 Chenyu Li1†
Chenyu Li1† Donglu Qin
Donglu Qin Yang Yang
Yang Yang Bilian Yu
Bilian Yu- 1Department of Cardiovascular Medicine, the Second Xiangya Hospital, Research Institute of Blood Lipid and Atherosclerosis, Central South University, Changsha, Hunan, China
- 2Department of Spine Surgery, the Second Xiangya Hospital, Central South University, Changsha, Hunan, China
The incidence of heart failure with preserved ejection fraction is increasing in patients with obesity, diabetes, hypertension, and in the aging population. However, there is a lack of adequate clinical treatment. Patients with obesity-related heart failure with preserved ejection fraction display unique pathophysiological and phenotypic characteristics, suggesting that obesity could be one of its specific phenotypes. There has been an increasing recognition that overnutrition in obesity causes adipose tissue expansion and local and systemic inflammation, which consequently exacerbates cardiac remodeling and leads to the development of obese heart failure with preserved ejection fraction. Furthermore, overnutrition leads to cellular metabolic reprogramming and activates inflammatory signaling cascades in various cardiac cells, thereby promoting maladaptive cardiac remodeling. Growing evidence indicates that the innate immune response pathway from the NLRP3 inflammasome, to interleukin-1 to interleukin-6, is involved in the generation of obesity-related systemic inflammation and heart failure with preserved ejection fraction. This review established the existence of obese heart failure with preserved ejection fraction based on structural and functional changes, elaborated the inflammation mechanisms of obese heart failure with preserved ejection fraction, proposed that NLRP3 inflammasome activation may play an important role in adiposity-induced inflammation, and summarized the potential therapeutic approaches.
Introduction
The incidence of heart failure (HF) is rapidly increasing. As of 2017, an estimated 64.3 million individuals have been diagnosed with HF worldwide (1). More than half of the patients with HF have HF with preserved ejection fraction (HFpEF), and the incidence rate of HFpEF is still increasing (2). HFpEF, which is characterized by an ejection fraction (EF) >50% with diastolic dysfunction, high filling pressures, and exercise intolerance (3), is a heterogeneous syndrome that exhibits different phenotypes due to various risk factors, such as obesity, hypertension, and diabetes.
The global number of people with obesity is increasing at an astonishing rate. By 2020, the populations of overweight and obesity worldwide have reached 1.9 billion and 650 million, respectively (4). Obesity is a prevalent and independent risk factor for HF, regardless of the ejection fraction. Many studies have demonstrated that patients with obesity have a greater risk of HFpEF than of HF with reduced ejection fraction (HFrEF), and excess weight and obesity are also prevalent in patients with HFpEF (83%) (5, 6). Compared to other types of HFpEF, obese HFpEF has unique hemodynamic and structural abnormalities, in which chronic inflammatory conditions play a crucial role in its emergence and development (7, 8). Therefore, this review identifies inflammation as a specific etiology of obese HFpEF, describes the pathophysiological mechanisms of inflammation in obese HFpEF, and proposes potential anti-inflammatory therapies.
Evidence supporting the existence of obese HFpEF
Anatomical structure and histological changes of the heart in obesity
Early autopsy reports have found increased heart weight and ventricular hypertrophy in patients with obesity, more commonly with left ventricular (LV) hypertrophy than the right (9, 10). Animal studies have also shown that the LV wall thickens, and its volume increases in the early stages of obesity (11). Therefore, the cardiac structural changes caused by obesity are mainly LV remodeling, with increased wall thickness more than dilatation. Two forms of LV hypertrophy, concentric and eccentric, have been found in individuals with obesity; however, concentric hypertrophy is more common (12, 13). It is noting that the different geometries of ventricular hypertrophy in individuals with obesity may be associated with different insulin sensitivity states and not all obese individuals develop LV hypertrophy. Sciacqua et al. (14) found that ventricular remodeling in metabolically unhealthy obese individuals showed more eccentric hypertrophy than that in metabolically healthy individuals, implying that obesity-related metabolic abnormalities play a role in ventricular hypertrophy. Another cross-sectional study also suggested that metabolic abnormalities, but not metabolically healthy obesity, were significantly associated with LV hypertrophy (15).
Although relatively rare, right ventricular (RV) thickness and volume can also increase in individuals with obesity. Preliminary studies have suggested that RV hypertrophy and dilatation are secondary to pulmonary hypertension due to obesity-induced LV failure, sleep apnea, and hypoventilation (16, 17). However, subsequent research (18) has demonstrated that body mass index (BMI) is the most relevant clinical factor for biventricular hypertrophy in individuals with obesity without pulmonary hypertension. Wong et al. (19) also found that increased BMI in subjects with overweight and obesity was associated with severity of RV dysfunction, independent of sleep apnea. These data suggested a direct correlation between obesity and RV hypertrophy.
In addition to macro-anatomical structures, the hearts of patients with obesity often undergo multiple morphological changes at the cellular level. Diffuse cardiomyocyte hypertrophy is the most specific histological feature of obesity-associated cardiac injury. Early clinical research found that compared with patients without obesity but with HF, cardiomyocyte hypertrophy is more evident in patients with obesity and HF (20). Despite cardiomyocyte remodeling, histological changes are different in concentric and eccentric hypertrophy. In contrast to concentric hypertrophy, in which cardiomyocytes grow only laterally, in eccentric hypertrophy, cardiomyocytes grow proportionally in both longitudinal and lateral directions (21).
Both animal and human studies have demonstrated microvascular rarefaction, interstitial fibrosis, and cellular infiltration in the myocardium of patients with obesity-related HFpEF (22, 23). Myocardial microvascular density is decreased in obesity-related HFpEF, which causes cardiac hypoperfusion owing to reduced myocardial oxygen delivery (24, 25). Furthermore, increased type I collagen and transforming growth factor (TGF)-β1 levels proved the interstitial fibrosis increase in the hearts of obese animal models (22, 26). Interstitial fibrosis mainly refers to the complex process of excessive proliferation of myocardial interstitial fibroblasts, abnormal distribution, and excessive deposition of collagen caused by various stresses, such as chronic hypoxia and increased oxidative stress (26, 27). Studies have also found that the expression of the pro-inflammatory macrophage markers monocyte chemoattractant protein-1 (MCP-1) and CD11c increased in the myocardial mRNA of obese mice, suggesting that obesity induces inflammatory cells and factors infiltration of cardiomyocytes (22), implying the role of inflammation in HFpEF.
Cardiac functional consequences associated with obesity
Studies have shown that LV diastolic dysfunction, rather than systolic dysfunction, represents the earliest sign of cardiac involvement in patients with obesity (28, 29). Diastolic dysfunction may precede systolic dysfunction or may occur simultaneously with systolic dysfunction. The typical diastolic dysfunction in patients with obesity is abnormal diastolic filling or decreased myocardial compliance, which manifests as higher left ventricular end-diastolic pressure (LVEDP) and lower mean mitral valve E/A ratio on echocardiography (28, 30). In addition, a load-dependent myocardial tissue imaging technique revealed increased myocardial stiffness in patients with obesity, further confirming abnormal myocardial relaxation (31, 32). However, there are inconsistent findings regarding the effect of obesity on LV systolic function in early studies. Several studies have shown no obvious difference in the mean LV systolic fraction between subjects with moderate to severe obesity and lean subjects (33, 34). In contrast, many studies have found that subjects with moderate obesity had a remarkably lower mean left ventricular ejection fraction (LVEF) than lean subjects (35, 36). Notably, despite the decline, the median LV systolic function in these studies was always within the normal range for obesity (33–36). Thus, the negative results in certain studies may be due to the load dependence and insensitivity of the criteria employed to assess the LV contractile function.
In recent years, methods to assess systolic function by quantifying myocardial strain have been proposed, including echocardiography to measure circumferential fiber-shortening velocity, tissue Doppler echocardiography, cardiac magnetic resonance tissue labeling, and feature tracking (37–39). Precise measurements using more sensitive approaches showed that the myocardial tissue strain index and velocity decrease proportionally with increasing obesity, even if the LVEF remains normal, suggesting that obesity is implicated in subclinical systolic dysfunction (40, 41). Obokata et al. (42) also found that, while LVEF was similar in both the obese HFpEF group and controls, LV systolic function was impaired in patients with obese HFpEF, as evidenced by decreased longitudinal strain.
Obesity not only leads to LV dysfunction but also RV dysfunction. Studies have found that the RV stroke volume and RV ejection fraction are lower in individuals with overweight and obesity, even if the LV parameters are adjusted, supporting the opinion that RV dysfunction in HFpEF may be parallel to LV dysfunction rather than a consequence of HF deterioration (42, 43). Thomas et al. (44) and Aschaueret et al. (45) confirmed that 19%–25% of patients with HFpEF present with RV dysfunction. Furthermore, RV dysfunction in HFpEF in these studies was primarily determined at rest. Recent studies have indicated that RV reserve is impaired during exercise in patients with HFpEF, even though RV systolic and diastolic functions may remain normal at rest (46), implying a higher incidence of RV dysfunction.
Difference between HFpEF with and without obesity
Compared with patients with HFpEF without obesity, patients with obese HFpEF have significantly different hemodynamics and a unique pattern of myocardial remodeling. At rest, patients with obese HFpEF show higher right atrium (RA) pressure and pulmonary capillary wedge pressure (PCWP). During exercise, obese HFpEF patients had higher left and right heart filling pressures and higher pulmonary artery pressures which induced more severe RV dysfunction. In addition to RV dysfunction, increased left ventricular mass and volume, and the increased ratio of the two indicated greater concentric remodeling in obese HFpEF (32, 42). Profound plasma volume overload, increased pericardial restraint caused by increased epicardial fat thickness, and enhanced ventricular interaction are contributors to the unique hemodynamic derangements and ventricular remodeling in HFpEF patients with obesity.
Despite more severe symptoms and hemodynamic abnormalities, brain natriuretic peptide (BNP) levels, which are a critical diagnostic tool for HF, are lower in patients with obesity than those without obesity. Multiple studies have found an inverse correlation between BMI and BNP levels (47, 48). Tarama et al. (49) further found that BNP levels were relatively lower in HF patients with overweight and obesity. In contrast, another study found that BNP and N-terminal pro-B-type natriuretic peptide (NT-proBNP) concentrations increase after weight loss in patients with obesity (50). Enhanced BNP degradation in the adipose tissue upon the action of adipocyte-derived neprilysin may contribute to the spuriously low BNP levels observed in patients with HF (51). In addition, the changes of signal natriuretic peptide receptor (NPR) - A and clearance of NPR-C receptor in adipose tissue may also be one of the reasons (52).
Low BNP levels in the obese HFpEF phenotype indicate that measurement of natriuretic peptides may be misleading in its diagnosis. Therefore, it is necessary to explore other specific biomarkers of the obese phenotype. Obese HFpEF has been associated with higher levels of inflammatory markers, such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and soluble growth stimulation expressed in gene 2 (53, 54). Based on differences in plasma biomarkers, response to spironolactone, echocardiography, and arterial tone, HFpEF has been classified into three types in the literature (55). Among them, phenotype group 3, prevalent in patients with obesity and diabetes mellitus, was characterized by systemic inflammation with elevated levels of inflammatory factors, especially TNF-α and renin, and a preferential response to spironolactone (55, 56). These results demonstrate that obese HFpEF differs from other types of HFpEF, and this difference suggests distinct underlying mechanisms across the clinically identifiable phenol groups of HFpEF.
Unique mechanisms of obese HFpEF
Various central pathophysiological anomalies have shown indications of being associated with the obese HFpEF phenotype, such as energy substrate metabolism changes, neurohormonal perturbations, and sodium retention (57). Inflammation has been extensively studied in recent years as a significant pathophysiological change in obese heart failure (54). Chronic nutrient overload, as the initiating factor of obesity, promotes adipose tissue expansion and activates inflammatory responses with the overproduction of pro-inflammatory cytokines and subsequent systemic inflammation and cardiac remodeling (58, 59) (Figure 1); on the other hand, overnutrition contributes to cellular metabolic reprogramming and exerts direct pro-inflammatory effects on different kinds of cardiac cells, which is also an important reason for HFpEF (59, 60) (Figure 2).
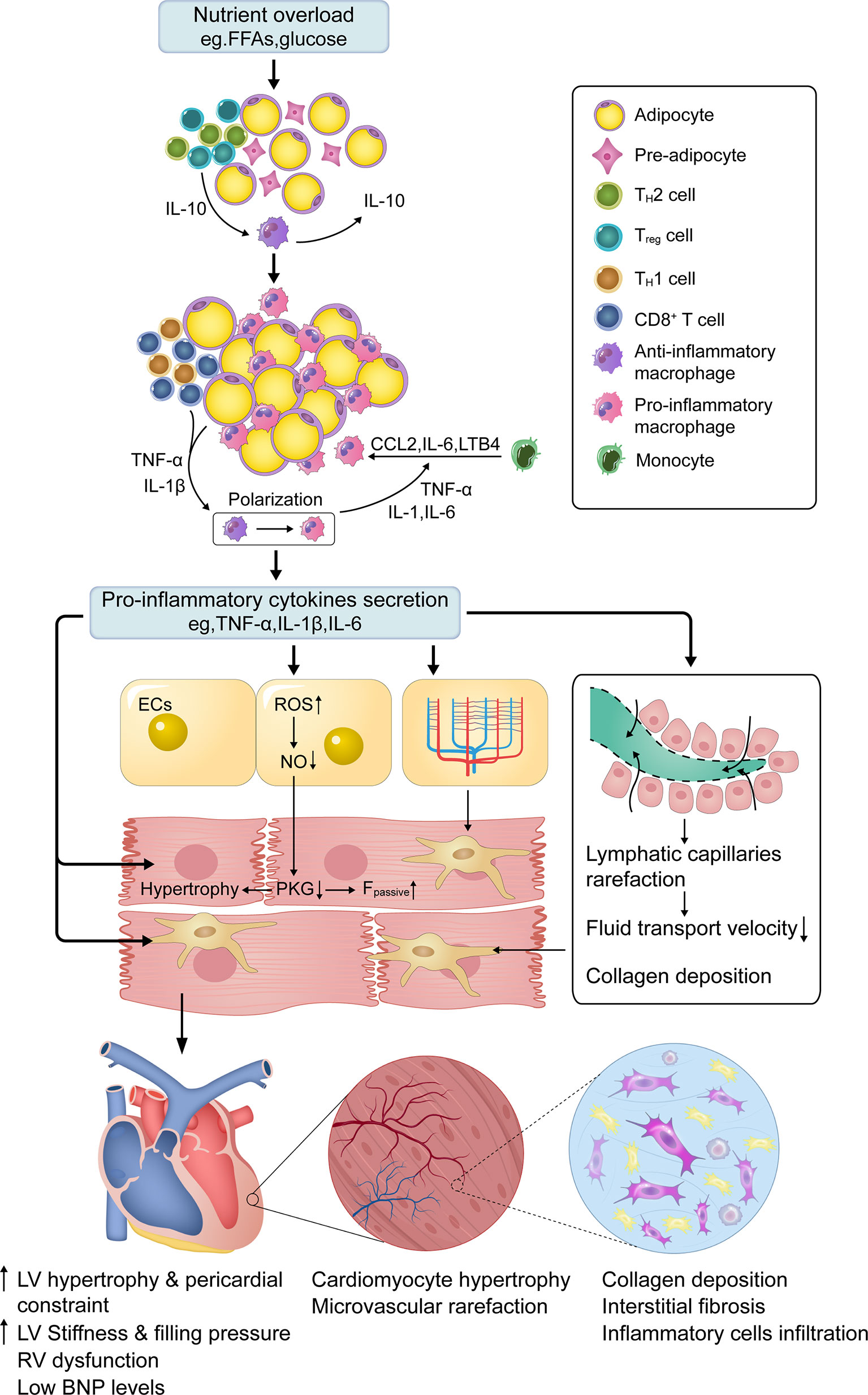
Figure 1 Central role of low-grade systemic inflammation in obese HFpEF. In normal adipose tissue, TH2 and Treg polarized T cells predominate in ATTs. These T cells secrete IL-10, which stimulates resident ATMs to secrete IL-10 to limit inflammation. Chronic nutrient overload induces adipose tissue expansion and a shift in ATTs such that CD8+ and TH1 polarized T cells dominate, which recruit circulating monocytes to adipose tissue through chemokines (e.g., CCL2). Under the stimulation of cytokines (e.g., TNF-α and IL-1β) secreted by ATTs and enlarged adipocytes, ATMs polarize towards a pro-inflammatory macrophage phenotype and attract additional monocytes by secreting pro-inflammatory cytokines (e.g., IL-6, IL-1, and TNF-α). This forms a vicious cycle in which adipose tissue becomes inflamed. High levels of circulating proinflammatory cytokines activate inflammatory cascades in different cardiac cells, which are associated with cellular dysfunction. Among them, coronary microvascular endothelial ROS overproduction and dysfunction drive cardiomyocyte hypertrophy and stiffening, as well as interstitial fibrosis, by lowering myocardial NO bioavailability and PKG activity, which is the critical mechanism for obese HFpEF. Microvascular and lymphatic capillary rarefaction also contributes to interstitial fibrosis through insufficient cardiac perfusion and poor lymphatic transport. FFAs—free fatty acids; ATTs—adipose tissue T cells; ATMs—adipose tissue macrophages; IL-1/1β/6/10/18—interleukin-1/1β/6/10/18; CCL2—C-C motif chemokine ligand 2; LTB4—leukotriene B4; TNF-α—Tumor necrosis factor-alpha; ECs—endothelial cells; NO—nitric oxide; PKG—protein kinase G; ROS—reactive oxygen species; Fpassive—cardiomyocyte resting tension; RV—right ventricle; IVS—interventricular septum; LV—left ventricle; HFpEF—heart failure with preserved ejection fraction; BNP, Brain Natriuretic Peptide.
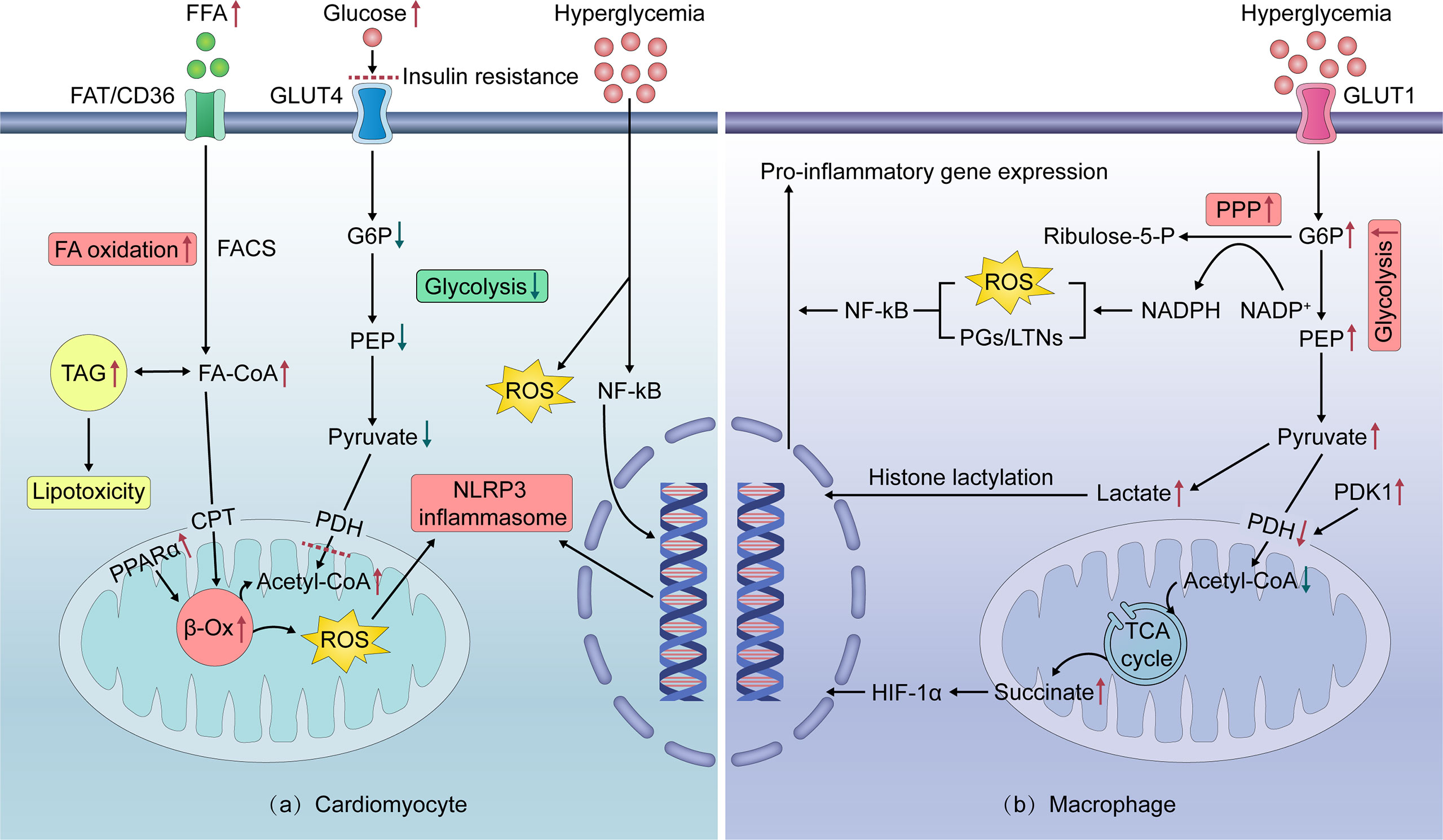
Figure 2 Intracellular mechanisms of cardiomyocyte and pro-inflammatory macrophage metabolic reprogramming in obesity. (A) In nutrient overloaded cardiomyocytes, excessive FFA intake activates PPARα, thereby enhancing FA β-oxidation. This leads to excess ROS production, which activates the NLRP3 inflammasome to promote the secretion of inflammatory cytokines. Excess FA is stored as TAGs, which can mediate lipotoxicity. Increased FA oxidation contributes to cardiomyocyte IR by inhibiting GLUT4-mediated glucose uptake and utilization, resulting in cardiomyocyte exposure to high glucose levels. Hyperglycemia can induce ROS overproduction and directly trigger a series of pro-inflammatory pathways in cardiomyocytes. (B) In nutrient overloaded macrophages, glucose intake via insulin-independent GLUT1 and metabolism reprogram to rapid aerobic glycolysis. The product of glycolysis, lactate, increases histone lactation, thereby inducing the pro-inflammatory polarization of macrophages. Glycolysis also generates NADPH through the PPP, which promotes the production of ROS and supports proinflammatory prostaglandin/leukotriene synthesis, thereby activating NF-kB. In addition, enhanced glycolysis induces a truncated tricarboxylic acid cycle and succinate accumulation via decreased pyruvate entry into the mitochondria, which promotes HIF-1α activation and pro-inflammatory gene expression. FA—fatty acid; FFAs—free fatty acids; FAD/CD36—fatty acid translocase; TAG—triacylglycerol; FACS—fatty acyl-coenzyme A synthetase; FA-CoA—fatty acyl-coenzyme A; CPT—carnitine palmitoyltransferase; PDH—pyruvate dehydrogenase complex; PDK1—pyruvate dehydrogenase kinase 1; Acetyl-CoA—Acetyl coenzyme A; PPARa—peroxisome proliferator-activated receptor alpha; β-Ox—beta-oxidation; G6P—glucose-6-phosphate; PEP—phosphoenolpyruvate; PPP—pentose phosphate pathway; NADPH—nicotinamide adenine dinucleotide phosphate; NADP+—oxidized form of nicotinamide adenine dinucleotide phosphate; PGs—prostaglandins; LTs/LTNs—leukotrienes; TCA cycle—tricarboxylic acid cycle; HIF-1α—hypoxia-inducible factor-1 alpha; Ribulose-5-P—ribulose-5-phosphate; PPARα—peroxisome proliferator-activated receptor α; ROS—reactive oxygen species; NLRP3—NOD-like receptor family, pyrin domain containing 3; TAGs—triacylglycerols; IR—insulin resistance; GLUT1/4—glucose transporters 1/4; IL-1β/6/12—interleukin-1β/6/12; TNF-α—Tumor necrosis factor-αlpha; NF-κB—nuclear factor kappa B.
Inflamed adipose tissue and low-grade systemic inflammation in obesity
Excessive intake of nutrients by the body increases insulin secretion and induces the transport and storage of adipose and muscle cells in the form of triglycerides and glycogen (61). Adipose tissue is a multifaceted fat depot and a cellular energy sensor. Adipocytes autonomously sense energy stores; accordingly, they initiate a feedback circuit by secreting adipokines that reduce food intake and activate the sympathetic nervous system to increase thermogenesis and lipolysis (62, 63). In a state of continuous positive energy balance, the constant deposition of triglycerides results in an increase in the size and number of adipocytes. However, these cellular adaptive changes will reach a threshold, and further anabolic pressure will not be regulated. Once this threshold is reached, adipocytes will be under pressure, and one of the responses to this pressure is to initiate the inflammatory process.
It is generally considered that the initial event of the inflammatory process is the imbalance of adipokine secretion from adipocytes, characterized by the reduced secretion of adiponectin and increased secretion of inflammatory mediators, including IL-6, leukotriene B4, and C-C motif chemokine ligand 2 (CCL2), which mediate circulating monocyte trafficking into the adipose tissue (59, 64, 65). Infiltrating monocytes and adipose tissue macrophages (ATMs) polarize into the pro-inflammatory subtype and attract additional monocytes by secreting their chemotactic and pro-inflammatory mediators (e.g., TNF-α, IL-1, and IL-6). The adipose tissue becomes inflamed, leading to chronic low-grade systemic inflammation (64, 66). Although the above process has been well-described, the cells that initiate the inflammatory cascade remain elusive. Several studies have shown that shifts in T cell composition in adipose tissue occur before monocyte-macrophage recruitment in obesity (67, 68). In lean mice, fat regulatory T (Tregs) and T helper type 2 (TH2) cells predominate in the adipose tissue and secrete IL-10 to block adipose tissue inflammation. There is a conversion in fat T cells during the onset of obesity, before the influx of macrophages allows CD8+ and TH1 adipose tissue T cells to dominate, and Treg and TH2 cells are depleted. This transformation may initiate the trafficking of macrophages to fat via chemokines such as CCL2, which promotes local and systemic inflammation.
In obesity, the degree of inflammation in the adipose tissue varies in different anatomic regions. The subcutaneous adipose tissue is the largest lipid reservoir that prevents fat accumulation in organs under physiological conditions (69). Since the subcutaneous adipose tissue cannot store excess fat, overloaded fat accumulates, particularly in the visceral adipose tissue (70). Thus, visceral adipose tissue is a prominent site that becomes dysfunctional and inflamed during obesity, and it plays a significant role in cardiac pathophysiology. Given its close anatomical proximity to the heart and shared vascular supply, epicardial adipose tissue (EAT) also plays an essential role in myocardial remodeling in obesity mainly through mechanical and metabolic effects (70, 71). Metabolically, since myocardial and EAT depend on the same microvascular system, EAT in patients with obesity secrete a variety of adipokines including TNF-α, MCP-1, IL-6, IL-1β, fibrinogen activator inhibitor-1 (PAI-1), which can act directly on myocardial tissue through paracrine and vascular secretion, producing a proinflammatory state that promotes cardiomyocyte stiffness and coronary endothelial dysfunction (72). Mechanically, the enlarged EAT volume in patients with obesity might result in pericardial constrain. Indeed, the right atrial pressure of obese HFpEF, the measurable pressure closest to the pericardial pressure, increased during rest and exercise, suggesting the existence of pericardial restraint in obese HFpEF (42). Pericardial constraint may also lead to enhanced ventricular interdependence, which manifests as a higher ratio of LV and RV filling pressure, increased pulmonary venous pressure, and a larger LV eccentricity index (42, 73).
Central role of low-grade systemic inflammation in obese HFpEF
Previous studies have suggested that hemodynamic changes and insulin resistance are leading causes of HFpEF (42, 57). However, in the recently proposed HFpEF paradigm, inflammation is considered the main pathophysiological factor of obese HFpEF (8, 54, 74). In cross-sectional studies of patients with HFpEF, higher circulating levels of TNF-α and IL-6 have been found (53, 54). Multiple studies have also demonstrated that inflammatory biomarkers such as TNF-α, IL-6, and hs-CRP are strongly associated with HFpEF incidence (53, 54, 75). Recently, a proteomic analysis to explore the inflammation paradigm in patients with HFpEF quantified 248 unique circulating proteins and showed that systemic inflammatory mediators were associated with echocardiographic indicators of worse hemodynamics and RV dysfunction. TNF-R1, UPAR, IGFBP-7, and GDF-15 were the top molecules that influenced the relationship between echocardiographic parameters and comorbidity burden (8). The obesity-induced systemic inflammatory status is implicated in several crucial processes in cardiac remodeling, including coronary microvascular and lymph-vessel dysfunction, cardiomyocyte hypertrophy and apoptosis, myocardial fibrosis, and consequently, HFpEF.
Coronary microcirculation disturbances are caused by the imbalance between endothelial-derived relaxing factors such as nitric oxide (NO) and contractile factors such as endothelin 1 (76). Pro-inflammatory cytokines are known to trigger the overproduction of endothelial reactive oxygen species (ROS) by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) (77), which induces increased expression of high nitrotyrosine and reduces NO bioavailability. Studies have found that plasma NO metabolite levels are lower in patients with HFpEF than in patients with HFrEF, confirming the reduced NO bioavailability in patients with HFpEF (78). Consequently, reduced NO bioavailability results in impaired NO-cyclic guanosine monophosphate-protein kinase G (NO-cGMP-PKG) signal transduction pathway, thereby causing the hypertrophy and stiffening of surrounding cardiomyocytes (54, 79). Along with the altered paracrine signaling between microvascular endothelium and cardiomyocytes, reduced myocardial microvascular density, known as rarefaction, is observed in patients with HFpEF. This impairs cardiac perfusion and myocardial oxygen delivery, leading to a deteriorated diastolic function in patients with HFpEF (24, 25).
Coronary microvascular endothelial dysfunction drives cardiomyocyte stiffening and hypertrophy by lowering myocardial NO bioavailability and PKG activity, which are critical mechanisms in obese HFpEF (54). In addition, robust evidence suggests that sustained stimulation of inflammatory cytokines has a direct detrimental impact on the heart (80). At the cellular level, inflammatory cytokines can directly bind to cardiomyocyte surface receptors, triggering the downstream signaling of cellular inflammation, hypertrophy, and apoptosis (80–82). TNF-α, for example, initiates the signaling cascade involved in cardiomyocyte hypertrophy from the cell membrane by binding to two different TNF receptors (83, 84). Moreover, IL-1β affects the progression of cardiomyocyte hypertrophy through insulin-like growth factor-1 (IGF-1) production, which negatively regulates c-Jun N-terminal kinase (JNK) signals (85). Consistently with the in vitro results, IL-1 receptor type I-deficient mice were protected from aging-exacerbated HFpEF, due to being unresponsive to IL-1 (86).
The extensive lymphatic network of the heart is critical for maintaining cardiac fluid balance, in which the lymphatic reflux is mainly driven by cardiac contraction (87). Lymphatic dysfunction and remodeling have been observed in various experimental models of HFpEF-related comorbidities (88, 89). Various immune cells were attracted to the lymphatic vessels, reducing the pumping rate of dermal lymphatic collection vessels and decreasing the density of lymphatic capillaries. This leads to reduced lymphatic transport in several preclinical obesity models (90, 91), implying a role of inflammation in lymphatic dysfunction.
The accumulation of excessive extracellular matrix and cardiac fibrosis are hallmarks of deleterious remodeling that occurs during HFpEF (24, 92, 93). Pro-inflammatory cytokine IL-16 and macrophage infiltration in the myocardium reportedly contribute to increased cardiac fibrosis and LV stiffness in HFpEF (94, 95). Furthermore, microvascular rarefaction is also associated with myocardial fibrosis in patients with HFpEF, suggesting that decreased cardiac perfusion may cause overproduction and accumulation of collagen (24). Interestingly, cardiac edema and inadequate lymphatic transport increase collagen production. In contrast, the reduction of cardiac edema by accelerating cardiac lymphangiogenesis alleviated interstitial fibrosis in a rodent model of myocardial infarction, implying a possible role of inflammation in regulating cardiac interstitial fibrosis through edema and poor lymphatic transport (96).
Direct pro-inflammatory effects of nutrients overload on theheart: The central role of cardiac metabolic reprogramming
In addition to circulating inflammatory factors, nutrient overload has recently been found to directly induce cardiac inflammation mainly through cellular metabolic reprogramming, particularly in myocytes (59). In normal hearts, over 95% of the energy is produced by mitochondrial oxidative phosphorylation. Free fatty acids (FFAs) are the primary fuel substrates for the myocardium, with the remaining 30% coming from glucose and other metabolites such as lactate (97). Normally, the heart exhibits remarkable fuel flexibility and switches between glucose and fatty acids, depending on the energy requirements of the heart and the body’s nutritional status. The glucose-fatty acid cycle of Randle controls the coordinated regulation of metabolic substrate utilization, which is important for the maintenance of the heart’s energy homeostasis (98).
In obesity, elevated FFA levels upregulate the expression of genes involved in fatty acid oxidation (FAO) in cardiomyocytes by activating the nuclear receptor peroxisome proliferator-activated receptor α. Once the enhanced FFA uptake exceeds the FAO capacity, excess FFAs are stored as triglycerides, which mediate the ectopic triglyceride accumulation in the heart and lipotoxicity (99). Increased FFA utilization and lipotoxicity favor ROS overproduction, and in turn, activate distinct cellular inflammatory signaling cascades, resulting in myocyte hypertrophy, apoptosis, and cardiac insufficiency (100). Furthermore, increased FAO inhibits insulin-dependent glucose transporter 4 (GLUT4)-mediated glucose uptake and utilization, which is associated with insulin resistance, thereby leading to the exposure of cardiomyocytes to high glucose levels. Hyperglycemia can directly induce a series of pro-inflammatory pathways in cardiomyocytes that converge toward NF-κB, including the activation of mitogen-activated protein kinase and JNK, which promotes the upregulation of TNF-α, IL-1β, IL-6, and IL-12 (101). In addition, the upregulation of histone H3 lysine 9 trimethyl at the IL-6 promoter with high-glucose stimulation also favors cardiomyocyte inflammation (102). Recently, a novel molecular pathway by which hyperglycemia triggers direct O-GlcNAcylation of Ser280 in Ca2+/calmodulin-dependent kinase II and the consequent activation of NOX was identified in cardiomyocytes (103). This leads to increased cytosolic ROS accumulation and the subsequent activation of cellular inflammation (103, 104).
In addition to cardiomyocytes, nutrient overload can directly induce inflammation in other types of non-cardiomyocytes, such as macrophages and endothelial cells. Resident cardiac macrophages play essential roles in maintaining myocardial equilibrium during steady-state and tissue repair in the injured heart. At a steady state, cardiac macrophages resemble alternatively activated anti-inflammatory macrophages and function in homeostasis by helping to remove senescent dead cells and defending against infection without inducing an immune response (105). These alternatively activated macrophages primarily undergo FAO and oxidative phosphorylation to produce ATP. In response to pro-inflammatory stimuli, macrophages reprogram their metabolism to rapid aerobic glycolysis because of the increased demand for energy (106).
In contrast to cardiomyocytes, macrophages are naturally insulin-resistant and take up glucose through insulin-independent GLUT1. Thus, in obesity-related insulin resistance, macrophages can sense glucose availability and dynamically reprogram their core metabolism by shifting toward cytosolic glycolysis and away from mitochondrial respiration, polarizing themselves away from the anti-inflammatory subtype and toward the pro-inflammatory subtype through glycolysis intermediates. For example, lactate has been shown to increase histone lactylation to induce homeostatic gene expression in macrophages (107). Moreover, glycolysis supplies metabolic substrates, such as glucose-6-phosphate, for the pentose phosphate pathway (PPP) to generate NADPH, which can be used by NOX to produce ROS and support the synthesis of inflammatory leukotrienes and prostaglandins, thereby triggering NF-kB signaling (59, 108). Enhanced glycolysis leads to suppression of the pyruvate funnel into the mitochondria, followed by a broken tricarboxylic acid cycle that results in succinate accumulation, which boosts hypoxia-inducible factor-1α (HIF-1α) activation of pro-inflammatory IL-1β gene expression (109).
The preference for substrate utilization in endothelial cells, particularly arterial endothelial cells, differs from that of cardiomyocytes and macrophages. Because of its low energy demand, glycolysis is the predominant glucose utilization pathway, whereas FFAs are not an important fuel in coronary endothelial cells in the normal state (110, 111). Recently, Wang et al. (112) demonstrated that metabolism in endothelial cells is reprogrammed during inflammatory stimuli, manifested as activated glycolysis, PPP, and mitochondrial respiration. While blocking the metabolic enhancement of glycolysis attenuates inflammation, inhibiting the activated PPP and mitochondrial respiration causes an aggravation of inflammation, implying the role of activated PPP and mitochondrial respiration as homeostatic anti-inflammatory mechanisms, which is different from that in macrophages.
Altogether, it is clear that the preference of different cells for metabolic substrates leads to different metabolic phenotypes in obese HFpEF. This phenomenon is important and should be considered in therapies targeting myocyte metabolism with unknown effects on other cardiac cells that continue to be prescribed.
NLR family pyrin domain containing 3 (NLRP3) inflammasome activation: A key culprit?
The innate immune system, which is the first line of defense of the host immune system, detects pathogens or host tissue damages, also known as pathogen-associated molecular patterns or damage-associated molecular patterns (DAMPs), respectively, mediated by pattern recognition receptors (PRRs). Upon recognition of DAMPs, some PRRs, including Nod-like receptors (NLRs) and Toll-like receptors (TLR), can assemble with other proteins to form multi-molecular complexes termed inflammasomes. Currently, the NLRP3 inflammasome is the most well-characterized inflammasome and is composed of the NLRP3, the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and pro-caspase-1 (113). The canonical NLRP3 inflammasome activation usually requires two steps: initial priming and subsequent inflammasome assembly (Figure 3). The first priming signal involves PRR or cytokine receptor activation to upregulate NLRP3 and pro–IL-1β/IL-18 transcription. NLRP3-specific stimulus (e.g., mitochondrial ROS, potassium efflux, long-chain saturated FAs, ceramide, and glucose) is implicated in NLRP3 activation (114, 115). Upon triggering, NLRP3 recruits the adapter molecule ASC to catalyze pro-caspase-1 maturation. Activated caspase-1 in turn cleaves pro-IL-1β/IL-18 and induces the release of their mature form (60, 116). IL-1 can induce the production and release of copious amounts of IL-6 from many cell types in an amplification loop (117).
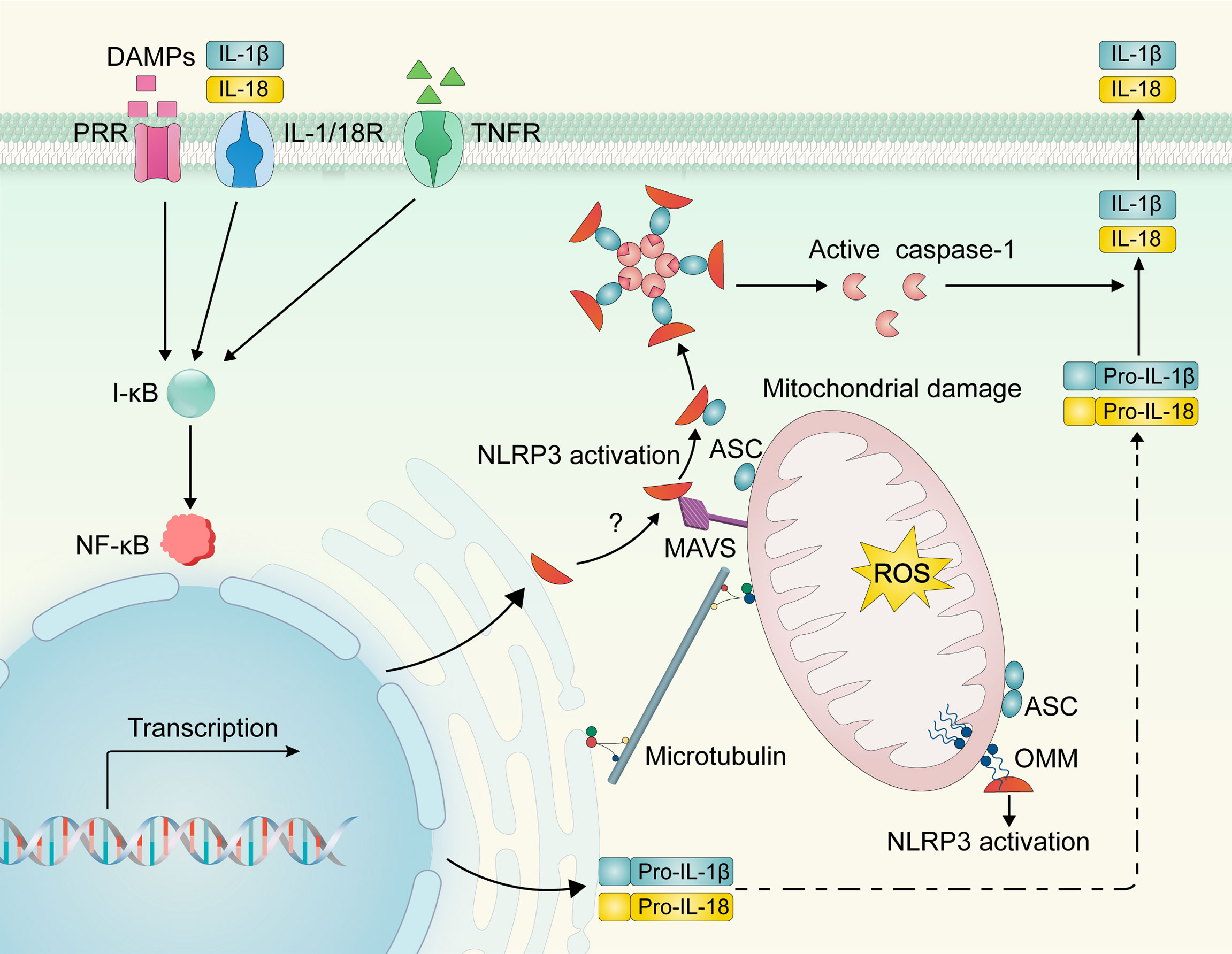
Figure 3 NLRP3 inflammasome activation. DAMPs and cytokines (e.g., IL-1β/18 and TNF-α) can bind to PRR and cytokine receptors, respectively, to activate the I-κB/NF-κB signaling pathway and induce NLRP3 and pro-IL-1β/IL-18 transcriptional upregulation. Upon activation, NLRP3 may be recruited from the ER to the mitochondria and interact with mitochondrial molecules (e.g., mitochondrial antiviral signaling protein and cardiolipin). Dynein-mediated mitochondria may regulate the interaction of NLRP3 and ASC via microtubule transport, resulting in inflammasome assembly, caspase-1 activation, and subsequent activation of IL-1β and IL-18. NLRP3—NOD-like receptor family, pyrin domain containing 3; DAMPs, damage-associated molecular patterns; PRR, pattern recognition receptor; IL-1/18R, interleukin-1/18 receptor; TNFR, Tumor necrosis factor receptor; NF-κB, nuclear factor kappa B; I-κB, inhibitory protein of NF-κB; pro-IL-1β/18, pro-interleukin-1β/18; ER, endoplasmic reticulum; OMM, outer mitochondrial membrane; ROS—reactive oxygen species; MAMs, mitochondria associate membranes; MAVS, mitochondrial antiviral signaling protein; CL, cardiolipin; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain.
Resting NLRP3 localizes to endoplasmic reticulum (ER) structures. However, a point of confusion is where the NLRP3 assembly occurs. Mitochondrial damage and ROS have been shown to act as upstream mechanisms of NLRP3 activation, implying a role of the mitochondria in NLRP3 inflammasome assembly. An informatics-based structure-function analysis predicted that NLRP3 localizes to mitochondria (118). Subsequently, several studies have shown that during NLRP3 inflammasome activation or overexpression, NLRP3 is recruited to the mitochondria and mitochondria-associated membranes, which are contact sites for the mitochondrial membrane and ER (119, 120). This recruitment requires a mitochondrial antiviral signaling protein whose interaction with NLRP3 depends on the N-terminal sequence of the latter molecule (121). Cardiolipin translocates from the inner mitochondrial membrane to the outer mitochondrial membrane and provides a docking site for NLRP3 in the impaired mitochondria (122).
There has been increasing recognition that NLRP3 inflammasome activation acts as a chief instigator of obesity, contributing to obesity-related systemic inflammation and insulin resistance. A Study by Stienstra et al. (123) found that the NLRP3 inflammasome contributes to the development of obesity and its comorbidities during high-fat diet (HFD) feeding in mice. Ablation of NLRP3 and deficiency of the inflammasome component, ASC or caspase-1, represses the inflammatory cascade response of inflamed adipose tissue and improves insulin signaling (123, 124). Mechanistically, it has been proposed that lipid messengers (e.g., palmitate and ceramides), which are elevated in both individuals with obesity and HFD-challenged mice, act as signals for NLRP3 inflammasome priming and activation (125–129). Previous studies have recognized that the elevation of palmitate in the context of obesity may induce NLRP3 inflammasome activation in macrophages, leading to the overproduction of inflammatory cytokines and subsequent insulin resistance in mice (125, 126). Vandanmagsar et al. (116) showed that the NLRP3 inflammasome senses ceramide in order to induce caspase-1 cleavage in macrophages and adipose tissue, thereby contributing to obesity-related inflammation and comorbidities.
In addition to the production of systemic inflammation originating from inflamed adipose tissue, recent research has shown that activation of the NLRP3 inflammasome is closely related to the severity of the HFpEF phenotype. Mice with HFpEF show NLRP3 inflammasome activation and overproduction of cytokines IL-1β/IL-18 in the heart (60, 130). Cardiac muscle mitochondrial abnormalities and ROS overproduction, which are important factors contributing to the pathophysiology of HFpEF, play a key role in activating the NLRP3 inflammasome (131, 132). Mitochondrial protein hyperacetylation in obese HFpEF increases the amount of ASC in mitochondria, exacerbating inflammation by facilitating NLPR3 inflammasome assembly in mitochondria and subsequent release of IL-1β/IL-18 and cardiac fibrosis (60). Collectively, these results could point to a key role of NLRP3 inflammasome in the pathophysiology of obese HFpEF and indicate that it should be inhibited to improve prognosis.
Targeting drug therapy of HFpEF: the road ahead and an exciting future
NLRP3 inflammasome, which is an important mediator in the development of obese HFpEF, is regarded as a promising therapeutic target due to its important role. This section focuses on drugs that directly or indirectly suppress inflammasome assembly and activation.
IL-1R/IL-1 inhibitors
Therapeutic targeting of the NLRP3 inflammasome in patients with HF has commonly used the IL-1 receptor antagonist (IL-1Ra) anakinra, thereby inhibiting signaling of both IL-1α and IL-1β. The D-HART pilot trial in patients with HFpEF showed that short-term IL-1 blockage with anakinra led to a significant reduction in systemic inflammatory mediators and improvement in peak oxygen consumption (VO2) and aerobic exercise capacity (133). Subsequently, the D-HART2 trial, in which 12 weeks of treatment resulted in a significant decline in NTpro-BNP concentrations compared with placebo, further confirmed the benefits of anakinra in the treatment of HFpEF (134).
Although these two trials suggested that blocking IL-1 is a promising treatment strategy for HFpEF with systemic inflammation, it also raises concerns regarding its safety because it interferes with immune homeostasis. Canakinumab, a monoclonal antibody that specifically neutralizes IL-1β and does not affect other IL-1 family cytokines, may be safer than anakinra. Although no clinical trials with canakinumab included patients with HFpEF, in a small substudy of the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) trial, 150 mg canakinumab treatment improved peak VO2 and increased LVEF in patients with HFrEF, implying the potential of canakinumab in HF treatment (135).
In addition to IL-1, IL-6 blockade in mice through genetic deletion is associated with decreased cardiac hypertrophy and fibrosis following angiotensin II stimulation (136). Although no clinical studies have explored anti-IL-6 therapeutics in patients with HFpEF, the findings regarding IL-6 and HFpEF development in the preclinical model warrant further exploration to investigate whether IL-6 is an appropriate therapeutic target.
Colchicine
Colchicine, the major alkaloid in Colchicum autumnale, is a potent drug with anti-inflammatory properties. It acts on inflammation through different mechanisms, with the inhibition of spindle microtubule assembly via copolymer formation being the best characterized (137). Recently, colchicine has been identified as a non-specific IL-1β inhibitor that blocks the activation of the cellular NLRP3 inflammasome, thus reducing the release of pro-inflammatory IL-1β/18 (138–143). Although colchicine is currently recommended for the treatment of acute gout attacks and familial Mediterranean fever, it has historically demonstrated benefits in various cardiovascular diseases, including postpericardiotomy syndrome, acute and recurrent pericarditis, postoperative atrial fibrillation, and chronic HF (144). In addition, clinical evidence of its action in coronary artery disease (CAD) has yielded encouraging results.
The low-dose colchicine (LoDoCo) pilot study and LoDoCo2 trial conducted by Nidorf et al. (145, 146) established a significant benefit of low-dose colchicine in preventing major adverse cardiovascular events in patients with stable CAD. Subsequently, the large-scale Colchicine Cardiovascular Outcomes Trial (COLCOT) tested low-dose colchicine in patients who experienced a recent myocardial infarction (MI) and served as a strong validation for the potential benefits of low-dose
colchicine in acute coronary syndrome (147). In addition to its effectiveness, the use of colchicine at 0.5–1.0 mg daily in cardiovascular trials has been proven safe in the absence of severe liver or renal disease. The most common side effects are gastrointestinal problems, which limit its usefulness in approximately 10% of patients; however, approximately 90% of patients tolerate the long-term continuous treatment well (148).
The effectiveness of low-dose colchicine in atherosclerotic cardiovascular disease, a chronic inflammatory disease, raises the question of whether inhibiting NLRP3 inflammasome-related inflammation with colchicine is effective in the treatment of obese HFpEF. Currently, evidence that colchicine can reduce the risk of HF is limited to a single randomized controlled trial that showed colchicine reduced the circulating inflammatory biomarker levels and had favorable effects on LV remodeling in patients with HFrEF. However, this did not translate into an impact on clinical outcomes, exercise capacity, or improvements in mortality (149). Considering the importance of chronic inflammation in the pathogenesis of obese HFpEF, the effectiveness of colchicine in HFpEF is worth exploring.
β-hydroxybutyrate
β-hydroxybutyrate (BHB) is a ketone metabolite tested by Youm et al. (150) for NLRP3 inflammasome blockade. Mechanistically, BHB inhibits the activation of the NLRP3 inflammasome by preventing the efflux of potassium and reducing ASC oligomerization and speck formation. Recently, in an HFpEF mouse model, by combining age, long-term HFD, and desoxycorticosterone pivalate challenge, Deng Y et al. (60) further proved that increasing the abundance of BHB blocked NLPR3 inflammasome formation and alleviated pro-inflammatory IL-1β/18-triggered mitochondrial dysfunction and myocardial fibrosis. These findings suggest that dietary or pharmacological attempts to increase BHB levels may be effective treatments for HFpEF. Caloric restriction, which increases BHB abundance, prevents inflammatory processes, thereby improving myocardial remodeling and LV diastolic function in both preclinical models and humans (23, 151, 152).
Sodium-dependent glucose co-transporter 2 (SGLT-2) inhibitors, which have been found to increase systemic and tissue BHB levels, thereby attenuating NLRP3 inflammasome activation and the development of HFpEF (153, 154), are another promising intervention for patients with obese HFpEF. In a multicenter randomized trial that evaluated the effect of SGLT2 inhibitor dapagliflozin on the Kansas City Cardiomyopathy Questionnaire Clinical Summary Score (KCCQ-CS) (155) in HFpEF patients, found that after 12 weeks of treatment with dapagliflozin, the KCCQ total symptom score, physical restriction score, and 6-minute walk test were improved, suggesting that SGLT2 inhibitor treatment improved the symptoms, physical restriction and motor function of patients with HFpEF. The recently concluded DELIVER trial which investigated the effects of dapagliflozin on major adverse cardiovascular events in patients with HFpEF, and reached a statistically significant and clinically meaningful reduction in the primary composite endpoint of cardiovascular CV death or worsening HF (156). In addition to dapagliflozin, the recently published EMPEROR-Preserved clinical trial explored the effect of empagliflozin (another SGLT2 inhibitor) on the primary composite endpoint of HF hospitalization or cardiovascular death in patients with HF and an EF of >40% (157). Empagliflozin produced a significant, early, and sustained reduction in the combined risk, regardless of the presence or absence of diabetes. These trials definitively defined the role of SGLT-2 inhibitors in patients with HFpEF (158).
The challenges from HFpEF animal models
As previously mentioned, although some therapeutic agents that directly or indirectly inhibit the NLRP3-IL-1/IL-6 pathway could turn out to be promising strategies against obese HFpEF, some have intolerable side effects and still need clinical trials. Future studies should focus on the discovery of drugs with specific targets and improved pharmacokinetic properties. Valuable HFpEF animal models that mimic the clinically obese HFpEF phenotype are essential for the successful development of novel drugs. Currently, the lack of suitable preclinical obese HFpEF models that adequately reflect the disease’s clinical complexity has led to the inability to determine the mechanistic models and develop new therapeutic agents.
The development of obese HFpEF in humans is the result of multiple comorbidities, including obesity, prediabetes, and pulmonary and systemic arterial hypertension, which are difficult to reproduce in one animal model. Recently, two diagnostic algorithms (H2FPEF and HFA-PEFF scores) have been proposed to standardize the diagnosis of human HFpEF (3, 159). Withaar et al. (160) evaluated the translational value of HFpEF mouse models in the context of these two clinical scores and found that most HFpEF animal models do not meet the clinical diagnostic criteria for HFpEF, although some multi-hit models simulate human HFpEF to a reasonable extent. Several typical obese HFpEF animal models and their pathophysiological characteristics are shown in Table 1.
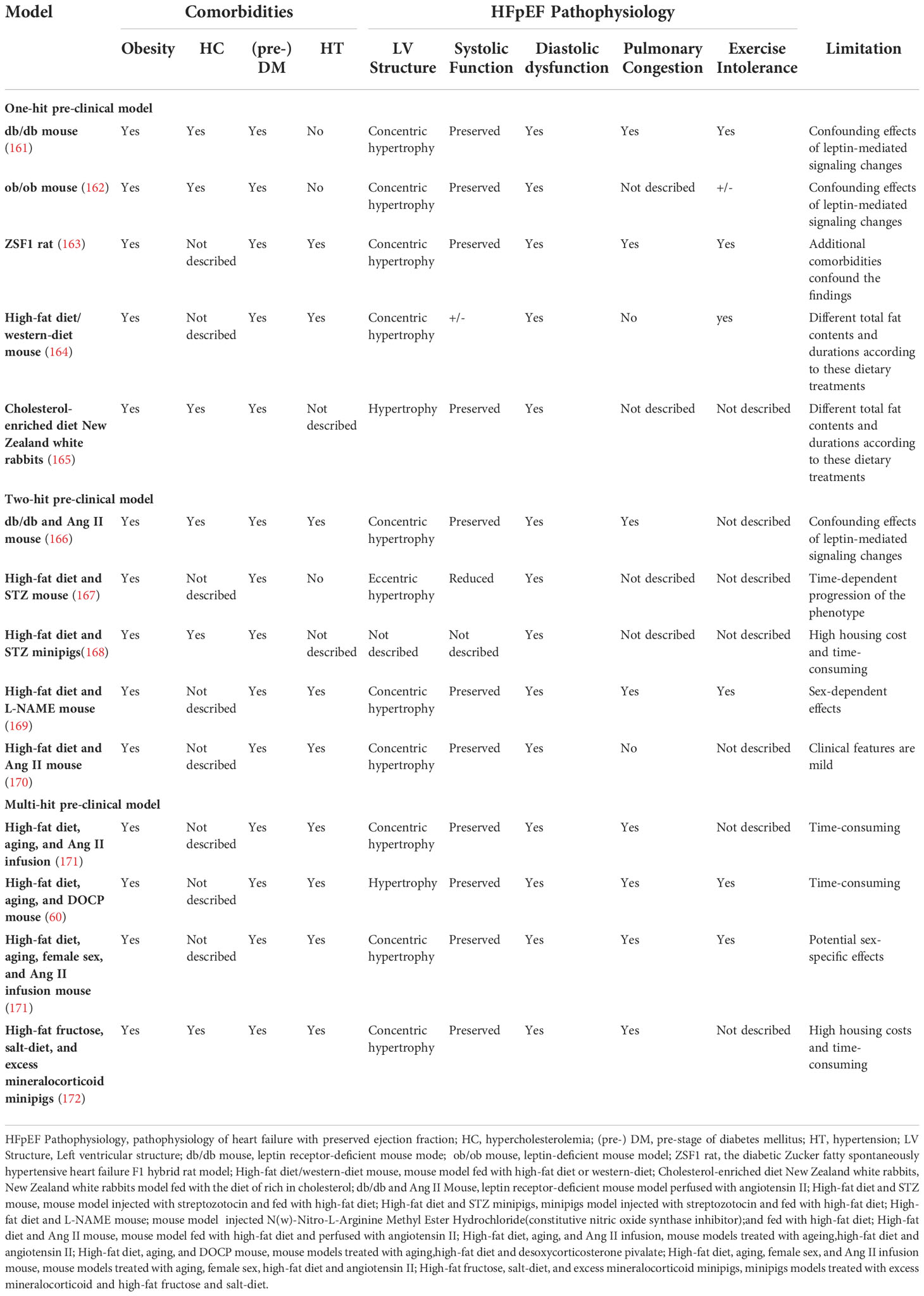
Table 1 Pathophysiology characteristics of Obese HFpEF animal models.
Several methods can be used to induce obesity in laboratory animals, including the use of genetically modified mice. Obese mouse models lacking the leptin gene (ob/ob) and leptin receptor (db/db) show signs of HFpEF, including concentric hypertrophy, exercise intolerance, and pulmonary hypertension (173). Obesity is characterized by leptin resistance in humans; however, the mouse model relies on leptin deficiency and does not accurately recapitulate the clinical obese HFpEF phenotype (174, 175). Similarly, the obese ZSF1 (Zucker fatty spontaneously hypertensive heart failure F1 hybrid) rat, which was developed by crossing rat strains with two separate leptin receptor mutations, also exhibited signs of HFpEF (elevated LVEDP, elevated E/e, and preserved LVEF) (163, 176). Notably, in addition to obesity and metabolic abnormalities, the ZSF1 model presents other comorbidities, such as spontaneous time-dependent chronic kidney disease or hypertension, which may be considered to mimic human HFpEF in many scenarios. However, if the aim is to discern the contribution of obesity to HFpEF, then the additional chronic kidney disease would confound the findings (173).
To avoid potential perturbations based on altered leptin expression and signaling in genetically modified models, numerous dietary treatment regimens have been used to induce obesity in rodents, which has become a standard approach in preclinical studies. HFD usually contains a total fat content of up to 60%, whereas a Western diet typically has a high-fat content and commonly uses sucrose. Cardiac abnormalities in the diet-induced obesity model vary depending on the total fat content and the duration of these dietary treatments, thereby limiting pre-clinical utility (177). For example, mice challenged with HFD with 60% fat content developed systolic dysfunction after 10 weeks of feeding and had an increase in mortality (178). In contrast, exposure to a Western diet leads solely to diastolic dysfunction, while preserving systolic function (164). To overcome the potential low penetrance of diabetes development in diet-induced obesity, several studies have used HFD plus low-dose streptozotocin to induce β-cell dysfunction and diabetes. Additional treatment with streptozotocin in HFD-challenged mice promoted eccentric cardiac hypertrophy and LV diastolic dysfunction (167, 179, 180).
Given that the development of obese HFpEF in humans is the result of several comorbidities, combinatory models of HFD and other factors have been evaluated. Schiattarella et al. (169) combined HFD and hypertension induced by inhibition of nitric oxide synthase to produce a 2-hit preclinical mouse model that resembles human obese HFpEF. However, sex-dependent effects, which are an obstacle to preclinical application, have been reported (181). The combination of HFD and angiotensin II (Ang II)-dependent hypertension also resulted in HFpEF. However, symptoms, signs, and hemodynamic features appear to be mild, since the effect on exercise capacity is unknown and lung congestion in young animals is absent. Recently, Deng et al. (60) developed a 3-Hit strategy consisting of a 13-month HFD and 16 months of aging, followed by an intraperitoneal injection of a bolus of desoxycorticosterone pivalate for 1 month. By the end of the 13 months, 3-hit-treated mice manifested pathological myocardial hypertrophy and fibrosis, diastolic dysfunction, as well as pulmonary congestion, and elevated natriuretic peptides, which recapitulated the typical pathological phenotype of obese HFpEF. However, generating this model is time-consuming and not conducive to testing potential new therapeutic agents. Because female sex was independently associated with the presence of diastolic dysfunction and worse clinical outcomes in HFpEF patients (182), Withaar et al. (171) developed a multi-hit model which combined advanced age, female sex, and an HFD with Ang II infusion. Although this HFpEF model doesn’t represent the entire spectrum of human HFpEF, it provides a sex-specific approach for exploring the pathophysiology of HFpEF.
In addition to the limitations mentioned above, the hearts of rodent models are not comparable in size, structure, and function to the hearts of humans. Therefore, there is an urgent need for large-animal HFpEF models that can recapitulate the complex features of obese HFpEF in humans. The currently available methods for generating large animal models of HFpEF are summarized in detail elsewhere (174, 175). A recent study proposed an HFpEF model in Göttingen minipigs, which induced hypertension and obesity through mineralocorticoid excess and high cholesterol, fat, fructose, and salt diet (172). This novel “multi-hit” minipig model of HF is free from the confounding effects inherent in hypertension and end-organ damage induced by invasive surgical procedures or artificial drug administration and can be a promising model used for molecular mechanisms and evaluation of potential therapeutic approaches to HFpEF.
In vitro cardiomyocyte models of humans can overcome the limitations of animal models and species differences. However, in vitro studies of cardiomyocytes have many technical challenges. Current in vitro cardiomyocyte models that can be used to simulate HFpEF include induced pluripotent stem cells (iPSCs), primary cells, and cardiac organoid models (174). The iPSC-derived cardiomyocytes (iPSC-CMs) usually dedifferentiate from fibroblasts or monocytes and differentiate into cardiomyocytes under different in vitro epigenetic and environmental regulations, which are unable to produce a mature phenotype that is representative of human adult ventricular cardiomyocytes. Primary cell culture can obtain pathological changes in cardiomyocytes from HFpEF patients, but it is difficult to obtain and the cells rapidly lose their phenotypic structure in a relatively short period after extraction (183). The cardiac organoid model can overcome the limitations of single-molecule culture. However, this model is still artificially constructed and the resulting phenotype is very different from the intact human myocardium (184).
Recently, a new human functional myocardial slicing method was proposed, in which isolated myocardial tissue was prepared into ultrathin slices (150–350 μm) of ventricular myocardium and cultured under mechanical tension and electric field stimulation (174). This method preserves the structure of the failing myocardium and its molecular characteristics and can be used to study the function of the failing myocardium, energy-substrate turnover, and molecular properties, as well as the cardiac response to drug treatment.
In summary, based on the advantages and disadvantages of each model, we advocate that future HFpEF studies that explore molecular mechanisms and screen for potential new drugs should consider the use of multiple HFpEF animal models to narrow the gaps in HFpEF pathophysiology and promote the study of molecular mechanisms and the development of HFpEF therapeutics.
Conclusion
Obesity is an independent risk factor for myocardial dysfunction and HF. Obese HFpEF has a unique cardiac remodeling pattern and pericardial restraint mechanics. Inflammation is considered the main pathophysiological factor in obese HFpEF. We propose inflammation-related mechanisms of obese HFpEF, as well as potential therapeutic approaches. Currently, treatment options for HFpEF are limited, and the pharmacological inhibition of NLRP3 inflammasome assembly and activation may provide a novel treatment strategy targeting obese HFpEF. However, the discovery of novel therapeutics is hindered by the lack of appropriate HFpEF animal models that recapitulate this complex, comorbidity-laden, heterogeneous disease. Successfully validated large animal models or in vitro models of human cardiomyocytes could provide a more comprehensive understanding of the complex paradigm of HFpEF and potentially serve as reliable preclinical platforms for testing its therapeutic potential.
Author contributions
YB directed the writing and revision of the thesis. CL and DQ wrote the article and generated figures and tables. DH, JH and YY contributed to the mechanisms of the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in whole or in part by the National Natural Science Foundation of China (No.82170483 to BY and No.82100496 to DH) and the Natural Science Foundation of Hunan Province of China (No.2020JJ4786 to JH).
Acknowledgments
All authors are grateful to Zhenyu Tian for illustrating our manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD 2017 Disease and Injury Incidence and Prevalence CollaboratorsGlobal, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet (London England) (2018) 392(10159):1789–858. doi: 10.1016/s0140-6736(18)32279-7
2. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, et al. Phenotype-specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation (2016) 134(1):73–90. doi: 10.1161/CIRCULATIONAHA.116.021884
3. Pieske B, Tschöpe C, de Boer RA, Fraser AG, Anker SD, Donal E, et al. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the heart failure association (HFA) of the European society of cardiology (ESC). Eur Heart J (2019) 40(40):3297–317. doi: 10.1093/eurheartj/ehz641
4. World Health Organization. Obesity and overweight facts Available at: https://www.who.int/zh/news-room/fact-sheets/detail/obesity-and-overweight.
5. Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, et al. Systolic and diastolic heart failure in the community. JAMA (2006) 296(18):2209–16. doi: 10.1001/jama.296.18.2209
6. Haass M, Kitzman DW, Anand IS, Miller A, Zile MR, Massie BM, et al. Body mass index and adverse cardiovascular outcomes in heart failure patients with preserved ejection fraction: results from the irbesartan in heart failure with preserved ejection fraction (I-PRESERVE) trial. Circ Heart Failure (2011) 4(3):324–31. doi: 10.1161/circheartfailure.110.959890
7. Sabbah MS, Fayyaz AU, de Denus S, Felker GM, Borlaug BA, Dasari S, et al. Obese-inflammatory phenotypes in heart failure with preserved ejection fraction. Circ Heart Fail Aug (2020) 13(8):e006414. doi: 10.1161/CIRCHEARTFAILURE.119.006414
8. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, et al. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: Results from the PROMIS-HFpEF study. Circulation (2020) 142(21):2029–44. doi: 10.1161/circulationaha.120.045810
9. Amad KH, Brennan JC, Alexander JK. The cardiac pathology of chronic exogenous obesity. Circulation (1965) 32(5):740–5. doi: 10.1161/01.cir.32.5.740
10. Duflou J, Virmani R, Rabin I, Burke A, Burke A, Smialek J. Sudden death as a result of heart disease in morbid obesity. American Heart J (1995) 130(2):306–13. doi: 10.1016/0002-8703(95)90445-x
11. Loganathan R, Bilgen M, Al-Hafez B, Alenezy MD, Smirnova IV. Cardiac dysfunction in the diabetic rat: quantitative evaluation using high resolution magnetic resonance imaging. Cardiovasc Diabetology (2006) 5:7. doi: 10.1186/1475-2840-5-7
12. Abhayaratna WP, Marwick TH, Smith WT, Becker NG. Characteristics of left ventricular diastolic dysfunction in the community: an echocardiographic survey. Heart (British Cardiac Society) (2006) 92(9):1259–64. doi: 10.1136/hrt.2005.080150
13. Ren J, Wu NN, Wang S, Sowers JR, Zhang Y. Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol Rev (2021) 101(4):1745–807. doi: 10.1152/physrev.00030.2020
14. Sciacqua A, Cimellaro A, Mancuso L, Cassano V, Perticone M, et al. Different patterns of left ventricular hypertrophy in metabolically healthy and insulin-resistant obese subjects. Nutrients (2020) 12(2):412. doi: 10.3390/nu12020412
15. Zhang N, Chen Y, Guo X, Sun G, Dai D, Sun Y. Metabolic abnormalities, but not metabolically healthy obesity, are associated with left ventricular hypertrophy. Heart Lung Circulation (2017) 26(3):251–7. doi: 10.1016/j.hlc.2016.06.1212
16. Guidry UC, Mendes LA, Evans JC, Levy D, O'Connor GT, Larson MG, et al. Echocardiographic features of the right heart in sleep-disordered breathing: the framingham heart study. Am J Respir Crit Care Med (2001) 164(6):933–8. doi: 10.1164/ajrccm.164.6.2001092
17. Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation (2013) 128(18):2016–25, 1-10. doi: 10.1161/circulationaha.113.001873
18. Masaidi M, Cuspidi C, Negri F, Giudici V, Sala C, Zanchetti A, et al. Left and right ventricular structural changes in obese hypertensives. Blood Pressure (2009) 18(1-2):23–9. doi: 10.1080/08037050902850226
19. Wong CY, O'Moore-Sullivan T, Leano R, Hukins C, Jenkins C, TH M. Association of subclinical right ventricular dysfunction with obesity. J Am Coll Cardiol (2006) 47(3):611–6. doi: 10.1016/j.jacc.2005.11.015
20. Kasper EK, Hruban RH, Baughman KL. Cardiomyopathy of obesity: a clinicopathologic evaluation of 43 obese patients with heart failure. Am J Cardiol (1992) 70(9):921–4. doi: 10.1016/0002-9149(92)90739-l
21. Calderone A, Takahashi N, Izzo NJ Jr, Thaik CM, Colucci WS. Pressure- and volume-induced left ventricular hypertrophies are associated with distinct myocyte phenotypes and differential induction of peptide growth factor mRNAs. Circulation (1995) 92(9):2385–90. doi: 10.1161/01.cir.92.9.2385
22. Bostick B, Habibi J, DeMarco VG, Jia G, Domeier TL, Lambert MD, et al. Mineralocorticoid receptor blockade prevents Western diet-induced diastolic dysfunction in female mice. Am J Physiol Heart Circulatory Physiol (2015) 308(9):H1126–35. doi: 10.1152/ajpheart.00898.2014
23. Alex L, Russo I, Holoborodko V, Frangogiannis NG. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circulatory Physiol (2018) 315(4):H934–49. doi: 10.1152/ajpheart.00238.2018
24. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation (2015) 131(6):550–9. doi: 10.1161/circulationaha.114.009625
25. van Empel VP, Mariani J, Borlaug BA, Kaye DM. Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J Am Heart Assoc (2014) 3(6):e001293. doi: 10.1161/jaha.114.001293
26. Warbrick I, Rabkin SW. Hypoxia-inducible factor 1-alpha (HIF-1α) as a factor mediating the relationship between obesity and heart failure with preserved ejection fraction. Obes Reviews: An Off J Int Assoc Study Obes (2019) 20(5):701–12. doi: 10.1111/obr.12828
27. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation (2015) 131(14):1247–59. doi: 10.1161/circulationaha.114.013215
28. Shah AS, Khoury PR, Dolan LM, Ippisch HM, Urbina EM, Daniels SR, et al. The effects of obesity and type 2 diabetes mellitus on cardiac structure and function in adolescents and young adults. Diabetologia (2011) 54(4):722–30. doi: 10.1007/s00125-010-1974-7
29. Rider OJ, Ajufo E, Ali MK, Petersen SE, Nethononda R, Francis JM, et al. Myocardial tissue phase mapping reveals impaired myocardial tissue velocities in obesity. Int J Cardiovasc Imaging (2015) 31(2):339–47. doi: 10.1007/s10554-014-0548-z
30. Russo C, Jin Z, Homma S, Rundek T, Elkind MS, Sacco RL, et al. Effect of obesity and overweight on left ventricular diastolic function: a community-based study in an elderly cohort. J Am Coll Cardiol (2011) 57(12):1368–74. doi: 10.1016/j.jacc.2010.10.042
31. Wohlfahrt P, Redfield MM, Lopez-Jimenez F, Melenovsky V, Kane GC, Rodeheffer RJ, et al. Impact of general and central adiposity on ventricular-arterial aging in women and men. JACC Heart Failure (2014) 2(5):489–99. doi: 10.1016/j.jchf.2014.03.014
32. Lam CS, Roger VL, Rodeheffer RJ, Bursi F, Borlaug BA, Ommen SR, et al. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted county, Minnesota. Circulation (2007) 115(15):1982–90. doi: 10.1161/circulationaha.106.659763
33. Koehler B, Małecka-Tendera E, Drzewiecka B, Gasior Z, Wackerman-Ramos A, Majer K, et al. Evaluation of the cardiovascular system in children with simple obesity. part II. echocardiographic assessment. Materia Med Polona Polish J Med Pharm (1989) 21(2):131–3. doi: 10.1159/000072588
34. Veille JC, Hanson R. Obesity, pregnancy, and left ventricular functioning during the third trimester. Am J Obstetrics Gynecology (1994) 171(4):980–3. doi: 10.1016/0002-9378(94)90018-3
35. Stoddard MF, Tseuda K, Thomas M, Dillon S, Kupersmith J. The influence of obesity on left ventricular filling and systolic function. Am Heart J (1992) 124(3):694–9. doi: 10.1016/0002-8703(92)90280-9
36. Scaglione R, Dichiara MA, Indovina A, Lipari R, Ganguzza A, Parrinello G, et al. Left ventricular diastolic and systolic function in normotensive obese subjects: influence of degree and duration of obesity. Eur Heart J (1992) 13(6):738–42. doi: 10.1093/oxfordjournals.eurheartj.a060249
37. Zerhouni EA, Parish DM, Rogers WJ, Yang A, Shapiro EP. Human heart: tagging with MR imaging–a method for noninvasive assessment of myocardial motion. Radiology (1988) 169(1):59–63. doi: 10.1148/radiology.169.1.3420283
38. Sutherland GR, Stewart MJ, Groundstroem KW, et al. Color Doppler myocardial imaging: a new technique for the assessment of myocardial function. J Am Soc Echocardiography Off Publ Am Soc Echocardiogr (1994) 7(5):441–58. doi: 10.1016/s0894-7317(14)80001-1
39. Claus P, Omar AMS, Pedrizzetti G, Sengupta PP, Nagel E. Tissue tracking technology for assessing cardiac mechanics: Principles, normal values, and clinical applications. JACC Cardiovasc Imaging (2015) 8(12):1444–60. doi: 10.1016/j.jcmg.2015.11.001
40. Amzulescu MS, De Craene M, Langet H, Pasquet A, Vancraeynest D, Pouleur AC, et al. Myocardial strain imaging: review of general principles, validation, and sources of discrepancies. Eur Heart J Cardiovasc Imaging (2019) 20(6):605–19. doi: 10.1093/ehjci/jez041
41. Wong CY, O'Moore-Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation (2004) 110(19):3081–7. doi: 10.1161/01.Cir.0000147184.13872.0f
42. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation (2017) 136(1):6–19. doi: 10.1161/circulationaha.116.026807
43. Chahal H, McClelland RL, Tandri H, Jain A, Turkbey EB, Hundley WG, et al. Obesity and right ventricular structure and function: the MESA-right ventricle study. Chest (2012) 141(2):388–95. doi: 10.1378/chest.11-0172
44. Gorter TM, Hoendermis ES, van Veldhuisen DJ, Voors AA, Lam CS, Geelhoed B, et al. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail (2016) 18(12):1472–87. doi: 10.1002/ejhf.630
45. Aschauer S, Kammerlander AA, Zotter-Tufaro C, Ristl R, Pfaffenberger S, Bachmann A, et al. The right heart in heart failure with preserved ejection fraction: insights from cardiac magnetic resonance imaging and invasive haemodynamics. Eur J Heart Fail (2016) 18(1):71–80. doi: 10.1002/ejhf.418
46. Borlaug BA, Kane GC, Melenovsky V, Olson TP. Abnormal right ventricular-pulmonary artery coupling with exercise in heart failure with preserved ejection fraction. Eur Heart J (2016) 37(43):3293–302. doi: 10.1093/eurheartj/ehw241
47. Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Wilson PW, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation (2004) 109(5):594–600. doi: 10.1161/01.Cir.0000112582.16683.Ea
48. Mehra MR, Uber PA, Park MH, Scott RL, Ventura HO, Harris BC, et al. Obesity and suppressed b-type natriuretic peptide levels in heart failure. J Am Coll Cardiol (2004) 43(9):1590–5. doi: 10.1016/j.jacc.2003.10.066
49. Horwich TB, Hamilton MA, Fonarow GC. B-type natriuretic peptide levels in obese patients with advanced heart failure. J Am Coll Cardiol (2006) 47(1):85–90. doi: 10.1016/j.jacc.2005.08.050
50. van Kimmenade R, van Dielen F, Bakker J, Nijhuis J, Crijns H, Buurman W, et al. Is brain natriuretic peptide production decreased in obese subjects? J Am College Cardiology (2006) 47:886–7. doi: 10.1016/j.jacc.2005.11.022
51. Packer M. Leptin-Aldosterone-Neprilysin axis: Identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation (2018) 137(15):1614–31. doi: 10.1161/circulationaha.117.032474
52. Gentili A, Frangione MR, Albini E, Vacca C, Ricci MA, De Vuono S, et al. Modulation of natriuretic peptide receptors in human adipose tissue: molecular mechanisms behind the "natriuretic handicap" in morbidly obese patients. Transl Res Aug (2017) 186:52–61. doi: 10.1016/j.trsl.2017.06.001
53. Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, et al. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Failure (2011) 13(10):1087–95. doi: 10.1093/eurjhf/hfr079
54. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol (2013) 62(4):263–71. doi: 10.1016/j.jacc.2013.02.092
55. Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, et al. Clinical phenogroups in heart failure with preserved ejection fraction: Detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Failure (2020) 8(3):172–84. doi: 10.1016/j.jchf.2019.09.009
56. Cohen JB. Hypertension in obesity and the impact of weight loss. Curr Cardiol Rep (2017) 19(10):98. doi: 10.1007/s11886-017-0912-4
57. Harada T, Obokata M. Obesity-related heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and potential therapies. Heart failure clinics (2020) 16(3):357–68. doi: 10.1016/j.hfc.2020.02.004
58. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res (2017) 113(4):389–98. doi: 10.1093/cvr/cvx012
59. Wenzl FA, Ambrosini S, Mohammed SA, Kraler S, Lüscher TF, Costantino S, et al. Inflammation in metabolic cardiomyopathy. Front Cardiovasc Med (2021) 8:742178. doi: 10.3389/fcvm.2021.742178
60. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res (2021) 128(2):232–45. doi: 10.1161/circresaha.120.317933
61. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature (2001) 414(6865):799–806. doi: 10.1038/414799a
62. Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med (2008) 14(6):667–75. doi: 10.1038/nm1775
63. Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol (1998) 275(2):E259–64. doi: 10.1152/ajpendo.1998.275.2.E259
64. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med (2012) 18(3):363–74. doi: 10.1038/nm.2627
65. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med (2015) 21(3):239–47. doi: 10.1038/nm.3800
66. Castoldi A, Naffah de Souza C, Câmara NO, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol (2015) 6:637. doi: 10.3389/fimmu.2015.00637
67. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
68. Kiran S, Kumar V, Murphy EA, Enos RT, Singh UP. High fat diet-induced CD8 + T cells in adipose tissue mediate macrophages to sustain low-grade chronic inflammation. Front Immunol (2021) 12:680944. doi: 10.3389/fimmu.2021.680944
69. Neeland IJ, Poirier P, Després JP. Cardiovascular and metabolic heterogeneity of obesity: Clinical challenges and implications for management. Circulation (2018) 137(13):1391–406. doi: 10.1161/circulationaha.117.029617
70. González N, Moreno-Villegas Z, González-Bris A, Egido J, Lorenzo Ó. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc Diabetol (2017) 16(1):44. doi: 10.1186/s12933-017-0528-4
71. Elsanhoury A, Nelki V, Kelle S, Van Linthout S, Tschöpe C. Epicardial fat expansion in diabetic and obese patients with heart failure and preserved ejection fraction-a specific HFpEF phenotype. Front Cardiovasc Med (2021) 8:720690. doi: 10.3389/fcvm.2021.720690
72. Baker AR, Silva NF, Quinn DW, Harte AL, Pagano D, Bonser RS, et al. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc Diabetology (2006) 5:1. doi: 10.1186/1475-2840-5-1
73. Gorter TM, Obokata M, Reddy YNV, Melenovsky V. Borlaug BA. exercise unmasks distinct pathophysiologic features in heart failure with preserved ejection fraction and pulmonary vascular disease. Eur Heart J (2018) 39(30):2825–35. doi: 10.1093/eurheartj/ehy331
74. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Failure (2011) 4(1):44–52. doi: 10.1161/circheartfailure.109.931451
75. Sanders-van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, et al. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail (2015) 17(10):1006–14. doi: 10.1002/ejhf.414
76. Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol (2009) 157(4):527–36. doi: 10.1111/j.1476-5381.2009.00240.x
77. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res (2000) 86(5):494–501. doi: 10.1161/01.res.86.5.494
78. Chirinos JA, Akers SR, Trieu L, Ischiropoulos H, Doulias PT, Tariq A, et al. Heart failure, left ventricular remodeling, and circulating nitric oxide metabolites. J Am Heart Assoc (2016) 5(10):e004133. doi: 10.1161/jaha.116.004133
79. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/circresaha.117.311401
80. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res (2002) 91(11):988–98. doi: 10.1161/01.res.0000043825.01705.1b
81. Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest (1995) 95(6):2555–64. doi: 10.1172/jci117956
82. Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res (2009) 82(1):59–66. doi: 10.1093/cvr/cvp040
83. Schulz R, Heusch G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and yang in myocardial infarction? Circulation (2009) 119(10):1355–7. doi: 10.1161/circulationaha.108.846105
84. Condorelli G, Morisco C, Latronico MV, Claudio PP, Dent P, Tsichlis P, et al. TNF-alpha signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-alpha receptor. FASEB J Off Publ Fed Am Societies Exp Biol (2002) 16(13):1732–7. doi: 10.1096/fj.02-0419com
85. Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, et al. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting akt and JNK pathways. Circ Res (2009) 105(11):1149–58. doi: 10.1161/circresaha.109.208199
86. Narayan P, Trikantzopoulos E, Mezzaroma E, Mauro AG, Vohra H, Abbate A, et al. The interleukin-1 receptor type I promotes the development of aging-associated cardiomyopathy in mice. Cytokine (2022) 151:155811. doi: 10.1016/j.cyto.2022.155811
87. Mehlhorn U, Geissler HJ, Laine GA, Allen SJ. Myocardial fluid balance. Eur J cardio-thoracic Surg Off J Eur Assoc Cardio-thoracic Surgery (2001) 20(6):1220–30. doi: 10.1016/s1010-7940(01)01031-4
88. Conway WC, Faries MB, Nicholl MB, Terando AM, Glass EC, Sim M, et al. Age-related lymphatic dysfunction in melanoma patients. Ann Surg Oncol (2009) 16(6):1548–52. doi: 10.1245/s10434-009-0420-x
89. Zawieja SD, Gasheva O, Zawieja DC, Muthuchamy M. Blunted flow-mediated responses and diminished nitric oxide synthase expression in lymphatic thoracic ducts of a rat model of metabolic syndrome. Annals of Surgical Oncol (2016) 310(3):H385–93. doi: 10.1152/ajpheart.00664.2015
90. Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest (2005) 115(2):247–57. doi: 10.1172/jci22037
91. Noon JP, Walker BR, Webb DJ, Shore AC, Holton DW, Edwards HV, et al. Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest (1997) 99(8):1873–9. doi: 10.1172/jci119354
92. Cuijpers I, Simmonds SJ, van Bilsen M, Czarnowska E, González Miqueo A, Heymans S, et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol (2020) 115(4):39. doi: 10.1007/s00395-020-0798-y
93. Querejeta R, López B, González A, Sánchez E, Larman M, Martínez Ubago JL, et al. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation (2004) 110(10):1263–8. doi: 10.1161/01.Cir.0000140973.60992.9a
94. Tamaki S, Mano T, Sakata Y, Ohtani T, Takeda Y, Kamimura D, et al. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLos One (2013) 8(7):e68893. doi: 10.1371/journal.pone.0068893
95. Shen JL, Xie XJ. Insight into the pro-inflammatory and profibrotic role of macrophage in heart failure with preserved ejection fraction. J Cardiovasc Pharmacol (2020) 76(3):276–85. doi: 10.1097/fjc.0000000000000858
96. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Lévy F, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation (2016) 133(15):1484–97. doi: 10.1161/circulationaha.115.020143
97. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res (2013) 113(6):709–24. doi: 10.1161/circresaha.113.300376
98. RANDLE PJ, GARLAND PB, HALES CN, NEWSHOLME EA. The glucose fatty-acid cycle. its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (London England) (1963) 1(7285):785–9. doi: 10.1016/s0140-6736(63)91500-9
99. Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the translational research committee of the heart failure association-European society of cardiology. Eur Heart J (2018) 39(48):4243–54. doi: 10.1093/eurheartj/ehy596
100. Carnicer R, Suffredini S, Liu X, Reilly S, Simon JN, Surdo NC, et al. The subcellular localisation of neuronal nitric oxide synthase determines the downstream effects of NO on myocardial function. Cardiovasc Res (2017) 113(3):321–31. doi: 10.1093/cvr/cvx002
101. Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang Y, et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes (2014) 63(10):3497–511. doi: 10.2337/db13-1577
102. Yu XY, Geng YJ, Liang JL, Zhang S, Lei HP, Zhong SL, et al. High levels of glucose induce "metabolic memory" in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep (2012) 39(9):8891–8. doi: 10.1007/s11033-012-1756-z
103. Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, et al. Hyperglycemia acutely increases cytosolic reactive oxygen species via O -linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res (2020) 126(10):e80–96. doi: 10.1161/circresaha.119.316288
104. Zhang H, Chen X, Zong B, Yuan H, Wang Z, Wei Y, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med (2018) 22(9):4437–48. doi: 10.1111/jcmm.13743
105. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells (2020) 10(1):51. doi: 10.3390/cells10010051
106. Russo S, Kwiatkowski M, Govorukhina N, Bischoff R, Melgert BN. Meta-inflammation and metabolic reprogramming of macrophages in diabetes and obesity: The importance of metabolites. Front Immunol (2021) 12:746151. doi: 10.3389/fimmu.2021.746151
107. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
108. Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: Novel roles of immunometabolism in macrophage activation and inflammation. Circ Res (2020) 126(6):789–806. doi: 10.1161/circresaha.119.312321
109. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
110. Krützfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol (1990) 22(12):1393–404. doi: 10.1016/0022-2828(90)90984-a
111. Spahr R, Krützfeldt A, Mertens S, Siegmund B, Piper HM. Fatty acids are not an important fuel for coronary microvascular endothelial cells. Mol Cell Biochem (1989) 88(1-2):59–64. doi: 10.1007/bf00223424
112. Xiao W, Oldham WM, Priolo C, Pandey AK, Loscalzo J. Immunometabolic endothelial phenotypes: Integrating inflammation and glucose metabolism. Circ Res (2021) 129(1):9–29. doi: 10.1161/circresaha.120.318805
113. Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol (2013) 132(2):287–94. doi: 10.1016/j.jaci.2013.06.022
114. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res (2018) 122(12):1722–40. doi: 10.1161/circresaha.118.311362
115. Olsen MB, Gregersen I, Sandanger Ø, Yang K, Sokolova M, Halvorsen BE, et al. Targeting the inflammasome in cardiovascular disease. JACC Basic to Trans science (2022) 7(1):84–98. doi: 10.1016/j.jacbts.2021.08.006
116. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17(2):179–88. doi: 10.1038/nm.2279
117. Libby P. Targeting inflammatory pathways in cardiovascular disease: The inflammasome, interleukin-1, interleukin-6 and beyond. Cells (2021) 10(4):951. doi: 10.3390/cells10040951
118. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
119. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev (2015) 265(1):35–52. doi: 10.1111/imr.12286
120. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
121. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol (Baltimore Md 1950) (2013) 191(8):4358–66. doi: 10.4049/jimmunol.1301170
122. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
123. Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci United States America (2011) 108(37):15324–9. doi: 10.1073/pnas.1100255108
124. Sehgal A, Behl T, Kaur I, Singh S, Sharma N, Aleya L. Targeting NLRP3 inflammasome as a chief instigator of obesity, contributing to local adipose tissue inflammation and insulin resistance. Environ Sci pollut Res Int (2021) 28(32):43102–13. doi: 10.1007/s11356-021-14904-4
125. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12(5):408–15. doi: 10.1038/ni.2022
126. Finucane OM, Lyons CL, Murphy AM, Reynolds CM, Klinger R, Healy NP, et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes (2015) 64(6):2116–28. doi: 10.2337/db14-1098
127. Weir JM, Wong G, Barlow CK, Greeve MA, Kowalczyk A, Almasy L, et al. Plasma lipid profiling in a large population-based cohort. J Lipid Res (2013) 54(10):2898–908. doi: 10.1194/jlr.P035808
128. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab (2014) 20(4):687–95. doi: 10.1016/j.cmet.2014.09.015
129. Bergman BC, Brozinick JT, Strauss A, Bacon S, Kerege A, Bui HHc, et al. Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia (2016) 59(4):785–98. doi: 10.1007/s00125-015-3850-y
130. Yang HJ, Kong B, Shuai W, Zhang JJ, Huang H. Knockout of MD1 contributes to sympathetic hyperactivity and exacerbates ventricular arrhythmias following heart failure with preserved ejection fraction via NLRP3 inflammasome activation. Exp Physiol (2020) 105(6):966–78. doi: 10.1113/ep088390
131. Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab (2017) 26(1):71–93. doi: 10.1016/j.cmet.2017.06.018
132. Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation (2019) 139(11):1435–50. doi: 10.1161/circulationaha.118.036259
133. Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the d-HART pilot study). Am J Cardiol (2014) 113(2):321–7. doi: 10.1016/j.amjcard.2013.08.047
134. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, et al. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Failure (2018) 11(8):e005036. doi: 10.1161/circheartfailure.118.005036
135. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
136. Smart N, Mojet MH, Latchman DS, Marber MS, Duchen MR, Heads RJ. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res (2006) 69(1):164–77. doi: 10.1016/j.cardiores.2005.08.017
137. Sternlicht H, Ringel I. Colchicine inhibition of microtubule assembly via copolymer formation. J Biol Chem (1979) 254(20):10540–50. doi: 10.1016/s0021-9258(19)86741-8
138. Demidowich AP, Davis AI, Dedhia N, Yanovski JA. Colchicine to decrease NLRP3-activated inflammation and improve obesity-related metabolic dysregulation. Med Hypotheses (2016) 92:67–73. doi: 10.1016/j.mehy.2016.04.039
139. Martínez GJ, Celermajer DS, Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis (2018) 269:262–71. doi: 10.1016/j.atherosclerosis.2017.12.027
140. Spartalis M, Spartalis E, Tzatzaki E, Tsilimigras DI, Moris D, Kontogiannis C, et al. The beneficial therapy with colchicine for atherosclerosis via anti-inflammation and decrease in hypertriglyceridemia. Cardiovasc Hematol Agents Med Chem (2018) 16(2):74–80. doi: 10.2174/1871525717666181211110332
141. Nikolaidis N, Polyzos SA, Giouleme O, Katsinelos P, Eugenidis N, Zeglinas C, et al. Colchicine to decrease inflammation and fibrosis in patients with metabolic dysregulation. Med Hypotheses (2016) 95:34. doi: 10.1016/j.mehy.2016.08.006
142. Whayne TF Jr. Inflammation may be the future of cardiovascular risk reduction: Does colchicine have a current indication? Am J Cardiovasc Drugs drugs devices other interventions (2021) 21(1):1–10. doi: 10.1007/s40256-020-00408-y
143. Levine JA, Sarrafan-Chaharsoughi Z, Patel TP, Brady SM, Chivukula KK, Miller E, et al. Effects of colchicine on lipolysis and adipose tissue inflammation in adults with obesity and metabolic syndrome. Obes (Silver Spring Md) (2022) 30(2):358–68. doi: 10.1002/oby.23341
144. D'Amario D, Cappetta D, Cappannoli L, Princi G, Migliaro S, Diana G, et al. Colchicine in ischemic heart disease: the good, the bad and the ugly. Clin Res Cardiol Off J German Cardiac Society (2021) 110(10):1531–42. doi: 10.1007/s00392-021-01828-9
145. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. New Engl J Med (2020) 383(19):1838–47. doi: 10.1056/NEJMoa2021372
146. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol (2013) 61(4):404–10. doi: 10.1016/j.jacc.2012.10.027
147. Bouabdallaoui N, Tardif JC, Waters DD, Pinto FJ, Maggioni AP, Diaz R, et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the colchicine cardiovascular outcomes trial (COLCOT). European Heart J (2020) 41::4092–4099. doi: 10.1093/eurheartj/ehaa659
148. Imazio M, Nidorf M. Colchicine and the heart. Eur Heart J (2021) 42(28):2745–60. doi: 10.1093/eurheartj/ehab221
149. Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, et al. Anti-inflammatory treatment with colchicine in stable chronic heart failure: a prospective, randomized study. JACC Heart Failure (2014) 2(2):131–7. doi: 10.1016/j.jchf.2013.11.006
150. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
151. Takatsu M, Nakashima C, Takahashi K, Murase T, Hattori T, Ito H, et al. Calorie restriction attenuates cardiac remodeling and diastolic dysfunction in a rat model of metabolic syndrome. Hypertension (Dallas Tex 1979) (2013) 62(5):957–65. doi: 10.1161/hypertensionaha.113.02093
152. Meyer TE, Kovács SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol (2006) 47(2):398–402. doi: 10.1016/j.jacc.2005.08.069
153. Lee CJ, Park S. Hypertension and heart failure with preserved ejection fraction. Heart Fail Clin (2021) 17(3):337–43. doi: 10.1016/j.hfc.2021.02.002
154. Kim JH, Lee M, Kim SH, Kim SR, Lee BW, Kang ES, et al. Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes Metab (2019) 21(4):801–11. doi: 10.1111/dom.13577
155. Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med (2021) 27(11):1954–60. doi: 10.1038/s41591-021-01536-x
156. Farxiga met primary endpoint in DELIVER phase III trial, reducing risk of cardiovascular death or worsening heart failure in patients with preserved ejection fraction (2022). Available at: https://www.astrazeneca.com/content/astraz/media-centre/press-releases/2022/farxiga-hfpef-phase-iii-trial-met-primary-endpoint.html.
157. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. New Engl J Med (2021) 385(16):1451–61. doi: 10.1056/NEJMoa2107038
158. Pabel S, Hamdani N, Singh J, Sossalla S. Potential mechanisms of SGLT2 inhibitors for the treatment of heart failure with preserved ejection fraction. Frontiers in Physiology (2021) 12:752370. doi: 10.3389/fphys.2021.752370
159. Reddy YNV, Carter RE, Obokata M, Redfield MM, Borlaug BA. A simple, evidence-based approach to help guide diagnosis of heart failure with preserved ejection fraction. Circulation (2018) 138(9):861–70. doi: 10.1161/circulationaha.118.034646
160. Withaar C, Lam CSP, Schiattarella GG, Schiattarella GG, Meems LMG. Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J (2021) 42(43):4420–30. doi: 10.1093/eurheartj/ehab389
161. Van den Bergh A, Flameng W, Herijgers P. Type II diabetic mice exhibit contractile dysfunction but maintain cardiac output by favourable loading conditions. Eur J Heart Failure (2006) 8(8):777–83. doi: 10.1016/j.ejheart.2006.03.001
162. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Sci (New York NY) (1995) 269(5223):540–3. doi: 10.1126/science.7624776
163. Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Failure (2013) 6(6):1239–49. doi: 10.1161/circheartfailure.113.000539
164. Qin F, Siwik DA, Luptak I, Hou X, Wang L, Higuchi A, et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation (2012) 125(14):1757–64, S1-6. doi: 10.1161/circulationaha.111.067801
165. Nachar W, Merlet N, Maafi F, Shi Y, Mihalache-Avram T, Mecteau M, et al. Cardiac inflammation and diastolic dysfunction in hypercholesterolemic rabbits. PLos One (2019) 14(8):e0220707. doi: 10.1371/journal.pone.0220707
166. Barandiarán Aizpurua A, Schroen B, van Bilsen M, van Empel V. Targeted HFpEF therapy based on matchmaking of human and animal models. Am J Physiol Heart Circulatory Physiol (2018) 315(6):H1670–83. doi: 10.1152/ajpheart.00024.2018
167. Mali VR, Ning R, Chen J, Yang XP, Xu J, Palaniyandi SS. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp Biol Med (Maywood NJ) (2014) 239(5):610–8. doi: 10.1177/1535370213520109
168. van den Dorpel MMP, Heinonen I, Snelder SM, Snelder SM, Vos HJ, Sorop O, van Domburg RT, et al. Early detection of left ventricular diastolic dysfunction using conventional and speckle tracking echocardiography in a large animal model of metabolic dysfunction. Int J Cardiovasc Imaging (2018) 34(5):743–9. doi: 10.1007/s10554-017-1287-8
169. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature (2019) 568(7752):351–6. doi: 10.1038/s41586-019-1100-z
170. Du W, Piek A, Schouten EM, van de Kolk CWA, Mueller C, Mebazaa A, et al. Plasma levels of heart failure biomarkers are primarily a reflection of extracardiac production. Theranostics (2018) 8(15):4155–69. doi: 10.7150/thno.26055
171. Withaar C, Meems LMG, Markousis-Mavrogenis G, Boogerd CJ, Silljé HHW, Schouten EM, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res (2021) 117(9):2108–24. doi: 10.1093/cvr/cvaa256
172. Sharp TE 3rd, Scarborough AL, Li Z, Polhemus DJ, Hidalgo HA, Schumacher JD, et al. Novel göttingen miniswine model of heart failure with preserved ejection fraction integrating multiple comorbidities. JACC Basic to Trans science (2021) 6(2):154–70. doi: 10.1016/j.jacbts.2020.11.012
173. Valero-Muñoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a "Fishing expedition". JACC Basic to Trans Sci (2017) 2(6):770–89. doi: 10.1016/j.jacbts.2017.07.013
174. Fusco-Allison G, Li DK, Hunter B, Jackson D, Bannon PG, Lal S, et al. Optimizing the discovery and assessment of therapeutic targets in heart failure with preserved ejection fraction. ESC Heart Failure (2021) 8(5):3643–55. doi: 10.1002/ehf2.13504
175. Conceição G, Heinonen I, Lourenço AP, Duncker DJ, Falcão-Pires I. Animal models of heart failure with preserved ejection fraction. Netherlands Heart J monthly J Netherlands Soc Cardiol Netherlands Heart Foundation (2016) 24(4):275–86. doi: 10.1007/s12471-016-0815-9
176. Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Männel A, et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Failure (2020) 7(5):2123–34. doi: 10.1002/ehf2.12915
177. Riehle C, Bauersachs J. Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Res Cardiol (2018) 114(1):2. doi: 10.1007/s00395-018-0711-0
178. Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest (2012) 122(3):1109–18. doi: 10.1172/jci60329
179. Wei M, Ong L, Smith MT, Ross FB, Schmid K, Hoey AJ, et al. The streptozotocin-diabetic rat as a model of the chronic complications of human diabetes. Heart Lung Circ (2003) 12(1):44–50. doi: 10.1046/j.1444-2892.2003.00160.x
180. Joffe II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, et al. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. J Am Coll Cardiol (1999) 34(7):2111–9. doi: 10.1016/s0735-1097(99)00436-2
181. Tong D, Schiattarella GG, Jiang N, May HI, Lavandero S, Gillette TG, et al. Female sex is protective in a preclinical model of heart failure with preserved ejection fraction. Circulation (2019) 140:1769–71. doi: 10.1161/circulationaha.119.042267
182. Sotomi Y, Hikoso S, Nakatani D, Mizuno H, Okada K, Dohi T, et al. Sex differences in heart failure with preserved ejection fraction. J Am Heart Assoc Feb (2021) 10(5):e018574. doi: 10.1161/JAHA.120.018574
183. Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol (2011) 51(3):288–98. doi: 10.1016/j.yjmcc.2011.06.012
Keywords: obesity, HFPEF, inflamed adipose tissue, NLRP3 inflammasome, inflammation
Citation: Li C, Qin D, Hu J, Yang Y, Hu D and Yu B (2022) Inflamed adipose tissue: A culprit underlying obesity and heart failure with preserved ejection fraction. Front. Immunol. 13:947147. doi: 10.3389/fimmu.2022.947147
Received: 18 May 2022; Accepted: 03 November 2022;
Published: 22 November 2022.
Edited by:
Benoit Pourcet, Université de Lille, FranceReviewed by:
Vyacheslav Ryabov, Cardiology Research Institute, (RAS), RussiaYin Cai, Hong Kong Polytechnic University, Hong Kong SAR, China
Copyright © 2022 Li, Qin, Hu, Yang, Hu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bilian Yu, yubilian@csu.edu.cn
†These authors share first authorship