 Ian M. Cartwright1,2,3,4*
Ian M. Cartwright1,2,3,4* Sean P. Colgan1,2,3,4
Sean P. Colgan1,2,3,4- 1Division of Gastroenterology and Hepatology, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Department of Medicine, University of Colorado School of Medicine, Aurora, CO, United States
- 3Mucosal Inflammation Program, University of Colorado School of Medicine, Aurora, CO, United States
- 4Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, CO, United States
On the backdrop of all acute inflammatory processes lies the activation of the resolution response. Recent years have witnessed an emerging interest in defining molecular factors that influence the resolution of inflammation. A keystone feature of the mucosal inflammatory microenvironment is hypoxia. The gastrointestinal tract, particularly the colon, exists in a state of physiological hypoxia and during active inflammation, this hypoxic state is enhanced as a result of infiltrating leukocyte oxygen consumption and the activation of oxygen consuming enzymes. Most evidence suggests that mucosal hypoxia promotes the active resolution of inflammation through a variety of mechanisms, including extracellular acidification, purine biosynthesis/salvage, the generation of specialized pro-resolving lipid mediators (ie. resolvins) and altered chemokine/cytokine expression. It is now appreciated that infiltrating innate immune cells (neutrophils, eosinophils, macrophages) have an important role in molding the tissue microenvironment to program an active resolution response. Structural or functional dysregulation of this inflammatory microenvironment can result in the loss of tissue homeostasis and ultimately progression toward chronicity. In this review, we will discuss how inflammatory hypoxia drives mucosal inflammatory resolution and its impact on other microenvironmental factors that influence resolution.
Introduction
The gastrointestinal (GI) tract is a highly complex tissue lined by an epithelium that covers a surface area of approximately 300m2 (1). This mucosal surface has a unique role in regulating contact between the mucosal immune system and the external environment. The GI tract is home to a diverse and densely populated microbiota and the epithelium forms an important barrier which prevents unregulated exposure of luminal antigens with the lamina propria which houses the mucosal immune system (2, 3). In recent years the view of the epithelium as a static barrier has changed. The epithelium harbors intrinsic innate immunity, regulates antigen sensitization, and molds the microbiota (4–6).
It is widely accepted that the GI tract is in a constant state of low-grade inflammation (7). This state of low-grade inflammation is primed by the constant processing of luminal antigenic material and allows for rapid mobilization of the mucosal immune system to antigens and microbes that penetrate the barrier. The inflammatory process is an essential protective response to pathogens, foreign objects, and injury. This response includes increased vascular dilation, changes in capillary permeability, and leukocyte recruitment (8). As seen in other tissues, an essential component of the innate immune response in the GI tract is the recruitment of polymorphonuclear leukocytes (PMN) to the site of infection or injury.REF Along with the intact epithelium, PMN serve at the front line of defense against microbial pathogens. PMN comprise 50-60% of the circulating leukocytes, making them the most abundant leukocyte population in the blood (9, 10). Once in the tissue, PMN perform a variety of antimicrobial functions; including, degranulation and phagocytosis making them a critical component of the bodies first line of defense (11).
To prevent the development of chronic disease, it is essential for the inflammatory process to be highly regulated and that signals to resolve are present so the tissue can return to a healthy state following the inflammatory insult (12). A critical aspect of the resolution response is the clearance of apoptotic PMN, which is completed by monocytes and macrophages (13). PMN apoptosis is an essential component to productive inflammatory resolution. As a point of fact, a major source of tissue damage in inflammatory conditions, including asthma, rheumatoid arthritis, and inflammatory bowel disease (IBD), is attributed to uncleared PMN (14–16). To ensure proper clearance of PMN at sites of acute inflammation, the mammals have evolved complex and highly coordinated process involving anti-inflammatory cytokines, suppression of pro-inflammatory receptors, induction of pro-resolving mediators, and shifts in the inflammatory microenvironment that all benefit a return to homeostasis (17).
It is now well established that inflammatory hypoxia is a direct result of PMN rapid metabolism of O2 as the enter the tissue and activate. All tissue compartments are impacted by inflammatory hypoxia, including infiltrating PMN and other immune cells. In recent years there has been a growing appreciation for the role that PMN play in molding the inflammatory microenvironment and that many changes induced by PMN migration prime the microenvironment for resolution (18). PMN infiltration alters extracellular adenine nucleotide concentrations, drives inflammatory acidification, molds chemokine/cytokine signaling, and induces the expression of pro-resolution lipid mediators (19–22). There is a growing body of literature that indicates that effective mucosal responses to inflammation are driven by changes in the tissue microenvironment. Here, we review some of these changes in the inflammatory microenvironment and how resolution of inflammation is influenced by inflammatory hypoxia, which is driven primarily by PMN infiltration (23).
Hypoxia and oxygen homeostasis
The gastrointestinal tract presents a unique microenvironment in which there is a steep oxygen gradient between the anaerobic lumen and the highly vascularized and oxygenated subepithelium. At sea level the partial O2 pressure (pO2) of the air we breathe is ~145 mm Hg and within the alveolus of health lungs the pO2 has a range between 100-110 mmHg (24). In stark contrast, the lumen of a healthy intestine exists at a pO2 of ~10 mm Hg (25–27). Extensive work using 2-nitroimidazole dyes, a class of compounds which can be utilized to image low-O2 environments, has shown that in normal GI mucosa, most notably in the colon, the tissue resides in a state of physiological hypoxia (Figure 1). The use of these dyes has revealed that inflammatory lesions in mouse models of colitis show enhanced hypoxia, and even anoxia, and that such hypoxia extends deep into the mucosal tissue (Figure 1) (28). This inflammatory hypoxia results from a combination of factors including recruitment of inflammatory cells (PMN, eosinophils, and monocytes), elevated oxidative metabolism, and activation of oxygen-consuming enzymes (29). The use of surrogate markers of hypoxia (e.g. GLUT1) have demonstrated a strong correlation between PMN accumulation and hypoxia in human IBD tissue (Figure 1).
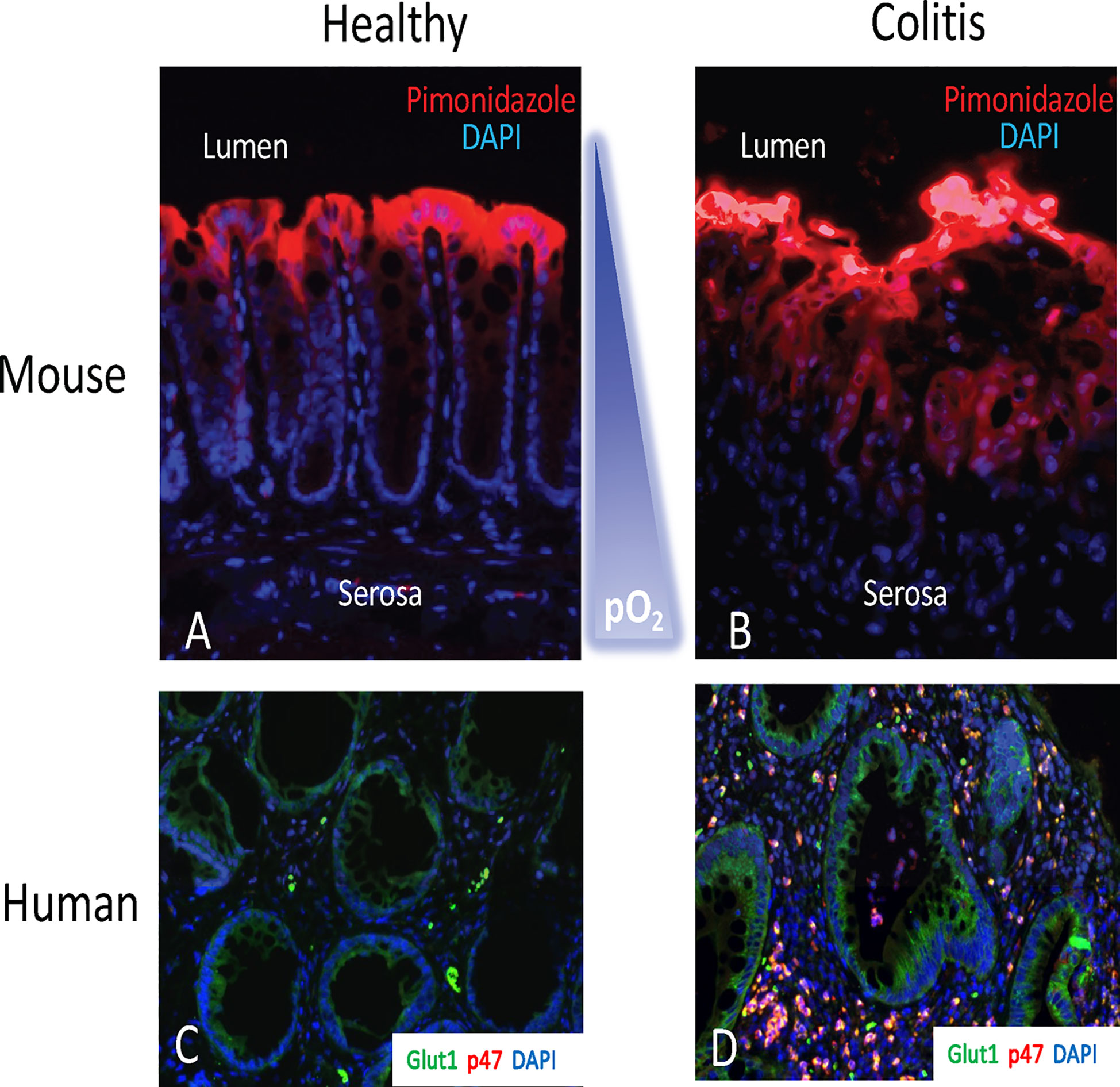
Figure 1 The hypoxic tissue microenvironment in the mucosa. Ulcerative colitis patients with crypt abscesses demonstrate hypoxia-dependent target induction. Panels (A, B) depict localization of hypoxic regions using pimonidazole staining (red) with nuclear counterstain (blue) in healthy mouse colon (A) and active murine coltis (B). Note the gradient of hypoxia (red) from lumen to serosa. In panels (C, D), colon biopsies from uninflamed margins (C) or inflamed regions with active crypt abscess (D) in patients with ulcerative colitis were processed for hypoxia-responsive Glut-1 (green), neutrophil p47phox (red) and nuclei (blue).
Given the physiological hypoxia and near anoxic conditions observed during inflammation, several studies have examined the impact of hypoxia-inducible factor (HIF) stabilization under low O2 conditions on epithelial gene expression, specifically promoting epithelial barrier function (27, 30, 31). There are three α isoforms of HIF (HIF-1α, HIF-2α, and HIF-3α). These isoforms are Per-ARNT-Sim (PAS) members of the basic helix-loop-helix (bHLH) family of transcriptions factors (32). Under hypoxic conditions, the α subunit is stabilized in the cytoplasm and following nuclear translocation, the α subunit forms a functional heterodimetric complex with the β subunit HIF-1β (also referred to as aryl hydrocarbon receptor nuclear translocator (ARNT)) (33, 34). In the setting of colitis, HIF-1α has been shown to be essential for regulating the inflammatory process. Epithelial specific loss of HIF-1α results in a significantly more severe colitis then observed in wild-type animals. HIF-1α deficient animals have increased weight loss, decreased colon length, and greatly increased intestinal permeability. In support of these observations, it was also observed that constitutively active intestinal epithelial HIF was protective in murine models of colitis (26). Further studies have demonstrated that HIF-1α stabilization in inflamed tissue promotes the expression of mucins, antimicrobial peptides, and tight junction proteins, all factors that play a pivotal role in limiting inflammation and promoting resolution (35).
PMN have been shown to have an important role in depleting the local O2 in inflamed tissue (3). As PMN transmigrate across the epithelium and become activated, they deplete the local O2. This depletion of O2 is driven by PMN NADPH oxidase and ultimately stabilizes HIF-1α within the epithelium. Studies examining the loss of neutrophil NADPH oxidase and depletion of PMN demonstrated that both the presence of PMN and PMN-induced hypoxia were required for mucosal protection during inflammation and resolution of inflammation (36, 37). The maintenance of barrier function is essential during inflammation and is pivotal to controlling the inflammatory response. In individuals with IBD, marked impairments in tight junction structure and function result in increased epithelial permeability (38). HIF-1α has a critical role in maintaining barrier function and mice deficient in HIF-1α have increase intestinal permeability and barrier function in the context of DSS colitis (26). It has been shown that one of the critical components of the intestinal epithelial tight junction complex, claudin-1, is a HIF-1α transcriptional target, where intestinal epithelial cells lacking HIF-1α manifest significant barrier defects (39). The role of HIF in barrier function during inflammation can also been shown in studies that have stabilized HIF-1α with the use of pharmacological inhibitors targeting polyl hydroxylase (PHD) or genetic deletion of the von Hippel-Lindau gene. In both situations, increased stabilization of HIF-1α results in increased barrier function in the context of colitis (26, 40, 41). Furthermore, HIF-1α stabilization has been demonstrated to induce the expression of several pro-resolution factors, including IL-33 and IL-10, and down regulation of pro-inflammatory cytokines and chemokines in both leukocytes and epithelial cells (42–44). The role of cytokines in the resolution response is discussed later in the review.
Extracellular adenosine generation and signaling
Another area of considerable interest is PMN-associated adenosine (Ado) signaling in inflammatory resolution (45). It is well established that PMN are a prominent reservoir for Ado precursors and that during inflammation PMN actively release ATP and ADP into the microenvironment (46). More recently, it has been shown that neutrophils are a significant source of diadenosine triphosphate (Ap3A), which is metabolized to ADP by ectonucleotide pyrophosphate/phosphodiesterase-1 (ENPP1) providing an additional source of nucleotides during inflammation (22). It is now widely accepted that ecto-nucleoside triphosphate diphosphohydrolase (NTPDase, CD39) and ecto-5’-nucleotidase (CD73) provide the major pathways for extracellular hydrolysis of ATP and ADP, allowing for the accumulation of extracellular Ado (31, 47, 48). Extracellular Ado can interact with cell surface Ado receptors on the intestinal epithelial cells (49). Currently, four subtypes of G protein-coupled Ado receptors have been identified, AA1R, AA2AR, AA2BR, and AA3R (49).
Several aspects of Ado signaling have been shown to be regulated by hypoxia. As previously discussed, PMN infiltrate is accompanied increases in extracellular adenine nucleotides. It has been shown that enzymes responsible for converting these adenine nucleotides, CD39 and CD73, are regulated by hypoxia. CD39 expression is induced under hypoxic conditions in a SP-1 dependent manner (50, 51). Likewise, CD73 is transcriptionally induced by the direct binding of HIF-1α to the promoter region of CD73, resulting in increased CD73 protein under hypoxic conditions (31, 52). This increase in the potential of intestinal epithelial cells to produce Ado from extracellular ATP and other adenine nucleotides is an important aspect of initiating the resolution response. ATP has been shown to be a proinflammatory molecule and that excessive levels of extracellular ATP can result in systemic inflammatory response syndromes, cytokine storms (53). In addition to enhancing the production of ado, it has also been shown that under hypoxic conditions the expression of the predominant Ado receptor in intestinal epithelial cells, AA2BR, is upregulated. Within the promoter of AA2BR there is a Hypoxia Response Element (HRE) and under hypoxic conditions HIF-1α binds to this site and increases AA2BR transcript expression. Further investigations determined that the increase in transcript was followed by increased protein expression and an overall increase in AA2BR function (54). These increases in Ado generation and signaling capacity are essential for the promotion of resolution in inflamed tissue.
Ado plays a critical role in maintaining tissue barrier function during inflammation. As PMN transmigrate across epithelium there is localized widening of the inter-junctional space which is associated with loosening of the tight junctional protein complex (55). As the PMN move into the luminal space they are actively releasing ATP and ADP, which when convert by epithelial cells into adenosine signals through the AA2BR receptor to stimulate the resealing of the epithelial barrier (56). This resealing is driven by increases in intracellular cyclic AMP (cAMP) following activation of AA2BR by adenosine. The increase in cAMP activates protein kinase A which phosphorylates vasodilator-stimulated phosphoprotein (VASP). Phospho-VASP localizes with ZO-1 and promotes barrier recovery following PMN transmigration (57, 58). Subsequent studies have revealed that VASP mutants with non-functional actin-binding domains results in diminished barrier recovery (58).
In addition to promoting barrier function during inflammation, it has also been reported that adenine nucleotides, including Ado, serve as a primitive defense against bacterial pathogens by promoting a mucosal flushing response. It was first reported in vitro that exposure to various adenine nucleotides metabolites induced the apical secretion of Cl- in cultured intestinal epithelial cells (59). These adenine nucleotides are converted to Ado which activates the AA2BR and increases intracellular cyclic adenosine monophosphate (cAMP). This increase in cAMP activates PKA which phosphorylates the cystic fibrosis transmembrane conductance regulator (CFTR) which pumps Cl- into the luminal space, promoting movement of H20 across the epithelial layer (60–62). Within the airway it has been established that loss of CFTR function results in chronic bacterial lung infections (63). Similar observations have been made in the intestines where it was shown that Escherichia coli and Salmonella enteritidis suppress Cl- secretion and inhibit fluid transport (64, 65). Interestingly, in mice infected with Salmonella enteritidis the CFTR was internalized in the crypts of the colon and overall ion transport was decreased, suggesting the observed diarrhea can be attributed to dysfunctions in the epithelial cells ability to absorb H20, not the active secretion of Cl- (65). Furthermore, it has been reported that intestinal epithelial cell specific CFTR promotes the outgrowth of pro-inflammatory bacterial species including Heliobacter typhlonius and Clostridium perfringens (66).
Finally, it has been shown that intestinal epithelial cells exposed to Ado have marked changes in gene expression. Ado signaling through AA2BR has been demonstrated to elicit pro-resolving responses by acting as an inhibitor of the NF-κB signaling pathway (67) through a number of mechanisms, including elevations in intracellular cyclic adenosine monophosphate (cAMP) (67, 68). More recently it was shown that Ado signaling through AA2BR activates CREB and induces the expression of SLC26A3, a major apical Cl-/HCO3- exchanger in intestinal epithelial cells. Increased expression of SLC26A3 buffered against PMN transepithelial migration associated acidification. This observation identifies SLC26A3 as an adaptive response in intestinal epithelial cells to maintain pH homeostasis (20).
Tissue acidification and pH regulation
An often-underappreciated aspect of tissue inflammation is extracellular acidification. Such “inflammatory acidification” (20) has been observed in numerous disease conditions including, cancer, rheumatoid arthritis, and IBD (69). Given the prevalence of inflammatory acidification, it should be considered a hallmark of inflammation. Extracellular acidification impacts cellular signaling and function in a wide range of cell types, including epithelial cells and immune cells (21, 70). At sites of inflammation, it is believed the decrease in extracellular pH is due to the infiltration and activation of inflammatory cells, resulting in an increase in glycolysis and lactic acid secretion (71, 72). Recently, it has been observed that PMN-epithelial interaction results marked inflammatory acidification that was dependent on PMN-epithelial contact, but independent of PMN transepithelial migration. The source of the acidification was identified to be the epithelial cells and is a result of increased lactic acid production and increased ROS generation during PMN-epithelial cell interactions (20, 73).
In addition to PMN-associated acidification, it is widely accepted that tissue hypoxia has a significant impact of tissue acidification. In the absence of cellular O2, the sharp increase in glycolysis results in pyruvate fermentation into lactate, which is transported out of the cell through monocarboxylate transporters (MCT) (74). In this context, lactate has been shown to have both pro- and anti-inflammatory properties (69). Lactate accumulates in chronically inflamed tissue and drives the production of the pro-inflammatory cytokine, IL-17, however, during an acute inflammatory response lactate promotes a resolution response in macrophages but inhibiting the inflammasome (75, 76). In the context of IBD, the role of lactate appears to be cell type-dependent. In vitro work demonstrated that overexpression of MCT4 in colonic epithelial cells resulted in of NF-κB activation and translocation to the nucleus which induced IL-6 expression and the dissociation of CREB from the ZO-1 promotor. In vivo extensions of the studies revealed that pharmaceutical inhibition of MCT4 increased barrier function in murine models of colitis by decreasing the expression of pro-inflammatory factors and promoting ZO-1 expression (77). Furthermore, It has been shown that there is an increase in the lactate transporter MCT4 and an increase in fecal lactate concentrations (78, 79). However, it has also been shown that loss of GPR81, a G-protein coupled receptor activated by lactate, increases sensitivity to colitis in a dendritic cell dependent pathway (80). There is additional evidence that acute treatment with lactate dampens the immune response by down-regulating glucose uptake and expression of IL-6 (81). In various tissues, it has been shown that lactate is rapidly cleared from tissue into the blood stream and disposed by the liver and kidneys (82, 83). Additionally, when the hypoxic microenvironment resolves, the remaining lactate is converted into pyruvate and utilized for aerobic respiration (84). Taken together, these findings suggest a complex role for lactate in the inflammatory response, where timing and duration of the lactate exposure as well as the ability to clear excess lactate determines the endpoint response.
In a healthy GI tract, a balance between lactic acid-producing and lactic acid-utilizing bacteria are kept low. However, in intestinal dysbiosis (such as IBD) an increase in lactic acid and opportunistic pathogens take advantage of the increase in lactic acid concentrations to out compete other commensals, further driving inflammation (85). Mice lacking lactate dehydrogenase A, the enzyme responsible for converting pyruvate to lactate, results in a significant decrease in inflammation in models of non-infectious colitis (86). Furthermore, it has been observed that mice colonized with lactate-utilizing bacteria experience decreased severity of DSS-induced colitis and that administration of these lactate-utilizing bacteria can reverse the dysbiosis associated with colitis (87).
Independent of lactate, there is a growing body of literature shown that an acidic microenvironment promotes inflammation and suppresses resolution. For example, exposure of PMN to mildly acidic environments (pH 6.5-7.0) results in a significant attenuation of apoptosis, increasing their functional life span. Extracellular acidosis activates the NF-κB pathway and significant accumulation of intracellular cAMP. Under acidic conditions, there is a disruption of the mitochondrial transmembrane potential and translocation of cytochrome c to the cytoplasm, inhibiting apoptosis and prolonging the inflammatory response (88). Furthermore, inflammatory acidification has been shown to negatively impact platelet function at sites of inflammation. Under acidic conditions, platelets show decreases in adhesion, spreading, activation of αIIbβ3 integrin, ATP release, and other functions. Acidic platelets increased neutrophil chemotaxis, activation, and survival, amplifying the neutrophil-mediated inflammatory response (89).
The full impact of inflammatory acidification on intestinal epithelial cell signaling and function remains unclear, however, in recent years there have been several studies that have highlighted the impact of extracellular acidification on epithelial cells. It has been well established that there are several G protein-coupled receptors (GPR 132, GPR4, GPR68, and GPR65), acid-sensitive ion channels (acid-sensing ion channels (ASIC) and transient receptor potential vannilloid-1 (TRPV1) expressed in a wide array of tissues in the body that sense and are activated by increased ion concentrations, the extent of their involvement in sensing pH by intestinal epithelial cells has not been well characterized (90). It was recently shown that intestinal epithelial cells sense extracellular acidosis through GPR31 in a CREB-dependent fashion. Activation of GPR31 by inflammatory acidosis induced an unique gene signature that was observed in both inflamed, acidic murine tissue and in tissue samples from patients with Crohn’s disease (21).
It appears that in the setting of chronic inflammation, the ability of intestinal epithelial cells to balance extracellular pH is diminished. In both murine models of colitis and patient samples from individuals with UC and CD the expression of SLC26A3 is significantly attenuated (20). Epithelial-specific SLC26A3 null mice the mucosal surfaces of the cecum and colon of are significantly more acidic then wild-type controls (91). These epithelial-specific SLC26A3 null mice offer a unique opportunity to investigate acidosis associated changes in the microbiome. It was observed that in SLC26A3 knockout mice there was an outgrowth of Bacteroidetes and Deferribacteres and a decrease in abundance of Actinobacteria and Firmicutes (91).
The role of pro-resolving lipid mediators
Without timely resolution of acute inflammation, chronic disease can develop. In recent years there has been a growing appreciation that specialized pro-resolving mediators (SPM) have a pivotal anti-inflammatory role in the tissue (92). SPM are a large family of mediators and including lipoxins, resolvins, protectins and maresins and are enzymatically derived from essential fatty acids during the acute inflammatory response by both epithelial and immune cells (93–96). SPM are immunoresolvent molecules, which are distinct from immunosuppressive molecules; meaning, they enhance the host defenses while dampening the inflammatory response. SPM signal through G-protein coupled receptors, which include N-formyl peptide receptor 2 (ALX/FPR2), chemokine-like receptor 1 (ChemR23), leukotrience B4 receptor 1 (BLT1) GRP18, and GPR32 and have distinct cell type-specific actions (97–100). For example, in intraepithelial lymphocytes (IEL), GPR18 has been shown to be required for intestinal homeostasis, where mice lacking GPR18 show a dysregulation of CD8 T cells accumulation in the intraepithelial and lamina propria compartment (101). In intestinal epithelial cells ALX/FPR2 and ChemR23 have been shown to be influenced by a number of microenvironmental factors and are important for resolution of inflammation. It has been shown that ALX/FPR2 is induced by Il-13 and interferon γ. Activation of ALX/FPR2 by lipoxins significantly inhibited the production of TNFα and Il-8 by intestinal epithelial cells (102). ChemR23 is highly expressed on apical surface of intestinal epithelial cells and activation by resolvin-E1 induces a wide array of inflammatory regulatory genes including alkaline phosphatase (ALPI). ALPI contributes to the detoxification of bacterial LPS and inhibits bacterial growth (103). In M1 macrophages RvE1 exposure induces the expression of the anti-inflammatory cytokine IL-10 (104). Using multiple murine models of colits, including DSS and TNBS colitis, it has been shown that treatment with both RvE1 and RvD2 improves colitis. RvE1 and RvD2 decrease leukocyte infiltrate and pro-inflammatory cytokines (105, 106). Furthermore, it has been shown that in IBD patients unresponsive to anti-TNFα, ChemR23 expression is increased. A recent study has shown that an agonist anti-ChemR23 antibody promoted resolution of both acute and chronic inflammation in murine models of colitis (107).
The expression of SPMs are highly dependent on the tissue microenvironment. Very quickly following tissue injury or pathogen invasion polyunsaturated fatty acids (PUFA) are released from membrane phospholipids (108). These early PUFA are rapidly metabolized into eicosanoids, which aid in the recruitment of PMN to the site of inflammation. Early in the inflammatory response, PMN recruitment and transmigration triggers lipid mediator class-switching resulting in the metabolism of PUFA into arachidonic acid and ultimately lipoxin, a pro-resolving mediator (109). This shift eicosanoids to lipoxin represses PMN recruitment; while, promoting influx of monocytes to the site of inflammation, stimulating the efferocytosis and promoting resolution and a return to tissue homeostasis (110). Recently, it has been shown that SPM not only impact the recruitment of PMN to site of inflammation, but also have a direct effect of PMN action in the tissue. In a murine E. coli lung inflammation model, it has been demonstrated that 15-epi-LAX4 and 17-epi-RvD1 signal through the ALX/FPR2 receptor on PMN to inhibit Toll-like receptor 9-mediated release of neutrophil elastase (NE) and proteinase 3 (PR3). NE and PR3 release down-regulates C5aR, resulting in the suppression of PMN phagocytosis and apoptosis, delaying the resolution of inflammation (111). Interestingly, several bacterial species have evolved to utilize SPM to their advantage. Staphylococcus aureus release α-hemolysin, which is a potent elicitor of SPM synthesis. This overproduction of SPM dampens the immune response, even in the presence of an ongoing infection (112).
Tissue hypoxia has a significant impact on the production and function of SPM. The influence of hypoxia on the production of SPM was initially observed in endothelial cells, where it was shown that hypoxic endothelial cells upregulate COX-2, an important enzyme in the production of E and D resolvins, and in the presence of Il-1β endothelia produce the precursor to the 17R series resolvin that is enzymatically activated by PMN via lipoxygenation and epoxidation. This bioactive 17R series resolvin was shown to inhibit PMN transmigration in vivo and diminish zymosan-induced peritonitis (113, 114). When M2 macrophages, the macrophage responsible for clearing apoptotic PMN from inflamed tissue, encounter an hypoxic environment they increase their production of an eicosapentaenoic acid-derived resolvin, RvE4, which promotes the clearance of both apoptotic PMN and senescent red blood cells (115). Another resolvin which strongly inhibits PMN infiltration, RvE2, has also been shown to be induced in PMN by hypoxic conditions (115). In addition to inhibiting PMN infiltration, RvE2 also increases macrophage phagocytosis and production of the anti-inflammatory cytokine, IL-10, promoting overall resolution (116).
Chemokine/cytokine signaling
Mucosal chemokines and cytokines are well-established in their roles of initiating and driving the inflammatory response; however, until recently their role in the resolution process has been underappreciated. Inflammatory insults are rapidly sensed by epithelial cells and tissue macrophages which triggers the release of numerous proinflammatory cytokines and chemokines (10). The foundation of the resolution response is the cessation of PMN infiltration and induction of apoptosis and eventual clearance of PMN (117). It is now well understood that this process is not passive, i.e dilution of chemokine gradient, but is in fact a highly organized and active process. An important aspect of the resolution response is the production of anti-inflammatory cytokines and chemokines. Prominent anti-inflammatory cytokines include IL-1 receptor antagonist (IL-1ra), IL-4, IL-6, IL-10, IL-11, IL-13, and TGF-β (118, 119). A key feature of these anti-inflammatory cytokines is that they act as immunomodulators that limit excess inflammation. The impact of the anti-inflammatory cytokines dependent on cell type, kinetics of their release, cytokine receptor density, and co-existence of other cytokine and chemokines at the site of inflammation (120).
IL-10 is considered one of the more important anti-inflammatory cytokines. IL-10 is primarily produced by T helper cells, monocytes, and macrophages; however, it has been reported that epithelial cells and granulocytes are also able to produce IL-10 in response to infection or tissue damage (121, 122). IL-10 binds to the heterodimeric IL-10 receptor (IL-10R1 and IL-10R2) which is expressed on a wide array of cell types, including, PMN, macrophages, and epithelial cells (123). It has been shown that IL-10 impacts a wide array of activated PMN functions, including recruitment, LPS-induced cytokine production, and ROS production (123, 124). In recent years it has become evident that IL-10 signaling in epithelial cells in essential for the resolution of inflammation. In response to INFγ inflamed intestinal epithelial cells upregulate IL-10R1 and IL-10 exposure promotes barrier function by repressing the expression of Claudin-2, a claudin often associated with “leaky” epithelial barrier (125, 126). Epithelial specific depletion of IL-10R1 greatly increases DSS colitis severity. Epithelial-specific IL10-R1 null mice have increased intestinal permeability, increased expression of pro-inflammatory cytokines, and a marked decrease in disease resolution (126). As with the other microenvironmental factors discussed in this review, IL-10 expression is influenced by hypoxia. Within B cells, stabilization of HIF under hypoxic conditions results in the induction of IL-10 transcript and secretion of IL-10 into the inflammatory microenvironment which promotes resolution of experimental colitis (44, 127).
Another important cytokine in the resolution response is TGF-β. TGF-β is produced by all leukocytes, epithelial cells, and T cells at sites of inflammation (128). Secreted TGF-β must be activated before it can bind to its receptors and trigger a signaling cascade within the cell. TGF-β can be activated by several extracellular factors, including metalloproteinases and reactive oxygen species (129–131). Within the GI tract, TGF-β suppresses local response to luminal antigens, increases secretion of IgA, and promotes barrier function. Mice lacking TGF-β or TGFβRII develop spontaneous colitis, as well as inflammation in a number of other organs (132–134). In murine models of colitis, it has been shown that induction of TGF-β by a dysregulated microbiota triggers the induction of FGF2, which in turns induces the secretion of IL-17 to promote repair of the epithelial layer and barrier function (135). However, like other anti-inflammatory cytokines, inappropriate activation of TGF-β can result in inappropriate wound healing and can lead to the development of fibrosis or cancers, highlighting the complexity of the resolution response (136).
Conclusions
In this review, we have outlined how various aspects of the mucosal inflammatory microenvironment are influenced by inflammatory hypoxia and how these alterations impact the resolution response. Both in vitro and in vivo studies have demonstrated that the resolution response is greatly impacted by the extracellular microenvironment and that tissue hypoxia plays a key role in initiating the resolution response by molding the inflammatory microenvironment. As PMN migrate into tissue they deplete the local oxygen and stabilize HIF-1α. This hypoxic environment is associated with increases in both ado generation and expression of Ado receptors, generation of SPM, and secrete anti-inflammatory cytokines, all of which promote the resolution response (Figure 2). As highlighted throughout this review, disruptions in the microenvironment can prevent the resolution of acute inflammation and drive the development of a chronic inflammatory lesion. The pharmacologic stabilization of HIF is already showing promise as an anti-inflammatory therapy in human patients (ClinicalTrials.gov: NCT04353791) (137). As highlighted in this review, it has been demonstrated targeting the inflammatory microenvironment can improve colitis in a variety of murine models, however the exact molecular mechanisms and pathways remain elusive. Given that over 50% of individuals with IBD will eventually encounter treatment failure, it is crucial that we indentify novel therapeutic targets (138). Furthering our understanding of how the microenvironment changes during inflammation and how various cell types respond to these changes will provide new insight into potential therapeutic targets for the treatment of human inflammatory diseases such as IBD.
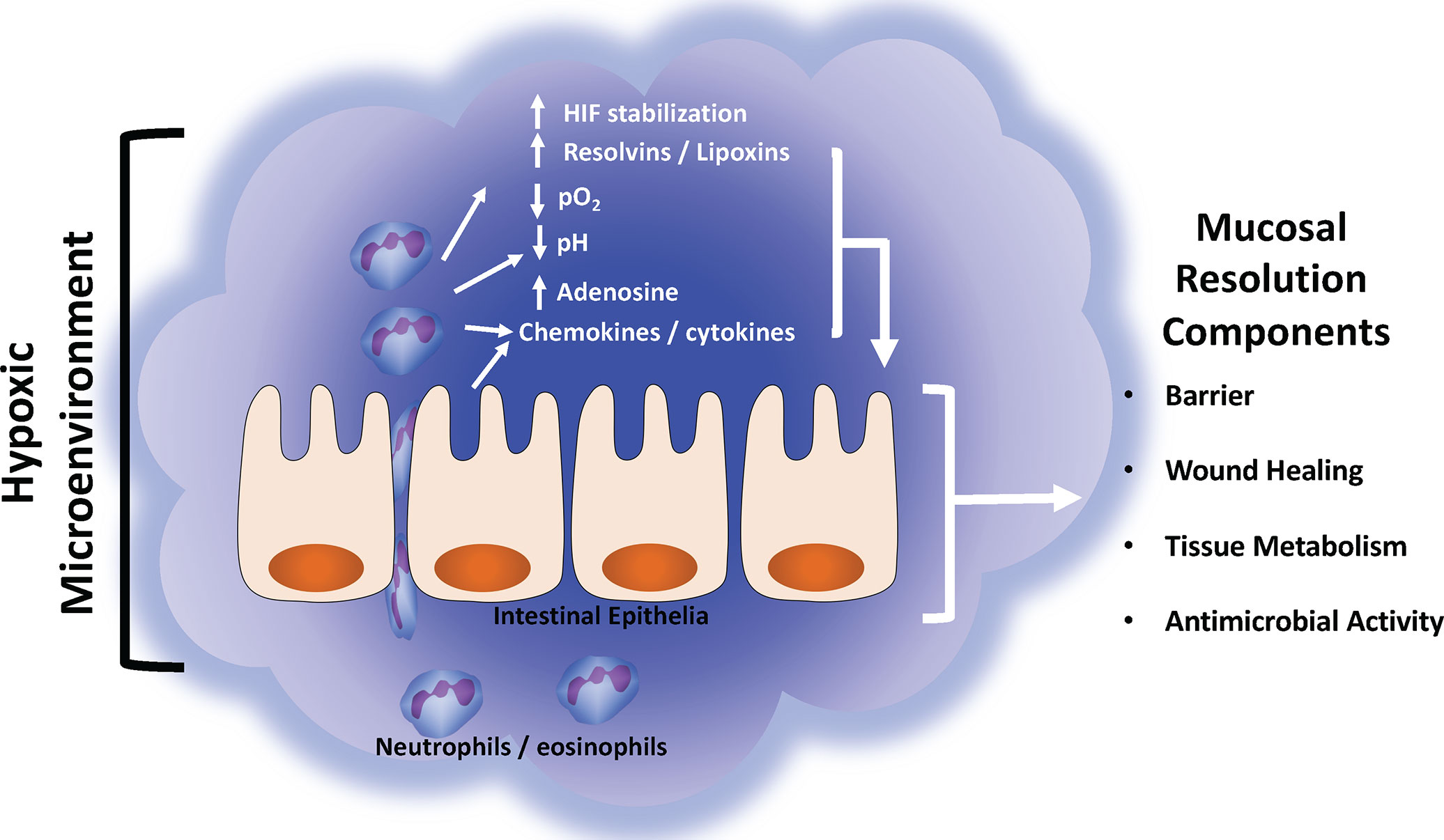
Figure 2 Summary of mucosal inflammatory resolution components driven by the hypoxic tissue microenvironment. During active inflammation, the normally hypoxic microenvironment (physiologic hypoxia) is enhanced (inflammatory hypoxia) by the infiltration and copious consumption local oxygen by innate immune cells (e.g., neutrophils and eosinophils). Such localized hypoxia drives a number of biochemical reactions that enhance the resolution response. See text for details of each resolution component and their respective response to hypoxia.
Author contributions
IC wrote the original draft. SC and IC reviewed and edited the manuscript. All authors contributed to the manuscript and approved the submission.
Funding
This work was supported by NIH grants DK104713, DK050189, DK122741, DK1200720, DK09549, VA Merit Award 1I01BX002182, and IK2BX005710.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lundquist P, Artursson P. Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv Drug Delivery Rev (2016) 106(Pt B):256–76. doi: 10.1016/j.addr.2016.07.007
2. McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Curr Opin Gastroenterol (2007) 23(6):647–54. doi: 10.1097/MOG.0b013e3282f0153b
3. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) (2007) 85(12):1295–300. doi: 10.1007/s00109-007-0277-z
4. Ali A, Tan H, Kaiko GE. Role of the intestinal epithelium and its interaction with the microbiota in food allergy. Front Immunol (2020) 11:604054. doi: 10.3389/fimmu.2020.604054
5. Hall AB, Tolonen AC, Xavier RJ. Human genetic variation and the gut microbiome in disease. Nat Rev Genet (2017) 18(11):690–9. doi: 10.1038/nrg.2017.63
6. Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, Lee S, et al. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut (2017) 66(6):1031–8. doi: 10.1136/gutjnl-2015-311326
7. Ginhoux F, Jung S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14(6):392–404. doi: 10.1038/nri3671
8. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget (2018) 9(6):7204–18. doi: 10.18632/oncotarget.23208
9. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
10. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5(4):354–66. doi: 10.1038/mi.2012.24
11. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: From mechanisms to disease. Annu Rev Immunol (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
12. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J (2017) 31(4):1273–88. doi: 10.1096/fj.201601222R
13. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat Rev Immunol (2002) 2(12):965–75. doi: 10.1038/nri957
14. Elliott SN, Wallace JL. Neutrophil-mediated gastrointestinal injury. Can J Gastroenterol (1998) 12(8):559–68. doi: 10.1155/1998/398384
15. Edwards SW, Hallett MB. Seeing the wood for the trees: The forgotten role of neutrophils in rheumatoid arthritis. Immunol Today (1997) 18(7):320–4. doi: 10.1016/S0167-5699(97)01087-6
16. Snelgrove RJ, Patel DF, Patel T, Lloyd CM. The enigmatic role of the neutrophil in asthma: Friend, foe or indifferent? Clin Exp Allergy (2018) 48(10):1275–85. doi: 10.1111/cea.13191
17. Barnig C, Bezema T, Calder PC, Charloux A, Frossard N, Garssen J, et al. Activation of resolution pathways to prevent and fight chronic inflammation: Lessons from asthma and inflammatory bowel disease. Front Immunol (2019) 10:1699. doi: 10.3389/fimmu.2019.01699
18. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol (2005) 6(12):1182–90. doi: 10.1038/ni1275
19. Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol (2007) 28(8):340–5. doi: 10.1016/j.it.2007.06.002
20. Cartwright IM, Curtis VF, Lanis JM, Alexeev EE, Welch N, Goldberg MS, et al. Adaptation to inflammatory acidity through neutrophil-derived adenosine regulation of SLC26A3. Mucosal Immunol (2020) 13(2):230–44. doi: 10.1038/s41385-019-0237-2
21. Cartwright IM, Dowdell AS, Lanis JM, Brink KR, Mu A, Kostelecky RE, et al. Mucosal acidosis elicits a unique molecular signature in epithelia and intestinal tissue mediated by GPR31-induced CREB phosphorylation. Proc Natl Acad Sci USA (2021) 118(20). doi: 10.1073/pnas.2023871118
22. Curtis VF, Cartwright IM, Lee JS, Wang RX, Kao DJ, Lanis JM, et al. Neutrophils as sources of dinucleotide polyphosphates and metabolism by epithelial ENPP1 to influence barrier function via adenosine signaling. Mol Biol Cell (2018) 29(22):2687–99. doi: 10.1091/mbc.E18-06-0377
23. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol (2017) 17(12):774–85. doi: 10.1038/nri.2017.103
24. Schaible B, Schaffer K, Taylor CT. Hypoxia, innate immunity and infection in the lung. Respir Physiol Neurobiol (2010) 174(3):235–43. doi: 10.1016/j.resp.2010.08.006
25. Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. (2014) 147(5):1055–63 e8. doi: 10.1053/j.gastro.2014.07.020
26. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest (2004) 114(8):1098–106. doi: 10.1172/JCI21086
27. Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med (2001) 193(9):1027–34. doi: 10.1084/jem.193.9.1027
28. Colgan SP, Campbell EL, Kominsky DJ. Hypoxia and mucosal inflammation. Annu Rev Pathol (2016) 11:77–100. doi: 10.1146/annurev-pathol-012615-044231
29. Colgan SP, Furuta GT, Taylor CT. Hypoxia and innate immunity: Keeping up with the HIFsters. Annu Rev Immunol (2020) 38:341–63. doi: 10.1146/annurev-immunol-100819-121537
30. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res (2002) 62(12):3387–94.
31. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110(7):993–1002. doi: 10.1172/JCI15337
32. Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol (2011) 76:347–53. doi: 10.1101/sqb.2011.76.010678
33. Semenza GL. HIF-1, O(2), and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell (2001) 107(1):1–3. doi: 10.1016/s0092-8674(01)00518-9
34. Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol (2001) 13(2):167–71. doi: 10.1016/s0955-0674(00)00194-0
35. Pral LP, Fachi JL, Correa RO, Colonna M, Vinolo MAR. Hypoxia and HIF-1 as key regulators of gut microbiota and host interactions. Trends Immunol (2021) 42(7):604–21. doi: 10.1016/j.it.2021.05.004
36. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40(1):66–77. doi: 10.1016/j.immuni.2013.11.020
37. Colgan SP, Dzus AL, Parkos CA. Epithelial exposure to hypoxia modulates neutrophil transepithelial migration. J Exp Med (1996) 184(3):1003–15. doi: 10.1084/jem.184.3.1003
38. Clayburgh DR, Shen L, Turner JR. A porous defense: The leaky epithelial barrier in intestinal disease. Lab Invest (2004) 84(3):282–91. doi: 10.1038/labinvest.3700050
39. Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell (2015) 26(12):2252–62. doi: 10.1091/mbc.E14-07-1194
40. Kim YI, Yi EJ, Kim YD, Lee AR, Chung J, Ha HC, et al. Local stabilization of hypoxia-inducible factor-1alpha controls intestinal inflammation via enhanced gut barrier function and immune regulation. Front Immunol (2020) 11:609689. doi: 10.3389/fimmu.2020.609689
41. Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. (2008) 134(1):156–65. doi: 10.1053/j.gastro.2007.10.012
42. Chen J, He Y, Tu L, Duan LH. Dual immune functions of IL-33 in inflammatory bowel disease. Histol Histopathol (2020) 35(2):137–46. doi: 10.14670/Hh-18-149
43. Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med (2019) 25(1):33–46. doi: 10.1016/j.molmed.2018.10.006
44. Meng XY, Grotsch B, Luo YB, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1 alpha is a critical transcription factor for IL-10-producing b cells in autoimmune disease. Nat Commun (2018) 9. doi: 10.1038/s41467-017-02683-x
45. Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol (2012) 74:153–75. doi: 10.1146/annurev-physiol-020911-153230
46. Eltzschig HK, Eckle T, Mager A, Kuper N, Karcher C, Weissmuller T, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res (2006) 99(10):1100–8. doi: 10.1161/01.RES.0000250174.31269.70
47. Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J Exp Med (2003) 198(5):783–96. doi: 10.1084/jem.20030891
48. Gendron FP, Benrezzak O, Krugh BW, Kong Q, Weisman GA, Beaudoin AR. Purine signaling and potential new therapeutic approach: Possible outcomes of NTPDase inhibition. Curr Drug Targets (2002) 3(3):229–45. doi: 10.2174/1389450023347713
49. Linden J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol (2001) 41:775–87. doi: 10.1146/annurev.pharmtox.41.1.775
50. Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol (2010) 184(7):4017–24. doi: 10.4049/jimmunol.0901851
51. Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. (2009) 113(1):224–32. doi: 10.1182/blood-2008-06-165746
52. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcila JC, et al. Crucial role for ecto-5-' nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med (2004) 200(11):1395–405. doi: 10.1084/Jem.20040915
53. Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis (2014) 5. doi: 10.1038/cddis.2014.70
54. Kong TQ, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J (2006) 20(13):2242–50. doi: 10.1096/fj.06-6419com
55. Chin AC, Parkos CA. Pathobiology of neutrophil transepithelial migration: Implications in mediating epithelial injury. Annu Rev Pathol (2007) 2:111–43. doi: 10.1146/annurev.pathol.2.010506.091944
56. Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5'-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med (1998) 188(8):1433–43. doi: 10.1084/jem.188.8.1433
57. Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J (1992) 11(6):2063–70. doi: 10.1002/j.1460-2075.1992.tb05264.x
58. Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol Cell Physiol (2002) 282(6):C1235–45. doi: 10.1152/ajpcell.00288.2001
59. Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, et al. 5'-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest (1993) 91(5):2320–5. doi: 10.1172/JCI116462
60. Huang P, Trotter K, Boucher RC, Milgram SL, Stutts MJ. PKA holoenzyme is functionally coupled to CFTR by AKAPs. Am J Physiol Cell Physiol (2000) 278(2):C417–22. doi: 10.1152/ajpcell.2000.278.2.C417
61. Sun F, Hug MJ, Bradbury NA, Frizzell RA. Protein kinase a associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J Biol Chem (2000) 275(19):14360–6. doi: 10.1074/jbc.275.19.14360
62. Strohmeier GR, Reppert SM, Lencer WI, Madara JL. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J Biol Chem (1995) 270(5):2387–94. doi: 10.1074/jbc.270.5.2387
63. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway pseudomonas infections of cystic fibrosis patients. J Clin Invest (2002) 109(3):317–25. doi: 10.1172/JCI13870
64. Li Z, Elliott E, Payne J, Isaacs J, Gunning P, O'Loughlin EV. Shiga toxin-producing escherichia coli can impair T84 cell structure and function without inducing attaching/effacing lesions. Infect Immun (1999) 67(11):5938–45. doi: 10.1128/IAI.67.11.5938-5945.1999
65. Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, Klinkenberg R, et al. Altered expression and localization of ion transporters contribute to diarrhea in mice with salmonella-induced enteritis. Gastroenterology. (2013) 145(6):1358–68 e1-4. doi: 10.1053/j.gastro.2013.08.054
66. Young S, Ericsson A, Woode R, Clarke L. Intestinal epithelial cell-specific cftr knockout mice exhibit fecal microbial dysbiosis. Gastroenterology. (2022) 162(3):S68–S9.
67. Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest (2007) 117(3):703–11. doi: 10.1172/JCI30049
68. Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, Brummer T. Adenosine and cAMP are potent inhibitors of the NF-kappa b pathway downstream of immunoreceptors. Eur J Immunol (2005) 35(1):31–41. doi: 10.1002/eji.200425524
69. Pucino V, Bombardieri M, Pitzalis C, Mauro C. Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur J Immunol (2017) 47(1):14–21. doi: 10.1002/eji.201646477
70. Rajamaki K, Nordstrom T, Nurmi K, Akerman KE, Kovanen PT, Oorni K, et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem (2013) 288(19):13410–9. doi: 10.1074/jbc.M112.426254
71. Menkin V. Biology of inflammation; chemical mediators and cellular injury. Science. (1956) 123(3196):527–34. doi: 10.1126/science.123.3196.527
72. Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, et al. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol (2009) 182(12):7974–81. doi: 10.4049/jimmunol.0804216
73. Cartwright IM, Dowdell AS, Hanson C, Kostelecky RE, Welch N, Steiner CA, et al. Contact-dependent, polarized acidification response during neutrophil-epithelial interactions. J Leukoc Biol (2022) 112(6):1543–53. doi: 10.1002/JLB.3MA0422-742R
74. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol-London (2021) 599(1):23–37. doi: 10.1113/Jp280572
75. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4(+) T cell metabolic rewiring. Cell Metab (2019) 30(6):1055. doi: 10.1016/j.cmet.2019.10.004
76. Manosalva C, Quiroga J, Hidalgo AI, Alarcon P, Anseoleaga N, Hidalgo MA, et al. Role of lactate in inflammatory processes: Friend or foe. Front Immunol (2022) 12:808799. doi: 10.3389/fimmu.2021.808799
77. Zhang S, Xu W, Wang H, Cao M, Li M, Zhao J, et al. Inhibition of CREB-mediated ZO-1 and activation of NF-kappaB-induced IL-6 by colonic epithelial MCT4 destroys intestinal barrier function. Cell Prolif (2019) 52(6):e12673. doi: 10.1111/cpr.12673
78. Hove H, Mortensen PB. Influence of intestinal inflammation (Ibd) and small and Large-bowel length on fecal short-chain fatty-acids and lactate. Digest Dis Sci (1995) 40(6):1372–80. doi: 10.1007/Bf02065554
79. He LY, Wang HL, Zhang YH, Geng LL, Yang M, Xu ZH, et al. Evaluation of monocarboxylate transporter 4 in inflammatory bowel disease and its potential use as a diagnostic marker. Dis Markers (2018) 2018. doi: 10.1155/2018/2649491
80. Ranganathan P, Shanmugam A, Swafford D, Suryawanshi A, Bhattacharjee P, Hussein MS, et al. GPR81, a cell-surface receptor for lactate, regulates intestinal homeostasis and protects mice from experimental colitis. J Immunol (2018) 200(5):1781–9. doi: 10.4049/jimmunol.1700604
81. Iraporda C, Romanin DE, Bengoa AA, Errea AJ, Cayet D, Foligne B, et al. Local treatment with lactate prevents intestinal inflammation in the TNBS-induced colitis model. Front Immunol (2016) 7:651. doi: 10.3389/fimmu.2016.00651
82. Bellomo R. Bench-to-bedside review: Lactate and the kidney. Crit Care (2002) 6(4):322–6. doi: 10.1186/cc1518
83. Cohen RD, Woods HF. Lactic acidosis revisited. Diabetes. (1983) 32(2):181–91. doi: 10.2337/diab.32.2.181
84. Feron O. Pyruvate into lactate and back: From the warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol (2009) 92(3):329–33. doi: 10.1016/j.radonc.2009.06.025
85. Gillis CC, Hughes ER, Spiga L, Winter MG, Zhu WH, de Carvalho TF, et al. Dysbiosis-associated change in host metabolism generates lactate to support salmonella growth. Cell Host Microbe (2018) 23(1):54. doi: 10.1016/j.chom.2017.11.006
86. Taylor SJ, Winter MG, Gillis CC, Silva LAD, Dobbins AL, Muramatsu MK, et al. Colonocyte-derived lactate promotes e. coli fitness in the context of inflammation-associated gut microbiota dysbiosis. Microbiome. (2022) 10(1):200. doi: 10.1186/s40168-022-01389-7
87. Chen LR, Li R, Wang ZG, Zhang ZW, Wang J, Qiao YB, et al. Lactate-utilizing bacteria ameliorates DSS-induced colitis in mice. Life Sci (2022) 288. doi: 10.1016/j.lfs.2021.120179
88. El Kebir D, de Oliveira Lima Dos Santos E, Mansouri S, Sekheri M, Filep JG. Mild acidosis delays neutrophil apoptosis via multiple signaling pathways and acts in concert with inflammatory mediators. J Leukoc Biol (2017) 102(6):1389–400. doi: 10.1189/jlb.3A0117-041R
89. Etulain J, Negrotto S, Carestia A, Pozner RG, Romaniuk MA, D'Atri LP, et al. Acidosis downregulates platelet haemostatic functions and promotes neutrophil proinflammatory responses mediated by platelets. Thromb Haemost (2012) 107(1):99–110. doi: 10.1160/TH11-06-0443
90. Holzer P. Acid-sensitive ion channels and receptors. Handb Exp Pharmacol (2009) 194):283–332. doi: 10.1007/978-3-540-79090-7_9
91. Kini A, Singh AK, Riederer B, Yang I, Tan X, di Stefano G, et al. Slc26a3 deletion alters pH-microclimate, mucin biosynthesis, microbiome composition and increases the TNFalpha expression in murine colon. Acta Physiol (Oxf) (2020) 230(2):e13498. doi: 10.1111/apha.13498
92. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510(7503):92–101. doi: 10.1038/nature13479
93. Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta (2015) 1851(4):397–413. doi: 10.1016/j.bbalip.2014.08.006
94. Serhan CN, Savill J. Resolution of inflammation: The beginning programs the end. Nat Immunol (2005) 6(12):1191–7. doi: 10.1038/ni1276
95. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8(5):349–61. doi: 10.1038/nri2294
96. Chiurchiu V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: Managing the fire within. Front Immunol (2018) 9:38. doi: 10.3389/fimmu.2018.00038
97. Chiang N, Gronert K, Clish CB, O'Brien JA, Freeman MW, Serhan CN. Leukotriene B4 receptor transgenic mice reveal novel protective roles for lipoxins and aspirin-triggered lipoxins in reperfusion. J Clin Invest (1999) 104(3):309–16. doi: 10.1172/JCI7016
98. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med (2005) 201(5):713–22. doi: 10.1084/jem.20042031
99. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med (2017) 58:114–29. doi: 10.1016/j.mam.2017.03.005
100. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med (2002) 8(11):1296–302. doi: 10.1038/nm786
101. Wang XM, Sumida H, Cyster JG. GPR18 is required for a normal CD8 alpha alpha intestinal intraepithelial lymphocyte compartment. J Exp Med (2014) 211(12):2351–9. doi: 10.1084/jem.20140646
102. Gronert K, Gewirtz A, Madara JL, Serhan CN. Identification of a human enterocyte lipoxin A(4) receptor that is regulated by interleukin (IL)-13 and interferon gamma and inhibits tumor necrosis factor alpha-induced IL-8 release. J Exp Med (1998) 187(8):1285–94. doi: 10.1084/jem.187.8.1285
103. Campbell EL, MacManus CF, Kominsky DJ, Keely S, Glover LE, Bowers BE, et al. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc Natl Acad Sci USA (2010) 107(32):14298–303. doi: 10.1073/pnas.0914730107
104. Herova M, Schmid M, Gemperle C, Hersberger M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J Immunol (2015) 194(5):2330–7. doi: 10.4049/jimmunol.1402166
105. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. P Natl Acad Sci USA (2005) 102(21):7671–6. doi: 10.1073/pnas.0409271102
106. Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol (2011) 187(4):1957–69. doi: 10.4049/jimmunol.1101305
107. Trilleaud C, Gauttier V, Biteau K, Girault I, Belarif L, Mary C, et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci Adv (2021) 7(14). doi: 10.1126/sciadv.abd1453
108. Kasuga K, Yang R, Porter TF, Agrawal N, Petasis NA, Irimia D, et al. Rapid appearance of resolvin precursors in inflammatory exudates: Novel mechanisms in resolution. J Immunol (2008) 181(12):8677–87. doi: 10.4049/jimmunol.181.12.8677
109. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat Immunol (2001) 2(7):612–9. doi: 10.1038/89759
110. Serhan CN. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol Aspects Med (2017) 58:1–11. doi: 10.1016/j.mam.2017.03.001
111. Sekheri M, El Kebir D, Edner N, Filep JG. 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci U S A. (2020) 117(14):7971–80. doi: 10.1073/pnas.1920193117
112. Jordan PM, Gerstmeier J, Pace S, Bilancia R, Rao ZG, Borner F, et al. Staphylococcus aureus-derived alpha-hemolysin evokes generation of specialized pro-resolving mediators promoting inflammation resolution. Cell Rep (2020) 33(2). doi: 10.1016/j.celrep.2020.108247
113. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192(8):1197–204. doi: 10.1084/jem.192.8.1197
114. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med (2002) 196(8):1025–37. doi: 10.1084/jem.20020760
115. Norris PC, Libreros S, Serhan CN. Resolution metabolomes activated by hypoxic environment. Sci Adv (2019) 5(10). doi: 10.1126/sciadv.aax4895
116. Oh SF, Dona M, Fredman G, Krishnamoorthy S, Irimia D, Serhan CN. Resolvin E2 formation and impact in inflammation resolution. J Immunol (2012) 188(9):4527–34. doi: 10.4049/jimmunol.1103652
117. Reville K, Crean JK, Vivers S, Dransfield I, Godson C. Lipoxin A(4) redistributes myosin IIA and Cdc42 in macrophages: Implications for phagocytosis of apoptotic leukocytes. J Immunol (2006) 176(3):1878–88. doi: 10.4049/jimmunol.176.3.1878
118. Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. (2000) 117(4):1162–72. doi: 10.1378/chest.117.4.1162
119. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi: 10.1038/nri3661
120. Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol (1998) 16(5-6):457–99. doi: 10.3109/08830189809043005
121. Jung M, Sabat R, Kratzschmar J, Seidel H, Wolk K, Schonbein C, et al. Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur J Immunol (2004) 34(2):481–93. doi: 10.1002/eji.200324323
122. Iyer SS, Cheng GH. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol (2012) 32(1):23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
123. Elbim C, Reglier H, Fay M, Delarche C, Andrieu V, El Benna J, et al. Intracellular pool of IL-10 receptors in specific granules of human neutrophils: Differential mobilization by proinflammatory mediators. J Immunol (2001) 166(8):5201–7. doi: 10.4049/jimmunol.166.8.5201
124. Sun L, Guo RF, Newstead MW, Standiford TJ, Macariola DR, Shanley TP. Effect of IL-10 on neutrophil recruitment and survival after pseudomonas aeruginosa challenge. Am J Resp Cell Mol (2009) 41(1):76–84. doi: 10.1165/rcmb.2008-0202OC
125. Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, et al. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J Immunol (2017) 199(8):2976–84. doi: 10.4049/jimmunol.1700105
126. Kominsky DJ, Campbell EL, Ehrentraut SF, Wilson KE, Kelly CJ, Glover LE, et al. IFN-gamma-Mediated induction of an apical IL-10 receptor on polarized intestinal epithelia. J Immunol (2014) 192(3):1267–76. doi: 10.4049/jimmunol.1301757
127. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Tedder TF, Sato S. IL-10-Producing regulatory B10 cells inhibit intestinal injury in a mouse model. Am J Pathology (2011) 178(2):735–43. doi: 10.1016/j.ajpath.2010.10.022
128. Prud'homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest (2007) 87(11):1077–91. doi: 10.1038/labinvest.3700669
129. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Gene Dev (2000) 14(2):163–76.
130. BarcellosHoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol (1996) 10(9):1077–83. doi: 10.1210/me.10.9.1077
131. Norgaard P, Hougaard S, Poulsen HS, Spangthomsen M. Transforming growth-Factor-Beta and cancer. Cancer Treat Rev (1995) 21(4):367–403. doi: 10.1016/0305-7372(95)90038-1
132. Gorelik L, Flavell RA. Abrogation of TGF beta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. (2000) 12(2):171–81. doi: 10.1016/S1074-7613(00)80170-3
133. Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh CG, et al. Transforming growth-Factor-Beta-1 null mice - an animal-model for inflammatory disorders. Am J Pathology (1995) 146(1):264–75.
134. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin MY, et al. Targeted disruption of the mouse transforming growth factor-Beta-1 gene results in multifocal inflammatory disease. Nature. (1992) 359(6397):693–9. doi: 10.1038/359693a0
135. Song XY, Dai D, He X, Zhu S, Yao YK, Gao HC, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. (2015) 43(3):488–501. doi: 10.1016/j.immuni.2015.06.024
136. Steiner CA, Cartwright IM, Taylor CT, Colgan SP. Hypoxia-inducible factor as a bridge between healthy barrier function, wound healing, and fibrosis. Am J Physiol Cell Physiol (2022) 323(3):C866–C78. doi: 10.1152/ajpcell.00227.2022
137. Levesque BG, Meadows KT, Buch A, Flynn M, Peters K, Olson A, et al. Gb004, a novel prolyl hydroxylase inhibitor for inflammatory bowel disease, leads to gut-targeted hif-1alpha pathway engagement in a multiple dose study in heathy subjects. Gastroenterology. (2020) 158(6):S1202–S. doi: 10.1093/ecco-jcc/jjz203.668
138. Gordon JP, McEwan PC, Maguire A, Sugrue DM, Puelles J. Characterizing unmet medical need and the potential role of new biologic treatment options in patients with ulcerative colitis and crohn's disease: A systematic review and clinician surveys. Eur J Gastroen Hepat (2015) 27(7):804–12. doi: 10.1097/Meg.0000000000000378
Keywords: mucosal inflammation, hypoxia, neutrophil, inflammatory resolution, acidification, adenine nucleotides
Citation: Cartwright IM and Colgan SP (2023) The hypoxic tissue microenvironment as a driver of mucosal inflammatory resolution. Front. Immunol. 14:1124774. doi: 10.3389/fimmu.2023.1124774
Received: 15 December 2022; Accepted: 06 January 2023;
Published: 18 January 2023.
Edited by:
Janos G. Filep, Montreal University, CanadaReviewed by:
Nan Chiang, Brigham and Women’s Hospital and Harvard Medical School, United StatesJennifer Catherine Brazil, University of Michigan, United States
Copyright © 2023 Cartwright and Colgan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian M. Cartwright, Ian.Cartwright@cuanschutz.edu