 Joana M. Pereira
Joana M. Pereira Shuying Xu
Shuying Xu John M. Leong
John M. Leong Sandra Sousa
Sandra Sousa- 1i3S-Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto, Portugal
- 2Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal
- 3Molecular and Cellular (MC) Biology PhD Program, ICBAS - Instituto de Ciência Biomédicas Abel Salazar, University of Porto, Porto, Portugal
- 4Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, MA, United States
- 5Graduate Program in Immunology, Tufts Graduate School of Biomedical Sciences, Boston, MA, United States
Pneumolysin (PLY) is a pore-forming toxin produced by the human pathobiont Streptococcus pneumoniae, the major cause of pneumonia worldwide. PLY, a key pneumococcal virulence factor, can form transmembrane pores in host cells, disrupting plasma membrane integrity and deregulating cellular homeostasis. At lytic concentrations, PLY causes cell death. At sub-lytic concentrations, PLY triggers host cell survival pathways that cooperate to reseal the damaged plasma membrane and restore cell homeostasis. While PLY is generally considered a pivotal factor promoting S. pneumoniae colonization and survival, it is also a powerful trigger of the innate and adaptive host immune response against bacterial infection. The dichotomy of PLY as both a key bacterial virulence factor and a trigger for host immune modulation allows the toxin to display both “Yin” and “Yang” properties during infection, promoting disease by membrane perforation and activating inflammatory pathways, while also mitigating damage by triggering host cell repair and initiating anti-inflammatory responses. Due to its cytolytic activity and diverse immunomodulatory properties, PLY is integral to every stage of S. pneumoniae pathogenesis and may tip the balance towards either the pathogen or the host depending on the context of infection.
1 Introduction
Streptococcus pneumoniae (Sp or pneumococcus) is an extracellular Gram-positive bacterium that asymptomatically colonizes the upper respiratory tract of 5–10% healthy adults and 20–40% of children (1). Sp was considered the major cause of lower respiratory infections (LRI) with a global incidence of 2.7% worldwide, resulting in more than a million deaths per year and thus ranking among the deadliest bacteria (2). In the United States alone, pneumococcal pneumonia leads to over 150,000 annual hospitalizations. Other forms of Sp infectious complications such as otitis media, bacteremia, and meningitis are also significant healthcare burdens, leading to an average of 5 000 000, 4 000, and 2 000 cases/year respectively (source CDC: https://www.cdc.gov/pneumococcal/clinicians/clinical-features.html). Elderly and children under 5 are particularly susceptible to severe disease, with an invasive pulmonary disease (IPD) incidence of 7%. A major health concern and economic burden, Sp infection results in over $17 billion in direct medical costs annually in the US (3, 4).
Sp is naturally transformable and displays high genome plasticity, contributing to the rapid emergence of antibiotic resistance and evasion of vaccine-mediated immunity (1, 5). Effective Sp prevention and treatment is complicated by the appearance multidrug-resistant infections, non-vaccine serotypes and aging of the world’s population (4, 6). Approximately 30% of Sp cases involved isolates resistant to one or more antibiotics (https://www.cdc.gov/drugresistance/pdf/threats-report/strep-pneumoniae-508.pdf). Worldwide, pneumococci resistant to penicillin, erythromycin, and trimethoprim-sulfamethoxazole are on the rise (7). To a lower extent, resistance to tetracycline, chloramphenicol and fluoroquinolone have also been identified (8). While in the United States, multi-drug resistant pneumococci prevalence has been reduced since introduction of pneumococcal vaccines, the risk remains high in susceptible populations, especially individuals aged 65 and over (9). Vaccination has been the cornerstone of pneumococcal disease prevention. However, currently, the two classes of pneumococcal vaccines, the 23-valent pneumococcal polysaccharide vaccine (PPSV23) and the pneumococcal vaccines based on protein-conjugated polysaccharides (PCV13/PCV23), only protect against a subset of over 90 different pneumococcal capsular variants (capsular serotypes) (10). Through both the expansion of pre-existing non-vaccine pneumococcal serotypes and serotype ‘switching’, an exchange of capsular polysaccharide genes through transformation, infectious strains not covered by standard vaccination are on the rise (11). As the efficacy of traditional antibiotics and vaccines become compromised, understanding the mechanisms of action of pneumococcal virulence determinants is of critical importance for the development of new therapeutics.
Sp transmits predominantly via aerosol as the bacteria is harbored in the nasopharynx. Asymptomatic Sp carriage can lead to localized infection of tissues contiguous with the nasopharynx, causing sinusitis, otitis media, and pneumonia (1). The bacterium is also capable of subsequent systemic spread to the heart and brain upon access to the bloodstream, causing serious diseases such as cardiac dysfunction and meningitis (1) (Figure 1). The versatility of Sp is reflected by the diversity of interactions with its host depending on the site of infection and degree of disease. To colonize the host nasopharynx, Sp forms biofilms on the mucosa of the upper respiratory tract (12). This causes mucosal inflammation which promotes bacterial shedding in secretions leading to transmission (13). Long-term asymptomatic nasopharyngeal carriage supports inflammation-induced transmission and predisposes the host to develop disease (Figure 1A). Sp can then spread from the nasopharynx to neighboring tissues, such as the sinusoidal cavities to cause sinusitis, the middle ear to cause otitis media, or the eye to cause keratitis (Figures 1B, C), all of which can be recurrent infections especially in immunocompromised patients (14). Sp aspiration into the lower respiratory tract causes pneumonia, where infection damages alveolar epithelial and endothelial cells promoting tissue permeability and induces thrombotic events (Figure 1D). If the immune response mounted in the lungs is not sufficient to eliminate the bacteria, Sp may gain access to the bloodstream and disseminate to cause IPD (Figures 1E–G). During IPD, Sp may invade the spleen (15), the heart (16–18), and/or may cross the blood-brain barrier (19). In the heart, Sp forms biofilms and damages both cardiomyocytes and immune cells, leading to myocardial dysfunction and long-term pneumonia-associated adverse cardiac events (PACE; Figure 1F). Sp-induced remodeling of brain tissue during meningitis likely accounts for permanent neurological damage reported in about 50% of the survivors (Figure 1G).
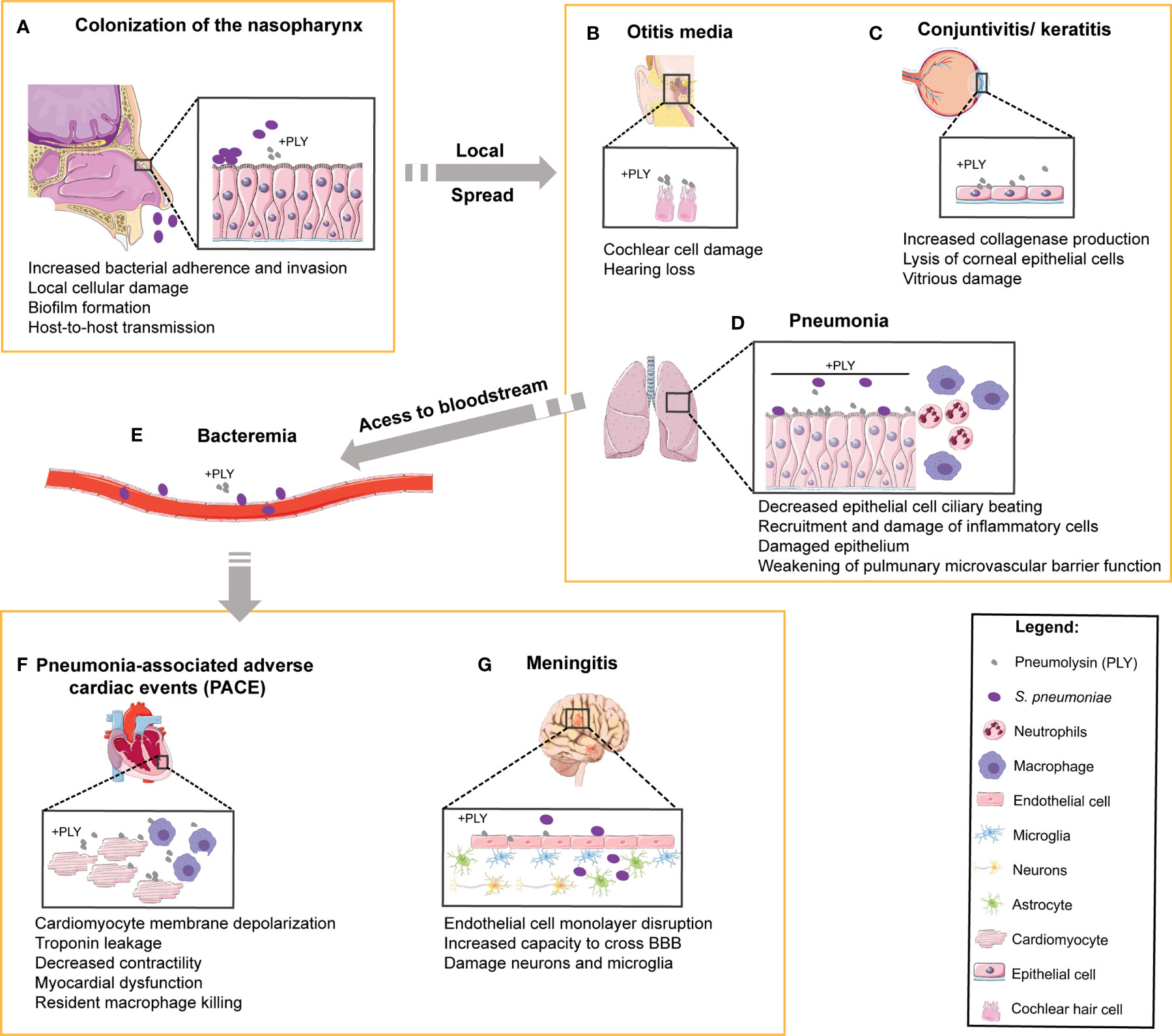
Figure 1 Role of PLY in human pneumococcal pathogenesis and in the targeting of multiple organs. In the multiple steps of pathogenesis during human infection by S. pneumoniae, the target organs and the cell types involved are indicated and the reported functions of PLY are listed below each step. (A) S. pneumoniae colonizes the human upper respiratory tract. Prolonged colonization of the nasopharynx may favor bacterial spread to neighboring tissues such as (B) the middle ear, causing otitis media and massive damage of cochlear hair cells, and (C) the eyes, leading to the development of keratitis and endophthalmitis. Upon aspiration, S. pneumoniae can reach (D) the lower respiratory tract and cause pneumonia. In the lungs, PLY causes the dysfunction of epithelial barrier, facilitating bacterial access to (E) the bloodstream, where PLY diminishes phagocytosis and disrupts endothelial/epithelial cells to promote tissue invasion. The bacteria are then able to infect (F) the heart, causing pneumonia-associated adverse cardiac events (PACE) by targeting the cardiomyocytes and resident macrophages, and (G) the brain, causing damage of both neurons and microglia.
Integral to all stages of Sp pathogenesis is the host-pathogen interaction mediated by pneumolysin (PLY), a key virulence factor produced by almost all clinical isolates and currently known Sp serotypes (20). PLY can promote asymptomatic carriage, colonization, transmission, immune evasion and dissemination, highlighting the multifaceted functions of this bacterial toxin. As a member of the cholesterol-dependent cytolysin (CDC) family, PLY is a pore-forming toxin (PFT) that at lytic concentrations disrupts host cell plasma membrane (PM) and promotes the uncontrolled influx and efflux of ions, small molecules, and proteins (21). PLY-induced pore formation interferes with a plethora of cellular signal transduction pathways. Depending on PLY concentration, which dictates the extent of PM damage and intracellular Ca2+ overload, host cells respond by activation of either cell death or cell survival and damage repair pathways. At lytic levels, PLY-induced pores can overwhelm cellular homeostatic mechanisms, triggering both cellular proinflammatory signaling pathways and irreversible cellular injury that results in the release of molecules heightening inflammation. The ensuing tissue damage is integral to pneumococcal disease (22). For example, in the lung, exacerbated inflammation promotes tissue invasion and systemic spread (21, 23). In contrast, at the sub-lytic concentrations likely present during the early stages of infection, PLY stimulates cell-autonomous repair mechanisms that overcome PM damage, restore cell integrity, and promote cell survival (24, 25), curtailing inflammation. In addition, depending on infection dose, site, disease stage, target cell, and the immune state of the host, PLY may display either “Yin” or “Yang” components, each with potentially opposing effects on Sp pathogenesis. The Yin of PLY promotes disease by promoting cell damage either directly or by excessive inflammation. The Yang of PLY mitigates the damage by triggering host repair mechanisms and modulating inflammation, allowing the carriage of Sp without symptoms. The remarkable ability of PLY to trigger both “Yin” and “Yang” responses depending on infection context is showcased in several in vitro and in vivo infection models (26, 27). Interestingly, heterogeneous expression of PLY within individual bacteria of an isogenic Sp population favors the appearance of subpopulations expressing different levels of PLY. Recent studies showed that genetically modified PLY expressing Sp strains producing different levels of PLY triggered different outcomes during infection. Specifically, high-PLY producing bacteria induced extensive autophagosome damage allowing efficient clearance of Sp from the host cells, yet, low-PLY producing bacteria facilitated evasion of host defense mechanism and promoted the cross of the blood-brain barrier (28, 29). Furthermore, rapid and high levels of PLY release drives hypervirulence in infection by serotype 1 pneumococci (30, 31), showcasing that the plasticity of PLY expression during Sp pathogenesis is fundamental and can influence the outcome of the infection.
Here we provide a comprehensive review of PLY functions and interactions with host cells by describing the mechanism of pore formation, its multiple targets and effects on host cells, and its antipodal interactions with the host immune system. We summarize the available data and recent discoveries that highlight the “Yin” and “Yang” perspectives of PLY activity contributing to Sp disease progression, unveiling the myriad and sometimes conflicting properties of this fascinating bacterial toxin.
2 Structural and Functional Insights of Pneumolysin
2.1 PLY Export
Secreted PLY is key for infection; however, the localization and export mechanism of PLY is still debated (32). PLY can be detected in the bacterial cytosol (32, 33), non-covalently attached to the cell wall (32), in culture supernatants (32), and in circulation during Sp infection (34). Given the lack of a canonical N-terminal signal peptide that promotes protein secretion, PLY was thought to be released solely by autolysis, an idea supported by experimental data showing reduced PLY release in the presence of non-bacteriolytic antibiotics (35). Nevertheless, autolysis may not be the only mechanism for PLY secretion (32, 36). In fact, although attenuated for virulence, Sp strains lacking autolysin release wild-type levels of PLY when cultured in vitro (37, 38). In addition, a functional SecA2 system is required for the non-covalent association of PLY to cell wall peptidoglycan (39), a structure that can hinder PLY delivery to the bacterial surface (40). Indeed, enzymatic digestion of Sp peptidoglycan promotes PLY release and is detrimental for virulence (40). Thus, PLY secretion is likely determined by several mechanisms that may cooperate to foster disease (31).
2.2 Mechanism of PLY-Mediated Membrane Permeabilization
PLY shares structural and functional properties with other bacterial PFTs belonging to the CDC family, such as listeriolysin (LLO), perfringolysin (PFO), and streptolysin (SLO) (41). It binds to cholesterol residues at the PM of host cells, oligomerizes, and undergoes conformational changes to form stable pores with well-defined sizes (Figure 2).

Figure 2 Mechanisms of PLY-mediated host plasma membrane permeabilization and conformational changes associated to pore formation. (A) PLY pore-formation is a multi-step process. PLY is released by S. pneumoniae as a water-soluble monomer (1) which specifically bind to cholesterol residues on the host cell plasma membrane (2). PLY monomers oligomerize by interacting with each other to form the early pre pore complex (3), which protrudes into the membrane surface establishing the late pre-pore (4). Finally, PLY inserts hairpins HP1 and HP2 across the membrane forming an open transmembrane channel, which allows the uncontrolled influx and efflux of ions and small molecules (5). (B) Soluble PLY. The 3D crystal structure of PLY monomer as it is released from S. pneumoniae is shown. The 4 major domains, from D1 to D4, as well as Helix Bundles (HB) 1 and 2 and cholesterol binding loop are indicated. The arrow indicates residues T459 and L460 in the D4 Trp-rich loop which are essential for cholesterol recognition. (C) PLY in pre-pore complex. Structure of PLY upon cholesterol binding via the conserved D4 Trp-rich loop. Interaction with cholesterol induces a 90° rotation of D2 (in yellow, indicated by the curved arrow) bringing D1and D3 towards the host plasma membrane. HB1 and HB2 are positioned just above the host membrane. This structural organization is maintained in the pre-pore stage. (D) PLY in the transmembrane pore. The 3D structure of PLY when inserted in the host cell plasma membrane is depicted. HB1 and HB2 refold into 85 Å β -hairpins HP1and HP2 (shown by the curved arrow), which insert (red circle) and cross the hydrophobic membrane to form an open transmembrane pore. Adapted from (42).
To generate pores, soluble PLY interacts with the PM, multimerizes into two successive “prepore” complexes, and then inserts into the PM to generate a large pore (Figure 2A). Soluble PLY, in its monomeric state, has been shown by crystallographic methods to be an asymmetric molecule composed of 4 major domains (D1 to D4), including α-helices and β-sheets (Figure 2B). The N-terminal D1 domain, although not essential for cell binding, stabilizes overall protein structure and enables hemolytic activity (42, 43). The non-contiguous D1 and D3 are adjacent in the 3D crystal structure and linked to the C-terminal of D4 via D2 (44) (Figure 2B). Blocking monoclonal antibodies and the analysis of binding-defective point mutants indicate that D4 promotes membrane binding (21, 45–49). In particular, two residues (T459 and L460) in a conserved D4 undecapeptide that comprises a Trp-rich loop are essential for cholesterol recognition (21, 41, 50, 51) (Figure 2B, arrow). In addition, the recombinant PLY toxoid B “PdB” harboring a mutation in Trp 433 in D4 is unable to undergo the conformational changes required for membrane insertion and strains expressing this mutant show reduced virulence, further reinforcing the role of D4 in membrane binding and pore formation (13, 34).
PLY monomers multimerize, forming transient pre-pore asymmetric structures with variable diameters and composed of 40 to 44 monomers (Figure 2A) (44, 46, 52–56). D2 undergoes a 90° rotation, bringing D1 and D3 towards the PM, compacting the overall structure and establishing the late pre-pore (Figures 2A, C) (21, 53). PM insertion occurs when the D3 helix bundles (HB1 and 2) are refolded into 85A° β-hairpins (HP1 and HP2) that perforate the hydrophobic membrane and assemble into an approximately 260A° diameter transmembrane channel (Figure 2D) (52, 53). Mutations in D3 residues Y150 and T172 are found in a naturally occurring non-hemolytic variant of PLY (45, 57), pointing to their critical role in the conformational alterations required for transmembrane hairpin formation, PM insertion and subsequent pore-formation (45). Although it is unclear what advantage Sp derives from loss of PLY hemolytic activity, serotypes harboring naturally occurring non-hemolytic PLY are commonly associated with non-lethal respiratory tract infections (57) pointing to roles played by non-pore formation dependent mechanisms of PLY during Sp pathogenesis.
Additionally, it has been reported that PLY monomers may be trapped in arcs or slit-shaped oligomers (21) that have the ability to generate pores (58, 59), possibly with variable sizes, different permeabilities and distinct functional roles (55, 60). At low PLY concentrations, likely occurring at early stages of infection, complete prepore ring structures are expected to form less efficiently than arcs, and so pore formation may rely on the efficiency of the conversion of arc oligomers to pores. At high PLY concentrations, potentially reached later in infection, binding of soluble PLY to PM is efficiently followed by assembly of full rings, so cell lysis mainly depends on the PLY affinity for cholesterol (60). PM disruption following arc or full ring assembly possibly dictates different host responses according to the phase of infection and could influence the balance of Yin and Yang outcomes.
2.3 PLY as a Ligand for Host Molecules
In addition to binding cholesterol, PLY interacts with several other host molecules. Such interactions contribute to its functions as a both pro- and anti-inflammatory agent, further discussed in Section 4 below. First, through its D4 domain, PLY binds the mannose receptor C type 1 (MRC-1), expressed at the cell surface of multiple immune cell types in the airways (61). MRC-1 acts as an internalization receptor, allowing Sp to invade MRC-1-expressing dendritic cells (DCs) and alveolar macrophages. At low infectious doses when immune stimulation by bacterial PAMPs is limited, MRC-1-PLY interaction and the consequent Sp phagocytosis modulate inflammation (discussed in section 4.2.2) and promote intracellular bacterial survival in the lungs in a murine pneumococcal pneumonia model (61). Second, PLY also has the capacity to modulate the interaction of Sp with complement by functioning as a molecular decoy, binding the Fc portion of human IgG and triggering C1q recruitment and complement cascade activation (62, 63) (reviewed in section 4.2.4). This interaction is dependent on specific residues within short non-contiguous PLY sequences that are homologous to the human C-reactive protein (CRP), which also activates the complement (63). In vivo, diversion of complement proteins by PLY promotes both pulmonary and systemic infection, as PLY-deficient Sp become more virulent in complement-deficient mice (64). Lastly, PLY was suggested to bind host cell glycans such as sialylated fucosylated glycan divalent-LewisX (sLeX) (65), and computational docking studies suggested the D4 and the interface between D3 and D4 as putative binding sites for LeX and sLeX, respectively (46). Nevertheless, interactions detected in vitro utilized high sLeX : PLY ratios and thus appear to be of low affinity, and PLY:sLeX co-crystals have never been observed (46), indicating the need for further studies are needed to explore potentially biologically interactions interaction PLY and sLeX.
2.4 PLY-Mediated Pathogenesis and Targeted Cells
Due to its ability to bind to cholesterol commonly found in mammalian PMs, PLY is able to target and modulate the function of virtually all cell types and thus plays key roles in many modes of infection, such as asymptomatic carriage, local disease, or life-threatening systemic disease (66) (Figure 1).
Early in infection, PLY has vital roles in colonization of the nasopharynx and in host-to-host transmission (13, 67, 68) (Figure 1A). In vitro, PLY-deficient mutants are impaired in adherence (69) and show lower bacterial burden in the nasopharynx of intranasally infected mice (67, 68). Sp grown in vitro as a biofilm show increased ply transcription and produced high levels of PLY (70, 71), suggesting that Sp biofilms developed in the respiratory mucosa might also produce high levels of PLY. The PLY-induced mucosal inflammation promotes increased bacterial shedding in secretions and promotes transmission in an infant mouse model of Sp infection (13).
PLY is also a key molecule in the progression of Sp infection from the nasopharynx to neighboring tissues. A PLY-deficient mutant causes mild histopathological changes and lower middle ear bacterial loads in chinchilla OM model (72, 73). In guinea pig infection models, PLY directly damages cochlear hair cells (74, 75). In addition, PLY intracochlear perfusion induces a strong cytotoxic effect possibly related to pore formation at the PM of inner hair cells (76, 77) (Figure 1B). PLY also plays a role in pneumococcal ocular infections causing endophthalmitis and keratitis (78, 79). Intravitreally injection of PLY causes inflammation and tissue damage (80, 81), likely related to PLY-induced corneal epithelial cell lysis (82) (Figure 1C).
In the lungs, PLY targets pivotal cell types during acute injury at the early phases of pneumonia and becomes a potent inducer of inflammation, facilitating damage of the respiratory epithelial barrier and host tissue penetration (Figure 1D). Purified PLY damages bronchial and alveolar epithelium and slows human ciliary beating in vitro (83); it damages alveolar epithelial and endothelial cells impairing barrier function (84, 85); and causes platelet destruction thus inhibiting platelet thrombus formation (86, 87). Anti-PLY polyvalent antibodies were reported to inhibit PLY-mediated platelet annihilation and were proposed to be of pharmacological interest to treat Sp community-acquired pneumonia (87). In addition, PLY can compromise effective immune response in the lungs through several mechanisms: it induces the necroptosis of alveolar macrophages (88–90), triggers caspase-dependent cell death of dendritic cells (91, 92), promotes neutrophil transepithelial cell migration and elastase release, thus impairing macrophage phagocytosis, and damages epithelial cells (93–95). The pro-inflammatory role of PLY is discussed in detail in section 4.
If not controlled in the lungs, the bacteria can reach the bloodstream and cause IPD (Figure 1E). In the heart, sub-lytic doses of PLY disrupt the PM of cardiomyocytes leading to the influx of Ca2+, membrane depolarization and induction of ER stress, which together cause alterations in cardiac rhythm and depression in contractile force. At higher concentrations, PLY causes cardiomyocyte cell death, inhibiting cell contractility and promoting extensive cardiac damage which may lead to microlesion formation and cardiac remodeling likely associated with long-term adverse cardiac events described in IPD patients (34, 96, 97) (Figure 1F). Sp biofilms, established in the heart of infected mice, release PLY causing rapid macrophage killing and impairing cytokine production (98).
During brain infection, PLY induces neuronal cell death (99, 100) and targets astrocytes, rearranging cytoskeleton and altering astrocyte cell shape, which leads to remodeling of brain tissue, astrocytic retraction, and cortical astroglial reorganization (101–103) (Figure 1G). PLY-dependent astrocyte cell death is mediated by connexin 43 (Cx43), a gap junction protein forming hemichannels, that amplifies ATP release and cytosolic Ca2+ influx, ultimately leading to astrocyte depletion and blood-brain barrier destabilization (100). PLY also targets other cells in the brain promoting endothelial cell disruption (104), limiting microglia motility (105), and decreasing cilia beating frequency in ependymal cells (106–108), which correlates with loss of cilia and damage of ependymal ultrastructure described in rat meningitis model (109).
3 PLY Triggers Multiple Cellular Responses: Irreversible Damage or Repair
Upon interaction with cells, PLY interferes with a plethora of signal transduction pathways to induce multiple and antipodal cellular responses that define the pathogenesis in a whole organism (Figure 3). Such responses are cell-type specific and are dependent on PLY concentration, which correlates with the extent of the PM damage and of the intracellular Ca2+ overload. Activation of different cellular responses can tip the balance towards activation of cell death or cell survival pathways, thus promoting tissue damage or repair, respectively. At lytic concentrations, PLY triggers irreversible damage and cell death by causing massive mitochondrial damage, excessive inflammation, and tissue injury. In contrast, at sub-lytic amounts, PLY activates repair pathways triggering PM remodeling, cytoskeleton reorganization, and, by transiently activating MAPK, cell survival.
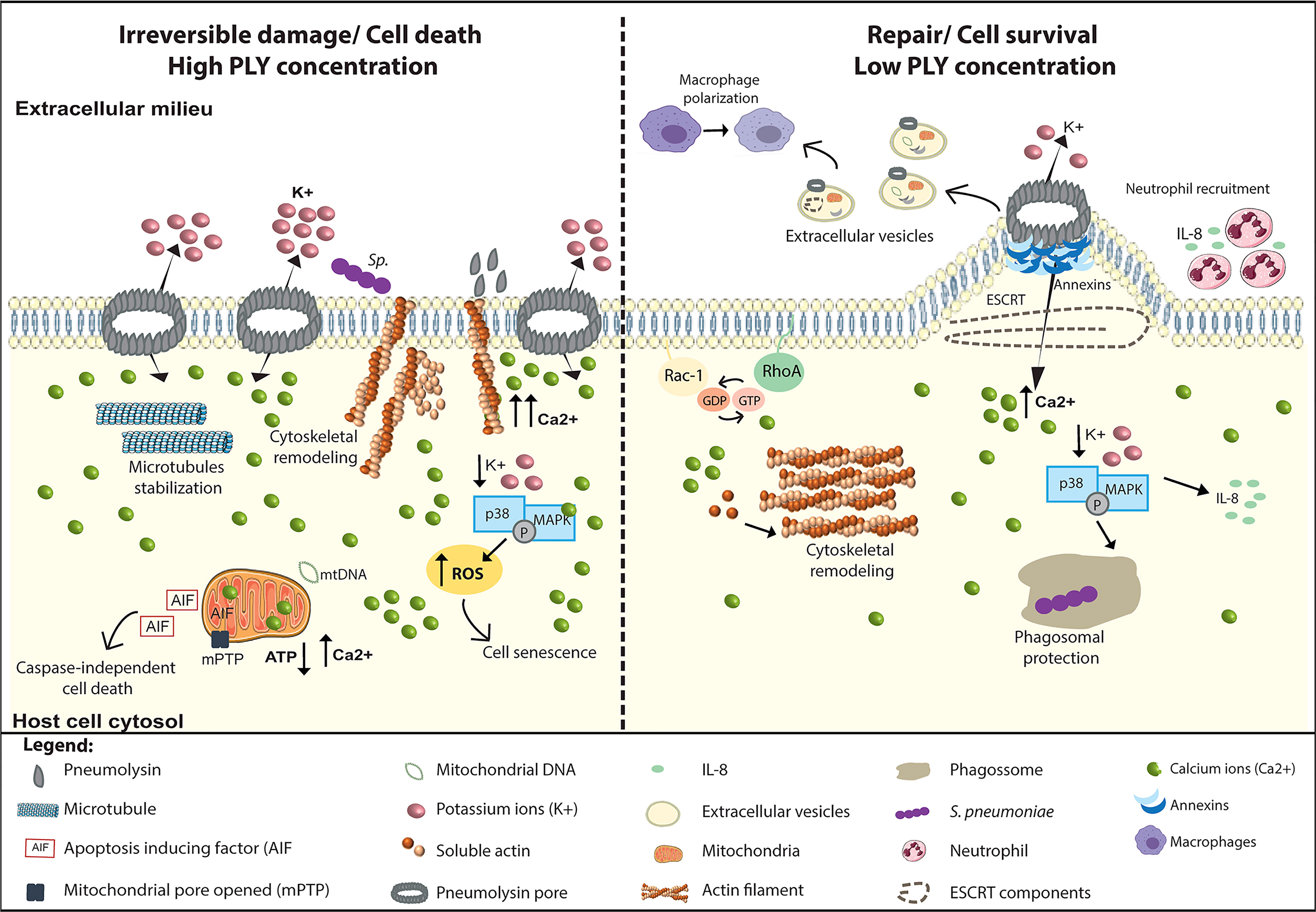
Figure 3 PLY is a trigger for multiple cellular responses. PLY interacts with cells and, depending on PLY concentration and the intracellular Ca2+ levels, induces a variety of antipodal cellular responses that can lead to irreversible damage or the induction of cellular repair mechanisms. Left Panel: At lytic amounts, the overwhelming increase in intracellular Ca2+ levels induces the surface exposure of actin which facilitate Sp adhesion and invasion, increasing cell death. In addition, PLY-mediated microtubule stabilization may perturb axonal transport, likely contributing neuronal damage. Also, in neuronal cells, p38/MAPK activation is detrimental for the host cell as it increases ROS production and induces senescence. High PLY concentrations cause irreversible mitochondrial damage by inducing swelling, loss of mitochondrial membrane potential, and morphologic and metabolic alterations. Concomitantly with Ca 2+ overload, mitochondrial permeability increases, the ATP levels decrease, and mitochondrial DNA is released into the cytosol. Following these events, the mitochondrial apoptosis-induced factor (AIF) reaches the cytoplasm and activates caspase-independent cell death. Right Panel: At sub-lytic amounts, the influx of limited amounts of extracellular Ca2+ triggers the sequential recruitment of annexins to the damaged sites where they assemble in 3D arrays to clog the PM pore. Increased intracellular Ca2+ also induces cytoskeleton remodeling through the activation of small GTPases Rac1 and RhoA and triggers PM rearrangements culminating in PM blebbing and ESCRT-mediated release of microvesicles containing PLY, annexins, actin-binding and Ca2+ regulated proteins, ESCRT components and mitochondrial DNA among others. Released microvesicles promote survival by eliminating the pore, transporting danger signals and enhancing immune responses. In response to K+ efflux cell survival pathways such p38/MAPK are activated and stimulate the production of pro-inflammatory cytokines such as IL-8, promoting neutrophil recruitment, and enhancing phagosomal integrity, thus limiting the release of toxic bacterial components into the cytosol.
3.1 Mitochondrial Damage
The role of PLY in mitochondrial damage was first described in primary rat neurons infected with Sp or incubated with culture supernatants, which both induce the Ca2+-dependent mitochondrial release of apoptosis-inducing factor (AIF), a likely manifestation of massive mitochondrial damage (Figure 3, left) (110). In addition, PLY was shown to trigger mitochondrial swelling, loss of mitochondrial membrane potential, and impairment in mitochondrial metabolism (111, 112). Electron microscopy analysis of PLY-intoxicated neurons show direct binding of PLY to the mitochondrial membrane (111). In human alveolar epithelial cells, PLY also induces dramatic morphological alterations of the mitochondria, which are accompanied by cytosolic Ca2+ overload, reduction in ATP levels, membrane depolarization, increased mitochondria permeability and release of mitochondrial DNA (mtDNA) into the cytosol (Figure 3, left) (112). mtDNA can then be released extracellularly through microvesicle shedding and act as a danger signal, thus contributing to inflammation (Figure 3, right) (112). In macrophages, cytosolic mtDNA released by PLY-damaged mitochondria is recognized by STING and upregulates the expression and secretion of IFN-β, demonstrated both in vitro and in vivo in lungs of Sp infected mice (113).
3.2 Interactions With the Host Cytoskeleton
PLY was reported to bind and promote actin polymerization in vitro (114) and was shown to interact with actin filaments underneath the PM of astrocytes (115) or exposed at the surface of damaged neuronal cells (99) (Figure 3, left). In fact, PLY D4 domain interacting with cholesterol in the PM of neurons enables β-actin exposure to the outer surface of the cell, facilitating Sp adhesion and invasion and increasing cell death (99). In addition, in those cells, PLY induces cytoskeleton instability depolymerizing intracellular actin filaments likely due to increased Ca2+ levels (99). How these data relate to the PLY intrinsic ability to polymerize actin in vitro needs further investigation, and an understanding of the consequence of PLY-mediated Sp interactions with the actin cytoskeleton may require kinetic analysis of the infection in relevant models.
PLY also bundles and stabilizes host cell microtubules during intoxication of neuronal cells and during pneumococcal meningitis in rabbit infection models (Figure 3, left) (102). Consistent with increased microtubule stabilization, high levels of acetylated tubulin were found in both the cell culture and animal model (102). PLY-triggered microtubule stabilization is comparable to pharmacological microtubule-stabilizing agents (e.g., taxol) and has been suggested to perturb axonal transport, likely contributing to neuronal damage during pneumococcal meningitis (102). PLY-induced microtubule perturbations are dependent on cholesterol binding and require Src kinase activity but are independent of Ca2+ influx and actin remodeling (102).
Several reports, primarily in neuronal cells, suggest that at low concentrations, PLY interaction with host PM causes rapid cytoskeleton remodeling which translates into cell shape changes (Figure 3, right). Alterations in the brain structure detected in infected rats were associated with PLY-mediated astrocytes retraction and reshaping of focal adhesions, which are underlined by cytoskeleton reorganization (101, 103). In primary mouse astrocytes and neuronal cells, sub-lytic concentrations of PLY binding to cholesterol promotes the formation of actin stress fibers, filopodia, and lamellipodia through the activation of RhoA and Rac1 GTPases (Figure 3, right) (114, 115).
3.3 Activation of Cell Survival Pathways
PLY binding to the PM triggers activation of cell survival pathways such as the mitogen-activated protein kinase p38 (p38/MAPK) (116, 117), which is a conserved response to sub-lytic doses of PFTs dependent on K+ efflux (118), that can stimulate cell survival pathways (117). In epithelial cells, activation of p38/MAPK by sub-lytic concentrations of PLY (119, 120) triggers the production of pro-inflammatory cytokines (e.g., IL-8) to attract neutrophils, thus promoting an effective immune response early in infection when bacterial numbers are low (Figure 3, right) (119). In macrophages, PLY-induced p38/MAPK activation and cytokine production take place at the PM upon pore formation (119, 120) and has been suggested to promote Sp clearance (121). Later in infection, following Sp uptake by macrophages, PLY induces the trafficking of pneumococcal cell wall components to the host cell cytosol, presumably through phagosomal damage (122). If p38/MAPK activation is blocked, leakage of Sp cell wall components to the host cell cytosol is exacerbated and results in macrophage cell death. Thus, PLY-induced p38/MAPK activation protects phagosomal integrity and limits the release of bacterial components, likely modulating the recognition of pathogen-associated molecular patterns by the host surveillance systems (Figure 3, right) (122, 123).
The timing of the MAPK response to PFTs can influence tissue injury. Although MAPK activation can promote cell survival responses upon intoxication by PFTs, its subsequent modulation by protein phosphatases PP1 and PP2A, observed in epithelial cells (120), can prevent excessive inflammatory responses that could lead to irreversible tissue damage. Conversely, the transient nature of p38/MAPK activation might not be sufficient to block cell death triggered by the recognition of Sp components in the macrophage cytosol (122).
Finally, in some cell types, activation of p38/MAPK is detrimental. In SH-SY5Y neuronal cells, p38/MAPK is associated with increased neuronal cell death and neurotoxic effects (124), and in microglial, PLY-mediated MAPK activation increases ROS production and promotes senescence (Figure 3, left) (125).
3.4 Activation of Plasma Membrane Repair Mechanisms
Supernatants from stationary phase cultures of Sp produce sufficient amounts of PLY to permeabilize cells (126); however, during infection, the majority of these perforated cells are able to recover from damage and survive. PLY pore assembly renders host cell PM permeable to ions and small molecules (116, 127, 128). An increase of Ca2+ concentrations to above 20 μM impairs host-cell signaling and engages cell death pathways (116, 129). However, an initial increase in intracellular Ca2+ levels act as a danger signal and activates PM repair mechanisms to prevent cell lysis (119, 124, 130–132). Thus, at otherwise sub-lytic PLY concentrations, reduced extracellular Ca2+ enhances PLY toxicity and result in lysis (133, 134). In the presence of extracellular Ca2+, PLY-induced Ca2+ influx triggers the recruitment of annexins, cytoplasmic Ca2+ responsive proteins that bind to negatively charged phospholipids at the sites of PM injury (135). Annexin A2, which displays the highest Ca2+ sensitivity, is the first to translocate to the site of damage, followed by annexin A6 and A1 (Figure 3, right). Annexin translocation fosters the formation of plasmalemmal nanotubes at the pore site that culminates in the release of microvesicles enriched in PLY, annexins, actin-binding and Ca2+ regulated proteins, and ESCRT components (Figure 3, right) (126, 134), thus shedding the PLY pore and modulating the rise in intracellular Ca2+ (129). This repair process has potential immunological implications for Sp infection, as PLY-containing microvesicles induce macrophage polarization that enhances the immune response towards molecular patterns of Gram-positive bacteria (136). By contrast, in alveolar epithelial cells, human lung explants and infected mice, PLY mediates the release of microvesicles containing mitochondrial cargo that, when uptake by neutrophils suppress their ability to release ROS and thus impair efficient immune response against Sp (137). The differential PLY sensitivity of immune cells is thought to be due to differential efficiency in PM repair (138). Myeloid cells, the first-line defenders, show enhanced shedding of PLY-containing microvesicles and increased resistance to PLY. In contrast, lymphoid cells are enriched in cholesterol-containing lipid rafts, which facilitate PLY binding and pore formation, and are impaired in microvesicles formation and PM resealing activity, leading to high PLY susceptibility (138).
4 PLY Interactions With the Immune System: Pro- or Anti- Inflammatory Responses
PLY can either exacerbate or mitigate damage during infection depending on the state of the immune system as well as the infection site, dose, timing, and interacting host cell type (26, 139). It can trigger inflammation-mediated tissue damage and promote bacterial dissemination (18, 140, 141). However, PLY can elicit host-protective responses in the innate and adaptive branches of the immune system, including activation and stimulation of cytokine production by macrophages, neutrophils, endothelial, epithelial, and dendritic cells (22, 142–144). The multifaceted nature of PLY-induced immune responses may account for some of the disparate outcomes of infection by PLY-deficient strains in different infection models (Figure 4 and Table 1). This also reflects the dual relationship of Sp with host cells, sometimes invasive and inducing severe disease, and other times remaining localized as an asymptomatic colonizer (as discussed in sections 1 and 2.4.).
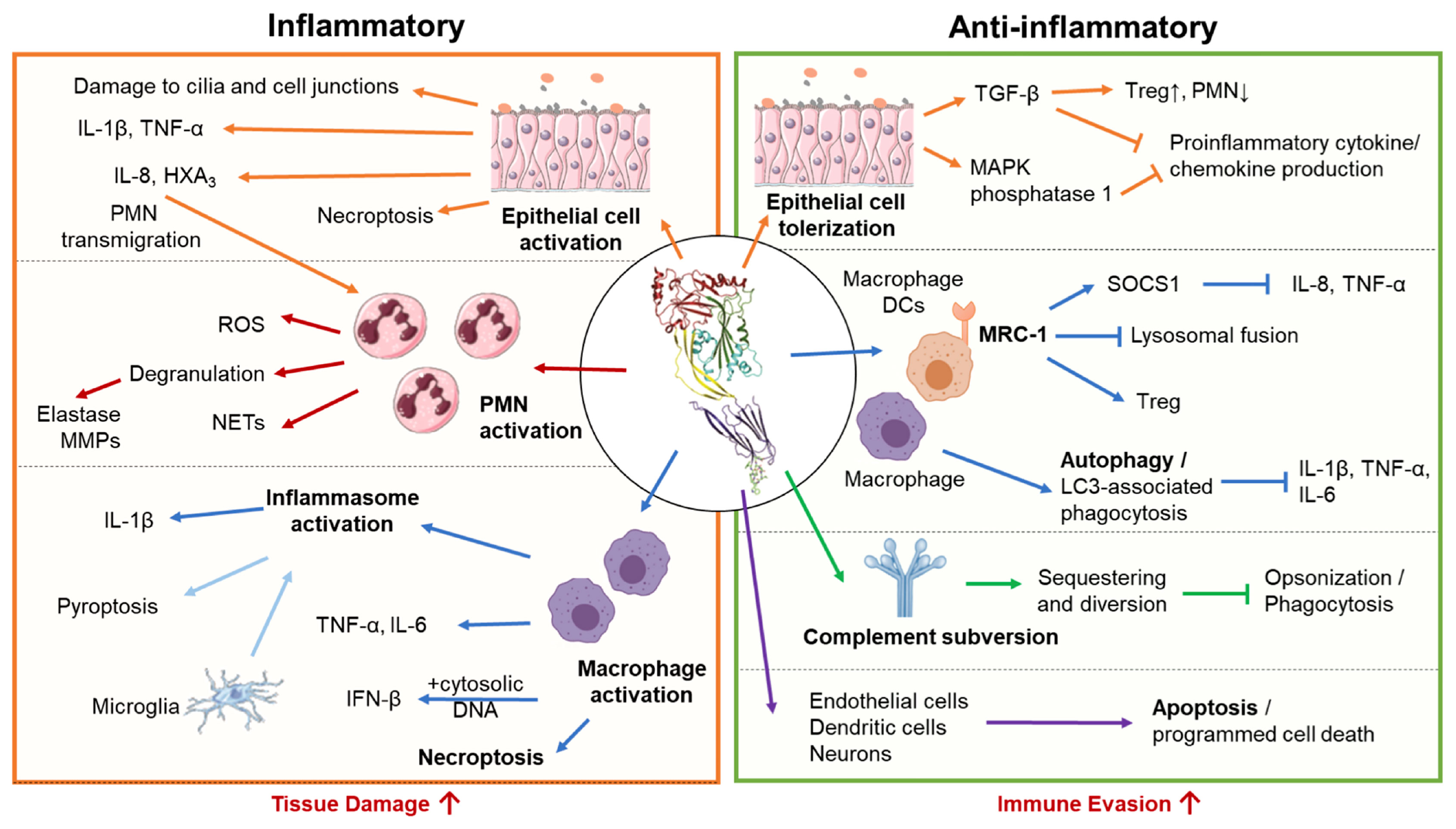
Figure 4 Pro-inflammatory and anti-inflammatory host responses to PLY. S. pneumoniae triggers both pro-inflammatory and anti-inflammatory responses depending on interacting host cell type and infection context. Left Panel: In excess, pro-inflammatory actions of PLY enhance tissue damage and promote bacterial spread. In epithelial cells, PLY induces production of pro-inflammatory cytokines and chemokines, promoting neutrophil transmigration and compromising epithelial barrier function. PLY-stimulated neutrophils engage in a wide range of effector functions, including degranulation, reactive oxygen species (ROS) production, and neutrophil extracellular trap (NET) release, many of which propagate inflammatory tissue damage and are associated with severe pathology in the lung. PLY can activate macrophages independently or in conjunction with other co-stimulants to cause the release of pro-inflammatory cytokines and chemokines. Macrophages can also be activated by PLY-dependent inflammasome activation, which in bone marrow-derived macrophages leads to IL-1β -mediated inflammation, and in microglia, pyroptotic cell death. PLY is also a potent inducer of macrophage necroptosis, often leading to acute tissue injury. Right Panel: Anti-inflammatory activities of PLY downregulate immune responses and may aid bacterial evasion. During pneumococcal colonization and early stages of lung infection, PLY suppresses inflammatory cytokine production by airway epithelial cells and enhances recruitment ofT regulatory cells, promoting unchecked bacterial colonization. Internalization of PLY by alveolar macrophages and dendritic cells via the mannose receptor MRC-1, or, in bone marrow derived macrophages, PLY triggered LC3- associated phagocytosis, suppress the production of inflammatory cytokines. To avoid complement mediated detection and opsonization, PLY acts as a decoy molecule to sequester complement proteins. Finally, PLY triggers apoptosis in a wide range of cell types, including endothelial cells, neurons, and dendritic cells, allowing for non-inflammatory removal of these cell types.
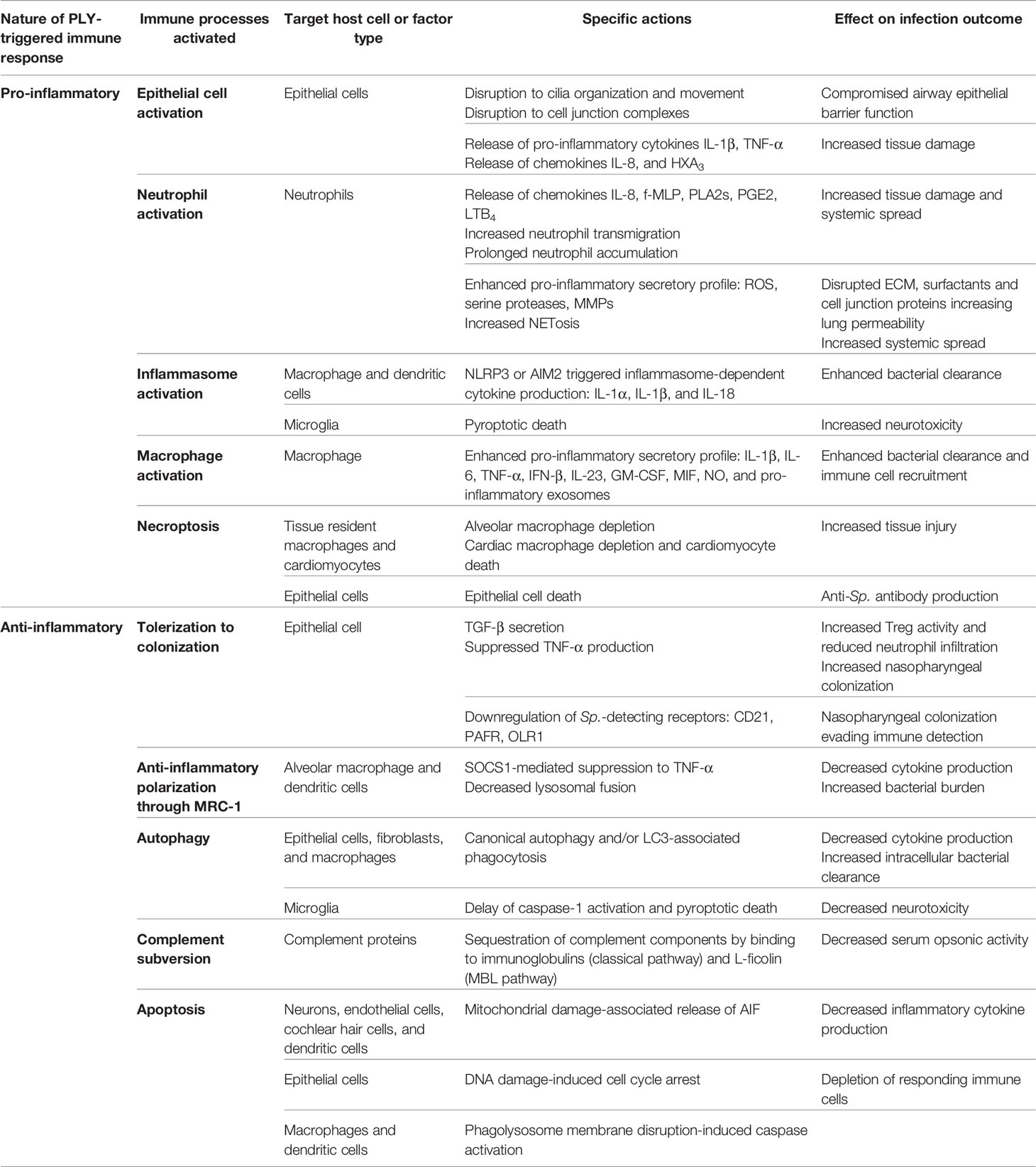
Table 1 PLY-triggered pro- and anti-inflammatory immune modulations.
4.1 Pro-Inflammatory Properties
4.1.1 Epithelial Cell Activation
Epithelial cells are a first line of defense against Sp, in part by functioning as a physical barrier against invasion and, in the lower respiratory tract, maintaining the mucociliary elevator that removes microbes from the lung. PLY both disrupts epithelial cell junctions, compromising this barrier, and interfering with the number and organization of cilia in human and mouse airway epithelium-derived air-liquid interface organoid cultures (145, 146), crippling the mucociliary elevator. In addition, as the sentinels for infection, the mucosal epithelium produces a range of antimicrobial peptides and pro-inflammatory signaling molecules to eliminate invading pathogens and alert immune cells to response to invasion. PLY induces β-defensin 2 secretion in A549 human lung epithelial cells and in human middle-ear cells (147). PLY, is also a potent activator of cytokine and chemoattractant secretion through pore forming insult to airway epithelial cells (94, 119, 148) (Figure 4, left). PLY induces epithelial cell release of IL-1β, TNF-α (148), and the neutrophil chemoattractants IL-8 (119) and hepoxilin A3 (HXA3) (94, 149, 150). Accumulation of inflammatory cytokines and chemokines facilitate the recruitment of leukocytes responding to and clearing Sp. Nevertheless, in both a lung epithelial tissue culture and a mouse pulmonary infection model, HXA3-mediated basolateral-to-apical neutrophil transmigration exacerbates damage to epithelial barrier and promotes lethal Sp systemic spread (151, 152).
4.1.2 Neutrophil Activation
As described above, neutrophil infiltration and the associated epithelial damage promote bacterial dissemination into the bloodstream, resulting in lethal systemic disease (151). While neutrophil recruitment to the lungs during early stages of infection aids Sp clearance, overtime, reduction in adenosine production by lowered CD73 expression correlated with loss of neutrophil antimicrobial efficacy (153). Extracellular ATP is shown to neutralize PLY-mediated neutrophil activation and may explain this shift in neutrophil response profile (154). Excessive neutrophil activities can be harmful to the host (140). Indeed, the prolonged accumulation of PMNs in a murine model of pneumococcal pneumonia results in increased burden and high tissue damage (153). Upon extravasation and encountering invading bacteria, exposure of PMNs to PLY can trigger the release of additional neutrophil-recruiting signals, including Ca2+-dependent increases in IL-8, f-MLP, phospholipase A, A2, prostaglandin E2, and leukotriene B4 (155, 156), reinforcing neutrophil-direction inflammation. Neutrophils at the site of infection can be further activated by Sp and PLY to produce reactive oxygen species (ROS) (93, 142, 157), degranulate, and form neutrophil extracellular traps (NETs) (Figure 4, left).
ROS have multiple immune-modulatory functions during bacterial infection: in addition to direct antimicrobial activity and signaling to modulate immune cell function, ROS can also cause damage to host cells and tissues (158). Interestingly, pretreating neutrophils with PLY promotes sensitivity to f-MLP (157), which conditions neutrophils to release higher and more sustained levels of ROS, and become more prone to degranulation (Figure 4, left) (142).
During degranulation, neutrophils secrete proteolytic enzymes capable of propagating tissue damage. These include serine proteases neutrophil elastase (NE), cathepsin G and proteinase-3, and neutrophil metalloproteinases (MMPs). PLY can trigger NE release either by degranulation of primary granules or by direct cell lysis (93, 142). Although NE degrades pneumococcal cell wall-localized aminopeptidase N and can facilitate opsonophagocytic killing in the presence of an antiphagocytic capsule (93, 159–161), excessive NE activity is detrimental to the host (162). NE has potent catalytic activity against a range of extracellular matrix proteins, including elastin, proteoglycan, fibronectin, and several collagen types (163), surfactant proteins (162), and alveolar epithelial cell junction protein such as E-cadherin (164). In addition, NE stimulates the lung epithelium to release proinflammatory cytokines and induces epithelial apoptosis (165–167). Severe pneumonia in human patients and experimental animals is associated with increased NE levels in lung bronchoalveolar lavage fluid (168, 169) and plasma (170). NE may thus be a critical inducer of epithelial permeability during pneumococcal pneumonia, compromising lung epithelial barrier and promoting bacterial dissemination (171). Cathepsin G and proteinase-3, which like NE are stored in azurophilc granules, also contribute to infection-related lung injury, albeit to a much lesser degree than NE (172). Metalloproteinases (MMPs) are neutrophil granule components that are also released following the increase in cytosolic Ca2+ levels resulting from PLY-mediated pore formation and can be augmented by concurrent stimulation by f-MLP (173). MMPs, especially MMP-8 and -9, have been correlated with tissue injury in pneumococcal pneumonia and meningitis (174–176). Thus, the PLY-mediated release of neutrophil granule components contributes to inflammation and pathogenesis, especially during severe forms of Sp disease.
Finally, pneumococcal capsule and PLY work in synergy to promote neutrophil extracellular trap (NET) production (162), a vital neutrophil effector function for trapping and killing extracellular microbes (Figure 4, left). While NETs exhibit significant antibacterial activity against Sp (177), their efficacy is counteracted by Sp-secreted endonucleases EndA and TatD that digest NETs and facilitate escape (162). NETs are intercalated with NE and other proteases that can damage host cells just as they can a pathogen, and excessive NETosis against Sp is implicated in promoting lung injury (178), sepsis (179), and increasing mortality (180). PLY is thus a potent activator of multiple neutrophil effector functions, most of which can propagate inflammatory tissue damage in the lung and are associated with severe pathology.
4.1.3 Inflammasome Activation
PLY induces inflammasome-dependent IL-1β secretion and initiates the pro-inflammatory cascade associated with Sp infection (181–183). PLY-mediated K+ efflux, which is sufficient to trigger NLRP3 activation (184), results in caspase-1 cleavage and the consequent production and secretion of IL-1β (181, 185). An alternative inflammasome activation pathway aided by PLY is the AIM2 inflammasome pathway. AIM2 senses double-stranded DNA from lysed bacteria, which can localize to host cell cytosol after PLY-mediated phagosomal membrane disruption (186, 187). PLY-deficient Sp induces less inflammasome-dependent cytokines production (including IL-1α, IL-1β, and IL-18), which influence downstream cytokine and chemokine responses to Sp. Indeed, inflammasome-dependent cytokines promote secretion of IL-17A, IFN-γ, and neutrophil chemoattractants, each of which aids bacterial clearance (123, 182, 188). Inflammasome activation is host protective in mouse and human macrophages ex vivo and in mouse pneumococcal infection models (182, 188, 189). Sp strains impaired in inflammasome activation, including some serotype 1, serotype 8, serotype 7F, and strains expressing the nonhemolytic allele 5 of PLY, tend to cause more invasive disease (189–191) and chronic infection (17). In contrast, in pneumococcal meningitis models, PLY-dependent inflammasome activation increasing neurotoxicity and pathology (192), possibly because PLY-triggered inflammasome activation results in rapid pyroptotic death in microglial cells (193). Thus, while inflammasome activation is in general a protective response against Sp infection, tissue types such as the brain can be susctiple to pathologies due to pyroptotic death (Figure 4, left).
4.1.4 Macrophage Activation
PLY activates macrophages to release of IL-1β, IL-6, TNF-α, IFN-β, IL-23, granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage migration inhibitory factor (MIF), and nitric oxide (NO) (194–197). Many of these pro-inflammaroty and chemotactic responses rely on p38 or inflammasome activation. Some PLY-induced macrophages responses require additional Sp factors as co-stimulants (198). For instance, phagosomal membrane disruption by PLY allows for pneumococcal DNA to enter the cell cytoplasm, triggerring cytosolic DNA sensors for IFN-β production (113). PLY can also modulate macrophage activation when sequestered into vesicles shed during membrane repair; interaction with PLY-containing vesicles increases macrophage IL-1β, TNFα, CCL5, CCL8, and CCL1 (136).
In the monocyte-derived THP-1 cell line, PLY is responsible for the majority of gene modulations upon exposure to Sp (194). Upregulated genes include those encoding proinflammatory molecules such as IL-8 and monocyte chemotactic protein 3 (MCP-3), and cell surface receptors that impact inflammation, such as macrophage inflammatory protein 1β (MIP-1β), IL-2 receptor β (IL-2Rβ), IL-15 receptor α (IL-15Rα), and interferon receptor 2, promoting pro-inflammatory cytokine propagation (Figure 4, left).
4.1.5 Necroptosis of Multiple Cell Types
PLY induces necroptosis, an inflammatory pathway of programmed cell death, in the lungs (90) and heart (199). In the lungs, PLY-mediated necroptosis of alveolar macrophages (90) and epithelial cells (90) is triggered in response to loss of ion homeostasis, ATP depletion, and ROS generation (90), and depends on the phosphorylation of MLKL (200), a master effector of necroptosis. The rapid depletion of alveolar macrophages by necroptosis may greatly contribute to extensive lung damage (Figure 4, left). In the heart, PLY-mediated necroptosis occurs in macrophages that infiltrate the infected myocardium and in cardiomyocytes, thereby suppressing the anti-Sp immune response at that site (199) and contributing to cardiac injury (201). The use of pharmacological inhibitors of necroptosis reduces acute injury in the lungs and heart during Sp infection, providing a novel therapeutic target to overcome infection (201). Interestingly, PLY-induced necroptosis was also reported in nasopharyngeal epithelial cells during Sp asymptomatic colonization and was associated with the increased production of anti-pneumococcal antibodies (202), suggesting a role in the development of protective immunity against Sp and highlighting the infection contex dependent nature of PLY-host interactions.
4.2 Anti-Inflammatory Properties
4.2.1 Epithelial Cell Tolerization
As a prerequisite to becoming a deadly pathogen, Sp asymptomatically colonizes the human nasopharynx. This immune-silent colonization and long-term Sp carriage are thought to be sustained by anti-inflammatory responses, which prevent the disruption of the epithelial barrier and the infiltration of neutrophils (203). After nasopharyngeal inoculation of 105 Sp in mice, a dose that results in asymptomatic carriage, the production of PLY is associated with higher levels of the immunosuppressive cytokine TGF-β1 and immunomodulatory T regulatory cells (Tregs) and lower levels of neutrophil recruitment (Figure 4, right). Sp infection of human nasopharyngeal epithelial cells and fibroblasts resulted in PLY-dependent secretion of TGF-β1. Treatment of these cells with purified PLY likewise results in TGF-β1 secretion (203). In contrast, compared to the 105 dose, nasopharyngeal inoculation of 107 Sp, which leads to bacterial clearance, results in the production of lower levels of TGF-β1 and Tregs, and higher levels of IFN-γ and neutrophil infiltration (203). These findings indicate that PLY is capable of fostering nasopharyngeal carriage by limiting proinflammatory responses.
Interestingly, compared to other bacterial respiratory pathogens, such as Haemophilus influenzae, Sp induced cytokine production by epithelial cells in vitro appears delayed (148), consistent with the low numbers of infiltrated neutrophils detected in early stages of lobular pneumonia in human patients, despite bacterial load (204). At these early infection stages, PLY promotes the expression of MAPK phosphatase 1, which dephosphorylates p38 to suppress TNF-α production (205) (Figure 4, right). Later during infection, secretion of pro-inflammatory cytokines by epithelial cells increase (148). This delay in the initiation of epithelial cell-mediated inflammation may delay immune cell infiltration and provide Sp with a window of unchecked growth to establish infection. In addition, PLY downregulated many binding receptors that aid Sp detection by macrophages, including complement component receptor 2/CD21, platelet-activating factor acetylhydrolase, and oxidized low-density lipoprotein receptor 1 (OLR1). Thus, on top of directly targeting epithelial cells for tolerization, PLY evades surveilling immune cells by downregulating critical receptors for Sp recognition.
4.2.2 MRC-1 as a Mediator of Anti-Inflammatory Response
As mentioned, PLY was recently described to directly interact with MRC-1, which is expressed by DCs and alveolar macrophages, two cell types that represent a first line of defense mounted against Sp in the lungs (61). The PLY-MRC1 interaction impairs inflammatory response, limiting inflammatory cytokine secretion through the cytokine suppressor SOCS1, as well as neutrophil infiltration (Figure 4, right) (61). Consistent with its role as a phagocytic receptor, MRC-1 expressed on DCs binds to sub-lytic concentrations of PLY and promotes internalization of PLY-producing Sp. In mouse alveolar macrophages, internalized PLY-producing Sp colocalizes with MRC-1, whereas PLY-deficient Sp colocalizes with lysosomes (Figure 4, right). Upon pulmonary challenge of mice, PLY production is associated with lower levels of TNF-α and greater numbers of bacteria in lavage fluid. Similarly, genetic ablation or antibody inhibition of MRC-1 results in higher levels of TNF-α and lower bacterial load (61).
These findings opened new therapeutic strategies to fight pneumococcus infection. Indeed, molecular docking approaches identified MRC-1-derived peptides that neutralize PLY-MRC-1 interaction that impair Sp internalization and promote pathogen killing by autophagy (206). Furthermore, MRC-1-derived peptides were shown to inhibit PLY-driven cell lysis, inflammation, and lung epithelium damage by limiting PLY interaction with cells and decreasing IL-8 and TNF-α secretion (206). In zebrafish and mouse models of pneumococcal infection, MRC-1-derived peptides reduce disease development, promote host survival, and decrease bacterial burden (206).
4.2.3 Triggering of Autophagic Processes
Autophagy is a natural self-degradative mechanism that is activated in epithelial and immune cells to degrade cytoplasmic content. Autophagy also plays major roles in pathogen elimination controlling both inflammation and adaptive immune response (207). Sp activates autophagy, which functions as a host protective mechanism by promoting Sp clearance (208). As anticipated by a membrane-damaging agent, PLY was shown to initiate autophagy in a variety of cells such as human alveolar epithelial cells (208), murine microglia (193), and osteoblast cells (209). ROS generation triggered by PLY leads to the inhibition of the PI3K/AKT/mTOR pathway and the consequent activation of canonical autophagy (193, 208), which degrades intracellular Sp and limits infection in host cells (208). In murine microglia, autophagy activated by Sp transiently blocks caspase-1 activation and IL-1β secretion, delaying pyroptosis, the caspase-1-dependent inflammatory cell death pathway (193). In these cells, Sp infection increases the expression of autophagy-related genes in the early phase of infection as a host protective mechanism before pyroptosis and concomitant extensive tissue damage can occur (Figure 4, right). However, at later stages of infection, sustained PLY-mediated high-level ROS generation activates caspase-1 and microglia pyroptosis (193). In osteoblasts, PLY-mediated activation of autophagy impairs differentiation by regulating the expression of differentiation-related genes, which require mTOR signaling (209), a finding with potential relevance to Sp osteomyelitis. Finally, PLY induces selective autophagy promoting the delivery of Sp entrapped in autophagosomes to lysosomes for further degradation (210). In fibroblasts, PLY triggers (LC3)-associated phagocytosis (LAP), followed by canonical autophagy activation, suggesting a hierarchical autophagy activation process leading to Sp clearance (211). In fibroblasts, the activation of canonical autophagy is required for Sp degradation, but in bone-marrow-derived macrophages (BMDMs), PLY-induced LAP is sufficient for bacterial clearance, allowing degradation to occur much more rapidly (212). In aged BMDMs, LAP is compromised, and cells display reduced Sp killing capacity and increased expression of proinflammatory cytokines (Figure 4, right) (212).
4.2.4 Complement Subversion
Complement activation has a crucial role in host protection against pathogens, but Sp utilizes PLY-mediated complement activation to sequester complement components away from Sp surface, thus protecting Sp from host defenses and facilitating bacterial spread and survival. Purified PLY activates the human complement cascade via the classical pathway independently of its lytic activity (62, 213). Homology domains shared with CRP, mediate the PLY binding to the Fc portion of immunoglobulins, which in turn recruit and activate C1q, and are required for PLY-triggered complement activation (62, 63). While in serum from C1q KO mice PLY fails to trigger complement activation, in human C1q-depleted serum, PLY still activates the complement and C3b deposition is observed (214), suggesting that in humans, PLY may stimulate complement through C1q-independent pathways that directly target C3. PLY was further shown to trigger lectin pathway by binding to L-ficolin with high affinity (214). However, PLY-triggered complement activation decreased the serum opsonic activity for Sp both in vitro and in vivo in mouse models, reducing bacterial uptake by neutrophils and impairing the recruitment of T cells to the sites of infection in the lung (62, 64, 213).
4.2.5 Induction of Apoptosis
Pathogen-induced apoptosis, a non-inflammatory programmed cell death pathway, plays an important role in tissue damage caused by infectious diseases and constitutes an important mechanism of protection from invasive disease. PLY cytolytic activity can prompt cells to engage in apoptosis by different mechanisms depending on PLY concentration and the cell type involved (Figure 4, right). One of the first studies connecting PLY to apoptosis showed that in neurons, PLY pore-forming activity triggers Ca2+ influx and mitochondrial damage which results in the release of pro-apoptotic factor (AIF) and induces apoptosis in a caspase-independent manner (110, 111). The same mechanism was described in brain microvascular endothelial cells, inner cochlear hair cells, and DCs (77, 91, 215). In addition, intracellular PLY induces caspase-dependent apoptosis in Sp-infected human dendritic cells, whereby blocking their maturation and the production of inflammatory cytokines, thus inhibiting dendritic cell-mediated inflammatory responses (92). Caspase-dependent apoptosis, triggered by PLY-induced phagolysosomal membrane permeabilization, also occurs in human monocyte‐derived macrophages (216, 217). In endothelial cells, PLY-triggered caspase activation and apoptosis depend on the activation of p38/MAPK and suppression of extracellular signaling regulation kinase (ERK)1/2 (218). Further supporting PLY’s ability to trigger apoptosis, in human epithelial lung alveolar cells PLY causes double-stranded DNA breaks, which led to cell cycle arrest followed by non-homologous end joining DNA repair or, if DNA damage persists, by engagement in apoptosis (219). Clinical isolates deficient for PLY are unable to induce DCs apoptosis and trigger a strong proinflammatory response leading to excessive lung inflammation (92). Thus, the deleterious effect of PLY on immune cells, blocking their maturation, inhibiting the secretion of inflammatory cytokines, and inducing apoptosis, promotes bacterial evasion from immune detection and allows for immune-silent colonization.
5 Future Perspectives
A better understanding of the complex “Yin” and “Yang” properties of PLY will facilitate the translation of critical Sp pathogenesis mechanisms into clinical intervention strategies. PLY stands as an attractive therapeutic target to complement classical antibiotic therapy, which, besides fostering the development of resistance, can trigger bacterial lysis and consequent release of PLY that alone can have deleterious effects on cells and the immune system (25). Several repurposed drugs and PLY-neutralizing compounds have been investigated. Statins, which inhibit cholesterol production, administered prior to infection both in vitro or in vivo confer significant resistance to PLY cytotoxicity by impairing binding (220, 221). Also, some natural compounds were shown to target the oligomerization process by binding to specific D3 and 4 residues, resulting in reduced cytolytic activity (222–226). The use of artificial liposomes was also suggested as a way to sequester PLY. The administration of liposomes prior to pneumococcal infection reduced septicemia and invasive disease in a mouse model (171). Recent efforts have been made to find novel therapeutic tools to inhibit PLY release, and recently, a study demonstrated that clarithromycin downregulates ply transcription in vitro and in vivo and consequently reduces PLY production by bacteria (227), proposing a new strategy to treat pneumococcal disease. The clinical potential of current PLY-neutralizing therapeutic strategies and their limitations have been extensively reviewed elsewhere (22, 25).
Since PLY is also a potent trigger for anti-Sp immune responses, harnessing the immunogenic potentials of PLY has long been of therapeutic interest. PLY-immunized mice displayed significantly increased survival upon infection, and thus suggested that PLY should be considered for inclusion in a human vaccine (228). Further studies using different animal models aimed to obtain an active immunization using a genetic toxoid derivative of PLY, alone or in combination with other proteins (229–234). In fact, PLY toxoid demonstrated potential effectiveness as an immunogenic component and in controlling bacteremia and bacterial colonization (229–235). Phase I clinical trials concerning the use of PLY toxoid vaccine formulations were performed in adults and children, and results demonstrated that it is well-tolerated and immunogenic when administered as individual protein vaccines or combined with capsule polysaccharide conjugates (230, 236, 237). Experimental evolution studies revealed the emergence of variants that produce low levels of PLY showing decreased virulence but increased persistence (238). Such lineages have been proposed to serve as starting point to the development of live-attenuated pneumococcal vaccines.
To use PLY-targeting therapies and treatment tailored to the stage and severity of pneumococcal infection, requires a better elucidation of the role of PLY in pathogenesis. As highlighted in this review, PLY induces multiple contradicting responses. The parameters that dictate the exact mechanisms triggered in the host cells during pore-formation are still elusive. It is known that PLY is present in circulation during early pneumococcus infection and sub-lytic doses induced toxicity and modulated host immune response; furthermore, host cells can trigger PM repair mechanisms to recover from damage and get rid of the toxin (116, 127, 130, 239). However, it is still not clear which molecular mechanisms and host proteins are involved during the process of cell survival and the importance of these events in the context of infection. Cells can expel the toxin through vesicle shedding, but whether they can function as a danger signal to neighboring cells or as a modulator of the immune response remains unclear (126, 129, 136, 138). PLY can also have several effects in intracellular signaling and modulate host cytoskeleton but remains uncertain how these events can help cells to survive and repair the damage (101, 103, 114, 115, 240). Furthermore, the use of complex models like 3D cell culture or in vivo models should now be considered to clarify the crosstalk between different host cell types and their microenvironment. Insights into the mechanistic control for cellular response to PLY pores may also help resolve the apparent contradictory inflammatory and anti-inflammatory implications under different infection contexts. The discovery of new mechanisms of host PM repair and survival may bring new insights to the development of therapies against the injurious actions of PLY during pneumococcus infection.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
SS research receives funds from FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operational Programme for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funds through FCT—Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior in the framework of the project POCI-01-0145-FEDER-030863 (PTDC/BIA-CEL/30863/2017). Research in JL’s laboratory is supported by NIH R21 AG071268-01. JMP is the recipient of an FCT fellowship (SFRH/BD/143940/2019). SS received support from FCT in the framework of CEEC-Institutional 2017 program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Weiser JN, Ferreira DM, Paton JC. Streptococcus Pneumoniae: Transmission, Colonization and Invasion. Nat Rev Microbiol (2018) 16:355–67. doi: 10.1038/s41579-018-0001-8
2. Troeger C, Blacker BF, Khalil IA, Rao PC, Cao S, Zimsen SRM, et al. Estimates of the Global, Regional, and National Morbidity, Mortality, and Aetiologies of Lower Respiratory Infections in 195 Countries, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis (2018) 18:1191–210. doi: 10.1016/S1473-3099(18)30310-4
3. Tong S, Amand C, Kieffer A, Kyaw MH. Trends in Healthcare Utilization and Costs Associated With Pneumonia in the United States During 2008–2014. BMC Health Serv Res (2018) 18:715. doi: 10.1186/s12913-018-3529-4
4. Zhang D, Petigara T, Yang X. Clinical and Economic Burden of Pneumococcal Disease in US Adults Aged 19–64 Years With Chronic or Immunocompromising Diseases: An Observational Database Study. BMC Infect Dis (2018) 18:436. doi: 10.1186/s12879-018-3326-z
5. Keller LE, Robinson DA, McDaniel LS. Nonencapsulated Streptococcus Pneumoniae: Emergence and Pathogenesis. mBio (2016) 7:e01792. doi: 10.1128/mBio.01792-15
6. Wroe PC, Finkelstein JA, Ray GT, Linder JA, Johnson KM, Rifas-Shiman S, et al. Aging Population and Future Burden of Pneumococcal Pneumonia in the United States. J Infect Dis (2012) 205:1589–92. doi: 10.1093/infdis/jis240
7. Negash AA, Asrat D, Abebe W, Aseffa A, Vaneechoutte M. Phenotypic and Molecular Characterization of Penicillin and Macrolide-Resistant Streptococcus Pneumoniae Serotypes Among Pediatric Patients in Addis Ababa, Ethiopia. Infect Drug Resist (2021) 14:1765–72. doi: 10.2147/IDR.S309876
8. Jones RN, Sader HS, Mendes RE, Flamm RK. Update on Antimicrobial Susceptibility Trends Among Streptococcus Pneumoniae in the United States: Report of Ceftaroline Activity From the SENTRY Antimicrobial Surveillance Program (1998–2011). Diagn Microbiol Infect Dis (2013) 75:107–9. doi: 10.1016/j.diagmicrobio.2012.08.024
9. Nava JM, Bella F, Garau J, Lite J, Morera M-A, Martí C, et al. Predictive Factors for Invasive Disease Due to Penicillin-Resistant Streptococcus Pneumoniae: A Population-Based Study. Clin Infect Dis (1994) 19:884–90. doi: 10.1093/clinids/19.5.884
10. Masomian M, Ahmad Z, Ti Gew L, Poh CL. Development of Next Generation Streptococcus Pneumoniae Vaccines Conferring Broad Protection. Vaccines (2020) 8:132. doi: 10.3390/vaccines8010132
11. Cremers AJH, Mobegi FM, de Jonge MI, van Hijum SAFT, Meis JF, Hermans PWM, et al. The Post-Vaccine Microevolution of Invasive Streptococcus Pneumoniae. Sci Rep (2015) 5:14952. doi: 10.1038/srep14952
12. Subramanian K, Henriques-Normark B, Normark S. Emerging Concepts in the Pathogenesis of the Streptococcus Pneumoniae: From Nasopharyngeal Colonizer to Intracellular Pathogen. Cell Microbiol (2019) 21:e13077. doi: 10.1111/cmi.13077
13. Zafar MA, Wang Y, Hamaguchi S, Weiser JN. Host-To-Host Transmission of Streptococcus Pneumoniae Is Driven by Its Inflammatory Toxin, Pneumolysin. Cell Host Microbe (2017) 21:73–83. doi: 10.1016/j.chom.2016.12.005
14. Brooks LRK, Mias GI. Streptococcus Pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01366
15. Ercoli G, Fernandes VE, Chung WY, Wanford JJ, Thomson S, Bayliss CD, et al. Intracellular Replication of Streptococcus Pneumoniae Inside Splenic Macrophages Serves as a Reservoir for Septicaemia. Nat Microbiol (2018) 3:600–10. doi: 10.1038/s41564-018-0147-1
16. Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, et al. The Role of Pneumolysin and Autolysin in the Pathology of Pneumonia and Septicemia in Mice Infected With a Type 2 Pneumococcus. J Infect Dis (1995) 172:119–23. doi: 10.1093/infdis/172.1.119
17. Benton KA, Everson MP, Briles DE. A Pneumolysin-Negative Mutant of Streptococcus Pneumoniae Causes Chronic Bacteremia Rather Than Acute Sepsis in Mice. Infect Immun (1995) 63:448–55. doi: 10.1128/iai.63.2.448-455.1995
18. Anderson R, Nel JG, Feldman C. Multifaceted Role of Pneumolysin in the Pathogenesis of Myocardial Injury in Community-Acquired Pneumonia. Int J Mol Sci (2018) 19:1147–68. doi: 10.3390/ijms19041147
19. Yau B, Hunt NH, Mitchell AJ, Too LK. Blood−Brain Barrier Pathology and CNS Outcomes in Streptococcus Pneumoniae Meningitis. Int J Mol Sci (2018) 19:3555–76. doi: 10.3390/ijms19113555.
20. Tettelin H, Chancey S, Mitchell T, Denapaite D, Schähle Y, Rieger M, et al. Chapter 5 - Genomics, Genetic Variation, and Regions of Differences. In: Brown J, Hammerschmidt S, Orihuela C, editors. Streptococcus Pneumoniae. Amsterdam: Academic Press (2015). p. 81–107.
21. van Pee K, Mulvihill E, Müller DJ, Yildiz Ö. Unraveling the Pore-Forming Steps of Pneumolysin From Streptococcus Pneumoniae. Nano Lett (2016) 16:7915–24. doi: 10.1021/acs.nanolett.6b04219
22. Nishimoto AT, Rosch JW, Tuomanen EI. Pneumolysin: Pathogenesis and Therapeutic Target. Front Microbiol (2020) 11:1543. doi: 10.3389/fmicb.2020.01543
23. Babiychuk EB, Draeger A. Defying Death: Cellular Survival Strategies Following Plasmalemmal Injury by Bacterial Toxins. Semin Cell Dev Biol (2015) 45:39–47. doi: 10.1016/j.semcdb.2015.10.016
24. Johnson MK, Hobden JA, Hagenah M, O'Callaghan RJ, Hill JM, Chen S. The Role of Pneumolysin in Ocular Infections With Streptococcus Pneumoniae. Curr Eye Res (1990) 9:1107–14. doi: 10.3109/02713689008997584
25. Anderson R, Feldman C. Pneumolysin as a Potential Therapeutic Target in Severe Pneumococcal Disease. J Infect (2017) 74:527–44. doi: 10.1016/j.jinf.2017.03.005
26. Marriott HM, Mitchell TJ, Dockrell DH. Pneumolysin: A Double-Edged Sword During the Host-Pathogen Interaction. Curr Mol Med (2008) 8:497–509. doi: 10.2174/156652408785747924
27. Rabes A, Suttorp N, Opitz B. Inflammasomes in Pneumococcal Infection: Innate Immune Sensing and Bacterial Evasion Strategies. Curr Top Microbiol Immunol (2016) 397:215–27. doi: 10.1007/978-3-319-41171-2_11
28. Surve MV, Banerjee A. Cell-To-Cell Phenotypic Heterogeneity in Pneumococcal Pathogenesis. Future Microbiol (2019) 14:647–51. doi: 10.2217/fmb-2019-0096
29. Surve MV, Bhutda S, Datey A, Anil A, Rawat S, Pushpakaran A, et al. Heterogeneity in Pneumolysin Expression Governs the Fate of Streptococcus Pneumoniae During Blood-Brain Barrier Trafficking. PloS Pathog (2018) 14:e1007168. doi: 10.1371/journal.ppat.1007168
30. Panagiotou S, Chaguza C, Yahya R, Audshasai T, Baltazar M, Ressel L, et al. Hypervirulent Pneumococcal Serotype 1 Harbours Two Pneumolysin Variants With Differential Haemolytic Activity. Sci Rep (2020) 10:17313. doi: 10.1038/s41598-020-73454-w
31. Jacques LC, Panagiotou S, Baltazar M, Senghore M, Khandaker S, Xu R, et al. Increased Pathogenicity of Pneumococcal Serotype 1 is Driven by Rapid Autolysis and Release of Pneumolysin. Nat Commun (2020) 11:1892. doi: 10.1038/s41467-020-15751-6
32. Price KE, Camilli A. Pneumolysin Localizes to the Cell Wall of Streptococcus Pneumoniae. J Bacteriol (2009) 191:2163–8. doi: 10.1128/JB.01489-08
33. Johnson MK. Cellular Location of Pneumolysin. FEMS Microbiol Lett (1977) 2:243–5. doi: 10.1111/j.1574-6968.1977.tb00951.x
34. Alhamdi Y, Neill DR, Abrams ST, Malak HA, Yahya R, Barrett-Jolley R, et al. Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury During Pneumococcal Infection. PloS Pathog (2015) 11:e1004836. doi: 10.1371/journal.ppat.1004836
35. Spreer A, Kerstan H, Böttcher T, Gerber J, Siemer A, Zysk G, et al. Reduced Release of Pneumolysin by Streptococcus Pneumoniae In Vitro and In Vivo After Treatment With Nonbacteriolytic Antibiotics in Comparison to Ceftriaxone. Antimicrob Agents Chemother (2003) 47:2649–54. doi: 10.1128/AAC.47.8.2649-2654.2003
36. Price KE, Greene NG, Camilli A. Export Requirements of Pneumolysin in Streptococcus Pneumoniae. J Bacteriol (2012) 194:3651–60. doi: 10.1128/JB.00114-12
37. Berry AM, Lock RA, Hansman D, Paton JC. Contribution of Autolysin to Virulence of Streptococcus Pneumoniae. Infect Immun (1989) 57:2324–30. doi: 10.1128/iai.57.8.2324-2330.1989
38. Balachandran P, Hollingshead SK, Paton JC, Briles DE. The Autolytic Enzyme LytA of Streptococcus Pneumoniae is Not Responsible for Releasing Pneumolysin. J Bacteriol (2001) 183:3108–16. doi: 10.1128/JB.183.10.3108-3116.2001
39. Bandara M, Skehel JM, Kadioglu A, Collinson I, Nobbs AH, Blocker AJ, et al. The Accessory Sec System (SecY2A2) in Streptococcus Pneumoniae is Involved in Export of Pneumolysin Toxin, Adhesion and Biofilm Formation. Microbes Infect (2017) 19:402–12. doi: 10.1016/j.micinf.2017.04.003
40. Greene NG, Narciso AR, Filipe SR, Camilli A. Peptidoglycan Branched Stem Peptides Contribute to Streptococcus Pneumoniae Virulence by Inhibiting Pneumolysin Release. PloS Pathog (2015) 11:e1004996. doi: 10.1371/journal.ppat.1004996
41. Billington SJ, Jost BH, Songer JG. Thiol-Activated Cytolysins: Structure, Function and Role in Pathogenesis. FEMS Microbiol Lett (2000) 182:197–205. doi: 10.1016/S0378-1097(99)00536-4
42. Hill J, Andrew PW, Mitchell TJ. Amino Acids in Pneumolysin Important for Hemolytic Activity Identified by Random Mutagenesis. Infect Immun (1994) 62:757–8. doi: 10.1128/iai.62.2.757-758.1994
43. Morgan PJ, Harrison G, Freestone PPE, Crane D, Rowe AJ, Mitchell TJ, et al. Structural and Functional Characterisation of Two Proteolytic Fragments of the Bacterial Protein Toxin, Pneumolysin. FEBS Lett (1997) 412:563–7. doi: 10.1016/S0014-5793(97)00838-7
44. Marshall JE, Faraj BH, Gingras AR, Lonnen R, Sheikh MA, El-Mezgueldi M, et al. The Crystal Structure of Pneumolysin at 2.0 Å Resolution Reveals the Molecular Packing of the Pre-Pore Complex. Sci Rep (2015) 5:13293. doi: 10.1038/srep13293
45. Badgujar DC, Anil A, Green AE, Surve MV, Madhavan S, Beckett A, et al. Structural Insights Into Loss of Function of a Pore Forming Toxin and its Role in Pneumococcal Adaptation to an Intracellular Lifestyle. PloS Pathog (2020) 16:e1009016. doi: 10.1371/journal.ppat.1009016
46. Lawrence SL, Feil SC, Morton CJ, Farrand AJ, Mulhern TD, Gorman MA, et al. Crystal Structure of Streptococcus Pneumoniae Pneumolysin Provides Key Insights Into Early Steps of Pore Formation. Sci Rep (2015) 5:14352. doi: 10.1038/srep14352
47. Owen RHG, Boulnois GJ, Andrew PW, Mitchell TJ. A Role in Cell-Binding for the C-Terminus of Pneumolysin, the Thiol-Activated Toxin of Streptococcus Pneumoniae. FEMS Microbiol Lett (1994) 121:217–21. doi: 10.1111/j.1574-6968.1994.tb07101.x
48. Baba H, Kawamura I, Kohda C, Nomura T, Ito Y, Kimoto T, et al. Essential Role of Domain 4 of Pneumolysin From Streptococcus Pneumoniae in Cytolytic Activity as Determined by Truncated Proteins. Biochem Biophys Res Commun (2001) 281:37–44. doi: 10.1006/bbrc.2001.4297
49. de los Toyos JR, Méndez FJ, Aparicio JF, Vázquez F, Del Mar García Suárez M, Fleites A, et al. Functional Analysis of Pneumolysin by Use of Monoclonal Antibodies. Infect Immun (1996) 64:480–4. doi: 10.1128/iai.64.2.480-484.1996
50. Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK. Only Two Amino Acids are Essential for Cytolytic Toxin Recognition of Cholesterol at the Membrane Surface. Proc Natl Acad Sci USA (2010) 107:4341–6. doi: 10.1073/pnas.0911581107
51. Nöllmann M, Gilbert R, Mitchell T, Sferrazza M, Byron O. The Role of Cholesterol in the Activity of Pneumolysin, a Bacterial Protein Toxin. Biophys J (2004) 86:3141–51. doi: 10.1016/S0006-3495(04)74362-3
52. Tilley SJ, Orlova EV, Gilbert RJC, Andrew PW, Saibil HR. Structural Basis of Pore Formation by the Bacterial Toxin Pneumolysin. Cell (2005) 121:247–56. doi: 10.1016/j.cell.2005.02.033
53. van Pee K, Neuhaus A, D'Imprima E, Mills DJ, Kühlbrandt W, Yildiz Ö. CryoEM Structures of Membrane Pore and Prepore Complex Reveal Cytolytic Mechanism of Pneumolysin. eLife (2017) 6:e23644. doi: 10.7554/eLife.23644
54. Vögele M, Bhaskara RM, Mulvihill E, van Pee K, Yildiz Ö., Kühlbrandt W, et al. Membrane Perforation by the Pore-Forming Toxin Pneumolysin. Proc Natl Acad Sci USA (2019) 116:13352–7. doi: 10.1073/pnas.1904304116
55. Sonnen AFP, Plitzko JM, Gilbert RJC. Incomplete Pneumolysin Oligomers Form Membrane Pores. Open Biol (2014) 4:140044–4. doi: 10.1098/rsob.140044
56. Gilbert RJ, Jiménez JL, Chen S, Tickle IJ, Rossjohn J, Parker M, et al. Two Structural Transitions in Membrane Pore Formation by Pneumolysin, the Pore-Forming Toxin of Streptococcus Pneumoniae. Cell (1999) 97:647–55. doi: 10.1016/S0092-8674(00)80775-8
57. Jefferies JMC, Johnston CHG, Kirkham L-AS, Cowan GJM, Ross KS, Smith A, et al. Presence of Nonhemolytic Pneumolysin in Serotypes of Streptococcus Pneumoniae Associated With Disease Outbreaks. J Infect Dis (2007) 196:936–44. doi: 10.1086/520091
58. Leung C, Dudkina NV, Lukoyanova N, Hodel AW, Farabella I, Pandurangan AP, et al. Stepwise Visualization of Membrane Pore Formation by Suilysin, a Bacterial Cholesterol-Dependent Cytolysin. Elife (2014) 3:e04247. doi: 10.7554/eLife.04247
59. Gilbert RJ, Mikelj M, Dalla Serra M, Froelich CJ, Anderluh G. Effects of MACPF/CDC Proteins on Lipid Membranes. Cell Mol Life Sci (2013) 70:2083–98. doi: 10.1007/s00018-012-1153-8
60. Gilbert RJC, Sonnen AFP. Measuring Kinetic Drivers of Pneumolysin Pore Structure. Eur Biophys J (2016) 45:365–76. doi: 10.1007/s00249-015-1106-x
61. Subramanian K, Neill DR, Malak HA, Spelmink L, Khandaker S, Dalla Libera Marchiori G, et al. Pneumolysin Binds to the Mannose Receptor C Type 1 (MRC-1) Leading to Anti-Inflammatory Responses and Enhanced Pneumococcal Survival. Nat Microbiol (2019) 4:62–70. doi: 10.1038/s41564-018-0280-x
62. Paton JC, Rowan-Kelly B, Ferrante A. Activation of Human Complement by the Pneumococcal Toxin Pneumolysin. Infect Immun (1984) 43:1085–7. doi: 10.1128/iai.43.3.1085-1087.1984
63. Mitchell TJ, Andrew PW, Saunders FK, Smith AN, Boulnois GJ. Complement Activation and Antibody Binding by Pneumolysin via a Region of the Toxin Homologous to a Human Acute-Phase Protein. Mol Microbiol (1991) 5:1883–8. doi: 10.1111/j.1365-2958.1991.tb00812.x
64. Yuste J, Botto M, Paton JC, Holden DW, Brown JS. Additive Inhibition of Complement Deposition by Pneumolysin and PspA Facilitates Streptococcus Pneumoniae Septicemia. J Immunol (2005) 175:1813–9. doi: 10.4049/jimmunol.175.3.1813
65. Shewell LK, Harvey RM, Higgins MA, Day CJ, Hartley-Tassell LE, Chen AY, et al. The Cholesterol-Dependent Cytolysins Pneumolysin and Streptolysin O Require Binding to Red Blood Cell Glycans for Hemolytic Activity. Proc Natl Acad Sci (2014) 111:E5312–20. doi: 10.1073/pnas.1412703111
66. Hirst RA, Kadioglu A, O'callaghan C, Andrew PW. The Role of Pneumolysin in Pneumococcal Pneumonia and Meningitis. Clin Exp Immunol (2004) 138:195–201. doi: 10.1111/j.1365-2249.2004.02611.x
67. Kadioglu A, Taylor S, Iannelli F, Pozzi G, Mitchell TJ, Andrew PW. Upper and Lower Respiratory Tract Infection by Streptococcus Pneumoniae is Affected by Pneumolysin Deficiency and Differences in Capsule Type. Infect Immun (2002) 70:2886–90. doi: 10.1128/IAI.70.6.2886-2890.2002
68. Hotomi M, Yuasa J, Briles DE, Yamanaka N. Pneumolysin Plays a Key Role at the Initial Step of Establishing Pneumococcal Nasal Colonization. Folia Microbiol (Praha) (2016) 61:375–83. doi: 10.1007/s12223-016-0445-z
69. Rubins JB, Janoff EN. Pneumolysin: A Multifunctional Pneumococcal Virulence Factor. J Lab Clin Med (1998) 131:21–7. doi: 10.1016/S0022-2143(98)90073-7
70. Allegrucci M, Hu FZ, Shen K, Hayes J, Ehrlich GD, Post JC, et al. Phenotypic Characterization of Streptococcus Pneumoniae Biofilm Development. J Bacteriol (2006) 188:2325–35. doi: 10.1128/JB.188.7.2325-2335.2006
71. Vidal JE, Ludewick HP, Kunkel RM, Zähner D, Klugman KP. The LuxS-Dependent Quorum-Sensing System Regulates Early Biofilm Formation by Streptococcus Pneumoniae Strain D39. Infect Immun (2011) 79:4050–60. doi: 10.1128/IAI.05186-11
72. Schachern PA, Tsuprun V, Goetz S, Cureoglu S, Juhn SK, Briles DE, et al. Viability and Virulence of Pneumolysin, Pneumococcal Surface Protein A, and Pneumolysin/Pneumococcal Surface Protein A Mutants in the Ear. JAMA Otolaryngol–Head Neck Surg (2013) 139:937–43. doi: 10.1001/jamaoto.2013.4104
73. Keller LE, Bradshaw JL, Pipkins H, McDaniel LS. Surface Proteins and Pneumolysin of Encapsulated and Nonencapsulated Streptococcus Pneumoniae Mediate Virulence in a Chinchilla Model of Otitis Media. Front Cell Infect Microbiol (2016) 6:55. doi: 10.3389/fcimb.2016.00055
74. Comis SD, Osborne MP, Stephen J, Tarlow MJ, Hayward TL, Mitchell TJ, et al. Cytotoxic Effects on Hair Cells of Guinea Pig Cochlea Produced by Pneumolysin, the Thiol Activated Toxin of Streptococcus Pneumoniae. Acta Otolaryngol (1993) 113:152–9. doi: 10.3109/00016489309135784
75. Winter AJ, Comis SD, Osborne MP, Tarlow MJ, Stephen J, Andrew PW, et al. A Role for Pneumolysin But Not Neuraminidase in the Hearing Loss and Cochlear Damage Induced by Experimental Pneumococcal Meningitis in Guinea Pigs. Infect Immun (1997) 65:4411–8. doi: 10.1128/iai.65.11.4411-4418.1997
76. Skinner LJ, Beurg M, Mitchell TJ, Darrouzet V, Aran JM, Dulon D. Intracochlear Perfusion of Pneumolysin, a Pneumococcal Protein, Rapidly Abolishes Auditory Potentials in the Guinea Pig Cochlea. Acta Otolaryngol (2004) 124:1000–7. doi: 10.1080/00016480410017125
77. Beurg M, Hafidi A, Skinner L, Cowan G, Hondarrague Y, Mitchell TJ, et al. The Mechanism of Pneumolysin-Induced Cochlear Hair Cell Death in the Rat. J Physiol (2005) 568:211–27. doi: 10.1113/jphysiol.2005.092478
78. Recchia FM, Busbee BG, Pearlman RB, Carvalho-Recchia CA, Ho AC. Changing Trends in the Microbiologic Aspects of Postcataract Endophthalmitis. Arch Ophthalmol (2005) 123:341–6. doi: 10.1001/archopht.123.3.341
79. Norcross EW, Sanders ME, Moore QC3, Marquart ME. Pathogenesis of A Clinical Ocular Strain of Streptococcus Pneumoniae and the Interaction of Pneumolysin With Corneal Cells. J Bacteriol Parasitol (2011) 2:108–8. doi: 10.4172/2155-9597.1000108
80. Ng EW, Samiy N, Rubins JB, Cousins FV, Ruoff KL, Baker AS, et al. Implication of Pneumolysin as a Virulence Factor in Streptococcus Pneumoniae Endophthalmitis. Retina (1997) 17:521–9. doi: 10.1097/00006982-199711000-00006
81. Sanders ME, Norcross EW, Moore QC3, Fratkin J, Thompson H, Marquart ME. Immunization With Pneumolysin Protects Against Both Retinal and Global Damage Caused by Streptococcus Pneumoniae Endophthalmitis. J Ocul Pharmacol Ther (2010) 26:571–7. doi: 10.1089/jop.2010.0077
82. Taylor SD, Sanders ME, Tullos NA, Stray SJ, Norcross EW, McDaniel LS, et al. The Cholesterol-Dependent Cytolysin Pneumolysin From Streptococcus Pneumoniae Binds to Lipid Raft Microdomains in Human Corneal Epithelial Cells. PloS One (2013) 8:e61300. doi: 10.1371/journal.pone.0061300
83. Steinfort C, Wilson R, Mitchell T, Feldman C, Rutman A, Todd H, et al. Effect of Streptococcus Pneumoniae on Human Respiratory Epithelium In Vitro. Infect Immun (1989) 57:2006–13. doi: 10.1128/iai.57.7.2006-2013.1989
84. Rubins JB, Duane PG, Clawson D, Charboneau D, Young J, Niewoehner DE. Toxicity of Pneumolysin to Pulmonary Alveolar Epithelial Cells. Infect Immun (1993) 61:1352–8. doi: 10.1128/iai.61.4.1352-1358.1993
85. Rubins JB, Duane PG, Charboneau D, Janoff EN. Toxicity of Pneumolysin to Pulmonary Endothelial Cells In Vitro. Infect Immun (1992) 60:1740–6. doi: 10.1128/iai.60.5.1740-1746.1992
86. Nel JG, Durandt C, Mitchell TJ, Feldman C, Anderson R, Tintinger GR. Pneumolysin Mediates Platelet Activation In Vitro. Lung (2016) 194:589–93. doi: 10.1007/s00408-016-9900-5
87. Jahn K, Handtke S, Palankar R, Weißmüller S, Nouailles G, Kohler TP, et al. Pneumolysin Induces Platelet Destruction, Not Platelet Activation, Which can be Prevented by Immunoglobulin Preparations In Vitro. Blood Adv (2020) 4:6315–26. doi: 10.1182/bloodadvances.2020002372
88. Maus UA, Srivastava M, Paton JC, Mack M, Everhart MB, Blackwell TS, et al. Pneumolysin-Induced Lung Injury is Independent of Leukocyte Trafficking Into the Alveolar Space. J Immunol (2004) 173:1307–12. doi: 10.4049/jimmunol.173.2.1307
89. García-Suárez Mdel M, Flórez N, Astudillo A, Vázquez F, Villaverde R, Fabrizio K, et al. The Role of Pneumolysin in Mediating Lung Damage in a Lethal Pneumococcal Pneumonia Murine Model. Respir Res (2007) 8:3. doi: 10.1186/1465-9921-8-3
90. González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-Forming Toxins Induce Macrophage Necroptosis During Acute Bacterial Pneumonia. PloS Pathog (2015) 11:e1005337. doi: 10.1371/journal.ppat.1005337
91. Colino J, Snapper CM. Two Distinct Mechanisms For Induction of Dendritic Cell Apoptosis in Response to Intact Streptococcus Pneumoniae. J Immunol (2003) 171:2354–65. doi: 10.4049/jimmunol.171.5.2354
92. Littmann M, Albiger B, Frentzen A, Normark S, Henriques-Normark B, Plant L. Streptococcus Pneumoniae Evades Human Dendritic Cell Surveillance by Pneumolysin Expression. EMBO Mol Med (2009) 1:211–22. doi: 10.1002/emmm.200900025
93. Domon H, Oda M, Maekawa T, Nagai K, Takeda W, Terao Y. Streptococcus Pneumoniae Disrupts Pulmonary Immune Defence via Elastase Release Following Pneumolysin-Dependent Neutrophil Lysis. Sci Rep (2016) 6:38013. doi: 10.1038/srep38013
94. Adams W, Bhowmick R, Bou Ghanem EN, Wade K, Shchepetov M, Weiser JN, et al. Pneumolysin Induces 12-Lipoxygenase-Dependent Neutrophil Migration During Streptococcus Pneumoniae Infection. J Immunol (2020) 204:101–11. doi: 10.4049/jimmunol.1800748
95. Moreland JG, Bailey G. Neutrophil Transendothelial Migration In Vitro to Streptococcus Pneumoniae is Pneumolysin Dependent. Am J Physiol-Lung Cell Mol Physiol 290 (2006) 5:L833–40. doi: 10.1152/ajplung.00333.2005
96. Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, et al. Streptococcus Pneumoniae Translocates Into the Myocardium and Forms Unique Microlesions That Disrupt Cardiac Function. PloS Pathog (2014) 10:e1004383. doi: 10.1371/journal.ppat.1004383
97. Brissac T, Shenoy AT, Patterson LA, Orihuela CJ. Cell Invasion and Pyruvate Oxidase-Derived H(2)O(2) Are Critical for Streptococcus Pneumoniae-Mediated Cardiomyocyte Killing. Infect Immun 86 (2018) 86:e00569-17. doi: 10.1128/IAI.00569-17
98. Shenoy AT, Brissac T, Gilley RP, Kumar N, Wang Y, Gonzalez-Juarbe N, et al. Streptococcus Pneumoniae in the Heart Subvert the Host Response Through Biofilm-Mediated Resident Macrophage Killing. PloS Pathog (2017) 13:e1006582. doi: 10.1371/journal.ppat.1006582
99. Tabusi M, Thorsdottir S, Lysandrou M, Narciso AR, Minoia M, Srambickal CV, et al. Neuronal Death in Pneumococcal Meningitis is Triggered by Pneumolysin and RrgA Interactions With β-Actin. PloS Pathog (2021) 17:e1009432. doi: 10.1371/journal.ppat.1009432
100. Bello C, Smail Y, Sainte-Rose V, Podglajen I, Gilbert A, Moreira V, et al. Role of Astroglial Connexin 43 in Pneumolysin Cytotoxicity and During Pneumococcal Meningitis. PloS Pathog (2020) 16:e1009152. doi: 10.1371/journal.ppat.1009152
101. Hupp S, Heimeroth V, Wippel C, Förtsch C, Ma J, Mitchell TJ, et al. Astrocytic Tissue Remodeling by the Meningitis Neurotoxin Pneumolysin Facilitates Pathogen Tissue Penetration and Produces Interstitial Brain Edema. Glia (2012) 60:137–46. doi: 10.1002/glia.21256
102. Iliev AI, Djannatian JR, Opazo F, Gerber J, Nau R, Mitchell TJ, et al. Rapid Microtubule Bundling and Stabilization by the Streptococcus Pneumoniae Neurotoxin Pneumolysin in a Cholesterol-Dependent, non-Lytic and Src-Kinase Dependent Manner Inhibits Intracellular Trafficking. Mol Microbiol (2009) 71:461–77. doi: 10.1111/j.1365-2958.2008.06538.x
103. Förtsch C, Hupp S, Ma J, Mitchell TJ, Maier E, Benz R, et al. Changes in Astrocyte Shape Induced by Sublytic Concentrations of the Cholesterol-Dependent Cytolysin Pneumolysin Still Require Pore-Forming Capacity. Toxins (Basel) (2011) 3:43–62. doi: 10.3390/toxins3010043
104. Zysk G, Schneider-Wald BK, Hwang JH, Bejo L, Kim KS, Mitchell TJ, et al. Pneumolysin is the Main Inducer of Cytotoxicity to Brain Microvascular Endothelial Cells Caused by Streptococcus Pneumoniae. Infect Immun (2001) 69:845–52. doi: 10.1128/IAI.69.2.845-852.2001
105. Hupp S, Grandgirard D, Mitchell TJ, Leib SL, Hathaway LJ, Iliev AI. Pneumolysin and the Bacterial Capsule of Streptococcus Pneumoniae Cooperatively Inhibit Taxis and Motility of Microglia. J Neuroinflamm (2019) 16:105. doi: 10.1186/s12974-019-1491-7
106. Hirst RA, Rutman A, Sikand K, Andrew PW, Mitchell TJ, O'Callaghan C. Effect of Pneumolysin on Rat Brain Ciliary Function: Comparison of Brain Slices With Cultured Ependymal Cells. Pediatr Res (2000) 47:381–4. doi: 10.1203/00006450-200003000-00016
107. Mohammed BJ, Mitchell TJ, Andrew PW, Hirst RA, O'Callaghan C. The Effect of the Pneumococcal Toxin, Pneumolysin on Brain Ependymal Cilia. Microbial Pathog (1999) 27:303–9. doi: 10.1006/mpat.1999.0306
108. Hirst RA, Sikand KS, Rutman A, Mitchell TJ, Andrew PW, O'Callaghan C. Relative Roles of Pneumolysin and Hydrogen Peroxide From Streptococcus Pneumoniae in Inhibition of Ependymal Ciliary Beat Frequency. Infect Immun (2000) 68:1557–62. doi: 10.1128/IAI.68.3.1557-1562.2000
109. Hirst RA, Gosai B, Rutman A, Andrew PW, O'Callaghan C. Streptococcus Pneumoniae Damages the Ciliated Ependyma of the Brain During Meningitis. Infect Immun (2003) 71:6095–100. doi: 10.1128/IAI.71.10.6095-6100.2003
110. Braun JS, Sublett JE, Freyer D, Mitchell TJ, Cleveland JL, Tuomanen EI, et al. Pneumococcal Pneumolysin and H(2)O(2) Mediate Brain Cell Apoptosis During Meningitis. J Clin Invest (2002) 109:19–27. doi: 10.1172/JCI12035
111. Braun JS, Hoffmann O, Schickhaus M, Freyer D, Dagand E, Bermpohl D, et al. Pneumolysin Causes Neuronal Cell Death Through Mitochondrial Damage. Infect Immun (2007) 75:4245–54. doi: 10.1128/IAI.00031-07
112. Nerlich A, Mieth M, Letsiou E, Fatykhova D, Zscheppang K, Imai-Matsushima A, et al. Pneumolysin Induced Mitochondrial Dysfunction Leads to Release of Mitochondrial DNA. Sci Rep (2018) 8:182. doi: 10.1038/s41598-017-18468-7
113. Hu X, Peng X, Lu C, Zhang X, Gan L, Gao Y, et al. Type I IFN Expression is Stimulated by Cytosolic MtDNA Released From Pneumolysin-Damaged Mitochondria via the STING Signaling Pathway in Macrophages. FEBS J (2019) 286:4754–68. doi: 10.1111/febs.15001
114. Hupp S, Förtsch C, Wippel C, Ma J, Mitchell TJ, Iliev AI. Direct Transmembrane Interaction Between Actin and the Pore-Competent, Cholesterol-Dependent Cytolysin Pneumolysin. J Mol Biol (2013) 425:636–46. doi: 10.1016/j.jmb.2012.11.034
115. Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS. Cholesterol-Dependent Actin Remodeling via RhoA and Rac1 Activation by the Streptococcus Pneumoniae Toxin Pneumolysin. Proc Natl Acad Sci USA (2007) 104:2897–902. doi: 10.1073/pnas.0608213104
116. Brito C, Cabanes D, Sarmento Mesquita F, Sousa S. Mechanisms Protecting Host Cells Against Bacterial Pore-Forming Toxins. Cell Mol Life Sci (2019) 76:1319–39. doi: 10.1007/s00018-018-2992-8
117. Porta H, Cancino-Rodezno A, Soberón M, Bravo A. Role of MAPK P38 in the Cellular Responses to Pore-Forming Toxins. Peptides (2011) 32:601–6. doi: 10.1016/j.peptides.2010.06.012
118. Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, et al. Pore-Forming Toxins Activate MAPK P38 by Causing Loss of Cellular Potassium. Biochem Biophys Res Commun (2009) 385:503–6. doi: 10.1016/j.bbrc.2009.05.121
119. Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. Epithelial Cells are Sensitive Detectors of Bacterial Pore-Forming Toxins. J Biol Chem (2006) 281:12994–8. doi: 10.1074/jbc.M511431200
120. Aguilar JL, Kulkarni R, Randis TM, Soman S, Kikuchi A, Yin Y, et al. Phosphatase-Dependent Regulation of Epithelial Mitogen-Activated Protein Kinase Responses to Toxin-Induced Membrane Pores. PloS One (2009) 4:e8076. doi: 10.1371/journal.pone.0008076
121. Das R, LaRose MI, Hergott CB, Leng L, Bucala R, Weiser JN. Macrophage Migration Inhibitory Factor Promotes Clearance of Pneumococcal Colonization. J Immunol (2014) 193:764–72. doi: 10.4049/jimmunol.1400133
122. Lemon JK, Weiser JN. Degradation Products of the Extracellular Pathogen Streptococcus Pneumoniae Access the Cytosol via its Pore-Forming Toxin. mBio 6 (2015) 6:e02110–14. doi: 10.1128/mBio.02110-14
123. Lemon JK, Miller MR, Weiser JN. Sensing of Interleukin-1 Cytokines During Streptococcus Pneumoniae Colonization Contributes to Macrophage Recruitment and Bacterial Clearance. Infect Immun (2015) 83:3204–12. doi: 10.1128/IAI.00224-15
124. Stringaris AK, Geisenhainer J, Bergmann F, Balshüsemann C, Lee U, Zysk G, et al. Neurotoxicity of Pneumolysin, a Major Pneumococcal Virulence Factor, Involves Calcium Influx and Depends on Activation of P38 Mitogen-Activated Protein Kinase. Neurobiol Dis (2002) 11:355–68. doi: 10.1006/nbdi.2002.0561
125. Kwon IS, Kim J, Rhee DK, Kim BO, Pyo S. Pneumolysin Induces Cellular Senescence by Increasing ROS Production and Activation of MAPK/NF-κb Signal Pathway in Glial Cells. Toxicon (2017) 129:100–12. doi: 10.1016/j.toxicon.2017.02.017
126. Wolfmeier H, Radecke J, Schoenauer R, Koeffel R, Babiychuk VS, Drücker P, et al. Active Release of Pneumolysin Prepores and Pores by Mammalian Cells Undergoing a Streptococcus Pneumoniae Attack. Biochim Biophys Acta (2016) 1860:2498–509. doi: 10.1016/j.bbagen.2016.07.022
127. Bischofberger M, Iacovache I, van der Goot FG. Pathogenic Pore-Forming Proteins: Function and Host Response. Cell Host Microbe (2012) 12:266–75. doi: 10.1016/j.chom.2012.08.005
128. Dal Peraro M, van der Goot FG. Pore-Forming Toxins: Ancient, But Never Really Out of Fashion. Nat Rev Microbiol (2016) 14:77–92. doi: 10.1038/nrmicro.2015.3
129. Wolfmeier H, Schoenauer R, Atanassoff AP, Neill DR, Kadioglu A, Draeger A, et al. Ca2+-Dependent Repair of Pneumolysin Pores: A New Paradigm for Host Cellular Defense Against Bacterial Pore-Forming Toxins. Biochim Biophys Acta (2015) 1853:2045–54. doi: 10.1016/j.bbamcr.2014.09.005
130. Jimenez AJ, Perez F. Plasma Membrane Repair: The Adaptable Cell Life-Insurance. Curr Opin Cell Biol (2017) 47:99–107. doi: 10.1016/j.ceb.2017.03.011
131. Bouillot S, Reboud E, Huber P. Functional Consequences of Calcium Influx Promoted by Bacterial Pore-Forming Toxins. Toxins (2018) 10:387. doi: 10.3390/toxins10100387
132. Etxaniz A, González-Bullón D, Martín C, Ostolaza H. Membrane Repair Mechanisms Against Permeabilization by Pore-Forming Toxins. Toxins (2018) 10:234. doi: 10.3390/toxins10060234
133. Wippel C, Förtsch C, Hupp S, Maier E, Benz R, Ma J, et al. Extracellular Calcium Reduction Strongly Increases the Lytic Capacity of Pneumolysin From Streptococcus Pneumoniae in Brain Tissue. J Infect Dis (2011) 204:930–6. doi: 10.1093/infdis/jir434
134. Maurer J, Hupp S, Pillich H, Mitchell TJ, Chakraborty T, Iliev AI. Missing Elimination via Membrane Vesicle Shedding Contributes to the Diminished Calcium Sensitivity of Listeriolysin O. Sci Rep (2018) 8:15846. doi: 10.1038/s41598-018-34031-4
135. Potez S, Luginbühl M, Monastyrskaya K, Hostettler A, Draeger A, Babiychuk EB. Tailored Protection Against Plasmalemmal Injury by Annexins With Different Ca2+ Sensitivities. J Biol Chem (2011) 286:17982–91. doi: 10.1074/jbc.M110.187625
136. Koffel R, Wolfmeier H, Larpin Y, Besancon H, Schoenauer R, Babiychuk VS, et al. Host-Derived Microvesicles Carrying Bacterial Pore-Forming Toxins Deliver Signals to Macrophages: A Novel Mechanism of Shaping Immune Responses. Front Immunol (2018) 9:1688. doi: 10.3389/fimmu.2018.01688
137. Letsiou E, Teixeira Alves LG, Fatykhova D, Felten M, Mitchell TJ, Müller-Redetzky HC, et al. Microvesicles Released From Pneumolysin-Stimulated Lung Epithelial Cells Carry Mitochondrial Cargo and Suppress Neutrophil Oxidative Burst. Sci Rep (2021) 11:9529. doi: 10.1038/s41598-021-88897-y
138. Larpin Y, Besançon H, Iacovache MI, Babiychuk VS, Babiychuk EB, Zuber B, et al. Bacterial Pore-Forming Toxin Pneumolysin: Cell Membrane Structure and Microvesicle Shedding Capacity Determines Differential Survival of Cell Types. FASEB J (2020) 34:1665–78. doi: 10.1096/fj.201901737RR
139. Neill DR, Mitchell TJ, Kadioglu A. Pneumolysin. Streptococcus Pneumoniae Elsevier (2015) 257–75. doi: 10.1016/B978-0-12-410530-0.00014-4
140. Pechous RD. With Friends Like These: The Complex Role of Neutrophils in the Progression of Severe Pneumonia. Front Cell Infect Microbiol (2017) 7:160. doi: 10.3389/fcimb.2017.00160
141. Mitchell TJ, Dalziel CE. The Biology of Pneumolysin. Subcell Biochem (2014) 80:145–60. doi: 10.1007/978-94-017-8881-6_8
142. Cockeran R, Theron AJ, Steel HC, Matlola NM, Mitchell TJ, Feldman C, et al. Proinflammatory Interactions of Pneumolysin With Human Neutrophils. J Infect Dis (2001) 183:604–11. doi: 10.1086/318536
143. Thapa R, Ray S, Keyel PA. Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival. Toxins (Basel) 12 (2020) 12:531–62 doi: 10.3390/toxins12090531
144. Rubins JB, Mitchell TJ, Andrew PW, Niewoehner DE. Pneumolysin Activates Phospholipase A in Pulmonary Artery Endothelial Cells. Infect Immun (1994) 62:3829–36. doi: 10.1128/iai.62.9.3829-3836.1994
145. Rayner CF, Jackson AD, Rutman A, Dewar A, Mitchell TJ, Andrew PW, et al. Interaction of Pneumolysin-Sufficient and -Deficient Isogenic Variants of Streptococcus Pneumoniae With Human Respiratory Mucosa. Infect Immun (1995) 63:442–7. doi: 10.1128/iai.63.2.442-447.1995
146. Fliegauf M, Sonnen AF, Kremer B, Henneke P. Mucociliary Clearance Defects in a Murine In Vitro Model of Pneumococcal Airway Infection. PloS One (2013) 8:e59925. doi: 10.1371/journal.pone.0059925
147. Kim Y-J, Shin H-S, Lee J-H, Jung YW, Kim H-B, Ha U-H. Pneumolysin-Mediated Expression of β-Defensin 2 is Coordinated by P38 MAP Kinase-MKP1 in Human Airway Cells. J Microbiol (2013) 51:194–9. doi: 10.1007/s12275-013-2579-x
148. Yoo IH, Shin HS, Kim YJ, Kim HB, Jin S, Ha UH. Role of Pneumococcal Pneumolysin in the Induction of an Inflammatory Response in Human Epithelial Cells. FEMS Immunol Med Microbiol (2010) 60:28–35. doi: 10.1111/j.1574-695X.2010.00699.x
149. Bhowmick R, Clark S, Bonventre JV, Leong JM, McCormick BA. Cytosolic Phospholipase a(2)α Promotes Pulmonary Inflammation and Systemic Disease During Streptococcus Pneumoniae Infection. Infect Immun 85 (2017) 85:e00280-17 doi: 10.1128/IAI.00280-17
150. Zukauskas A, Mrsny RJ, Cortés Barrantes P, Turner JR, Leong JM, McCormick BA. Transporters MRP1 and MRP2 Regulate Opposing Inflammatory Signals To Control Transepithelial Neutrophil Migration During Streptococcus Pneumoniae Lung Infection. mSphere 3 (2018) 3:e00303-18 doi: 10.1128/mSphere.00303-18
151. Bhowmick R, Maung N, Hurley BP, Ghanem EB, Gronert K, McCormick BA, et al. Systemic Disease During Streptococcus Pneumoniae Acute Lung Infection Requires 12-Lipoxygenase-Dependent Inflammation. J Immunol (2013) 191:5115–23. doi: 10.4049/jimmunol.1300522
152. Bhowmick R, Clark S, Bonventre JV, Leong JM, McCormick BA. Cytosolic Phospholipase A2alpha Promotes Pulmonary Inflammation and Systemic Disease During Streptococcus Pneumoniae Infection. Infect Immun 85 (2017) 85:e00280-17. doi: 10.1128/IAI.00280-17
153. Bou Ghanem EN, Clark S, Roggensack SE, McIver SR, Alcaide P, Haydon PG, et al. Extracellular Adenosine Protects Against Streptococcus Pneumoniae Lung Infection by Regulating Pulmonary Neutrophil Recruitment. PloS Pathog (2015) 11:e1005126. doi: 10.1371/journal.ppat.1005126
154. Cuypers F, Klabunde B, Gesell Salazar M, Surabhi S, Skorka SB, Burchhardt G, et al. Adenosine Triphosphate Neutralizes Pneumolysin-Induced Neutrophil Activation. J Infect Dis (2020) 222:1702–12. doi: 10.1093/infdis/jiaa277
155. Fickl H, Cockeran R, Steel HC, Feldman C, Cowan G, Mitchell TJ, et al. Pneumolysin-Mediated Activation of NFkappaB in Human Neutrophils is Antagonized by Docosahexaenoic Acid. Clin Exp Immunol (2005) 140:274–81. doi: 10.1111/j.1365-2249.2005.02757.x
156. Cockeran R, Steel HC, Mitchell TJ, Feldman C, Anderson R. Pneumolysin Potentiates Production of Prostaglandin E(2) and Leukotriene B(4) by Human Neutrophils. Infect Immun (2001) 69:3494–6. doi: 10.1128/IAI.69.5.3494-3496.2001
157. Martner A, Dahlgren C, Paton JC, Wold AE. Pneumolysin Released During Streptococcus Pneumoniae Autolysis is a Potent Activator of Intracellular Oxygen Radical Production in Neutrophils. Infect Immun (2008) 76:4079–87. doi: 10.1128/IAI.01747-07
158. Segal AW. How Neutrophils Kill Microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
159. Nganje CN, Haynes SA, Qabar CM, Lent RC, Bou Ghanem EN, Shainheit MG. PepN is a non-Essential, Cell Wall-Localized Protein That Contributes to Neutrophil Elastase-Mediated Killing of Streptococcus Pneumoniae. PloS One (2019) 14:e0211632. doi: 10.1371/journal.pone.0211632
160. Standish AJ, Weiser JN. Human Neutrophils Kill Streptococcus Pneumoniae via Serine Proteases. J Immunol (2009) 183:2602–9. doi: 10.4049/jimmunol.0900688
161. Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, et al. Mice Lacking Neutrophil Elastase Reveal Impaired Host Defense Against Gram Negative Bacterial Sepsis. Nat Med (1998) 4:615–8. doi: 10.1038/nm0598-615
162. Domon H, Terao Y. The Role of Neutrophils and Neutrophil Elastase in Pneumococcal Pneumonia. Front Cell Infect Microbiol (2021) 11:615959. doi: 10.3389/fcimb.2021.615959
163. Watanabe H, Hattori S, Katsuda S, Nakanishi I, Nagai Y. Human Neutrophil Elastase: Degradation of Basement Membrane Components and Immunolocalization in the Tissue. J Biochem (1990) 108:753–9. doi: 10.1093/oxfordjournals.jbchem.a123277
164. Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, et al. Neutrophil Elastase Cleaves Epithelial Cadherin in Acutely Injured Lung Epithelium. Respir Res (2016) 17:129. doi: 10.1186/s12931-016-0449-x
165. Witherden IR, Vanden Bon EJ, Goldstraw P, Ratcliffe C, Pastorino U, Tetley TD. Primary Human Alveolar Type II Epithelial Cell Chemokine Release: Effects of Cigarette Smoke and Neutrophil Elastase. Am J Respir Cell Mol Biol (2004) 30:500–9. doi: 10.1165/rcmb.4890
166. Chen HC, Lin HC, Liu CY, Wang CH, Hwang T, Huang TT, et al. Neutrophil Elastase Induces IL-8 Synthesis by Lung Epithelial Cells via the Mitogen-Activated Protein Kinase Pathway. J BioMed Sci (2004) 11:49–58. doi: 10.1007/BF02256548
167. Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP. Leukocyte Elastase Induces Lung Epithelial Apoptosis via a PAR-1-, NF-kappaB-, and P53-Dependent Pathway. Am J Respir Cell Mol Biol (2009) 41:742–55. doi: 10.1165/rcmb.2008-0157OC
168. Hagio T, Kishikawa K, Kawabata K, Tasaka S, Hashimoto S, Hasegawa N, et al. Inhibition of Neutrophil Elastase Reduces Lung Injury and Bacterial Count in Hamsters. Pulm Pharmacol Ther (2008) 21:884–91. doi: 10.1016/j.pupt.2008.10.002
169. Wilkinson TS, Conway Morris A, Kefala K, O'Kane CM, Moore NR, Booth NA, et al. Ventilator-Associated Pneumonia is Characterized by Excessive Release of Neutrophil Proteases in the Lung. Chest (2012) 142:1425–32. doi: 10.1378/chest.11-3273
170. Tagami T, Kushimoto S, Tosa R, Omura M, Yonezawa K, Akiyama G, et al. Plasma Neutrophil Elastase Correlates With Pulmonary Vascular Permeability: A Prospective Observational Study in Patients With Pneumonia. Respirology (2011) 16:953–8. doi: 10.1111/j.1440-1843.2011.01997.x
171. Ginzberg HH, Cherapanov V, Dong Q, Cantin A, McCulloch CA, Shannon PT, et al. Neutrophil-Mediated Epithelial Injury During Transmigration: Role of Elastase. Am J Physiol Gastrointest Liver Physiol 281 (2001) 3:G705–17. doi: 10.1152/ajpgi.2001.281.3.G705
172. Moraes TJ, Zurawska JH, Downey GP. Neutrophil Granule Contents in the Pathogenesis of Lung Injury. Curr Opin Hematol (2006) 13:21–7. doi: 10.1097/01.moh.0000190113.31027.d5
173. Cockeran R, Mitchell TJ, Feldman C, Anderson R. Pneumolysin Induces Release of Matrix Metalloproteinase-8 and -9 From Human Neutrophils. Eur Respir J (2009) 34:1167–70. doi: 10.1183/09031936.00007109
174. Hartog CM, Wermelt JA, Sommerfeld CO, Eichler W, Dalhoff K, Braun J. Pulmonary Matrix Metalloproteinase Excess in Hospital-Acquired Pneumonia. Am J Respir Crit Care Med (2003) 167:593–8. doi: 10.1164/rccm.200203-258OC
175. Nguyen HX, O'Barr TJ, Anderson AJ. Polymorphonuclear Leukocytes Promote Neurotoxicity Through Release of Matrix Metalloproteinases, Reactive Oxygen Species, and TNF-Alpha. J Neurochem (2007) 102:900–12. doi: 10.1111/j.1471-4159.2007.04643.x
176. Schaaf B, Liebau C, Kurowski V, Droemann D, Dalhoff K. Hospital Acquired Pneumonia With High-Risk Bacteria is Associated With Increased Pulmonary Matrix Metalloproteinase Activity. BMC Pulm Med (2008) 8:12. doi: 10.1186/1471-2466-8-12
177. Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S. Alpha-Enolase of Streptococcus Pneumoniae Induces Formation of Neutrophil Extracellular Traps. J Biol Chem (2012) 287:10472–81. doi: 10.1074/jbc.M111.280321
178. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
179. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil Extracellular Traps Induce Organ Damage During Experimental and Clinical Sepsis. PloS One (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
180. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of Neutrophil Extracellular Traps Predict Adverse Outcome in Community-Acquired Pneumonia: Secondary Analysis of a Randomised Controlled Trial. Eur Respir J 51 (2018) 51:1701389–99. doi: 10.1183/13993003.01389-2017
181. Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, et al. Critical Roles of ASC Inflammasomes in Caspase-1 Activation and Host Innate Resistance to Streptococcus Pneumoniae Infection. J Immunol (2011) 187:4890–9. doi: 10.4049/jimmunol.1100381
182. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin Activates the NLRP3 Inflammasome and Promotes Proinflammatory Cytokines Independently of TLR4. PloS Pathog (2010) 6:e1001191. doi: 10.1371/journal.ppat.1001191
183. Greaney AJ, Leppla SH, Moayeri M. Bacterial Exotoxins and the Inflammasome. Front Immunol (2015) 6:570. doi: 10.3389/fimmu.2015.00570
184. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) Efflux is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
185. Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, et al. Neutrophil IL-1β Processing Induced by Pneumolysin is Mediated by the NLRP3/ASC Inflammasome and Caspase-1 Activation and is Dependent on K+ Efflux. J Immunol (2015) 194:1763–75. doi: 10.4049/jimmunol.1401624
186. LaRock CN, Nizet V. Inflammasome/IL-1beta Responses to Streptococcal Pathogens. Front Immunol (2015) 6:518. doi: 10.3389/fimmu.2015.00518
187. Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, et al. Streptococcus Pneumoniae Stimulates a STING- and IFN Regulatory Factor 3-Dependent Type I IFN Production in Macrophages, Which Regulates RANTES Production in Macrophages, Cocultured Alveolar Epithelial Cells, and Mouse Lungs. J Immunol (2012) 188:811–7. doi: 10.4049/jimmunol.1004143
188. Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, et al. Critical Involvement of Pneumolysin in Production of Interleukin-1alpha and Caspase-1-Dependent Cytokines in Infection With Streptococcus Pneumoniae In Vitro: A Novel Function of Pneumolysin in Caspase-1 Activation. Infect Immun (2008) 76:1547–57. doi: 10.1128/IAI.01269-07
189. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 Inflammasome is Differentially Activated by Pneumolysin Variants and Contributes to Host Defense in Pneumococcal Pneumonia. J Immunol (2011) 187:434–40. doi: 10.4049/jimmunol.1003143
190. Geldhoff M, Mook-Kanamori BB, Brouwer MC, Troost D, Leemans JC, Flavell RA, et al. Inflammasome Activation Mediates Inflammation and Outcome in Humans and Mice With Pneumococcal Meningitis. BMC Infect Dis (2013) 13:358. doi: 10.1186/1471-2334-13-358
191. Weinberger DM, Harboe ZB, Sanders EA, Ndiritu M, Klugman KP, Ruckinger S, et al. Association of Serotype With Risk of Death Due to Pneumococcal Pneumonia: A Meta-Analysis. Clin Infect Dis (2010) 51:692–9. doi: 10.1086/655828
192. Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, et al. The NLRP3 Inflammasome Contributes to Brain Injury in Pneumococcal Meningitis and is Activated Through ATP-Dependent Lysosomal Cathepsin B Release. J Immunol (2011) 187:5440–51. doi: 10.4049/jimmunol.1100790
193. Kim J-Y, Paton JC, Briles DE, Rhee D-K, Pyo S. Streptococcus Pneumoniae Induces Pyroptosis Through the Regulation of Autophagy in Murine Microglia. Oncotarget 6 (2015) 6:44161–78. doi: 10.18632/oncotarget.6592
194. Rogers PD, Thornton J, Barker KS, McDaniel DO, Sacks GS, Swiatlo E, et al. Pneumolysin-Dependent and -Independent Gene Expression Identified by cDNA Microarray Analysis of THP-1 Human Mononuclear Cells Stimulated by Streptococcus Pneumoniae. Infect Immun (2003) 71:2087–94. doi: 10.1128/IAI.71.4.2087-2094.2003
195. Braun JS, Novak R, Gao G, Murray PJ, Shenep JL. Pneumolysin, a Protein Toxin of Streptococcus Pneumoniae, Induces Nitric Oxide Production From Macrophages. Infect Immun (1999) 67:3750–6. doi: 10.1128/IAI.67.8.3750-3756.1999
196. Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, et al. Streptococcus Pneumoniae DNA Initiates Type I Interferon Signaling in the Respiratory Tract. mBio (2011) 2:e00016–11. doi: 10.1128/mBio.00016-11
197. Houldsworth S, Andrew PW, Mitchell TJ. Pneumolysin Stimulates Production of Tumor Necrosis Factor Alpha and Interleukin-1 Beta by Human Mononuclear Phagocytes. Infect Immun (1994) 62:1501–3. doi: 10.1128/iai.62.4.1501-1503.1994
198. Harvey RM, Hughes CE, Paton AW, Trappetti C, Tweten RK, Paton JC. The Impact of Pneumolysin on the Macrophage Response to Streptococcus Pneumoniae is Strain-Dependent. PloS One (2014) 9:e103625. doi: 10.1371/journal.pone.0103625
199. Gilley RP, González-Juarbe N, Shenoy AT, Reyes LF, Dube PH, Restrepo MI, et al. Infiltrated Macrophages Die of Pneumolysin-Mediated Necroptosis Following Pneumococcal Myocardial Invasion. Infect Immun (2016) 84:1457–69. doi: 10.1128/IAI.00007-16
200. González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, et al. Pore-Forming Toxin-Mediated Ion Dysregulation Leads to Death Receptor-Independent Necroptosis of Lung Epithelial Cells During Bacterial Pneumonia. Cell Death Differ (2017) 24:917–28. doi: 10.1038/cdd.2017.49
201. Beno SM, Riegler AN, Gilley RP, Brissac T, Wang Y, Kruckow KL, et al. Inhibition of Necroptosis to Prevent Long-Term Cardiac Damage During Pneumococcal Pneumonia and Invasive Disease. J Infect Dis (2020) 222:1882–93. doi: 10.1093/infdis/jiaa295
202. Riegler AN, Brissac T, Gonzalez-Juarbe N, Orihuela CJ. Necroptotic Cell Death Promotes Adaptive Immunity Against Colonizing Pneumococci. Front Immunol 10 (2019) 615. doi: 10.3389/fimmu.2019.00615
203. Neill DR, Coward WR, Gritzfeld JF, Richards L, Garcia-Garcia FJ, Dotor J, et al. Density and Duration of Pneumococcal Carriage is Maintained by Transforming Growth Factor β1 and T Regulatory Cells. Am J Respir Crit Care Med (2014) 189:1250–9. doi: 10.1164/rccm.201401-0128OC
204. Pahal P, Rajasurya V, Sharma S. Typical Bacterial Pneumonia. Treasure Island (FL: StatPearls (2021).
205. Shin HS, Yoo IH, Kim YJ, Kim HB, Jin S, Ha UH. MKP1 Regulates the Induction of Inflammatory Response by Pneumococcal Pneumolysin in Human Epithelial Cells. FEMS Immunol Med Microbiol (2010) 60:171–8. doi: 10.1111/j.1574-695X.2010.00733.x
206. Subramanian K, Iovino F, Tsikourkitoudi V, Merkl P, Ahmed S, Berry SB, et al. Mannose Receptor-Derived Peptides Neutralize Pore-Forming Toxins and Reduce Inflammation and Development of Pneumococcal Disease. EMBO Mol Med (2020) 12:e12695. doi: 10.15252/emmm.202012695
207. Deretic V, Saitoh T, Akira S. Autophagy in Infection, Inflammation and Immunity. Nat Rev Immunol (2013) 13:722–37. doi: 10.1038/nri3532
208. Li P, Shi J, He Q, Hu Q, Wang YY, Zhang LJ, et al. Streptococcus Pneumoniae Induces Autophagy Through the Inhibition of the PI3K-I/Akt/mTOR Pathway and ROS Hypergeneration in A549 Cells. PloS One (2015) 10:e0122753. doi: 10.1371/journal.pone.0122753
209. Kim J, Lee H-W, Rhee DK, Paton JC, Pyo S. Pneumolysin-Induced Autophagy Contributes to Inhibition of Osteoblast Differentiation Through Downregulation of Sp1 in Human Osteosarcoma Cells. Biochim Biophys Acta (BBA) - Gen Subj (2017) 1861:2663–73. doi: 10.1016/j.bbagen.2017.07.008
210. Ogawa M, Matsuda R, Takada N, Tomokiyo M, Yamamoto S, Shizukuishi S, et al. Molecular Mechanisms of Streptococcus Pneumoniae-Targeted Autophagy via Pneumolysin, Golgi-Resident Rab41, and Nedd4-1-Mediated K63-Linked Ubiquitination. Cell Microbiol (2018) 20:e12846. doi: 10.1111/cmi.12846
211. Ogawa M, Takada N, Shizukuishi S, Tomokiyo M, Chang B, Yoshida M, et al. Streptococcus Pneumoniae Triggers Hierarchical Autophagy Through Reprogramming of LAPosome-Like Vesicles via NDP52-Delocalization. Commun Biol (2020) 3:25. doi: 10.1038/s42003-020-0753-3
212. Inomata M, Xu S, Chandra P, Meydani SN, Takemura G, Philips JA, et al. Macrophage LC3-Associated Phagocytosis is an Immune Defense Against Streptococcus Pneumoniae That Diminishes With Host Aging. Proc Natl Acad Sci (2020) 117:33561–9. doi: 10.1073/pnas.2015368117
213. Jounblat R, Kadioglu A, Mitchell TJ, Andrew PW. Pneumococcal Behavior and Host Responses During Bronchopneumonia are Affected Differently by the Cytolytic and Complement-Activating Activities of Pneumolysin. Infect Immun (2003) 71:1813–9. doi: 10.1128/IAI.71.4.1813-1819.2003
214. Ali YM, Kenawy HI, Muhammad A, Sim RB, Andrew PW, Schwaeble WJ. Human L-Ficolin, a Recognition Molecule of the Lectin Activation Pathway of Complement, Activates Complement by Binding to Pneumolysin, the Major Toxin of Streptococcus Pneumoniae. PloS One (2013) 8:e82583. doi: 10.1371/journal.pone.0082583
215. Bermpohl D, Halle A, Freyer D, Dagand E, Braun JS, Bechmann I, et al. Bacterial Programmed Cell Death of Cerebral Endothelial Cells Involves Dual Death Pathways. J Clin Invest (2005) 115:1607–15. doi: 10.1172/JCI23223
216. Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MKB, Dockrell DH. Nitric Oxide Levels Regulate Macrophage Commitment to Apoptosis or Necrosis During Pneumococcal Infection. FASEB J (2004) 18:1126–8. doi: 10.1096/fj.03-1450fje
217. Bewley MA, Naughton M, Preston J, Mitchell A, Holmes A, Marriott HM, et al. Pneumolysin Activates Macrophage Lysosomal Membrane Permeabilization and Executes Apoptosis by Distinct Mechanisms Without Membrane Pore Formation. mBio (2014) 5:e01710–14. doi: 10.1128/mBio.01710-14
218. Zhou A, Wang H, Lan K, Zhang X, Xu W, Yin Y, et al. Apoptosis Induced by Pneumolysin in Human Endothelial Cells Involves Mitogen-Activated Protein Kinase Phosphorylation. Int J Mol Med (2012) 29:1025–30. doi: 10.3892/ijmm.2012.946
219. Rai P, He F, Kwang J, Engelward BP, Chow VTK. Pneumococcal Pneumolysin Induces DNA Damage and Cell Cycle Arrest. Sci Rep (2016) 6:22972. doi: 10.1038/srep22972
220. Rosch JW, Boyd AR, Hinojosa E, Pestina T, Hu Y, Persons DA, et al. Statins Protect Against Fulminant Pneumococcal Infection and Cytolysin Toxicity in a Mouse Model of Sickle Cell Disease. J Clin Invest (2010) 120:627–35. doi: 10.1172/JCI39843
221. Statt S, Ruan JW, Hung LY, Chang CY, Huang CT, Lim JH, et al. Statin-Conferred Enhanced Cellular Resistance Against Bacterial Pore-Forming Toxins in Airway Epithelial Cells. Am J Respir Cell Mol Biol (2015) 53:689–702. doi: 10.1165/rcmb.2014-0391OC
222. Zhao X, Li H, Wang J, Guo Y, Liu B, Deng X, et al. Verbascoside Alleviates Pneumococcal Pneumonia by Reducing Pneumolysin Oligomers. Mol Pharmacol (2016) 89:376–87. doi: 10.1124/mol.115.100610
223. Zhao X, Liu B, Liu S, Wang L, Wang J. Anticytotoxin Effects of Amentoflavone to Pneumolysin. Biol Pharm Bull (2017) 40:61–7. doi: 10.1248/bpb.b16-00598
224. Li H, Zhao X, Wang J, Dong Y, Meng S, Li R, et al. β-Sitosterol Interacts With Pneumolysin to Prevent Streptococcus Pneumoniae Infection. Sci Rep (2015) 5:17668–8. doi: 10.1038/srep17668
225. Li H, Zhao X, Deng X, Wang J, Song M, Niu X, et al. Insights Into Structure and Activity of Natural Compound Inhibitors of Pneumolysin. Sci Rep (2017) 7:42015. doi: 10.1038/srep42015
226. Lv Q, Zhang P, Quan P, Cui M, Liu T, Yin Y, et al. Quercetin, a Pneumolysin Inhibitor, Protects Mice Against Streptococcus Pneumoniae Infection. Microb Pathog (2020) 140:103934. doi: 10.1016/j.micpath.2019.103934
227. Domon H, Isono T, Hiyoshi T, Tamura H, Sasagawa K, Maekawa T, et al. Clarithromycin Inhibits Pneumolysin Production via Downregulation of Ply Gene Transcription Despite Autolysis Activation. Microbiol Spectr (2021) 9:e0031821. doi: 10.1128/Spectrum.00318-21
228. Paton JC, Lock RA, Hansman DJ. Effect of Immunization With Pneumolysin on Survival Time of Mice Challenged With Streptococcus Pneumoniae. Infect Immun (1983) 40:548–52. doi: 10.1128/iai.40.2.548-552.1983
229. Mann B, Thornton J, Heath R, Wade KR, Tweten RK, Gao G, et al. Broadly Protective Protein-Based Pneumococcal Vaccine Composed of Pneumolysin Toxoid-CbpA Peptide Recombinant Fusion Protein. J Infect Dis (2014) 209:1116–25. doi: 10.1093/infdis/jit502
230. Alexander JE, Lock RA, Peeters CC, Poolman JT, Andrew PW, Mitchell TJ, et al. Immunization of Mice With Pneumolysin Toxoid Confers a Significant Degree of Protection Against at Least Nine Serotypes of Streptococcus Pneumoniae. Infect Immun (1994) 62:5683–8. doi: 10.1128/iai.62.12.5683-5688.1994
231. Kirkham L-AS, Kerr AR, Douce GR, Paterson GK, Dilts DA, Liu D-F, et al. Construction and Immunological Characterization of a Novel Nontoxic Protective Pneumolysin Mutant for Use in Future Pneumococcal Vaccines. Infect Immun (2006) 74:586. doi: 10.1128/IAI.74.1.586-593.2006
232. Ogunniyi AD, Woodrow MC, Poolman JT, Paton JC. Protection Against Streptococcus Pneumoniae Elicited by Immunization With Pneumolysin and CbpA. Infect Immun (2001) 69:5997–6003. doi: 10.1128/IAI.69.10.5997-6003.2001
233. Denoël P, Philipp MT, Doyle L, Martin D, Carletti G, Poolman JT. A Protein-Based Pneumococcal Vaccine Protects Rhesus Macaques From Pneumonia After Experimental Infection With Streptococcus Pneumoniae. Vaccine (2011) 29:5495–501. doi: 10.1016/j.vaccine.2011.05.051
234. Nel JG, Theron AJ, Durandt C, Tintinger GR, Pool R, Mitchell TJ, et al. Pneumolysin Activates Neutrophil Extracellular Trap Formation. Clin Exp Immunol (2016) 184:358–67. doi: 10.1111/cei.12766
235. Verhoeven D, Perry S, Pichichero ME. Contributions to Protection From Streptococcus Pneumoniae Infection Using the Monovalent Recombinant Protein Vaccine Candidates PcpA, PhtD, and PlyD1 in an Infant Murine Model During Challenge. Clin Vaccine Immunol (2014) 21:1037–45. doi: 10.1128/CVI.00052-14
236. Leroux-Roels G, Maes C, De Boever F, Traskine M, Rüggeberg JU, Borys D. Safety, Reactogenicity and Immunogenicity of a Novel Pneumococcal Protein-Based Vaccine in Adults: A Phase I/II Randomized Clinical Study. Vaccine (2014) 32:6838–46. doi: 10.1016/j.vaccine.2014.02.052
237. Prymula R, Pazdiora P, Traskine M, Rüggeberg JU, Borys D. Safety and Immunogenicity of an Investigational Vaccine Containing Two Common Pneumococcal Proteins in Toddlers: A Phase II Randomized Clinical Trial. Vaccine (2014) 32:3025–34. doi: 10.1016/j.vaccine.2014.03.066
238. Green AE, Howarth D, Chaguza C, Echlin H, Langendonk RF, Munro C, et al. Pneumococcal Colonization and Virulence Factors Identified Via Experimental Evolution in Infection Models. Mol Biol Evol (2021) 38:2209–26. doi: 10.1093/molbev/msab018
239. Christie MP, Johnstone BA, Tweten RK, Parker MW, Morton CJ. Cholesterol-Dependent Cytolysins: From Water-Soluble State to Membrane Pore. Biophys Rev (2018) 10:1337–48. doi: 10.1007/s12551-018-0448-x
Keywords: Streptococcus pneumoniae, pore-forming toxin, cholesterol-dependent cytolysin, plasma membrane, pneumonia, pro- and anti-inflammatory immune responses, pneumolysin
Citation: Pereira JM, Xu S, Leong JM and Sousa S (2022) The Yin and Yang of Pneumolysin During Pneumococcal Infection. Front. Immunol. 13:878244. doi: 10.3389/fimmu.2022.878244
Received: 17 February 2022; Accepted: 23 March 2022;
Published: 22 April 2022.
Edited by:
Alice Prince, Columbia University, United StatesReviewed by:
Elaine I. Tuomanen, St. Jude Children’s Research Hospital, United StatesGee W. Lau, University of Illinois at Urbana-Champaign, United States
Sven Hammerschmidt, University of Greifswald, Germany
Karthik Subramanian, Rajiv Gandhi Centre for Biotechnology, India
Anirban Banerjee, Indian Institute of Technology Bombay, India
Copyright © 2022 Pereira, Xu, Leong and Sousa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Sousa, srsousa@ibmc.up.pt
†These authors share first authorship