 Huanhuan Ning1†
Huanhuan Ning1† Jian Kang1†
Jian Kang1† Yanzhi Lu1†
Yanzhi Lu1† Xuan Liang1,2
Xuan Liang1,2 Jie Zhou3
Jie Zhou3 Rui Ren1Shan Zhou4Yong Zhao5
Rui Ren1Shan Zhou4Yong Zhao5 Yanling Xie1,6
Yanling Xie1,6 Lu Bai1,6Linna Zhang7Yali Kang1,7Xiaojing Gao1,7Mingze Xu1
Lu Bai1,6Linna Zhang7Yali Kang1,7Xiaojing Gao1,7Mingze Xu1 Yanling Ma2
Yanling Ma2 Fanglin Zhang1*
Fanglin Zhang1* Yinlan Bai1*
Yinlan Bai1*- 1Department of Microbiology and Pathogen Biology, School of Preclinical Medicine, Air Force Medical University, Xi’an, China
- 2College of Life Sciences, Northwest University, Xi’an, China
- 3Department of Endocrinology, Xijing Hospital, Air Force Medical University, Xi’an, China
- 4Department of Clinical Laboratory, Xijing Hospital, Air Force Medical University, Xi’an, China
- 5Laboratory Animal Center, Air Force Medical University, Xi’an, China
- 6School of Life Sciences, Yan’an University, Yan’an, China
- 7Department of Physiology, Basic Medical School, Ningxia Medical University, Yinchuan, China
Bacillus Calmette-Guérin (BCG) is a licensed prophylactic vaccine against tuberculosis (TB). Current TB vaccine efforts focus on improving BCG effects through recombination or genetic attenuation and/or boost with different vaccines. Recent years, it was revealed that BCG could elicit non-specific heterogeneous protection against other pathogens such as viruses through a process termed trained immunity. Previously, we constructed a recombinant BCG (rBCG-DisA) with elevated c-di-AMP as endogenous adjuvant by overexpressing di-adenylate cyclase of Mycobacterium tuberculosis DisA, and found that rBCG-DisA induced enhanced immune responses by subcutaneous route in mice after M. tuberculosis infection. In this study, splenocytes from rBCG-DisA immunized mice by intravenous route (i.v) elicited greater proinflammatory cytokine responses to homologous and heterologous re-stimulations than BCG. After M. tuberculosis infection, rBCG-DisA immunized mice showed hallmark responses of trained immunity including potent proinflammatory cytokine responses, enhanced epigenetic changes, altered lncRNA expressions and metabolic rewiring in bone marrow cells and other tissues. Moreover, rBCG-DisA immunization induced higher levels of antibodies and T cells responses in the lung and spleen of mice after M. tuberculosis infection. It was found that rBCG-DisA resided longer than BCG in the lung of M. tuberculosis infected mice implying prolonged duration of vaccine efficacy. Then, we found that rBCG-DisA boosting could prolong survival of BCG-primed mice over 90 weeks against M. tuberculosis infection. Our findings provided in vivo experimental evidence that rBCG-DisA with c-di-AMP as endogenous adjuvant induced enhanced trained immunity and adaptive immunity. What’s more, rBCG-DisA showed promising potential in prime-boost strategy against M. tuberculosis infection in adults.
Introduction
Bacillus Calmette-Guérin (BCG) is a live attenuated vaccine for the prevention of tuberculosis (TB), which is caused by Mycobacterium tuberculosis infection. Usually, BCG is inoculated to newborns after birth, of which protection efficiency against meningitis and miliary TB is over 70% (1). Whereas its protection efficacy varies from 0% to 80% against pulmonary TB in adults (2). Vaccinology to improve the efficacy of canonical BCG shows good application prospects, which is necessary to control TB epidemic.
BCG was obtained from virulent Mycobacterium bovis, which could elicit heterologous protective immune response against M. tuberculosis infection. Additionally, BCG has been found to elicit non-specific protection against non-mycobacterial bacteria infections (3), viral infections (4), tumors (5), and autoimmune diseases (3). This phenomenon of BCG-induced heterologous protection has been referred to as trained immunity (6). The hallmarks of trained immunity include elevated levels of proinflammatory cytokine responses, myeloid cells and macrophage reprogramming towards inflammatory, activated phenotypes, epigenetic modifications, and metabolomic changes towards glycolysis (7–9). BCG vaccination shows long-term effects on innate immunity, which lasts for at least one year and then wanes in mice model (10). It is speculated that increasing morbidity of TB adults in BCG vaccinated population may be related to the waning efficiency of BCG (11, 12). Furthermore, BCG vaccination could promote protective monocyte responses against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (13, 14), though its protective effect on SARS-CoV-2 remains controversial (15, 16). Therefore, it is reasonable to increase the protection of BCG by exploring new BCG strains or booster vaccines to enhance BCG-induced trained immunity.
Cyclic di-AMP (c-di-AMP) is a ubiquitous bacterial second messenger (17, 18), which regulates various bacterial physiological processes including bacterial virulence. What’s more, there is growing evidence that c-di-AMP as a pathogen associated molecular pattern (PAMP) is engaged in host innate immune response including type I IFN response, autophagy, as well as inflammasome activation (19–21). Our previous work initially confirmed that Rv3586 is the only diadenylate cyclase (DacA, or DisA) (22), and Rv2837c is a cyclic nucleotide phosphodiesterase (CnpB) as a c-di-AMP hydrolyzing enzyme in M. tuberculosis (23). Either overexpression of DisA or deletion of CnpB could promote the accumulation of c-di-AMP in M. tuberculosis (23–25), BCG (24, 26, 27), as well as M. smegmatis (28–30). Further, we constructed a c-di-AMP elevated BCG with disA overexpressing (rBCG-DisA) and found that it induced stronger immune responses compared with BCG after M. tuberculosis infection in mice, with elevated cytokine responses of IL-6, TNF-α and enrichment of tri-methylation of lysine 4 on histone H3 (H3K4me3), which showed an enhanced trained immunity induced by rBCG-DisA (27).
However, we found that rBCG-DisA immunization by subcutaneous (s.c) route did not provide extra protection against M. tuberculosis in the mice model of i.v infection (27). While a similar rBCG-DisA with MT3692 (M. tuberculosis CDC1551 DisA, 100% similarity with Rv3586) overexpression has been shown to provide enhanced protection against aerosol M. tuberculosis infection compared to BCG in the guinea pig model (31). Moreover, rBCG-DisA induced improved antitumor efficacy by intravesical instillation in a rat model of bladder cancer, which was identified as an improved trained immunity compared with BCG in primary human and murine macrophages in vitro (32). It has been proved that BCG vaccination by i.v route is superior to induce trained immunity than that of s.c route (33). In addition, administration of BCG by i.v route conferred protection against M. tuberculosis challenge in non-human primate (34). In this study, we further explored immune responses induced by i.v rBCG-DisA especially trained immunity in vivo, and evaluated its protection efficiency and as a boosting vaccine for BCG-primed mice against M. tuberculosis intranasal (i.n) infection in mice model.
Materials and methods
Mice, bacterial strains, and cell lines
Six- to eight-week C57BL/6J male mice were purchased from Animal Center of Air Force Medical University. Recombinant BCG overexpressing DisA (termed as rBCG-DisA) was constructed in our previous work (27). M. tuberculosis H37Ra, BCG were grown in Middlebrook 7H9 broth (BD) supplemented with 0.5% (v/v) glycerol, 0.05% Tween 80 and 10% oleic acid albumin-dextrose-catalase (OADC) (BD) at 37°C. rBCG-DisA was grown in complete 7H9 medium with 25µg/mL kanamycin. All mycobacteria were plated on 7H10 agar (BD) supplemented with 10% OADC for colony forming units (CFUs) enumeration. Staphylococcus aureus was grown in LB medium at 37°C. Murine macrophage cell line RAW264.7 was used for in vitro experiments.
Vaccination and infection of mice
Mice were anesthetized with an intraperitoneal injection of 50mg/kg pentobarbital sodium. Then, mice were vaccinated with 1×106 CFU single bacterial suspension of BCG or rBCG-DisA in 200µL PBS at the base of the tail by i.v route, respectively (33). After 12 weeks, all vaccinated mice were challenged with 2×105 CFU M. tuberculosis H37Ra in 50µL PBS by i.n route. Normal mice (Naïve) and M. tuberculosis H37Ra infected mice without vaccination (UN) were used as control. At 12 weeks post vaccination and 6 weeks post infection, mice were euthanatized for immunological assays, histopathological analysis and CFUs measurements.
Antigen-specific antibody detection by ELISA
Sera from all mice were collected at indicated time points after vaccination or infection respectively. Enzyme linked immunosorbent assay (ELISA) microplates were coated with BCG or DisA antigen at 10µg/mL. Sera were diluted with PBS at indicated dilutions. Antigen-specific IgG level was measured by ELISA. The absorbance was detected at 450 nm by microplate reader (BioTek).
Splenocytes preparation, stimulation, and detection of proliferation in vitro
Spleen single cell suspensions were prepared and 1×106 splenocytes were seeded and stimulated by BCG proteins (25µg/mL) for 72h. Cell proliferation was determined by CFSE (Sigma) reagent according to previous study (27, 35). For cytokines measurement, 1×106 cells were seeded in 96-well microplates stimulated with BCG proteins (25µg/mL), M. tuberculosis proteins (25µg/mL), S. aureus proteins (25µg/mL), E. coli O111:B4 lipopolysaccharide (LPS) (100ng/mL) (InvivoGen) for 72h, respectively. Cells were incubated at 37°C with 5% CO2 and supernatants were collected for cytokines assays.
Determination of cytokines production by ELISA
Cytokines measurement in the cell supernatants was performed using commercial ELISA kits. Cytokines detection of IFN-γ, IL-2, IL-10, IL-17, TNF-α, IL-1β, IL-6 (from Thermo Fisher), CXCL9, CXCL10 and CXCL11 (CLOUD-CLONE CORP, China) were performed according to the instructions. Cell supernatants and diluted standards were added 100µL/well, then incubated at room temperature for 2h. Finally, ELISA plates were monitored at 450 nm using a microplate reader (BioTek).
Flow cytometry
Splenocytes were washed with PBS containing 2.5% FBS. Cells were resuspended in 100μL Staining Buffer (BioLegend) containing TruStain Fc PLUS (anti-mouse CD16/32) (BioLegend) and incubated for 10min on ice. Cells were stained with fluorochrome-labeled antibodies of CD3 (17A2), CD4 (RM4-5), CD8 (53-6.7), CD19 (6D5), CD49 (DX5), CD11b (M1/70), Ly-6G (1A8) (BioLegend) for 30min at 4°C, then washed with PBS containing 2.5% FBS twice. Finally, cells were resuspended in 500µL Staining Buffer for flow cytometry analysis (BD FACSCanto). The frequency of specific cell subpopulation was analyzed using FlowJo V10 (Tree Star Inc.).
Generation of BMDMs
After 4 weeks of vaccination, bone marrow from both femurs and tibiae was harvested in RPMI 1640. Cells were subsequently resuspended in RPMI 1640 supplemented with 10% FBS, 100U/mL penicillin, 100mg/mL streptomycin and 100ng/mL M-CSF (PeproTech). Cells were seeded in Petri dishes (100 mm). After 3 days of incubation at 37°C with 5% CO2, the medium was removed and replaced with fresh medium. Cells were cultured for another 2 days allowing to differentiate into macrophages. On day 5, cells were harvested and resuspended in complete RPMI 1640 medium for in vitro assay.
Macrophage infection
RAW264.7 macrophages and BMDMs of 2×105 were seeded in 24-well plates supplemented with complete RPMI 1640 without penicillin/streptomycin, and incubated at 37°C with 5% CO2 overnight. Cells were infected with M. tuberculosis attenuated strain H37Ra (MOI = 1), BCG and rBCG-DisA (MOI = 10). Cells were incubated for 4h at 37°C with 5% CO2. Subsequently, cells were washed with cold sterile PBS for 3 times, and then incubated in complete RPMI 1640 without penicillin/streptomycin. This time point was termed as 0 d post infection (dpi). Intracellular bacteria CFUs from lysed cells by 0.025% SDS were enumerated on plates at day 1, 3 and 5 post infection.
Quantitative RT-PCR
Total RNA of mice tissues and bone marrow (BM) cells was extracted using the RNeasy kit (Omega Bio-Tek) following the manufacturer’s instruction, and quantified by microplate reader (BioTek). Next, elimination of genomic DNA and cDNA synthesis of 500 ng RNA were performed by HiScript Reverse Transcriptase Kit (Vazyme). Quantitative RT-PCR assay was performed using SYBR qPCR Master Mix (Vazyme). Fold changes of mRNA expression of indicated genes was calculated according to 2-ΔΔCt, gene of gapdh was used as expression normalization. Primers used in this study were synthesized by Tsingke Biological Technology and listed in Table S1.
Histopathology and immunohistochemistry
For histopathology, upper lobes of left lungs and 1/3 of spleen tissue in infected mice was fixed in 10% buffered formalin. The tissue was dehydrated and embedded in paraffin wax. Sections (5µm) were cut and transferred onto glass slides. Hematoxylin/eosin (H&E) staining for pathohistological analysis was performed by the Department of Histopathology (Air Force Medical University, China), and results were quantified by Image J software.
The counts of CD4 and CD8 T cells and expression levels of H3 mono-, or tri-methylated at lysine 4 (H3K4me1, or H3K4me3) and H3K27 acetylation (H3K27ac) in the lung were determined by immunohistochemistry (IHC). Antibodies used in IHC assays were rabbit mAb anti-CD4 (D7D2Z) (CST), rabbit mAb CD8α (D4W2Z) (CST), rabbit mAb anti-Histone H3 (mono methyl K4) (Abcam), rabbit mAb anti-Histone H3 (acetyl K27) (Abcam), rabbit mAb anti-Histone H3 (tri methyl K4) (Abcam). Secondary antibody used was HRP-conjugated anti-rabbit antibody (Jackson Immunoresearch Laboratories). IHC assay was performed by Chengdu Lilai Biotechnology Company (Chengdu, China). IHC images were quantitatively analyzed with Image J software.
Sera collection and extraction for metabolomics analysis
Whole blood samples from C57BL/6J mice were drawn at 6 weeks post M. tuberculosis infection. Sera were separated by centrifugation at 3 000 rpm for 5min after blood samples were incubated at 37°C for 30min. For LC-MS/MS analysis, 100µL serum was thoroughly mixed with 100µL pre-cooled water and 400µL cold methanol acetonitrile (v/v, 1:1) through vortex. Then, the mixture was processed with sonication for 1h on ice, and incubated at -20°C for 1h. The mixture was centrifuged at 4°C for 20min with 14 000g. The supernatants were harvested and dried under vacuum. 100 µL acetonitrile water solution (v/v, 1:1) was added to redissolve the samples, then centrifuged at 16 000g at 4°C for 20min. The supernatant was harvested for further analysis. Extraction of samples was performed by Bioprofile (Shanghai, China).
Liquid chromatography tandem mass spectrometry (LC-MS/MS) based untargeted metabolomics analysis
For hydrophilic interaction liquid chromatography (HILIC) separation, samples were analyzed using a 2.1mm×100mm ACQUIY UPLC BEH Amide 1.7µm column (Waters, Ireland) by SHIMADZU-LC30 ultra-high performance liquid chromatography system (UHPLC, Shimadzu). Both electro-spray ionization (ESI) positive mode and negative mode were applied for MS data acquisition by QE Plus mass spectrometer (Thermo Fisher). The raw MS data were processed using MS-DIAL for peak alignment, retention time correction and peak area extraction. The metabolites were identified by accuracy mass (mass tolerance <0.01Da) and MS/MS data (mass tolerance <0.02Da) which were matched with Human Metabolome Database (HMDB) and Massbank databases. For the extracted data, ion peaks with the missing values >50% were deleted from the group. The total peak areas of positive and negative ion data were normalized, positive and negative ion peaks were integrated, and R software was used for pattern recognition. After the data were pretreated by Unit variance Scaling, subsequent data analysis was performed. LC-MS/MS based untargeted metabolomics analysis and data analysis were performed by Bioprofile (Shanghai, China).
CFU enumeration and genotypic identification of bacilli by PCR
Lungs and spleens of mice were aseptically removed and homogenized through a 40µm cell strainer in 4mL RPIM 1640 medium. Then the tissue homogenates were serially diluted and plated on 10% OADC supplemented 7H10 agar plates containing polymyxin B sulfate (80µg/mL) and azlocillin sodium (5µg/mL) to avoid other bacterial contamination. Bacterial colonies were counted following 3-4 weeks of incubation at 37°C. The number of single colony of bacilli was recorded, and CFUs data are represented as Log10CFU. In order to distinguish M. tuberculosis H37Ra and BCG/rBCG-DisA, colonies on the plate were amplified by PCR with ag85b (present in both BCG and H37Ra) and cfp10-esat-6 operon (only present in H37Ra) fragments (Table S1).
Strategy of prime-boost and M. tuberculosis challenge
Female BALB/c mice were vaccinated with 1×107 CFU of BCG by s.c route. At 58-week after vaccination, mice received a second boosting vaccination with the same dose and route of rBCG-DisA. Six weeks post boost, mice were challenged with 1×105 CFU of M. tuberculosis H37Ra by i.n route. After infection, mice body weights were monitored weekly and survivals were recorded.
Statistical analysis
Results were represented as mean values ± SEM. Statistical analysis of data performed by Graphpad Prism 9.0 Software (Graphpad Software, USA). Statistical significances were determined by two-tailed Student’s t test or for multiple comparisons by one-way ANOVA. For analysis of survival curves, we used log-rank test. The results were significant when *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as given in the figure legends unless otherwise specified.
Results
rBCG-DisA elicited greater proinflammatory cytokine responses to homologous and heterologous re-stimulations than BCG
Following exposure to infectious agents or vaccines, trained immunity can mount a faster and greater response against secondary challenge with homologous or even heterologous pathogens (8). Proinflammatory cytokines and chemokines have been reported as primary indicators of trained immunity, mainly including TNF-α, IL-1β, IL-6, CXCL9, CXCL10 and CXCL11 (36, 37). Both in vitro and in vivo experiments have shown that BCG immunization could induce increased extensive cytokine responses of IL-1β, TNF-α and IL-6 after training with BCG (33, 36, 38). It has been demonstrated that rBCG-DisA could elicit greater proinflammatory cytokines including IFN-β, IL-1β, IL-6 and TNF-α than that of BCG in primary human and murine macrophages (31, 32). In this study, splenocytes of immunized mice were re-stimulated with BCG, M. tuberculosis, S. aureus and E. coli LPS respectively (Figure 1A). BCG immunization by i.v route induced more IL-1β, IL-6, TNF-α, CXCL9 and CXCL11 releases to homologous stimulus re-stimulation in splenocytes of mice (Figure 1B). Our previous study found that rBCG-DisA immunization by s.c route only induced elevated IL-6 production in splenocytes of mice (27). Here, rBCG-DisA immunization induced elevated proinflammatory cytokines and chemokines of IL-1β, IL-6, TNF-α, CXCL9 and CXCL11 releases than that of BCG in splenocytes of mice responding to homologous antigens, and CXCL10 was under the limit of detection (Figure 1B).
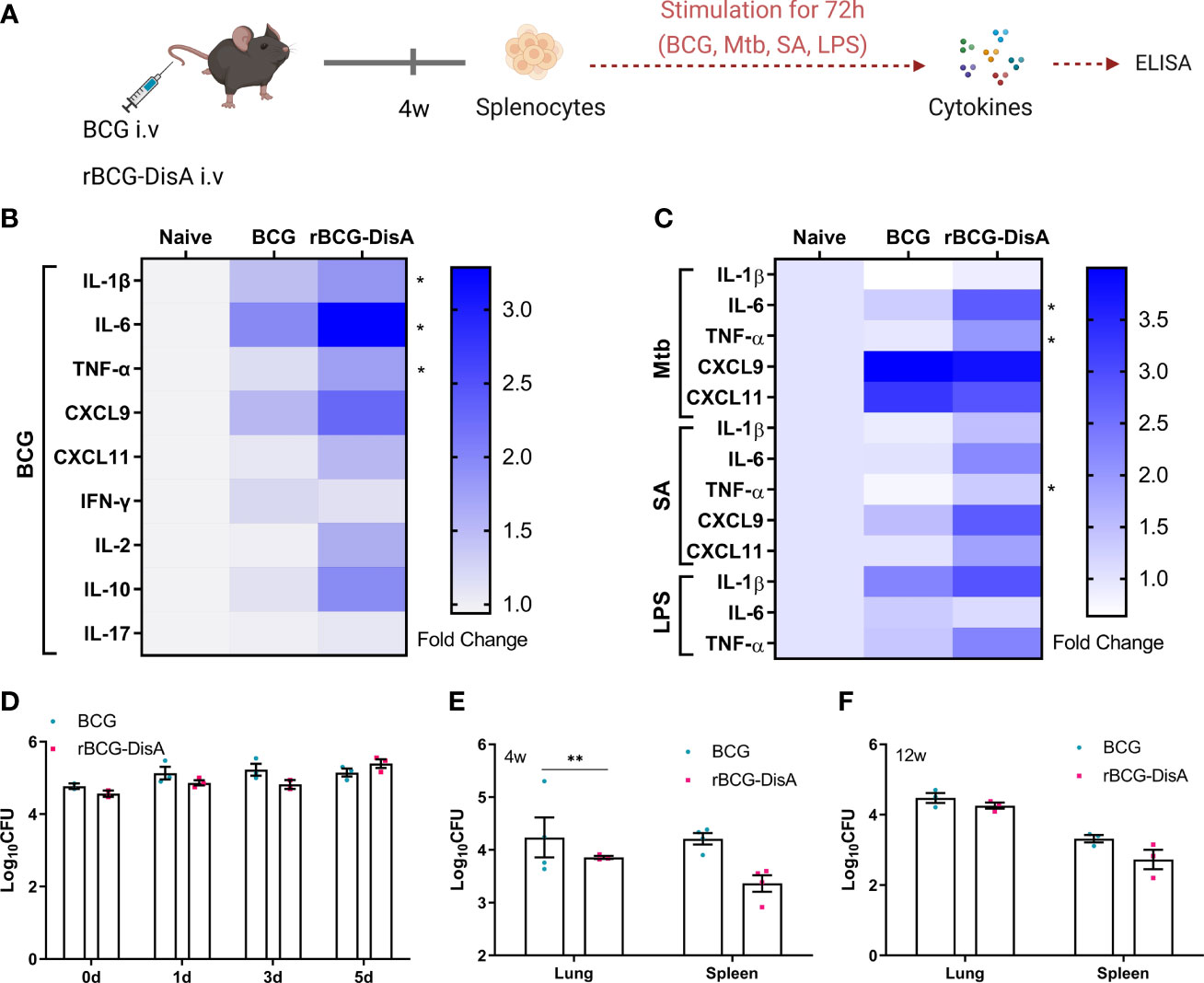
Figure 1 Inflammatory cytokine levels of rBCG-DisA i.v vaccinated mice and intracellular survival of rBCG-DisA in macrophages and mice. (A) Schematic diagram of homologous or heterologous stimulation of splenocytes in vitro (created with BioRender.com). After 4 weeks of BCG and rBCG-DisA i.v vaccination, splenocytes were isolated and re-stimulated with BCG protein extracts (BCG) (25μg/mL), M. tuberculosis (Mtb) (25μg/mL), S. aureus (SA) (25μg/mL), and E. coli O111:B4 LPS (LPS) (100ng/mL) for 72h in vitro. Normal mice (Naïve) were used as control. Supernatants were collected to measure cytokine secretion level by ELISA. (B, C) Cytokines and chemokines production of IL-1β, IL-6, TNF-α, CXCL9, CXCL11, IFN-γ, IL-2, IL-10 and IL-17 in supernatants were determined after re-stimulations by ELISA. The results are expressed as relative folds of each cytokine concentration (ng/mL) compared to the Naïve group (n=3). (D) Intracellular survival of BCG and rBCG-DisA in RAW264.7 macrophages at 0, 1, 3, and 5 days post infection (MOI=10). (E, F) The number of bacilli in the lung and spleen of mice was measured after 4 (E) and 12 (F) weeks of i.v immunization, respectively (n=3). (Panels B, C, *P < 0.05 stands for rBCG-DisA v.s BCG group. Panel E, **P < 0.01.).
Besides, BCG immunization increased the heterologous IFN-γ production and induced long lasting heterologous IL-17 responses in mice (39). Previously, we showed that splenocytes of rBCG-DisA s.c immunized mice produced Th1/Th2 cytokines of IFN-γ, IL-2 and IL-10 as that of BCG to homologous proteins (27). In this study, BCG immunization by i.v route induced slightly elevated IFN-γ and IL-10 production, while rBCG-DisA i.v immunization induced elevated IL-2 and IL-10 production to homologous proteins in splenocytes of mice (Figure 1B). It was found that BCG and rBCG-DisA did not induce IL-17 production to homologous proteins in splenocytes of mice after 4 weeks of immunization by i.v route (Figure 1B). rBCG-DisA immunization induced comparable levels of IFN-γ and IL-17 secretion after re-stimulated by homologous stimulus as that of BCG, but higher levels of IL-2 and IL-10 than that of BCG (Figure 1B).
It was proved that BCG trained innate immune cells showed specific responses according to different stimuli (37). Then splenocytes of immunized mice were re-stimulated with M. tuberculosis, S. aureus, and E.coli LPS respectively (Figure 1A). BCG immunization induced an enhanced IL-1β release to LPS, but not to M. tuberculosis and S. aureus (Figure 1C). rBCG-DisA immunization caused elevated releases of TNF-α to three bacterial components than that of BCG immunized mice, and IL-6 to M. tuberculosis and S. aureus (Figure 1C). Splenocytes from BCG and rBCG-DisA immunized mice produced significant chemokines of CXCL9 and CXCL11 responded to M. tuberculosis re-stimulation (Figure 1C). These data suggested that rBCG-DisA with c-di-AMP as endogenous adjuvant elicited enhanced proinflammatory cytokine responses in mice to homologous and heterologous stimuli.
Further, intracellular survival of BCG and rBCG-DisA in RAW264.7 macrophages were counted on plates. rBCG-DisA showed a slight decline on survival within 3 days, and increased at 5 days after infection, but no differences were found between two strains (Figure 1D). It was reported that a similar rBCG-DisA increased the levels of phagocytosis and autophagic processing within macrophages than BCG (32). After 4 weeks of immunization by i.v route, rBCG-DisA loads decreased in organs, especially in the lung of mice, but showed no difference compared with that of BCG in the lung and a slight decrease in the spleen after 12 weeks (Figures 1E, F). A latest study demonstrated that the similar rBCG-DisA based on another strain of Tice BCG proliferated to a lower degree in the lung of mice through aerosol inhalation (32). These results implied that enhanced cytokine responses of rBCG-DisA were not caused by different persistence of bacteria in host.
rBCG-DisA immunization triggered similar cytokine responses in the spleen of mice with BCG did after M. tuberculosis challenge
After 12 weeks of i.v immunization, mice were re-stimulated with M. tuberculosis H37Ra infection by i.n route (Figure S1A). At 6 weeks post M. tuberculosis challenge, spleen tissues were separated for detecting the transcription levels of cytokines and chemokines. As showed in Figure 2A, rBCG-DisA immunization induced similar transcription levels of almost all of proinflammatory cytokines and chemokines than un-immunized and BCG in the spleen after M. tuberculosis infection. Significant CXCL15 (IL-8) was reported before (36, 37, 40), which was not obvious in splenocytes of rBCG-DisA group. rBCG-DisA immunized mice showed an increasing trend on the transcription levels of IFN-γ, IL-2 and IL-10 in the spleen after M. tuberculosis infection, though no significant differences compared with that of UN and BCG immunized mice (Figure 2A). We speculated that splenocytes may not fully stimulated by i.n M. tuberculosis in vivo compared with proteins doses used in vitro. Further, we stimulated splenocytes from M. tuberculosis infected mice with homologous and heterologous stimuli. We found that rBCG-DisA immunization induced higher IL-6, IFN-γ and IL-10 secretions than BCG to homologous but not heterologous stimuli after M. tuberculosis infection (Figures 2B–E; S1B–E). We also showed that splenocytes from rBCG-DisA immunized mice and M. tuberculosis infected mice did not induce IL-1β to LPS stimulation, as UN and BCG groups did (Figure 2D).
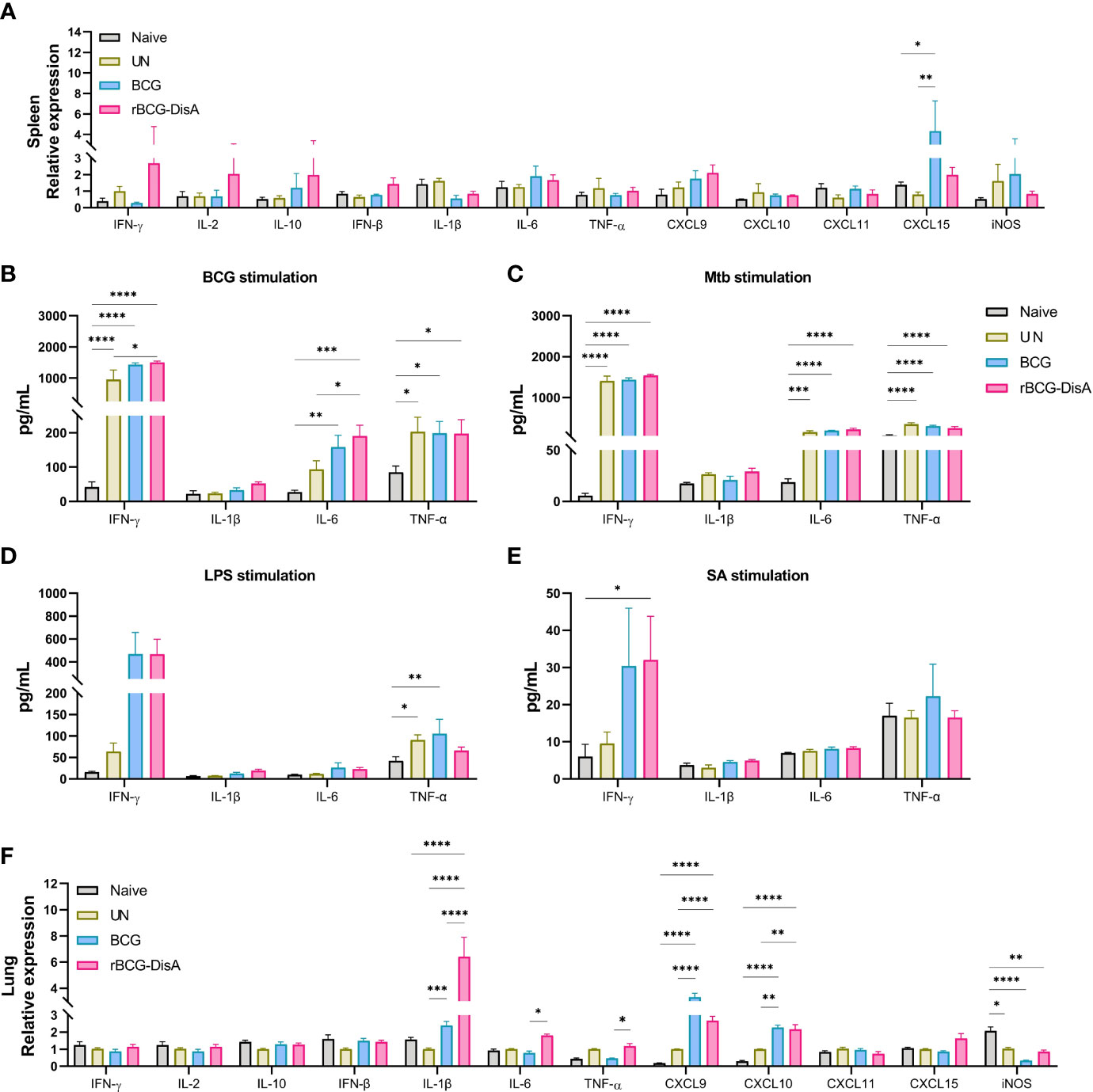
Figure 2 Inflammatory cytokines expression in the spleen and lung of vaccinated mice after M. tuberculosis infection. Mice were vaccinated by i.v BCG and rBCG-DisA for 12 weeks. Then, mice were infected with M. tuberculosis by i.n route, normal mice (Naïve) and M. tuberculosis infected mice without immunization (UN) were used as control. (A) At 6 weeks post infection, transcription level of indicated genes in the spleen of mice was assayed by quantitative RT-PCR (n=3). (B–E) At 6 weeks post infection, splenocytes were isolated and re-stimulated with BCG protein extracts (BCG) (25μg/mL) (B), M. tuberculosis (Mtb) (25μg/mL) (C), E. coli O111:B4 LPS (LPS) (100ng/mL) (D), and S. aureus (SA) (25μg/mL) (E) for 72h in vitro. Cytokines production of IFN-γ, IL-1β, IL-6 and TNF-α in supernatants were determined by ELISA (n=4). (F) At 6 weeks post infection, transcription level of indicated genes in the lung of mice was assayed by quantitative RT-PCR (n=3). (*P < 0.05, **P < 0.01, ***P < 0.001,****P < 0.0001.).
rBCG-DisA elicited greater inflammatory cytokines in the lung of mice after M. tuberculosis intranasal challenge
In this study, mice were immunized and then re-stimulated with M. tuberculosis infection by i.n route (Figure S1A). Lung tissues were also separated for detecting the transcription levels of cytokines and chemokines. rBCG-DisA i.v immunization significantly up-regulated proinflammatory cytokines of IL-1β, IL-6, and TNF-α in the lung than BCG i.v immunization after 6 weeks of M. tuberculosis challenge (Figure 2F). For chemokines, BCG and rBCG-DisA immunization promoted a similar increasing of CXCL9 and CXCL10, and rBCG-DisA immunization induced a slight increasing trend of CXCL15 expression in the lung after M. tuberculosis infection (Figure 2F). However, the results showed that the transcription levels of IFN-γ, IL-2 and IL-10 in the lung were no changes after M. tuberculosis infection in the lung of all groups (Figure 2F). These data suggested that rBCG-DisA immunization induced higher level of trained immunity in the lung, and were in line with such recombinant BCG that could trigger proinflammatory cytokine responses in vitro of BMDMs and macrophage cell line (31, 32).
rBCG-DisA enhanced epigenetic changes than BCG in mice after M. tuberculosis challenge
More data showed BCG is an inducer of trained immunity through increased transcription of proinflammatory cytokine and chemokine genes by epigenetic modifications (33, 41). It has been proved that epigenetic signatures of H3K4me3 and H3K27ac would decrease gradually with BCG inoculation time, and H3K4me1 persisted for long time to form immune memory (42, 43). Previously, we had identified that rBCG-DisA immunization by s.c route induced an enhanced H3K4me3 in the lung of mice after 4 weeks of M. tuberculosis infection (27). In this study, mice were immunized by i.v route for 12 weeks and challenged with M. tuberculosis (Figure S1A). Above, we had already established that rBCG-DisA acted as a more potent inducer of TNF-α and IL-6 production than BCG after re-stimulations in vitro (Figures 1B, C) and in the lung of mice (Figure 2F). Further, we detected epigenetic signatures of BCG-induced trained immunity including H3K4me3, H3K4me1 and H3K27ac by IHC in the lung of M. tuberculosis infected mice. It was shown that rBCG-DisA immunization by i.v route could obviously elicit enhanced epigenetic changes of H3K4me1 and H3K4me3, and a mild increasing in H3K27ac (Figure 3A), which suggested that rBCG-DisA elicited extended epigenetic changes compared with BCG, especially H3K4me1 for long time of immune memory.
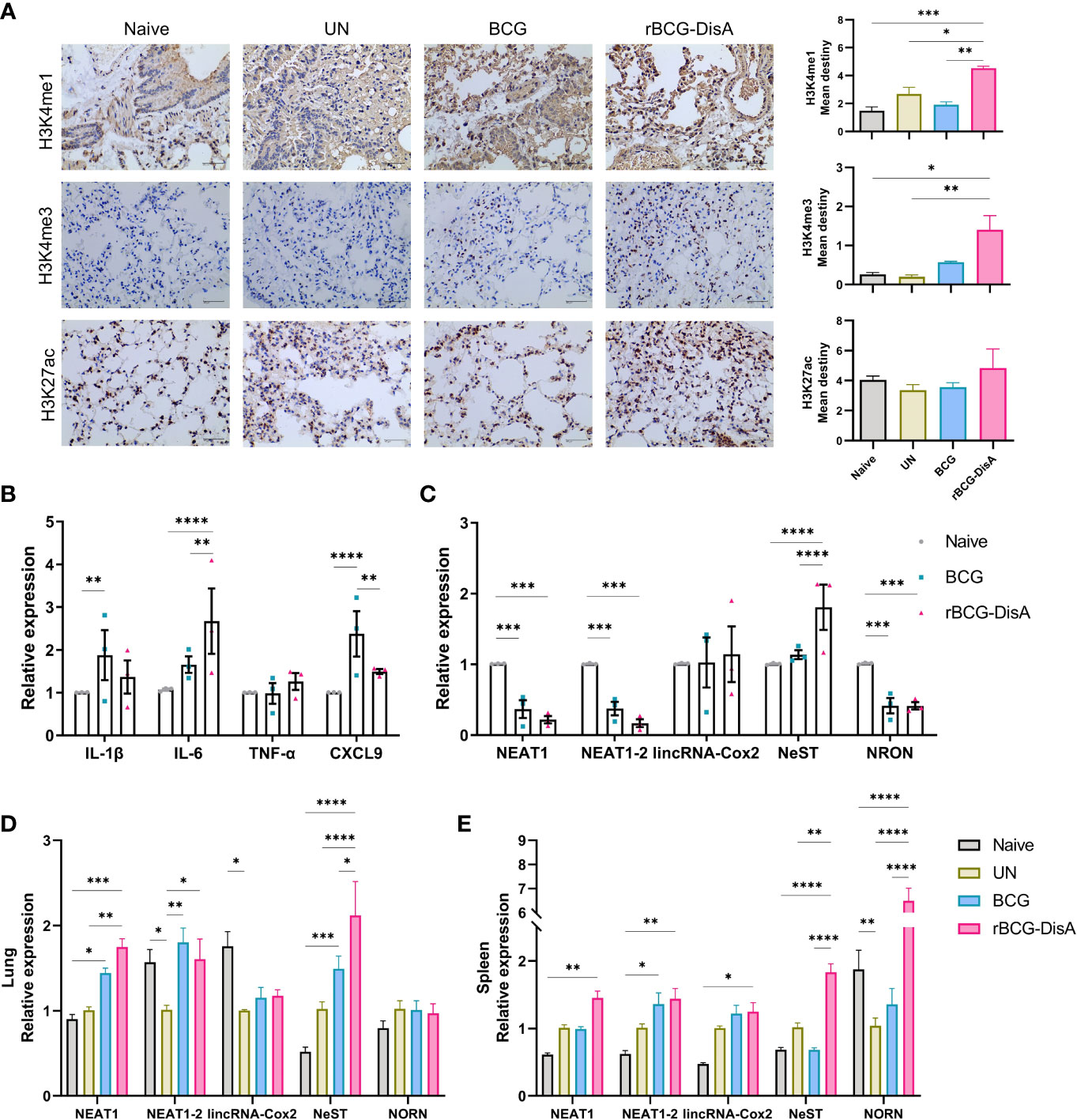
Figure 3 Expression of lncRNA and histone modifications in BM cells, lung and spleen of mice. Mice were vaccinated by i.v BCG and rBCG-DisA for 8 weeks (for BM cells isolation) and 12 weeks (for infection). Then, mice were infected with M. tuberculosis by i.n route, normal mice (Naïve) and M. tuberculosis infected mice without immunization (UN) were used as control. (A) At 6 weeks post infection, immunohistochemical analysis of H3K4me1, H3K4me3, and H3K27ac expression in the lung tissue (left). Quantitative analysis of H3K4me1, H3K4me3, and H3K27ac expression by Image J (right) (n=3). (B, C) At 8 weeks after BCG and rBCG-DisA i.v immunization, the expression of indicated cytokines (B) and non-coding RNA (C) in BM cells was detected by quantitative RT-PCR (n=3). (D, E) At 6 weeks post infection, immune responses were detected. Levels of indicated noncoding RNA in the lung (D) and spleen (E) were assayed by quantitative RT-PCR (n=3). (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.).
rBCG-DisA caused lncRNA expression changes in BM cells as well as in the lung of mice after M. tuberculosis challenge as BCG
It has been reported that i.v BCG could access bone marrow to expand hematopoietic stem cells (HSCs) and promotes myelopoiesis, and educated HSCs to generate trained monocytes/macrophages (33). To further characterize the trained immunity-inducing potential of rBCG-DisA versus BCG, we investigated the cytokine and chemokine expressions of BM cells from mice after 8 weeks i.v immunization. It was found that enhanced IL-6 expression was induced in BM cells of rBCG-DisA immunized mice compared with Naïve and BCG (Figure 3B). Whereas, rBCG-DisA immunization did not trained BM cells to express more IL-1β and CXCL9 as BCG did (Figure 3B). There were no significant differences in expressions of other cytokines and chemokines including IFN-γ, IFN-β, CXCL10, CXCL11, CXCL15 and iNOS (Figure S2). These results suggested BCG and rBCG-DisA immunization elicited different trained immune responses in central immune organ in mice.
Long noncoding RNAs (lncRNAs) are increasingly appreciated as regulators of cell-specific gene expression, which may involve in regulation of trained immunity. It has been identified that a lncRNA, named as stream master lncRNA of the inflammatory chemokine locus (UMLILO), could be brought proximal to immune genes (CXCL1, CXCL2, CXCL3, and CXCL15) prior to their activation and involve in innate immune defense (44). Since UMLILO is absent in mice, we detected some candidates lncRNAs that mediates expressions of immune response genes. It was found that lncRNA cyclooxygenase-2 (cox-2) (lincRNA-Cox2) was significantly increased in patients and macrophages with M. tuberculosis H37Ra infection (45). Both BCG and rBCG-DisA just induced a slight increase of lincRNA-Cox2 expression in BM cells from immunized mice (Figure 3C). After M. tuberculosis challenge, lincRNA-Cox2 expression level in the lung decreased, while the expression level in the spleen remained unchanged (Figures 3D, E).
It was reported that lncRNA NEAT1 (nuclear paraspeckle assembly transcript 1) was highly expressed in PBMCs and granulomatous tissue from TB patients, as well as in M. tuberculosis infected macrophages, and declined gradually with treatment (46). LncRNA NEAT1 appears in two isoforms of NEAT1-1 (short isoform) and NEAT1-2 (long isoform). We found that both BCG and rBCG-DisA seemed to inhibit NEAT1 expression in BM cells of immunized mice (Figure 3C). NEAT1 expression levels were elevated in the lung of BCG and rBCG-DisA immunized mice after M. tuberculosis infection (Figure 3D). Noticeably, rBCG-DisA immunization induced significant NEAT1 expression in the lung and spleen compared with that of BCG (Figures 3D, E). And there were no differences of NEAT1-2 expression levels between mice with BCG and rBCG-DisA immunizations (Figures 3D, E).
LncRNA NeST (Tmevpg1, abbreviates Nettoie Salmonella pas Theiler’s) is located in the nucleus, which is an enhancer lncRNA. In activated CD8+ T cells, NeST could recruit mixed lineage leukemia protein (MLL) and transactivate IFN-γ by binding to the adaptor protein WD repeat-containing protein 5 (WDR5) (47). It was observed that rBCG-DisA immunization elicited significant NeST transcription in BM cells of mice than that of BCG (Figure 3C). Similar increases were observed in the lung and spleen of rBCG-DisA i.v mice post M. tuberculosis infection (Figures 3D, E).
LncRNA NRON (noncoding repressor of nuclear factor of activates T cells [NFAT]) is highly expressed in resting CD4+ T lymphocytes, which potently suppresses the latent HIV-1 transcription (48). Our data revealed that both BCG and rBCG-DisA i.v vaccination repressed the expression of NRON in BM cells (Figure 3C). After M. tuberculosis infection, rBCG-DisA increased the level of NRON than BCG in the spleen but not in the lung (Figures 3D, E).
rBCG-DisA vaccination leads to an altered metabolic profile after M. tuberculosis challenge
Metabolic rewiring is the major signature of BCG-induced trained immunity. We collected sera at 8 weeks post infection for untargeted metabolomics analysis based on LC-MS (Figure 4A). To reveal the changes of metabolites in BCG or rBCG-DisA immunized mice, supervised multivariate orthogonal partial least squares discriminate analysis (OPLS-DA) models were constructed to discriminate the differentiating variables. The results showed that rBCG-DisA group was obviously separated from the BCG group (Figure 4B), indicating that these two groups had different metabolic profiles in sera. Among these differentially expressed metabolites of rBCG-DisA v.s BCG, first proportion (26.67%) belonged to lipids or lipid-like molecules, and the subsequent proportions were organic acids or organic nitrogen molecules, respectively (Figures 4C, D). Through KEGG analysis, it was found that metabolites were mainly enriched in metabolic pathways such as lipolysis, aldosterone synthesis, protein digestion and absorption, etc. (Figure 4E). As shown in Figure 4F, the level of acetylcarnitine was up-regulated in rBCG-DisA immunized mice after M. tuberculosis infection, which helps long-chain fatty acyl-CoA to enter mitochondria, indicating that fatty acid oxidation and energy metabolism were promoted (49). Arachidonic acid, which inhibits fat mobilization, was also down-regulated in rBCG-DisA-immunized mice (Figure 4F). Thus, rBCG-DisA immunization promoted fat mobilization and lipolysis in mice.

Figure 4 Metabonomics analysis of mice vaccinated with BCG or rBCG-DisA after M. tuberculosis infection. Mice were vaccinated by BCG and rBCG-DisA for 6 weeks. Then, mice were infected with M. tuberculosis by i.n route, normal mice (Naïve) and M. tuberculosis infected mice without immunization (UN) were used as control. At 8 weeks post infection, sera were harvested for metabonomics analysis (n=4). (A) A scheme diagram of detection on sera of mice vaccine with BCG or rBCG-DisA (created with BioRender.com). (B) OPLS-DA score plots of positive and negative ion modes of BCG and rBCG-DisA groups (n=4). (C) Heatmap of differential expressed metabolites of rBCG-DisA v.s BCG (n=4). (D) Circle diagram of metabolites classification of rBCG-DisA v.s BCG (n=4). (E) KEGG pathway enrichment of metabolites of rBCG-DisA v.s BCG (n=4). (F) Heatmap of differential expressed metabolites regarding lipid, cholesterol and amino acids metabolism of rBCG-DisA v.s BCG (n=4).
rBCG-DisA repressed specific IgG and CD4+ T cells by intravenous immunization
It is concluded that the heterologous protection conferred by BCG is likely the result of two mechanisms that synergize to induce protection: heterologous T cell immunity and trained immunity (3, 39). Previously, we found that rBCG-DisA s.c administration induces comparable humoral and cellular immune responses as BCG did in mice (27). We further analyzed the level of adaptive immune response in mice vaccinated with i.v BCG and rBCG-DisA strains. During the whole experiment, mice showed similar general behaviors such as spontaneous behavior, arousal behavior, grooming behavior, fecal traits among Naïve, BCG and rBCG-DisA groups. The body weights of BCG group mice increased steadily comparable to Naïve mice within 12 weeks after vaccination (Figure 5A). rBCG-DisA vaccinated mice showed lower weight gain than control and BCG mice (Figure 5A).
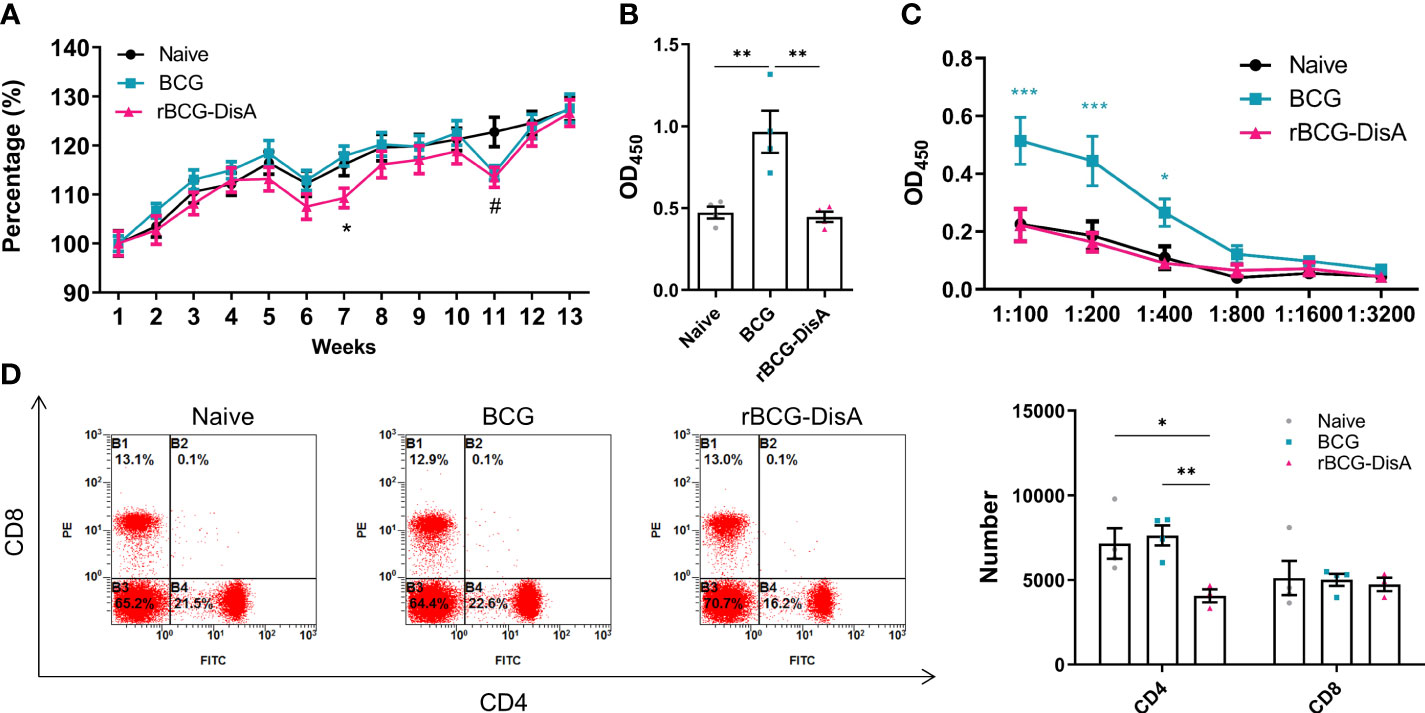
Figure 5 Adaptive immune response induced by rBCG-DisA vaccination. Male C57BL/6J mice were vaccinated with 1×106 CFU BCG and rBCG-DisA by i.v route. At 4- and 12-week post vaccination, immune responses were determined. Normal mice (Naïve) were used as control. (A) Dynamic monitoring of mice body weights at each week after immunization. Data were presented as a percentage of body weight to initial body weight (n=3). (B) After 4 weeks of i.v immunization, BCG-specific IgG levels in mice sera (1:200) were assayed by ELISA (n=4). (C) After 12 weeks of i.v immunization, BCG-specific IgG titers in sera of mice at indicated dilutions were assayed by ELISA (n=4). (D) After 4 weeks of i.v immunization, CD4 and CD8 T cells in splenocytes were analyzed by flow cytometry of immunized mice (left). Numbers of CD4 and CD8 T cells in splenocytes were measured by flow cytometry (n=4) (right). (Panel A, *P < 0.05 stands for rBCG-DisA v.s BCG, #P < 0.05 stands for rBCG-DisA v.s Naïve. Panel C, *P < 0.05 and ***P < 0.001 stand for rBCG-DisA v.s BCG/Naïve. Panel D, *P < 0.05, **P < 0.01.).
BCG administrated by i.v route induced higher mycobacteria-specific humoral immune response (Figures 5B, C; S3A). Whereas, rBCG-DisA i.v vaccination may repress the increasing of IgG than BCG 4-week and 12-week post vaccination (Figures 5B, C, S3A). BCG vaccination induced long-lasting effects of Th1/Th17 responses, and T-cell subpopulations did not show major shifts in CD4 and CD8 lymphocytes for at least one year in healthy volunteers (39). The proliferation of mice splenocytes were comparable in both two vaccinated groups compared to that of Naïve mice after re-stimulated by mycobacteria antigens by CFSE staining (Figure S3B). We found that rBCG-DisA immunization by s.c route increased the proportion of CD4+ but not CD8+ T cells (27). Whereas, rBCG-DisA vaccination by i.v route reduced the number and proportion of CD4+ T cells compared to both control and BCG groups (Figure 5D, S3D), and no significant changes on CD8+ T cells (Figure 5D, S3C).
rBCG-DisA vaccination increased the CD4+ and CD8+ T cells after M. tuberculosis infection
After 12 weeks of i.v immunization, mice were infected with M. tuberculosis H37Ra by i.n route (Figure S1A). Six weeks post M. tuberculosis challenge, BCG i.v immunized mice exhibited low mycobacteria-specific IgG as Naïve and M. tuberculosis H37Ra i.n infection (UN) mice (Figure 6A). rBCG-DisA vaccination elicited stronger BCG- and DisA-specific IgG than that of BCG (Figures 6A, B). To our surprise, all infected mice showed similar percentages of T cell, B cell, NK cell, macrophage and neutrophil with Naïve mice (Figure S4). Further analysis of T cell subsets found that M. tuberculosis H37Ra i.n infection (UN) had negligible effect on proportions of CD4+ T cells, but caused a significant decline of CD8+ T cells in splenocytes (Figure 6C). BCG i.v immunized mice showed a decline of CD4+ T cells compared with UN group, but rBCG-DisA didn’t (Figure 6C). Both BCG and rBCG-DisA immunization resisted the reduce of CD8+ T cells caused by M. tuberculosis infection, and BCG showed a significant increase compared with UN group (Figure 6C). rBCG-DisA immunization kept the percentage of CD8+ T cells in splenocytes after M. tuberculosis infection closed to that of Naïve mice (Figure 6C).

Figure 6 Analysis of specific antibodies and T cells in the spleen and lung of vaccinated mice after M. tuberculosis infection. Mice were vaccinated by i.v BCG and rBCG-DisA for 12 weeks. Then, mice were infected with M. tuberculosis by i.n route, normal mice (Naïve) and M. tuberculosis infected mice without immunization (UN) were used as control. (A) At 6 weeks post infection, BCG-specific IgG titers in sera were determined by ELISA at indicated dilutions (n=4). (B) At 6 weeks post infection, DisA-specific IgG levels in sera was determined by ELISA at dilution of 1:200 (n=4). (C) At 6 weeks post infection, proportions of CD4 and CD8 T cells in splenocytes were assayed by flow cytometry (n=4). (D) At 6 weeks post infection, immunohistochemical analysis of CD4 and CD8 T cells in the lung of mice (left). Quantitative analysis of CD4 and CD8 T cells by Image J (right) (n=3). (*P < 0.05, **P < 0.01.).
Above results showed that rBCG-DisA i.v vaccinated mice exhibited greater innate immune inflammatory responses in the lung after M. tuberculosis infection (Figure 2F). Further, adaptive immune response in the lung was observed through the distribution of CD4+ and CD8+ T cells in the lung by IHC after M. tuberculosis infection by i.n route. As illustrated in Figure 6D, BCG i.v immunization caused similar T cells distribution after M. tuberculosis infection with UN group. More importantly, it was clearly showed that increased of CD4+ and CD8+ T cells in the lung of rBCG-DisA immunized mice after M. tuberculosis i.n infection (Figure 6D) implying significant T cell infiltration against infection.
rBCG-DisA caused similar pathological changes as BCG after M. tuberculosis intranasal challenge
Since trained immunity confers broad immunological protection, it was worried that enhanced immune response of reprogrammed innate immune cells might result in the development or persistence of chronic metabolic, autoimmune or neuro-inflammatory disorders (50). rBCG-DisA induced stronger innate and adaptive immune responses, and did not cause excessive immunopathological damages by s.c inoculating in mice or intradermally route in guinea pig model (27, 31). Recently, the latest research demonstrates that a similar rBCG-DisA with different BCG strain was less pathogenic in immunocompromised SCID mice (32). As shown in Figure 7A, we did not observe the characteristics of tuberculosis such as tubercle and caseous necrosis in both lung and spleen tissues of infected mice. In the lung, M. tuberculosis infected mice exhibited obviously inflammatory cells infiltration (Figure 7A). In the lung of BCG and rBCG-DisA immunized mice, the alveolar structures were intact basically, accompanied with occasional thickening of alveolar mesenchyme, inflammatory cell infiltration, erythrocyte and histological fluid exudation (Figure 7A). Additionally, spleen of two immunized groups showed inflammatory cell infiltration as un-immunized mice after M. tuberculosis infection (Figure 7A). Overall, the inflammatory manifestations in the lung were comparable between rBCG-DisA and BCG vaccinated mice after M. tuberculosis challenge.
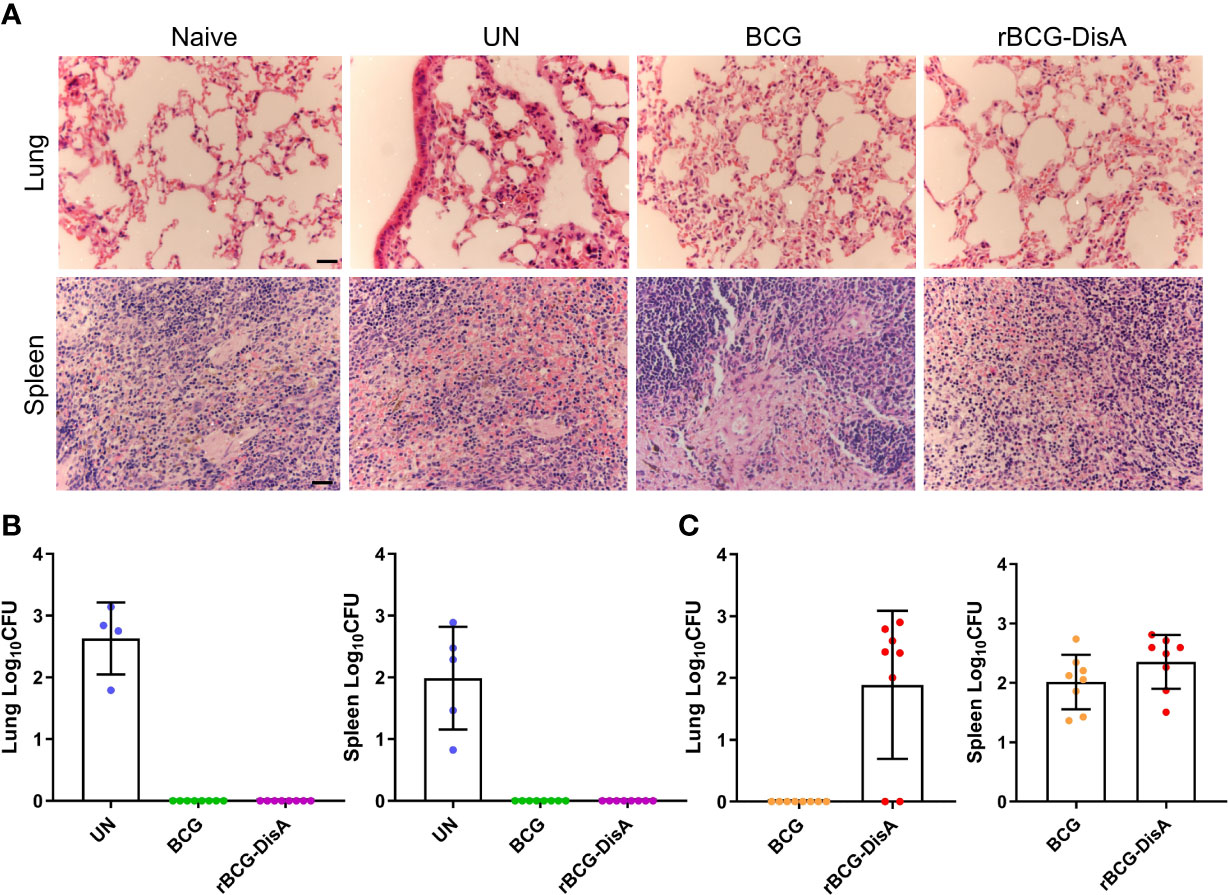
Figure 7 Histopathological changes and bacilli number of mice after M. tuberculosis infection. Mice were vaccinated by i.v BCG and rBCG-DisA for 12 weeks. Then, mice were infected with M. tuberculosis by i.n route, normal mice (Naïve) and M. tuberculosis infected mice without immunization (UN) were used as control. (A) At 6 weeks post infection, representative images of H&E-stained lung tissue sections at ×40 magnification (scale bar=100 μm). (B, C) At 6 weeks post infection, bacilli burdens in the lung and spleen were incubated at 37°C for 3-4 weeks (n=4~8). The colonies on the plates were determined according to the time of colony formation, morphology and genotype verification to determine whether the clone was M. tuberculosis [ag85B (+), cfp10-esat-6 (+)] (B) or BCG/rBCG-DisA [ag85B (+), cfp10-esat-6 (-)] (C).
rBCG-DisA provided similar protection against M. tuberculosis intranasal infection and persisted longer in the lung than BCG
After 12 weeks of immunization by i.v route, mice were infected with M. tuberculosis H37Ra infection by i.n route (Figure S1A). Six weeks post M. tuberculosis H37Ra i.n infection, the number of bacilli in the spleen and lung of mice was enumerated on plates. Considering that bacilli colonies could be M. tuberculosis, BCG or rBCG-DisA, we randomly selected numbers of bacterial clones for identification with specific primers (Table S1). The results showed that M. tuberculosis H37Ra were not identified under our detection method in the spleen and lung of BCG and rBCG-DisA immunized mice (Figure 7B). While, genotype identification of bacillus showed that the clones in two immunized groups were BCG or rBCG-DisA strains (Figure 7C). Further, we found BCG below the limit of detection in the lung after M. tuberculosis infection, but only two of eight mice showed non-sterilized of rBCG-DisA (Figure 7C). While the distribution of BCG and rBCG-DisA were similar in the spleen after H37Ra challenge (Figure 7C). These results suggested that rBCG-DisA immunization by i.v route could provide long-term protection than BCG after M. tuberculosis infection, which may be resulted from longtime survival of rBCG-DisA in the lung.
rBCG-DisA boosting prolonged the lifespan of BCG-primed mice against M. tuberculosis intranasal infection
BCG-induced trained immunity gradually declines over time and vaccination protection declines with time, so a booster vaccine may be given in adolescence or adults when the effects of BCG may start to wane (11, 12). In this study, BALB/c mice were primed by s.c BCG for 58 weeks, then mice were boosted with rBCG-DisA at the same route (Figure 8A). After 6 weeks, mice were challenged by M. tuberculosis H37Ra i.n infection (Figure 8A). rBCG-DisA boosted mice showed a weight maintenance within 9 weeks infection, compared with weight loss in BCG-primed with or without M. tuberculosis infected mice (Figure 8B). We observed a significant longevity in rBCG-DisA boosted mice compared to only BCG primed mice after M. tuberculosis i.n challenge (Figure 8B). BCG primed mice all died within 14 weeks after M. tuberculosis infection, and all non-infected mice died within 87 weeks (Figure 8C). Surprisingly, until now 66% of rBCG-DisA boosted mice were alive over 90 weeks (Figure 8C). Thus, rBCG-DisA may be used as a candidate vaccine for BCG prime-boost regimens against M. tuberculosis infection in adults.
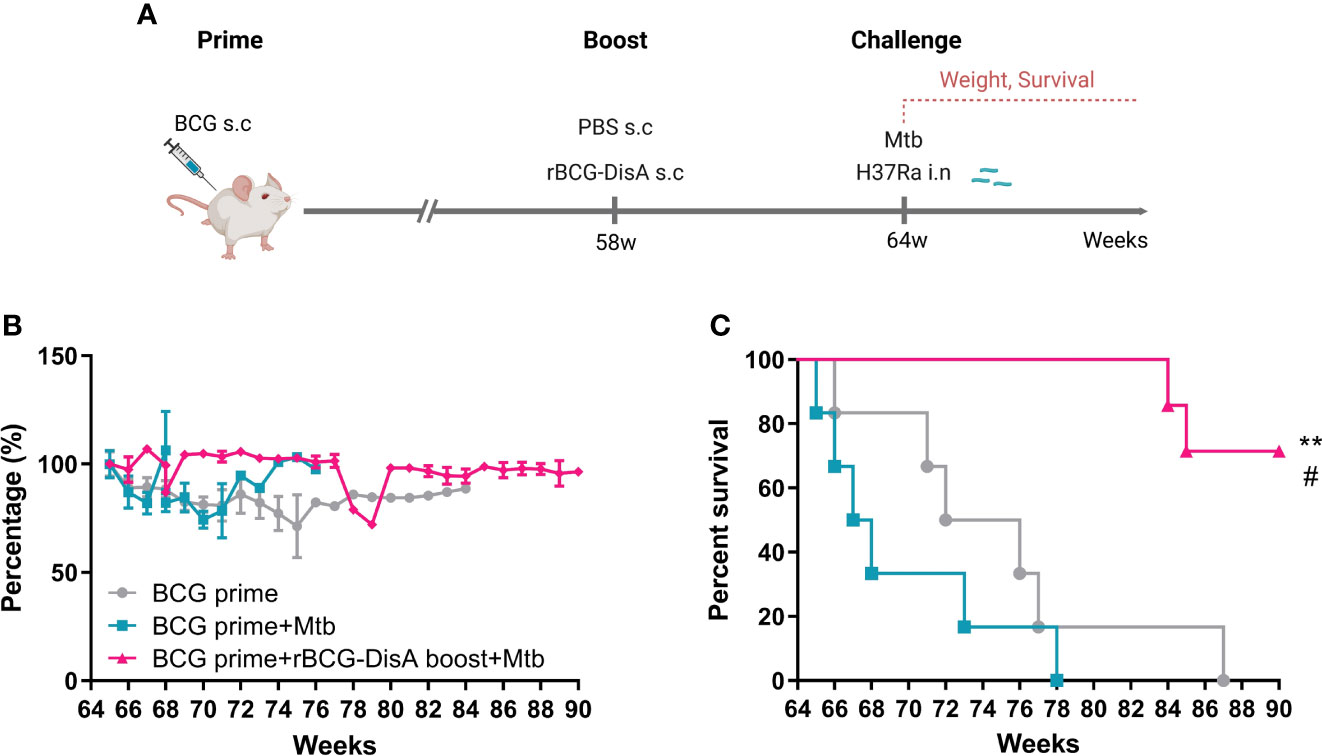
Figure 8 Body weight and survival in BCG prime following rBCG-DisA boost mice after M. tuberculosis infection. (A) BALB/c mice were primed by s.c BCG for 58-week following s.c rBCG-DisA boosting for 6-week. Subsequently, mice infected with M. tuberculosis by i.n route. Body weight and survival of mice were continuously monitored post M. tuberculosis infection. The schematic diagram of prime-boost strategy was created with BioRender.com. (B) Dynamic monitoring of mouse body weight at each week after M. tuberculosis infection. Results are presented as a percentage of body weight to initial body weight. (C) Survival of mice after M. tuberculosis infection (n=5~6). (Panel C, **P < 0.01 stands for rBCG-DisA boosting group v.s BCG primed group. #P < 0.05 stands for rBCG-DisA boosting group v.s and BCG primed+Mtb group.).
Discussion
In the previous study, we found that rBCG-DisA by s.c route induced stronger immune responses compared with BCG in mice, which suggested an enhanced trained immunity was induced in mice (27). It has been proved that BCG accesses to BM and educate HSCs to generate epigenetically modified macrophages, and provide protective innate immunity against M. tuberculosis through trained immunity (33). In this study, we provided in vivo experimental evidence that rBCG-DisA with c-di-AMP as endogenous adjuvant could induce enhanced trained immunity and concomitant activated adaptive immune response, which showed a significant protection against M. tuberculosis infection in prime-boost strategy in adult mice.
Trained monocytes and macrophages display functional and epigenetic reprogramming, leading to increased production of cytokines and chemokines, which are viewed as primary indicators of trained immunity (37), and improved phagocytosis and killing capacity (51, 52). c-di-AMP has been identified to be a potent inducer of several proinflammatory cytokines and chemokines such as IL-1β, IL-6, and TNF-α in macrophages (27, 31, 32, 53). These proinflammatory cytokines are also considered as mediators of trained immunity (8, 9, 54, 55). It has been reported that rBCG-DisA could elicit greater proinflammatory cytokines than that of BCG in primary human and murine macrophages (27, 31, 32). Our data showed that BCG and rBCG-DisA elicited proinflammatory cytokines and chemokines transcriptions with different models in splenocytes of immunized mice, according to homologous or heterologous stimuli (Figures 1B, C). Remarkably, rBCG-DisA immunization induced enhanced proinflammatory cytokines, chemokines and Th1/Th2 cytokines transcriptions to re-stimulations in vitro than BCG in splenocytes of immunized mice (Figure 1B), and significant proinflammatory cytokines transcriptions in the lung to re-stimulation of M. tuberculosis i.n infection (Figure 3F). However, rBCG-DisA did not improve phagocytosis and killing capacity of macrophages than that of BCG as other inducer of trained immunity (Figure 1D) (51).
Generally, the induction of trained immunity is accompanied with the epigenetic modification at the promoters of cytokines such as IL-1β, IL-6, and TNF-α (56). Accumulations of H3K4me1 and H3K4me3 caused by rBCG-DisA may contribute to higher IL-6 than BCG in BM cells (Figures 2F, 3A). However, the molecular mechanism of histones regulating the expression of inflammation-related genes have not yet been elucidated. H3K4me3 is directed to specific promoters in the genome by the presence of a class of lncRNAs, called immune gene-proximal lncRNAs (IPLs), which to be brought proximal to immune genes prior to their activation (44). UMLILO is the first reported IPL, which acts in cis to direct the WDR5MLL1 complex across the chemokine promoters (44). Surprisingly, an enhancer lncRNA of NeST, which binding to the adaptor protein WDR5 (47), was greater up-regulated in BM cells of rBCG-DisA i.v immunized mice than that of BCG, as well as in the lung and spleen after M. tuberculosis i.n infection (Figures 3C–E). IPLs are transcribed in a NFAT. Silencing of IPLs or abrogation of NFAT signaling results in loss of H3K4me3 accumulation at trained immune genes (44). We found that lncRNA NRON was repressed in BM cells of both BCG and rBCG-DisA i.v immunized mice, and showed no change in the lung but up-regulated in the spleen of rBCG-DisA i.v immunized mice to re-stimulation of M. tuberculosis infection (Figures 3C–E). Then we found that the expression of NEAT1 was down-regulated in BM cells of both BCG and rBCG-DisA immunized mice, but up-regulated significantly in the organs of rBCG-DisA immunized mice after M. tuberculosis challenge (Figures 3C–E). A growing body of research has revealed abnormally expressed lncRNAs in macrophages of TB patients (57). Thus, these lncRNAs that screened in this study may play roles in the regulation of the trained immunity, which should be investigated further.
Studies have demonstrated that immunological signals, metabolic rewiring of cell metabolism, and epigenetic reprogramming are integrated, representing the molecular substrates for induction of trained immunity (52). A recently research reported that another DisA overexpressing BCG (BCG Tice strain) elicited increased glycolytic metabolites, reduced kynurenine accumulation and itaconate production than wild type BCG in human or murine macrophages (32). However, these observations usually were generated in vitro model with different types of macrophages, which are known to arise from distinct cell lineages emerging at different stages in embryonic development. Thus, in vivo reality is of much greater nuance and complexity than can be accommodated by a macrophages model. In this study, we checked the metabolic profiles of mice immunized by BCG and rBCG-DisA after M. tuberculosis infection by metabolomic analysis, and found that increased fatty acid oxidation, fat mobilization, lipolysis and energy metabolism, as well as mTOR activation (Figure 4). It has been confirmed that induction of trained immunity accompanied by activated STING with enhanced mTOR-HIF-1α pathway activation and concomitant elevation in glucose transporter levels (58–60), which implied that rBCG-DisA promoted glycolysis in mice post M. tuberculosis infection. Besides, mTOR has been implicated in both the breakdown and synthesis of fatty acids, and rBCG-DisA immunized mice presented fatty acid oxidation trend. Thus, rBCG-DisA vaccination increased the catabolic signatures of fat mobilization and lipolysis than BCG in mice after M. tuberculosis infection. Cholesterol metabolism is a part of lipid metabolism. BCG-induced activation of TCA cycle promotes acetyl-CoA production, causes mevalonate accumulation, which promotes cholesterol synthesis (61). rBCG-DisA induced down-regulation of taurochenodeoxycholic acid, serine-cholic acid, and corticosterone, while the expression of mevalonic acid lactone inhibiting mevalonic acid synthesis was up-regulated (Figures 4C, F), suggesting that cholesterol synthesis in rBCG-DisA immunized mice was inhibited compared with BCG. Besides, rBCG-DisA induced up-regulation of essential amino acids and their metabolites such as methionine, valine betaine, L-norvaline valine, etc. Meanwhile, the expression of non-essential amino acid and its metabolites like L-proline, 4-(beta-acetylaminoethyl) imidazole (also called N-acetylhistamine) increased. The level of essential amino acids is positively correlated with activation of immune system induced by M. tuberculosis (62). And N-acetylhistamine may be related to anaphylaxis (63). Thus, shifts in metabolite levels may be a plausible mechanism behind the integration of immuno-metabolic and epigenetic programs for the enhanced trained immunity induced by rBCG-DisA immunization in mice.
Trained immunity is mediated by innate immune cells such as monocytes, macrophages, or NK cells (64), which produce heterologous lymphocyte activation, resulting in enhanced proinflammatory cytokines production, macrophage activity, T cell responses, and antibody titers (52). Activation of surface molecules such as pattern recognition receptor (PRR) and signaling pathways leading to the induction of Th1/Th2/Th17 response during trained immunity (39, 64). BCG could drive a robust mycobacteria specific antibody response in plasma and lungs, including IgG1, IgA, and IgM by i.v route, which could provide protection to an attenuated strain of M. tuberculosis (ΔsigH in the M. tuberculosis CDC1551) in macaques (65). Present study revealed that rBCG-DisA i.v immunization led to lower specific IgG than BCG (Figures 5B, C, S3A), which was inconsistent with our previous research by s.c route (27). While, anti-DisA antibody levels raised than that of BCG after M. tuberculosis infection (Figure 6B), which further confirmed strong immunogenicity of DisA as our previous work (27). Here, i.v rBCG-DisA led to the increasing of CD4 T cells in the spleen of C57BL/6J mice compared with BCG after M. tuberculosis infection (Figure 6C), as that of by s.c route in BALB/c mice (27). Unexpectedly, rBCG-DisA immunized mice showed a significant increase CD4 T cells in the lung after M. tuberculosis infection, which suggested that enhanced cellular immune response induced in the lung of mice. Besides, our recent work showed that elevated c-di-AMP regulated expressions of several immune-associated proteins, and increased immunogenicity of Mycobacterium smegmatis by s.c route in mice (30). Hence, immune responses was induced by rBCG-DisA i.v immunization may be the results of the dual functions of overexpressed DisA and c-di-AMP regulated proteins.
Above, we had showed that rBCG-DisA i.v immunization could induce enhanced trained immunity as well as adaptive immunity in mice, especially accumulated T cells in the lung (Figure 6D). We found that rBCG-DisA i.v immunization could retain the body weight compared with BCG in mice (Figure 5A), but did not observe adverse reactions during the experiment, and pathological damage by H&E staining (Figure 7A). As well, H37Ra can be used as a surrogate for studying M. tuberculosis virulence in biosafety level 2 (BSL2) facilities (66). Previous, we showed that M. tuberculosis H37Ra and H37Rv were capable to induce comparable levels of humoral and cellular immunity by i.v route (67). The protection efficiency data showed that rBCG-DisA i.v vaccination promoted the clearance of M. tuberculosis H37Ra to undetectable in the lung and spleen of mice as BCG (Figure 7B). It was noted that rBCG-DisA rather than BCG was capable of long-term persistence in the lung after M. tuberculosis infection (Figure 7C). The longer resident of rBCG-DisA in the lung of mice may explain elevated T cell responses (Figure 6D, 7D) and enhanced epigenetic modification, especially immune memory related H3K4me1 (Figure 3A). While, BMDMs from both rBCG-DisA and BCG immunized mice showed similar proinflammatory cytokine responses and protection against M. tuberculosis H37Ra infection (Figures S5A–D). It has been elucidated that BMDMs from BCG vaccinated mice could inhibit intracellular survival of virulent H37Rv (33). We speculate that the virulence and MOI of M. tuberculosis strain caused differences in results. Together, these results showed that rBCG-DisA may provide longer protective after M. tuberculosis infection, which was attributed to enhanced T cell immunity as well as trained immunity (3, 39).
It was reported that the protection efficacy of BCG vaccination at birth rarely persists beyond 15-20 years in TB epidemic regions, but highly variable in adults (68, 69). Currently, several new mycobacterial vaccine designs that aimed to improve the efficacy over BCG have been evaluated in animal models and some in humans, such as boosting BCG with homologous BCG or heterogeneous vaccines such as subunit vaccine of H4:IC31 (70). In view of the waned protective efficiency after one year of BCG vaccination (10, 71), we had a try to boost BCG-primed mice of 58 weeks with rBCG-DisA by s.c route (Figure 8A). Surprisingly, we found that rBCG-DisA boosting could help mice resist weight loss and significantly prolonged survival of mice after M. tuberculosis i.n challenge (Figures 8B, C). Regrettably, our prime-boost experiment was lack of a control group of un-primed mice. Epidemiological data suggest that BCG vaccination is safe and decreases the infections of all causes in the elderly, especially respiratory tract infections (7, 72). Hence, our results suggested a promising potential of rBCG-DisA in prime-boost strategy, especially in adults.
In present study, enhanced immune responses including trained immunity and adaptive immune response induced by rBCG-DisA i.v immunization were investigated. rBCG-DisA induced innate immunity including potent proinflammatory cytokine responses, epigenetic modification, altered lncRNA and metabolic profile. These results suggested that rBCG-DisA induced enhanced trained immunity, which may be stored within BM progenitor cells against infection over long periods. Additionally, vaccination to enhance trained immunity is a promising strategy which will be applied to adults or immunocompromised individuals due to defective adaptive immunity (52, 52, 54).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. The metabolome sequencing data presented in the study are deposited in the MetaboLights repository (URL https://www.ebi.ac.uk/metabolights/MTBLS5496), accession number MTBLS5496.
Ethics statement
The animal study was reviewed and approved by The Institutional Ethics Committee of Second Affiliated Hospital of Air Force Medical University, using the recommendations from the Guide for the Care and Use of Laboratory Animals of the Institute (approval no. TDLL-20190213).
Author contributions
HN, JK, YL, XL, RR, YZ, YX, LB, YK, XG, and MX performed experiments and analyzed data. JZ, SZ, LZ, and YM contributed cells, reagents and expertise. HN, YL, and YB wrote the manuscript. YB and FZ conceived and designed experiments. YB supervised this work. All authors have read and agreed with the data.
Funding
This study was funded by National Natural Science Foundation (No. 81971560, 81371774, 81671638), National Major Special Projects of 13th Five-year Plan (No. 2018ZX10302302002004), Provincial Natural Science Foundation of Shaanxi Province (No. 2022ZDLSF01-07, 2019ZDLSF02-04), and Ningxia Natural Science Foundation (No. 2021AAC03124).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GD declared a shared affiliation, with no collaboration, with some of the authors, LZ, YK, XG, to the handling editor at the time of the review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.943667/full#supplementary-material
References
1. Ahmed A, Rakshit S, Adiga V, Dias M, Dwarkanath P, D'Souza G, et al. A century of BCG: Impact on tuberculosis control and beyond. Immunol Rev (2021) 301(1):98–121. doi: 10.1111/imr.12968
2. Brewer TF. Preventing tuberculosis with bacillus calmette-guerin vaccine: a meta-analysis of the literature. Clin Infect Dis (2000) 31 (Suppl 3):S64–67. doi: 10.1086/314072
3. Singh AK, Netea MG, Bishai WR. BCG Turns 100: its nontraditional uses against viruses, cancer, and immunologic diseases. J Clin Invest (2021) 131(11):e148291. doi: 10.1172/JCI148291
4. Moorlag S, Arts RJW, van Crevel R, Netea MG. Non-specific effects of BCG vaccine on viral infections. Clin Microbiol Infect (2019) 25(12):1473–8. doi: 10.1016/j.cmi.2019.04.020
5. Jordan B, Meeks JJ. T1 bladder cancer: current considerations for diagnosis and management. Nat Rev Urol (2019) 16(1):23–34. doi: 10.1038/s41585-018-0105-y
6. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi: 10.1016/j.chom.2011.04.006
7. Giamarellos-Bourboulis EJ, Tsilika M, Moorlag S, Antonakos N, Kotsaki A, Domínguez-Andrés J, et al. Activate: Randomized clinical trial of bcg vaccination against infection in the elderly. Cell (2020) 183(2):315–323.e9. doi: 10.1016/j.cell.2020.08.051
8. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
9. Soto JA, Galvez NMS, Andrade CA, Ramirez MA, Riedel CA, Kalergis AM, et al. BCG Vaccination induces cross-protective immunity against pathogenic microorganisms. Trends Immunol (2022) 43(4):322–35. doi: 10.1016/j.it.2021.12.006
10. McShane H. Tuberculosis vaccines: beyond bacille calmette-guerin. Philos Trans R Soc Lond B Biol Sci (2011) 366(1579):2782–9. doi: 10.1098/rstb.2011.0097
11. Dalmia N, Ramsay AJ. Prime-boost approaches to tuberculosis vaccine development. Expert Rev Vaccines (2012) 11(10):1221–33. doi: 10.1586/erv.12.94
12. Nguipdop-Djomo P, Heldal E, Rodrigues LC, Abubakar I, Mangtani P. Duration of BCG protection against tuberculosis and change in effectiveness with time since vaccination in Norway: a retrospective population-based cohort study. Lancet Infect Dis (2016) 16(2):219–26. doi: 10.1016/S1473-3099(15)00400-4
13. White AD, Sibley L, Sarfas C, Morrison AL, Bewley K, Churchward C, et al. Influence of aerosol delivered BCG vaccination on immunological and disease parameters following SARS-CoV-2 challenge in rhesus macaques. Front Immunol (2021) 12:801799. doi: 10.3389/fimmu.2021.801799
14. Ahmed SM, Nasr MA, Elshenawy SE, Hussein AE, El-Betar AH, Mohamed RH, et al. BCG Vaccination and the risk of COVID 19: A possible correlation. Virology (2022) 565:73–81. doi: 10.1016/j.virol.2021.10.003
15. Hilligan KL, Namasivayam S, Clancy CS, O’Mard D, Oland SD, Robertson SJ, et al. Intravenous administration of BCG protects mice against lethal SARS-CoV-2 challenge. J Exp Med (2022) 219(2):e20211862. doi: 10.1084/jem.20211862
16. Kaufmann E, Khan N, Tran KA, Ulndreaj A, Pernet E, Fontes G, et al. BCG Vaccination provides protection against IAV but not SARS-CoV-2. Cell Rep (2022) 38(10):110502. doi: 10.1016/j.celrep.2022.110502
17. Witte G, Hartung S, Buttner K, Hopfner KP. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol Cell (2008) 30(2):167–78. doi: 10.1016/j.molcel.2008.02.020
18. Kalia D, Merey G, Nakayama S, Zheng Y, Zhou J, Luo Y, et al. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem Soc Rev (2013) 42(1):305–41. doi: 10.1039/c2cs35206k
19. Fu T, Zhao Y, Xi J. A new second messenger: Bacterial c-di-AMP. Crit Rev Eukaryot Gene Expr (2016) 26(4):309–16. doi: 10.1615/CritRevEukaryotGeneExpr.2016016642
20. Fahmi T, Port GC, Cho KH. C-di-AMP: An essential molecule in the signaling pathways that regulate the viability and virulence of gram-positive bacteria. Genes (Basel) (2017) 8(8):197. doi: 10.3390/genes8080197
21. Devaux L, Kaminski P-A, Trieu-Cuot P, Firon A. Cyclic di-AMP in host–pathogen interactions. Curr Opin Microbiol (2018) 41:21–8. doi: 10.1016/j.mib.2017.11.007
22. Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, et al. Two DHH subfamily 1 proteins in streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol (2013) 195(22):5123–32. doi: 10.1128/JB.00769-13
23. Yang J, Bai Y, Zhang Y, Gabrielle VD, Jin L, Bai G. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol Microbiol (2014) 93(1):65–79. doi: 10.1111/mmi.12641
24. Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med (2015) 21(4):401–6. doi: 10.1038/nm.3813
25. Dey RJ, Dey B, Zheng Y, Cheung LS, Zhou J, Sayre D, et al. Inhibition of innate immune cytosolic surveillance by an m. tuberculosis phosphodiesterase. Nat Chem Biol (2017) 13(2):210–7. doi: 10.1038/nchembio.2254
26. Zhang Y, Yang J, Bai G. Cyclic di-AMP-mediated interaction between mycobacterium tuberculosis DeltacnpB and macrophages implicates a novel strategy for improving BCG vaccination. Pathog Dis (2018) 76(2):fty008. doi: 10.1093/femspd/fty008
27. Ning H, Wang L, Zhou J, Lu Y, Kang J, Ding T, et al. Recombinant BCG with bacterial signaling molecule cyclic di-AMP as endogenous adjuvant induces elevated immune responses after mycobacterium tuberculosis infection. Front Immunol (2019) 10:1519. doi: 10.3389/fimmu.2019.01519
28. Zhang L, Li W, He ZG. DarR, a TetR-like transcriptional factor, is a cyclic di-AMP-responsive repressor in mycobacterium smegmatis. J Biol Chem (2013) 288(5):3085–96. doi: 10.1074/jbc.M112.428110
29. Tang Q, Luo Y, Zheng C, Yin K, Ali MK, Li X, et al. Functional analysis of a c-di-AMP-specific phosphodiesterase MsPDE from mycobacterium smegmatis. Int J Biol Sci (2015) 11(7):813–24. doi: 10.7150/ijbs.11797
30. Ning H, Liang X, Xie Y, Bai L, Zhang W, Wang L, et al. C-di-AMP accumulation regulates growth, metabolism, and immunogenicity of mycobacterium smegmatis. Front Microbiol (2022) 13:865045. doi: 10.3389/fmicb.2022.865045
31. Dey RJ, Dey B, Singh AK, Praharaj M, Bishai W. Bacillus calmette-guerin overexpressing an endogenous stimulator of interferon genes agonist provides enhanced protection against pulmonary tuberculosis. J Infect Dis (2020) 221(7):1048–56. doi: 10.1093/infdis/jiz116
32. Singh AK, Praharaj M, Lombardo KA, Yoshida T, Matoso A, Baras AS, et al. Re-engineered BCG overexpressing cyclic di-AMP augments trained immunity and exhibits improved efficacy against bladder cancer. Nat Commun (2022) 13(1):878. doi: 10.1038/s41467-022-28509-z
33. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG Educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell (2018) 172(1-2):176–90.e119. doi: 10.1016/j.cell.2017.12.031
34. Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth M, Hughes TK, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature (2020) 577(7788):95–102. doi: 10.1038/s41586-019-1817-8
35. Lu Y, Kang J, Ning H, Wang L, Xu Y, Xue Y, et al. Immunological characteristics of mycobacterium tuberculosis subunit vaccines immunized through different routes. Microb Pathog (2018) 125:84–92. doi: 10.1016/j.micpath.2018.09.009
36. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe (2018) 23(1):89–100 e105. doi: 10.1016/j.chom.2017.12.010
37. Joosten SA, van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE, et al. Mycobacterial growth inhibition is associated with trained innate immunity. J Clin Invest (2018) 128(5):1837–51. doi: 10.1172/JCI97508
38. Arts RJ, Blok BA, Aaby P, Joosten LA, de Jong D, van der Meer JW, et al. Long-term in vitro and in vivo effects of gamma-irradiated BCG on innate and adaptive immunity. J Leukoc Biol (2015) 98(6):995–1001. doi: 10.1189/jlb.4MA0215-059R
39. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun (2014) 6(2):152–8. doi: 10.1159/000355628
40. Smith SG, Kleinnijenhuis J, Netea MG, Dockrell HM. Whole blood profiling of bacillus calmette-Guérin-Induced trained innate immunity in infants identifies epidermal growth factor, IL-6, platelet-derived growth factor-AB/BB, and natural killer cell activation. Front Immunol (2017) 8:644. doi: 10.3389/fimmu.2017.00644
41. van Puffelen JH, Keating ST, Oosterwijk E, van der Heijden AG, Netea MG, Joosten LA, et al. Trained immunity as a molecular mechanism for BCG immunotherapy in bladder cancer. Nat Rev Urol (2020) 17(9):513–25. doi: 10.1038/s41585-020-0346-4
42. Arts RJW, Netea MG. Adaptive characteristics of innate immune responses in macrophages. Microbiol Spectr (2016) 4(4):MCHD-0023-2015. doi: 10.1128/microbiolspec.MCHD-0023-2015
43. Netea MG, Schlitzer A, Placek K, Joosten LAB, Schultze JL. Innate and adaptive immune memory: an evolutionary continuum in the host's response to pathogens. Cell Host Microbe (2019) 25(1):13–26. doi: 10.1016/j.chom.2018.12.006
44. Fanucchi S, Fok ET, Dalla E, Shibayama Y, Borner K, Chang EY, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet (2019) 51(1):138–50. doi: 10.1038/s41588-018-0298-2
45. Li D, Gao C, Zhao L, Zhang Y. Inflammatory response is modulated by lincRNACox2 via the NFkappaB pathway in macrophages infected by mycobacterium tuberculosis. Mol Med Rep (2020) 21(6):2513–21. doi: 10.3892/mmr.2020.11053
46. Huang S, Huang Z, Luo Q, Qing C. The expression of lncRNA NEAT1 in human tuberculosis and its antituberculosis effect. BioMed Res Int (2018) 2018:9529072. doi: 10.1155/2018/9529072
47. Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell (2013) 152(4):743–54. doi: 10.1016/j.cell.2013.01.015
48. Li J, Chen C, Ma X, Geng G, Liu B, Zhang Y, et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat Commun (2016) 7:11730. doi: 10.1038/ncomms11730
49. Adeva-Andany MM, Carneiro-Freire N, Seco-Filgueira M, Fernández-Fernández C, Mouriño-Bayolo DJM. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion (2019) 46:73–90. doi: 10.1016/j.mito.2018.02.009
50. Włodarczyk M, Druszczyńska M, Fol M. Trained innate immunity not always amicable. Int J Mol Sci (2019) 20(10):2565. doi: 10.3390/ijms20102565
51. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
52. Khader SA, Divangahi M, Hanekom W, Hill PC, Maeurer M, Makar KW, et al. Targeting innate immunity for tuberculosis vaccination. J Clin Invest (2019) 129(9):3482–91. doi: 10.1172/jci128877
53. Mahmoud L, Abdulkarim AS, Kutbi S, Moghrabi W, Altwijri S, Khabar KSA, et al. Post-transcriptional inflammatory response to intracellular bacterial c-di-AMP. Front Immunol (2019) 10:3050. doi: 10.3389/fimmu.2019.03050
54. Covian C, Fernandez-Fierro A, Retamal-Diaz A, Diaz FE, Vasquez AE, Lay MK, et al. BCG-Induced cross-protection and development of trained immunity: Implication for vaccine design. Front Immunol (2019) 10:2806. doi: 10.3389/fimmu.2019.02806
55. O'Neill LAJ, Netea MG. BCG-Induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol (2020) 20(6):335–7. doi: 10.1038/s41577-020-0337-y
56. Koeken V, Verrall AJ, Netea MG, Hill PC, van Crevel R. Trained innate immunity and resistance to mycobacterium tuberculosis infection. Clin Microbiol Infect (2019) 25(12):1468–72. doi: 10.1016/j.cmi.2019.02.015
57. Wei L, Liu K, Jia Q, Zhang H, Bie Q, Zhang B. The roles of host noncoding RNAs in mycobacterium tuberculosis infection. Front Immunol (2021) 12:664787. doi: 10.3389/fimmu.2021.664787
58. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
59. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep (2016) 17(10):2562–71. doi: 10.1016/j.celrep.2016.11.011
60. Gomes MTR, Guimarães ES, Marinho FV, Macedo I, Aguiar E, Barber GN, et al. STING regulates metabolic reprogramming in macrophages via HIF-1α during brucella infection. PloS Pathog (2021) 17(5):e1009597. doi: 10.1371/journal.ppat.1009597
61. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell (2018) 172(1-2):135–146 e139. doi: 10.1016/j.cell.2017.11.025
62. Crowther RR, Qualls JE. Metabolic regulation of immune responses to mycobacterium tuberculosis: A spotlight on l-arginine and l-tryptophan metabolism. Front Immunol (2020) 11:628432. doi: 10.3389/fimmu.2020.628432
63. Xu Y, Guo N, Dou D, Ran X, Liu C. Metabolomics analysis of anaphylactoid reaction reveals its mechanism in a rat model. Asian Pac J Allergy Immunol (2017) 35(4):224–32. doi: 10.12932/AP0845
64. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Jacobs C, Xavier RJ, et al. BCG-Induced trained immunity in NK cells: Role for non-specific protection to infection. Clin Immunol (2014) 155(2):213–9. doi: 10.1016/j.clim.2014.10.005
65. Irvine EB, O'Neil A, Darrah PA, Shin S, Choudhary A, Li W, et al. Robust IgM responses following intravenous vaccination with bacille calmette-guerin associate with prevention of mycobacterium tuberculosis infection in macaques. Nat Immunol (2021) 22(12):1515–23. doi: 10.1038/s41590-021-01066-1
66. Yang SJ, Chen YY, Hsu CH, Hsu CW, Chang CY, Chang JR, et al. Activation of M1 macrophages in response to recombinant TB vaccines with enhanced antimycobacterial activity. Front Immunol (2020) 11:1298. doi: 10.3389/fimmu.2020.01298
67. Ning H, Lu Y, Kang J, Xu Y, Wang L, Wang Y, et al. Establishment of mouse models of persistent tuberculosis and characteristics of that infection. J Pathogen Biol (2017) 12(03):219–23. doi: 10.13350/j.cjpb.170306
68. Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PE, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis (2014) 58(4):470–80. doi: 10.1093/cid/cit790
69. Dockrell HM, Smith SG. What have we learnt about BCG vaccination in the last 20 Years? Front Immunol (2017) 8:1134. doi: 10.3389/fimmu.2017.01134
70. Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, et al. Prevention of m. tuberculosis infection with H4:IC31 vaccine or BCG revaccination. N Engl J Med (2018) 379(2):138–49. doi: 10.1056/NEJMoa1714021
71. Keyser A, Troudt JM, Taylor JL, Izzo AA. BCG Sub-strains induce variable protection against virulent pulmonary mycobacterium tuberculosis infection, with the capacity to drive Th2 immunity. Vaccine (2011) 29(50):9308–15. doi: 10.1016/j.vaccine.2011.10.019
Keywords: Bacillus Calmette-Guérin, cyclic di-AMP, trained immunity, adjuvant, Mycobacterium tuberculosis
Citation: Ning H, Kang J, Lu Y, Liang X, Zhou J, Ren R, Zhou S, Zhao Y, Xie Y, Bai L, Zhang L, Kang Y, Gao X, Xu M, Ma Y, Zhang F and Bai Y (2022) Cyclic di-AMP as endogenous adjuvant enhanced BCG-induced trained immunity and protection against Mycobacterium tuberculosis in mice. Front. Immunol. 13:943667. doi: 10.3389/fimmu.2022.943667
Received: 14 May 2022; Accepted: 13 July 2022;
Published: 23 August 2022.
Edited by:
Wenping Gong, The 8th Medical Center of PLA General Hospital, ChinaReviewed by:
Anuradha Rajamanickam, International Centers for Excellence in Research (ICER), IndiaGuangcun Deng, Ningxia University, China
Erwan Pernet, McGill University Health Center, Canada
Oscar Taboga, Instituto Nacional de Tecnología Agropecuaria, Argentina
Copyright © 2022 Ning, Kang, Lu, Liang, Zhou, Ren, Zhou, Zhao, Xie, Bai, Zhang, Kang, Gao, Xu, Ma, Zhang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinlan Bai, yinlanbai@fmmu.edu.cn; Fanglin Zhang, flzhang@fmmu.edu.cn
†These authors have contributed equally to this work