 Jian Yang
Jian Yang Jialuo He
Jialuo He Yiting Feng
Yiting Feng Ming Xiang
Ming Xiang- Department of Pharmacology, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
It is generally recognized that the initiation of obesity-related hepatocellular carcinoma (HCC) is closely associated with hepatic inflammation. However, the paradoxical role of inflammation in the initiation and progression of HCC is highlighted by the fact that the inflammatory HCC is accompanied by significant immune effector cells infiltration compared to non-inflammatory HCC and HCC with enhanced immune response exhibits better survival. Importantly, the cancer progression has been primarily attributed to the immunosuppression, which can also be induced by obesity. Furthermore, the increased risk of viral infection and thus viral-HCC in obese individuals supports the view that obesity contributes to HCC via immunosuppression. Here, we have reviewed the various mechanisms responsible for obesity-induced tumor immune microenvironment and immunosuppression in obesity-related HCC. We highlight that the obesity-induced immunosuppression originates from lipid disorder as well as metabolic reprogramming and propose potential therapeutic strategy for HCC based on the current success of immunotherapy.
1 Introduction
Obesity, usually caused by an imbalance between energy intake and expenditure, was defined as an epidemic by WHO in 1997 and is widely prevalent both in developed and developing countries (1). Based on an abnormal metabolic environment, obesity results in altered immune functions, initially in the form of low-grade chronic systemic inflammation, and can develop into immune dysfunction (1, 2). Obesity is an independent risk factor for many types of cancer, and predominantly in liver and pancreatic cancer (3). Liver cancer is the fifth most commonly occurring cancer in the world and the third leading cause of mortality, and hepatocellular carcinoma (HCC) accounts for 80% of liver cancers (4). In a cohort study including 900,000 US people, male with a BMI ≥35 kg/m2 had 4.5 times the lethal risk of liver cancer than those with a normal BMI (≤25 kg/m2, 3). In addition, findings of another study, which consisted of 7 million participants indicated that even every 5 kg/m2 increase in BMI upregulated the risk rate of liver cancer by 24%, underscoring the potential association between obesity and HCC development (5).
Generally speaking, obesity contributes to the initiation of HCC in three ways. 1) Excessive fat deposition in adipose tissue can lead to upregulation of pro-inflammatory adipokines and downregulation of anti-inflammatory adipokines, thereby resulting in chronic inflammation. Chronic and mild inflammation can induce local or systemic insulin resistance (IR), which in turn induces high levels of insulin and IGF-1, thus stimulating abnormal hepatocyte proliferation (6, 7). 2) Chronic inflammation and hepatic lipid infiltration can cause cellular damage and oxidative stress in hepatocytes through multiple mechanisms, and chronic oxidative stress promotes DNA damage and compensatory repair processes, thus facilitating gene mutation and oncogenesis (8). 3) Obesity is directly related to the occurrence of non-alcohol fatty liver disease (NAFLD), which will progress into non-alcohol steatohepatitis (NASH), hepatic fibrosis or even hepatic cirrhosis if the symptom is not relieved (9). Hence, chronic liver injury leads to a cycle of cell death-repair-fibrosis, in which pre-HCC cells undergo malignant transformation and lead to tumorigenesis (10). These predisposing factors can occur both individually or simultaneously during obesity.
Inflammatory HCC, usually in the early stage, characterized by infiltration of CD8+ T cells as well as M1 macrophages, implies strong antitumor immunity, and HCC with enhanced immune response exhibits better survival (11). Conversely, non-inflammatory HCC, characterized by infiltration of Treg cells and M2 macrophages, is more prone to TP53 mutations and thus recruitment of immunosuppressive cells (11). Considering that the liver is rich in immune cell infiltration which usually indicates an effective antitumor effect, shifting from early inflammation to lately immunosuppressive environment in obesity-related HCC is critical for HCC progression (12). Since the function of immune cells largely depends on their metabolic pattern, which can be easily disrupted by metabolic dysfunction, revealing the possible impacts of obesity-induced lipid disorder on immune function is meaningful (13). Hence, this review focuses on the crucial role of obesity in HCC initiation, tumor immune microenvironment (TIME) formation, immunosuppression, and immunotherapy during HCC progression, and aims to provide potential immunotherapeutic targets and treatment strategies.
2 Obesity remodels TIME
TIME is essential for both HCC development and immunosuppression (14). The close proximity of the liver to visceral adipose tissue facilitates the transportation of the adipose metabolites to liver via the portal vein and lymphatic vessels, and thus the HCC TIME can be easily influenced by adipose tissue (15, 16). As the neovascular system is unable to keep pace with the rapid adipose expansion and results in reduced adipose tissue blood flow, adipocytes can rapidly reach the limit of oxygen diffusion, triggering adipose tissue hypoxia, which is an early determinant of adipose tissue dysfunction and may lay foundation for local hypoxia in liver via disturbing hepatic oxygen gradients (17–19). Correspondingly, high-fat diet increases hypoxia-inducible factor 1α (HIF-1α) and vascular endothelial growth factor (VEGF) expression, thus leading to increased lactate production and lactate accumulation in adipocytes (20). Excessive fat accumulation inhibits lactate dehydrogenase β (LDHβ), thereby blocking the conversion of lactate to pyruvate and contributing to lactate accumulation in adipose tissue (21). Adipocyte lactate production is often accompanied by glucose consumption, which is further promoted under conditions of obesity-induced increase in HCC glycolysis. Although obesity usually implies excessive calorie intake, carbohydrates tend to be taken up by adipocytes and HCC cells for glycolysis, leading to nutrient limitation in TIME (22–24). As a result, there is a lack of sufficient nutrients in TIME to support rapid proliferation of immune cells for antitumor immunity. Glucose deprivation can also induce T cell exhaustion and immune escape (25). For instance, low glucose environment facilitates the conversion of effector T cells to Treg cells through inducing forkhead box P3 (FOXP3) expression and activation (26). The function of NK and dendritic cells is markedly impaired by low glucose, and reduced HIF-1α-mediated secretion of pro-inflammatory cytokines is responsible for it (27). In addition, lactate inhibits T cell functions by suppressing T cell proliferation as well as interferon γ (IFN-γ) production (28). Moreover, lactate uptake is not necessary for peripheral Tregs but found to be required for intratumor Tregs for its role in metabolic support (29). A recent study has shown that lactate in high glycolytic environment upregulated programmed cell death protein 1 (PD-1) levels in Tregs, and anti-PD-1 therapy reactivates Tregs, thus leading to therapy failure (30). In addition, lactate drives M2 polarization in TIME via the mitochondrial pyruvate metabolism (31).
Thus, obesity involves in remodeling immunosuppressive tumor microenvironment by modulating adipose tissue metabolism. Under these conditions, liver-infiltrating immune effector cells exhibit a restricted proliferative capacity and tend to be immunosuppressive due to metabolic adaptations.
3 Obesity interrupts adipokine balance and facilitates HCC initiation
Adipokines are a group of cytokines secreted by the adipose tissue, and exhibit pleiotropic effects on the metabolism, signal transduction and inflammatory pathways. HCC initiation could be primarily attributed to the adipokine imbalance, which is directly related to the systemic inflammation, metabolic disorder and immune dysfunction (Table 1).
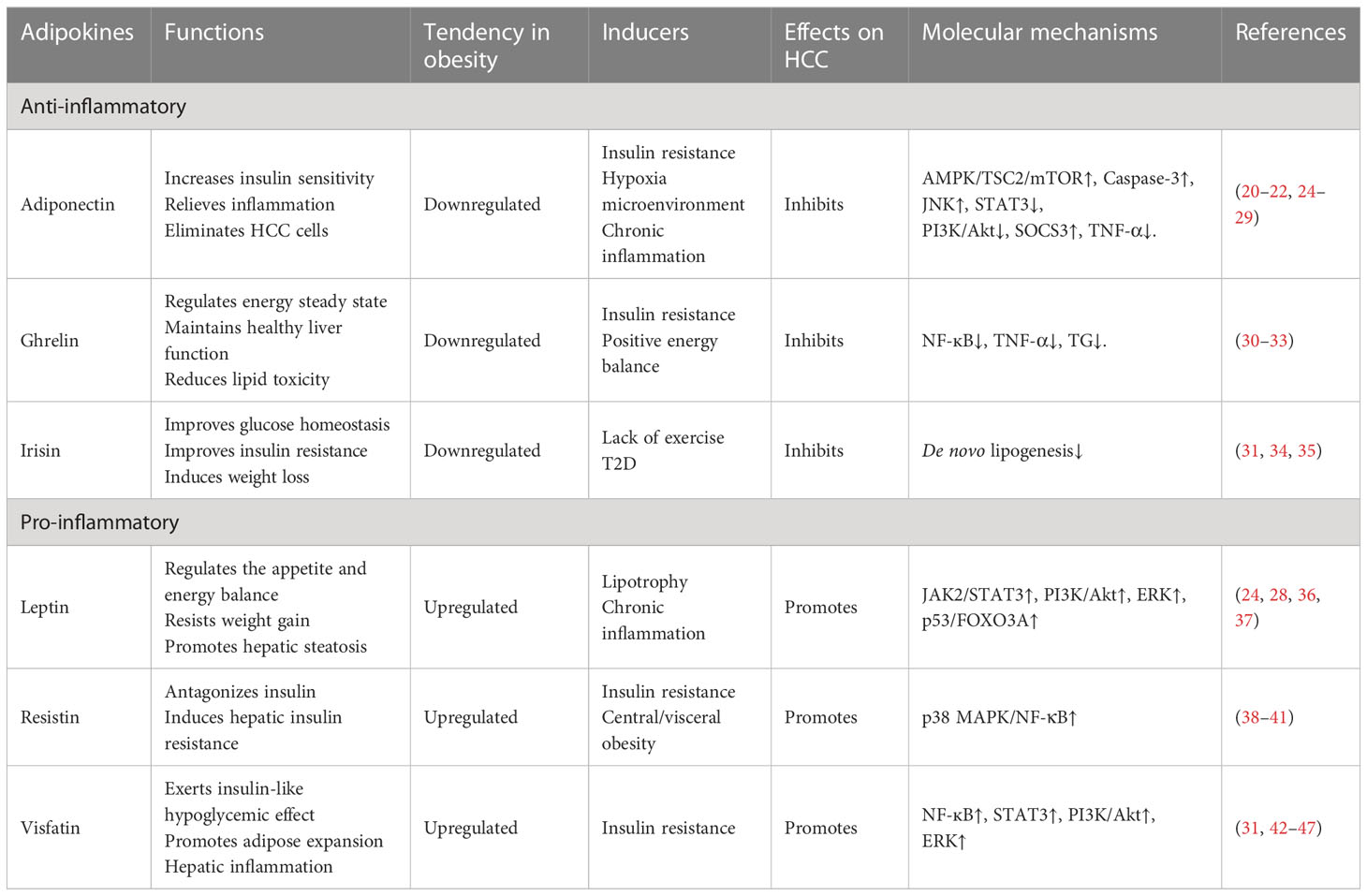
Table 1 Mechanisms responsible for obesity-induced adipokines dysfunction and their roles in obesity-related HCC.
3.1 Anti-inflammatory adipokines
3.1.1 Adiponectin
Adiponectin is primarily produced in the adipose tissue of lean subjects, and its secretion is usually inversely related to BMI in obesity (32). Mechanistically, obesity induces IR and thus inhibits adiponectin secretion through PI3K/FoxO1 pathway (33). In addition, lipid deposition in adipose tissue induces a hypoxic microenvironment, which inhibits adiponectin transcription through HIF-1α pathway (34). Chronic inflammation caused by obesity leads to increased secretion of tumor necrosis factor-α (TNF-α), IL-6, IL-18, and other Th1 cytokines that can also inhibit adiponectin (35). As an anti-inflammatory adipokine, adiponectin levels are positively related to Th2 cytokines such as IL-10 (48). Adiponectin suppresses the HCC progression in vivo by inhibiting cell proliferation and inducing cell apoptosis, and antagonizes carcinogenic effects of leptin (36). In addition, adiponectin activates tuberous sclerosis complex 2 (TSC2) protein through receptor-mediated phosphorylation of 5’- adenosine monophosphate-activated protein kinase (AMPK), thus attenuating the phosphorylation of mammalian target of rapamycin (mTOR), and directly protecting against HCC (37, 49). Adiponectin also eliminates HCC cells by activating caspase-3 and increasing the phosphorylation of c-Jun N-terminal kinase (JNK) (50). Moreover, adiponectin regulates ceramide metabolism, promotes insulin sensitivity, reduces inflammation, supports hepatocyte survival and inhibits tumor growth (51). Adiponectin attenuates leptin-induced signal transducer and activator of transcription 3 (STAT3) and protein kinase B (Akt) activation by upregulating suppressor of cytokine signaling 3 (SOCS3), a physiological negative regulator of leptin signal transduction, thus inhibiting leptin-induced hepatoma cell proliferation (38). However, low adiponectin levels restrict inhibitory effect of TNF-α on tumor cell proliferation by inhibiting the production of TNF-α in macrophages (49). Above all, adiponectin plays a vital role in maintaining homeostasis and its downregulation predicts elevated risk of obesity-related HCC.
3.1.2 Ghrelin
Ghrelin is upregulated in under-nourished states, such as anorexia nervosa, and is downregulated in conditions linked with positive energy balance, such as obesity (39). Obesity-induced IR is associated with decreased ghrelin levels (40). Sufficient serum ghrelin levels play vital roles in maintaining healthy liver function. Ghrelin induces immunosuppression via facilitating M2 and Treg phenotypes (41). Thus, ghrelin decreases the levels of Th1 cytokines including TNF-α, IFN-γ, IL-1β, IL-6 and increases Th2 cytokines including IL-10, IL-4, transforming growth factor (TGF-β), thereby inhibiting hepatic inflammation and HCC initiation (41). In mouse models, ghrelin inhibits HCC progression by reducing TG content and cytokines such as TNF-α as well as IL-6, and alleviates lipotoxicity by stimulating autophagy and inhibiting nuclear factor-κB (NF-ĸB) pathway (40). Notably, circulating ghrelin levels in obese people were found not to be reduced by a meal (52).
3.1.3 Irisin
Irisin was initially regarded as a hormone secreted by skeletal muscles in response to the tremor and movement stimuli, and further studies revealed that irisin was also present in other healthy tissues (42). It plays a critical role in the metabolism regulation, improves glucose homeostasis, IR and induces weight loss (40). Thus, irisin exhibits a positive effect on obesity, hyperlipidemia and hyperglycemia caused by the metabolism dysfunction. In addition, irisin acts as an important negative regulator of cancer and inhibits the proliferation, migration and invasion of cancer cells (43). In HCC cells, fibronectin type III domain-containing protein 5 (FNDC5), a precursor of irisin, was found to regulate gene expressions involved in lipogenesis, tumorigenesis as well as inflammation, and increased irisin levels might restrict HCC development via inhibiting de novo lipogenesis (42). Importantly, irisin levels have been shown to decrease in more advanced HCC (44). Moreover, circulating irisin levels have been observed to increase in individuals who engage in exercise-inducing activities, but they gradually decrease in those who are sedentary and lack exercise, suggesting that exercise could be a promising prevention for obesity-related HCC (45). However, irisin secretion is significantly hampered in obesity and Type 2 diabetes mellitus (T2D), explaining underlying cause for increased risk of obesity-related HCC (53).
3.2 Pro-inflammatory adipocytokines
3.2.1 Leptin
Leptin is a multifunctional adipokine that regulates appetite and energy balance (46). Although leptin is supposed to resist the weight gain, obesity usually leads to increased secretion of leptin (47). Inflammatory activation increases leptin synthesis and release (54). In turn, leptin contributes to IR, hepatic steatosis, and fibrosis, thus playing a vital role in regulating immune responses, glucose homeostasis, and angiogenesis (55). Studies have reported that leptin is a key regulator of HCC development and ectopic serum leptin levels are regarded as a hallmark of metabolic disorders leading to HCC (36). Leptin promotes the proliferation and inhibits the apoptosis of hepatoma cells (51). Moreover, it can enhance mitosis, invasion, and metastatic potential of HCC cells by activating the JAK2/STAT3, PI3K/Akt, and ERK pathways (36). Besides, leptin induces autophagy through affecting the p53/FOXO3A axis, thereby eliminating apoptosis (56). Interestingly, studies have also shown that leptin-leptin receptor (OBR) is involved in the angiogenesis of HCC, thus facilitating the progression of NASH to HCC (57). Moreover, leptin acts on monocytes/macrophages by inducing the synthesis of eicosanoid, NO, and several Th1 cytokines (58). In addition, leptin induces neutrophil chemotaxis and stimulates the release of oxygen free radicals as part of the immune response and host defense mechanisms. A recent study reported that inhibition of ATX-LPA-Lpar2-p38-leptin axis in the mouse HCC model can inhibit tumor growth (59). Thus, obesity, characterized by hyperleptinemia and central leptin resistance, directly increases the risk of HCC.
3.2.2 Resistin
Resistin is a pro-inflammatory adipokine mediating hepatic insulin resistance, and named for its role in antagonizing insulin (60). Resistin has been associated with the progression, angiogenesis, chemical resistance and increased risk of metastasis in various cancer models (61). Activation of p38 MAPK/NF-κB signaling pathway is responsible for resistin-mediated declined HCC cell adhesion and thus metastasis (62). In addition, meta-analysis results have suggested that high resistin levels are related to an increased risk of obesity-related cancer (63). Since serum resistin levels were positively correlated to the central/visceral obesity (but not BMI) and IR, resistin may be involved in driving obesity-related HCC, though further investigations are needed (64).
3.2.3 Visfatin
Visfatin is a cytokine mainly secreted by the visceral adipose tissue and functions both as an extracellular factor and an intracellular enzyme (65). It exerts insulin-like hypoglycemic effect through binding to the insulin receptor and promotes adipose tissue differentiation and proliferation (66). Visfatin is upregulated by cytokines which promote IR such as TNF-α and IL-6, and thus elevated in T2D and IR (40, 67). Increased visfatin levels in turn induce IR and hepatic inflammation via NF-ĸB and STAT3 pathway (68). Interestingly, visfatin levels were also associated with the severity of hepatic steatosis and fibrosis, thus facilitating HCC progression. It has been reported that visfatin increases miR-21 expression to promote HCC migration (69). Moreover, visfatin also plays a crucial role in hepatocyte proliferation (70). Recent studies have also shown that enhanced invasion in liver cancer cells caused by visfatin could be attributed to PI3K/Akt and ERK signaling cascades (71). Hence, obesity results in upregulated serum visfatin levels and thus potentially increases the risk of HCC (72).
4 Obesity induces dysfunction of immune effector cells
Immune effector cells engaged in anti-cancer immunity primarily include CD4/8+ T cells, macrophages, B cells and nature killer (NK) cells. However, these cells undergo function loss and numeric decrease owning to the metabolic dysfunction and lipotoxicity (Table 2).

Table 2 Mechanisms responsible for altered functions of immune cells in obesity-related HCC.
4.1 CD8+ T cells
CD8+ T cells are the major component against tumor progression among anti-tumor immunity system. Once activated, the increased requirement for biomass and energy results in metabolic shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, and thus robust proliferation (73). Since obesity is usually accompanied by IR and thus increased levels of blood glucose, this fills the need for glucose and thus facilitates glycolysis in CD8+ T cells (74). Moreover, increased leptin levels also promote glycolysis in CD8+ T cells via PI3K/Akt/mTOR pathway, which partly accounts for obesity-induced chronic inflammation (75). The metabolic shift reduces reliance of CD8+ T cells on oxygen for energy acquisition and thus renders them to retain their immunological function even after migrating into environments with poor oxygen, such as hypoxic TIME within HCC. Importantly, the reliance on glycolysis causes CD8+ T cells become more sensitive to glucose deprivation, which leads to decreased production of IFN-γ, granzyme B and perforin in CD8+ T cells (109). Moreover, increased leptin levels and availability to fatty acid (FA) also results in robust STAT3 signaling, which in turn promotes fatty acid β oxidation (FAO) and inhibits glycolysis due to glucose deprivation in TIME, leading to disability of CD8+ T cells to restrict tumor proliferation (110). Hence, obesity remodels TIME and promotes migration of CD8+ T cells into TIME partly via metabolic shift, thus leading to suppressed immunity. Many studies have indicated that obesity induces harmful effects on T cell compartment, including decreased frequency of T cell progenitors in the thymus, thymic involution as well as restricted TCR diversity, and results in increased infiltration of PD-1+ exhausted CD8+ T cells in adipose tissue and liver (76–78). In mice with HFD, CD8+ T cells exhibited downregulated expression of Ki-67, inducible co-stimulator (ICOS) and granzyme B when compared to mice with chow diet, and altered FA partitioning could be responsible for the reduced function of CD8+ T cells (111). The function of CD8+ T cells could be also impaired by deposition of unsaturated fatty acids (such as cholesterol) via lipid peroxidation (112). Moreover, a recent study has also reported that caloric restriction delayed immune senescence and increased levels of hepatic CD4+ and CD8+ T cells in obesity-related HCC via shaping gut microbiome (113).
4.2 CD4+ T cells
Many prior studies have reported that CD4+ T cells inhibit HCC initiation and restrain tumor progression (79, 114). Obesity usually increases circulating CD4+ T cell levels via leptin-mediated upregulation of major histocompatibility complexes-II (MHC-II, 80). However, CD4+ T cells derived from obese mice displayed inhibited proliferation and cytokine production when stimulated ex vivo, and same results were obtained in humans (78). This may be attributed to increased expression of exhaustion markers such as PD-1, LAG-3 and Tim-3. CD4+ T cells possess greater mitochondrial mass compared to CD8+ T cells and thus produce more mitochondria-derived ROS, which meant that CD4+ T cells could be easily impaired by mitochondrial dysfunction (79, 81). Hence, increased levels of FA especially linoleic acid, can cause mitochondrial oxidative stress and mediate selective loss of CD4+ T cells in the liver, thus inducing liver carcinogenesis (81). Besides, increased availability to FA also activates peroxisome proliferator-activated receptor-α (PPAR-α) pathway and leads to carnitine palmitoyltransferase upregulation, which in turn facilitates influx of FA into mitochondria and leads to greater apoptosis of CD4+ T cells via ROS burst (82). Moreover, reduced tumor infiltration of CD4+ T cells caused by ROS burst might also impair immunotherapy targeting liver tumors, suggesting that overcoming FA-induced impairment could be a potential strategy for obesity-related HCC (115).
4.3 B cells
During obesity, activation of B cells is directly related to systemic inflammation via antigen presentation and pro-inflammatory cytokine secretion (116). Increasing evidence indicates that intrahepatic B cells play key roles in NAFLD progression (83). In several experimental models and patients, NASH is characterized by B cell infiltration, suggesting that obesity may facilitate B cell activation and thus exacerbate NAFLD or NASH, which is important risk factor for obesity-related HCC (84, 85). However, obesity also impairs B cell biology. Mice with HFD exhibit decreased frequency of B cells in bone marrow (86). Mechanistically, adipocytes secrete soluble factors and facilitate MDSCs development, thereby inhibiting B lymphopoiesis (117). Although B cells from obese subjects secrete more pro-inflammatory cytokines, they also exhibit higher senescence-associated secretory phenotype (SASP) markers (87, 88). Moreover, the persistent high inflammation in obesity restrains proper regulation of B cell responses to novel antigens, such as cancer cell markers (89). Increased inflammation and persistent immune activation induced by obesity also accelerate age defects in B cells (88). Importantly, infiltration of B cells positively correlates with HCC progression in chronic liver injury, and B cells from mice fed with HFD disrupted antitumor immunity in NASH-driven HCC (84, 90). The possible reason is that fatty acids caused by enhanced lipolysis promote survival of regulatory B cells (Bregs), which could accelerate HCC progression (91, 92). However, it doesn’t explain the reduction of Bregs in obesity, and further studies about the roles of B cells in obesity-related HCC are needed (118).
4.4 NK cells
Circulating NK cells in obese populations display increased CD69 expression, which indicates chronic activation, ultimately leading to diminished NK cell cytotoxic activity (119). Liver infiltrated-NK cells during obesity also tend to possess less cytotoxic ILC-1 phenotype and may explain increased risk of HCC in obese populations (120). Dominik Pfister et al. indicated that thirty percent of mice with choline-deficient high-fat diet developed HCC with similar genetic alterations of human NAFLD-HCC, but anti-PD-1 therapy failed to regress tumor burden in spite of increased infiltration of immune effector cells, and this effect could be attributed to impaired function of CD8+ and NK cells (121). Lipidomic profiling revealed the accumulation of long-chain acylcarnitines (LCACs) and aberrant lipid metabolism in HCC tissues, which finally led to senescence of invariant natural killer T cells (iNKT) in TIME (93). Similarly, lipid accumulation in NK cells cause metabolic shift based on lipid-rich environment in obesity, thus hampering NK cell functions (94). Increased uptake of FA and cholesterol might be also responsible for impairment of NK cells in HCC and blocking transport of lipids to mitochondria could effectively restore NK cell cytotoxicity and restrain tumor growth (95). However, the role of cholesterol could be controversial since a prior study has also reported that cholesterol accumulation in NK cells stimulates effector functions and thus inhibits HCC progression (96).
5 Obesity enhances function of immunosuppressive cells
In contrast of immune effector cells, immunosuppressive cells including myeloid-derived suppressive cells (MDSCs), tumor-associated macrophages (TAMs) and regulatory T cells (Tregs) are able to adjust to lipid-rich environment as well as TIME, and thereby can facilitate immunosuppression. FAO plays a vital role in this process.
5.1 MDSCs
MDSCs are a highly heterogeneous group of immature myeloid immune cells, which include the naïve macrophages, granulocytes and dendritic cells (122). Inflammatory-cell cycle-related kinase (CCRK) circuitry under obese circumstance drives immunosuppressive metabolic reprogramming and enhances polymorphonuclear MDSCs recruitment as well as tumorigenicity, thereby facilitating obesity-related HCC progression (123). Although lipid deposition leads to metabolic dysfunction in immune effector cells and thus immunosuppression, FA availability in TIME enhances the function of MDSC. Enhanced FAO facilitates infiltration of MDSCs via secreting complement C3, whereas suppression of FAO results in MDSCs inhibition (124). Moreover, in lipid-rich environment, MDSCs can reprogram their metabolism and transfer from aerobic glycolysis to FAO to reduce their reliance on glucose and thereby increase the utilization of FA, thus meeting high energy demand and sustaining suppressive function in TIME, whereas genetic depletion of CD36 can hamper lipid uptake and inhibit MDSCs (97–99). Obesity promotes hepatic inositol requiring enzyme 1 α (IRE1α) activation and X-box binding protein 1 (XBP1)-drived production of cholesterol, which in turn elicits immunosuppression by enhancing the functions of MDSCs (100). In addition, obesity elevates CXCL1 levels in TIME and promotes the infiltration of MDSCs in tumor sites, thereby inducing cell death of CD8+ T cells (125). However, increased cholesterol accumulation under oxidative stress status also stimulates the generation of the different cholesterol derivatives. These cholesterol derivatives, especially oxysterols, could activate liver X receptor (LXR)-ApoE axis and block MDSCs survival and abundance (126). Hence, caution is needed if MDSCs metabolism is targeted for therapeutic purposes.
5.2 TAMs
Generally, TAMs are mixed intratumoral macrophage populations with different phenotypes and are generally categorized into M1/M2 macrophages, though more precise classification is needed (127). Many studies have indicated that glucose transporter 1 (GLUT1) is upregulated in TAMs and leads to increased glycolysis (101, 102). It is not surprising that hyperglycemia in obesity facilitates glycolysis in TAMs and results in immunosuppression via inducing chemokine and programmed cell death protein ligand 1 (PD-L1) expression (103). However, glucose may be also scarce in TIME as highlighted above, suggesting that glycolysis in TAMs may be restrained. Under this condition, obesity-induced M2-like TAMs are largely dependent on increased availability to FA. Lipid droplets originated from FA are essential organelles in TAMs for FA metabolism and sustain immunosuppressive phenotype (104). FAO is critical for maintaining the function of TAMs since its inhibition can impair IL-4-induced M2 activation (105). Enhanced FAO can also promote mitochondrial OXPHOS and ROS production, leading to STAT6 activation and transactivation of genes related to TAM generation and function (106). However, a recent study based on HCC model indicated that decreased FAO caused by fatty acid binding protein 5 (FABP5) leads to accumulation of lipids in macrophages and fosters immune tolerance, suggesting that FABP5 may act as a potential biomarker of TAMs (128). Importantly, increased lipid deposition also facilitates ROS production via lipid peroxidation, thus in turn promoting FAO via ROS-Caspase1-PPAR pathway and leading to liver carcinogenesis (129). Hence, declined FAO may potentially serve as a bad marker of M1 macrophages since it provides signaling molecules and sufficient lipids for TAM initiation. A recent study highlighted the vital role of GSK3β in TAMs, since GSK3β deficiency in macrophage can restrict HCC progression by inhibiting M2 phenotype and enhancing the sensitivity of anti-PD-1 immunotherapy (130). Overall, considering that obesity is usually accompanied by increased serum insulin levels, which are known to activate GSK3β, there is still a possibility that obesity can facilitate M2-like phenotype and thereby promote HCC progression in certain circumstances.
5.3 Tregs
Tregs are a common obstacle against anti-tumor therapies. While Tregs are usually enriched in lean visceral adipose tissue, obese populations exhibit diminished frequency of Tregs (131). However, intratumoral Tregs can be distinct with peripheral Tregs. Tregs show a tendency towards FAO and exhibit resistance to lipotoxicity (107). In the obese, intratumoral Tregs can upregulate their surface FA transporter CD36 to obtain sufficient biomass and adapt to the lipid-rich environment (108). Increased FA uptake can then induce PPAR-dependent lipid metabolism and thus enhance Tregs viability as well as function. Moreover, a recent study indicated that neutrophil extracellular traps are abundant in livers with steatohepatitis and promote Treg differentiation through metabolic reprogramming, thereby affecting the balance of Th17/Treg cells and contributing to liver carcinogenesis (132). FOXP3 reprograms T cell metabolism to enhance OXPHOS and inhibit glycolysis, thus rendering Tregs advantageous in obesity-induced TIME (133). Insulin signaling plays a dual role in regulating the functions of Tregs. For example, although insulin was found to drive Tregs in a HIF-1α–dependent manner, obesity-induced hyperinsulinemia can impair the ability of Tregs to secrete IL-10 (134, 135). However, increased levels of IL-6, IL-1β and TGF-β can impair T cells proliferation and induce their polarization to Tregs.
6 Obesity affects cytokine secretion to facilitate HCC progression
Cytokines are critical for obesity-related pathology. However, most of them play dual roles in regulating inflammation and HCC. We here introduce four distinct cytokines including TNF-α, interleukins, TGF-β and chemokines. All of them are secreted abnormally in obesity due to dysfunction of adipose and immune cells, thus facilitating inflammation in early stage but also promoting HCC progression via inducing immunosuppression.
6.1 TNF-α
The elevated serum TNF-α levels are generally attributed to increased secretion by macrophages as well as immune cells (136). In obesity, excessive lipid deposition in adipocytes leads to increased cell apoptosis and causes increased accumulation of macrophages around dying adipocytes, regarded as crown-like structure, thus secreting TNF-α (137). TNF-α activates JNK pathway to impair insulin signaling and interacts with NF-κB to upregulate genes involved in regulating apoptosis, proliferation, inflammation, angiogenesis. Hence, increased TNF-α secretion is associated with inflammation and increased risk of HCC (137). Although increased inflammatory environment may cause damage to HCC cells, leptin has been shown to counteract toxicity exerted by TNF-α (138). Importantly, TNF-α can also promote lipolysis in adipocytes and facilitate release of FA, which plays a key role in development of lipid disorder and obesity-induced immunosuppression (139). However, TNF-α levels may be decreased in TIME due to impaired function of CD8+ T cells and macrophages.
6.2 Interleukins
Interleukins associated with tumorigenesis belong to an extremely large family which is still increasing as the number of papers exploring the roles of new members are being published. Among them, IL-6 and IL-1β are crucial for obesity-related HCC. Like TNF-α, IL-6 is related to the progression from steatosis to HCC, and its increased levels in obesity is related to several ways (140, 141). IL-6 facilitates cell proliferation and differentiation via promoting STAT3 activation, which is crucial for HCC development (142). Moreover, a recent study has reported that adipocyte-derived IL-6 sensitized macrophages to IL-4 signaling and thus facilitated M2 phenotype (143). Besides, IL-6 can also increase infiltration of Tregs in TIME and thus facilitate HCC progression, which could be suppressed by cystathionine β-synthase-mediated STAT3 inhibition (144). IL-1β is also associated with increased HCC risk and poor prognosis (145). Increased IL-1β levels are attributed to NF-κB activation and NOD-like receptor family, pyrin domain containing 3 (NLRP3) activity, which in turn could be stimulated in obesity by several factor such as FA, cholesterol, ROS and hyperglycemia (146). Although known as inflammatory factor, IL-1β can also lead to immunosuppression and IL-1β deficiency results in tumor regression (147). Blocking IL-1β enhanced effector immune cells and inhibited immunosuppressive cells, thus facilitating checkpoint inhibition therapy.
6.3 TGF-β
Increased TGF-β secretion can originate from high number of adipose-derived stem cells, which has been associated with adipose tissue expansion (148, 149). In obesity, TGF-β is an underlying contributor to IR via inducing cell hypertrophy and reducing the functions of islet β cells (150, 151). Besides, a recent study has also highlighted that TGF-β signaling in hepatocyte can inhibit white adipose tissue browning and thus facilitate obesity and NAFLD (152). In HCC cells, increased TGF-β levels induce epithelial-mesenchymal transition and reprogram lipid metabolism, thus promoting adaption of HCC to lipid-rich environment (153). In addition, TGF-β signaling works as a key regulator in immune cell differentiation, proliferation as well as survival and thus contributes to NAFLD and NAFLD-HCC (154). TGF-β can also induce immunosuppression through distinct mechanisms. For instance, in CD8+ T cells, enhanced TGF-β signaling can suppress IFN-γ expression to inhibit its cytotoxicity effects (155). TGF-β can suppress NK cells and thus inhibit the recruitment of immune effector cells to tumor. In addition, TGF-β drives M2 phenotype and Treg phenotype, leading to immunosuppression in HCC (156). Acid metabolites like lactate in TIME could also enhance TGF-β and its downstream signaling in Tregs to promote tumorigenesis (157). Importantly, TGF-β promotes PD-1 expression in antigen-specific T cells via SMAD3 activation, suggesting that increased levels of TGF-β in obesity could be a potential biomarker for anti-PD-1 therapy (158).
6.4 Chemokines
Chemokines can induce leukocyte chemotaxis and thus affect tumor behavior via immune regulation. CCL2 is mainly secreted by preadipocytes and adipocytes during obesity (159, 160). Increased CCL2 levels can lead to macrophage differentiation and accumulation in adipose tissue, thus promoting inflammation and inducing IR as well as hepatic steatosis. However, elevated CCL2 expression has been linked to loss of Kupffer cells and can cause increase of immature macrophages in liver, thus contributing to immunosuppressive microenvironment in liver cancer (161). Similarly, a recent study has also shown that CCL2 activation could also promote TAMs recruitment in TIME and facilitate HCC progression (162). CXCL-8 (also known as IL-8), another chemokine known to increase with the development of hepatic steatosis, is also involved in immunosuppression via MDSCs and TAMs recruitment, and contributes to the formation of TIME (163). Moreover, CCL22 has been reported to be induced by IL-6 and TNF-α, thereby stimulating Treg chemotaxis and accumulation (164). A recent study also indicated that excess serum lipid levels downregulate CXCR3 expression which promotes transfer of T cells into the tumor sites, thereby resulting in decrease of T cell infiltration in tumors (165).
7 Obesity impacts immunotherapy for HCC
Systemic therapy such as sorafenib is used as first-line therapy for HCC. In recent years, several immunotherapy regimens including immune checkpoint inhibitors (ICIs) and adoptive cell therapy have shown strong anti-tumor effects for HCC. Despite of the prevalence of obesity-related HCC, there are few studies that have focused on the potential impacts of obesity on immunotherapy of HCC.
7.1 ICIs therapy
Obesity appears to play a positive role in immunotherapy (mainly including ICIs therapy and adoptive cell therapy) of various cancers. ICIs therapy (such as anti-PD-1 and anti-CTLA4 antibodies) works by targeting depressed immune effector cells and enhancing their function, resulting in robust immune activation. In patients with NSCLC, high BMI was found to be independently associated with a better prognosis after atezolizumab treatment (166). A multicenter study also indicated that overweight predicted a better prognosis in a variety of cancers after ICIs therapy (167). Anti-PD-L1 & anti-angiogenic immunotherapy (atezolizumab in combination with bevacizumab) is currently the first-line treatment for advanced HCC. A subgroup analysis of survival outcomes based on the trials evaluating the efficacy of ICIs as first-line therapy showed that viral-HCC could benefit from ICIs (168). However, for non-viral-HCC patients, ICIs were not observed to be superior to sorafenib, which suggested that obesity may be not a positive marker for ICI therapy in HCC. However, all current clinical trials were not able to distinguish the subgroups of nonviral-HCC, thus implying that they also included cases of HCC related to NASH, obesity, alcohol-related, autoimmune hepatitis etc. Hence, the detailed classification of clinical cases of nonviral-HCC is necessary. In fact, a recent case with obesity and T2D showed that the efficacy of the regimen of atezolizumab combined with bevacizumab remained guaranteed and could overcome nivolumab tolerance (169). Notably, high BMI was related to the better prognosis upon anti-PD-1 therapy (including nivolumab, pembrolizumab, sintilimab, and toripalimab, 170). Patients in advanced HCC with BMI <25 had a worse median OS when treated with anti-PD-1 therapy in comparison to those with BMI ≥25 (171). This may be because, compared to healthy individuals, obesity predicts higher T-cell PD-1 expression, which may be associated with upregulated leptin (78). Thus, while obesity-induced T-cell PD-1 upregulation may predict systemic immunosuppression, this might imply greater sensitivity to anti-PD-1 therapy. However, a recent study highlighted that GSK3β activation in macrophage restricted anti-PD-1 immunotherapy in HCC (130). Since obesity is usually accompanied by hyperinsulinemia, obesity could also serve as a poor marker for anti-PD1 immunotherapy. Of note, obesity appears to increase the risk of side effects after ICIs therapy. Since ICIs enhance body’s immune system, they can often lead to autoimmune reactions. A recent study, which included 13,480 cases showed that obesity predicted a higher risk of colitis (172). Another study also revealed that obesity was related to an increased risk of immune-related adverse effects (irAEs) after nivolumab treatment (173). For patients treated with pembrolizumab, elevated BMI was associated with an increased risk of irAEs (174). However, irAEs can also predict a stronger immune response. In another study, Jacobo Rogado et al. reported that in patients with advanced cancer treated with single-agent anti-PD-1 antibody nivolumab or pembrolizumab immunotherapy, overweight was associated with greater outcomes, and the observed treatment benefit was significantly enhanced when irAEs were present in the overweight population (175). Importantly, due to the limitation of BMI in descripting the detailed information related to obesity, people should be more concerned about the impact of body composition on immunotherapy. For example, people with myosteatosis caused by excessive lipid accumulation in the muscle are associated with higher risk of failure after immunotherapy as well as a poor prognosis in HCC (176). Likewise, myosteatosis can predict strong toxicity of nivolumab.
7.2 Adoptive cell therapy
Adoptive cell therapy works by modifying immune effector cells (such as T cells) to express specific immune effector marker (such as chimeric antigen receptor), resulting in enhanced anti-tumor immunity in patients. Currently, studies about the effects of obesity on adoptive cell therapy for HCC are lacking. Wenshu Tang et al. observed that obesity-induced hypercholesterolemia can effectively impair NK cell function, whereas Wenhao Qin et al. indicated that high cholesterol increased NK cell lipid accumulation thereby enhancing their function (95, 96). Thus, the effect of obesity on allogeneic NK cell adoptive therapy may be positive or negative. However, there is a lack of sufficient data to determine the extent of cholesterol accumulation and the duration of cholesterol accumulation, which may account for the differential effects of cholesterol on NK cells. However, a recent study suggested that NK cell therapy could display potential efficacy in obesity-related cancer (177). Recently it has been found that GPC3-CAR-T cell therapy based on the GPC3 target of HCC cells was effective in clinical HCC treatment. Manuel Garcia-Jaramillo et al. reported that western diet increased hepatic GPC3 expression in the male mice (178). In addition, IR was also associated with increased levels of hepatic GPC3 expression, suggesting that obesity may predict better response to GPC3-CAR-T cell therapy (179).
8 Conclusion
Overall, based on the general understanding, obesity-related HCC is primarily attributed to NAFLD and IR. However, there are few studies that have focused on the inhibitory effects of obesity-induced metabolic disorder on antitumor immunity. In this review, we have presented a comprehensive overview about the distinct mechanisms through which obesity facilitates HCC progression via remodeling TIME and inducing immunosuppression (Figure 1).
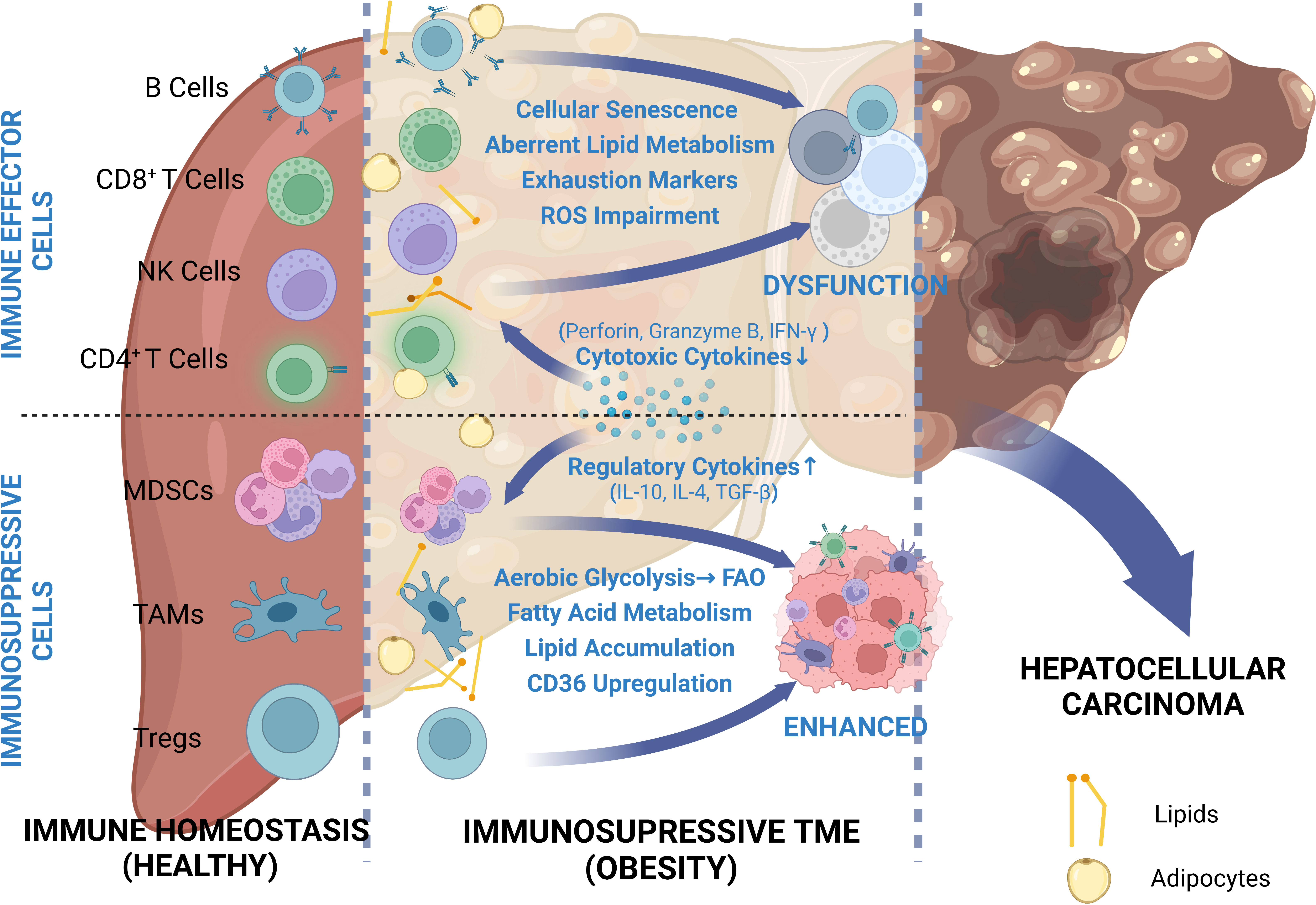
Figure 1 Comprehensive review of depressed cellular immunity in obesity-related HCC. Due to the proximity of the liver to visceral adipose tissue, the HCC TME can be easily influenced by adipose metabolites. In order to adapt to this abnormal metabolic environment, immune cells undergo metabolic shift and thus phenotypic change. While immune effector cells are depressed due to restricted glycolysis and exhausted marker expressions, immunosuppressive cells take full advantage of increased availability to lipids and exhibit enhanced functions.
Obesity-related immunosuppression is mechanistically related to the disrupted balance between adipocytes, hepatocytes, and immune cells. Excessive lipid deposition in visceral adipose tissue is the trigger for obesity-related HCC, and can directly lead to adipose tissue expansion (180). Thus, the body possess decreased anti-inflammatory adipokines levels and increased pro-inflammatory adipokines levels, thereby facilitating local or systemic inflammation, which is crucial for NAFLD-driven HCC (37). In this process, anti-inflammatory adipokines exert anti-tumor effects by inducing apoptosis or inhibiting expression of pro-tumor cytokines, whereas pro-inflammatory adipokines promote HCC progression via facilitating autophagy, angiogenesis and proliferation. Obesity-related inflammation and IR can also promote the conversion of HCC cells to the glycolytic pathway through upregulation of the different growth factors (insulin, IGF1) and ROS (179). Excessive adipose tissue expansion leads to reduced blood supply and induces a hypoxic environment, which in turn promotes adipose tissues and HCC cells glycolysis, leading to increased glucose consumption and lactate production. Thus, the excessive carbohydrate intake is mainly utilized by adipose tissue and HCC cells, sparing only a small portion for immune cells. This hypoxic, low-glucose, high-lactate environment inhibits liver-infiltrating immune effector cell function and proliferation, thus promoting the conversion of immune effector cells to immune suppressor cells. Obesity-induced inflammation not only facilitates oncogenesis via promoting a cycle of cell death-repair-fibrosis, but also leads to aging of immune effector cells such as T cells and B cells. Moreover, obesity also facilitates immune dysfunction via inducing lipid disorder. Cholesterol deposition in NK cells directly leads to their functional inhibition, thus producing less IFN-γ and expressing low levels of granzyme B as well as perforin (95). Increased FA uptake and FAO have been tightly related to enhanced immunosuppression in MDSC (99). Upregulated expression of CD36 as well as SREBP can result in augmented immunosuppressive function of Tregs, whereas lipid metabolism in TAMs was found to be involved in regulation of its immunosuppressive function (105, 108). The altered immune cell phenotype can directly lead to cytokine disruption, which in turn promotes tumor growth as well as immunosuppression.
Conventional therapy including surgery resection and systemic therapy is widely used in HCC treatment. The impact of obesity on HCC resection remains controversial. Many studies indicated that obesity has shown no effects on lethality rate after HCC resection, whereas several studies had shown that severe obesity increased lethality rate after HCC resection (181, 182). Besides, obesity appears to promote a range of complications after HCC resection (183, 184). Multi-kinase inhibitors (especially sorafenib) are widely used for the treatment of advanced HCC. The effect of obesity on sorafenib treatment has varied across clinical trials, which may be attributes to different obesity-related symptoms (185–187). In addition, body composition may affect the efficacy of sorafenib treatment. In brief, obesity upregulates overall survival rate after sorafenib when muscle mass is unaffected, whereas mortality increased when obesity leads to muscle loss, such as sarcopenia obesity (187–189). Considering that few studies have highlighted the effect of obesity on sorafenib efficacy during the past 16 years (since sorafenib was approved for HCC treatment in 2007), obesity may not be an important factor to predict responses to sorafenib. Recently, the emergency of immunotherapy suggests that people have better understanding of cancer progression. Although immunotherapy may be not significantly superior to sorafenib, it undoubtedly offers a new treatment strategy for HCC and provides an alternative treatment option when sorafenib tolerance occurs. Based on the fact that obesity can induce the onset of immunosuppression through affecting multiple pathways, the impact of obesity on immunotherapy appears to be paradoxical. Obesity predicts a better response to ICI regimens, perhaps because obesity can lead to increased levels of target marker proteins (which is also responsible for obesity-promoted HCC). Importantly, it remains unclear whether obesity is an independent factor that favors immunotherapy because most relative studies are clinical trials and mechanism studies are needed. Besides, enhanced responses to immunotherapy in obesity-related HCC may be attributed to relatively healthy state (when compared to patients with severe cancer-related disorders, such as cachexia, which is characterized by low BMI). In this circumstance, obesity may be just a marker of effective immunotherapy rather than independent factor. Since the immunosuppressive microenvironment caused by obesity undoubtedly hinders the function of immune effector cells, selective improvement of the obesity status according to application of the different immunotherapies (ICIs or adoptive cell therapy) might be a guaranteed therapeutic modality.
Above all, obesity can facilitate HCC progression via remodeling TIME and inducing immunosuppression, all of which mainly originate from the inflammatory environment but can exert pro-tumor effects once HCC has occurred. Given the tolerance to conventional therapy and the continued success of immunotherapy for HCC, more emphasis should be placed on obesity-induced immunosuppression. Investigations about obesity-related HCC progression from an immunosuppressive perspective can provide the basis for biomarker discovery and facilitate diagnostic evaluation as well as therapeutic development. However, it is unclear when the inflammatory environment might shift to immunosuppressive state, which may be an important direction for future insights into the development of obesity-related HCC.
Author contributions
JY designed and drafted the manuscript. JH and YF revised the manuscript. MX designed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research was supported by the National Natural Science Foundation of China (No.82071749).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Collaboration NCDRF. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19. 2 Million Participants Lancet (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
2. Naik A, Monjazeb AM, Decock J. The obesity paradox in cancer, tumor immunology, and immunotherapy: potential therapeutic implications in triple negative breast cancer. Front Immunol (2019) 10:1940. doi: 10.3389/fimmu.2019.01940
3. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U. S Adults N Engl J Med (2003) 348(17):1625–38. doi: 10.1056/NEJMoa021423
4. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology (2007) 132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061
5. Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer (2007) 97(7):1005–8. doi: 10.1038/sj.bjc.6603932
6. Gallagher EJ, LeRoith D. Minireview: igf, insulin, and cancer. Endocrinology (2011) 152(7):2546–51. doi: 10.1210/en.2011-0231
7. Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol (2011) 4:30. doi: 10.1186/1756-8722-4-30
8. Ioannou GN. Epidemiology and risk-stratification of nafld-associated hcc. J Hepatol (2021) 75(6):1476–84. doi: 10.1016/j.jhep.2021.08.012
9. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: from pathophysiology to therapeutics. Metabolism (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014
10. Vanni E, Bugianesi E. Obesity and liver cancer. Clin Liver Dis (2014) 18(1):191–203. doi: 10.1016/j.cld.2013.09.001
11. Montironi C, Castet F, Haber PK, Pinyol R, Torres-Martin M, Torrens L, et al. Inflamed and non-inflamed classes of hcc: a revised immunogenomic classification. Gut (2023) 72(1):129–40. doi: 10.1136/gutjnl-2021-325918
12. Wang H, Xi Z, Deng L, Pan Y, He K, Xia Q. Macrophage polarization and liver ischemia-reperfusion injury. Int J Med Sci (2021) 18(5):1104–13. doi: 10.7150/ijms.52691
13. Chapman NM, Chi H. Metabolic adaptation of lymphocytes in immunity and disease. Immunity (2022) 55(1):14–30. doi: 10.1016/j.immuni.2021.12.012
14. Foerster F, Gairing SJ, Ilyas SI, Galle PR. Emerging immunotherapy for hcc: a guide for hepatologists. Hepatology (2022) 75(6):1604–26. doi: 10.1002/hep.32447
15. Cornide-Petronio ME, Jimenez-Castro MB, Gracia-Sancho J, Peralta C. New insights into the liver-visceral adipose axis during hepatic resection and liver transplantation. Cells (2019) 8(9):1100. doi: 10.3390/cells8091100
16. Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab (2000) 11(9):351–6. doi: 10.1016/s1043-2760(00)00323-4
17. Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab (2013) 18(4):470–7. doi: 10.1016/j.cmet.2013.06.016
18. Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J (2009) 417(1):183–93. doi: 10.1042/BJ20080868
19. Lefere S, Van Steenkiste C, Verhelst X, Van Vlierberghe H, Devisscher L, Geerts A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease. Cell Mol Life Sci (2016) 73(18):3419–31. doi: 10.1007/s00018-016-2222-1
20. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers hif-1alpha, causing inflammation and insulin resistance in obesity. Cell (2014) 157(6):1339–52. doi: 10.1016/j.cell.2014.05.012
21. Wang T, Chen K, Yao W, Zheng R, He Q, Xia J, et al. Acetylation of lactate dehydrogenase b drives nafld progression by impairing lactate clearance. J Hepatol (2021) 74(5):1038–52. doi: 10.1016/j.jhep.2020.11.028
22. Speakman JR, Hall KD. Carbohydrates, insulin, and obesity. Science (2021) 372(6542):577–8. doi: 10.1126/science.aav0448
23. Paskeh MDA, Ghadyani F, Hashemi M, Abbaspour A, Zabolian A, Javanshir S, et al. Biological impact and therapeutic perspective of targeting Pi3k/Akt signaling in hepatocellular carcinoma: promises and challenges. Pharmacol Res (2023) 187:106553. doi: 10.1016/j.phrs.2022.106553
24. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer (2021) 20(1):28. doi: 10.1186/s12943-021-01316-8
25. Sukumar M, Roychoudhuri R, Restifo NP. Nutrient competition: a new axis of tumor immunosuppression. Cell (2015) 162(6):1206–8. doi: 10.1016/j.cell.2015.08.064
26. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for Cd4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
27. Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, et al. Glucose represses dendritic cell-induced T cell responses. Nat Commun (2017) 8:15620. doi: 10.1038/ncomms15620
28. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
29. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
30. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes pd-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell (2022) 40(2):201–18 e9. doi: 10.1016/j.ccell.2022.01.001
31. Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv (2021) 7(46):eabi8602. doi: 10.1126/sciadv.abi8602
32. Nri-Ezedi CA, Ulasi T, Chukwuka J, Okpara H, Ofiaeli O, Nwaneli E, et al. Serum total adiponectin in healthy pre-pubertal Nigerian school children. Niger J Clin Pract (2021) 24(6):821–7. doi: 10.4103/njcp.njcp_427_20
33. Parida S, Siddharth S, Sharma D. Adiponectin, obesity, and cancer: clash of the bigwigs in health and disease. Int J Mol Sci (2019) 20(10):2519. doi: 10.3390/ijms20102519
34. Zappala G, Rechler MM. Igfbp-3, hypoxia and tnf-alpha inhibit adiponectin transcription. Biochem Biophys Res Commun (2009) 382(4):785–9. doi: 10.1016/j.bbrc.2009.03.112
35. Liu M, Liu F. Transcriptional and post-translational regulation of adiponectin. Biochem J (2009) 425(1):41–52. doi: 10.1042/BJ20091045
36. Duan XF, Tang P, Li Q, Yu ZT. Obesity, adipokines and hepatocellular carcinoma. Int J Cancer (2013) 133(8):1776–83. doi: 10.1002/ijc.28105
37. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol (2012) 56(3):704–13. doi: 10.1016/j.jhep.2011.09.020
38. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology (2010) 52(5):1713–22. doi: 10.1002/hep.23892
39. Cui H, Lopez M, Rahmouni K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat Rev Endocrinol (2017) 13(6):338–51. doi: 10.1038/nrendo.2016.222
40. Kim H, Lee DS, An TH, Park HJ, Kim WK, Bae KH, et al. Metabolic spectrum of liver failure in type 2 diabetes and obesity: from nafld to Nash to hcc. Int J Mol Sci (2021) 22(9):4495. doi: 10.3390/ijms22094495
41. Pereira J, da Silva FC, de Moraes-Vieira PMM. The impact of ghrelin in metabolic diseases: an immune perspective. J Diabetes Res (2017) 2017:4527980. doi: 10.1155/2017/4527980
42. Pinkowska A, Podhorska-Okolow M, Dziegiel P, Nowinska K. The role of irisin in cancer disease. Cells (2021) 10(6):1479. doi: 10.3390/cells10061479
43. Waseem R, Shamsi A, Mohammad T, Hassan MI, Kazim SN, Chaudhary AA, et al. Fndc5/Irisin: physiology and pathophysiology. Molecules (2022) 27(3):1118. doi: 10.3390/molecules27031118
44. Pazgan-Simon M, Zuwala-Jagiello J, Menzyk T, Bator M, Derra A, Lekstan A, et al. Serum betatrophin and irisin levels in hepatocellular carcinoma. J Physiol Pharmacol (2020) 71(1):113–23. doi: 10.26402/jpp.2020.1.11
45. Arhire LI, Mihalache L, Covasa M. Irisin: a hope in understanding and managing obesity and metabolic syndrome. Front Endocrinol (Lausanne (2019) 10:524. doi: 10.3389/fendo.2019.00524
46. Hammond JA, Bennett KA, Walton MJ, Hall AJ. Molecular cloning and expression of leptin in Gray and harbor seal blubber, bone marrow, and lung and its potential role in marine mammal respiratory physiology. Am J Physiol Regul Integr Comp Physiol (2005) 289(2):R545–R53. doi: 10.1152/ajpregu.00203.2004
47. Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, inflammation, and cancer. Annu Rev Pathol (2016) 11:421–49. doi: 10.1146/annurev-pathol-012615-044359
48. Choi KM, Ryu OH, Lee KW, Kim HY, Seo JA, Kim SG, et al. Serum adiponectin, interleukin-10 levels and inflammatory markers in the metabolic syndrome. Diabetes Res Clin Pract (2007) 75(2):235–40. doi: 10.1016/j.diabres.2006.06.019
49. Kelesidis I, Kelesidis T, Mantzoros CS. Adiponectin and cancer: a systematic review. Br J Cancer (2006) 94(9):1221–5. doi: 10.1038/sj.bjc.6603051
50. Saxena NK, Fu PP, Nagalingam A, Wang J, Handy J, Cohen C, et al. Adiponectin modulates c-jun n-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology (2010) 139(5):1762–73, 73 e1-5. doi: 10.1053/j.gastro.2010.07.001
51. Rajesh Y, Sarkar D. Association of adipose tissue and adipokines with development of obesity-induced liver cancer. Int J Mol Sci (2021) 22(4):2163. doi: 10.3390/ijms22042163
52. English PJ, Ghatei MA, Malik IA, Bloom SR, Wilding JP. Food fails to suppress ghrelin levels in obese humans. J Clin Endocrinol Metab (2002) 87(6):2984. doi: 10.1210/jcem.87.6.8738
53. Kurdiova T, Balaz M, Vician M, Maderova D, Vlcek M, Valkovic L, et al. Effects of obesity, diabetes and exercise on Fndc5 gene expression and irisin release in human skeletal muscle and adipose tissue: In vivo and in vitro studies. J Physiol (2014) 592(5):1091–107. doi: 10.1113/jphysiol.2013.264655
54. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol (2005) 115(5):911–9. doi: 10.1016/j.jaci.2005.02.023
55. Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med (2016) 67:103–17. doi: 10.1146/annurev-med-090514-013832
56. Nepal S, Kim MJ, Hong JT, Kim SH, Sohn DH, Lee SH, et al. Autophagy induction by leptin contributes to suppression of apoptosis in cancer cells and xenograft model: involvement of P53/Foxo3a axis. Oncotarget (2015) 6(9):7166–81. doi: 10.18632/oncotarget.3347
57. Ribatti D, Belloni AS, Nico B, Di Comite M, Crivellato E, Vacca A. Leptin-leptin receptor are involved in angiogenesis in human hepatocellular carcinoma. Peptides (2008) 29(9):1596–602. doi: 10.1016/j.peptides.2008.05.011
58. Wang SN, Lee KT, Ker CG. Leptin in hepatocellular carcinoma. World J Gastroenterol (2010) 16(46):5801–9. doi: 10.3748/wjg.v16.i46.5801
59. Ren G, Guo JH, Feng CL, Ding YW, Dong B, Han YX, et al. Berberine inhibits carcinogenesis through antagonizing the atx-Lpa-Lpar2-P38-Leptin axis in a mouse hepatoma model. Mol Ther Oncolytics (2022) 26:372–86. doi: 10.1016/j.omto.2022.08.001
60. McTernan PG, Kusminski CM, Kumar S. Resistin. Curr Opin Lipidol (2006) 17(2):170–5. doi: 10.1097/01.mol.0000217899.59820.9a
61. Deb A, Deshmukh B, Ramteke P, Bhati FK, Bhat MK. Resistin: a journey from metabolism to cancer. Transl Oncol (2021) 14(10):101178. doi: 10.1016/j.tranon.2021.101178
62. Yang CC, Chang SF, Chao JK, Lai YL, Chang WE, Hsu WH, et al. Activation of amp-activated protein kinase attenuates hepatocellular carcinoma cell adhesion stimulated by adipokine resistin. BMC Cancer (2014) 14:112. doi: 10.1186/1471-2407-14-112
63. Gong WJ, Zheng W, Xiao L, Tan LM, Song J, Li XP, et al. Circulating resistin levels and obesity-related cancer risk: a meta-analysis. Oncotarget (2016) 7(36):57694–704. doi: 10.18632/oncotarget.11034
64. Huang X, Yang Z. Resistin's, obesity and insulin resistance: the continuing disconnect between rodents and humans. J Endocrinol Invest (2016) 39(6):607–15. doi: 10.1007/s40618-015-0408-2
65. Wang YY, Hung AC, Lo S, Yuan SF. Adipocytokines visfatin and resistin in breast cancer: clinical relevance, biological mechanisms, and therapeutic potential. Cancer Lett (2021) 498:229–39. doi: 10.1016/j.canlet.2020.10.045
66. Radzicka S, Pietryga M, Iciek R, Brazert J. The role of visfatin in pathogenesis of gestational diabetes (Gdm). Ginekol Pol (2018) 89(9):518–21. doi: 10.5603/GP.a2018.0088
67. Ognjanovic S, Bao S, Yamamoto SY, Garibay-Tupas J, Samal B, Bryant-Greenwood GD. Genomic organization of the gene coding for human pre-B-Cell colony enhancing factor and expression in human fetal membranes. J Mol Endocrinol (2001) 26(2):107–17. doi: 10.1677/jme.0.0260107
68. Heo YJ, Choi SE, Jeon JY, Han SJ, Kim DJ, Kang Y, et al. Visfatin induces inflammation and insulin resistance Via the nf-kappab and Stat3 signaling pathways in hepatocytes. J Diabetes Res (2019) 2019:4021623. doi: 10.1155/2019/4021623
69. Liang N, Chen Y, Yang L, He S, Liu T. Visfatin increases mir-21 to promote migration in hcc. Cell Mol Biol (Noisy-le-grand (2018) 64(6):48–52. doi: 10.14715/cmb/2018.64.6.9
70. Ninomiya S, Shimizu M, Imai K, Takai K, Shiraki M, Hara T, et al. Possible role of visfatin in hepatoma progression and the effects of branched-chain amino acids on visfatin-induced proliferation in human hepatoma cells. Cancer Prev Res (Phila (2011) 4(12):2092–100. doi: 10.1158/1940-6207.CAPR-11-0340
71. Miethe C, Torres L, Zamora M, Price RS. Inhibition of Pi3k/Akt and erk signaling decreases visfatin-induced invasion in liver cancer cells. Horm Mol Biol Clin Investig (2021) 42(4):357–66. doi: 10.1515/hmbci-2021-0011
72. Gan L, Liu Z, Sun C. Obesity linking to hepatocellular carcinoma: a global view. Biochim Biophys Acta Rev Cancer (2018) 1869(2):97–102. doi: 10.1016/j.bbcan.2017.12.006
73. Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science (2021) 371(6527):405–10. doi: 10.1126/science.abb2683
74. Turbitt WJ, Buchta Rosean C, Weber KS, Norian LA. Obesity and Cd8 T cell metabolism: implications for anti-tumor immunity and cancer immunotherapy outcomes. Immunol Rev (2020) 295(1):203–19. doi: 10.1111/imr.12849
75. Gerriets VA, Danzaki K, Kishton RJ, Eisner W, Nichols AG, Saucillo DC, et al. Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol (2016) 46(8):1970–83. doi: 10.1002/eji.201545861
76. Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood (2009) 114(18):3803–12. doi: 10.1182/blood-2009-03-213595
77. Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises tcr repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol (2010) 185(3):1836–45. doi: 10.4049/jimmunol.1000021
78. Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and pd-1 checkpoint blockade. Nat Med (2019) 25(1):141–51. doi: 10.1038/s41591-018-0221-5
79. Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. Cd4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell (2010) 18(5):485–98. doi: 10.1016/j.ccr.2010.10.002
80. Pecht T, Gutman-Tirosh A, Bashan N, Rudich A. Peripheral blood leucocyte subclasses as potential biomarkers of adipose tissue inflammation and obesity subphenotypes in humans. Obes Rev (2014) 15(4):322–37. doi: 10.1111/obr.12133
81. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. Nafld causes selective Cd4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature (2016) 531(7593):253–7. doi: 10.1038/nature16969
82. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces Cd4(+) T cell apoptosis promoting hcc development. Cell Death Dis (2018) 9(6):620. doi: 10.1038/s41419-018-0687-6
83. Barrow F, Khan S, Wang H, Revelo XS. The emerging role of b cells in the pathogenesis of nafld. Hepatology (2021) 74(4):2277–86. doi: 10.1002/hep.31889
84. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced iga+ cells dismantle anti-liver cancer immunity. Nature (2017) 551(7680):340–5. doi: 10.1038/nature24302
85. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of nafld. Nat Rev Gastroenterol Hepatol (2020) 17(2):81–92. doi: 10.1038/s41575-019-0210-2
86. Kosaraju R, Guesdon W, Crouch MJ, Teague HL, Sullivan EM, Karlsson EA, et al. B cell activity is impaired in human and mouse obesity and is responsive to an essential fatty acid upon murine influenza infection. J Immunol (2017) 198(12):4738–52. doi: 10.4049/jimmunol.1601031
87. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U.S.A. (2013) 110(13):5133–8. doi: 10.1073/pnas.1215840110
88. Frasca D, Diaz A, Romero M, Garcia D, Blomberg BB. B cell immunosenescence. Annu Rev Cell Dev Biol (2020) 36:551–74. doi: 10.1146/annurev-cellbio-011620-034148
89. Oleinika K, Slisere B, Catalan D, Rosser EC. B cell contribution to immunometabolic dysfunction and impaired immune responses in obesity. Clin Exp Immunol (2022) 210(3):263–72. doi: 10.1093/cei/uxac079
90. Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology (2018) 67(5):1970–85. doi: 10.1002/hep.29636
91. Shao Y, Lo CM, Ling CC, Liu XB, Ng KT, Chu AC, et al. Regulatory b cells accelerate hepatocellular carcinoma progression Via Cd40/Cd154 signaling pathway. Cancer Lett (2014) 355(2):264–72. doi: 10.1016/j.canlet.2014.09.026
92. Nishimura S, Manabe I, Takaki S, Nagasaki M, Otsu M, Yamashita H, et al. Adipose natural regulatory b cells negatively control adipose tissue inflammation. Cell Metab (2013) 18(5):759–66. doi: 10.1016/j.cmet.2013.09.017
93. Cheng X, Tan X, Wang W, Zhang Z, Zhu R, Wu M, et al. Long-chain acylcarnitines induce senescence of invariant natural killer T cells in hepatocellular carcinoma. Cancer Res (2022) 83(4):582–94. doi: 10.1158/0008-5472.CAN-22-2273
94. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol (2018) 19(12):1330–40. doi: 10.1038/s41590-018-0251-7
95. Tang W, Zhou J, Yang W, Feng Y, Wu H, Mok MTS, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol (2022) 19(7):834–47. doi: 10.1038/s41423-022-00872-3
96. Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, et al. High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology (2020) 158(6):1713–27. doi: 10.1053/j.gastro.2020.01.028
97. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3(11):1236–47. doi: 10.1158/2326-6066.CIR-15-0036
98. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology (2017) 6(10):e1344804. doi: 10.1080/2162402X.2017.1344804
99. Hofer F, Di Sario G, Musiu C, Sartoris S, De Sanctis F, Ugel S. A complex metabolic network confers immunosuppressive functions to myeloid-derived suppressor cells (Mdscs) within the tumour microenvironment. Cells (2021) 10(10):2700. doi: 10.3390/cells10102700
100. Yang Z, Huo Y, Zhou S, Guo J, Ma X, Li T, et al. Cancer cell-intrinsic Xbp1 drives immunosuppressive reprogramming of intratumoral myeloid cells by promoting cholesterol production. Cell Metab (2022) 34(12):2018–35 e8. doi: 10.1016/j.cmet.2022.10.010
101. Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab (2016) 24(5):701–15. doi: 10.1016/j.cmet.2016.09.008
102. Ning WR, Jiang D, Liu XC, Huang YF, Peng ZP, Jiang ZZ, et al. Carbonic anhydrase xii mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest (2022) 132(7):e153110. doi: 10.1172/JCI153110
103. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates hif-1alpha activity and il-1beta induction and is a critical determinant of the warburg effect in lps-activated macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
104. Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J, et al. Targeting lipid metabolism to overcome emt-associated drug resistance Via integrin Beta3/Fak pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics (2019) 9(1):265–78. doi: 10.7150/thno.27246
105. Menegaut L, Thomas C, Lagrost L, Masson D. Fatty acid metabolism in macrophages: a target in cardio-metabolic diseases. Curr Opin Lipidol (2017) 28(1):19–26. doi: 10.1097/MOL.0000000000000370
106. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res (2020) 80(7):1438–50. doi: 10.1158/0008-5472.CAN-19-2994
107. Howie D, Ten Bokum A, Cobbold SP, Yu Z, Kessler BM, Waldmann H. A novel role for triglyceride metabolism in Foxp3 expression. Front Immunol (2019) 10:1860. doi: 10.3389/fimmu.2019.01860
108. Prendeville H, Lynch L. Diet, lipids, and antitumor immunity. Cell Mol Immunol (2022) 19(3):432–44. doi: 10.1038/s41423-021-00781-x
109. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in Cd8+ T cells. Eur J Immunol (2008) 38(9):2438–50. doi: 10.1002/eji.200838289
110. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. Stat3 activation-induced fatty acid oxidation in Cd8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab (2020) 31(1):148–61 e5. doi: 10.1016/j.cmet.2019.10.013
111. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell (2020) 183(7):1848–66 e26. doi: 10.1016/j.cell.2020.11.009
112. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces Cd8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–56 e5. doi: 10.1016/j.cmet.2019.04.002
113. Sbierski-Kind J, Grenkowitz S, Schlickeiser S, Sandforth A, Friedrich M, Kunkel D, et al. Effects of caloric restriction on the gut microbiome are linked with immune senescence. Microbiome (2022) 10(1):57. doi: 10.1186/s40168-022-01249-4
114. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature (2011) 479(7374):547–51. doi: 10.1038/nature10599
115. Heinrich B, Brown ZJ, Diggs LP, Vormehr M, Ma C, Subramanyam V, et al. Steatohepatitis impairs T-Cell-Directed immunotherapies against liver tumors in mice. Gastroenterology (2021) 160(1):331–45 e6. doi: 10.1053/j.gastro.2020.09.031
116. Winer DA, Winer S, Chng MH, Shen L, Engleman EG. B lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci (2014) 71(6):1033–43. doi: 10.1007/s00018-013-1486-y
117. Kennedy DE, Knight KL. Inhibition of b lymphopoiesis by adipocytes and il-1-Producing myeloid-derived suppressor cells. J Immunol (2015) 195(6):2666–74. doi: 10.4049/jimmunol.1500957
118. Capasso M, Rashed Alyahyawi A, Spear S. Metabolic control of b cells: more questions than answers. Front Immunol (2015) 6:80. doi: 10.3389/fimmu.2015.00080
119. Lynch LA, O'Connell JM, Kwasnik AK, Cawood TJ, O'Farrelly C, O'Shea DB. Are natural killer cells protecting the metabolically healthy obese patient? Obes (Silver Spring (2009) 17(3):601–5. doi: 10.1038/oby.2008.565
120. Cuff AO, Sillito F, Dertschnig S, Hall A, Luong TV, Chakraverty R, et al. The obese liver environment mediates conversion of nk cells to a less cytotoxic Ilc1-like phenotype. Front Immunol (2019) 10:2180. doi: 10.3389/fimmu.2019.02180
121. Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. Nash Limits anti-tumour surveillance in immunotherapy-treated hcc. Nature (2021) 592(7854):450–6. doi: 10.1038/s41586-021-03362-0
122. Ilkovitch D, Lopez DM. The liver is a site for tumor-induced myeloid-derived suppressor cell accumulation and immunosuppression. Cancer Res (2009) 69(13):5514–21. doi: 10.1158/0008-5472.CAN-08-4625
123. Sun H, Yang W, Tian Y, Zeng X, Zhou J, Mok MTS, et al. An inflammatory-ccrk circuitry drives Mtorc1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun (2018) 9(1):5214. doi: 10.1038/s41467-018-07402-8
124. Wang N, Tan HY, Lu Y, Chan YT, Wang D, Guo W, et al. Piwil1 governs the crosstalk of cancer cell metabolism and immunosuppressive microenvironment in hepatocellular carcinoma. Signal Transduct Target Ther (2021) 6(1):86. doi: 10.1038/s41392-021-00485-8
125. Woodall MJ, Neumann S, Campbell K, Pattison ST, Young SL. The effects of obesity on anti-cancer immunity and cancer immunotherapy. Cancers (Basel (2020) 12(5):1230. doi: 10.3390/cancers12051230
126. Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. Lxr/Apoe activation restricts innate immune suppression in cancer. Cell (2018) 172(4):825–40 e18. doi: 10.1016/j.cell.2017.12.026
127. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
128. Liu J, Sun B, Guo K, Yang Z, Zhao Y, Gao M, et al. Lipid-related Fabp5 activation of tumor-associated monocytes fosters immune privilege Via pd-L1 expression on treg cells in hepatocellular carcinoma. Cancer Gene Ther (2022) 29(12):1951–60. doi: 10.1038/s41417-022-00510-0
129. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. Ripk3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res (2020) 8(5):710–21. doi: 10.1158/2326-6066.CIR-19-0261
130. Sun G, Liu H, Zhao J, Zhang J, Huang T, Sun G, et al. Macrophage Gsk3beta-deficiency inhibits the progression of hepatocellular carcinoma and enhances the sensitivity of anti-Pd1 immunotherapy. J Immunother Cancer (2022) 10(12):e005655. doi: 10.1136/jitc-2022-005655
131. Li C, Wang G, Sivasami P, Ramirez RN, Zhang Y, Benoist C, et al. Interferon-Alpha-Producing plasmacytoid dendritic cells drive the loss of adipose tissue regulatory T cells during obesity. Cell Metab (2021) 33(8):1610–23 e5. doi: 10.1016/j.cmet.2021.06.007
132. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol (2021) 75(6):1271–83. doi: 10.1016/j.jhep.2021.07.032
133. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93 e7. doi: 10.1016/j.cmet.2016.12.018
134. Li Y, Lu Y, Lin SH, Li N, Han Y, Huang Q, et al. Insulin signaling establishes a developmental trajectory of adipose regulatory T cells. Nat Immunol (2021) 22(9):1175–85. doi: 10.1038/s41590-021-01010-3
135. Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits il-10-Mediated regulatory T cell function: implications for obesity. J Immunol (2014) 192(2):623–9. doi: 10.4049/jimmunol.1302181
136. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis (2011) 70 Suppl 1:i104–8. doi: 10.1136/ard.2010.140145
137. Engin AB. Adipocyte-macrophage cross-talk in obesity. Adv Exp Med Biol (2017) 960:327–43. doi: 10.1007/978-3-319-48382-5_14
138. Takahashi N, Waelput W, Guisez Y. Leptin is an endogenous protective protein against the toxicity exerted by tumor necrosis factor. J Exp Med (1999) 189(1):207–12. doi: 10.1084/jem.189.1.207-a
139. Gasic S, Tian B, Green A. Tumor necrosis factor alpha stimulates lipolysis in adipocytes by decreasing gi protein concentrations. J Biol Chem (1999) 274(10):6770–5. doi: 10.1074/jbc.274.10.6770
140. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing il-6 and tnf expression. Cell (2010) 140(2):197–208. doi: 10.1016/j.cell.2009.12.052
141. Ghosh S, Ashcraft K. An il-6 link between obesity and cancer. Front Biosci (Elite Ed (2013) 5(2):461–78. doi: 10.2741/e628
142. Hodge DR, Hurt EM, Farrar WL. The role of il-6 and Stat3 in inflammation and cancer. Eur J Cancer (2005) 41(16):2502–12. doi: 10.1016/j.ejca.2005.08.016
143. Luan D, Dadpey B, Zaid J, Bridge-Comer PE, DeLuca JH, Xia W, et al. Adipocyte-secreted il-6 sensitizes macrophages to il-4 signaling. Diabetes (2022) 72(3):367–74. doi: 10.2337/db22-0444
144. Zhou YF, Song SS, Tian MX, Tang Z, Wang H, Fang Y, et al. Cystathionine beta-synthase mediated Prrx2/Il-6/Stat3 inactivation suppresses tregs infiltration and induces apoptosis to inhibit hcc carcinogenesis. J Immunother Cancer (2021) 9(8):e003031. doi: 10.1136/jitc-2021-003031
145. Xu J, Yin Z, Cao S, Gao W, Liu L, Yin Y, et al. Systematic review and meta-analysis on the association between il-1b polymorphisms and cancer risk. PloS One (2013) 8(5):e63654. doi: 10.1371/journal.pone.0063654
146. Sharma BR, Kanneganti TD. Nlrp3 inflammasome in cancer and metabolic diseases. Nat Immunol (2021) 22(5):550–9. doi: 10.1038/s41590-021-00886-5
147. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking il-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-Pd-1 for tumor abrogation. Proc Natl Acad Sci U.S.A. (2019) 116(4):1361–9. doi: 10.1073/pnas.1812266115
148. Freese KE, Kokai L, Edwards RP, Philips BJ, Sheikh MA, Kelley J, et al. Adipose-derived stems cells and their role in human cancer development, growth, progression, and metastasis: a systematic review. Cancer Res (2015) 75(7):1161–8. doi: 10.1158/0008-5472.CAN-14-2744
149. Bellows CF, Zhang Y, Simmons PJ, Khalsa AS, Kolonin MG. Influence of bmi on level of circulating progenitor cells. Obes (Silver Spring (2011) 19(8):1722–6. doi: 10.1038/oby.2010.347
150. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of tgf-Beta/Smad3 signaling. Cell Metab (2011) 14(1):67–79. doi: 10.1016/j.cmet.2011.04.013
151. Wu L, Derynck R. Essential role of tgf-beta signaling in glucose-induced cell hypertrophy. Dev Cell (2009) 17(1):35–48. doi: 10.1016/j.devcel.2009.05.010
152. Zhao J, Hu L, Gui W, Xiao L, Wang W, Xia J, et al. Hepatocyte tgf-beta signaling inhibiting wat browning to promote nafld and obesity is associated with let-7b-5p. Hepatol Commun (2022) 6(6):1301–21. doi: 10.1002/hep4.1892
153. Soukupova J, Malfettone A, Bertran E, Hernandez-Alvarez MI, Penuelas-Haro I, Dituri F, et al. Epithelial-mesenchymal transition (Emt) induced by tgf-beta in hepatocellular carcinoma cells reprograms lipid metabolism. Int J Mol Sci (2021) 22(11):5543. doi: 10.3390/ijms22115543
154. Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. Tgf-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst (2014) 106(2):djt369. doi: 10.1093/jnci/djt369
155. Sun X, Bernhardt SM, Glynn DJ, Hodson LJ, Woolford L, Evdokiou A, et al. Attenuated tgfb signalling in macrophages decreases susceptibility to dmba-induced mammary cancer in mice. Breast Cancer Res (2021) 23(1):39. doi: 10.1186/s13058-021-01417-8
156. Chen J, Gingold JA, Su X. Immunomodulatory tgf-beta signaling in hepatocellular carcinoma. Trends Mol Med (2019) 25(11):1010–23. doi: 10.1016/j.molmed.2019.06.007
157. Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating moesin lactylation and enhancing tgf-beta signaling in regulatory T cells. Cell Rep (2022) 39(12):110986. doi: 10.1016/j.celrep.2022.110986
158. Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. Tgfbeta1-mediated Smad3 enhances pd-1 expression on antigen-specific T cells in cancer. Cancer Discovery (2016) 6(12):1366–81. doi: 10.1158/2159-8290.CD-15-1347
159. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. Ccr2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116(1):115–24. doi: 10.1172/JCI24335
160. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116(6):1494–505. doi: 10.1172/JCI26498
161. Thomann S, Weiler SME, Wei T, Sticht C, de la Torre C, Toth M, et al. Yap-induced Ccl2 expression is associated with a switch in hepatic macrophage identity and vascular remodelling in liver cancer. Liver Int (2021) 41(12):3011–23. doi: 10.1111/liv.15048
162. Chen J, Ji K, Gu L, Fang Y, Pan M, Tian S. Hmga1 promotes macrophage recruitment Via activation of nf-Kappab-Ccl2 signaling in hepatocellular carcinoma. J Immunol Res (2022) 2022:4727198. doi: 10.1155/2022/4727198
163. Han ZJ, Li YB, Yang LX, Cheng HJ, Liu X, Chen H. Roles of the Cxcl8-Cxcr1/2 axis in the tumor microenvironment and immunotherapy. Molecules (2021) 27(1):137. doi: 10.3390/molecules27010137
164. Newman G, Gonzalez-Perez RR. Leptin-cytokine crosstalk in breast cancer. Mol Cell Endocrinol (2014) 382(1):570–82. doi: 10.1016/j.mce.2013.03.025
165. Dyck L, Prendeville H, Raverdeau M, Wilk MM, Loftus RM, Douglas A, et al. Suppressive effects of the obese tumor microenvironment on Cd8 T cell infiltration and effector function. J Exp Med (2022) 219(3):e20210042. doi: 10.1084/jem.20210042
166. Kichenadasse G, Miners JO, Mangoni AA, Rowland A, Hopkins AM, Sorich MJ. Association between body mass index and overall survival with immune checkpoint inhibitor therapy for advanced non-small cell lung cancer. JAMA Oncol (2020) 6(4):512–8. doi: 10.1001/jamaoncol.2019.5241
167. Cortellini A, Bersanelli M, Buti S, Cannita K, Santini D, Perrone F, et al. A multicenter study of body mass index in cancer patients treated with anti-Pd-1/Pd-L1 immune checkpoint inhibitors: when overweight becomes favorable. J Immunother Cancer (2019) 7(1):57. doi: 10.1186/s40425-019-0527-y
168. Costante F, Airola C, Santopaolo F, Gasbarrini A, Pompili M, Ponziani FR. Immunotherapy for nonalcoholic fatty liver disease-related hepatocellular carcinoma: lights and shadows. World J Gastrointest Oncol (2022) 14(9):1622–36. doi: 10.4251/wjgo.v14.i9.1622
169. Swed B, Ryan K, Gandarilla O, Shah MA, Brar G. Favorable response to second-line atezolizumab and bevacizumab following progression on nivolumab in advanced hepatocellular carcinoma: a case report demonstrating that anti-vegf therapy overcomes resistance to checkpoint inhibition. Med (Baltimore (2021) 100(25):e26471. doi: 10.1097/MD.0000000000026471
170. Chen J, Lu L, Qu C, AG, Deng F, Cai M, et al. Body mass index, as a novel predictor of hepatocellular carcinoma patients treated with anti-Pd-1 immunotherapy. Front Med (Lausanne (2022) 9:981001. doi: 10.3389/fmed.2022.981001. A G.
171. Akce M, Liu Y, Zakka K, Martini DJ, Draper A, Alese OB, et al. Impact of sarcopenia, bmi, and inflammatory biomarkers on survival in advanced hepatocellular carcinoma treated with anti-Pd-1 antibody. Am J Clin Oncol (2021) 44(2):74–81. doi: 10.1097/COC.0000000000000787
172. Farha N, Alkhayyat M, Lindsey A, Mansoor E, Saleh MA. Immune checkpoint inhibitor induced colitis: a nationwide population-based study. Clin Res Hepatol Gastroenterol (2022) 46(1):101778. doi: 10.1016/j.clinre.2021.101778
173. McQuade JL, Hammers H, Furberg H, Engert A, Andre T, Blumenschein G Jr., et al. Association of body mass index with the safety profile of nivolumab with or without ipilimumab. JAMA Oncol (2023) 9(1):102–11. doi: 10.1001/jamaoncol.2022.5409
174. Eun Y, Kim IY, Sun JM, Lee J, Cha HS, Koh EM, et al. Risk factors for immune-related adverse events associated with anti-Pd-1 pembrolizumab. Sci Rep (2019) 9(1):14039. doi: 10.1038/s41598-019-50574-6
175. Rogado J, Romero-Laorden N, Sanchez-Torres JM, Ramos-Levi AM, Pacheco-Barcia V, Ballesteros AI, et al. Effect of excess weight and immune-related adverse events on the efficacy of cancer immunotherapy with anti-Pd-1 antibodies. Oncoimmunology (2020) 9(1):1751548. doi: 10.1080/2162402X.2020.1751548
176. Hirsch L, Bellesoeur A, Boudou-Rouquette P, Arrondeau J, Thomas-Schoemann A, Kirchgesner J, et al. The impact of body composition parameters on severe toxicity of nivolumab. Eur J Cancer (2020) 124:170–7. doi: 10.1016/j.ejca.2019.11.003
177. Mylod E, Lysaght J, Conroy MJ. Natural killer cell therapy: a new frontier for obesity-associated cancer. Cancer Lett (2022) 535:215620. doi: 10.1016/j.canlet.2022.215620
178. Garcia-Jaramillo M, Spooner MH, Lohr CV, Wong CP, Zhang W, Jump DB. Lipidomic and transcriptomic analysis of Western diet-induced nonalcoholic steatohepatitis (Nash) in female ldlr -/- mice. PloS One (2019) 14(4):e0214387. doi: 10.1371/journal.pone.0214387
179. De Minicis S, Agostinelli L, Rychlicki C, Sorice GP, Saccomanno S, Candelaresi C, et al. Hcc development is associated to peripheral insulin resistance in a mouse model of Nash. PloS One (2014) 9(5):e97136. doi: 10.1371/journal.pone.0097136
180. Haslam DW, James WP. Obesity. Lancet (2005) 366(9492):1197–209. doi: 10.1016/S0140-6736(05)67483-1
181. Hobeika C, Ronot M, Beaufrere A, Paradis V, Soubrane O, Cauchy F. Metabolic syndrome and hepatic surgery. J Visc Surg (2020) 157(3):231–8. doi: 10.1016/j.jviscsurg.2019.11.004
182. Balzan S, Nagarajan G, Farges O, Galleano CZ, Dokmak S, Paugam C, et al. Safety of liver resections in obese and overweight patients. World J Surg (2010) 34(12):2960–8. doi: 10.1007/s00268-010-0756-1
183. Goonawardena J, Ratnayake C, Cheung KT, Fox A. Should bariatric surgery be offered for hepatocellular adenomas in obese patients? Surg Obes Relat Dis (2020) 16(12):2117–24. doi: 10.1016/j.soard.2020.06.043
184. Li Q, Wang Y, Ma T, Lv Y, Wu R. Clinical outcomes of patients with and without diabetes mellitus after hepatectomy: a systematic review and meta-analysis. PloS One (2017) 12(2):e0171129. doi: 10.1371/journal.pone.0171129
185. Howell J, Samani A, Mannan B, Hajiev S, Motedayen Aval L, Abdelmalak R, et al. Impact of nafld on clinical outcomes in hepatocellular carcinoma treated with sorafenib: an international cohort study. Therap Adv Gastroenterol (2022) 15:17562848221100106. doi: 10.1177/17562848221100106
186. Nault JC, Pigneur F, Nelson AC, Costentin C, Tselikas L, Katsahian S, et al. Visceral fat area predicts survival in patients with advanced hepatocellular carcinoma treated with tyrosine kinase inhibitors. Dig Liver Dis (2015) 47(10):869–76. doi: 10.1016/j.dld.2015.07.001
187. Imai K, Takai K, Miwa T, Taguchi D, Hanai T, Suetsugu A, et al. Rapid depletions of subcutaneous fat mass and skeletal muscle mass predict worse survival in patients with hepatocellular carcinoma treated with sorafenib. Cancers (Basel (2019) 11(8):1206. doi: 10.3390/cancers11081206
188. Labeur TA, van Vugt JLA, Ten Cate DWG, Takkenberg RB, JNM IJ, Groot Koerkamp B, et al. Body composition is an independent predictor of outcome in patients with hepatocellular carcinoma treated with sorafenib. Liver Cancer (2019) 8(4):255–70. doi: 10.1159/000493586
Keywords: obesity, tumor immune microenvironment, immune dysfunction, metabolic shift, fatty acid, immunotherapy
Citation: Yang J, He J, Feng Y and Xiang M (2023) Obesity contributes to hepatocellular carcinoma development via immunosuppressive microenvironment remodeling. Front. Immunol. 14:1166440. doi: 10.3389/fimmu.2023.1166440
Received: 15 February 2023; Accepted: 05 May 2023;
Published: 17 May 2023.
Edited by:
Eva Surmacz, Allysta Pharmaceuticals, Inc., United StatesReviewed by:
Min Yang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaMaria Azrad, University of Alabama, United States
Guilherme Zweig Rocha, State University of Campinas, Brazil
Copyright © 2023 Yang, He, Feng and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Xiang, xiangming@tjmu.edu.cn