 Wanjin Li
Wanjin Li Junying Yuan
Junying Yuan- Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China
Receptor-Interacting Serine/Threonine-Protein Kinase 1 (RIPK1) is a master regulator of TNFR1 signaling in controlling cell death and survival. While the scaffold of RIPK1 participates in the canonical NF-κB pathway, the activation of RIPK1 kinase promotes not only necroptosis and apoptosis, but also inflammation by mediating the transcriptional induction of inflammatory cytokines. The nuclear translocation of activated RIPK1 has been shown to interact BAF-complex to promote chromatin remodeling and transcription. This review will highlight the proinflammatory role of RIPK1 kinase with focus on human neurodegenerative diseases. We will discuss the possibility of targeting RIPK1 kinase for the treatment of inflammatory pathology in human diseases.
Introduction
Inflammation is a common pathological aspect of human diseases, including neurodegenerative diseases. Neuroinflammation has been well-established to be involved in the onset and progression of multiple human chronic neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease, amyotrophic lateral sclerosis and multiple sclerosis (1, 2). The critical issue is how to target neuroinflammation safely and effectively. The nonsteroidal anti-inflammatory drugs (NSAIDs), commonly used in the treatment of peripheral inflammatory diseases such as rheumatoid arthritis, had shown some early promises in delaying the onset of dementia in AD; however, large clinical studies of NSAIDs in AD demonstrated no efficacy and unfavorable side-effects (3–6). In animal and cell models, NSAIDs are known to induce apoptosis (7–9). In this review, we will highlight the role of RIPK1 kinase as a key mediator of sterile inflammation in the CNS and can be safely targeted in mouse models and in humans.
Receptor-Interacting Serine/Threonine-Protein Kinase 1 (RIPK1) has been established as a master regulator of TNFR1 signaling and an important therapeutic target (10). In TNF stimulated cells, RIPK1 controls the cell fate decision whether to promote cell survival or cell death. The activation of RIPK1 kinase activity is involved in mediating pathological cell death and inflammation in a wide arrange of major human diseases from peripheral rheumatoid arthritis and Crohn’s disease to neurodegenerative disorders such as amyotrophic lateral sclerosis and Alzheimer’s disease (10–13). Multiple lines of RIPK1 kinase-dead knockin mutant mice, including D138N, K45A, K584R, S166A and S25D, have been shown to be normal and highly resistant to TNF, while TNF induces cell death and animal lethality in WT mice (14–18). Structurally RIPK1 kinase has a hydrophobic pocket associated with its activation segment that is highly amenable for developing small molecule inhibitors which can stabilize its inactive conformation and allows BBB passage (19–22). Human clinical trials of small molecule RIPK1 inhibitors have demonstrated safety of targeting RIPK1 kinase (23–25). These studies demonstrated the feasibility and safety of targeting RIPK1 in humans.
RIPK1 is a key mediator of TNFR1 signaling
RIPK1 contains a N-terminal kinase domain, a C-terminal death domain (DD) and an intermediate domain that includes RHIM domain (Figure 1). RIPK1 is recruited specifically to the TNFR1 upon its activation by TNF (26, 27) (Figure 2). The interaction of RIPK1 with TNFR1 is mediated by homotypic interaction of its DD with the DD in the C-terminus of TNFR1. Another DD-containing adaptor molecule, TRADD, is also recruited to TNFR1 to form TNF-RSC (complex I). TRADD in turn recruits TRAF2 and the E3 ubiquitin ligases cIAP1/2 into complex I to modulate RIPK1 by K63 ubiquitination. K63 ubiquitination of RIPK1 recruits the LUBAC complex containing HOIP, HOIL1, and SHARPIN to perform M1 ubiquitination of RIPK1 which plays an important role in simultaneously mediating the activation of NF-κB pathway by recruiting the ubiquitin binding proteins, such as NEMO, ABIN1, TBK1 and OPTN, as well as IKK complex, and suppressing the activation of RIPK1 (12, 28–30). The polyubiquitin chains in complex I also recruits the ubiquitin binding adaptors TAB2/TAB3 which in turn recruit and activate TAK1, a key kinase involved in the activation of NF-κB pathway (31). These different components of complex I perform extensive ubiquitinating modifications of RIPK1, on as many as 34 lysine residues (32) (Figure 1), which collectively decide if the cell should survive by activating NF-κB, or die. The scaffold of RIPK1 has an important function in supporting cell survival by promoting the NF-κB activation, while the activation of RIPK1 kinase activity, regulated by its modifiers in complex I, is pro-death and proinflammation (26, 32–37). The ubiquitination of RIPK1 in complex I serves as the checkpoint I within minutes of TNF stimulation to control the activation of RIPK1 kinase, which may promote proinflammatory cytokine production or cell death including RIPK1 dependent apoptosis (RDA) and necroptosis (Figure 2). Dysregulated ubiquitination of RIPK1 in complex I results in the activation of its kinase activity to mediate apoptosis or necroptosis, depending the presence of downstream signaling components such as caspase-8 and RIPK3/MLKL by forming the alternative complex IIa or complex IIb (30, 38–40). While necroptosis as a form of regulated necrosis has been recognized to trigger inflammation (12, 41–44), the activation of RIPK1 kinase activity has been found to promote the transcriptional induction of proinflammatory cytokines but the mechanism was unclear (45–52)(Figure 2). A recent report showed that RIPK1 might regulate inflammation by modulating chromatin remodeling directly (53).
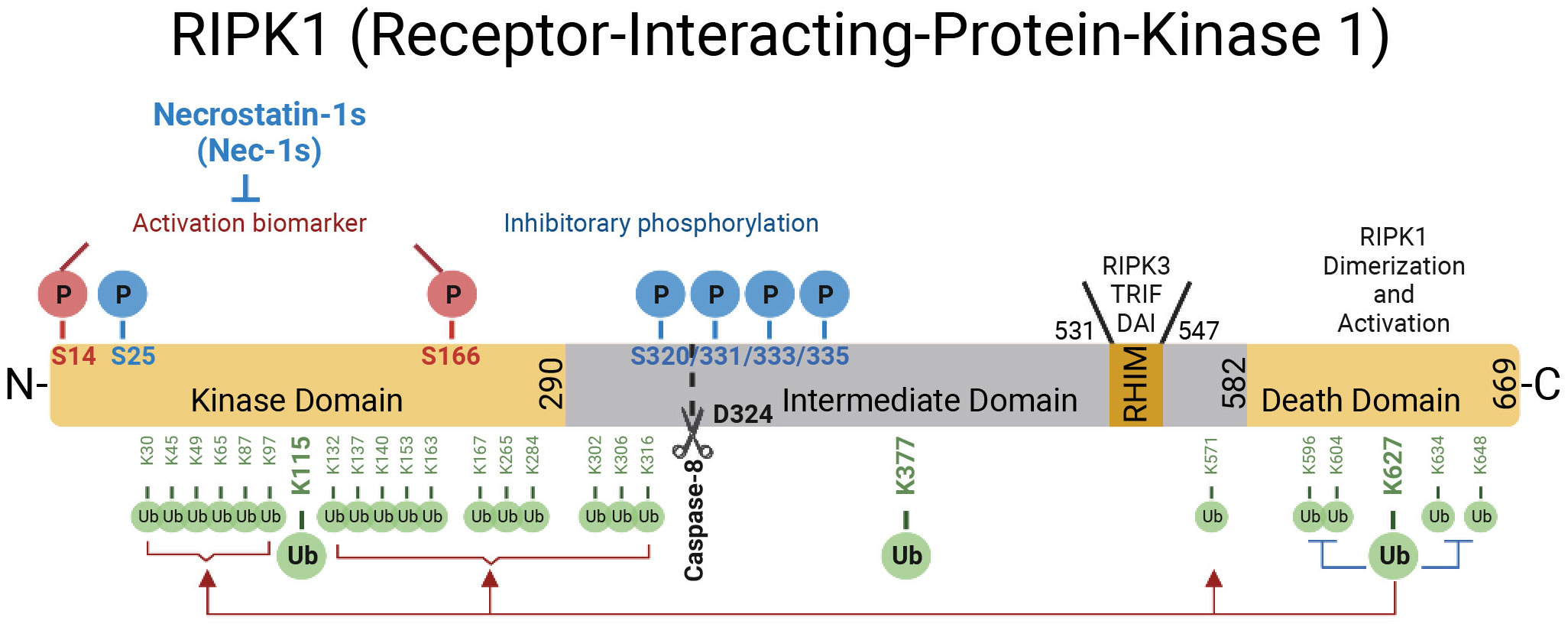
Figure 1 The domains of RIPK1. RIPK1 is a 76-kDa protein with an N-terminal kinase domain, an intermediate domain with a RIP homotypic interaction motif (RHIM), and a C-terminal death domain (DD). The RHIM is required for binding with RIPK3 to mediate necroptosis and other RHIM-containing proteins. The DD is not only crucial for mediating RIPK1 heterodimerization with other DD-containing proteins but also required for RIPK1 activation by mediating RIPK1 dimerization during the transition from complex I to complex II. The top of the RIPK1 kinase depicts the phosphorylation sites in RIPK1. Autophosphorylation of Ser166 is a biomarker for RIPK1 activation. The small molecule Nec-1s is caged in a hydrophobic pocket between the N and C lobes of the kinase domain and forms an H bond between its nitrogen atom and the hydroxyl oxygen of Ser161 on the activation loop to inhibit the activation of RIPK1. The cleavage of RIPK1 after Asp324 by caspase-8 or the phosphorylation on Ser320/331/333/335 in human RIPK1 and Ser321/332/334/336 in murine RIPK1 by TAK1 or MK2 leads to the suppression of RIPK1 activation. As depicted below the RIPK1 kinase, RIPK1 is extensively modulated by ubiquitination with upto 34 ubiquitination sites, especially in the kinase domain and death domain.
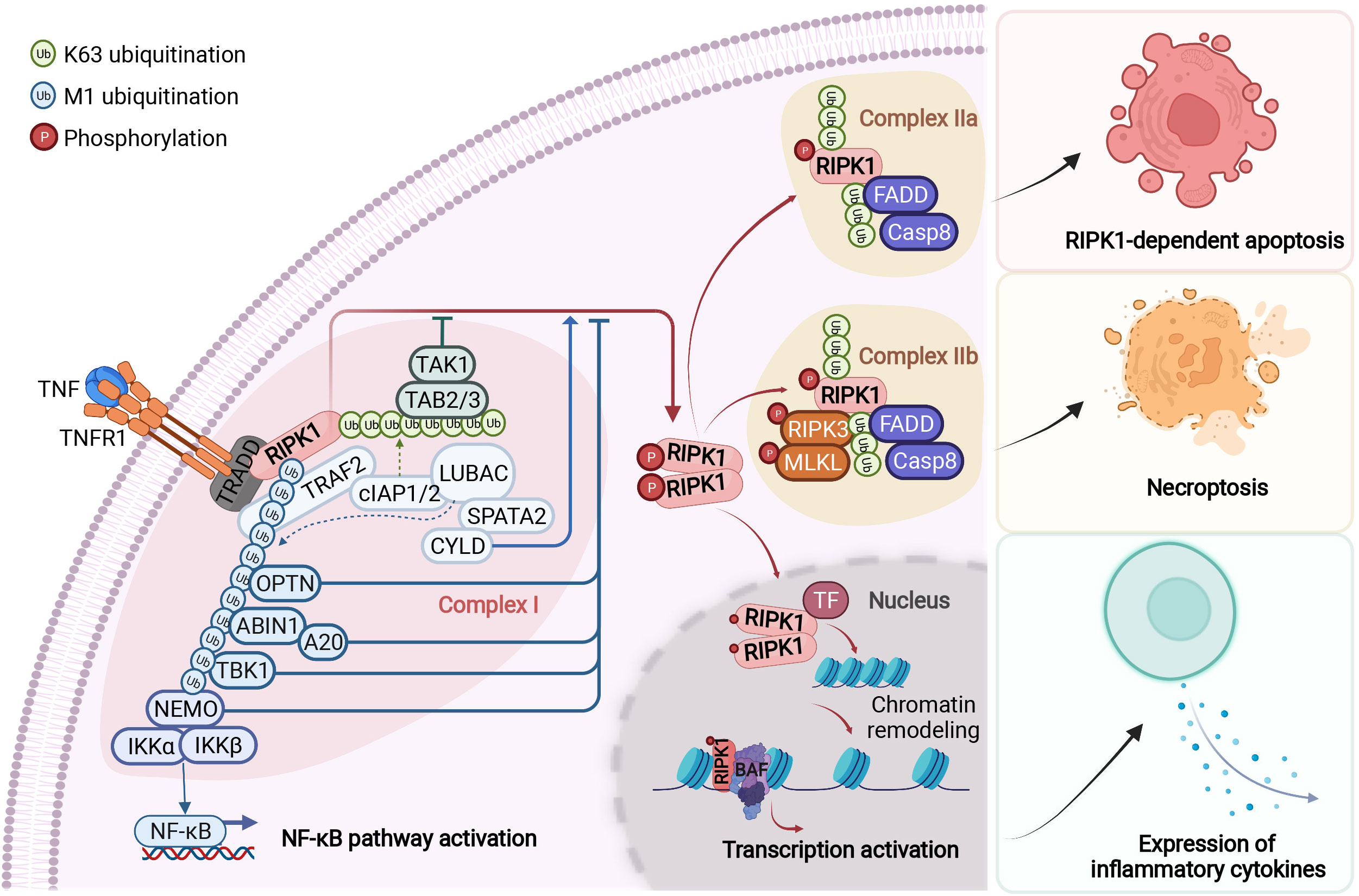
Figure 2 RIPK1-mediated signaling events downstream of TNFR1. Upon binding to tumour necrosis factor (TNF), the cytoplasmic death domain of trimerized TNF receptor 1 (TNFR1) recruits a membrane-associated complex, named complex I. This complex comprises the adaptor protein TNFR1-associated death domain protein (TRADD), the death domain-containing protein kinase receptor-interacting protein 1 (RIPK1) and several ubiquitin E3 ligases, including TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein 1 (cIAP1), cIAP2 and the linear ubiquitin chain assembly complex (LUBAC). In complex I, RIPK1 is rapidly polyubiquitylated by Lys63-linked and linear Met1-linked ubiquitin chains, which mediate the recruitment and activation of the TGFβ-activated kinase 1 (TAK1) and IκB kinase (IKK) complexes. The phosphorylation and subsequent ubiquitin–proteasome system (UPS)-mediated degradation of inhibitor of κB (IκB) leads to the activation of nuclear factor- κB (NF-κB). Subsequently, the activity of the deubiquitylation enzymes A20 and cylindromatosis (CYLD) disassembles complex I by deubiquitylating its components, including RIPK1 and TRAF2, within 10–15 minutes after TNF stimulation. This leads to the formation of one of the two alternative cytosolic complexes, complex IIa or complex IIb. Complex IIa includes the adaptor FAS-associated death domain protein (FADD), caspase-8 and RIPK1. The formation of complex IIa mediates the activation of caspase-8. This, in turn, activates downstream caspases, such as caspase-3 (not shown), ultimately leading to apoptosis. When the activation of caspase-8 is inhibited, RIPK1 kinase is activated and binds to RIPK3 to form complex IIb, which mediates the downstream events of necroptosis. The transition from complex I to complex II is an important regulatory step. After disassembled from complex I, RIPK1 is able to translocate to the nucleus and form a RIPK1/BAF complex by directly interacting with two components of BAF, SMARCC2 and BRG1. RIPK1/BAF complex is recruited to the target genomic regions by specific transcription factors upon TNF stimulation, which mediates the chromatin remodeling and transcription activation of proinflammatory genes. NEMO, NF-κB essential modulator; TAB, TAK1-binding protein; TF, transcription factor. Figure is created with BioRender.com.
The activation of NF-κB pathway suppresses the activation of RIPK1 and cell death mediated by at least two of its transcriptional targets, TNFAIP3 and CFLAR (CASP8 and FADD Like Apoptosis Regulator, also called c-FLIP) (54, 55). In cells stimulated by TNF, TNFAIP3, transcriptionally induced by the NF-κB pathway, encodes the ubiquitin editing enzyme A20, which acts an upstream regulator of RIPK1 by modulating its ubiquitination pattern (56). Thus, A20 serves as a key ubiquitinating modifying enzyme in the checkpoint I that controls the activation of RIPK1 (Figure 2). On the other hand, CFLAR encodes multiple c-FLIP isoforms which share similarity in amino acid sequence with caspase-8 but lack the caspase activity, and at least some of the c-FLIP isoforms can directly bind to caspase-8 to inhibit its activation (57). Since the cleavage of human RIPK1 at D324 and murine RIPK1 at D325 controls the activation of the kinase by separating it from the C-terminal DD which can dimerize to promote the N-terminal kinase activation (16, 58) (Figure 1), caspase-8 is a downstream negative regulator of RIPK1 kinase. Thus, the cleavage of RIPK1 mediated by caspase-8 may serve as the checkpoint II in TNF stimulated cells that controls the activation of RIPK1 (Figure 2). Expression of RIPK1 mutants D325V or D325H, which block the cleavage of RIPK1 by caspase-8, sensitize cells to RIPK1 activation-mediated apoptosis and necroptosis, as well as induce the production of pro-inflammatory cytokines such as IL-6 and TNF (52, 59). Thus, the activation of RIPK1 and NF-κB may cooperate to mediate inflammatory response.
RIPK1 regulates inflammatory response in human diseases
While the role of RIPK1 in regulating cell death has been well-established, the recent studies highlighted its role in mediating proinflammatory responses. Heterozygous mutations that altered Asp324 of RIPK1 in human lead to early-onset periodic fever syndrome, defined as cleavage-resistant RIPK1-induced autoinflammatory syndrome (CRIA) (46, 48, 52). In particular, the expression of RIPK1 noncleavable variants, such as D324V and D324H, in humans, which blocks the cleavage by caspase-8, leads to an autoinflammation clinically characterized by recurrent fevers and lymphadenopathy (46, 52). Overproduction of inflammatory cytokines and chemokines were detected in the blood of these patients and the recurrent fevers in the patients respond partially to the treatment of anti-IL6. The expression of the caspase-8 cleavage-resistant RIPK1 (Ripk1D325A/D325A) resulted in embryonic lethality around E10.5 (46, 48). Though Mlkl−/−Fadd−/− or Ripk3−/−caspase-8−/− mice are able to fully rescue Ripk1D325A/D325A animals, there were characteristic autoimmune lymphoproliferative syndrome (ALPS) observed in the surviving mice, suggesting the role of activated RIPK1 in mediating inflammation independent of necroptosis (46, 48, 49, 60, 61). In animal models and human pathological studies, the activation of RIPK1 has also been implicated in mediating inflammation in major human chronic inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease and psoriasis (10, 62–65).
M1 ubiquitination of RIPK1 mediated by LUBAC complex plays a critical role in restraining RIPK1 kinase activity in skin inflammation. LUBAC is a key regulator of TNFR1 mediated cell death and inflammation (29, 66–69). Mice deficient for the gene encoding the LUBAC regulatory subunit SHARPIN, known as the chronic proliferative dermatitis mice (cpdm) (Sharpincpdm/cpdm mice), suffer from severe multi-organ inflammation particularly in the skin that resembles atopic dermatitis and psoriasis in humans (15, 70). Inhibition of RIPK1 by GNE684 or RIPK1D138N/D138N protects against skin inflammation in cpdm mice (15, 44, 71). OTULIN is a deubiquitinating enzyme that plays a critical role in regulating LUBAC activity (72). Biallelic hypomorphic mutations in OTULIN in humans lead to a severe form of autoinflammatory disease, known as OTULIPENIA or ORAS (73, 74). Mice with epidermis-specific loss of OTULIN develop inflammatory skin lesions that can be inhibited by TNFR1 deficiency and inhibition of RIPK1 kinase (75).
The activation of RIPK1 has also been implicated in mediating inflammatory responses in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and Alzheimer’s disease. Pathological evidence for neuroinflammation in ALS, a progressive neurodegenerative disease that affects motor neurons in the brain and the spinal cord, include changes in circulating immune cell populations and cytokines, which can be noted in the early stage of the disease (76). Loss of OPTN, a monogenic cause of ALS, leads to RIPK1-dependent microglial activation, dysmyelination and necroptosis of motor neurons in the spinal cords of OPTN-/- mice, which was rescued by genetic and pharmacological inactivation of RIPK1 kinase (77). OPTN deficient microglia present an activated inflammatory state which can be suppressed upon inhibition of RIPK1. A single-cell RNA-seq study identified a class of microglia, termed RIPK1-Regulated Inflammatory Microglia (RRIMs), in the early stage of ALS that show significant up-regulation of classical proinflammatory pathways in RIPK1-dependent manner (78).
MS is an autoimmune disease of the CNS that involves RIPK1 activation (79). TNFα is strongly linked with the etiology of MS (80, 81). The loss of myelinating oligodendrocytes, which normally insulate the axons of neurons, is a hallmark of MS. Necroptosis is the primary mode of cell death for oligodendrocytes in the presence of TNFα stimulation alone (79). TNFAIP3 has been identified as a risk gene in a large genetic association study highlighting the contribution of peripheral immune cells and microglia to MS (82). Down-regulation of caspase-8 levels has been noted in the cortical pathological samples from MS patients (79). Thus, the pathology of MS may include the defects in both checkpoints I and II that control the activation of RIPK1. Genetic (RIPK1D138N/D138N) and pharmacological (Nec-1s) inhibition of RIPK1 kinase ameliorated disease pathology, improved animal behavior, attenuated the production of proinflammatory cytokines, and decreased recruitment of immune cells in multiple animal models of MS (79, 83).
RIPK1 may also play an important role in driving neuroinflammation in AD (84). In microglia of mouse models of AD, the activation of RIPK1 drives transcriptional induction of Cst7, which encodes an endosomal/lysosomal cathepsin inhibitor named Cystatin F, a biomarker for disease-associated microglia (DAM) present in spatial proximity to Aβ plaques in both postmortem human AD brain samples and in an AD mouse model (85). Inhibition of RIPK1 reduces neuroinflammation and cognitive deficits in the APP/PS1 amyloid β-driven mouse model of AD (84).
RIPK1-mediated inflammatory response has also been shown to be involved in severe cases of Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (86). SARS-CoV-2 poses substantial challenges to public health worldwide mainly due to its ability to promote life-threatening systemic inflammatory response in a subset of COVID-19 patients. Activated RIPK1 was found in human COVID-19 lung pathological samples, respiratory tract epithelial cells from COVID-19 patients, cultured human lung organoids and ACE2 transgenic mice infected by SARS-CoV-2 (86, 87). Inhibition of RIPK1 using multiple small-molecule inhibitors reduced the viral load in human lung organoids infected by SARS-CoV-2. The SARS-CoV2 3C-like protease (3CLpro) can cleave NEMO and promote RIPK1-mediated endothelial cell death and BBB-damage in COVID-19 (88, 89). The viral load in lung and mortality and is reduced by the treatment of RIPK1 therapeutic inhibitor Nec-1s, blocking SARS-CoV-2 manifestation in the CNS of ACE2 transgenic mice (86). NSP12 is an RNA-dependent RNA polymerase of SARS-CoV-2, which is important in mediating coronaviral transcription and replication. The C14408T variant of SARS-CoV-2, first reported in Italy and present in all subsequently evolved strains including Delta and Omicron, encodes NSP12 323L variant. This 323L variant of NSP12 induced higher activation level of RIPK1 compared to wildtype NSP12. Notably, the transcriptional expression level of host factors promoting viral entry such as EGFR and ACE2 as well as proinflammatory genes can be downregulated with inhibition of RIPK1. Thus, SARS-CoV-2 may be able to hijack the RIPK1-mediated host defense response to its advantage and that inhibition of RIPK1 may provide a therapeutic option for the treatment of severe COVID-19 to help ending this public health emergency.
Activated RIPK1 mediates sustained inflammatory response involving NF-κB
TNFα-induced necroptosis leads to two waves of cell-autonomous proinflammatory cytokine production (51). The first wave, transient and weak in nature, is activated in response to TNFα alone; whereas the second wave, stronger and sustained, depends upon the necroptotic signaling and the activation of RIPK1. In contrast, the level of cytokine production promoted by a direct oligomerization of MLKL-induced necroptosis is much lower than that by TNFα-induced necroptosis. Thus, the activation of RIPK1 can also mediate inflammatory response during necroptosis.
The activation of inflammation by lipopolysaccharide (LPS) may also involve such RIPK1-mediated biphasic production of inflammatory response (90). Upon inhibition of caspases in cells stimulated by LPS, a biphasic production of proinflammatory cytokines has been noted. With caspase inhibition, the early production of LPS stimulated proinflammation cytokines is independent of RIPK1 kinase activity but dependent upon the scaffold function of RIPK1 involving in the NF-κB pathway. Another TNFR1-associated complex containing key mediators of NF-κB, FADD, activated RIPK1 and caspase-8 is formed upon the stimulation of autocrine TNFα produced from the early phase. In contrast with the early phase, the second phase of proinflammatory cytokine production is dependent of RIPK1 kinase activity as it can be blocked with pharmacological inhibition of RIPK1 kinase. Interestingly, such biphasic mode of proinflammatory cytokine production has also been previously reported in cells stimulated by TRAIL, another member of TNF family (91). Therefore, the scaffold function of RIPK1 is promoted by its kinase activation to regulate the proinflammatory cytokine induction mediated by NF-κB signaling with inhibition of caspases. Since the cleavage of RIPK1 after the kinase domain at D324 hRIPK1 and D325 mRIPK1 by caspase-8 negatively regulates its kinase activation, the periodic fever episodes of rare human carriers of non-cleavable RIPK1 may involve their interaction with TLR ligands which may cause minimal inflammation normally inconsequential in normal individuals (46, 52).
cIAP1/2 mediated-ubiquitination of RIPK1 recruits adaptors TAB1/2 to support TAK1 (Transforming growth factor-α-activated kinase 1, also called MAP3K7) activation (31). Activated TAK1 then mediates the phosphorylation IKK complex consisting of IKKα/β/γ (NEMO) to support its formation (92). Interestingly, RIPK1 is a phosphorylation substrate of TAK1 as well as IKKs (93, 94). IKKs mediated S25 phosphorylation on RIPK1 negatively regulates the activation of RIPK1 kinase activity and RIPK1-dependent cell death. TAK1 mediates the phosphorylation of multiple sites in the intermediate domain of RIPK1 including S321 (93). Phospho-Ser321 (S321) RIPK1 is a biomarker for phosphorylation of RIPK1 by TAK1. While the S321A mutation per se has no effect on the NF-κB activation, the loss of TAK1-mediated phosphorylation of RIPK1 promotes its binding to FADD and RIPK1-dependent apoptosis (RDA); furthermore, sustained TAK1-mediated phosphorylation of RIPK1 promotes its interaction with RIPK3 to mediate necroptosis (93, 95). Thus, there are intriguing interactions of upstream activators of NF-κB, TAK1 and IKKs, with RIPK1, whose scaffold also functions in mediating the activation of NF-κB pathway. The intertwined relationship between RIPK1 and NF-κB pathway may promote inflammatory responses in a temporal specific manner: the activation of NF-κB pathway initiates an early inflammatory response while the activation of RIPK1 is important for mediating sustained chronic inflammation.
TBK1 (TANK binding kinase 1) is known as a key regulator of type I IFN, NF-κB and TNF-mediated RIPK1-dependent cell death (96). Rare human individuals with homozygous loss-of-function mutations in TBK1 suffer from chronic systemic autoinflammation (97). Interestingly, the loss of TBK1 in humans leads to reduced IFN-I production but nevertheless adequate antiviral response, and nearly normal NF-κB-mediated IL6 production, suggesting that the role of TBK1 in regulating IFN-I and NF-κB signaling may be insufficient to explain the autoinflammatory phenotype. Biallelic loss of TBK1 in mice is embryonic lethal which can be effectively suppressed by knockout of TNF or TNFR1, and by genetic inactivation of RIPK1 (30, 98). TBK1 can directly inhibit the activation of RIPK1 kinase by preforming inhibitory phosphorylation on RIPK1 (29, 30). The loss of TBK1 sensitized to RIPK1-dependent cell death in both mouse and human cells which may be responsible for the autoinflammatory pathology in TBK1 deficient individuals (30, 97). In addition, another recent study reported the role of RIPK1/RIPK3-mediated inflammatory cascade of alloreactive T cell responses in promoting graft-versus-host disease (GVHD) (99). Inhibition of RIPK1 was able to reduce the expression of proinflammatory cytokines and MHC class II molecules in intestinal epithelial cells and arrest GVHD without compromising the graft-versus-leukemia (GVL) effect. Therefore, targeting RIPK1/RIPK3 in IECs provides a strategy for GVHD treatment without compromising the normal reaction of immune system. These studies suggest the contribution of necroptosis to inflammatory responses in different human pathological conditions.
Heterozygousity in TBK1 has been linked with the onset of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) in aging (100–102). Genetic interaction of specific mutations with aging is associated with a variety of neurodegenerative diseases. Since both TBK1 and TAK1 preform inhibitory phosphorylation on RIPK1, the interaction of TAK1 and TBK1 in aging was explored (30, 93). The reduction of TAK1 expression in aging human brains was shown to cooperate with heterozygous loss-of-TBK1 to promote late onset ALS/FTD-like pathology mediated by decreased RIPK1 inhibition. Reduced expression of TAK1 in aging human brains may promote RIPK1-mediated inflammatory response to cooperate with additional pre-existing conditions to mediate the onset of neurodegeneration. Thus, inhibiting RIPK1 kinase may provide an effective strategy for reducing chronic inflammation that underlies a variety of aging-related human diseases.
Chromatin remodeling in inflammatory responses
Activation of inflammatory responses involves very elaborated mechanisms to ensure a coordinated program that regulates the expression of induced genes in a temporally and spatially controlled manner. Regulation of inflammatory responses by NF-κB, which has been extensively studied, provides such an example. The interaction of master regulators of NF-κB pathway with chromatin is a key step in driving the complex and diverse gene expression programs (103). Mammalian genomic DNA is tightly packed into chromatins (104, 105). Chromatin remodeling complexes were found to be important for granting dynamic access to the highly packaged DNA and to remodel the nucleosome composition on their target regions. Chromatin remodeling allows accessibility and transcription of specific genes, by precisely controlling the dynamics of chromatin (106–111). Thus, it is not surprising that chromatin remodeling is involved in the transcriptional induction of proinflammatory factors.
BRG1/BRM-associated factor (BAF) chromatin-remodeling complex, the mammalian homolog of the SWI/SNF complex in yeast, is an important regulator of gene expression. It regulates higher-order chromatin organization that modulate chromatin accessibility by displacing the positions of nucleosomes in an ATP-dependent manner in response to specific developmental and environmental signals (111, 112). While SWI/SNF complex has been demonstrated to be involved in tumorigenesis (113, 114) and in neural development and disorders (105), recent studies have showed the role of SWI/SNF complex in the regulation of inflammatory responses. SWI/SNF complex is shown to be recruited to the promoters of IFN-γ induced genes (115–117) and is critically important in the transcriptional induction of LPS-induced inflammatory genes (118–122). LPS induces histone modifications and chromatin remodeling to allow NF-κB to gain access to the IL-12 promoter and other NF-κB-dependent promoters (123, 124). BRG1, the catalytic subunit of SWI/SNF, is recruited by MALAT1 to the promoter region of IL-6 and CXCL8 and interacts with NF-κB to promote LPS-induced inflammatory factors production in HCC cells (125). BRG1 is also involved in LPS or endotoxin tolerance. In LPS tolerant macrophages, TLR inducible recruitment of BRG1 and chromatin remodeling are lost on the promoters of proinflammatory genes which are not responsive to re-stimulation with LPS (126). BRG1 plays an important role in T cell development, differentiation and transcription of cytokines (127–133). The enrichment of BRG1 on chromatin dynamically changes during T cell activation and varies in different types of T cells. BRG1 promotes inflammation in CD4 cells while the Treg-specific deletion of BRG1 impairs Treg activation and results in early-onset fatal inflammation (134). The recruitment of SWI/SNF complex to the promoters or enhancers of proinflammatory genes can be mediated by transcriptional activators (135, 136), long noncoding RNAs (lncRNAs) (137–139), histone modifications (140, 141) or the mediator complex (122, 141–143). Thus, the binding specificity of SWI/SNF complex in different tissue or cells upon various stimulation is directed by the factors recruiting them.
Nuclear RIPK1 promotes inflammatory response by mediating chromatin remodeling
The extensive involvement of RIPK1 in mediating inflammatory responses raises the question how activation of RIPK1 may promote the expression of pro-inflammatory cytokines. Using p-S166-RIPK1 as a biomarker of RIPK1 activation (21, 79), activated RIPK1 is detected in both nucleus and cytoplasm of MEFs upon stimuli causing RDA or necroptosis which is associated with the induction of pro-inflammatory cytokines (51, 52). Nuclear RIPK1 activation is also observed in spinal cords of ALS patients (53). RIPK1 has also been detected in nucleus during TNF-induced necroptosis (144, 145) and H2O2-induced oxidative cell death (146). Interestingly, blocking necroptosis by RIPK3 knockout or MLKL knockout could not inhibit the nuclear translocation of activated RIPK1 or RIPK1-mediated induction of inflammatory gene expression, suggesting that RIPK1 mediated cell death can be mechanistically separated from that of RIPK1-regulated inflammation (53).
The genomic targets of activated RIPK1 in RIPK1 D325A MEFs, as well as in spinal cords from human ALS patients and Tbk1+/-; Tak1ΔM/+ ALS mouse model (30, 53), are highly enriched in inflammatory responses, suggesting that nuclear RIPK1 directly participates in the regulation of specific gene transcription. The nuclear interactome of RIPK1 includes a striking collection of transcriptional activators/coactivators, suggesting that nuclear RIPK1 may be directly involved in mediating transcriptional activation. Interestingly, the nuclear interacting partners of RIPK1 includes p65 (RELA), the key transcription factor in the canonical NF-κB pathway and other transcription factors that have been found to promote inflammation, such as SP1, JUNB (147–151), suggesting that the activated RIPK1 interacts with multiple transcriptional factors in nucleus to mediate inflammation.
Targeting BAF complex to specific genomic regions during development is known to involve the interactions with transcriptional factors including the members of NF-κB family and JUN (152). Activated nuclear RIPK1 was found to interact with almost all components of BAF complex, including BRG1 and SMARCC2. Thus, the binding of activated RIPK1 with the transcriptional factors and BAF complex integrates the external TNF signaling to the specific genomic regions on chromatin in nucleus to modulate chromatin dynamics for activating an inflammatory response.
Phosphorylation of BAF complex is known to modulate its activity in response to specific signaling. ATM kinase, when activated by DNA damage, can phosphorylate BRG1 to stimulate BRG1 nucleosome binding activity by enhancing its affinity for H3K14ac (153). In TNF stimulated condition, nuclear RIPK1 directly phosphorylates Ser306 of SMARCC2, a key scaffolding subunit of BAF complex. p-S166 RIPK1, BRG1 and SMARCC2 co-occupation was found on the key TNF induced accessible genomic regions of proinflammatory genes (53). The DNA accessibility and co-occupancy of BRG1 and SMARCC2 upon TNF in RIPK1 D325A mutant MEFs at p-S166 RIPK1 binding sites are both reduced upon inhibition of RIPK1 kinase. Thus, activated RIPK1 in the nucleus of TNF stimulated cells is recruited by transcription factors to interact with BAF and form a RIPK1/BAF complex to regulate chromatin remodeling activity and the accessibility to specific genomic DNA regions to promote the transcription of proinflammatory genes (Figure 3).
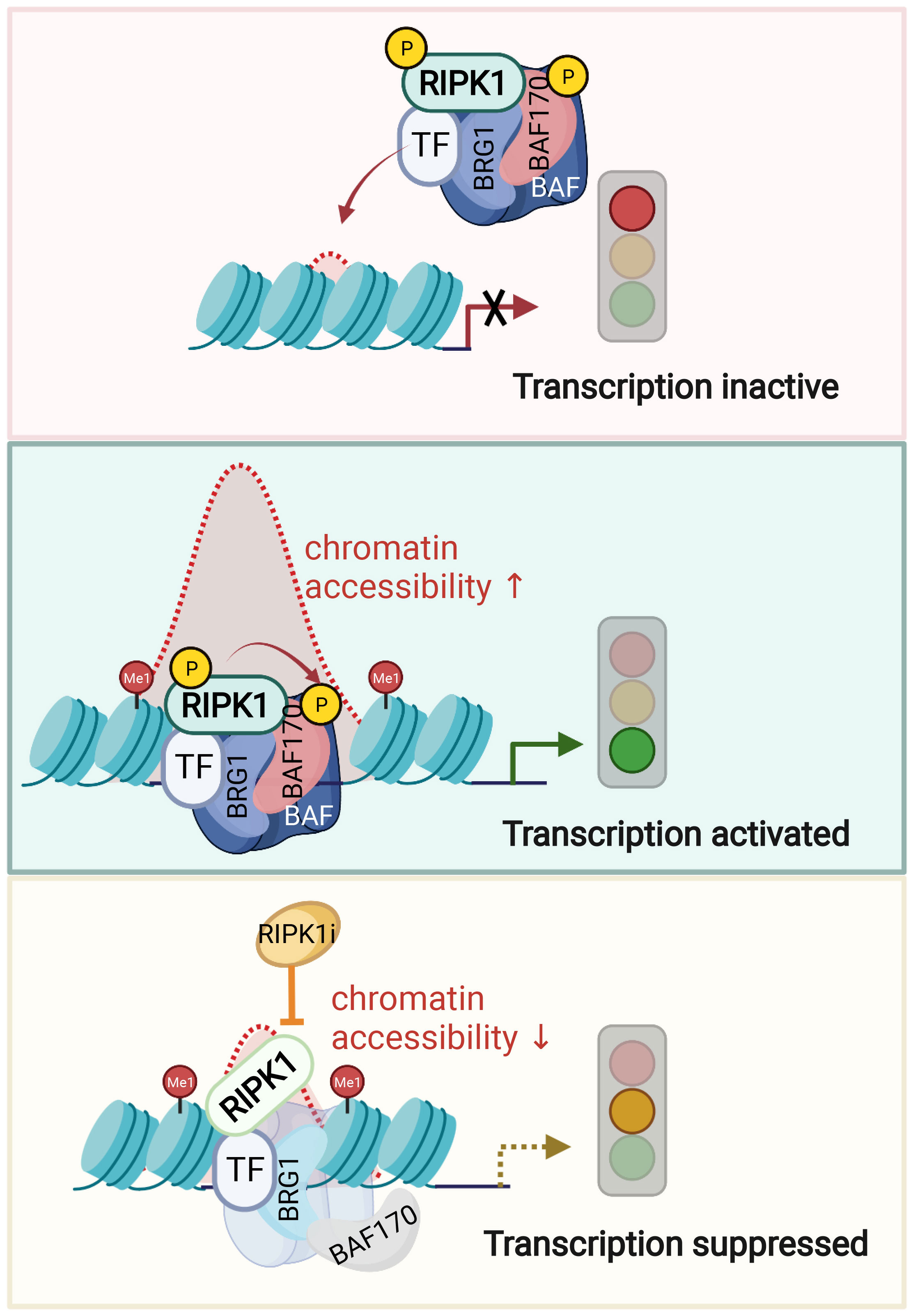
Figure 3 Nuclear RIPK1 promotes transcription of inflammatory cytokines by modulating chromatin remodeling activity. A model of RIPK1 kinase activity dependent transcriptional activation by regulating chromatin modeling activity. Upon TNFα stimulation, transcription factors recruit RIPK1/BAF complex to the genomic target sites. RIPK1 interacts with BRG1 and SMARCC2 directly. Activated RIPK1 mediates the phosphorylation of SMARCC2, which is important in promoting the BAF complex-medicated chromatin remodeling. Consequently, the targeting enhancer DNA accessibility is increased, and the transcription of target genes is activated. Inhibition of RIPK1 suppresses the phosphorylation of SMARCC2, which disrupts the co-occupancy of SMARCC2 and BRG1 on the target sites and as a result, suppresses the transcription of proinflammatory factors by reducing the enhancer DNA accessibility. Figures are created with BioRender.com.
Conclusions and remaining questions
The past two decades of research have provided ever expanding knowledge on RIPK1 biology, which led to the appreciation on the importance of RIPK1 activation in the cell types that are key mediators of proinflammatory response in a variety of pathological conditions, such as macrophages and microglia. Activation of RIPK1 has been shown to mediate not only apoptosis and necroptosis in TNF-stimulated condition but also the activation of proinflammatory responses (51, 90). While necroptosis has been shown to be proinflammatory, the functional consequence of RIPK1 activation in mediating transcriptional induction of inflammatory response independent of cell death may be mechanistically separated from circumstances where cell death is present. Recent studies have begun to demonstrate the mechanism by which nuclear RIPK1 directly participates in the transcriptions upon recruitment by specific transcriptional factors and chromatin remodeling BAF complexes independent of cell death (53). We suggest that since inflammation represents a reversible cellular state, unlike that of cell death which can lead to permanent loss or reduction of a certain cell population, targeting RIPK1 kinase for inhibiting its nuclear function in modulating BAF-mediated chromatin remodeling may represent a clinically important paradigm where inflammation is the dominant pathology. Human clinical studies using small molecule inhibitors of RIPK1 are currently underway for the treatment of human conditions that involve cell death and inflammation, e.g. ALS, AD and MS etc. (10). Importantly, the nuclear function of RIPK1 in driving inflammatory response suggests that small molecule inhibitors of RIPK1 should be considered for the treatment of human conditions that are inflammatory in nature and may not include cell death. However, many questions remain to be answered by future studies. For example, what might be the mechanism that regulates the translocation of RIPK1 from cytoplasm to nucleus? If and how the nuclear translocation of RIPK1 is coupled with its activation? What might be the substrates of RIPK1 in the nucleus? Can we manipulate the subcellular location of activated RIPK1 in order to modulate the process of RIPK1-dependent cell death and inflammation precisely? The answers to these questions will help us to further understand the mechanism by which RIPK1 regulates cell death and inflammation and better develop the therapeutic strategies for the treatment of human inflammatory and neurodegenerative diseases.
Author contributions
This review was written by JY and WL. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Natural Science Foundation of China (82188101, 91849204, 21837004, 92049303 and 32170755), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39030200), the Shanghai Municipal Science and Technology Major Project (2019SHZDZX02), the National Natural Science Youth Foundation of China (31801163), the Science and Technology Commission of Shanghai Municipality (18JC1420500, 22JC1410400) and the Shanghai Key Laboratory of Aging Studies (19DZ2260400).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
2. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med (2017) 23:1018–27. doi: 10.1038/nm.4397
3. Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of alzheimer's disease: Systematic review and meta-analysis of observational studies. Bmj (2003) 327:128. doi: 10.1136/bmj.327.7407.128
4. Hoozemans JJ, O'Banion MK. The role of COX-1 and COX-2 in alzheimer's disease pathology and the therapeutic potentials of non-steroidal anti-inflammatory drugs. Curr Drug Targets CNS Neurol Disord (2005) 4:307–15. doi: 10.2174/1568007054038201
5. in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of alzheimer's disease. N Engl J Med (2001) 345:1515–21. doi: 10.1056/NEJMoa010178
6. Villarejo-Galende A, Gonzalez-Sanchez M, Blanco-Palmero VA, Llamas-Velasco S, Benito-Leon J. Non-steroidal anti-inflammatory drugs as candidates for the prevention or treatment of alzheimer's disease: Do they still have a role? Curr Alzheimer Res (2020) 17:1013–22. doi: 10.2174/1567205017666201127163018
7. Cifone MG, Migliorati G, Parroni R, Marchetti C, Millimaggi D, Santoni A, et al. Dexamethasone-induced thymocyte apoptosis: Apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase c, acidic sphingomyelinase, and caspases. Blood (1999) 93:2282–96. doi: 10.1182/blood.V93.7.2282
8. Garvy BA, King LE, Telford WG, Morford LA, Fraker PJ. Chronic elevation of plasma corticosterone causes reductions in the number of cycling cells of the b lineage in murine bone marrow and induces apoptosis. Immunology (1993) 80:587–92.
9. Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, et al. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev (2011) 10:305–10. doi: 10.1016/j.autrev.2010.11.009
10. Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discovery (2020) 19:553–71. doi: 10.1038/s41573-020-0071-y
11. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
12. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol (2013) 14:727–36. doi: 10.1038/nrm3683
13. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science (2016) 352:aaf2154. doi: 10.1126/science.aaf2154
14. Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol (2014) 193:1539–43. doi: 10.4049/jimmunol.1400590
15. Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol (2014) 192:5476–80. doi: 10.4049/jimmunol.1400499
16. Meng H, Liu Z, Li X, Wang H, Jin T, Wu G, et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci USA (2018) 115:E2001–9. doi: 10.1073/pnas.1722013115
17. Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun (2020) 11:1747. doi: 10.1038/s41467-020-15466-8
18. Blanchett S, Dondelinger Y, Barbarulo A, Bertrand MJM, Seddon B. Phosphorylation of RIPK1 serine 25 mediates IKK dependent control of extrinsic cell death in T cells. Front Immunol (2022) 13:1067164. doi: 10.3389/fimmu.2022.1067164
19. Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, et al. Structural basis of RIP1 inhibition by necrostatins. Structure (2013) 21:493–9. doi: 10.1016/j.str.2013.01.016
20. Chen L, Zhang X, Ou Y, Liu M, Yu D, Song Z, et al. Advances in RIPK1 kinase inhibitors. Front Pharmacol (2022) 13:976435. doi: 10.3389/fphar.2022.976435
21. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol (2008) 4:313–21. doi: 10.1038/nchembio.83
22. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA (2019) 116:9714–22.
23. Vissers M, Heuberger J, Groeneveld GJ, Oude Nijhuis J, De Deyn PP, Hadi S, et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: Randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin Transl Sci (2022) 15:2010–23. doi: 10.1111/cts.13317
24. Weisel K, Scott N, Berger S, Wang S, Brown K, Powell M, et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol (2021) 8:1–9. doi: 10.1136/bmjgast-2021-000680
25. Grievink HW, Heuberger J, Huang F, Chaudhary R, Birkhoff WAJ, Tonn GR, et al. DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther (2020) 107:406–14. doi: 10.1002/cpt.1615
26. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes Dev (2018) 32:327–40. doi: 10.1101/gad.312561.118
27. Wertz IE, Dixit VM. Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev (2008) 19:313–24. doi: 10.1016/j.cytogfr.2008.04.014
28. Peltzer N, Darding M, Walczak H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol (2016) 26:445–61. doi: 10.1016/j.tcb.2016.01.006
29. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol (2018) 20:1389–99. doi: 10.1038/s41556-018-0229-6
30. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell (2018) 174:1477–1491 e19. doi: 10.1016/j.cell.2018.07.041
31. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
32. Li X, Zhang M, Huang X, Liang W, Li G, Lu X, et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat Commun (2020) 11:6364. doi: 10.1038/s41467-020-19935-y
33. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell (2014) 157:1189–202. doi: 10.1016/j.cell.2014.04.018
34. Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PloS One (2013) 8:e76841.
35. Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol (2011) 12:439–52. doi: 10.1038/nrm3143
36. Li X, Zhong CQ, Wu R, Xu X, Yang ZH, Cai S, et al. RIP1-dependent linear and nonlinear recruitments of caspase-8 and RIP3 respectively to necrosome specify distinct cell death outcomes. Protein Cell (2021) 12:858–76. doi: 10.1007/s13238-020-00810-x
37. Varfolomeev E, Vucic D. RIP1 post-translational modifications. Biochem J (2022) 479:929–51. doi: 10.1042/BCJ20210725
38. Xu D, Zhao H, Jin M, Zhu H, Shan B, Geng J, et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature (2020) 587:133–8. doi: 10.1038/s41586-020-2757-z
39. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
40. Kist M, Komuves LG, Goncharov T, Dugger DL, Yu C, Roose-Girma M, et al. Impaired RIPK1 ubiquitination sensitizes mice to TNF toxicity and inflammatory cell death. Cell Death Differ (2021) 28:985–1000. doi: 10.1038/s41418-020-00629-3
41. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. doi: 10.1038/nature14191
42. Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem (2016) 85:743–63. doi: 10.1146/annurev-biochem-060815-014830
43. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18:127–36. doi: 10.1038/nrm.2016.149
44. Patel S, Webster JD, Varfolomeev E, Kwon YC, Cheng JH, Zhang J, et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ (2020) 27:161–75. doi: 10.1038/s41418-019-0347-0
45. Christofferson DE, Li Y, Zhou W, Hitomi J, Upperman C, Zhu H, et al. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis (2012) 3:e320.
46. Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature (2020) 577:103–8. doi: 10.1038/s41586-019-1828-5
47. Muendlein HI, Sarhan J, Liu BC, Connolly WM, Schworer SA, Smirnova I, et al. Constitutive interferon attenuates RIPK1/3-mediated cytokine translation. Cell Rep (2020) 30:699–713 e4. doi: 10.1016/j.celrep.2019.12.073
48. Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature (2019) 574:428–31. doi: 10.1038/s41586-019-1548-x
49. Speir M, Lawlor KE. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin Cell Dev Biol (2021) 109:114–24. doi: 10.1016/j.semcdb.2020.07.011
50. Zelic M, Pontarelli F, Woodworth L, Zhu C, Mahan A, Ren Y, et al. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep (2021) 35:109112. doi: 10.1016/j.celrep.2021.109112
51. Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X, et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis (2018) 9:500. doi: 10.1038/s41419-018-0524-y
52. Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature (2020) 577:109–14. doi: 10.1038/s41586-019-1830-y
53. Li W, Shan B, Zou C, Wang H, Zhang MM, Zhu H, et al. Nuclear RIPK1 promotes chromatin remodeling to mediate inflammatory response. Cell Res (2022) 32:621–37. doi: 10.1038/s41422-022-00673-3
54. Martin F, Dixit VM. A20 edits ubiquitin and autoimmune paradigms. Nat Genet (2011) 43:822–3. doi: 10.1038/ng.916
55. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol (2001) 21:5299–305. doi: 10.1128/MCB.21.16.5299-5305.2001
56. Wertz I, Dixit V. A20–a bipartite ubiquitin editing enzyme with immunoregulatory potential. Adv Exp Med Biol (2014) 809:1–12. doi: 10.1007/978-1-4939-0398-6_1
58. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev (1999) 13:2514–26. doi: 10.1101/gad.13.19.2514
59. Zhang Y, Huang K, Zhang Y, Han T, Li L, Ruan C, et al. A unique death pathway keeps RIPK1 D325A mutant mice in check at embryonic day 10.5. PloS Biol (2021) 19:e3001304.
60. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature (2011) 471:368–72. doi: 10.1038/nature09857
61. Zhang X, Dowling JP, Zhang J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis (2019) 10:245. doi: 10.1038/s41419-019-1490-8
62. Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity (2011) 35:908–18. doi: 10.1016/j.immuni.2011.09.020
63. Garcia-Carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL, et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc Natl Acad Sci USA (2018) 115:E9192–200. doi: 10.1073/pnas.1810584115
64. Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat Cell Biol (2019) 21:731–42. doi: 10.1038/s41556-019-0324-3
65. Weisel K, Berger S, Papp K, Maari C, Krueger JG, Scott N, et al. Response to inhibition of receptor-interacting protein kinase 1 (RIPK1) in active plaque psoriasis: A randomized placebo-controlled study. Clin Pharmacol Ther (2020) 108:808–16. doi: 10.1002/cpt.1852
66. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
67. Peltzer N, Darding M, Montinaro A, Draber P, Draberova H, Kupka S, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature (2018) 557:112–7. doi: 10.1038/s41586-018-0064-8
68. Peltzer N, Walczak H. Cell death and inflammation - a vital but dangerous liaison. Trends Immunol (2019) 40:387–402. doi: 10.1016/j.it.2019.03.006
69. Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature (2011) 471:633–6. doi: 10.1038/nature09815
70. HogenEsch H, Gijbels MJ, Offerman E, van Hooft J, van Bekkum DW, Zurcher C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am J Pathol (1993) 143:972–82.
71. Webster JD, Kwon YC, Park S, Zhang H, Corr N, Ljumanovic N, et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J Leukoc Biol (2020) 107:941–52. doi: 10.1002/JLB.3MA1219-398R
72. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell (2013) 153:1312–26. doi: 10.1016/j.cell.2013.05.014
73. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell (2016) 166:1215–1230 e20. doi: 10.1016/j.cell.2016.07.019
74. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA (2016) 113:10127–32.
75. Schunke H, Gobel U, Dikic I, Pasparakis M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat Commun (2021) 12:5912. doi: 10.1038/s41467-021-25945-1
76. McCauley ME, Baloh RH. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol (2019) 137:715–30. doi: 10.1007/s00401-018-1933-9
77. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (2016) 353:603–8. doi: 10.1126/science.aaf6803
78. Mifflin L, Hu Z, Dufort C, Hession CC, Walker AJ, Niu K, et al. A RIPK1-regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc Natl Acad Sci USA (2021) 118. doi: 10.1073/pnas.2025102118
79. Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, et al. Activation of necroptosis in multiple sclerosis. Cell Rep (2015) 10:1836–49. doi: 10.1016/j.celrep.2015.02.051
80. Pegoretti V, Baron W, Laman JD, Eisel ULM. Selective modulation of TNF-TNFRs signaling: Insights for multiple sclerosis treatment. Front Immunol (2018) 9:925. doi: 10.3389/fimmu.2018.00925
81. Gregory AP, Dendrou CA, Attfield KE, Haghikia A, Xifara DK, Butter F, et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature (2012) 488:508–11. doi: 10.1038/nature11307
82. International Multiple Sclerosis Genetics C. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science (2019) 365.
83. Yoshikawa M, Saitoh M, Katoh T, Seki T, Bigi SV, Shimizu Y, et al. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 h-pyrazolo[3,4- c]pyridine derivatives as potent, orally available, and brain-penetrating receptor interacting protein 1 (RIP1) kinase inhibitors: Analysis of structure-kinetic relationships. J Med Chem (2018) 61:2384–409. doi: 10.1021/acs.jmedchem.7b01647
84. Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, et al. RIPK1 mediates a disease-associated microglial response in alzheimer's disease. Proc Natl Acad Sci USA (2017) 114:E8788–97. doi: 10.1073/pnas.1714175114
85. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of alzheimer's disease. Cell (2017) 169:1276–1290 e17. doi: 10.1016/j.cell.2017.05.018
86. Xu G, Li Y, Zhang S, Peng H, Wang Y, Li D, et al. SARS-CoV-2 promotes RIPK1 activation to facilitate viral propagation. Cell Res (2021) 31:1230–43. doi: 10.1038/s41422-021-00578-7
87. Riebeling T, Jamal K, Wilson R, Kolbrink B, von Samson-Himmelstjerna FA, Moerke C, et al. Primidone blocks RIPK1-driven cell death and inflammation. Cell Death Differ (2021) 28:1610–26. doi: 10.1038/s41418-020-00690-y
88. Wenzel J, Lampe J, Muller-Fielitz H, Schuster R, Zille M, Muller K, et al. The SARS-CoV-2 main protease m(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci (2021) 24:1522–33. doi: 10.1038/s41593-021-00926-1
89. Hameedi MA, Prates ET, Garvin MR, Mathews II, Amos BK, Demerdash O, et al. Structural and functional characterization of NEMO cleavage by SARS-CoV-2 3CLpro. Nat Commun (2022) 13:5285. doi: 10.1038/s41467-022-32922-9
90. Huang X, Tan S, Li Y, Cao S, Li X, Pan H, et al. Caspase inhibition prolongs inflammation by promoting a signaling complex with activated RIPK1. J Cell Biol (2021) 220. doi: 10.1083/jcb.202007127
91. Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem (2005) 280:40599–608. doi: 10.1074/jbc.M509560200
92. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412:346–51. doi: 10.1038/35085597
93. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8:359. doi: 10.1038/s41467-017-00406-w
94. Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun (2019) 10:1729. doi: 10.1038/s41467-019-09690-0
95. Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, et al. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol (2014) 204:607–23. doi: 10.1083/jcb.201305070
96. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med (2007) 13:460–9. doi: 10.1016/j.molmed.2007.09.002
97. Taft J, Markson M, Legarda D, Patel R, Chan M, Malle L, et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell (2021) 184:4447–4463 e20. doi: 10.1016/j.cell.2021.07.026
98. Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J (2000) 19:4976–85. doi: 10.1093/emboj/19.18.4976
99. Yu X, Ma H, Li B, Ji Y, Du Y, Liu S, et al. A novel RIPK1 inhibitor reduces GVHD in mice via a non-immunosuppressive mechanism that restores intestinal homeostasis. Blood (2022). doi: 10.1182/blood.2022017262
100. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347:1436–41. doi: 10.1126/science.aaa3650
101. Freischmidt A, Muller K, Ludolph AC, Weishaupt JH, Andersen PM. Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol (2017) 74:110–3. doi: 10.1001/jamaneurol.2016.3712
102. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol (2015) 130:77–92. doi: 10.1007/s00401-015-1436-x
103. Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol (2005) 6:439–45. doi: 10.1038/ni1196
104. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res (2011) 21:396–420. doi: 10.1038/cr.2011.32
105. Sokpor G, Xie Y, Rosenbusch J, Tuoc T, Chromatin Remodeling BAF. (SWI/SNF) complexes in neural development and disorders. Front Mol Neurosci (2017) 10:243. doi: 10.3389/fnmol.2017.00243
106. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem (2009) 78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223
107. Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci (2009) 34:71–7. doi: 10.1016/j.tibs.2008.10.010
108. Lai AY, Wade PA. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat Rev Cancer (2011) 11:588–96. doi: 10.1038/nrc3091
109. Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol (2000) 20:1899–910. doi: 10.1128/MCB.20.6.1899-1910.2000
110. Yen K, Vinayachandran V, Pugh BF. SWR-c and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell (2013) 154:1246–56. doi: 10.1016/j.cell.2013.08.043
111. Ho L, Crabtree GR. Chromatin remodelling during development. Nature (2010) 463:474–84. doi: 10.1038/nature08911
112. He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, et al. Structure of nucleosome-bound human BAF complex. Science (2020) 367:875–81. doi: 10.1126/science.aaz9761
113. Baranov E, McBride MJ, Bellizzi AM, Ligon AH, Fletcher CDM, Kadoch C, et al. A novel SS18-SSX fusion-specific antibody for the diagnosis of synovial sarcoma. Am J Surg Pathol (2020) 44:922–33. doi: 10.1097/PAS.0000000000001447
114. McBride MJ, Pulice JL, Beird HC, Ingram DR, D'Avino AR, Shern JF, et al. The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell (2018) 33:1128–1141 e7. doi: 10.1016/j.ccell.2018.05.002
115. Pattenden SG, Klose R, Karaskov E, Bremner R. Interferon-gamma-induced chromatin remodeling at the CIITA locus is BRG1 dependent. EMBO J (2002) 21:1978–86. doi: 10.1093/emboj/21.8.1978
116. Pray-Grant MG, Daniel JA, Schieltz D, Yates J, Grant PA. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature (2005) 433:434–8. doi: 10.1038/nature03242
117. Lee HS, Park MH, Yang SJ, Jung HY, Byun SS, Lee DS, et al. Gene expression analysis in human gastric cancer cell line treated with trichostatin a and s-adenosyl-L-homocysteine using cDNA microarray. Biol Pharm Bull (2004) 27:1497–503. doi: 10.1248/bpb.27.1497
118. Tartey S, Takeuchi O. Chromatin remodeling and transcriptional control in innate immunity: Emergence of Akirin2 as a novel player. Biomolecules (2015) 5:1618–33. doi: 10.3390/biom5031618
119. Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell (2009) 138:114–28. doi: 10.1016/j.cell.2009.04.020
120. Ramirez-Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN, et al. Selective and antagonistic functions of SWI/SNF and mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev (2006) 20:282–96. doi: 10.1101/gad.1383206
121. Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, et al. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell (1998) 95:625–36. doi: 10.1016/S0092-8674(00)81633-5
122. Feoktistov AV, Georgieva SG, Soshnikova NV. [Role of the SWI/SNF chromatin remodeling complex in regulation of inflammation gene expression]. Mol Biol (Mosk) (2022) 56:244–58. doi: 10.1134/S0026893322020054
123. Weinmann AS, Mitchell DM, Sanjabi S, Bradley MN, Hoffmann A, Liou HC, et al. Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, rel-independent event. Nat Immunol (2001) 2:51–7. doi: 10.1038/83168
124. Saccani S, Pantano S, Natoli G. p38-dependent marking of inflammatory genes for increased NF-kappa b recruitment. Nat Immunol (2002) 3:69–75. doi: 10.1038/ni748
125. Huang M, Wang H, Hu X, Cao X. lncRNA MALAT1 binds chromatin remodeling subunit BRG1 to epigenetically promote inflammation-related hepatocellular carcinoma progression. Oncoimmunology (2019) 8:e1518628. doi: 10.1080/2162402X.2018.1518628
126. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi: 10.1038/nature05836
127. Wurster AL, Pazin MJ. BRG1-mediated chromatin remodeling regulates differentiation and gene expression of T helper cells. Mol Cell Biol (2008) 28:7274–85. doi: 10.1128/MCB.00835-08
128. Letimier FA, Passini N, Gasparian S, Bianchi E, Rogge L. Chromatin remodeling by the SWI/SNF-like BAF complex and STAT4 activation synergistically induce IL-12Rbeta2 expression during human Th1 cell differentiation. EMBO J (2007) 26:1292–302. doi: 10.1038/sj.emboj.7601586
129. Zhang F, Boothby M. T Helper type 1-specific Brg1 recruitment and remodeling of nucleosomes positioned at the IFN-gamma promoter are Stat4 dependent. J Exp Med (2006) 203:1493–505. doi: 10.1084/jem.20060066
130. Chi TH, Wan M, Lee PP, Akashi K, Metzger D, Chambon P, et al. Sequential roles of brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity (2003) 19:169–82. doi: 10.1016/S1074-7613(03)00199-7
131. Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Littman DR, et al. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature (2002) 418:195–9. doi: 10.1038/nature00876
132. Jani A, Wan M, Zhang J, Cui K, Wu J, Preston-Hurlburt P, et al. A novel genetic strategy reveals unexpected roles of the swi-snf-like chromatin-remodeling BAF complex in thymocyte development. J Exp Med (2008) 205:2813–25. doi: 10.1084/jem.20080938
133. Wan M, Zhang J, Lai D, Jani A, Prestone-Hurlburt P, Zhao L, et al. Molecular basis of CD4 repression by the Swi/Snf-like BAF chromatin remodeling complex. Eur J Immunol (2009) 39:580–8. doi: 10.1002/eji.200838909
134. Chaiyachati BH, Jani A, Wan Y, Huang H, Flavell R, Chi T. BRG1-mediated immune tolerance: Facilitation of treg activation and partial independence of chromatin remodelling. EMBO J (2013) 32:395–408. doi: 10.1038/emboj.2012.350
135. Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature (2004) 430:218–22. doi: 10.1038/nature02738
136. Tando T, Ishizaka A, Watanabe H, Ito T, Iida S, Haraguchi T, et al. Requiem protein links RelB/p52 and the brm-type SWI/SNF complex in a noncanonical NF-kappaB pathway. J Biol Chem (2010) 285:21951–60. doi: 10.1074/jbc.M109.087783
137. Hu G, Gong AY, Wang Y, Ma S, Chen X, Chen J, et al. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J Immunol (2016) 196:2799–808. doi: 10.4049/jimmunol.1502146
138. Kawaguchi T, Tanigawa A, Naganuma T, Ohkawa Y, Souquere S, Pierron G, et al. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc Natl Acad Sci USA (2015) 112:4304–9.
139. Zhu Y, Rowley MJ, Bohmdorfer G, Wierzbicki AT. A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol Cell (2013) 49:298–309. doi: 10.1016/j.molcel.2012.11.011
140. Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep (2018) 24:1722–9. doi: 10.1016/j.celrep.2018.07.041
141. Park J, Wood MA, Cole MD. BAF53 forms distinct nuclear complexes and functions as a critical c-myc-interacting nuclear cofactor for oncogenic transformation. Mol Cell Biol (2002) 22:1307–16. doi: 10.1128/MCB.22.5.1307-1316.2002
142. Fukasawa R, Tsutsui T, Hirose Y, Tanaka A, Ohkuma Y. Mediator CDK subunits are platforms for interactions with various chromatin regulatory complexes. J Biochem (2012) 152:241–9. doi: 10.1093/jb/mvs065
143. Cho H, Orphanides G, Sun X, Yang XJ, Ogryzko V, Lees E, et al. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol Cell Biol (1998) 18:5355–63. doi: 10.1128/MCB.18.9.5355
144. Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol (2018) 1:6. doi: 10.1038/s42003-017-0007-1
145. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ (2016) 23:253–60. doi: 10.1038/cdd.2015.92
146. Jang KH, Jang T, Son E, Choi S, Kim E. Kinase-independent role of nuclear RIPK1 in regulating parthanatos through physical interaction with PARP1 upon oxidative stress. Biochim Biophys Acta Mol Cell Res (2018) 1865:132–41. doi: 10.1016/j.bbamcr.2017.10.004
147. Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell (2014) 56:219–31. doi: 10.1016/j.molcel.2014.08.024
148. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood (2003) 101:2652–60. doi: 10.1182/blood-2002-05-1300
149. Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC. JUNB is a key transcriptional modulator of macrophage activation. J Immunol (2015) 194:177–86. doi: 10.4049/jimmunol.1401595
150. Fujihara M, Muroi M, Muroi Y, Ito N, Suzuki T. Mechanism of lipopolysaccharide-triggered junB activation in a mouse macrophage-like cell line (J774). J Biol Chem (1993) 268:14898–905. doi: 10.1016/S0021-9258(18)82418-8
151. Tak PP, Firestein GS. NF-kappaB: A key role in inflammatory diseases. J Clin Invest (2001) 107:7–11. doi: 10.1172/JCI11830
152. Ho PJ, Lloyd SM, Bao X. Unwinding chromatin at the right places: How BAF is targeted to specific genomic locations during development. Development (2019) 146. doi: 10.1242/dev.178780
Keywords: RIPK1, inflammation, chromatin remodeling, caspase, necroptosis, apoptosis
Citation: Li W and Yuan J (2023) Targeting RIPK1 kinase for modulating inflammation in human diseases. Front. Immunol. 14:1159743. doi: 10.3389/fimmu.2023.1159743
Received: 06 February 2023; Accepted: 27 February 2023;
Published: 08 March 2023.
Edited by:
Liming Sun, Shanghai Institute of Biochemistry and Cell Biology (CAS), ChinaReviewed by:
Xin Chen, Xiamen University, ChinaSudan He, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2023 Li and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wanjin Li, wanjinli@sioc.ac.cn; Junying Yuan, junying_yuan@sioc.ac.cn