low oxygen levels) and in vitro conditions (with high glucose and high oxygen levels) inherently lead to different functionalities of matured T cells. Specifically, in vivo-differentiated Th17 cells exhibit a higher dependence on OXPHOS, whereas in vitro-differentiated Th17 cells rely more on aerobic glycolysis. Consequently, in vivo-differentiated Th17 cells appear to be more sensitive to complex V inhibitor, oligomycin, treatment (60). In line with this, oligomycin treatment significantly reduces Th1 proliferation (149), IL-17α secreting CD3+ T cells, and thereby attenuates TNBS-induced colitis animal model comparable to the group treated with RORγt inhibitor ursolic acid (60) (Figure 6; Table 3).
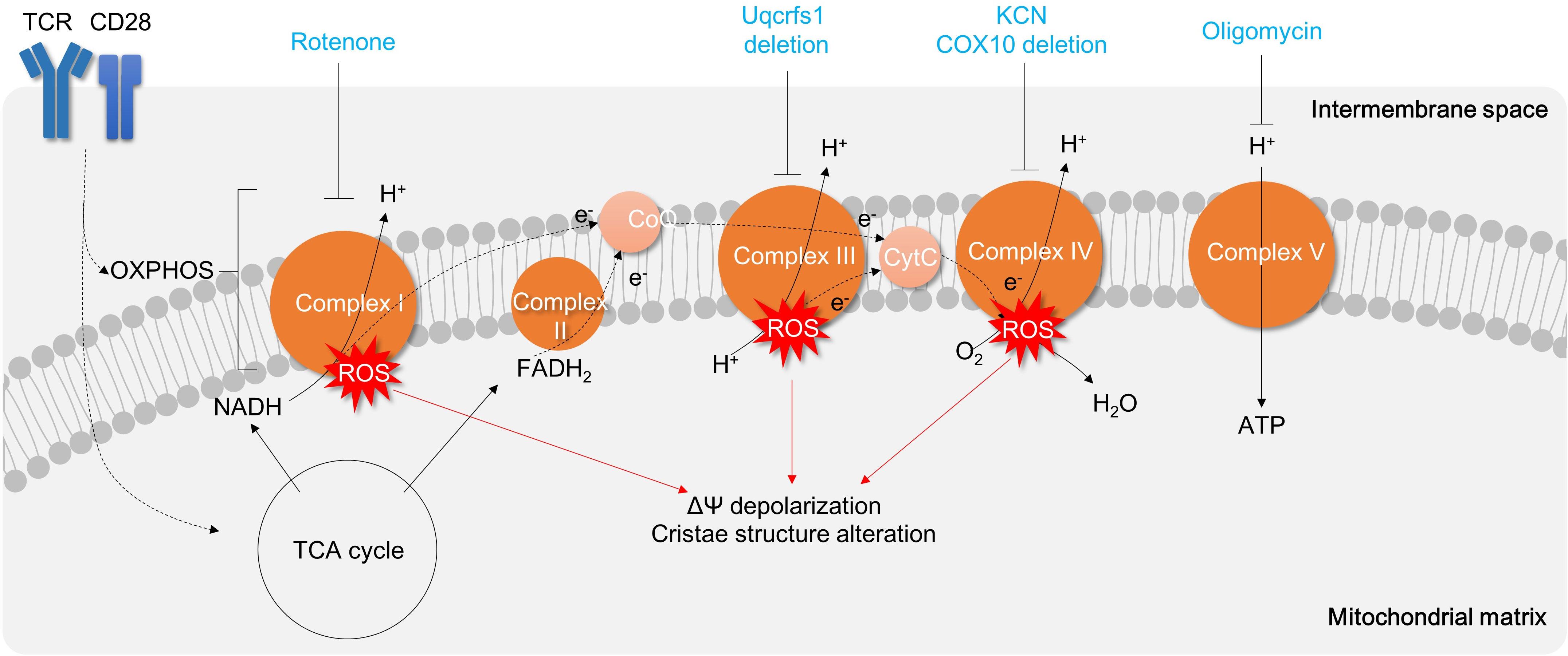
Figure 6 Targeting Mitochondrial Respiration Complexes for Modulating T Cell Activation and Inflammation. Activation of T cells results in upregulation of the TCA cycle and OXPHOS, which leads to the generation of oxidative stress. Inhibition of Complex V (such as oligomycin) hinders mitochondrial respiration and subsequently suppresses T cell activation and proliferation. Other mitochondrial respiratory complexes are also being explored as potential targets for drugs aimed at reducing T cell inflammation. TCR, T cell receptor; OXPHOS, oxidative phosphorylation; TCA cycle, tricarboxylic acid cycle;ROS, reactive oxygen species; CytC, Cytochrome C.
Other mitochondrial respiratory chain complexes I, III, or IV are also critical in T cell activation and differentiation. Treatment of rotenone, a complex I inhibitor, significantly attenuates Th1 polarization in CD4+ T cells (149) (Figure 6). Uqcrfs1, a subunit of complex III, is required to promote excessive mROS production and IL-2 cytokine secretion in CD4+ T cells (66) (Figure 6). Moreover, the inhibition of complex IV by CD4+ T cell-specific deletion of COX10 or treatment of complex IV inhibitor potassium cyanide impairs mitochondrial respiration, mitochondrial cristae structure, T cell activation, proliferation, Th1 (13) and Th17 differentiation and results in apoptosis (13, 150) (Figure 6). Therefore, mitochondrial oxidative respiration is critical in T cell activation, differentiation, and effector functions, and targeting mitochondrial oxidative phosphorylation against IBD may have therapeutic benefits.
3.3 Mitochondrial calcium homeostasis
T cell activation triggers calcium influx that orchestrates approximately three-quarters of the transcription associated with cell cycle, signal transduction, apoptosis, and effector function (151). Previous literature has utilized blocking or modulating plasma membrane calcium channels such as transient receptor potential ankyrin (TRPA), transient receptor potential vanilloid (TRPV), calcium release-activated calcium modulator (ORAI), and calcium-activated potassium channel (KCa) in mouse colitis models (152–161). Moreover, immunosuppressive drugs, cyclosporine A (162) or FK506 (163), classical inhibitors of calcium-dependent calcineurin, induce clinical responses in patients with IBD. In addition, calcium response in human CD45RO+ CD4+ central/effector memory T cells is greater than in CD45RA+ CD4+ naïve T cells (164). While it is strongly predictable that manipulating calcium signaling in T cells could have therapeutic applications, it has yet to be proven whether maintaining mitochondrial calcium homeostasis in T cells has any therapeutic benefit for treating IBD.
Mitochondria have been reported to relocate to the immune synapse (IS) in T cells at early stage of T cell activation (165). When T cells encounter antigen-presenting cells, they require a large quantity of ATP to maintain immunological synapse (IS) architecture as a cellular substrate for immune activities such as kinase action, motor proteins, and degranulation. In addition, the mitochondria at the IS support robust calcium channel opening at the plasma membrane by clearing excess calcium, and thus amplify the calcium signaling pathway in T cells (166). Nonetheless, this mitochondrial calcium governs mitochondrial ATP and mROS production (167) (Figure 7). Three dehydrogenase enzymes consisting of TCA-cycle (PDH, α-KG dehydrogenase (KGDH), and isocitrate dehydrogenase (IDH)) are well-known enzymes directly regulated by mitochondrial calcium (168–170) (Figure 7). In some cases, mitochondrial calcium plays a role as a weak uncoupler, perhaps due to the pH gradient (ΔpH) and membrane potentials (ΔΨ) across the inner membrane (167). The production of mROS and the maintenance of mitochondrial membrane potential are essential for effector function at the early T cell activation. Treatment of mitochondrial-targeted antioxidant mitovitamin E or mitochondrial membrane potential depolarizing agent, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), successfully attenuates IL-2 secretion in CD4+ T cells (66) (Figure 7). Thus, mitochondrial calcium unquestionably plays an important role in regulating T cell activation and their effector functions.
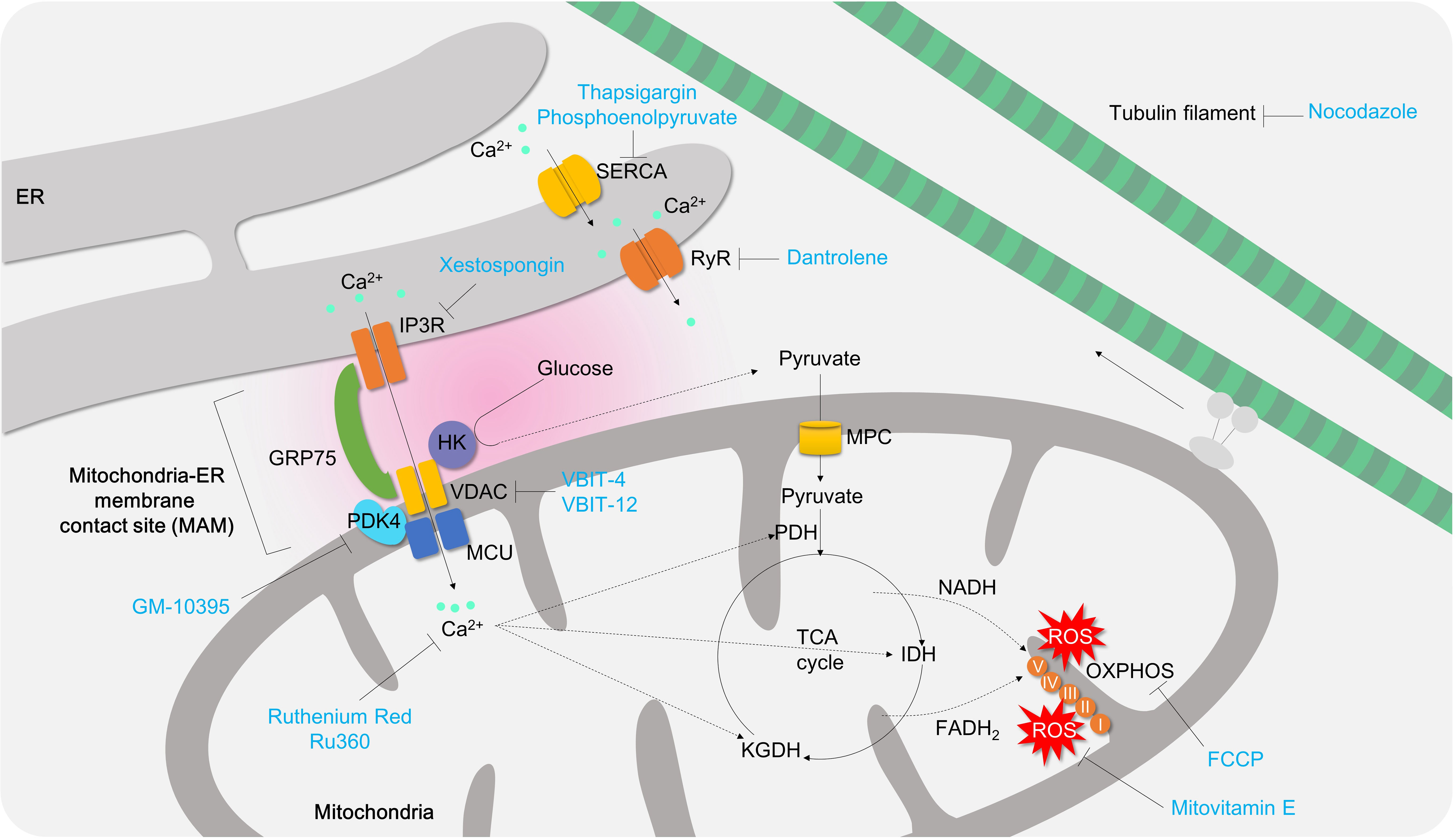
Figure 7 The role of mitochondrial calcium signaling and MAM in controlling T cell activation and differentiation. Under normal physiological conditions, mitochondria play a critical role in maintaining redox balance and energy metabolism by absorbing local calcium ions. However, during T cell activation, mitochondria take up cytosolic or ER calcium through the mitochondria-ER associated membrane contact site (MAM), which promotes TCA cycle, OXPHOS, and oxidative stress production. If mitochondrial calcium is depleted (e.g., using Ru360 or Ruthenium Red), or MAM formation is inhibited (e.g. using GM-10395, Xestospongin, VBIT-4, VBIT-12, nocodazole), it can suppress T cell activation, proliferation, and differentiation into effector T cells. ER, endoplasmic reticulum; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase;IP3R, inositol 1,4,5-trisphosphate receptor;HK, hexokinase;GRP75, glucose-regulated protein 75;PDK4, pyruvate dehydrogenase kinase 4; PDH, pyruvate dehydrogenase; MPC, mitochondria pyruvate carrier; VDAC, voltage-dependent anion channel; RyR, Ryanodine receptor; FCCP, Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; IDH, isocitrate dehydrogenase;KGDH, α-ketoglutarate dehydrogenase.
Mitochondrial calcium instability is contributed by endoplasmic reticulum (ER) calcium deficiency, which transports approximately 25 to 50 percent of ER calcium release triggered by caffeine or thapsigargin to nearby mitochondria in mast cells or myocytes (171). There are two options to control ER calcium for mitochondrial calcium homeostasis: (i) inhibiting ER calcium importer to limit the ER-Ca2+ pool or (ii) inhibiting ER calcium exporter to mitochondria. However, the former approach may not be helpful to treat colitis. Sarco/endoplasmic Reticulum Calcium ATPase (SERCA) is a calcium ATPase which transfers the cytosol calcium into ER. Thapsigargin, an irreversible SERCA inhibitor, is known to cause ER calcium shortage and markedly enhance Th17 differentiation, likely due to ER stress (172) and amplified Ca2+/NFAT signaling (64). Non-canonical SERCA inhibitor phosphoenolpyruvate augments Ca2+/NFAT signaling in Th1 cells as well (64). Furthermore, ER stress and unfolded protein responses have been associated with IBD (173). Therefore, mitochondrial calcium controlling requires other strategies without alleviating ER stress.
Ryanodine receptor (RyR) and IP3R mediate calcium release from ER to cytosol or mitochondria. Inhibition of RyR by dantrolene or IP3R by xestospongin C has been shown to restore thapsigargin-induced ER stress in hepatocytes (174) or islet cells (175) (Figure 7). Although there are no animal colitis studies or clinical trials conducted to date, the applications of these inhibitors for colitis might be worth a try. In T cells, RyR inhibition by ryanodine or dantrolene successfully attenuates store-operated calcium entry (SOCE), T cell proliferation and IL-2 production (176, 177) (Figure 7). IP3R inhibition by xestospongin significantly inhibits differentiation of naive T cells to pro-inflammatory Th9 cells (178) (Figure 7).
However, targeting mitochondrial calcium is also a promising approach. Treatment of mitochondrial calcium uniporter blockers, Ru360 and Ruthenium red (179), significantly inhibits mROS production in CD4+ T cells (66) and IFN secretion and cell proliferation (180). In in vivo TNBS-induced colitis animal model treatment of ruthenium red remarkably attenuates intestinal inflammations. Therefore, targeting mitochondrial calcium is sufficient to decrease T cell effector function and intestinal inflammation (56) (Figure 7; Table 3).
3.3.1 Mitochondrial calcium uniporter
Mitochondria contain a number of calcium transport channels, including the mitochondrial calcium uniporter (MCU), which is sub-localized in the inner membrane of mitochondria (181). Mitochondrial-mediated Ca2+ uptake plays an important role in cytosolic Ca2+ buffering in mast cells. Studies have shown that in MCU-deficient mast cells, antigen-induced beta-hexominidase release was suppressed, suggesting a role for MCU-mediated mitochondrial Ca2+ flux in mast cell degranulation (182). Furthermore, knockdown of mitochondrial MCU has been shown to reduce cytoplasmic calcium fluctuation and SOCE in mast cells (183). However, the pathological role of both MCU and MICU1 (a gatekeeper of MCU-mediated mitochondrial Ca2+ uptake) in T cells remains poorly understood. Loss of MICU1 has been shown to generate excessive ROS, delay proliferation, and impair migration in HeLa cells (184), Thus, blocking MCU or MICU1 in CD4+ T cells is likely to result in mitochondrial Ca2+ instability and ROS generation, leading to suppression of cell proliferation and gut penetration in pathogenic CD4+ T cells (Figure 7).
3.3.2 Voltage-dependent anion channel
Voltage-dependent anion channel (VDAC) is a mitochondrial outer membrane transporter, and has been associated with mitochondrial calcium transport, cell death, and lupus-like autoimmunity (185). Upon 24-hour T cell activation, VDAC1 expression increases along with the Glut1 and HK2 in PBMC (186), which are activation signatures of T cells. In addition, PBMC from patients with coronavirus-A displays mitochondrial dysfunctions characterized by fragmented mitochondrial morphology and apoptotic signaling (186).
The efficacy of VBIT-4 and VBIT-12, VDAC inhibitors, has been documented in treating IBD using DSS- or TNBS-induced colitis animal models (57–59) (Figure 7; Table 3). Although the detailed mechanism behind VDAC inhibition has been shown to be associated with MAVS and inflammasome activation on intestinal epithelial cells (58), it may normalize mitochondrial calcium imbalance and attenuate effector functions in pro-inflammatory T cells (186).
3.3.3 Mitochondria-associated membrane
Mitochondria and ER are physically connected to form a junction called mitochondria-associated membrane (MAM), which provide a platform for various cellular processes including calcium homeostasis, autophagy, lipid metabolism, and apoptosis. The MAMs contain several tethering molecules such as IP3R, VDAC, and GRP75. In 2018, Bantug and colleagues demonstrated the critical role of MAMs in early activation of effector/memory CD8+ T cells (Figure 7), which controls effector functions in memory CD8+ T cells through an immunometabolic reprogramming that involves mitochondrial respiration, HK binding to VDAC, mTOR/AKT/GSK3b signaling. Inhibition of MAM formation using nocodazole significantly reduces mitochondrial respiration, HK expression, mTOR/AKT/GSK3b signaling pathway, and cytokine secretion (Figure 7). However, inhibition of mitochondrial calcium does not affect cytokine production in CD8+ T cells (187).
PDK4 has been identified as a novel modulator of MAM integrity and mitochondrial quality control (97). Genetic deletion of PDK4 in activated CD4+ T cells significantly diminishes MAM formation, SOCE, and mitochondrial calcium transfer compared to control cells. A novel PDK4 inhibitor, GM-10395, also ameliorates SOCE, mitochondrial calcium, and T cell activation. The anti-inflammatory effects of PDK4 suppression have been demonstrated in DSS-induced colitis animal models and naïve T cell transfer colitis models in vivo (49) (Figure 7; Table 3).
4 Future perspectives and conclusion
Over the past few decades, a wealth of scientific knowledge has been generated about the pathophysiology of inflammatory bowel disease (IBD). While the exact mechanisms behind the onset of IBD are complex and subject to debate, immune dysregulation between effector T and regulatory T cells appears to be a major contributing factor.
Experimental studies in IBD have characterized colitogenic T cells, which undergo rapid metabolic reprogramming when exposed to an inflammatory environment. This metabolic adaptation, also known as the Warburg effect, was first observed in cancer cells, but recent research has shown that cellular metabolism is closely linked to the activation, differentiation, and functions of CD4+ T cells. Additionally, mitochondria play a vital role not only in ATP production, but also in redox balance and Ca2+ signaling in T cells.
In this review, we suggest that metabolic regulation, specifically targeting the mitochondrial fitness of CD4+ T cells, has the potential to be a next-generation therapy for autoimmune diseases, such as IBD. Supporting this approach, preclinical evidence from animal studies shows that healthy mitochondria from mesenchymal stem cells can redirect the fate of T cells from Th17 effector to Foxp3+ Treg cell and can also display immunosuppressive effects on animal models of graft-versus-host disease (188, 189).
Presently, the only clinical trials targeting mitochondria are the MARVEL studies (Mitochondrial Anti-oxidant Therapy to Resolve Inflammation in Ulcerative Colitis), NCT04276740, and NCT05539625, which utilize mitoQ to inhibit mitochondrial ROS. However, other preclinical studies mentioned in this review, which have demonstrated successful results, should also be considered for clinical trials.
Emerging evidence shows that gut microbiota signaling to mitochondria plays a pivotal role in maintaining intestinal homeostasis through metabolic regulation and T-cell activation. Bacterial metabolites, such as short-chain fatty acids and hydrogen sulfide, communicate with colonic epithelial and immune cells, and impact on their metabolic, epigenetic, and genetic functions. However, this topic is not covered here as it is beyond the scope of the present review.
However, autoimmune diseases like IBD are complex and multifactorial, and identifying their etiology can be challenging due to highly variable symptoms and underlying mechanisms. Despite the pathological importance of T cells, other immune cells such as monocytes, macrophages, neutrophils, dendritic cells, and innate lymphoid cells are also implicated in IBD (190). Metabolic reprogramming also plays a significant role in their activation and differentiation (191). Thus, it is important not to overlook the clinical therapeutic approach towards modulating the metabolic regulation of these distinct immune cell populations. In addition, personal genetic backgrounds can also present significant obstacles to developing effective IBD therapeutics (192). Therefore, future studies should focus on developing reliable biomarkers for mitochondrial quality and translating these findings into personalized medication. Moreover, because mitochondrial fitness is believed to be crucial to metabolic homeostasis and energy status, combinatorial strategies using non-drug-based (e.g. diet or lifestyle) or drug-based medications for metabolic syndromes in conjunction with current IBD medications could have a profound impact on patients with IBD, especially those who have reached a plateau in drug efficacy. With continued research in this area, we can look forward to a future where patients with IBD have more effective and personalized treatment options.
Author contributions
All authors wrote the manuscript. HL wrote the first draft of the manuscript. J-HJ and E-SK wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Basic Science Research Program, through the National Research Foundation (NRF) of Korea government (MSIT) (NRF-2020R1C1C1009322, NRF-2017R1D1A1B03028512, NRF-2020R1C1C1012729, and NRF-2021R1A5A2021614).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee CH, Koh SJ, Radi ZA, Habtezion A. Animal models of inflammatory bowel disease: novel experiments for revealing pathogenesis of colitis, fibrosis, and colitis-associated colon cancer. Intestinal Res (2023) 21(3):295–305. doi: 10.5217/ir.2023.00029
2. Momozawa Y, Dmitrieva J, Theatre E, Deffontaine V, Rahmouni S, Charloteaux B, et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat Commun (2018) 9(1):2427. doi: 10.1038/s41467-018-04365-8
3. Huang H, Fang M, Jostins L, Umicevic Mirkov M, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature (2017) 547(7662):173–8. doi: 10.1038/nature22969
4. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380(9853):1590–605. doi: 10.1016/S0140-6736(12)60026-9
5. Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet (2012) 380(9853):1606–19. doi: 10.1016/S0140-6736(12)60150-0
6. Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S, et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat Commun (2021) 12(1):1921. doi: 10.1038/s41467-021-22164-6
7. Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol (2020) 5(50). doi: 10.1126/sciimmunol.abb4432
8. Corridoni D, Chapman T, Antanaviciute A, Satsangi J, Simmons A. Inflammatory bowel disease through the lens of single-cell RNA-seq technologies. Inflammatory bowel diseases (2020) 26(11):1658–68. doi: 10.1093/ibd/izaa089
9. Stronkhorst A, Radema S, Yong SL, Bijl H, ten Berge IJ, Tytgat GN, et al. CD4 antibody treatment in patients with active Crohn’s disease: a phase 1 dose finding study. Gut (1997) 40(3):320–7. doi: 10.1136/gut.40.3.320
10. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol (2019) 20(3):288–300. doi: 10.1038/s41590-018-0298-5
11. Raine T, Liu JZ, Anderson CA, Parkes M, Kaser A. Generation of primary human intestinal T cell transcriptomes reveals differential expression at genetic risk loci for immune-mediated disease. Gut (2015) 64(2):250–9. doi: 10.1136/gutjnl-2013-306657
12. Konjar S, Veldhoen M. Dynamic metabolic state of tissue resident CD8 T cells. Front Immunol (2019) 10:1683. doi: 10.3389/fimmu.2019.01683
13. Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity (2017) 46(3):488–503. doi: 10.1016/j.immuni.2017.02.010
14. Baixauli F, Acin-Perez R, Villarroya-Beltri C, Mazzeo C, Nunez-Andrade N, Gabande-Rodriguez E, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab (2015) 22(3):485–98. doi: 10.1016/j.cmet.2015.07.020
15. Teng X, Li W, Cornaby C, Morel L. Immune cell metabolism in autoimmunity. Clin Exp Immunol (2019) 197(2):181–92. doi: 10.1111/cei.13277
16. Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug discovery (2019) 18(9):669–88. doi: 10.1038/s41573-019-0032-5
17. Piranavan P, Bhamra M, Perl A. Metabolic targets for treatment of autoimmune diseases. Immunometabolism (2020) 2(2):51–57. doi: 10.20900/immunometab20200012
18. Desdin-Mico G, Soto-Heredero G, Mittelbrunn M. Mitochondrial activity in T cells. Mitochondrion (2018) 41:51–7. doi: 10.1016/j.mito.2017.10.006
19. Qin Y, Gao C, Luo J. Metabolism characteristics of th17 and regulatory T cells in autoimmune diseases. Front Immunol (2022) 13:828191. doi: 10.3389/fimmu.2022.828191
20. Novak EA, Mollen KP. Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol (2015) 3:62. doi: 10.3389/fcell.2015.00062
21. Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, Thompson-Snipes L, et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut (2020) 69(11):1928–38. doi: 10.1136/gutjnl-2019-319523
22. Khaloian S, Rath E, Hammoudi N, Gleisinger E, Blutke A, Giesbertz P, et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut (2020) 69(11):1939–51. doi: 10.1136/gutjnl-2019-319514
23. Zhang D, Jin W, Wu R, Li J, Park SA, Tu E, et al. High glucose intake exacerbates autoimmunity through reactive-oxygen-species-mediated TGF-beta cytokine activation. Immunity (2019) 51(4):671–81 e5. doi: 10.1016/j.immuni.2019.08.001
24. Laffin M, Fedorak R, Zalasky A, Park H, Gill A, Agrawal A, et al. A high-sugar diet rapidly enhances susceptibility to colitis via depletion of luminal short-chain fatty acids in mice. Sci Rep (2019) 9(1):12294. doi: 10.1038/s41598-019-48749-2
25. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
26. Jodeleit H, Al-Amodi O, Caesar J, Villarroel Aguilera C, Holdt L, Gropp R, et al. Targeting ulcerative colitis by suppressing glucose uptake with ritonavir. Dis Models Mech (2018) 11(11). doi: 10.1242/dmm.036210
27. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
28. Hochrein SM, Wu H, Eckstein M, Arrigoni L, Herman JS, Schumacher F, et al. The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming. Cell Metab (2022) 34(4):516–32 e11. doi: 10.1016/j.cmet.2022.02.015
29. Mehta MM, Weinberg SE, Steinert EM, Chhiba K, Martinez CA, Gao P, et al. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab (2018) 6:10. doi: 10.1186/s40170-018-0184-5
30. Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M, et al. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest (2013) 123(12):5165–78. doi: 10.1172/JCI69751
31. Lyons J, Ghazi PC, Starchenko A, Tovaglieri A, Baldwin KR, Poulin EJ, et al. The colonic epithelium plays an active role in promoting colitis by shaping the tissue cytokine profile. PloS Biol (2018) 16(3):e2002417. doi: 10.1371/journal.pbio.2002417
32. Ogino H, Nakamura K, Iwasa T, Ihara E, Akiho H, Motomura Y, et al. Regulatory T cells expanded by rapamycin in vitro suppress colitis in an experimental mouse model. J gastroenterol (2012) 47(4):366–76. doi: 10.1007/s00535-011-0502-y
33. Farkas S, Hornung M, Sattler C, Guba M, Steinbauer M, Anthuber M, et al. Rapamycin decreases leukocyte migration in vivo and effectively reduces experimentally induced chronic colitis. Int J colorectal disease (2006) 21(8):747–53. doi: 10.1007/s00384-005-0793-7
34. Guan Y, Zhang L, Li X, Zhang X, Liu S, Gao N, et al. Repression of mammalian target of rapamycin complex 1 inhibits intestinal regeneration in acute inflammatory bowel disease models. J Immunol (2015) 195(1):339–46. doi: 10.4049/jimmunol.1303356
35. Yin H, Li X, Zhang B, Liu T, Yuan B, Ni Q, et al. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology (2013) 139(4):494–502. doi: 10.1111/imm.12096
36. Zhao Q, Duck LW, Huang F, Alexander KL, Maynard CL, Mannon PJ, et al. CD4(+) T cell activation and concomitant mTOR metabolic inhibition can ablate microbiota-specific memory cells and prevent colitis. Sci Immunol (2020) 5(54). doi: 10.1126/sciimmunol.abc6373
37. Wu X, Dou Y, Yang Y, Bian D, Luo J, Tong B, et al. Arctigenin exerts anti-colitis efficacy through inhibiting the differentiation of Th1 and Th17 cells via an mTORC1-dependent pathway. Biochem Pharmacol (2015) 96(4):323–36. doi: 10.1016/j.bcp.2015.06.008
38. Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY, et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between th17/treg balance. PloS One (2015) 10(9):e0135858. doi: 10.1371/journal.pone.0135858
39. Higashiyama M, Hokari R, Hozumi H, Kurihara C, Ueda T, Watanabe C, et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J leukocyte Biol (2012) 91(6):901–9. doi: 10.1189/jlb.1011518
40. Lee JH, Elly C, Park Y, Liu YC. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42(6):1062–74. doi: 10.1016/j.immuni.2015.05.016
41. Flannigan KL, Agbor TA, Motta JP, Ferraz JG, Wang R, Buret AG, et al. Proresolution effects of hydrogen sulfide during colitis are mediated through hypoxia-inducible factor-1alpha. FASEB J (2015) 29(4):1591–602. doi: 10.1096/fj.14-266015
42. Tambuwala MM, Manresa MC, Cummins EP, Aversa V, Coulter IS, Taylor CT. Targeted delivery of the hydroxylase inhibitor DMOG provides enhanced efficacy with reduced systemic exposure in a murine model of colitis. J Control Release (2015) 217:221–7. doi: 10.1016/j.jconrel.2015.09.022
43. Halligan DN, Khan MN, Brown E, Rowan CR, Coulter IS, Doherty GA, et al. Hypoxia-inducible factor hydroxylase inhibition enhances the protective effects of cyclosporine in colitis. Am J Physiol Gastrointestinal liver Physiol (2019) 317(2):G90–G7. doi: 10.1152/ajpgi.00049.2019
44. Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology (2008) 134(1):145–55. doi: 10.1053/j.gastro.2007.09.033
45. Kim YI, Yi EJ, Kim YD, Lee AR, Chung J, Ha HC, et al. Local stabilization of hypoxia-inducible factor-1alpha controls intestinal inflammation via enhanced gut barrier function and immune regulation. Front Immunol (2020) 11:609689. doi: 10.3389/fimmu.2020.609689
46. Saravia J, Zeng H, Dhungana Y, Bastardo Blanco D, Nguyen TM, Chapman NM, et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Science Advances. (2020) 6(1):. doi: 10.1126/sciadv.aaw6443
47. Guo X, Guo R, Luo X, Zhou L. Ethyl pyruvate ameliorates experimental colitis in mice by inhibiting the HMGB1-Th17 and Th1/Tc1 responses. Int immunopharmacol (2015) 29(2):454–61. doi: 10.1016/j.intimp.2015.10.015
48. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/JCI76012
49. Lee H, Jeon JH, Lee YJ, Kim MJ, Kwon WH, Chanda D, et al. Inhibition of pyruvate dehydrogenase kinase 4 in CD4(+) T cells ameliorates intestinal inflammation. Cell Mol Gastroenterol Hepatol (2022) 15(2):439–61. doi: 10.1016/j.jcmgh.2022.09.016
50. Hou YC, Wu JM, Wang MY, Wu MH, Chen KY, Yeh SL, et al. Glutamine supplementation attenuates expressions of adhesion molecules and chemokine receptors on T cells in a murine model of acute colitis. Mediators inflammation (2014) 2014:837107. doi: 10.1155/2014/837107
51. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signaling (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
52. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of th17 and th1 cell differentiation by glutaminase-dependent metabolism. Cell (2018) 175(7):1780–95 e19. doi: 10.1016/j.cell.2018.10.001
53. Li J, Zuo L, Tian Y, He Y, Zhang Z, Guo P, et al. Spontaneous colitis in IL-10-deficient mice was ameliorated via inhibiting glutaminase1. J Cell Mol Med (2019) 23(8):5632–41. doi: 10.1111/jcmm.14471
54. Fu Z, Ye J, Dean JW, Bostick JW, Weinberg SE, Xiong L, et al. Requirement of mitochondrial transcription factor A in tissue-resident regulatory T cell maintenance and function. Cell Rep (2019) 28(1):159–71 e4. doi: 10.1016/j.celrep.2019.06.024
55. Tosiek MJ, Fiette L, El Daker S, Eberl G, Freitas AA. IL-15-dependent balance between Foxp3 and RORgammat expression impacts inflammatory bowel disease. Nat Commun (2016) 7:10888. doi: 10.1038/ncomms10888
56. Fichna J, Mokrowiecka A, Cygankiewicz AI, Zakrzewski PK, Malecka-Panas E, Janecka A, et al. Transient receptor potential vanilloid 4 blockade protects against experimental colitis in mice: a new strategy for inflammatory bowel diseases treatment? Neurogastroenterol Motil (2012) 24(11):e557–60. doi: 10.1111/j.1365-2982.2012.01999.x
57. Shoshan-Barmatz V. VDAC inhibitors for treating inflammatory bowel disease. U.S. Patent Publication No 20220023382. (2018). Washington, DC: U.S. Patent and Trademark Office.
58. Verma A, Pittala S, Alhozeel B, Shteinfer-Kuzmine A, Ohana E, Gupta R, et al. The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: a potential therapeutic target. Mol Ther (2021) 30(2):726–44. doi: 10.1016/j.ymthe.2021.06.024
59. Shoshan-Barmatz V, Shteinfer-Kuzmine A, Verma A. VDAC1 at the intersection of cell metabolism, apoptosis, and diseases. Biomolecules (2020) 10(11). doi: 10.3390/biom10111485
60. Franchi L, Monteleone I, Hao LY, Spahr MA, Zhao W, Liu X, et al. Inhibiting oxidative phosphorylation in vivo restrains th17 effector responses and ameliorates murine colitis. J Immunol (2017) 198(7):2735–46. doi: 10.4049/jimmunol.1600810
61. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
62. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183(10):6095–101. doi: 10.4049/jimmunol.0803510
63. Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol (2005) 174(8):4670–7. doi: 10.4049/jimmunol.174.8.4670
64. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
65. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep (2018) 22(6):1509–21. doi: 10.1016/j.celrep.2018.01.040
66. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
67. Galgani M, De Rosa V, Matarese G. T cell metabolism and susceptibility to autoimmune diseases. Mol Immunol (2015) 68(2 Pt C):558–63. doi: 10.1016/j.molimm.2015.07.035
68. Kappelman MD, Galanko JA, Porter CQ, Sandler RS. Association of paediatric inflammatory bowel disease with other immune-mediated diseases. Arch Dis childhood (2011) 96(11):1042–6. doi: 10.1136/archdischild-2011-300633
69. Binus AM, Han J, Qamar AA, Mody EA, Holt EW, Qureshi AA. Associated comorbidities in psoriasis and inflammatory bowel disease. J Eur Acad Dermatol Venereol: JEADV (2012) 26(5):644–50. doi: 10.1111/j.1468-3083.2011.04153.x
70. Din H, Anderson AJ, Ramos Rivers C, Proksell S, Koutroumpakis F, Salim T, et al. Disease characteristics and severity in patients with inflammatory bowel disease with coexistent diabetes mellitus. Inflammatory bowel diseases (2020) 26(9):1436–42. doi: 10.1093/ibd/izz305
71. Tseng CH. Metformin use is associated with a lower risk of inflammatory bowel disease in patients with type 2 diabetes mellitus. J Crohn’s colitis (2021) 15(1):64–73. doi: 10.1093/ecco-jcc/jjaa136
72. Qu D, Shen L, Liu S, Li H, Ma Y, Zhang R, et al. Chronic inflammation confers to the metabolic reprogramming associated with tumorigenesis of colorectal cancer. Cancer Biol Ther (2017) 18(4):237–44. doi: 10.1080/15384047.2017.1294292
73. Linke M, Fritsch SD, Sukhbaatar N, Hengstschlager M, Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS letters (2017) 591(19):3089–103. doi: 10.1002/1873-3468.12711
74. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12(5):325–38. doi: 10.1038/nri3198
75. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi: 10.1016/j.immuni.2009.04.014
76. Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev (2007) 21(9):1037–49. doi: 10.1101/gad.1529107
77. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
78. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
79. Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrugge L, Csizmadia E, et al. HIF-1alpha-induced xenobiotic transporters promote Th17 responses in Crohn’s disease. J Autoimmun (2018) 94:122–33. doi: 10.1016/j.jaut.2018.07.022
80. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai AC, et al. HIF-2alpha is indispensable for regulatory T cell function. Nat Commun (2020) 11(1):5005. doi: 10.1038/s41467-020-18731-y
81. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci United States America (2012) 109(41):E2784–93. doi: 10.1073/pnas.1202366109
82. Li HS, Zhou YN, Li L, Li SF, Long D, Chen XL, et al. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol (2019) 25:101109. doi: 10.1016/j.redox.2019.101109
83. Marchingo JM, Sinclair LV, Howden AJ, Cantrell DA. Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. eLife (2020) 9. doi: 10.7554/eLife.53725
84. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
85. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93 e7. doi: 10.1016/j.cmet.2016.12.018
86. Park S, Jeon JH, Min BK, Ha CM, Thoudam T, Park BY, et al. Role of the pyruvate dehydrogenase complex in metabolic remodeling: differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab J (2018) 42(4):270–81. doi: 10.4093/dmj.2018.0101
87. Jeon JH, Thoudam T, Choi EJ, Kim MJ, Harris RA, Lee IK. Loss of metabolic flexibility as a result of overexpression of pyruvate dehydrogenase kinases in muscle, liver and the immune system: Therapeutic targets in metabolic diseases. J Diabetes Invest (2021) 12(1):21–31. doi: 10.1111/jdi.13345
88. Min BK, Park S, Kang HJ, Kim DW, Ham HJ, Ha CM, et al. Pyruvate dehydrogenase kinase is a metabolic checkpoint for polarization of macrophages to the M1 phenotype. Front Immunol (2019) 10:944. doi: 10.3389/fimmu.2019.00944
89. Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell (2013) 12(1):49–61. doi: 10.1016/j.stem.2012.10.011
90. Ramstead AG, Wallace JA, Lee SH, Bauer KM, Tang WW, Ekiz HA, et al. Mitochondrial pyruvate carrier 1 promotes peripheral T cell homeostasis through metabolic regulation of thymic development. Cell Rep (2020) 30(9):2889–99 e6. doi: 10.1016/j.celrep.2020.02.042
91. Jun S, Mahesula S, Mathews TP, Martin-Sandoval MS, Zhao Z, Piskounova E, et al. The requirement for pyruvate dehydrogenase in leukemogenesis depends on cell lineage. Cell Metab (2021) 33(9):1777–92 e8. doi: 10.1016/j.cmet.2021.07.016
92. Koprivica I, Gajic D, Pejnovic N, Paunovic V, Saksida T, Stojanovic I. Ethyl pyruvate promotes proliferation of regulatory T cells by increasing glycolysis. Molecules (2020) 25(18). doi: 10.3390/molecules25184112
93. Koprivica I, Vujicic M, Gajic D, Saksida T, Stojanovic I. Ethyl pyruvate stimulates regulatory T cells and ameliorates type 1 diabetes development in mice. Front Immunol (2018) 9:3130. doi: 10.3389/fimmu.2018.03130
94. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8(+) T cells. Immunity (2019) 51(5):856–70 e5. doi: 10.1016/j.immuni.2019.09.003
95. Soriano-Baguet L, Grusdat M, Kurniawan H, Benzarti M, Binsfeld C, Ewen A, et al. Pyruvate dehydrogenase fuels a critical citrate pool that is essential for Th17 cell effector functions. Cell Rep (2023) 42(3):112153. doi: 10.1016/j.celrep.2023.112153
96. Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci Signaling (2020) 13(628). doi: 10.1126/scisignal.aaz6206
97. Thoudam T, Ha CM, Leem J, Chanda D, Park JS, Kim HJ, et al. PDK4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes (2019) 68(3):571–86. doi: 10.2337/db18-0363
98. Park BY, Jeon JH, Go Y, Ham HJ, Kim JE, Yoo EK, et al. PDK4 deficiency suppresses hepatic glucagon signaling by decreasing cAMP levels. Diabetes (2018) 67(10):2054–68. doi: 10.2337/db17-1529
99. Lee JM, Kim MJ, Lee SJ, Kim BG, Choi JY, Lee SM, et al. PDK2 deficiency prevents ovariectomy-induced bone loss in mice by regulating the RANKL-NFATc1 pathway during osteoclastogenesis. J Bone mineral Res (2021) 36(3):553–66. doi: 10.1002/jbmr.4202
100. Larrieu CM, Storevik S, Guyon J, Pagano Zottola AC, Bouchez CL, Derieppe MA, et al. Refining the role of pyruvate dehydrogenase kinases in glioblastoma development. Cancers (2022) 14(15). doi: 10.3390/cancers14153769
101. Kumar K, Wigfield S, Gee HE, Devlin CM, Singleton D, Li JL, et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J Mol Med (Berl) (2013) 91(6):749–58. doi: 10.1007/s00109-013-0996-2
102. Na YR, Jung D, Song J, Park JW, Hong JJ, Seok SH. Pyruvate dehydrogenase kinase is a negative regulator of interleukin-10 production in macrophages. J Mol Cell Biol (2020) 12(7):543–55. doi: 10.1093/jmcb/mjz113
103. Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, et al. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J basic Clin Physiol Pharmacol (2013) 24(4):271–6. doi: 10.1515/jbcpp-2013-0001
104. Eleftheriadis T, Sounidaki M, Pissas G, Antoniadi G, Liakopoulos V, Stefanidis I. In human alloreactive CD4(+) T-cells, dichloroacetate inhibits aerobic glycolysis, induces apoptosis and favors differentiation towards the regulatory T-cell subset instead of effector T-cell subsets. Mol Med Rep (2016) 13(4):3370–6. doi: 10.3892/mmr.2016.4912
105. Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q, et al. The role of low-level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol (2012) 302(3):L300–7. doi: 10.1152/ajplung.00221.2011
106. Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol (2016) 196(1):106–14. doi: 10.4049/jimmunol.1501766
107. Makita N, Ishiguro J, Suzuki K, Nara F. Dichloroacetate induces regulatory T-cell differentiation and suppresses Th17-cell differentiation by pyruvate dehydrogenase kinase-independent mechanism. J Pharm Pharmacol (2017) 69(1):43–51. doi: 10.1111/jphp.12655
108. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol (2016) 196(1):80–90. doi: 10.4049/jimmunol.1501537
109. Badr MM, Qinna NA, Qadan F, Matalka KZ. Dichloroacetate modulates cytokines toward T helper 1 function via induction of the interleukin-12-interferon-gamma pathway. OncoTargets Ther (2014) 7:193–201. doi: 10.2147/OTT.S56688
110. Weisshaar N, Madi A, Cui G. Early TCR signaling sweetens effector function through PDHK1. Trends Endocrinol metabolism: TEM (2018) 29(9):595–7. doi: 10.1016/j.tem.2018.03.016
111. Kono M, Yoshida N, Maeda K, Skinner NE, Pan W, Kyttaris VC, et al. Pyruvate dehydrogenase phosphatase catalytic subunit 2 limits Th17 differentiation. Proc Natl Acad Sci United States America (2018) 115(37):9288–93. doi: 10.1073/pnas.1805717115
112. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
113. Metzler B, Gfeller P, Guinet E. Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human T cells with high FOXP3 expression and regulatory properties. J Immunol (2016) 196(9):3618–30. doi: 10.4049/jimmunol.1501756
114. He J, Yang W, Cheng B, Ma L, Tursunjiang D, Ding Z, et al. Integrated metabolomic and transcriptomic profiling reveals the tissue-specific flavonoid compositions and their biosynthesis pathways in Ziziphora bungeana. Chin Med (2020) 15:73. doi: 10.1186/s13020-020-00354-6
115. Villa E, Ali ES, Sahu U, Ben-Sahra I. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers (2019) 11(5). doi: 10.3390/cancers11050688
116. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162(3):540–51. doi: 10.1016/j.cell.2015.07.016
117. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162(3):552–63. doi: 10.1016/j.cell.2015.07.017
118. Sener Z, Cederkvist FH, Volchenkov R, Holen HL, Skalhegg BS. T helper cell activation and expansion is sensitive to glutaminase inhibition under both hypoxic and normoxic conditions. PloS One (2016) 11(7):e0160291. doi: 10.1371/journal.pone.0160291
119. Kono M, Yoshida N, Maeda K, Suarez-Fueyo A, Kyttaris VC, Tsokos GC. Glutaminase 1 inhibition reduces glycolysis and ameliorates lupus-like disease in MRL/lpr mice and experimental autoimmune encephalomyelitis. Arthritis Rheumatol (2019) 71(11):1869–78. doi: 10.1002/art.41019
120. Kono M, Yoshida N, Maeda K, Tsokos GC. Transcriptional factor ICER promotes glutaminolysis and the generation of Th17 cells. Proc Natl Acad Sci United States America (2018) 115(10):2478–83. doi: 10.1073/pnas.1714717115
121. Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Crossman DK, et al. CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity (2017) 47(2):251–67 e7. doi: 10.1016/j.immuni.2017.07.015
122. Jones N, Cronin JG, Dolton G, Panetti S, Schauenburg AJ, Galloway SAE, et al. Metabolic adaptation of human CD4(+) and CD8(+) T-cells to T-cell receptor-mediated stimulation. Front Immunol (2017) 8:1516. doi: 10.3389/fimmu.2017.01516
123. D’Souza AD, Parikh N, Kaech SM, Shadel GS. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion (2007) 7(6):374–85. doi: 10.1016/j.mito.2007.08.001
124. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab (2016) 24(1):104–17. doi: 10.1016/j.cmet.2016.06.007
125. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun (2016) 7:13683. doi: 10.1038/ncomms13683
126. Akkaya B, Roesler AS, Miozzo P, Theall BP, Al Souz J, Smelkinson MG, et al. Increased mitochondrial biogenesis and reactive oxygen species production accompany prolonged CD4(+) T cell activation. J Immunol (2018) 201(11):3294–306. doi: 10.4049/jimmunol.1800753
127. Callender LA, Carroll EC, Bober EA, Akbar AN, Solito E, Henson SM. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell (2020) 19(2):e13067. doi: 10.1111/acel.13067
128. Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion (2015) 25:67–75. doi: 10.1016/j.mito.2015.10.001
129. Gerner MC, Niederstaetter L, Ziegler L, Bileck A, Slany A, Janker L, et al. Proteome analysis reveals distinct mitochondrial functions linked to interferon response patterns in activated CD4+ and CD8+ T cells. Front Pharmacol (2019) 10:727. doi: 10.3389/fphar.2019.00727
130. Desdin-Mico G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabande-Rodriguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science (2020) 368(6497):1371–76. doi: 10.1126/science.aax0860
131. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol basis disease (2017) 1863(6):1539–55. doi: 10.1016/j.bbadis.2017.02.017
132. Filograna R, Mennuni M, Alsina D, Larsson NG. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett (2021) 595(8):976–1002. doi: 10.1002/1873-3468.14021
133. Shim JA, Jo Y, Hwang H, Lee SE, Ha D, Lee JH, et al. Defects in aminoacyl-tRNA synthetase cause partial B and T cell immunodeficiency. Cell Mol Life Sci (2022) 79(2):87. doi: 10.1007/s00018-021-04122-z
134. Drake A, Kaur M, Iliopoulou BP, Phennicie R, Hanson A, Chen J. Interleukins 7 and 15 maintain human T cell proliferative capacity through STAT5 signaling. PloS One (2016) 11(11):e0166280. doi: 10.1371/journal.pone.0166280
135. DeGottardi MQ, Okoye AA, Vaidya M, Talla A, Konfe AL, Reyes MD, et al. Effect of anti-IL-15 administration on T cell and NK cell homeostasis in rhesus macaques. J Immunol (2016) 197(4):1183–98. doi: 10.4049/jimmunol.1600065
136. Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest (2015) 125(9):3477–90. doi: 10.1172/JCI81261
137. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. doi: 10.1016/j.cell.2016.05.035
138. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci United States America (2013) 110(35):14336–41. doi: 10.1073/pnas.1221740110
139. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi: 10.1016/j.immuni.2011.12.007
140. Miranda-Carus ME, Benito-Miguel M, Llamas MA, Balsa A, Martin-Mola E. Human T cells constitutively express IL-15 that promotes ex vivo T cell homeostatic proliferation through autocrine/juxtacrine loops. J Immunol (2005) 175(6):3656–62. doi: 10.4049/jimmunol.175.6.3656
141. Pandiyan P, Yang XP, Saravanamuthu SS, Zheng L, Ishihara S, O’Shea JJ, et al. The role of IL-15 in activating STAT5 and fine-tuning IL-17A production in CD4 T lymphocytes. J Immunol (2012) 189(9):4237–46. doi: 10.4049/jimmunol.1201476
142. Waickman AT, Ligons DL, Hwang S, Park JY, Lazarevic V, Sato N, et al. CD4 effector T cell differentiation is controlled by IL-15 that is expressed and presented in trans. Cytokine (2017) 99:266–74. doi: 10.1016/j.cyto.2017.08.004
143. Younes SA, Talla A, Pereira Ribeiro S, Saidakova EV, Korolevskaya LB, Shmagel KV, et al. Cycling CD4+ T cells in HIV-infected immune nonresponders have mitochondrial dysfunction. J Clin Invest (2018) 128(11):5083–94. doi: 10.1172/JCI120245
144. Brounais-Le Royer B, Pierroz DD, Velin D, Frossard C, Zheng XX, Lehr HA, et al. Effects of an interleukin-15 antagonist on systemic and skeletal alterations in mice with DSS-induced colitis. Am J pathol (2013) 182(6):2155–67. doi: 10.1016/j.ajpath.2013.02.033
145. Almeida L, Dhillon-LaBrooy A, Carriche G, Berod L, Sparwasser T. CD4(+) T-cell differentiation and function: Unifying glycolysis, fatty acid oxidation, polyamines NAD mitochondria. J Allergy Clin Immunol (2021) 148(1):16–32. doi: 10.1016/j.jaci.2021.03.033
146. Kaymak I, Luda KM, Duimstra LR, Ma EH, Longo J, Dahabieh MS, et al. Carbon source availability drives nutrient utilization in CD8(+) T cells. Cell Metab (2022) 34(9):1298–311 e6. doi: 10.1016/j.cmet.2022.07.012
147. Hasan F, Chiu Y, Shaw RM, Wang J, Yee C. Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight (2021) 6(10). doi: 10.1172/jci.insight.138970
148. Place TL, Domann FE, Case AJ. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic Biol Med (2017) 113:311–22. doi: 10.1016/j.freeradbiomed.2017.10.003
149. Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature (2019) 571(7765):403–7. doi: 10.1038/s41586-019-1311-3
150. Tarasenko TN, Pacheco SE, Koenig MK, Gomez-Rodriguez J, Kapnick SM, Diaz F, et al. Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab (2017) 25(6):1254–68 e7. doi: 10.1016/j.cmet.2017.05.007
151. Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol (2001) 2(4):316–24. doi: 10.1038/86318
152. Radhakrishnan VM, Ramalingam R, Larmonier CB, Thurston RD, Laubitz D, Midura-Kiela MT, et al. Post-translational loss of renal TRPV5 calcium channel expression, Ca(2+) wasting, and bone loss in experimental colitis. Gastroenterology (2013) 145(3):613–24. doi: 10.1053/j.gastro.2013.06.002
153. Ohya S, Fukuyo Y, Kito H, Shibaoka R, Matsui M, Niguma H, et al. Upregulation of KCa3.1 K(+) channel in mesenteric lymph node CD4(+) T lymphocytes from a mouse model of dextran sodium sulfate-induced inflammatory bowel disease. Am J Physiol Gastrointestinal liver Physiol (2014) 306(10):G873–85. doi: 10.1152/ajpgi.00156.2013
154. Ohya S. [Physiological role of K(+) channels in the regulation of T cell function]. Yakugaku zasshi: J Pharm Soc Japan (2016) 136(3):479–83. doi: 10.1248/yakushi.15-00246-4
155. Mencarelli A, Vacca M, Khameneh HJ, Acerbi E, Tay A, Zolezzi F, et al. Calcineurin B in CD4(+) T cells prevents autoimmune colitis by negatively regulating the JAK/STAT pathway. Front Immunol (2018) 9:261. doi: 10.3389/fimmu.2018.00261
156. McCarl CA, Khalil S, Ma J, Oh-hora M, Yamashita M, Roether J, et al. Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J Immunol (2010) 185(10):5845–58. doi: 10.4049/jimmunol.1001796
157. Kaufmann U, Kahlfuss S, Yang J, Ivanova E, Koralov SB, Feske S. Calcium signaling controls pathogenic th17 cell-mediated inflammation by regulating mitochondrial function. Cell Metab (2019) 29(5):1104–18 e6. doi: 10.1016/j.cmet.2019.01.019
158. Endo K, Kito H, Tanaka R, Kajikuri J, Tanaka S, Elboray EE, et al. Possible contribution of inflammation-associated hypoxia to increased K2P5.1 K(+) channel expression in CD4(+) T cells of the mouse model for inflammatory bowel disease. Int J Mol Sci (2019) 21(1). doi: 10.3390/ijms21010038
159. Bertin S, Aoki-Nonaka Y, Lee J, de Jong PR, Kim P, Han T, et al. The TRPA1 ion channel is expressed in CD4+ T cells and restrains T-cell-mediated colitis through inhibition of TRPV1. Gut (2017) 66(9):1584–96. doi: 10.1136/gutjnl-2015-310710
160. Bertin S, Aoki-Nonaka Y, de Jong PR, Nohara LL, Xu H, Stanwood SR, et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat Immunol (2014) 15(11):1055–63. doi: 10.1038/ni.3009
161. Letizia M, Wang YH, Kaufmann U, Gerbeth L, Sand A, Brunkhorst M, et al. Store-operated calcium entry controls innate and adaptive immune cell function in inflammatory bowel disease. EMBO Mol Med (2022) 14(9):e15687. doi: 10.15252/emmm.202215687
162. Laharie D, Bourreille A, Branche J, Allez M, Bouhnik Y, Filippi J, et al. Ciclosporin versus infliximab in patients with severe ulcerative colitis refractory to intravenous steroids: a parallel, open-label randomised controlled trial. Lancet (2012) 380(9857):1909–15. doi: 10.1016/S0140-6736(12)61084-8
163. Ogata H, Kato J, Hirai F, Hida N, Matsui T, Matsumoto T, et al. Double-blind, placebo-controlled trial of oral tacrolimus (FK506) in the management of hospitalized patients with steroid-refractory ulcerative colitis. Inflammatory bowel diseases (2012) 18(5):803–8. doi: 10.1002/ibd.21853
164. Arrol HP, Church LD, Bacon PA, Young SP. Intracellular calcium signalling patterns reflect the differentiation status of human T cells. Clin Exp Immunol (2008) 153(1):86–95. doi: 10.1111/j.1365-2249.2008.03677.x
165. Yog R, Barhoumi R, McMurray DN, Chapkin RS. n-3 polyunsaturated fatty acids suppress mitochondrial translocation to the immunologic synapse and modulate calcium signaling in T cells. J Immunol (2010) 184(10):5865–73. doi: 10.4049/jimmunol.0904102
166. Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem (2006) 281(52):40302–9. doi: 10.1074/jbc.M607896200
167. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol (2004) 287(4):C817–33. doi: 10.1152/ajpcell.00139.2004
168. Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol (2015) 593(16):3447–62. doi: 10.1113/JP270254
169. Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta (2009) 1787(11):1334–41. doi: 10.1016/j.bbabio.2009.05.011
170. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (2009) 1787(11):1309–16. doi: 10.1016/j.bbabio.2009.01.005
171. Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol (2000) 529 Pt 3:553–64. doi: 10.1111/j.1469-7793.2000.00553.x
172. Brucklacher-Waldert V, Ferreira C, Stebegg M, Fesneau O, Innocentin S, Marie JC, et al. Cellular stress in the context of an inflammatory environment supports TGF-beta-independent T helper-17 differentiation. Cell Rep (2017) 19(11):2357–70. doi: 10.1016/j.celrep.2017.05.052
173. Cao SS. Epithelial ER stress in crohn’s disease and ulcerative colitis. Inflammatory bowel diseases (2016) 22(4):984–93. doi: 10.1097/MIB.0000000000000660
174. Tamitani M, Yamamoto T, Yamamoto N, Fujisawa K, Tanaka S, Nakamura Y, et al. Dantrolene prevents hepatic steatosis by reducing cytoplasmic Ca(2+) level and ER stress. Biochem biophysics Rep (2020) 23:100787. doi: 10.1016/j.bbrep.2020.100787
175. Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes (2009) 58(2):422–32. doi: 10.2337/db07-1762
176. Dadsetan S, Zakharova L, Molinski TF, Fomina AF. Store-operated Ca2+ influx causes Ca2+ release from the intracellular Ca2+ channels that is required for T cell activation. J Biol Chem (2008) 283(18):12512–9. doi: 10.1074/jbc.M709330200
177. Thakur P, Dadsetan S, Fomina AF. Bidirectional coupling between ryanodine receptors and Ca2+ release-activated Ca2+ (CRAC) channel machinery sustains store-operated Ca2+ entry in human T lymphocytes. J Biol Chem (2012) 287(44):37233–44. doi: 10.1074/jbc.M112.398974
178. Shen Y, Song Z, Lu X, Ma Z, Lu C, Zhang B, et al. Fas signaling-mediated TH9 cell differentiation favors bowel inflammation and antitumor functions. Nat Commun (2019) 10(1):2924. doi: 10.1038/s41467-019-10889-4
179. Bae JH, Park JW, Kwon TK. Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem Biophys Res Commun (2003) 303(4):1073–9. doi: 10.1016/S0006-291X(03)00479-0
180. Feng J, Lu S, Ding Y, Zheng M, Wang X. Homocysteine activates T cells by enhancing endoplasmic reticulum-mitochondria coupling and increasing mitochondrial respiration. Protein Cell (2016) 7(6):391–402. doi: 10.1007/s13238-016-0245-x
181. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature (2004) 427(6972):360–4. doi: 10.1038/nature02246
182. Takekawa M, Furuno T, Hirashima N, Nakanishi M. Mitochondria take up Ca2+ in two steps dependently on store-operated Ca2+ entry in mast cells. Biol Pharm bulletin (2012) 35(8):1354–60. doi: 10.1248/bpb.b110576
183. Samanta K, Douglas S, Parekh AB. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PloS One (2014) 9(7):e101188. doi: 10.1371/journal.pone.0101188
184. Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell (2012) 151(3):630–44. doi: 10.1016/j.cell.2012.10.011
185. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science (2019) 366(6472):1531–6. doi: 10.1126/science.aav4011
186. Thompson EA, Cascino K, Ordonez AA, Zhou W, Vaghasia A, Hamacher-Brady A, et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep (2021) 34(11):108863. doi: 10.1016/j.celrep.2021.108863
187. Bantug GR, Fischer M, Grahlert J, Balmer ML, Unterstab G, Develioglu L, et al. Mitochondria-endoplasmic reticulum contact sites function as immunometabolic hubs that orchestrate the rapid recall response of memory CD8(+) T cells. Immunity (2018) 48(3):542–55 e6. doi: 10.1016/j.immuni.2018.02.012
188. Luz-Crawford P, Hernandez J, Djouad F, Luque-Campos N, Caicedo A, Carrere-Kremer S, et al. Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Stem Cell Res Ther (2019) 10(1):232. doi: 10.1186/s13287-019-1307-9
189. Court AC, Le-Gatt A, Luz-Crawford P, Parra E, Aliaga-Tobar V, Batiz LF, et al. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep (2020) 21(2):e48052. doi: 10.15252/embr.201948052
190. Kaluzna A, Olczyk P, Komosinska-Vassev K. The role of innate and adaptive immune cells in the pathogenesis and development of the inflammatory response in ulcerative colitis. J Clin Med (2022) 11(2). doi: 10.3390/jcm11020400
191. Zaiatz Bittencourt V, Jones F, Doherty G, Ryan EJ. Targeting immune cell metabolism in the treatment of inflammatory bowel disease. Inflammatory bowel diseases (2021) 27(10):1684–93. doi: 10.1093/ibd/izab024
Keywords: mitochondria, IBD - inflammatory bowel disease, immunometabolism, T cell, treatment, inflammation
Citation: Lee H, Jeon J-H and Kim ES (2023) Mitochondrial dysfunctions in T cells: focus on inflammatory bowel disease. Front. Immunol. 14:1219422. doi: 10.3389/fimmu.2023.1219422
Received: 09 May 2023; Accepted: 06 September 2023;
Published: 22 September 2023.
Edited by:
Murugaiyan Gopal, Harvard Medical School, United StatesReviewed by:
Seung-Hyo Lee, Korea Advanced Institute of Science and Technology (KAIST), Republic of KoreaGlauben Tamara Landskron, Universidad Finis Terrae, Chile
Copyright © 2023 Lee, Jeon and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eun Soo Kim, dandy813@hanmail.net
†These authors share first authorship