 Hillard M. Lazarus1*
Hillard M. Lazarus1* Katherine Pitts2
Katherine Pitts2 Tisha Wang3
Tisha Wang3 Elinor Lee3
Elinor Lee3 Elizabeth Buchbinder4,5,6
Elizabeth Buchbinder4,5,6 Michael Dougan4,5,7
Michael Dougan4,5,7 David G. Armstrong8
David G. Armstrong8 Robert Paine III9
Robert Paine III9 Carolyn E. Ragsdale2
Carolyn E. Ragsdale2 Timothy Boyd10Edwin P. Rock10
Timothy Boyd10Edwin P. Rock10 Robert Peter Gale11
Robert Peter Gale11- 1Department of Medicine, Division of Hematology and Oncology, Case Western Reserve University, Cleveland, OH, United States
- 2Medical Affairs, Partner Therapeutics, Inc., Lexington, MA, United States
- 3Division of Pulmonary, Critical Care, and Sleep Medicine, David Geffen School of Medicine at University of California, Los Angeles (UCLA), Los Angeles, CA, United States
- 4Department of Medicine, Harvard Medical School, Boston, MA, United States
- 5Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 6Department of Medicine, Brigham and Women’s Hospital, Boston, MA, United States
- 7Division of Gastroenterology, Department of Medicine, Massachusetts General Hospital, Boston, MA, United States
- 8Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
- 9Division of Respiratory, Critical Care, and Occupational Pulmonary Medicine, University of Utah, Salt Lake City, UT, United States
- 10Clinical Development, Partner Therapeutics, Inc., Lexington, MA, United States
- 11Hematology Centre, Department of Immunology and Inflammation, Imperial College, London, United Kingdom
Introduction: Endogenous granulocyte-macrophage colony-stimulating factor (GM-CSF), identified by its ability to support differentiation of hematopoietic cells into several types of myeloid cells, is now known to support maturation and maintain the metabolic capacity of mononuclear phagocytes including monocytes, macrophages, and dendritic cells. These cells sense and attack potential pathogens, present antigens to adaptive immune cells, and recruit other immune cells. Recombinant human (rhu) GM-CSF (e.g., sargramostim [glycosylated, yeast-derived rhu GM-CSF]) has immune modulating properties and can restore the normal function of mononuclear phagocytes rendered dysfunctional by deficient or insufficient endogenous GM-CSF.
Methods: We reviewed the emerging biologic and cellular effects of GM-CSF. Experts in clinical disease areas caused by deficient or insufficient endogenous GM-CSF examined the role of GM-CSF in mononuclear phagocyte disorders including autoimmune pulmonary alveolar proteinosis (aPAP), diverse infections (including COVID-19), wound healing, and anti-cancer immune checkpoint inhibitor therapy.
Results: We discuss emerging data for GM-CSF biology including the positive effects on mitochondrial function and cell metabolism, augmentation of phagocytosis and efferocytosis, and immune cell modulation. We further address how giving exogenous rhu GM-CSF may control or treat mononuclear phagocyte dysfunction disorders caused or exacerbated by GM-CSF deficiency or insufficiency. We discuss how rhu GM-CSF may augment the anti-cancer effects of immune checkpoint inhibitor immunotherapy as well as ameliorate immune-related adverse events.
Discussion: We identify research gaps, opportunities, and the concept that rhu GM-CSF, by supporting and restoring the metabolic capacity and function of mononuclear phagocytes, can have significant therapeutic effects. rhu GM-CSF (e.g., sargramostim) might ameliorate multiple diseases of GM-CSF deficiency or insufficiency and address a high unmet medical need.
1. Introduction
Granulocyte-macrophage colony-stimulating factor (GM-CSF) was identified in the 1960s as a myeloid growth factor, purified in the 1970s, molecularly-cloned in the 1980s, and clinically developed in the 1990s (1). Sargramostim (Leukine®; Partner Therapeutics, Inc., Lexington, MA) is a glycosylated, yeast-derived recombinant human granulocyte-macrophage colony-stimulating factor (rhu GM-CSF), FDA-approved for 6 disease indications based on its safe and efficacious hematopoietic growth factor function, differing from human GM-CSF by one amino acid at position 23, where leucine is substituted for arginine (2). Its primary licensed use is for myeloid reconstitution after autologous or allogeneic blood and bone marrow transplantation (2). It is also used to shorten time to neutrophil recovery induced by chemotherapy for acute myeloid leukemia and as a medical countermeasure to treat people exposed to sufficient radiation to cause severe myelosuppression (2, 3). Here, we review emerging pleiotropic effects and therapeutic uses of GM-CSF and highlight results of recent and ongoing sargramostim clinical trials.
The hematopoietic growth factor medication class includes both rhu GM-CSF (e.g., sargramostim) and rhu G-CSF; however, these products are not interchangeable. They differ in mechanism due to different receptors expressed on overlapping yet different target cells (4, 5). The G-CSF receptor is mainly expressed on neutrophils and bone marrow precursor cells, whereas the GM-CSF receptor is more broadly expressed on neutrophils, monocytes, eosinophils, and basophils.
Innovatively identifying and classifying diseases in terms of relative or absolute GM-CSF deficiency or insufficiency, and associated host cell dysfunction, have facilitated the recent investigations demonstrating the immunomodulatory functions of rhu GM-CSF on mononuclear phagocyte target cells (Table 1) (32). While beyond the scope of this paper, ongoing research in other disease states, such as neurodegenerative disorders, may also demonstrate potential effects of innate immune system modulation on patient outcomes (33–36). The mononuclear phagocyte system is a network of cells including monocytes, macrophages, and dendritic cells which are similar in their ability to sense and migrate to potential pathogens, cytotoxically engulf pathogens or dying cells, present antigens to adaptive immune cells, and secrete mediators to recruit additional immune cells (37). There is evidence that in diseases of GM-CSF deficiency and insufficiency, therapeutic use of exogenous rhu GM-CSF administration may augment mononuclear phagocyte function and correct for disease pathogenesis (28, 29, 38–67).

Table 1 Mechanisms and effects of GM-CSF deficiency or insufficiency disorders.
Three described rhu GM-CSF formulations differ in their glycosylation based on the expression system in which they’re produced (68, 69). Glycosylation in turn influences pharmacokinetics, biologic activity, and safety of each formulation. Molgramostim is produced in prokaryotic Escherichia coli, hence is not glycosylated, and regramostim is mammalian-derived from Chinese hamster ovary cells, hence has mammalian glycosylation; these two formulations are not commercially available (68, 69). Marketed sargramostim is yeast-derived with glycosylation similar to that of native GM-CSF (2). Of the three described rhu GM-CSFs, sargramostim glycosylation closely resembles that of native GM-CSF leading to comparable biologic activity, stability, resistance to degradation, tolerability, and immunogenicity (68, 69).
Sargramostim may be effective in multiple GM-CSF deficiency and insufficiency states. Sections to follow are organized by clinical disease area and information provided by experts in each therapeutic area who are studying sargramostim in clinical research. Sections include: emerging biology of GM-CSF, autoimmune pulmonary alveolar proteinosis (aPAP), infection, wound healing, and enhanced anti-cancer potential yet mitigation of immune checkpoint inhibitor immune-related adverse events.
2. Emerging biology of GM-CSF
In addition to myelopoietic actions, GM-CSF possesses anti-apoptotic effects and is reported to induce proliferation, mobilization, and activation of hematopoietic stem cells (70, 71), endothelial progenitor cells (6, 67), mesenchymal stromal cells (7, 8), pericytes (9), neural stem cells (72–76), and oligodendrocyte progenitor cells (76).
2.1. GM-CSF plays a crucial role in mitochondrial biogenesis and function
GM-CSF is crucial for mitochondrial maintenance in mononuclear phagocytes, as modeled in murine HIV studies (77). Furthermore, gene knockout animals reveal that GM-CSF influences mitochondrial turnover, function, and fatty acid β-oxidation (78). GM-CSF increases mitochondrial tricarboxylic acid cycle activity, oxidative phosphorylation, ATP production, and regulation of key metabolic pathways, such as glycolysis, pentose phosphate pathway, and amino acid synthesis. Magmas (mitochondria-associated granulocyte-macrophage CSF signaling molecule) is rapidly induced in vitro when murine myeloid-cell-line PGMD1 cells in culture are switched from IL-3 to GM-CSF in the medium (79). Also known as Tim16 in mammals, Pam16 in yeast, and Blp in drosophilia, Magmas is conserved across species, essential for cell growth, and anti-apoptotic when over-expressed (80–83). Magmas gene knockout mice die as embryos. RNAi-mediated knockdown of Blp resulted in mitochondrial membrane depolarization, 60% decreased ATP levels, 3.5-fold higher reactive oxygen species (ROS), cell-cycle arrest, autophagy activation, and 65% reduced cytochrome c oxidase activity in the mitochondrial electron transport chain (84). Magmas additionally functions as a ROS sensor and regulator, leading to reduced cellular ROS production (85).
2.2. GM-CSF supports efferocytosis
In addition to other effects that induce phagocyte populations (12, 86–90), GM-CSF supports efferocytosis, an energy intensive process in which macrophages engulf and digest apoptotic cells, such as short-lived tissue-infiltrating neutrophils, thereby preventing release or accumulation of necrotic inflammatory material (13). Decreased efferocytosis is associated with tissue necrosis and autoimmune disease (91). Opsonizing milk fat globule epidermal growth factor 8 (MFG–E8) bridges the “eat me” signal of phosphatidylserine displayed on apoptotic cell membranes with integrins αVβ5 and potentially αVβ3 on efferocytotic phagocytes (92). GM-CSF is required for expression of MFG-E8 by efferocytotic antigen-presenting cells (APCs) (93, 94) and induces integrins αVβ3 and αVβ5 (95, 96).
In addition, growth arrest-specific protein 6 (GAS6) bridges MerTK receptors on efferocytotic phagocytes to phosphatidylserine on the apoptotic cell external plasma membrane (91). Murine GM-CSF-induced bone marrow-derived macrophages express MerTK receptors and exhibit high phagocytic ability (97). Human macrophages differentiated with GM-CSF from healthy adult monocyte samples, and which were cultured either under growth/serum factor deficiency, or with subsequent treatment of IL-4 or incubation with apoptotic cells, result in M2-polarized macrophages that exhibit increased MerTK expression (98). Finally, activated peroxisome proliferator-activated receptor (PPAR)-γ and liver X receptor (LXR)-α drive anti-inflammatory macrophage engulfment of apoptotic cells (91). GM-CSF induces expression of LXRα in human blood mononuclear cells (99) and induces PPAR-γ expression in multiple myeloid cell types (100, 101).
2.3. GM-CSF modulates innate and adaptive immunity
GM-CSF broadly affects neutrophil biology via neutrophil induction (2), in particular by enhancing pro-survival effects (102). Oddly and reflective of cytokine pleiotropy, GM-CSF also facilitates auto-phagocyte-like neutrophil cell death (103). Also, GM-CSF downregulates chemotaxis via loss of signaling in response to Interleukin-8 (IL-8), the primary neutrophil chemotactic factor, whereas N-formyl-methyl-leucyl-phenylalanine (fMLP) chemotaxis is maintained (104). Finally, GM-CSF down-regulates neutrophil IL-8 receptor expression (105). In summary, GM-CSF might either facilitate or inhibit neutrophil chemotaxis depending on local environmental influences.
Separately, GM-CSF prevents blood neutrophil extravasation into tissues. L-Selectin mediates neutrophil trans-endothelial migration and is rapidly shed after activation and during the rolling phase of extravasation (106). ADAM17 (a disintegrin and metalloproteinase 17), also known as tumor necrosis factor-alpha-converting enzyme (TACE), is the principal “sheddase” that cleaves surface L-Selectin. Interestingly, ADAM17 sheddase activity acts on neutrophils but not monocytes. Consistent with its stimulation of ADAM17 expression, GM-CSF induces rapid, complete loss of L-Selectin, also known as leukocyte adhesion molecule-1 (LAM-1), from neutrophils, monocytes, and marrow cells but not lymphocytes (107).
Although its receptors are not expressed on lymphocytes, GM-CSF indirectly induces regulatory T cells (Tregs) in multiple autoimmune and chronic inflammatory disease models (108, 109). GM-CSF-deficient APCs exposed to MFG-E8-opsonized apoptotic cells produce altered cytokine profiles, resulting in decreased Tregs and increased inflammatory Th1 cells (93). GM-CSF also induces myeloid-derived suppressor cells (MDSCs) that suppress pro-inflammatory cytokine production, inhibit T cell proliferation, mediate chemotaxis, and activate Tregs (110–112).
In summary, GM-CSF influences a myriad of primarily myeloid cells, in part due to maturation and maintenance of metabolic capacity both systemically and locally, although the specific concentration that drives this change is yet to be determined (113). Follow-on effects include enhancement of phagocytosis and efferocytosis, as well as modulation of other immune cells including neutrophils and Tregs. Together, these data suggest that therapeutic rhu GM-CSF (e.g., sargramostim) might generate benefit in diseases characterized by mononuclear phagocyte dysfunction or dysregulation.
3. Autoimmune pulmonary alveolar proteinosis (aPAP): A GM-CSF deficiency state
3.1. aPAP pathophysiology
High titers of neutralizing GM-CSF autoantibodies in aPAP lead to multiple effects that drive pathophysiology of this disease. These effects include reduced alveolar macrophage cholesterol clearance, impaired surfactant homeostasis, dysfunctional immune defense, and in a subset of patients, pulmonary fibrosis and end stage lung disease (6, 7, 114, 115). As reviewed above in Emerging biology of GM-CSF, GM-CSF deficiency impairs the expression of PPAR-γ, a key cholesterol regulator, leading to surfactant lipid accumulation within foamy alveolar macrophages (116, 117). GM-CSF autoantibodies also diminish neutrophil phagocytic antimicrobial functions and may lessen alveolar epithelial cell-derived GM-CSF activation and recruitment of alveolar macrophages, dendritic cells, and T cells (118). Reduced immune cell signaling and impaired gas exchange from surfactant accumulation contribute to increased incidence (13-25%) of opportunistic infections from organisms including Aspergillus, Cryptococcus, Nocardia, or atypical mycobacteria (7, 115, 119, 120). Because aPAP is a very rare disorder, the true prevalence of infection in this patient population and its associated mortality remain unclear.
Fibrosis, an uncommon but severe complication of aPAP, probably results from multiple mechanisms (114). Type II alveolar epithelial cells produce GM-CSF that aids in alveolar epithelial cell repair, leading to epithelial proliferation and barrier restoration (11, 118). In the presence of neutralizing GM-CSF autoantibodies, these homeostatic processes are impaired (6). Also, absence of GM-CSF results in lipid composition changes within the alveolar space that may lead to reduced synthesis of antifibrotic prostaglandin PGE2, which may enhance fibrogenesis, especially in the presence of additional insults (121, 122). The relationship between GM-CSF deficiency and fibrogenesis has been studied in murine models to date; human studies are needed yet are challenging in this rare disease (122). With the progression of pulmonary fibrosis, patients may develop severe, irreversible lung dysfunction for which the only known effective treatment is lung transplantation.
3.2 aPAP clinical investigations and gaps
Inhaled rhu GM-CSF is a potentially disease-modifying therapy with promising applications in aPAP, a mononuclear phagocyte disease. The inhaled route of administration delivers high drug concentrations directly to the disease site in the lung (123, 124). Clinical studies of inhaled rhu GM-CSF in aPAP have focused on achieving disease control or slowing or preventing disease progression. Trial endpoints have included measures of lung gas exchange, in particular diffusing capacity of the lungs for carbon monoxide (DLCO), exercise capacity, symptoms, and health-related quality of life. Inhaled rhu GM-CSF has been reported in clinical trials to improve clinical outcomes (42, 125). Table 2 summarizes phase 2-3 studies of sargramostim and molgramostim that report benefit in achieving disease control for patients with aPAP. The adverse event data are reported in Supplement Table S1.
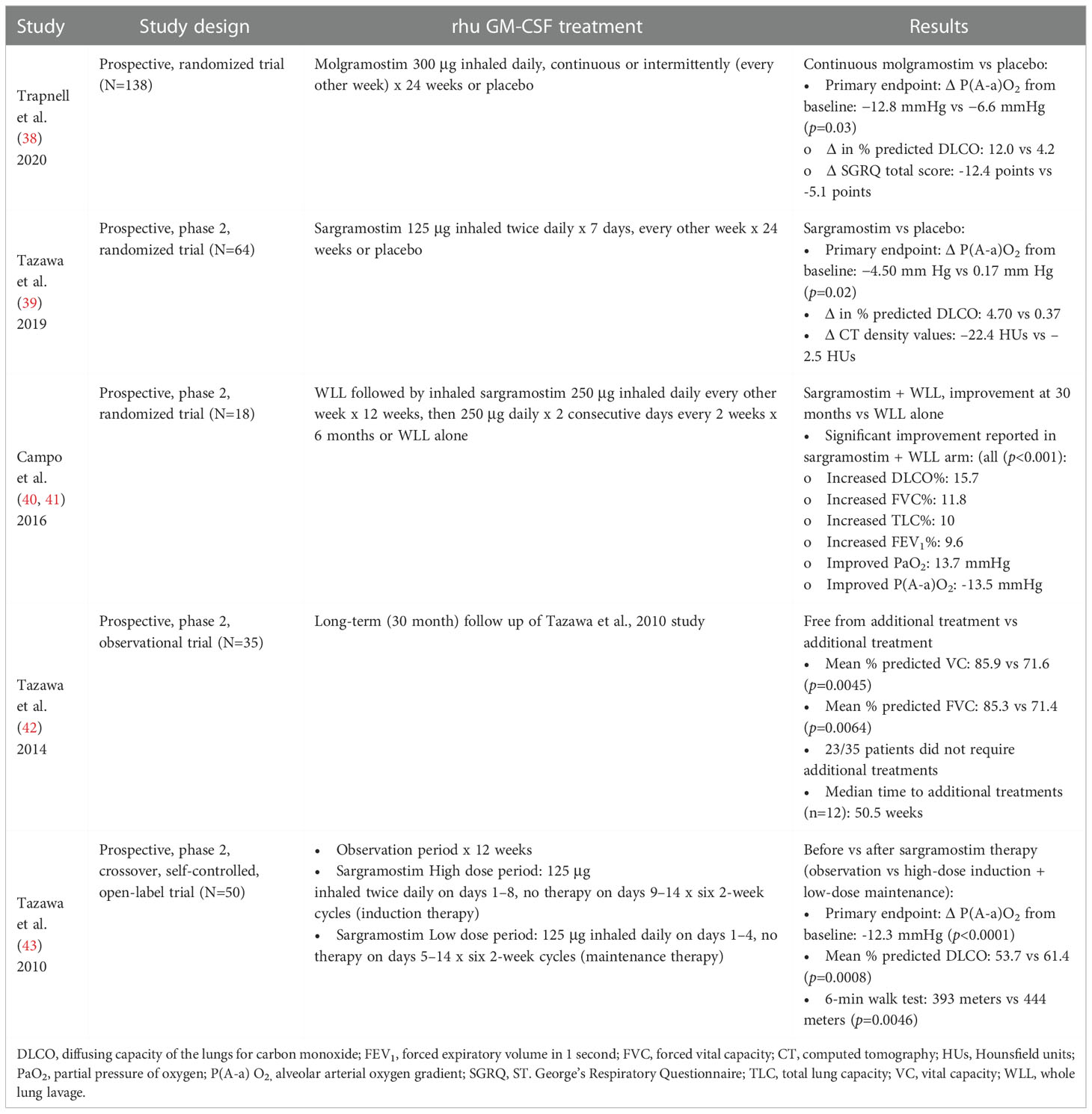
Table 2 Inhaled rhu GM-CSF phase 2-3 clinical studies in aPAP.
Trapnell et al. and Tazawa et al. reported benefits utilizing inhaled molgramostim and sargramostim, respectively, compared to placebo in aPAP patients (38, 39). Campo et al. reported that sargramostim combined with whole lung lavage (WLL; the current standard of care for therapy in aPAP) was safe and more effective than WLL alone (40, 41). Also, in a case series of 5 patients with aPAP, Ohkouchi et al. (126) reported that inhaled sargramostim given after WLL significantly improved disease severity score parameters. These parameters included biomarkers such as mucin-like glycoprotein KL-6, carcinoembryonic antigen (CEA), and lactate dehydrogenase (LDH), as well as markers of oxygenation including alveolar-arterial oxygen gradient (A-aDO2) and partial pressure of oxygen (PaO2). Sargramostim given only before WLL did not improve these parameters. Although optimal dosing and duration of therapy have yet to be established, results of current ongoing research and real-world evidence are eagerly awaited (127). A disease-modifying therapy for aPAP is a high unmet need to slow disease progression, reduce infectious complications, and prevent pulmonary fibrosis and death.
3.3. Potential future developments in aPAP
Of the three rhu GM-CSF formulations described in Introduction (sargramostim, molgramostim, and regramostim), sargramostim is the only form that is currently commercially available. Sargramostim can be sourced from the United States and obtained globally through a “named patient program” per each nation’s healthcare governing body (128). Based on data mentioned here and additional case reports (126), sargramostim use may decrease healthcare utilization in this rare lung disease population. In a study evaluating 15 million people in the US from 2008 to 2012, patients with PAP were determined to have more outpatient visits (17.30 ± 13.77 vs 10.40 ± 11.38; p < 0.01), more emergency room visits (1.49 ± 1.17 vs 1.08 ± 0.27; p = 0.014), and longer hospital stays (15.96 ± 20.71 vs 5.40 ± 5.07 inpatient days; p = 0.027), compared with non-PAP controls (129). Annual per-patient healthcare costs were also 5-fold higher (approximately $40,000 more annually) for PAP patients than for non-PAP controls. Increased costs were attributed to disease-related treatments, including prescriptions, hospitalizations, and outpatient visits. In another retrospective cohort study of 500 U.S. patients admitted with a primary diagnosis of PAP between 2012 and 2014, mean actual cost per admission was $29,932 (CI: 13,739-46,124) with an overall annual cost burden of approximately $5 million (130).
Timely, accurate aPAP diagnosis also remains an issue due to disease rarity, low physician awareness, and limited access to the blood test for serum GM-CSF antibodies, which is performed in few centers worldwide. Testing centers include the CAP/CLIA certified lab at Cincinnati Children’s Hospital, National Jewish Health, the National Institutes of Health, and Cincinnati Children’s Hospital Pulmonary Alveolar Proteinosis Clinical Research Lab (131–133). Additional testing centers can be found in Japan, Germany, and China. Similar to other rare diseases, a patient advocacy organization has emerged (www.papfoundation.org) with the goal to unite the patient community and to connect patients with the specialist physician community for access to appropriate testing and relevant clinical trials.
Sargramostim is not approved by the FDA for use in aPAP, which limits access, reimbursement, and manufacturer ability to provide label information on safe and effective use in this setting. Also, broader sargramostim use in aPAP is limited by the absence of aPAP clinical consensus guidelines. Important questions to be addressed include impact and timing of treatment for asymptomatic or mildly symptomatic patients, as well as ideal dose and treatment duration for those with more severe disease. An ongoing international, multi-center, placebo-controlled trial of molgramostim will provide more information on dosing, efficacy, and safety data for the rhu GM-CSF agent class (127). Other aPAP treatment agents that upregulate PPAR-γ (e.g., thiazolidinediones) or lower cholesterol (e.g., statins) show preclinical promise and could deploy additional repurposed therapeutics available with known safety (134–136). Answers to these clinical questions and more are critical to patients and providers and will hopefully be elucidated via continued investigation of the potential of sargramostim and other therapies to modulate disease and prevent infection and fibrosis.
4. Immune responses to infections and risk of GM-CSF insufficiency
4.1. Infection pathophysiology
Mononuclear phagocyte dysfunction due to GM-CSF insufficiency can contribute to disease (e.g., sepsis) precipitated by various events including trauma, major surgery, and hematopoietic cell transplant (HCT) (137, 138). Viral, bacterial, or fungal opportunistic infections all can cause sepsis and life-threatening organ dysfunction (139, 140). The focus of this section is viral respiratory pathogens and the delicate balance between an effective host response to eliminate respiratory viral infections versus an inadequate or even an overactive immune response to sepsis. The subset of patients who experience these inadequate or overactive immune responses may suffer serious or even fatal adverse events (8, 141, 142).
Insights garnered across dysfunctional mononuclear phagocyte disease states such as COVID-19, pneumonia, sepsis, and intensive care unit (ICU)-related critical illness may potentially be applied to many types of infections. Immunomodulatory agents (e.g., sargramostim) that orchestrate the immune system and behaviors of immune cells for optimal host immune response to different pathogens may be beneficial in improving outcomes for many patients (58, 61, 62, 64, 65). While the hematopoietic growth factor rhu G-CSF (discussed in the Introduction) is more widely prescribed and comprises more than 95% of recombinant growth factor use, its use in infection has not demonstrated a mortality benefit in pneumonia when used in combination with antibiotic therapy (1, 4, 5, 143). G-CSF recruits and increases the number of neutrophils, whereas GM-CSF orchestrates the behavior of many innate and adaptive immune cells to combat pathogens while avoiding an overwhelming systemic response (4, 5). Viral infections and sepsis can be viewed as examples of mononuclear phagocyte dysfunction sequelae and will serve as models for further investigation of pathology, immune responses, and novel treatment strategies across patient populations to decrease morbidity and mortality.
4.1.1. Respiratory viral infection
Respiratory pathogen transmission starts in the upper airway and occurs via direct physical contact, respiratory droplets, and/or airborne dissemination (141, 144). Specifically, viruses then incubate, replicate, and cause symptomatic infection. For immunocompetent patients, many acute respiratory infections are mild, self-limiting, and remain in the upper respiratory tract. For others, the infectious viral load can overwhelm and dysregulate the innate and adaptive immune systems, spread to the lower respiratory tract, and cause lung damage. This immune system dysregulation can ultimately cause life-threatening multiorgan dysfunction due to sequential failures in respiration, coagulation, liver function, cardiovascular status, and renal function (145).
In healthy lungs, alveolar macrophages, DCs, alveolar epithelial cells, and tissue-resident leukocytes continuously patrol and protect tissues from pathogens (118). Alveolar macrophages, which comprise more than 90% of lung leukocytes, are nurtured and controlled by alveolar epithelial cell signaling and phagocytose inhaled foreign particulates without triggering inflammation (8). During respiratory viral infection (likewise in bacterial and fungal infection), the microenvironment quickly changes to an inflammatory state (118). Alveolar macrophages and other lung-resident innate immune cells intercept the viral pathogen (8). Alveolar epithelial cells secrete chemokines and growth factors to recruit and activate neutrophils, monocytes, natural killer cells, and T cells for virus elimination. Lung-resident DCs are the main antigen-presenting cells (APCs) responsible for activating cytotoxic CD8+ and helper CD4+ T cells. Recruited short-lived neutrophils form and release neutrophil extracellular traps to capture viruses and halt viral spread. Neutrophils then undergo apoptosis and are removed by alveolar macrophages via efferocytosis, similar to neutrophil removal by tissue-resident macrophages as discussed in Wound healing and risk of GM-CSF deficiency (8, 17). Alveolar macrophages are often reduced in numbers in the lungs due to dysfunctional type II alveolar epithelial cells which are directly infected by both SARS-CoV-2 and influenza viruses (8, 9). Decreased alveolar macrophage function and numbers lead to dysregulated efferocytosis, prolonged inflammation, and tissue damage.
4.1.2. Sepsis
An overly pro-inflammatory immune response leads to systemic inflammatory response syndrome (SIRS) with clinical features of fever, tachycardia, tachypnea, capillary leakage, and diffuse alveolar damage histology (146, 147). Subsequently, within minutes to hours of the pro-inflammatory response, the compensatory anti-inflammatory response syndrome (CARS) is initiated, which slows the immune response via downregulation of intracellular signaling (including internalization of HLA-DR on monocytes), transitioning the immune system to a hyporesponsive, immunosuppressive state (146, 147). In an immunocompetent patient, simultaneous SIRS and CARS are considered normal complementary physiologic mechanisms that balance one another to restore homeostasis after infection onset (Figure 1) (147). However, complications or dysregulated immune systems can incite excessive SIRS or CARS, and skew the delicate balance (147). The result may include inducing acute respiratory distress syndrome (ARDS) that potentially can progress to damage in other vital organs (e.g., kidneys, heart, GI system, brain) leading to multiple organ dysfunction syndrome and death.
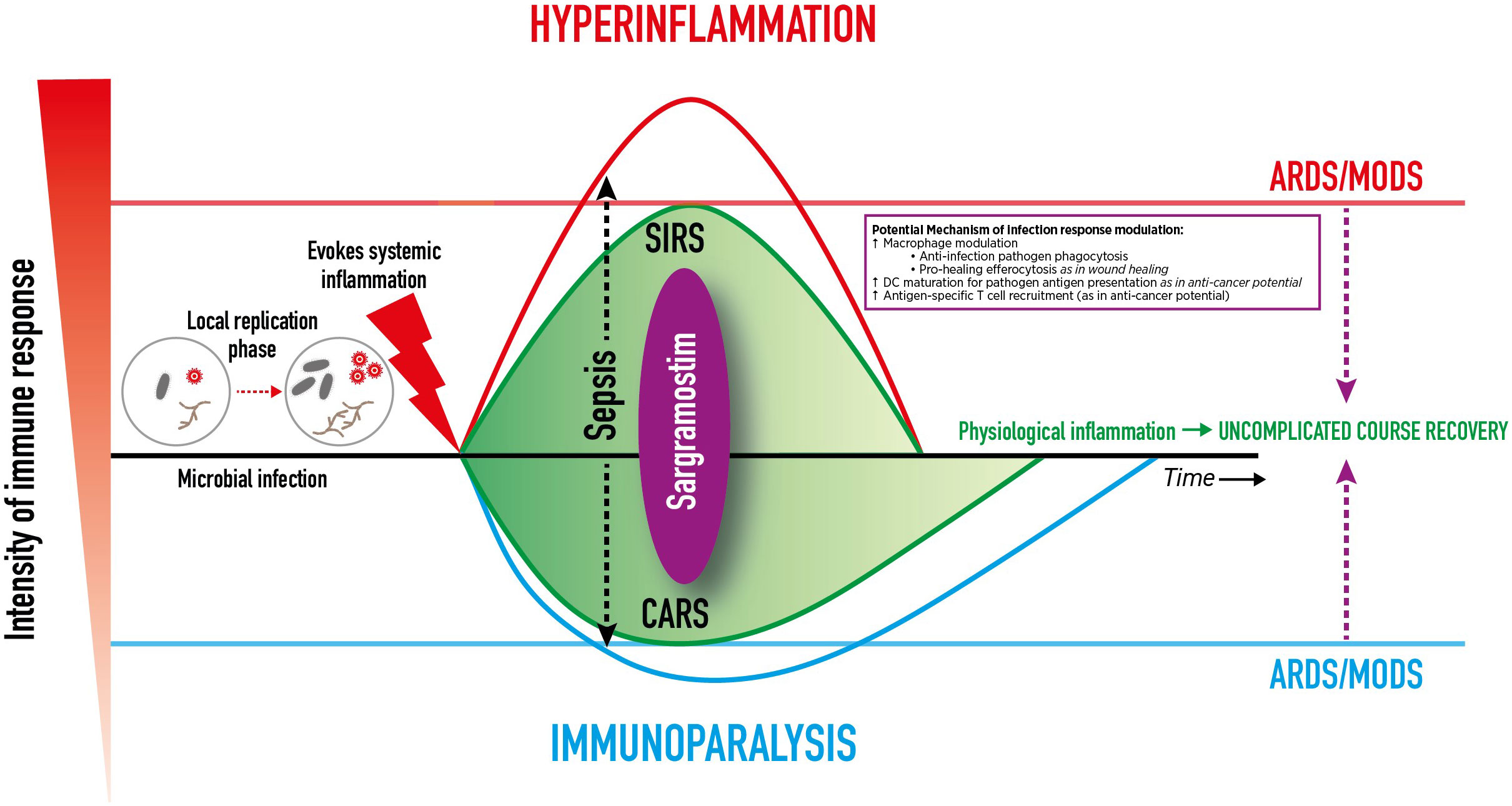
Figure 1 Dynamic Clinical Immune Response to Infection. Dynamic Clinical Immune Response to Infection and Potential Sargramostim Effect. After transmission, an infectious pathogen incubates, replicates, and induces systemic inflammation. A high pathogen load can overwhelm and dysregulate the innate and adaptive immune systems, spread, and cause life-threatening organ dysfunction. An overly pro-inflammatory immune response leads to systemic inflammatory response syndrome (SIRS).The compensatory anti-inflammatory response syndrome (CARS) slows the immune response. Simultaneous SIRS and CARS are considered normal complementary physiologic mechanisms that balance to restore homeostasis after infection onset. However, complications or dysregulated immune systems can incite excessive SIRS or CARS, skew the balance, and induce damage to vital organs (e.g., lungs, kidneys, heart, GI system, brain) and cause multiple organ dysfunction syndrome (MODS), and death. In the case of respiratory viral infections, damage to the lungs can result in acute respiratory distress syndrome (ARDS). Sargramostim (recombinant human granulocytemacrophage colony-stimulating factor) may mirror the effects of endogenous GM-CSF to modulate the immune response by alveolar macrophage activation, dendritic cell maturation, and antigen-specific T cell recruitment to aid in pathogen clearance. This may mitigate hyperinflammation and immunoparalysis to prevent ARDS and other organ damage.
Infection-induced mononuclear phagocyte dysfunction, SIRS or CARS, may result in either hyperinflammation or immunoparalysis which in turn can progress to ARDS as influenced by disease severity, patient characteristics, pathogen or insult, and the physiologic inflammatory state (146). ARDS typically develops within 7 days of pneumonia or sepsis onset (142). In ARDS, edematous fluid accumulates within the interstitium and alveoli which may activate epithelial and endothelial cells, injure the microvasculature, impair gas exchange, and cause hypoxemia (8, 142). Alveolar macrophages recruit additional neutrophils which may lead to unmitigated-neutrophil release of inflammatory mediators, reactive oxygen species, and extracellular traps. Dysregulated inflammatory neutrophil activity may lead to a loss of pulmonary basement membrane integrity to further disrupt the epithelial-endothelial barrier and may promote ongoing dysfunctional endothelial or epithelial cell inflammatory mediator release, propagating the proinflammatory state (as seen in SARS-CoV-2 or influenza infection) (8, 142). Meanwhile, severe and prolonged CARS can result in a paralyzed immune system, sometimes termed “immunoparalysis.” Definitions for immunoparalysis vary and include: HLA-DR levels less than 8,000 monoclonal antibodies per cell in CD14+ monocytes; less than 30% of monocytes expressing HLA-DR; or a markedly decreased mononuclear phagocyte function that produce TNF-α in response to ex vivo challenge with lipopolysaccharide (65, 146) Also during CARS, B cells and DCs undergo apoptosis, T cells enter an exhausted state, and Treg and MDSC numbers increase (148).
Immunoparalysis and hyperinflammation due to infection can each result in ARDS, organ failure, and death, but their processes differ (149). In immunoparalysis, the immune response is suppressed such that pathogens are allowed to replicate and spread without challenge from the host immune system, leading to host cell damage, organ failure, and/or death (146, 150). Conversely, in hyperinflammation, overly activated immune cells, stimulated in response to causative pathogens, damage host cells via infiltration and exaggerates pathogen-mediated toxic substance release which can lead to organ failure and/or death (147).
An example of a unique population with iatrogenic mononuclear phagocyte dysfunction is immunocompromised HCT recipients receiving extremely immunosuppressive myeloablative preparatory agents (151, 152). Important endogenous pleiotropic cytokines, that are involved in the differentiation, maturation, and proliferation of host immune cells, are released as a compensatory mechanism after the ablation of the marrow (153, 154). After myeloablation, the immune system then responds via an outpouring of cytokines, like GM-CSF, in an attempt to stimulate bone marrow neutrophil production (155, 156). This immunosuppression and other predisposing factors heighten the risk of opportunistic infection, ARDS, and death in immunocompromised patients (157, 158). Other risk factors for immunocompromised HCT recipients include prior infections (viral, parasitic, fungal), immunosuppressive graft-versus-host disease prophylaxis agents that impair viral clearance (calcineurin inhibitors, corticosteroids), metabolic alterations, barrier defects, and qualitative and quantitative blood dyscrasias (neutropenia, lymphopenia, monocytopenia) (157). If infected, HCT recipients experience prolonged viral shedding and higher rates of upper respiratory infections often progressing to the lower respiratory tract (159, 160).
4.1.3. GM-CSF in respiratory infection, ARDS, and sepsis-induced immunoparalysis
In healthy lungs, type II alveolar epithelial cells produce GM-CSF to aid in alveolar epithelial cell repair and restoration and to maintain surfactant homeostasis via alveolar macrophage cholesterol clearance (discussed in Autoimmune Pulmonary Alveolar Proteinosis (aPAP): a GM-CSF deficiency state and in the setting of checkpoint-induced pneumonitis in Anti-cancer potential and mitigation of immune checkpoint inhibitor immune-related adverse events and risk of GM-CSF insufficiency) (7, 11, 118, 125). GM-CSF is necessary for normal maturation and function of alveolar macrophages (6, 125). During lower respiratory viral infection, type II alveolar epithelial cells release GM-CSF to enhance the innate immune response of alveolar macrophage opsonophagocytosis of pathogens (as discussed in Emerging biology of GM-CSF) (11, 12). In studies in murine models, GM-CSF promotes adaptive immune responses via T cell, B cell, and DC maturation and activation that enable viral-specific antibodies production (161). After expansion and activation, GM-CSF facilitates lung-resident DCs’ migration to draining lymph nodes for additional antigen-specific T cell recruitment to improve viral clearance (162). Additional GM-CSF antiviral signaling may work in concert with interferon pathways (112).
Murine models have been very instructive for understanding the role of GM-CSF in respiratory viral infections. In transgenic mice lacking GM-CSF, survival after influenza infection was decreased due to impaired macrophage pathogen clearance (163). Conversely, transgenic mice with increased lung-GM-CSF expression experienced increased survival after influenza virus infection via enhanced alveolar macrophage activity (164). In another preclinical study, elevated alveolar GM-CSF concentrations in mice treated with intranasal recombinant murine GM-CSF increased alveolar macrophage numbers in bronchoalveolar lavage fluid (BALF) and improved survival after lethal influenza virus infection (165).
Release of antiviral pro-inflammatory immune response molecules into the systemic circulation can result in sepsis and can lead to ARDS (166). In animal models of post-viral ARDS, murine GM-CSF demonstrated immunomodulatory effects that improved the clinical response and symptoms associated with viral respiratory infections (167, 168). Increased airway GM-CSF expression and secretion in infected mice conferred a survival advantage in influenza-induced ARDS, attributed in part to the transition of pro-inflammatory M1 macrophages to the pro-healing M2 phenotype facilitated by GM-CSF (a similar transition discussed in Wound healing and risk of GM-CSF deficiency) (167). Inhaled recombinant murine GM-CSF improved locally-mediated murine-lung antibacterial resistance to systemic bacteremia during influenza infection (168). In adult patients with ARDS, elevated GM-CSF levels present in bronchoalveolar lavage fluid was associated with improved epithelial barrier integrity and survival (169).
In sepsis-induced immunoparalysis, impaired monocyte function leads to a diminished response to immune signaling, reduced pathogen phagocytosis, and reduced HLA-DR expression and thus a reduced ability to function as APCs (137, 170). As mentioned in Wound healing and risk of GM-CSF deficiency, HLA-DR is a class II MHC molecule typically found on APCs that links innate and adaptive immune responses via foreign antigen presentation to adaptive immune cells (e.g., T cells) (25). In in vitro studies, GM-CSF has been shown to reverse sepsis-induced monocyte hyporesponsiveness by normalizing monocyte HLA-DR expression and subsequently improving pathogen antigen presentation to adaptive immune response cells to restore immunocompetence (171, 172). Timing and GM-CSF concentration may impact the degree of immune response (113). A study evaluated effects of rhu GM-CSF (molgramostim) and rhu G-CSF on HLA-DR expression in neonates with sepsis (n=60) versus healthy controls (n=41) (173). HLA-DR expression was decreased across all neonates with sepsis which was then progressively restored over 5 days. Normal values of HLA-DR expression were observed as early as day 1 for patients treated with molgramostim therapy, yet not until day 3 for patients treated with G-CSF and placebo. Molgramostim and sargramostim are both rhu GM-CSFs discussed in more detail in Introduction and Autoimmune Pulmonary Alveolar Proteinosis (aPAP): a GM-CSF deficiency state.
4.2. Infection immune response clinical investigation and gaps
Immunomodulatory agents that boost host immune function against different pathogens may be beneficial to many patients by improving oxygenation, preventing ARDS, and reversing immunoparalysis. When given via various routes (inhalation, intravenous, and subcutaneous), rhu GM-CSF has been reported to improve outcomes for patients who are critically ill, immunocompromised, and suffering from respiratory infection (Table 3) (58–65, 67). The adverse event data are reported in Supplement Table S1. Optimal rhu GM-CSF dosing, route of administration, and duration of therapy, however, have yet to be established. Several authors report sargramostim therapy benefited patients with these conditions. The studies, however, were small, used varying routes of rhu GM-CSF administration, and included infections from multiple or unknown pathogens; hence further investigations are needed.
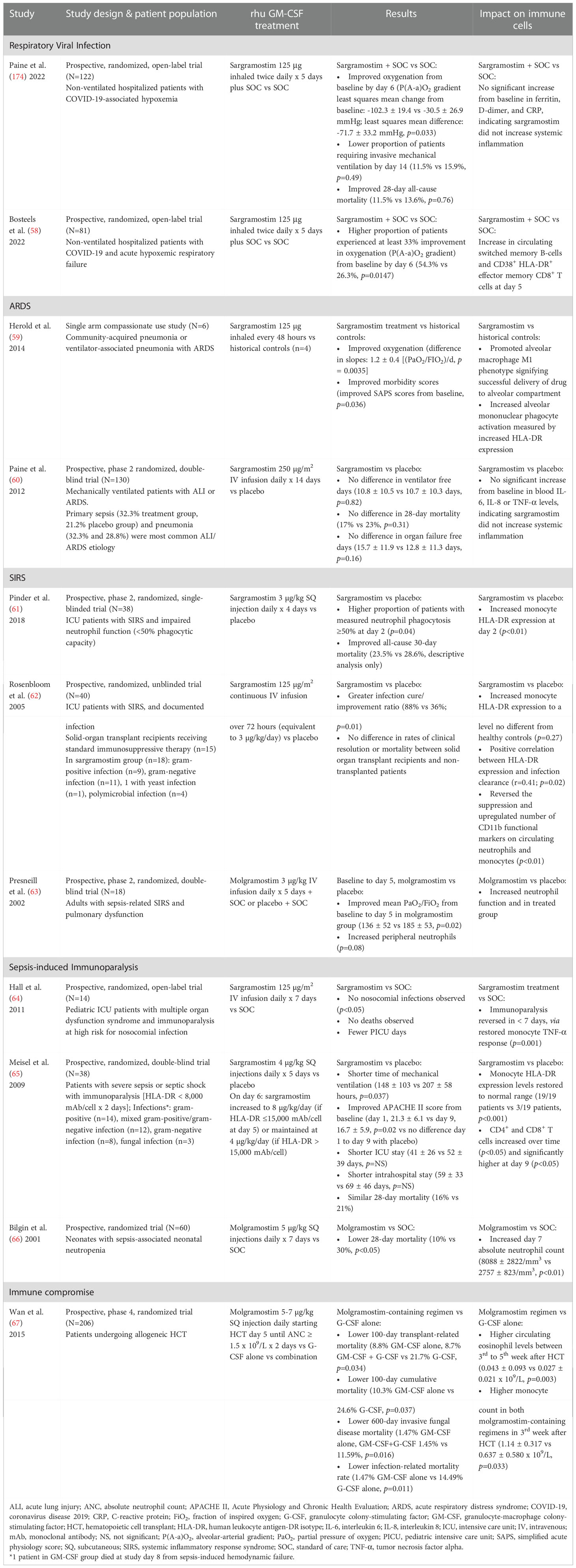
Table 3 Use of rhu GM-CSF in respiratory viral infection, ARDS, SIRS, sepsis-induced immunoparalysis, and immune compromise.
4.2.1. Respiratory viral infection, ARDS, and SIRS
Rosenbloom et al. (62) reported greater infection cure/improvement ratio for sargramostim over placebo for infectious pneumonia, intra-abdominal, central nervous system, or blood stream infections of various microbial etiologies (i.e., gram-positive, gram-negative, yeast, and polymicrobial). In the setting of SARS-CoV-2 infection, Bosteels et al. (58) showed inhaled sargramostim improved oxygenation and alveolar gas exchange and increased numbers of circulating class-switched B cells and effector COVID-19-specific CD8+ lymphocytes. Studies have suggested efficacy and safety of inhaled sargramostim for COVID-19 treatment in hospitalized patients (174, 175).
Herold et al. (59) reported improved oxygenation with inhaled sargramostim for ARDS in hospitalized patients experiencing ARDS-pneumonia. Paine et al. (60) reported sargramostim treatment was found to be safe in patients with acute lung injury (ALI) and ARDS. The authors concluded sargramostim should continue to be studied in ARDS.
In a study conducted in critically ill patients with SIRS, Pinder et al. (61) reported decreased all-cause 30-day mortality with sargramostim compared to placebo.
4.2.2. Sepsis-induced immunoparalysis and immunocompromised
Meisel et al. (65) reported decreased mechanical ventilation duration assessed at day 9, improved disease severity scores at day 9, and decreased length of ICU stay with subcutaneous sargramostim injections in patients manifesting immunoparalysis. CD4+ and CD8+ T cell numbers were increased, and HLA-DR levels were restored to normal levels as well. Additionally, Rosenbloom et al. (62) reported a positive correlation between HLA-DR expression and infection clearance after sargramostim therapy. Also, monocyte HLA-DR expression increased to levels no different from healthy controls. In pediatric ICU patients, Hall et al. (64) reported fewer pediatric ICU days, no deaths, and no nosocomial infections in patients who received sargramostim intravenous infusion.
In immunocompromised HCT recipients, Wan et al. (67) reported lower transplant-related mortality, lower cumulative mortality, lower invasive fungal disease mortality, and lower infection-related mortality in prophylactic molgramostim-containing regimens compared to granulocyte colony-stimulating factor (G-CSF). Additionally, therapeutic use of intranasal recombinant murine GM-CSF in immunosuppressed mice resulted in a decreased quantitative, PCR-assessed, fungal burden as compared to placebo (p=0.045) (176). In a new case series of invasive fungal disease in pediatric malignancy (n=15) and a systematic review of immunocompromised and immunocompetent patients (n=50), 92% and 82% of cases, respectively, showed a complete and/or partial response to invasive fungal disease when treated with adjunctive rhu GM-CSF in addition to standard of care (177).
Current and ongoing trials include important endpoints such as improvement in oxygen saturation, clinical indicators, PaO2/FiO2 ratio, and enhanced immunological effects, as well as improvements in major endpoints like reduction in mortality and days of hospitalization. Trials using an alternate route of rhu GM-CSF administration, specifically the inhaled route, may address availability concerns for respiratory treatments in both the inpatient and ambulatory settings (124). As learned from COVID-19 healthcare rationing, future investigations regarding rapid clinical responses using noninvasive direct pulmonary drug delivery with non-disease specific inhaled agents (e.g., sargramostim) may improve patient outcomes. Many disease-specific treatments such as monoclonal antibodies and antivirals, are virus- and/or variant-specific, limiting the potential patient population that could experience treatment benefit (178). The inhaled drug delivery technique, especially with versatile agents like sargramostim, may ultimately be demonstrated to be useful.
4.3. Potential future developments in infection immune response
There is enthusiasm for developing innovative therapies to improve patient outcomes after respiratory viral infections, ARDS, and sepsis-induced immunoparalysis. Beyond antimicrobial therapy, strategies include enhancing host defense by either replacing deficient cells (such as neutrophils in cases of cancer/chemotherapy-induced neutropenia) or potentially providing specially activated immune/inflammatory cells, akin to CAR-T cell therapy (179–181). Such treatments require enormous resources and in many instances (such as white blood cell transfusion for infection) have not shown clear benefit (182, 183). In contrast, sargramostim may safely target and modulate specific cells and cellular behavior to achieve an effective and efficient immune response (58, 59, 61, 62, 64, 65). Sargramostim’s actions on alveolar macrophages, dendritic cells, and T cells may allow for complementary immune response changes versus increases in cell numbers as seen with other agents such as rhu G-CSF (Figure 1) (5, 11, 12, 161, 162). Other novel products, such as hematopoietc stem cell-derived ex vivo-expanded myeloid progenitor cells and phenotypically typed functional granulocytes, have been shown to be efficacious in animal models (184, 185).
Biomarkers to predict and measure treatment effects in immunoparalysis, SIRS, CARS, and ARDS are urgently needed. Current surrogate biomarkers of inflammation (C-reactive protein [CRP], procalcitonin [PCT], ferritin) are used in practice, but further investigations of more sensitive and specific biomarkers are warranted (8). Definitions for immunoparalysis vary, for example monocyte HLA-DR levels less than 8,000 mAb/cell or TNF-α response assay to ex vivo stimulation results and should be standardized (64, 65). Standardized biomarkers would help stratify patients to better anticipate those at increased infection risk and hence should receive antimicrobial prophylaxis or treatment. Prospective immunophenotyping and/or patient stratification based on blood cell counts, immune function assays, cytokine levels, GM-CSF auto-antibody levels [e.g., in Cryptococcus gattii cryptococcosis (186)], or other markers of inflammation would help define optimal timing of drug administration (e.g., sargramostim) to prevent organ failure and death (9). Vulnerable patient populations such as immunocompromised HCT recipients, are at higher risk for all types of infection and should be a continued focus of future studies (138, 157).
Trials in pediatric sepsis [NCT03769844 (187), NCT05266001 (188)] aim to better understand the potential of sargramostim in modulating the immune system to enhance the pulmonary host defense capacity to eliminate pathogens, maintain alveolar homeostasis, and prevent disease progression. These trials will add to the existing evidence for sargramostim including attenuation of epithelial cell injury, epithelial repair, and improved barrier function and gas exchange in ARDS (59).
As the world continues to endure the COVID-19 pandemic, many trials investigating variant-independent treatment options are ongoing. Using single RNA-sequencing of MAFB and MAF transcription factors, 3 main lung macrophage populations expressing associated markers have been identified: FCN1 (ficolin-1; macrophages derived from circulating monocytes), SPP1 (secreted phosphoprotein 1; macrophage origin unknown), and FABP4 (fatty acid binding protein 4, an intracellular lipid chaperone and adipokine; found in GM-CSF-dependent alveolar macrophages) (118). These macrophage population ratios have shown to be altered in association with COVID-19 severity. Analysis of bronchoalveolar lavage fluid from patients infected with COVID-19 showed increased FCN1high and SPP1high macrophages and decreased FABP4high macrophages correlated with disease progression (189). Altering the ratios of these lung macrophage populations towards an increased proportion of the FABP4high macrophage subset, may promote pathogen clearance and epithelial repair while limiting an overly inflammatory response seen at the later stages of COVID-19 and other respiratory viral infections (189). Potential therapeutic intervention targeting the macrophage-activating upstream Jun N-terminal kinases (JNK) via MAPK inhibitors may alter macrophage population ratios, hence making these subsets both potential biomarkers and biopredictors (190).
The rapid SARS-CoV-2 genetic modifications created new variants that circumvented vaccine efforts and made it challenging to keep up with therapeutic targets due to changing resistance patterns. A trial in outpatients with COVID-19 is investigating using sargramostim to rebalance lung homeostasis to prevent disease progression to severe COVID-19 [NCT04707664 (191)]. Influenza shares many characteristics with SARS-CoV-2 as both pathogens invade and damage alveolar epithelial cells and have circumvented annual vaccine efforts, previously to an epidemic proportion. Although anticipated every year, seasonal influenza still causes significant morbidity and mortality, especially in high-risk HCT recipients (192). Prior to the COVID-19 pandemic, one study attributed influenza (seasonal A and B) to 30% of all respiratory viral infections in this population with up to 35% progressing to lower respiratory tract infections (193). In addition to HCT recipients, patients with other high risk factors (the very young, nursing home residence, chronic lung or heart disease, history of smoking) (194) contribute to the unmet need for further investigations to build upon the preclinical and clinical insights from sargramostim studies in COVID-19, pneumonia-associated lung injury, ARDS, and sepsis. Targeting these patient populations in future studies based on disease etiology, disease severity, and ideally key disease pathways per the individual patient will hone effective and efficient disease treatments and minimize side effects.
5. Wound healing and risk of GM-CSF insufficiency
5.1. Wound healing pathophysiology
Spontaneous acute wound healing in a normal host involves complex immune system interactions over time to restore the skin barrier after injury (17). Wound healing is comprised of four sequential and overlapping phases including hemostasis, inflammation, growth, and re-epithelization (Figure 2). Due to stresses both internal (e.g., aging, genetics, nutrition, chronic disease) and external (e.g., bacteria, medications), normal wound healing may be delayed or arrested at any stage (195). With concomitant diseases, such as diabetes, pathophysiologically inherent disease factors might further impair immune response, worsen peripheral arterial disease, and generate repetitive trauma due to neuropathic desensitization, all of which halt normal wound healing and contribute to development of chronic, non-healing wounds (i.e., diabetic foot ulcer) (16, 17). Diabetic foot ulcers typically stall at the inflammation phase, partly attributable to accumulation of advanced glycation end products (AGEs) (196). Presence of AGEs leads to increased oxidative stress and inflammation, stiffer skin, and reduced innate immune cell adhesion. Also, inhibition of immune-cell-signaling p38/MAPK pathway results in decreased damaged cell removal and reduced primary skin cell (keratinocyte) migration (197).
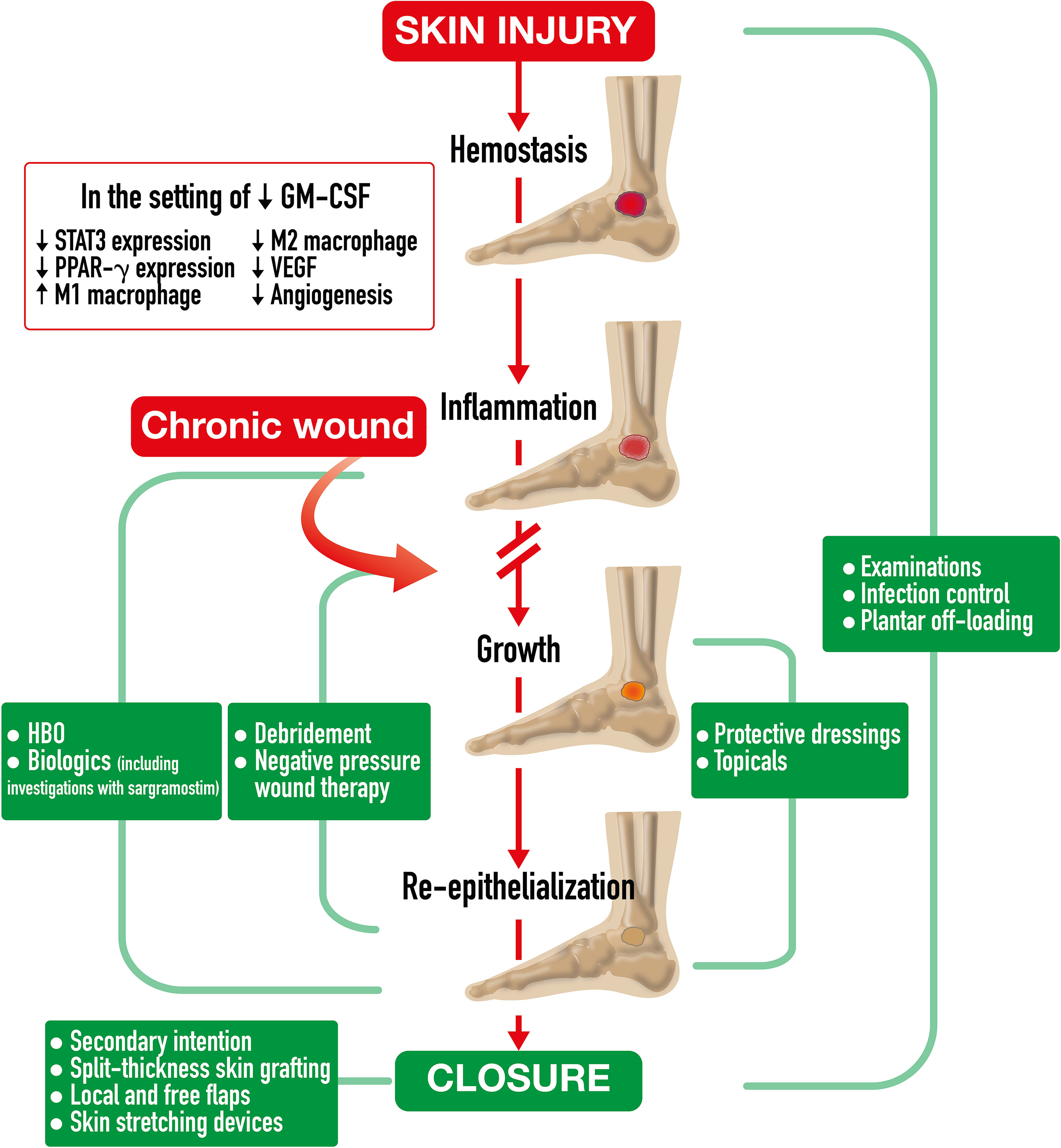
Figure 2 Chronic Wound Healing Process and Treatment Strategies. Chronic Wound Healing Process and Treatment Strategies. After spontaneous skin injury, the chronic wound healing process is comprised of four phases including hemostasis, inflammation, growth, and re-epithelization. This process often stalls at the inflammation stage in which GMCSF deficiency leads to reduced neutrophil and macrophage chemotaxis and infiltration, reduced signal transducer and activator of transcription 3 (STAT3) expression, insufficient macrophage differentiation, decreased efferocytotic function, impaired PPAR-g expression, and diminished pro-inflammatory M1 to pro-healing M2 transition. Insufficient macrophage actions impede granulation tissue formation, vascular endothelial growth factor (VEGF)-dependent angiogenesis, and contractile myofibroblast differentiation. All these factors ultimately delay wound closure. A continuum of treatment strategies for chronic lower extremity wounds is required for wound healing. Diverse strategies overlap to address key healing mechanisms. Strategies include wound bed preparation by debridement and negative pressure therapy, experimental immunologic modifications, and granulation tissue promotion with biologics such as sargramostim [recombinant human granulocytemacrophage colony-stimulating factor]), hyperbaric oxygen therapy (HBO), topicals, and protective dressings, as well as wound closure, including secondary intention, split thickness skin grafting, local and free flaps, and skin stretching devices. Examinations, antibiotic therapy, and plantar off-loading may be required at any phase.
Within hours of a spontaneous skin injury in an intact host, blood vessels constrict, and platelets form a fibrinogenic plug to stop bleeding to start the hemostasis phase (17). Local neutrophils and macrophages extravasate to the injury to defend against invading bacteria with cell recruitment increasing over 2 to 3 days (198, 199). Injured cells and mast cells release cytokines and other bioactive molecules to attract leukocytes, including Langerhans cells, dendritic cells, T cells, neutrophils, and monocytes (17, 195). Keratinocytes release endogenous GM-CSF that promotes local myeloid proliferation and supports additional inflammatory signaling (200, 201). A day after injury, neutrophils constitute half of all cells in the wound (17). In human diabetic foot ulcers, impairment in GM-CSF activation of signal transducer and activator of transcription 3 (STAT3) expression results in decreased immune cell recruitment (202). In a murine diabetes model, proinflammatory cytokines, such as interleukin 6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and GM-CSF, were reduced in wounds, compared to wounds in non-diabetic mice (15). Reduced signaling led to reduced neutrophil and macrophage recruitment and delaying healing. Mouse diabetic wound healing was almost completely restored by 2 weeks with perilesional exogenous rhu GM-CSF injected intradermally. In non-diabetic mice, exogenous rhu GM-CSF did not enhance wound healing.
In the inflammatory phase (3-20 days duration), immune cell recruitment continues, leading to pathogen, debris, and necrotic tissue removal (14). With an impaired wound healing environment, i.e. pressure ulcers in the elderly, decreased GM-CSF signaling leads to lower expression of nucleotide-binding domain-like receptor protein 3 (NLRP3) and reduced neutrophil interleukin-1 beta (IL-1β), resulting in impaired innate immune responses (203–205). In a normal host, GM-CSF signaling facilitates recruited monocyte differentiation into various immune response cells, including macrophages (206). Macrophages are often considered the most important immune cells in wound healing (19). They recognize and engulf pathogens, as well as eliminate expended neutrophils within 3 to 4 days of skin injury by efferocytosis (17). Decreased macrophage infiltration from impaired GM-CSF signaling in diabetes reduces neutrophil clearance, causing additional tissue damage from lysed neutrophils that prolongs the inflammatory phase. As mentioned in Emerging biology of GM-CSF, GM-CSF also induces PPAR-γ expression which is key to transitioning macrophages from a pro-inflammatory M1 phenotype to a pro-healing M2 phenotype (18). Impaired macrophage PPAR-γ activity and elevated M1 macrophages in diabetic wounds generate negative downstream effects. Decreased growth factor release, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), reduces granulation tissue formation and prolongs inflammation both ultimately resulting in delayed wound closure (18, 207).
Within days to weeks from injury in a normal host, the growth phase begins in which granulation tissue formation and neovascularization occur (17, 208). Pro-healing M2 macrophages deposit extracellular matrix components, induce myofibroblasts, and phagocytose excess cells/matrix (17, 18). VEGF released by GM-CSF-stimulated macrophages acts as a key growth factor in early angiogenesis to promote micro-vessel sprouting (17, 209). VEGF and PDGF promote proliferation of keratinocytes, fibroblasts, and epithelial cells to create granulation tissue (207). GM-CSF also promotes maturation and stabilization of newly developed micro-vessels (capillaries) to establish new tissue blood supply (15). In diabetic foot ulcers, the necessary M2 macrophage population is decreased due to a dysregulated M1 to M2 phenotype shift, thereby delaying granulation and blood vessel formation (18).
In weeks to months, normal host re-epithelization occurs, and the wound closes (208). Normal connective tissue replaces granulation tissue while epidermal stem cells give rise to epidermal layers, hair follicles, and glands (17). GM-CSF-dependent M2 macrophages induce fibroblast differentiation into contractile myofibroblasts for wound closure (17, 19, 20, 210). Conversely, for patients with diabetes, fewer M2 macrophages result in decreased fibroblast differentiation and slower wound closure.
5.2. Wound healing clinical investigations and gaps
Clinical studies to date of rhu GM-CSF (e.g., sargramostim or molgramostim) used diverse dosages, dosage forms, and durations of study therapy in small trials of diverse wound etiologies. Clinical studies of rhu GM-CSF in wound healing are summarized in Table 4. The adverse event data are reported in Supplement Table S1. Routes of administration have included both perilesional and topical, wound size and duration have varied considerably, and the affected subjects have varying degrees of immunocompromise. Hence, larger, randomized controlled trials that provide guidance on dose, route, frequency, and duration of therapy for definitive wound closure, specific to diabetes and other current etiologies, are needed to further confirm benefit of rhu GM-CSF.
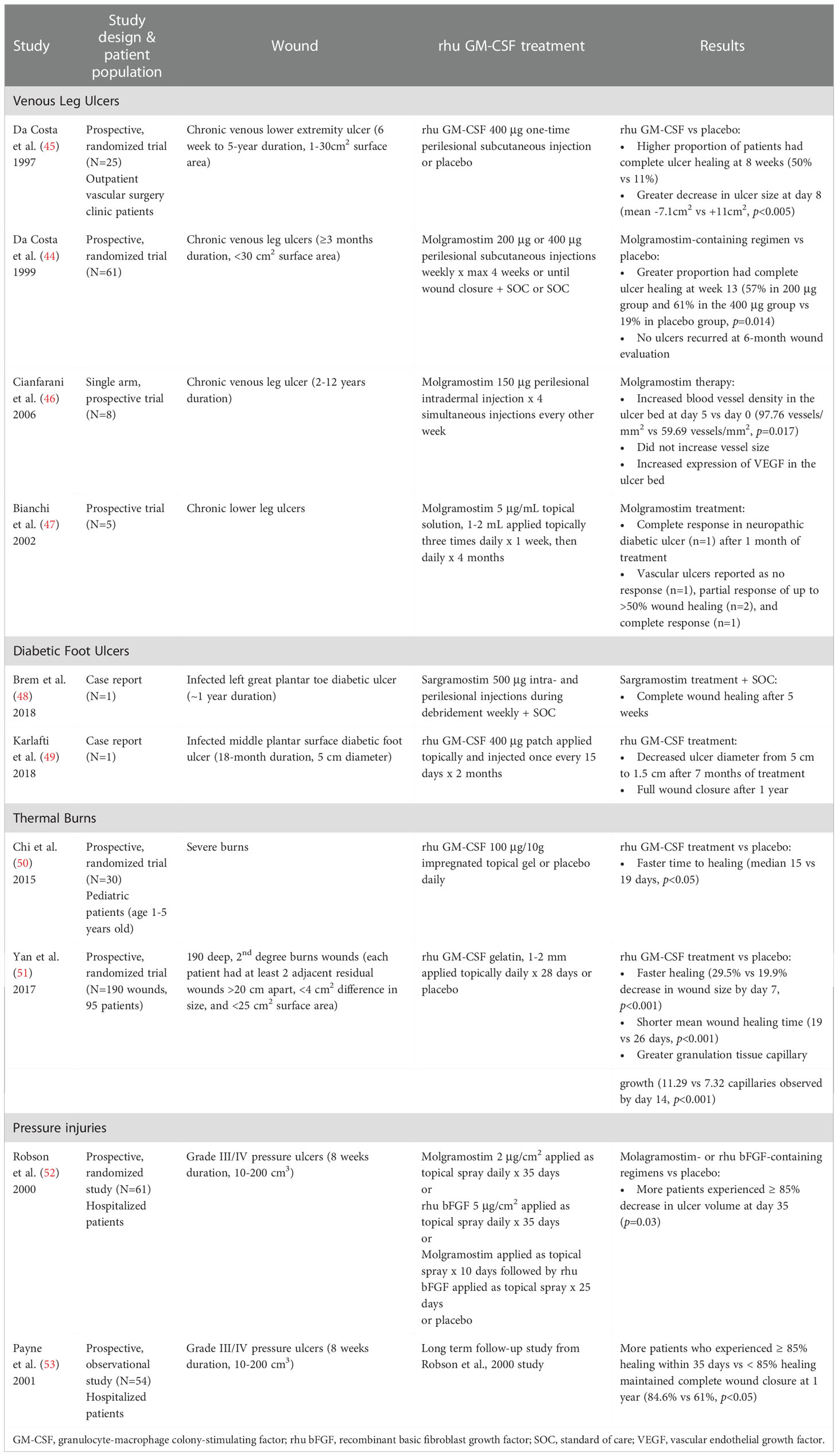
Table 4 Clinical studies of rhu GM-CSF in wound healing.
Other conditions have also been the subject of wound healing investigations. Case reports describe improved chronic wound healing with rhu GM-CSF treatment of patients with leukocyte or vascular dysfunction disorders, including glycogen storage disease (211), chronic granulomatous disease (211), common variable immunodeficiency (212), Klippel-Trénaunay-Weber syndrome (213), and cutaneous polyarteritis nodosa (214). Important issues to be addressed include optimal dose, route, schedule, and therapy duration within each wound etiology.
5.3. Potential future developments in wound healing
Deficiency of autocrine and paracrine GM-CSF activity in every stage of chronic wound healing illustrates a vital role for this cytokine. In a young, healthy host, endogenous GM-CSF is necessary for immune cell recruitment and maturation, phenotype shifts, keratinocyte proliferation, and angiogenesis. Successful wound repair and regeneration may well rely on timely activity of GM-CSF within healing wounds, especially its impact on macrophages, fibroblasts, and endothelial cells in rebuilding skin architecture.
Additional biomarkers to inform wound healing are needed. Vatankhah and colleagues (215) reported correlation of blood neutrophil-to-lymphocyte ratio with likelihood of diabetic foot ulcer nonhealing. Sawaya and associates (202) demonstrated that the diabetic foot ulcer immune cell landscape featured diminished GM-CSF activity with high and low proportions of monocytes and macrophages, respectively, indicating successful monocyte recruitment but deficient activation. Similar to blood monocytes, dermal stem cells of patients with diabetes mellitus also express low human leukocyte antigen-DR (HLA-DR), the receptor responsible for antigen presentation to CD4+ helper T lymphocytes and initiation of adaptive immune responses (216). As sargramostim is known to restore HLA-DR expression in post-surgery patients and sepsis-associated immunosuppression (as mentioned in Immune responses to infections and risk of GM-CSF insufficiency (65, 217), this receptor might become a clinically useful wound healing biomarker.
It soon may be possible to improve wound prevention of diverse etiologies, importantly including diabetic foot ulcers. Noninvasive Terahertz screening of diabetic foot skin dehydration estimates the amount of water that evaporates through skin to the external environment (218). Alternatively, thermometry (219) and point of care ultrasound imaging (220) are proposed to quantify risk of wound development. Topical barrier gels often serve as prophylaxis for potential post-spinal-cord injury pressure ulcers (221, 222). As mentioned in Emerging biology of GM-CSF, GM-CSF also may have protective neural cell effects (76, 223), hence a sargramostim gel could be a potentially interesting prophylactic approach.
Patients with chronic non-healing wounds have high morbidity and mortality, require multimodal healing treatment, and present an unmet need for novel therapies to improve outcomes (224). In the future, approaching all chronic wounds as a disease state, instead of as an underlying component of disease or the aging process, would establish a fresh viewpoint for management and prevention. Additionally, it is important to include elderly patients in future clinical trials as they are often excluded, as evidenced in cancer clinical trials (225, 226). Sargramostim’s immunotherapeutic potential should be further studied in at-risk patients and address wound healing outcomes that optimize ulcer-free, hospital-free, and activity-rich days. Sargramostim’s preventative therapeutic potential should be further studied, especially in combination with improved noninterventional diagnostic practices.
6. Anti-cancer potential and mitigation of immune checkpoint inhibitor immune-related adverse events and risk of GM-CSF insufficiency
6.1. Pathophysiology of antitumor effects
In a healthy immune system, immune checkpoint receptors dampen immune responses to prevent prolonged T cell activation and autoimmunity (227). Immune checkpoint inhibitors (ICIs) block checkpoint-originated immune suppression both in the tumor, to enable a desired antitumor T cell response, and potentially in healthy organ systems such as the gastrointestinal (GI) tract and lungs, causing unwanted tissue damage. Immune-related toxicity can occur at any time during treatment, is a frequent cause of ICI discontinuation, and may persist after cessation of therapy (228).The GI and pulmonary organ systems provide dual immune protection via physical barriers and cell-mediated immune responses. Immune-mediated colitis and checkpoint-induced pneumonitis can resemble infectious or spontaneous autoimmune disorders, and may have a delayed clinical presentation creating a diagnostic challenge for clinicians (228).
Immunotherapy aims to mobilize the immune system and restore antitumor immunity that is actively suppressed either by tumors cells themselves or by other immune cells in the tumor microenvironment (21). In an intact immune system, immune checkpoint receptors like CTLA-4, LAG-3, and PD-1 inhibit T cell activity at different steps of the immune response (229, 230). These receptors assist in limiting and preventing autoimmunity that could occur with unencumbered activated T cells. Blocking these receptors with ICIs inhibits inactivation, allowing the activated immune system to overcome cancer escape mechanisms and eliminate tumor cells (230–232).
Current research in cancer immunotherapy includes the activities of endogenous cytokines, such as GM-CSF, and their potential to influence an antitumor immune response (21). While sargramostim has been used for 30 years for other purposes, recent research has focused on its immunomodulatory properties as discussed in the preceding sections (1). Intralesional rhu GM-CSF therapy increased tumor-infiltrating CD4+ T cell numbers and localized tumoricidal macrophages (233). CD4+ T cells play a major role in CD8+ T cell-mediated responses. An expanded macrophage response may lead to increased tumor-cell phagocytosis, antigen presentation and T-cell stimulation. Proposed mechanisms for increased survival in clinical studies with sargramostim include increased cytotoxic CD8+ T cell and dendritic cell recruitment to the tumor and sentinel lymph node, respectively (22–24). In addition, GM-CSF stimulation of increased metabolic capacity of mononuclear phagocytes may counteract the immunosuppressive potential of tumor associated myeloid cells (25). Interactions among these cells may lead to enhanced antitumor T cell priming and activation via increased dendritic cell tumor-associated antigen presentation (Figure 3).
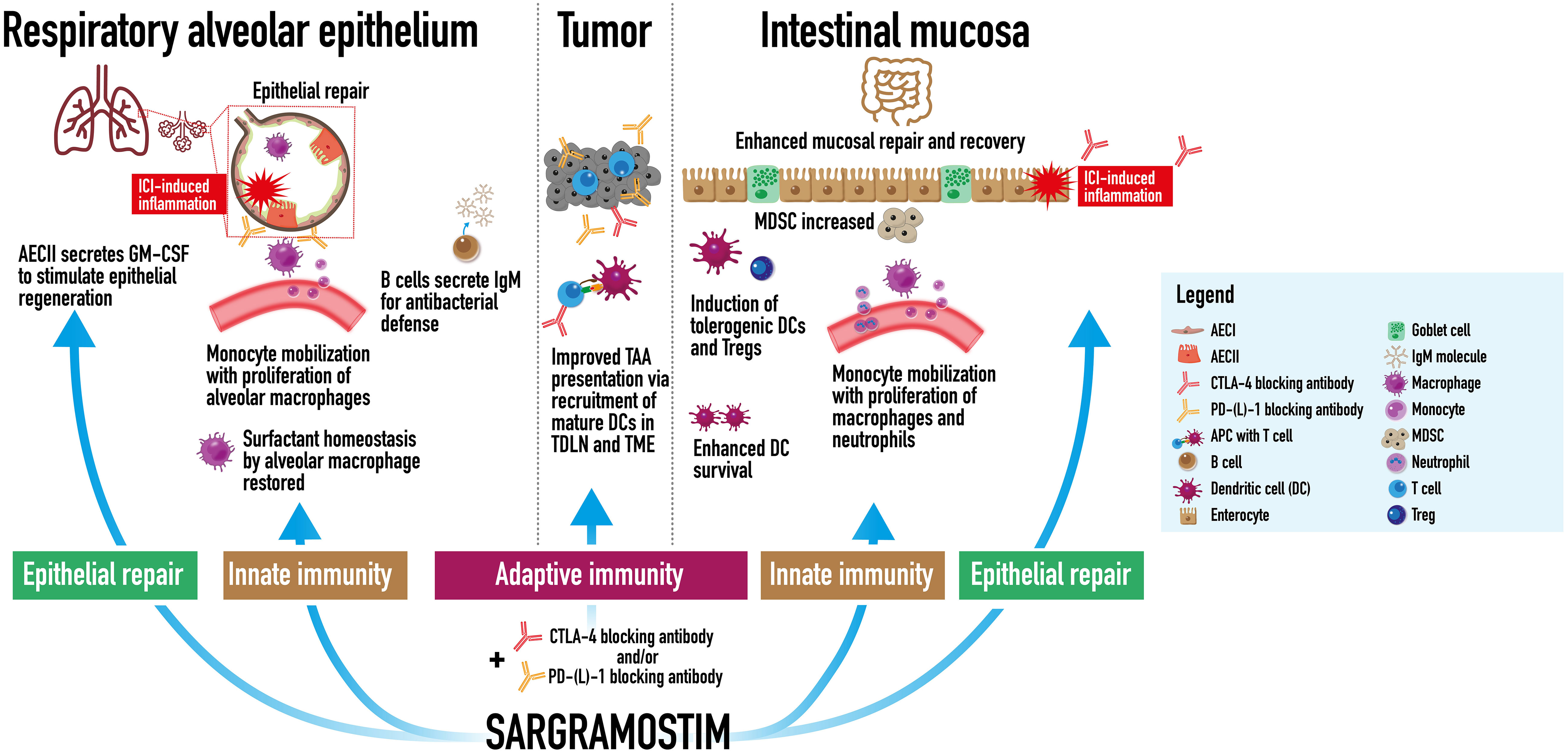
Figure 3 Possible antitumor and restorative activity of sargramostim in immune checkpoint inhibitor combination therapy. In checkpoint-induced pneumonitis, sargramostim (recombinant human granulocyte-macrophage colony-stimulating factor; rhu GM-CSF) may stimulate epithelial regeneration as modeled by endogenous GM-CSF-secreting Type II alveolar epithelial cell (AECII). Monocyte and alveolar macrophage mobilization and functional restoration of alveolar macrophages (which express programmed cell death protein 1 [PD-1] receptors) may contribute as well. Restoration of surfactant homeostasis by alveolar macrophages may support barrier renewal. Sargramostim may reinforce antibacterial defense via immunoglobulin M (IgM) secretion from B cells. Potential antitumor mechanisms of combination sargramostim and immune checkpoint inhibitor therapy may include improved tumor-associated antigen (TAA) presentation via recruitment of mature dendritic cells (DCs) in the tumor draining lymph node (TDLN) and tumor microenvironment (TME). In immune-mediated colitis, sargramostim mayenhance mucosal repair and recovery via induction of tolerogenic DCs and regulatory T cells (Tregs), and enhanced DC survival. Monocytes, macrophages, and neutrophils may be mobilized to bolster antibacterial defenses. Myeloid derived suppressor cells (MDSC) may be increased to enhance anti-inflammatory response. AECI, Type I alveolar epithelial cell; AECII, Type II alveolar epithelial cell; CTLA-4, cytotoxic T-lymphocyte associated protein-4; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICI, immune checkpoint inhibitor; IgM, immunoglobulin M; PD-1, programmed death-1; PD-L1, programmed death ligand-1.
ICI and sargramostim therapy link blockade of immune inhibitor mechanisms with increased immune cell activation which might boost antitumor responses in solid tumors (29). Correspondingly, in prostate cancer, sargramostim and ipilimumab combination therapy augmented tumor-reactive cytotoxic circulating CD8+ T cell responses (54). Furthermore, in melanoma, sargramostim and ipilimumab combination therapy increased overall survival (OS) and enhanced expression of inducible T-cell co-stimulator (ICOS) on CD4+ and CD8+ T cells over ipilimumab alone (28). Hence, addition of sargramostim appears to promote synergistic activation of key adaptive immune cells in antitumor immune responses.
6.2. Pathophysiology of immune-related adverse events
Until recently, the mechanisms of immune-mediated colitis were unclear. Biopsies from patients with colitis, in contrast to control patients, revealed a large population of proliferating, cytotoxic CD8+ T cells (234). Further analyses of inflamed tissue revealed that ICI colitis is characterized by expanded populations of IFN-γ- and granzyme B+-producing, cytotoxic CD8+ T cells, and to a lesser extent—expanded populations of Th1 skewed CD4+ T cells, and inflammatory macrophages (235–237). Based on clonal rearrangement analysis used to identify T cells and their progeny, these cytotoxic CD8+ T cells were determined to be derived from colon-resident memory cells, likely held in check by CLTA-4 and/or PD-1 receptors (238). These tissue-resident memory T cells are likely reactivated due to CLTA-4 and/or PD-1 blockade (237). It is hypothesized that reactivated tissue-resident memory T cells proliferate, become cytotoxic, and release IFN-γ. IFN-γ then may signal myeloid cells to amplify the inflammatory response and recruit T cells from the circulation, overwhelming Treg-mediated immunosuppression to damage colon tissue and impair barrier integrity.
In animal models, antitumor action of CTLA-4 blocking antibodies occurs via Treg depletion, cells that express high CTLA-4 levels (237). Human immune-mediated colitis biopsies indicate CTLA-4 Treg depletion is likely not a major mechanism of action of anti-CTLA-4 antibodies as the Treg population was preserved or expanded in post-CLTA-4 blockade biopsies.
Based on immune cells present in organs at homeostasis, the lungs share the same functional predictors of inflammatory toxicities as the GI tract (26). Both GI tract and lungs contain epithelial barriers colonized with microbiota, as well as a large population of tissue-resident memory T cells. Therefore, PD-1 pneumonitis may share mechanisms seen in colitis, including upregulation of inflammatory immune alveolar cells (TNF-α, IFN-γ, CD8+ cytotoxic T cells) and downregulation of important suppressor regulatory cells such as Tregs and alveolar macrophages that express PD-1 (27). Expression of CLTA-4 and PD-1 differs among different tissue cells and immune cells, which might explain why adverse event prevalence varies for immune checkpoint inhibitors between GI tract and lungs (229). Importantly, the addition of sargramostim to CTLA-4 blockade with ipilimumab reduced the incidence of GI and pulmonary adverse events, compared to ipilimumab alone in a phase 2 study of patients with metastatic melanoma (28).
6.2.1. GM-CSF in lung and GI tract inflammatory disease
GM-CSF effects on immune cells have been documented in inflammatory diseases of the lung (e.g., influenza virus and aPAP) and GI tract (e.g., Crohn’s) and may play a role in checkpoint-induced pneumonitis and immune-mediated colitis (6, 11, 30). In the lung, type II alveolar epithelial cells secrete endogenous GM-CSF to stimulate alveolar epithelial regeneration (11, 118). As noted in Emerging biology of GM-CSF, GM-CSF induces activity, stimulates production, or functionally restores lung immune cells, e.g., monocytes, alveolar macrophages, and B cells. As discussed in the aPAP section (Autoimmune Pulmonary Alveolar Proteinosis (aPAP): a GM-CSF deficiency state, alveolar macrophages play a major role in surfactant homeostasis, key to normal lung function (6). B cells secrete immunoglobulins, including IgM, as an antibacterial defense to protect the lung barrier (239). In the GI tract, GM-CSF has dual roles of increasing the number of MDSCs, and inducing monocytes, macrophages, neutrophils, and lamina propria dendritic cells (critical to Treg activation). Timing and extent of required or dysregulated immune response influence GM-CSF activity to either enhance anti-inflammatory activity or bolster antibacterial activity (30). Dendritic cell survival is also enhanced. Restoring or activating these pulmonary and GI cells after injury or infection (e.g., potentially caused by dysregulated immunity), may contribute to epithelial cell regeneration and bacterial defense resulting in mucosal healing and fortification (Figure 3) (1).
6.3. Anti-tumor effects and irAE clinical investigations and gaps
Clinical combination studies of ICIs and sargramostim have shown benefit in metastatic melanoma and prostate cancer (only melanoma studies detailed here) (28, 29, 54–56, 240). The adverse event data are reported in Supplement Table S1. There were 3 author-reported grade 5 adverse events (death) in these clinical trials. All studies are of sargramostim in combination with ICI therapy.
In a phase 2 study, Hodi et al. (28) reported longer survival (17.5 months vs 12.7 months, p=0.01) and reduced grade 3-5 adverse events (45% vs 58%, p=0.04), for patients with unresectable stage III or IV melanoma treated with ipilimumab 10mg/kg plus sargramostim 250 μg subcutaneously days 1 to 14 of 21-day cycles (n=123) vs ipilimumab alone (n=122). Efficacy results were similar in two much smaller, non-randomized ipilimumab/sargramostim melanoma treatment studies wherein most patients had poor prognostic characteristics such as brain and liver metastases (55, 56). Patients with brain metastases are often excluded from clinical trials, however, they were included in the following studies which reported combination treatment benefit. A single institution, retrospective analysis (N=32) examined combination ipilimumab 3 mg/kg and sargramostim 250 μg/day for 14 days in 21-day cycles for 4 cycles (55). These authors reported overall disease control ≥ 12 weeks in 50% by response evaluation criteria in solid tumors (RECIST) and 44% by immune-related response criteria. Median OS was 41 weeks with an overall incidence of immune-related adverse events of 31.3% with 9.4% grade 3-4 events. A separate prospective, single-arm phase 2 trial (N=22) examined ipilimumab 10 mg/kg with sargramostim 125 μg/m2/day for 14 days in 21-day cycles, followed by sargramostim alone, then maintenance therapy of ipilimumab with sargramostim every 3 months for up to 2 years (56). Authors reported disease control in 41% of patients at 24 weeks. Median OS was 21.1 months, and grade 3-4 adverse events occurred in 41% of patients.
The ongoing ECOG-ACRIN EA6141 phase 2-3 trial is evaluating the combination of ipilimumab, nivolumab and sargramostim, compared with ipilimumab and nivolumab, in patients with unresectable stage III and stage IV melanoma. The trial advanced to the phase 3 portion after meeting prespecified efficacy and safety thresholds in the lead-in phase 2 portion (57). Trials continue to demonstrate the benefit of combination immunotherapy in melanoma and other diseases. However, this effect comes at a high toxicity cost for patients, making this exploration of triplet therapy incredibly important to the oncology field.
In addition to efficacy, sargramostim can act as an immune modulator to potentially attenuate or avoid immune-related adverse events (Figure 3). Randomized controlled trials primarily focusing on immune-related adverse event prevention and/or treatment are lacking. In patients with dysregulated immunity of the GI tract or lung (Crohn’s disease or aPAP), sargramostim has been shown to be more effective than placebo at inducing disease control (39, 241). In the phase 2 trial in melanoma discussed above, Hodi et al. (28) reported combination sargramostim and ipilimumab therapy led to fewer GI and pulmonary toxicities and improved survival compared to ipilimumab alone. Treatment paradigms have shifted since completion of this study and ipilimumab doses less than 10mg/kg are now used due to treatment-limiting toxicity of higher ipilimumab doses; thus, additional studies are warranted to verify these findings with the current treatment regimens.
6.4. Potential future developments in anti-tumor effects and irAE
Guidelines are evolving to include more combination ICI therapy options. Investigators for the phase 3 CheckMate 067 trial recently reported 6.5-year OS data for patients with stage III/IV melanoma (242). 49% of patients treated with nivolumab/ipilimumab (n=314) versus 42% with nivolumab alone (n=316), or 23% with ipilimumab alone (n=315) achieved a durable OS response. Duration of response for study patients at 6.5 years for nivolumab/ipilimumab dramatically exceeded that of single agent use of ipilimumab or nivolumab (61.9-NR months vs 8.8,-47.4 months vs 45.7-NR, respectively). However, patients assigned to the nivolumab/ipilimumab arm experienced higher rates of autoimmune toxicities than patients treated with the single agent immunotherapy agents. The ongoing study EA6141 will assess if the addition of sargramostim to the nivolumab/ipilimumab combination corroborates and improves results obtained from CheckMate 067 (57). The study will assess the triplet combination of sargramostim/nivolumab/ipilimumab also in the alleviation of toxicities which may make therapy more tolerable and greatly expand the patient population able to benefit from this therapy.
EA6141 and CheckMate 067 exclude melanoma patients with poor prognostic factors like active or untreated brain metastases (57, 242). CheckMate 204 focused on the effects of nivolumab/ipilimumab combination therapy on melanoma brain metastases (243). Follow-up CheckMate 204 report noted a 3-year OS of 71.9% for asymptomatic (n=101) and 36% for symptomatic (n=18) patients (244). An important clinical question to answer for patients with poor prognostic factors would be the impact of adding sargramostim to combination ICI therapy (i.e., nivolumab/ipilimumab) on patient outcomes and rates of severe toxicity.
Investigations are underway to identify biomarkers with predictive utility for benefit from sargramostim. One strategy examines blood monocyte HLA-DR (human leukocyte antigen-DR isotype, a humanized major histocompatibility complex [MHC] II) expression on CD14+ immunosuppressive peripheral blood mononuclear cells, also known as MDSCs (25). This cell population is continuously renewed and reflects functional capacity of these cells, including their ability to phagocytose, to digest and present tumor antigens to T lymphocytes, as well as to support those lymphocytes with cytokines. Low HLA-DR expression has been proposed to be a surrogate marker of immunoparalysis in multiple clinical conditions, including sepsis where it is associated with worse patient outcomes (245). Equally, elevated monocytes with low HLA-DR expression have been correlated with worse outcomes for patients with melanoma treated with ipilimumab, among other cancers and therapies (246). In a number of different disorders as mentioned in Wound healing and risk of GM-CSF deficiency and Immune responses to infection and risk of GM-CSF insufficiency, sargramostim therapy increases HLA-DR expression and reverses immunoparalysis (65, 217) justifying further investigation in the context of cancer immunotherapy.
Additional trials are needed to evaluate immune-related adverse event risk factor prediction and prevention/reduction strategies, as well as the impact of immune-related adverse event management on ICI efficacy. Lozano et al. (247) reported, regardless of organ system, increased circulating activated CD4+ memory T cell numbers and increased T cell receptor diversity in melanoma correlate with an increased risk for severe immune-related adverse event development. Uncovering mechanisms responsible for organ-specific immune-related adverse events may enable expanded use of immunomodulatory agents in other disease states (e.g., GM-CSF autoantibodies in aPAP or Crohn’s disease) (125, 248). Future study designs might incorporate serial tissue biopsies to map toxicity development, identify potential biomarkers, and define the immune cell signaling involved in checkpoint-induced pneumonitis and colitis. These research goals could be incorporated into randomized controlled trials that measure immune-related adverse events with immunomodulatory agents like sargramostim.
Supported by Hodi et al. (28), the anti-cancer potential of sargramostim may be additive to or synergistic with ICIs to boost efficacy in patients with metastatic melanoma. Results from ICI combination studies are encouraging and may help answer the unmet need of low cure rates in melanoma and other cancers. Combination sargramostim and the PD-1 inhibitor, pembrolizumab, is being studied in melanoma [NCT04703426 (249)], biliary cancer [NCT02703714 (250)], and NSCLC [NCT04856176 (251)]. These trials and other investigations in renal cell carcinoma, and head and neck cancers aim to better understand the role that sargramostim plays in the generation of antitumor immune responses as well as attenuating toxicity.
7. Discussion
GM-CSF deficiency disease classification and correlation with mononuclear phagocyte dysfunction has led to investigations of sargramostim in aPAP, infection, wound healing, anti-cancer treatment, and irAE amelioration. Within these disease treatment paradigms, further investigations of biomarkers and/or biopredictors such as HLA-DR status in combination with sargramostim may allow for efficient patient stratification and individualized treatment approaches in order to improve outcomes (172, 173, 217, 245). Ongoing investigations in other disease states and infections [e.g. nontuberculosis mycobacteria (252), pulmonary aspergillus (176), C. gattii cryptococcosis (186), and respiratory syncytial virus (112, 253)] further highlight the wide potential role of immunomodulatory agents such as sargramostim. Specifically, for neurodegenerative diseases, early investigations suggest innate immune modulation and illustrate GM-CSF/sargramostim potential to ameliorate symptoms and pathology of Alzheimer’s and Parkinson’s Diseases, among others (33–36). We await results of such on-going clinical trials.
Author contributions
HL, conceptualization, methodology, data curation, supervision, writing- original draft and multiple revised drafts. KP and CR, methodology, data curation, supervision, writing- original draft and multiple revised drafts. TW, EL, EB, MD, DA, RP, and TB, writing-reviewing-editing. RG and ER, supervision and reviewing-editing. All authors contributed to the article and approved the submitted version.
Funding
Publication of this manuscript was supported by Partner Therapeutics, Inc. The funder, outside of employed authors, was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
We thank Dr. Drew Provan, MD (Herodotus Media) for figure creation.
Conflict of interest
HL is a paid consultant to Partner Therapeutics and has stock options. KP, TB, and CR, are employees of Partner Therapeutics and have stock options. ER at the time of drafting of this manuscript was an employee of Partner Therapeutics and has stock options. TW, EB, MD, RP, and RG received funds from Partner Therapeutics within the past 3 years, but none in relation to this publication. HL is a paid consultant to Pluri-Biotech, Inc. TW acknowledges support from Savara, Kiniksa, Kinevant (for participation on a DSMB or advisory board), the PAP Foundation (serves as the vice president and clinical director of the PAP Foundation) and is a paid consultant to IQVIA. EL acknowledges funding from the NIH, myCME, is a paid consultant to Guidepoint Global, and received support for travel from the PAP Foundation. EB acknowledges support from Genentech, Novartis, and Lilly, and received funds from Apexigen, Shionogi, Bristol Myers Squibb, Nektar, Instilbio, and Xilio for consulting within the past 3 years. MD received funds from Neolukin Therapeutics (and has stock options), Moderna, Aditum, OREC, Tillotts Pharma, SQZ Biotech, AzurRx, and Mallinckrodt for consulting within the past 3 years, and acknowledges support from UpToDate, Sandoz Academy, Experts at Your Fingertips, and WebMD. DA acknowledges funding from the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. RP acknowledges support to his institution from the US Department of Veterans Affairs and the NHLBI. RG is a consultant to NexImmune Inc., Ananexa Pharma Ascentage Pharm Group, Antengene Biotech LLC, and a Medical Director within FFF Enterprises Inc. and AZAC Inc. RG serves on the Board of Directors of the Russian Foundation for Cancer Research Support and is on the Scientific Advisory Board of StemRad Ltd.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1069444/full#supplementary-material
References
1. Lazarus HM, Ragsdale CE, Gale RP, Lyman GH. Sargramostim (Rhu GM-CSF) as cancer therapy (Systematic review) and an immunomodulator. A Drug before Its Time? Front Immunol (2021) 12:706186. doi: 10.3389/fimmu.2021.706186
2. Partner Therapeutics, Inc. Leukine®(Sargramostim) for injection, for subcuteous or intravenous use prescribing information.
3. Lazarus HM, McManus J, Gale RP. Sargramostim in acute radiation syndrome. Expert Opin Biol Ther (2022) 22(11):1345–52. doi: 10.1080/14712598.2022.2143261
4. Lazarus HM, Gale RP. G-CSF and GM-CSF are different. which one is better for COVID-19? Acta Haematol (2021) 144(4):355–9. doi: 10.1159/000510352
5. Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BS. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine (2015) 75(2):261–71. doi: 10.1016/j.cyto.2015.05.030
6. Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers (2019) 5(1):16. doi: 10.1038/s41572-019-0066-3
7. Ataya A KV, Carey B, Lee E, Tarling E, Wang T. The role of GM-CSF autoantibodies in infection and autoimmune pulmonary alveolar proteinosis: A concise review. Front Immunol (2021). doi: 10.3389/fimmu.2021.752856/full
8. Clementi N, Ghosh S, Santis MD, Castelli M, Criscuolo E, Zanoni I, et al. Viral respiratory pathogens and lung injury. Clin Microbiol Rev (2021) 34(3):e00103-20. doi: 10.1128/CMR.00103-20
9. Hall MW, Joshi I, Leal L, Ooi EE. Immune immunomodulation in coronavirus disease 2019 (COVID-19): Strategic considerations for personalized therapeutic intervention. Clin Infect Dis (2022) 74(1):144–8. doi: 10.1093/cid/ciaa904
10. Sturrock A, Mir-Kasimov M, Baker J, Rowley J, Paine R. Key role of microrna in the regulation of granulocyte macrophage colony-stimulating factor expression in murine alveolar epithelial cells during oxidative stress. J Biol Chem (2014) 289(7):4095–105. doi: 10.1074/jbc.m113.535922
11. Rosler B, Herold S. Lung epithelial GM-CSF improves host defense function and epithelial repair in influenza virus pneumonia-a new therapeutic strategy? Mol Cell Pediatr (2016) 3(1):29. doi: 10.1186/s40348-016-0055-5
12. Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, Via Pu.1, regulates alveolar macrophage fcgamma r-mediated phagocytosis and the IL-18/IFN-Gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood (2002) 100(12):4193–200. doi: 10.1182/blood-2002-04-1102
13. Kalinski AL, Yoon C, Huffman LD, Duncker PC, Kohen R, Passino R, et al. Analysis of the immune response to sciatic nerve injury identifies efferocytosis as a key mechanism of nerve debridement. Elife (2020) 9. doi: 10.7554/eLife.60223
14. Miricescu D, Badoiu SC, Stanescu-Spinu I-I, Totan AR, Stefani C, Greabu M. Growth factors, reactive oxygen species, and metformin–promoters of the wound healing process in burns? Int J Mol Sci (2021) 22(17):9512. doi: 10.3390/ijms22179512
15. Fang Y, Shen J, Yao M, Beagley KW, Hambly BD, Bao S. Granulocyte-macrophage colony-stimulating factor enhances wound healing in diabetes Via upregulation of proinflammatory cytokines. Br J Dermatol (2010) 162(3):478–86. doi: 10.1111/j.1365-2133.2009.09528.x
16. Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. New Engl J Med (2017) 376(24):2367–75. doi: 10.1056/NEJMra1615439
17. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: A cellular perspective. Physiol Rev (2019) 99(1):665–706. doi: 10.1152/physrev.00067.2017
18. Mirza RE, Fang MM, Novak ML, Urao N, Sui A, Ennis WJ, et al. Macrophage PPARgamma and impaired wound healing in type 2 diabetes. J Pathol (2015) 236(4):433–44. doi: 10.1002/path.4548
19. Willenborg S, Eming SA. Macrophages - sensors and effectors coordinating skin damage and repair. JDDG: J der Deutschen Dermatologischen Gesellschaft (2014) 12(3):214–21. doi: 10.1111/ddg.12290
20. Martin P. Wound healing–aiming for perfect skin regeneration. Science (1997) 276(5309):75–81. doi: 10.1126/science.276.5309.75
21. Desai R, Coxon AT, Dunn GP. Therapeutic applications of the cancer immunoediting hypothesis. Semin Cancer Biol (2022) 78:63–77. doi: 10.1016/j.semcancer.2021.03.002
22. Kurbacher CM, Kurbacher JA, Cramer EM, Rhiem K, Mallman PK, Reichelt R, et al. Continuous low-dose GM-CSF as salvage therapy in refractory recurrent breast or female genital tract carcinoma. Oncol (Williston Park) (2005) 19(4 Suppl 2):23–6.
23. Grotz TE, Kottschade L, Pavey ES, Markovic SN, Jakub JW. Adjuvant GM-CSF improves survival in high-risk stage iiic melanoma: A single-center study. Am J Clin Oncol (2014) 37(5):467–72. doi: 10.1097/COC.0b013e31827def82
24. Wei XX, Chan S, Kwek S, Lewis J, Dao V, Zhang L, et al. Systemic GM-CSF recruits effector T cells into the tumor microenvironment in localized prostate cancer. Cancer Immunol Res (2016) 4(11):948–58. doi: 10.1158/2326-6066.Cir-16-0042
25. Mengos AE, Gastineau DA, Gustafson MP. The Cd14+HLA-DRlo/Neg monocyte: An immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front Immunol (2019) 10:1147. doi: 10.3389/fimmu.2019.01147
26. Dougan M. Checkpoint blockade toxicity and immune homeostasis in the gastrointestinal tract. Front Immunol (2017) 8:1547(1547). doi: 10.3389/fimmu.2017.01547
27. Suresh K, Naidoo J, Zhong Q, Xiong Y, Mammen J, De Flores MV, et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J Clin Invest (2019) 129(10):4305–15. doi: 10.1172/jci128654
28. Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: A randomized clinical trial. JAMA (2014) 312(17):1744–53. doi: 10.1001/jama.2014.13943
29. Chen P, Chen F, Zhou B. Comparisons of therapeutic efficacy and safety of ipilimumab plus GM-CSF versus ipilimumab alone in patients with cancer: A meta-analysis of outcomes. Drug Design Dev Ther (2018) 12:2025–38. doi: 10.2147/dddt.s154258
30. Däbritz J. Granulocyte macrophage colony-stimulating factor and the intestinal innate immune cell homeostasis in crohn’s disease. Am J Physiology-Gastrointestinal Liver Physiol (2014) 306(6):G455–G65. doi: 10.1152/ajpgi.00409.2013
31. Kumar A, Taghi Khani A, Sanchez Ortiz A, Swaminathan S. GM-CSF: A double-edged sword in cancer immunotherapy. Front Immunol (2022) 13:901277. doi: 10.3389/fimmu.2022.901277
32. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukocyte Biol (2016) 100(3):481–9. doi: 10.1189/jlb.3ru0316-144r
33. Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical parkinson’s disease trial. NPJ Parkinson’s Dis (2017) 3(1). doi: 10.1038/s41531-017-0013-5
34. Olson KE, Namminga KL, Lu Y, Schwab AD, Thurston MJ, Abdelmoaty MM, et al. Safety, tolerability, and immune-biomarker profiling for year-long sargramostim treatment of parkinson’s disease. EBioMedicine (2021) 67:103380. doi: 10.1016/j.ebiom.2021.103380
35. Potter H, Woodcock JH, Boyd TD, Coughlan CM, O’Shaughnessy JR, Borges MT, et al. Safety and efficacy of sargramostim (GM-CSF) in the treatment of alzheimer’s disease. Alzheimers Dement (N Y) (2021) 7(1):e12158. doi: 10.1002/trc2.12158
36. Abdelmoaty MM, Machhi J, Yeapuri P, Shahjin F, Kumar V, Olson KE, et al. Monocyte biomarkers define sargramostim treatment outcomes for parkinson’s disease. Clin Trans Med (2022) 12(7). doi: 10.1002/ctm2.958
37. Lavoie PM, Levy O. Mononuclear phagocyte system. Fetal Neonatal Physiol (Fifth Edition) (2017), 1208–16.e3.
38. Trapnell BC, Inoue Y, Bonella F, Morgan C, Jouneau S, Bendstrup E, et al. Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med (2020) 383(17):1635–44. doi: 10.1056/NEJMoa1913590
39. Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med (2019) 381(10):923–32. doi: 10.1056/NEJMoa1816216
40. Campo I, Mariani F, Paracchini E, Kadija Z, Zorzetto M, Tinelli C, et al. Inhaled sargramostim and whole lung lavage (WLL) as therapy of autoimmune pulmonary alveolar proteinosis (Apap). Eur Respir J (2016) 48(suppl 60):PA3870. doi: 10.1183/13993003.congress-2016.PA3870
41. Campo I, Mariani F, Paracchini E, Kadija Z, Zorzetto M, Tinelli C, et al. Whole Lung Lavage Followed by Inhaled Sargramostim as Therapy of Autoimmune Pulmonary Alveolar Proteinosis. Am J Respir Crit Care Med (2016) 2016:A6438
42. Tazawa R, Inoue Y, Arai T, Takada T, Kasahara Y, Hojo M, et al. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte-macrophage colony-stimulating factor therapy. Chest (2014) 145(4):729–37. doi: 10.1378/chest.13-0603
43. Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled Granulocyte/Macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2010) 181(12):1345–54. doi: 10.1164/rccm.200906-0978OC
44. Da Costa RM, Ribeiro Jesus FM, Aniceto C, Mendes M. Randomized, double-blind, placebo-controlled, dose- ranging study of granulocyte-macrophage colony-stimulating factor in patients with chronic venous leg ulcers. Wound Repair Regener (1999) 7(1):17–25. doi: 10.1046/j.1524-475x.1999.00017.x
45. Da Costa RM, Jesus FM, Aniceto C, Mendes M. Double-blind randomized placebo-controlled trial of the use of granulocyte-macrophage colony-stimulating factor in chronic leg ulcers. Am J Surg (1997) 173(3):165–8. doi: 10.1016/s0002-9610(97)89589-x
46. Cianfarani F, Tommasi R, Failla CM, Viviano MT, Annessi G, Papi M, et al. Granulocyte/Macrophage colony-stimulating factor treatment of human chronic ulcers promotes angiogenesis associated with De novo vascular endothelial growth factor transcription in the ulcer bed. Br J Dermatol (2006) 154(1):34–41. doi: 10.1111/j.1365-2133.2005.06925.x
47. Bianchi L, Ginebri A, Hagman JH, Francesconi F, Carboni I, Chimenti S. Local treatment of chronic cutaneous leg ulcers with recombinant human granulocyte-macrophage colony-stimulating factor. J Eur Acad Dermatol Venereol (2002) 16(6):595–8. doi: 10.1046/j.1468-3083.2002.00526.x
48. Brem H, Howell R, Criscitelli T, Senderowicz A, Siegart N, Gorenstein S, et al. Practical application of granulocyte-macrophage colony-stimulating factor (GM-CSF) in patients with wounds. Surg Technol Int (2018) 32:61–6.
49. Karlafti E, Savopoulos C, Hatzitolios A, Didangelos T. Local use of granulocyte-macrophages colony stimulating factor in treatment of chronic diabetic neuropathic ulcer (Case review). Georgian Med News (2018) 277):21–7.
50. Chi YF, Chai JK, Luo HM, Zhang QX, Feng R. Safety of recombinant human granulocyte-macrophage colony-stimulating factor in healing pediatric severe burns. Genet Mol Res (2015) 14(1):2735–41. doi: 10.4238/2015.March.31.3
51. Yan D, Liu S, Zhao X, Bian H, Yao X, Xing J, et al. Recombinant human granulocyte macrophage colony stimulating factor in deep second-degree burn wound healing. Med (Baltimore) (2017) 96(22):e6881. doi: 10.1097/MD.0000000000006881
52. Robson MC, Hill DP, Smith PD, Wang X, Meyer-Siegler K, Ko F, et al. Sequential cytokine therapy for pressure ulcers: Clinical and mechanistic response. Ann Surg (2000) 231(4):600–11. doi: 10.1097/00000658-200004000-00020
53. Payne WG, Ochs DE, Meltzer DD, Hill DP, Mannari RJ, Robson LE, et al. Long-term outcome study of growth factor-treated pressure ulcers. Am J Surg (2001) 181(1):81–6. doi: 10.1016/S0002-9610(00)00536-5
54. Fong L, Kwek SS, O’Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA-4 blockade and GM-CSF. Cancer Res (2009) 69(2):609–15. doi: 10.1158/0008-5472.Can-08-3529
55. Luke JJ, Donahue H, Nishino M, Giobbie-Hurder A, Davis M, Bailey N, et al. Single institution experience of ipilimumab 3 Mg/Kg with sargramostim (GM-CSF) in metastatic melanoma. Cancer Immunol Res (2015) 3(9):986–91. doi: 10.1158/2326-6066.CIR-15-0066
56. Kwek SS, Kahn J, Greaney SK, Lewis J, Cha E, Zhang L, et al. GM-CSF and ipilimumab therapy in metastatic melanoma: Clinical outcomes and immunologic responses. Oncoimmunology (2016) 5(4):e1101204. doi: 10.1080/2162402X.2015.1101204
57. Clinicaltrials.Gov. A phase II/III trial of nivolumab, ipilimumab, and GM-CSF in patients with advanced melanoma (2021) (Accessed November 30, 2022).
58. Bosteels C, Van Damme KFA, De Leeuw E, Declercq J, Maes B, Bosteels V, et al. Loss of GM-CSF-Dependent instruction of alveolar macrophages in COVID-19 provides a rationale for inhaled GM-CSF treatment. Cell Rep Med (2022). doi: 10.1016/j.xcrm.2022.100833
59. Herold S, Hoegner K, Vadász I, Gessler T, Wilhelm J, Mayer K, et al. Inhaled Granulocyte/Macrophage colony–stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am J Respir Crit Care Med (2014) 189(5):609–11. doi: 10.1164/rccm.201311-2041le
60. Paine R 3rd, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med (2012) 40(1):90–7. doi: 10.1097/CCM.0b013e31822d7bf0
61. Pinder EM, Rostron AJ, Hellyer TP, Ruchaud-Sparagano MH, Scott J, Macfarlane JG, et al. Randomised controlled trial of GM-CSF in critically ill patients with impaired neutrophil phagocytosis. Thorax (2018) 73(10):918–25. doi: 10.1136/thoraxjnl-2017-211323
62. Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest (2005) 127(6):2139–50. doi: 10.1378/chest.127.6.2139
63. Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW. A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med (2002) 166(2):138–43. doi: 10.1164/rccm.2009005
64. Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med (2011) 37(3):525–32. doi: 10.1007/s00134-010-2088-x
65. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte–macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression. Am J Respir Crit Care Med (2009) 180(7):640–8. doi: 10.1164/rccm.200903-0363oc
66. Bilgin K, Yaramiş A, Haspolat K, Taş MA, Günbey S, Derman O. A randomized trial of granulocyte-macrophage colony-stimulating factor in neonates with sepsis and neutropenia. Pediatrics (2001) 107(1):36–41. doi: 10.1542/peds.107.1.36
67. Wan L, Zhang Y, Lai Y, Jiang M, Song Y, Zhou J, et al. Effect of granulocyte-macrophage colony-stimulating factor on prevention and treatment of invasive fungal disease in recipients of allogeneic stem-cell transplantation: A prospective multicenter randomized phase iv trial. J Clin Oncol (2015) 33(34):3999–4006. doi: 10.1200/jco.2014.60.5121
68. Dorr RT. Clinical properties of yeast-derived versus escherichia coli-derived granulocyte-macrophage colony-stimulating factor. Clin Ther (1993) 15(1):19–29; discussion 18.
69. Sola RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci (2009) 98(4):1223–45. doi: 10.1002/jps.21504
70. Gu L, Chiang KY, Zhu N, Findley HW, Zhou M. Contribution of STAT3 to the activation of survivin by GM-CSF in CD34+ cell lines. Exp Hematol (2007) 35(6):957–66. doi: 10.1016/j.exphem.2007.03.007
71. Sudo T, Motomura Y, Okuzaki D, Hasegawa T, Yokota T, Kikuta J, et al. Group 2 innate lymphoid cells support hematopoietic recovery under stress conditions. J Exp Med (2021) 218(5). doi: 10.1084/jem.20200817
72. Kim JK, Choi BH, Park HC, Park SR, Kim YS, Yoon SH, et al. Effects of GM-CSF on the neural progenitor cells. Neuroreport (2004) 15(14):2161–5. doi: 10.1097/00001756-200410050-00003
73. Kruger C, Laage R, Pitzer C, Schabitz WR, Schneider A. The hematopoietic factor GM-CSF (Granulocyte-macrophage colony-stimulating factor) promotes neuronal differentiation of adult neural stem cells in vitro. BMC Neurosci (2007) 8:88. doi: 10.1186/1471-2202-8-88
74. Choi JK, Kim KH, Park H, Park SR, Choi BH. Granulocyte macrophage-colony stimulating factor shows anti-apoptotic activity in neural progenitor cells Via Jak/Stat5-Bcl-2 pathway. Apoptosis (2011) 16(2):127–34. doi: 10.1007/s10495-010-0552-2
75. Bouhy D, Malgrange B, Multon S, Poirrier AL, Scholtes F, Schoenen J, et al. Delayed GM-CSF treatment stimulates axonal regeneration and functional recovery in paraplegic rats Via an increased bdnf expression by endogenous macrophages. FASEB J (2006) 20(8):1239–41. doi: 10.1096/fj.05-4382fje
76. Hayashi K, Ohta S, Kawakami Y, Toda M. Activation of dendritic-like cells and neural Stem/Progenitor cells in injured spinal cord by GM-CSF. Neurosci Res (2009) 64(1):96–103. doi: 10.1016/j.neures.2009.01.018
77. Staitieh BS, Auld SC, Ahmed M, Fan X, Smirnova N, Yeligar SM. Granulocyte macrophage-colony stimulating factor reverses HIV protein-induced mitochondrial derangements in alveolar macrophages. AIDS Res Hum Retroviruses (2021) 37(3):224–32. doi: 10.1089/aid.2020.0176
78. Wessendarp M, Watanabe-Chailland M, Liu S, Stankiewicz T, Ma Y, Kasam RK, et al. Role of GM-CSF in regulating metabolism and mitochondrial functions critical to macrophage proliferation. Mitochondrion (2021) 62:85–101. doi: 10.1016/j.mito.2021.10.009
79. Jubinsky PT, Messer A, Bender J, Morris RE, Ciraolo GM, Witte DP, et al. Identification and characterization of magmas, a novel mitochondria-associated protein involved in granulocyte-macrophage colony-stimulating factor signal transduction. Exp Hematol (2001) 29(12):1392–402. doi: 10.1016/s0301-472x(01)00749-4
80. Becker S, Gehrsitz A, Bork P, Buchner S, Buchner E. The black-pearl gene of drosophila defines a novel conserved protein family and is required for larval growth and survival. Gene (2001) 262(1-2):15–22. doi: 10.1016/s0378-1119(00)00548-5
81. Jubinsky PT, Short MK, Mutema G, Morris RE, Ciraolo GM, Li M. Magmas expression in neoplastic human prostate. J Mol Histol (2005) 36(1-2):69–75. doi: 10.1007/s10735-004-3840-8
82. Sinha D, Joshi N, Chittoor B, Samji P, D’Silva P. Role of magmas in protein transport and human mitochondria biogenesis. Hum Mol Genet (2010) 19(7):1248–62. doi: 10.1093/hmg/ddq002
83. Tagliati F, Gentilin E, Buratto M, Molè D, degli Uberti EC, Zatelli MC. Magmas, a gene newly identified as overexpressed in human and mouse ACTH-secreting pituitary adenomas, protects pituitary cells from apoptotic stimuli. Endocrinology (2010) 151(10):4635–42. doi: 10.1210/en.2010-0441
84. Roy S, Short MK, Stanley ER, Jubinsky PT. Essential role of drosophila black-pearl is mediated by its effects on mitochondrial respiration. FASEB J (2012) 26(9):3822–33. doi: 10.1096/fj.11-193540
85. Srivastava S, Sinha D, Saha PP, Marthala H, D’Silva P. Magmas functions as a ROS regulator and provides cytoprotection against oxidative stress-mediated damages. Cell Death Dis (2014) 5(8):e1394. doi: 10.1038/cddis.2014.355
86. Mylonas KJ, Anderson J, Sheldrake TA, Hesketh EE, Richards JA, Ferenbach DA, et al. Granulocyte macrophage-colony stimulating factor: A key modulator of renal mononuclear phagocyte plasticity. Immunobiology (2019) 224(1):60–74. doi: 10.1016/j.imbio.2018.10.007
87. Quan Y, Möller T, Weinstein JR. Regulation of fcgamma receptors and immunoglobulin G-mediated phagocytosis in mouse microglia. Neurosci Lett (2009) 464(1):29–33. doi: 10.1016/j.neulet.2009.08.013
88. Richardson MD, Brownlie CE, Shankland GS. Enhanced phagocytosis and intracellular killing of candida albicans by GM-CSF-Activated human neutrophils. J Med Vet Mycol (1992) 30(6):433–41.
89. Coleman DL, Chodakewitz JA, Bartiss AH, Mellors JW. Granulocyte-macrophage colony-stimulating factor enhances selective effector functions of tissue-derived macrophages. Blood (1988) 72(2):573–8. doi: 10.1182/blood.V72.2.573.573
90. Trapnell BC, Whitsett JA. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol (2002) 64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847
91. Doran AC, Yurdagul A Jr., Tabas I. Efferocytosis in health and disease. Nat Rev Immunol (2020) 20(4):254–67. doi: 10.1038/s41577-019-0240-6
92. Akakura S, Singh S, Spataro M, Akakura R, Kim JI, Albert ML, et al. The opsonin MFG-E8 is a ligand for the Alphavbeta5 integrin and triggers Dock180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res (2004) 292(2):403–16. doi: 10.1016/j.yexcr.2003.09.011
93. Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G. MFG-E8-Mediated uptake of apoptotic cells by apcs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest (2007) 117(7):1902–13. doi: 10.1172/jci30966
94. Miyasaka K, Hanayama R, Tanaka M, Nagata S. Expression of milk fat globule epidermal growth factor 8 in immature dendritic cells for engulfment of apoptotic cells. Eur J Immunol (2004) 34(5):1414–22. doi: 10.1002/eji.200424930
95. De Nichilo MO, Burns GF. Granulocyte-macrophage and macrophage colony-stimulating factors differentially regulate alpha V integrin expression on cultured human macrophages. Proc Natl Acad Sci U.S.A. (1993) 90(6):2517–21. doi: 10.1073/pnas.90.6.2517
96. Inoue M, Namba N, Chappel J, Teitelbaum SL, Ross FP. Granulocyte macrophage-colony stimulating factor reciprocally regulates alphav-associated integrins on murine osteoclast precursors. Mol Endocrinol (1998) 12(12):1955–62. doi: 10.1210/mend.12.12.0213
97. Na YR, Jung D, Gu GJ, Seok SH. GM-CSF grown bone marrow derived cells are composed of phenotypically different dendritic cells and macrophages. Mol Cells (2016) 39(10):734–41. doi: 10.14348/molcells.2016.0160
98. Sakhno LV, Shevela EY, Tikhonova MA, Maksimova AA, Tyrinova TV, Ostanin AA, et al. Efferocytosis modulates arginase-1 and tyrosine kinase mer expression in GM-CSF-Differentiated human macrophages. Bull Exp Biol Med (2021) 170(6):778–81. doi: 10.1007/s10517-021-05153-z
99. Kazawa T, Kawasaki T, Sakamoto A, Imamura M, Ohashi R, Jiang S, et al. Expression of liver X receptor alpha and lipid metabolism in granulocyte-macrophage colony-stimulating factor-induced human monocyte-derived macrophage. Pathol Int (2009) 59(3):152–60. doi: 10.1111/j.1440-1827.2009.02343.x
100. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-Γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15(11):1026–37. doi: 10.1038/ni.3005
101. Nieto C, Bragado R, Municio C, Sierra-Filardi E, Alonso B, Escribese MM, et al. The activin a-peroxisome proliferator-activated receptor gamma axis contributes to the transcriptome of GM-CSF-Conditioned human macrophages. Front Immunol (2018) 9:31. doi: 10.3389/fimmu.2018.00031
102. Yasui K, Sekiguchi Y, Ichikawa M, Nagumo H, Yamazaki T, Komiyama A, et al. Granulocyte macrophage-colony stimulating factor delays neutrophil apoptosis and primes its function through ia-type phosphoinositide 3-kinase. J Leukoc Biol (2002) 72(5):1020–6. doi: 10.1189/jlb.72.5.1020
103. von Gunten S, Schaub A, Vogel M, Stadler BM, Miescher S, Simon HU. Immunologic and functional evidence for anti-Siglec-9 autoantibodies in intravenous immunoglobulin preparations. Blood (2006) 108(13):4255–9. doi: 10.1182/blood-2006-05-021568
104. Shen L, Smith JM, Shen Z, Hussey SB, Wira CR, Fanger MW. Differential regulation of neutrophil chemotaxis to IL-8 and FMLP by GM-CSF: Lack of direct effect of oestradiol. Immunology (2006) 117(2):205–12. doi: 10.1111/j.1365-2567.2005.02280.x
105. Wolach B, van der Laan LJ, Maianski NA, Tool AT, van Bruggen R, Roos D, et al. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp Hematol (2007) 35(4):541–50. doi: 10.1016/j.exphem.2006.12.008
106. Tang J, Zarbock A, Gomez I, Wilson CL, Lefort CT, Stadtmann A, et al. Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood (2011) 118(3):786–94. doi: 10.1182/blood-2010-11-321406
107. Griffin JD, Spertini O, Ernst TJ, Belvin MP, Levine HB, Kanakura Y, et al. Granulocyte-macrophage colony-stimulating factor and other cytokines regulate surface expression of the leukocyte adhesion molecule-1 on human neutrophils, monocytes, and their precursors. J Immunol (1990) 145(2):576–84. doi: 10.4049/jimmunol.145.2.576
108. Zou T, Satake A, Ojha P, Kambayashi T. Cellular therapies supplement: The role of granulocyte macrophage colony-stimulating factor and dendritic cells in regulatory T-cell homeostasis and expansion. Transfusion (2011) 51 Suppl 4:160s–8s. doi: 10.1111/j.1537-2995.2011.03379.x
109. Hotta M, Yoshimura H, Satake A, Tsubokura Y, Ito T, Nomura S. GM-CSF therapy inhibits chronic graft-Versus-Host disease Via expansion of regulatory T cells. Eur J Immunol (2019) 49(1):179–91. doi: 10.1002/eji.201847684
110. Park MY, Lim BG, Kim SY, Sohn HJ, Kim S, Kim TG. GM-CSF promotes the expansion and differentiation of cord blood myeloid-derived suppressor cells, which attenuate xenogeneic graft-Vs.-Host disease. Front Immunol (2019) 10:183. doi: 10.3389/fimmu.2019.00183
111. Chou HS, Hsieh CC, Charles R, Wang L, Wagner T, Fung JJ, et al. Myeloid-derived suppressor cells protect islet transplants by B7-H1 mediated enhancement of T regulatory cells. Transplantation (2012) 93(3):272–82. doi: 10.1097/TP.0b013e31823ffd39
112. Petrina M, Martin J, Basta S. Granulocyte macrophage colony-stimulating factor has come of age: From a vaccine adjuvant to antiviral immunotherapy. Cytokine Growth Factor Rev (2021) 59:101–10. doi: 10.1016/j.cytogfr.2021.01.001
113. Hamilton JA. GM-CSF-Dependent inflammatory pathways. Front Immunol (2019) 10:2055. doi: 10.3389/fimmu.2019.02055
114. Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary alveolar proteinosis in adults: Pathophysiology and clinical approach. Lancet Respir Med (2018) 6(7):554–65. doi: 10.1016/S2213-2600(18)30043-2
115. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: Progress in the first 44 years. Am J Respir Crit Care Med (2002) 166(2):215–35. doi: 10.1164/rccm.2109105
116. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol (2003) 29(6):677–82. doi: 10.1165/rcmb.2003-0148OC
117. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med (2001) 7(1):53–8. doi: 10.1038/83348
118. McCormick TS, Hejal RB, Leal LO, Ghannoum MA. GM-CSF: Orchestrating the pulmonary response to infection. Front Pharmacol (2022) 12:735443. doi: 10.3389/fphar.2021.735443
119. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med (2007) 356(6):567–79. doi: 10.1056/NEJMoa062505
120. Lee E, Miller C, Ataya A, Wang T. Opportunistic infection associated with elevated GM-CSF autoantibodies: A case series and review of the literature. Open Forum Infect Dis (2022) 9(5):ofac146. doi: 10.1093/ofid/ofac146
121. Griese M, Bonella F, Costabel U, De Blic J, Tran N-B, Liebisch G. Quantitative lipidomics in pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2019) 200(7):881–7. doi: 10.1164/rccm.201901-0086oc
122. Moore BB, Coffey MJ, Christensen P, Sitterding S, Ngan R, Wilke CA, et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis Via a prostaglandin-dependent mechanism. J Immunol (2000) 165(7):4032–9. doi: 10.4049/jimmunol.165.7.4032
123. Sheng G, Chen P, Wei Y, Chu J, Cao X, Zhang H-L. Better approach for autoimmune pulmonary alveolar proteinosis treatment: Inhaled or subcutaneous granulocyte-macrophage colony-stimulating factor: A meta-analyses. Respir Res (2018) 19(1). doi: 10.1186/s12931-018-0862-4
124. Labiris NR, Dolovich MB. Pulmonary drug delivery. part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol (2003) 56(6):588–99. doi: 10.1046/j.1365-2125.2003.01892.x
125. McCarthy C, Carey B, Trapnell BC. Autoimmune pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2022) 205(9):1016–35. doi: 10.1164/rccm.202112-2742SO
126. Ohkouchi S, Akasaka K, Ichiwata T, Hisata S, Iijima H, Takada T, et al. Sequential granulocyte-macrophage colony-stimulating factor inhalation after whole-lung lavage for pulmonary alveolar proteinosis. A Rep Five Intractable Cases. Ann Am Thorac Soc (2017) 14(8):1298–304. doi: 10.1513/AnnalsATS.201611-892BC
127. Clinicaltrials.Gov. Clinical trial of inhaled molgramostim nebulizer solution in autoimmune pulmonary alveolar proteinosis (aPAP) (Impala-2) (2022) (Accessed November 30, 2022).
128. Tannerpharma Group. Ptx Partnership: Leaping Beyond Boundaries. (2021). Available at: https://Tannerpharma.Com/Our-Impact/Ptx-Partnership/.
129. McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis (2018) 13(1):129. doi: 10.1186/s13023-018-0846-y
130. Tome RG, McElyea C, Chang C-F. Incidence, mortality and characteristics of patients admitted with pulmonary alveolar proteinosis in the United States. Chest (2020) 158(4):A1043. doi: 10.1016/j.chest.2020.08.969
131. National Jewish Health, Advanced Diagnostic Laboratories. (2022). Available at: https://Www.Nationaljewish.Org/for-Professionals/Diagnostic-Testing/Adx/Tests/Anti-Gmcsf-Autoantibody.
132. Cincinnati Children’s, Cbdi Clinical Laboraties, Search for a Test. (2022). Available at: https://Www.Cincinnatichildrens.Org/Service/C/Cancer-Blood/Hcp/Clinical-Laboratories/Search-for-Test#Contentmaster_1_Pagemaincontent_1_Columnprimary_1_Rptaccordion_Faqcardwrap_13.
133. Salvaterra E, Campo I. Pulmonary alveolar proteinosis: From classification to therapy. Breathe (2020) 16(2):200018–. doi: 10.1183/20734735.0018-2020
134. Thomassen MJ, Barna BP, Malur AG, Bonfield TL, Farver CF, Malur A, et al. ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J Lipid Res (2007) 48(12):2762–8. doi: 10.1194/jlr.P700022-JLR200
135. Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep (2017) 7(1):10211. doi: 10.1038/s41598-017-10879-w
136. McCarthy C, Lee E, Bridges JP, Sallese A, Suzuki T, Woods JC, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Nat Commun (2018) 9(1):3127. doi: 10.1038/s41467-018-05491-z
137. Pfortmueller CA, Meisel C, Fux M, Schefold JC. Assessment of immune organ dysfunction in critical illness: Utility of innate immune response markers. Intensive Care Med Exp (2017) 5(1). doi: 10.1186/s40635-017-0163-0
138. Fontana L, Strasfeld L. Respiratory virus infections of the stem cell transplant recipient and the hematologic malignancy patient. Infect Dis Clin North Am (2019) 33(2):523–44. doi: 10.1016/j.idc.2019.02.004
139. Iuliano, Roguski, Chang, Muscatello, Palekar, Tempia, et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet (2018) 391(10127):1285–300. doi: 10.1016/s0140-6736(17)33293-2
140. Jean-Baptiste E. Cellular mechanisms in sepsis. J Intensive Care Med (2007) 22(2):63–72. doi: 10.1177/0885066606297123
141. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol (2022) 50(5):270–84. doi: 10.1038/s41579-022-00713-0
142. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. New Engl J Med (2017) 377(6):562–72. doi: 10.1056/NEJMra1608077
143. Cheng AC, Stephens DP, Currie BJ. Granulocyte-colony stimulating factor (G-CSF) as an adjunct to antibiotics in the treatment of pneumonia in adults. Cochrane Database Systematic Rev (2007) 2):CD004400. doi: 10.1002/14651858.CD004400.pub3
144. Brankston G, Gitterman L, Hirji Z, Lemieux C, Gardam M. Transmission of influenza a in human beings. Lancet Infect Diseases (2007) 7(4):257–65. doi: 10.1016/s1473-3099(07)70029-4
145. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315(8):801. doi: 10.1001/jama.2016.0287
146. Hall MW. Immune modulation in pediatric sepsis. J Pediatr Intensive Care (2019) 8(1):42–50. doi: 10.1055/s-0038-1676607
147. Teuben MPJ, Pfeifer R, Teuber H, De Boer LL, Halvachizadeh S, Shehu A, et al. Lessons learned from the mechanisms of posttraumatic inflammation extrapolated to the inflammatory response in COVID-19: A review. Patient Saf Surg (2020) 14(1). doi: 10.1186/s13037-020-00253-7
148. Gu X, Zhou F, Wang Y, Fan G, Cao B. Respiratory viral sepsis: Epidemiology, pathophysiology, diagnosis and treatment. Eur Respir Rev (2020) 29(157):200038. doi: 10.1183/16000617.0038-2020
149. Zhang Y, Chen Y, Meng Z. Immunomodulation for severe COVID-19 pneumonia: The state of the art. Front Immunol (2020) 11:577442. doi: 10.3389/fimmu.2020.577442
150. Monneret G, Lepape A, Voirin N, Bohe J, Venet F, Debard AL, et al. Persisting low monocyte human leukocyte antigen-Dr expression predicts mortality in septic shock. Intensive Care Med (2006) 32(8):1175–83. doi: 10.1007/s00134-006-0204-8
151. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood (2014) 124(3):344–53. doi: 10.1182/blood-2014-02-514778
152. Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the intensity of conditioning regimens: Working definitions. Biol Blood Marrow Tr (2009) 15(12):1628–33. doi: 10.1016/j.bbmt.2009.07.004
153. Melenhorst JJ, Tian X, Xu D, Sandler NG, Scheinberg P, Biancotto A, et al. Cytopenia and leukocyte recovery shape cytokine fluctuations after myeloablative allogeneic hematopoietic stem cell transplantation. Haematologica (2012) 97(6):867–73. doi: 10.3324/haematol.2011.053363
154. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood (2012) 119(15):3383–93. doi: 10.1182/blood-2011-11-370130
155. Copelan EA. Hematopoietic stem-cell transplantation. New Engl J Med (2006) 354(17):1813–26. doi: 10.1056/NEJMra052638
156. Copelan, Chojecki, Lazarus, Avalos. Allogeneic hematopoietic cell transplantation; the current renaissance. Blood Rev (2019) 34:34–44. doi: 10.1016/j.blre.2018.11.001
157. Chu S, McCormick TS, Lazarus HM, Leal LO, Ghannoum MA. Invasive fungal disease and the immunocompromised host including allogeneic hematopoietic cell transplant recipients: Improved understanding and new strategic approach with sargramostim. Clin Immunol (2021) 228:108731. doi: 10.1016/j.clim.2021.108731
158. Cortegiani A, Madotto F, Gregoretti C, Bellani G, Laffey JG, Pham T, et al. Immunocompromised patients with acute respiratory distress syndrome: Secondary analysis of the lung safe database. Crit Care (2018) 22(1). doi: 10.1186/s13054-018-2079-9
159. Chemaly RF, Shah DP, Boeckh MJ. Management of respiratory viral infections in hematopoietic cell transplant recipients and patients with hematologic malignancies. Clin Infect Dis (2014) 59(suppl_5):S344–S51. doi: 10.1093/cid/ciu623
160. De Lima CRA, Mirandolli TB, Carneiro LC, Tusset C, Romer CM, Andreolla HF, et al. Prolonged respiratory viral shedding in transplant patients. Transplant Infect Dis (2014) 16(1):165–9. doi: 10.1111/tid.12167
161. Chen Q, He F, Kwang J, Chan JKY, Chen J. GM-CSF and IL-4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J Immunol (2012) 189(11):5223–9. doi: 10.4049/jimmunol.1201789
162. Unkel B, Hoegner K, Clausen BE, Lewe-Schlosser P, Bodner J, Gattenloehner S, et al. Alveolar epithelial cells orchestrate dc function in murine viral pneumonia. J Clin Invest (2012) 122(10):3652–64. doi: 10.1172/JCI62139
163. Sever-Chroneos Z, Murthy A, Davis J, Florence JM, Kurdowska A, Krupa A, et al. GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res (2011) 92(2):319–28. doi: 10.1016/j.antiviral.2011.08.022
164. Huang FF, Barnes PF, Feng Y, Donis R, Chroneos ZC, Idell S, et al. GM-CSF in the lung protects against lethal influenza infection. Am J Respir Crit Care Med (2011) 184(2):259–68. doi: 10.1164/rccm.201012-2036OC
165. Subramaniam R, Hillberry Z, Chen H, Feng Y, Fletcher K, Neuenschwander P, et al. Delivery of GM-CSF to protect against influenza pneumonia. PloS One (2015) 10(4):e0124593. doi: 10.1371/journal.pone.0124593
166. Li W, Li D, Chen Y, Abudou H, Wang H, Cai J, et al. Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: New research progress and prospect. Dis Markers (2022) 2022:1–9. doi: 10.1155/2022/6362344
167. Halstead ES, Umstead TM, Davies ML, Kawasawa YI, Silveyra P, Howyrlak J, et al. GM-CSF overexpression after influenza A virus infection prevents mortality and moderates M1-like airway Monocyte/Macrophage polarization. Respir Res (2018) 19(1):3. doi: 10.1186/s12931-017-0708-5
168. Umstead TM, Hewage EK, Mathewson M, Beaudoin S, Chroneos ZC, Wang M, et al. Lower respiratory tract delivery, airway clearance, and preclinical efficacy of inhaled GM-CSF in a postinfluenza pneumococcal pneumonia model. Am J Physiol Lung Cell Mol Physiol (2020) 318(4):L571–L9. doi: 10.1152/ajplung.00296.2019
169. Matute-Bello G, Liles WC, Radella F 2nd, Steinberg KP, Ruzinski JT, Hudson LD, et al. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and Granulocyte/Macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med (2000) 28(1):1–7. doi: 10.1097/00003246-200001000-00001
170. Chousterman BG, Arnaud M. Is there a role for hematopoietic growth factors during sepsis? Front Immunol (2018) 9:1015. doi: 10.3389/fimmu.2018.01015
171. Hornell TM, Beresford GW, Bushey A, Boss JM, Mellins ED. Regulation of the Class II MHC pathway in primary human monocytes by granulocyte-macrophage colony-stimulating factor. J Immunol (2003) 171(5):2374–83. doi: 10.4049/jimmunol.171.5.2374
172. Perry SE, Mostafa SM, Wenstone R, Shenkin A, McLaughlin PJ. HLA-DR regulation and the influence of GM-CSF on transcription, surface expression and shedding. Int J Med Sci (2004) 1(3):126–36. doi: 10.7150/ijms.1.126
173. Drossou-Agakidou V, Kanakoudi-Tsakalidou F, Sarafidis K, Tzimouli V, Taparkou A, Kremenopoulos G, et al. In vivo effect of rhGM-CSF and rhG-CSF on monocyte HLA-DR expression of septic neonates. Cytokine (2002) 18(5):260–5. doi: 10.1006/cyto.2002.1037
174. Paine R, Chasse R, Halstead ES, Nfonoyim J, Park DJ, Byun T, et al. Inhaled sargramostim (Recombinant human granulocyte-macrophage colony-stimulating factor) for COVID-19-Associated acute hypoxemia: Results of the phase 2, randomized, open-label trial (Ileukpulm). Military Med (2022). doi: 10.1093/milmed/usac362
176. Quezada G, Koshkina NV, Zweidler-McKay P, Zhou Z, Kontoyiannis DP, Kleinerman ES. Intranasal granulocyte-macrophage colony-stimulating factor reduces the aspergillus burden in an immunosuppressed murine model of pulmonary aspergillosis. Antimicrobial Agents chemotherapy (2008) 52(2):716–8. doi: 10.1128/aac.00760-07
177. Chen TK, Batra JS, Michalik DE, Casillas J, Patel R, Ruiz ME, et al. Recombinant human granulocyte-macrophage colony-stimulating factor (Rhu GM-CSF) as adjuvant therapy for invasive fungal diseases. Open Forum Infect Dis (2022) 9(11). doi: 10.1093/ofid/ofac535
178. Triggle CR, Bansal D, Ding H, Islam MM, Farag EABA, Hadi HA, et al. A comprehensive review of viral characteristics, transmission, pathophysiology, immune response, and management of SARS-CoV-2 and COVID-19 as a basis for controlling the pandemic. Front Immunol (2021) 12:631139(338). doi: 10.3389/fimmu.2021.631139
179. Metlay JP, Waterer GW, Long AC, Anzueto A, Brozek J, Crothers K, et al. Diagnosis and treatment of adults with community-acquired pneumonia. an official clinical practice guideline of the American thoracic society and infectious diseases society of America. Am J Respir Crit Care Med (2019) 200(7):e45–67. doi: 10.1164/rccm.201908-1581st
180. Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Crit Care Med (2021) 49(11):e1063–e143. doi: 10.1097/ccm.0000000000005337
181. Brunck MEG, Nielsen LK. Concise review: Next-generation cell therapies to prevent infections in neutropenic patients. Stem Cells Trans Med (2014) 3(4):541–8. doi: 10.5966/sctm.2013-0145
182. Price, Boeckh, Harrison, McCullough, Ness, Strauss, et al. Efficacy of transfusion with granulocytes from G-CSF/Dexamethasone–treated donors in neutropenic patients with infection. Blood (2015) 126(18):2153–61. doi: 10.1182/blood-2015-05-645986
183. Gale RP, Schiffer CA, Lazarus HM. Granulocyte transfusions in haematopoietic cell transplants and leukaemia: The phoenix or beating a dead horse? Bone Marrow Transplant (2021) 56(9):2046–9. doi: 10.1038/s41409-021-01399-3
184. Domen J, Christensen JL, Gille D, Smith-Berdan S, Fong T, Brown JMY, et al. Cryopreserved ex vivo-expanded allogeneic myeloid progenitor cell product protects neutropenic mice from a lethal fungal infection. Cell Transplant (2016) 25(1):17–33. doi: 10.3727/096368915x687688
185. Trump LR, Nayak RC, Singh AK, Emberesh S, Wellendorf AM, Lutzko CM, et al. Neutrophils derived from genetically modified human induced pluripotent stem cells circulate and phagocytose bacteria in vivo. Stem Cells Trans Med (2019) 8(6):557–67. doi: 10.1002/sctm.18-0255
186. Wang S-Y, Lo Y-F, Shih H-P, Ho M-W, Yeh C-F, Peng J-J, et al. Cryptococcus gattii infection as the major clinical manifestation in patients with autoantibodies against granulocyte–macrophage colony-stimulating factor. J Clin Immunol (2022) 42(8):1730–41. doi: 10.1007/s10875-022-01341-2
187. Clinicaltrials.Gov. GM-CSF for reversal of immunoparalysis in pediatric sepsis-induced MODS study (GRACE) (2022) (Accessed November 30, 2022).
188. Clinicaltrials.Gov. GM-CSF for reversal of immunoparalysis in pediatric sepsis-induced mods (GRACE-2) (2022) (Accessed November 30, 2022).
189. Vega MA, Simón-Fuentes M, González de la Aleja A, Nieto C, Colmenares M, Herrero C, et al. Mafb and maf transcription factors as macrophage checkpoints for COVID-19 severity. Front Immunol (2020) 11:603507. doi: 10.3389/fimmu.2020.603507
190. Lee S, Rauch J, Kolch W. Targeting mapk signaling in cancer: Mechanisms of drug resistance and sensitivity. Int J Mol Sci (2020) 21(3):1102. doi: 10.3390/ijms21031102
191. Clinicaltrials.Gov. Sargramostim use in COVID-19 to recover patient health (SCOPE) (2022) (Accessed November 30, 2022).
192. Kmeid J, Vanichanan J, Shah DP, El Chaer F, Azzi J, Ariza-Heredia EJ, et al. Outcomes of influenza infections in hematopoietic cell transplant recipients: Application of an immunodeficiency scoring index. Biol Blood Marrow Tr (2016) 22(3):542–8. doi: 10.1016/j.bbmt.2015.11.015
193. Chemaly RF, Ghosh S, Bodey GP, Rohatgi N, Safdar A, Keating MJ, et al. Respiratory viral infections in adults with hematologic malignancies and human stem cell transplantation recipients: A retrospective study at a major cancer center. Medicine (2006) 85(5):278–87. doi: 10.1097/01.md.0000232560.22098.4e
194. Lina B, Georges A, Burtseva E, Nunes MC, Andrew MK, McNeil SA, et al. Complicated hospitalization due to influenza: Results from the global hospital influenza network for the 2017–2018 season. BMC Infect Dis (2020) 20(1). doi: 10.1186/s12879-020-05167-4
195. Gushiken LFS, Beserra FP, Bastos JK, Jackson CJ, Pellizzon CH. Cutaneous wound healing: An update from physiopathology to current therapies. Life (Basel) (2021) 11(7). doi: 10.3390/life11070665
196. Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in chronic wounds. Int J Mol Sci (2016) 17(12). doi: 10.3390/ijms17122085
197. Li L, Zhang J, Zhang Q, Zhang D, Xiang F, Jia J, et al. High glucose suppresses keratinocyte migration through the inhibition of P38 Mapk/Autophagy pathway. Front Physiol (2019) 10:24. doi: 10.3389/fphys.2019.00024
198. Wilgus TA, Roy S, McDaniel JC. Neutrophils and wound repair: Positive actions and negative reactions. Adv Wound Care (2013) 2(7):379–88. doi: 10.1089/wound.2012.0383
199. Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg (2004) 187(5A):11S–6S. doi: 10.1016/S0002-9610(03)00296-4
200. Breuhahn K, Mann A, Muller G, Wilhelmi A, Schirmacher P, Enk A, et al. Epidermal overexpression of granulocyte-macrophage colony-stimulating factor induces both keratinocyte proliferation and apoptosis. Cell Growth Differ (2000) 11(2):111–21.
201. Mann A, Breuhahn K, Schirmacher P, Blessing M. Keratinocyte-derived granulocyte-macrophage colony-stimulating factor accelerates wound healing: Stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol (2001) 117(6):1382–90. doi: 10.1046/j.0022-202x.2001.01600.x
202. Sawaya AP, Stone RC, Brooks SR, Pastar I, Jozic I, Hasneen K, et al. Deregulated immune cell recruitment orchestrated by Foxm1 impairs human diabetic wound healing. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-18276-0
203. Furuya MY, Asano T, Sumichika Y, Sato S, Kobayashi H, Watanabe H, et al. Tofacitinib inhibits granulocyte–macrophage colony-stimulating factor-induced Nlrp3 inflammasome activation in human neutrophils. Arthritis Res Ther (2018) 20(1). doi: 10.1186/s13075-018-1685-x
204. Kim HO, Kim H-S, Youn J-C, Shin E-C, Park S. Serum cytokine profiles in healthy young and elderly population assessed using multiplexed bead-based immunoassays. J Trans Med (2011) 9(1):113. doi: 10.1186/1479-5876-9-113
205. Stojadinovic O, Minkiewicz J, Sawaya A, Bourne JW, Torzilli P, De Rivero Vaccari JP, et al. Deep tissue injury in development of pressure ulcers: A decrease of inflammasome activation and changes in human skin morphology in response to aging and mechanical load. PloS One (2013) 8(8):e69223. doi: 10.1371/journal.pone.0069223
206. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood (1980) 56(6):947–58. doi: 10.1182/blood.V56.6.947.947
207. Larouche J, Sheoran S, Maruyama K, Martino MM. Immune regulation of skin wound healing: Mechanisms and novel therapeutic targets. Adv Wound Care (New Rochelle) (2018) 7(7):209–31. doi: 10.1089/wound.2017.0761
208. Armstrong DG, Gurtner GC. A histologically hostile environment made more hospitable? Nat Rev Endocrinol (2018) 14(9):511–2. doi: 10.1038/s41574-018-0073-6
209. Zhao J, Chen L, Shu B, Tang J, Zhang L, Xie J, et al. Granulocyte/Macrophage colony-stimulating factor influences angiogenesis by regulating the coordinated expression of vegf and the Ang/Tie system. PloS One (2014) 9(3):e92691. doi: 10.1371/journal.pone.0092691
210. Gabbiani G, Ryan GB, Majno G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia (1971) 27(5):549–50. doi: 10.1007/bf02147594
211. De Ugarte DA, Roberts RL, Lerdluedeeporn P, Stiehm ER, Atkinson JB. Treatment of chronic wounds by local delivery of granulocyte-macrophage colony-stimulating factor in patients with neutrophil dysfunction. Pediatr Surg Int (2002) 18(5-6):517–20. doi: 10.1007/s00383-002-0733-3
212. Hatab AZ, McDanel D, Ballas ZK. Perilesional GM-CSF therapy of a chronic leg ulcer in a patient with common variable immunodeficiency. J Allergy Clin Immunol (2005) 116(2):460–2. doi: 10.1016/j.jaci.2005.04.008
213. Siddiqui FH, Mokhashi MH, Boathman A. Recombinant granulocyte-macrophage colony-stimulating factor in the treatment of indolent ulcers with klippel-Trenaunay-Weber syndrome: A case report. J Pediatr Surg (2007) 42(3):558–60. doi: 10.1016/j.jpedsurg.2006.10.093
214. Tursen U, Api H, Kaya TI, Cinel L, Ikizoglu G. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony-stimulating factor therapy in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol (2006) 20(10):1341–3. doi: 10.1111/j.1468-3083.2006.01688.x
215. Vatankhah N, Jahangiri Y, Landry GJ, McLafferty RB, Alkayed NJ, Moneta GL, et al. Predictive value of neutrophil-to-Lymphocyte ratio in diabetic wound healing. J Vasc Surg (2017) 65(2):478–83. doi: 10.1016/j.jvs.2016.08.108
216. Ferroni L, Gardin C, Dalla Paola L, Campo G, Cimaglia P, Bellin G, et al. Characterization of dermal stem cells of diabetic patients. Cells (2019) 8(7). doi: 10.3390/cells8070729
217. Spies C, Luetz A, Lachmann G, Renius M, von Haefen C, Wernecke KD, et al. Influence of granulocyte-macrophage colony-stimulating factor or influenza vaccination on HLA-DR, infection and delirium days in immunosuppressed surgical patients: Double blind, randomised controlled trial. PloS One (2015) 10(12):e0144003. doi: 10.1371/journal.pone.0144003
218. Hernandez-Cardoso GG, Rojas-Landeros SC, Alfaro-Gomez M, Hernandez-Serrano AI, Salas-Gutierrez I, Lemus-Bedolla E, et al. Terahertz imaging for early screening of diabetic foot syndrome: A proof of concept. Sci Rep (2017) 7(1):42124. doi: 10.1038/srep42124
219. Salvo P, Dini V, Kirchhain A, Janowska A, Oranges T, Chiricozzi A, et al. Sensors and biosensors for c-reactive protein, temperature and pH, and their applications for monitoring wound healing: A review. Sensors (Basel) (2017) 17(12). doi: 10.3390/s17122952
220. Henshaw FR, Reid IB, Spencer AM, Turner DE. Point of care ultrasound imaging as a wound assessment tool in diabetic foot ulcers: A case series. J Wound Care (2020) 29(Sup8):S28–s34. doi: 10.12968/jowc.2020.29.Sup8.S28
221. Reddy M, Gill SS, Rochon PA. Preventing pressure ulcers: A systematic review. JAMA (2006) 296(8):974–84. doi: 10.1001/jama.296.8.974
222. Kruger EA, Pires M, Ngann Y, Sterling M, Rubayi S. Comprehensive management of pressure ulcers in spinal cord injury: Current concepts and future trends. J Spinal Cord Med (2013) 36(6):572–85. doi: 10.1179/2045772313y.0000000093
223. Thomaty S, Pezard L, Xerri C, Brezun JM. Acute granulocyte macrophage-colony stimulating factor treatment modulates neuroinflammatory processes and promotes tactile recovery after spinal cord injury. Neuroscience (2017) 349:144–64. doi: 10.1016/j.neuroscience.2017.02.035
224. Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: A unifying hypothesis. Plast Reconstructive Surg (2006) 117(7S Suppl):35S–41S. doi: 10.1097/01.prs.0000225431.63010.1b
225. Ludmir EB, Mainwaring W, Lin TA, Miller AB, Jethanandani A, Espinoza AF, et al. Factors associated with age disparities among cancer clinical trial participants. JAMA Oncol (2019) 5(12):1769–73. doi: 10.1001/jamaoncol.2019.2055
226. Sedrak MS, Freedman RA, Cohen HJ, Muss HB, Jatoi A, Klepin HD, et al. Older adult participation in cancer clinical trials: A systematic review of barriers and interventions. CA: A Cancer J Clin (2021) 71(1):78–92. doi: 10.3322/caac.21638
227. Brahmer JR, Abu-Sbeih H, Ascierto PA, Brufsky J, Cappelli LC, Cortazar FB, et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer (2021) 9(6). doi: 10.1136/jitc-2021-002435
228. Couey MA, Bell RB, Patel AA, Romba MC, Crittenden MR, Curti BD, et al. Delayed immune-related events (Dire) after discontinuation of immunotherapy: Diagnostic hazard of autoimmunity at a distance. J ImmunoTherapy Cancer (2019) 7(1):165. doi: 10.1186/s40425-019-0645-6
229. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/coc.0000000000000239
230. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding LAG-3 signaling. Int J Mol Sci (2021) 22(10):5282. doi: 10.3390/ijms22105282
231. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
232. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
233. Si Z, Hersey P, Coates AS. Clinical responses and lymphoid infiltrates in metastatic melanoma following treatment with intralesional GM-CSF. Melanoma Res (1996) 6(3):247–55. doi: 10.1097/00008390-199606000-00008
234. Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, et al. Molecular pathways of colon inflammation induced by cancer immunotherapy. Cell (2020) 182(3):655–71.e22. doi: 10.1016/j.cell.2020.06.001
235. Dougan M. Immune checkpoint inhibitor colitis: Resident memory unleashed. Gastroenterology (2021) 161(4):1106–8. doi: 10.1053/j.gastro.2021.07.007
236. Bello E, Dougan M. Elevated circulating memory T cells precede immunotherapy toxicities in melanoma. Trends Cancer (2022) 8(5):347–9. doi: 10.1016/j.trecan.2022.02.008
237. Dougan M, Luoma AM, Dougan SK, Wucherpfennig KW. Understanding and treating the inflammatory adverse events of cancer immunotherapy. Cell (2021) 184(6):1575–88. doi: 10.1016/j.cell.2021.02.011
238. Sasson SC, Slevin SM, Cheung VTF, Nassiri I, Olsson-Brown A, Fryer E, et al. Interferon-Gamma–producing Cd8+ tissue resident memory T cells are a targetable hallmark of immune checkpoint inhibitor–colitis. Gastroenterology (2021) 161(4):1229–44.e9. doi: 10.1053/j.gastro.2021.06.025
239. Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, et al. Pleural innate response activator B cells protect against pneumonia Via a GM-CSF-IgM axis. J Exp Med (2014) 211(6):1243–56. doi: 10.1084/jem.20131471
240. Kwek SS, Lewis J, Zhang L, Weinberg V, Greaney SK, Harzstark AL, et al. Preexisting levels of CD4 T cells expressing PD-1 are related to overall survival in prostate cancer patients treated with ipilimumab. Cancer Immunol Res (2015) 3(9):1008–16. doi: 10.1158/2326-6066.cir-14-0227
241. Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ. Sargramostim for active crohn’s disease. N Engl J Med (2005) 352(21):2193–201. doi: 10.1056/NEJMoa041109
242. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Checkmate 067: 6.5-year outcomes in patients (Pts) with advanced melanoma. J Clin Oncol (2021) 39(15_suppl):9506–. doi: 10.1200/JCO.2021.39.15_suppl.9506
243. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. New Engl J Med (2018) 379(8):722–30. doi: 10.1056/nejmoa1805453
244. Tawbi HA, Forsyth PA, Hodi FS, Algazi AP, Hamid O, Lao CD, et al. Long-term outcomes of patients with active melanoma brain metastases treated with combination nivolumab plus ipilimumab (Checkmate 204): Final results of an open-label, multicentre, phase 2 study. Lancet Oncol (2021) 22(12):1692–704. doi: 10.1016/S1470-2045(21)00545-3
245. Winkler MS, Rissiek A, Priefler M, Schwedhelm E, Robbe L, Bauer A, et al. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFα response: A diagnostic tool for immunosuppression? PloS One (2017) 12(8):e0182427. doi: 10.1371/journal.pone.0182427
246. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical significance of circulating CD33+CD11b+HLA-DR– myeloid cells in patients with stage iv melanoma treated with ipilimumab. Clin Cancer Res (2016) 22(23):5661–72. doi: 10.1158/1078-0432.ccr-15-3104
247. Lozano AX, Chaudhuri AA, Nene A, Bacchiocchi A, Earland N, Vesely MD, et al. T Cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat Med (2022) 28(2):353–62. doi: 10.1038/s41591-021-01623-z
248. Mortha A, Remark R, Del Valle DM, Chuang L-S, Chai Z, Alves I, et al. Neutralizing anti-GM-CSF autoantibodies recognize posttranslational glycosylations on GM-CSF years prior to diagnosis and predict complicated crohn’s disease. Gastroenterology (2022) 163(3):659–70. doi: 10.1053/j.gastro.2022.05.029
249. Clinicaltrials.Gov. A phase II/III trial of nivolumab, ipilimumab, and GM-CSF in patients with advanced melanoma (2022) (Accessed November 30, 2022).
250. Clinicaltrials.Gov. Pembrolizumab and GM-CSF in biliary cancer (2022) (Accessed Novmeber 30, 2022).
251. Clinicaltrials.Gov. A study of sargramostim plus pembrolizumab with or without pemetrexed in patients with advance non-small cell lung cancer after completion of chemoimmunotherapy (2022) (Accessed November 30, 2022).
252. Thomson RM, Waterer G, Loebinger MR, Ganslandt C. Use of inhaled GM-CSF in treatment-refractory ntm infection. an open-label, exploratory clinical trial. Eur Respir J (2021) 58(suppl 65):OA1603. doi: 10.1183/13993003.congress-2021.OA1603
253. Naessens, Schepens, Smet, Pollard, Van Hoecke, De Beuckelaer, et al. GM-CSF treatment prevents respiratory syncytial virus–induced pulmonary exacerbation responses in postallergic mice by stimulating alveolar macrophage maturation. J Allergy Clin Immunol (2016) 137(3):700–9.e9. doi: 10.1016/j.jaci.2015.09.031
Keywords: mononuclear phagocyte, sargramostim, immunomodulation, granulocyte-macrophage colony-stimulating factor, autoimmune pulmonary alveolar proteinosis (aPAP), COVID-19, wound healing, immune checkpoint inhibitors (ICI)
Citation: Lazarus HM, Pitts K, Wang T, Lee E, Buchbinder E, Dougan M, Armstrong DG, Paine R III, Ragsdale CE, Boyd T, Rock EP and Gale RP (2023) Recombinant GM-CSF for diseases of GM-CSF insufficiency: Correcting dysfunctional mononuclear phagocyte disorders. Front. Immunol. 13:1069444. doi: 10.3389/fimmu.2022.1069444
Received: 13 October 2022; Accepted: 05 December 2022;
Published: 05 January 2023.
Edited by:
Peter Brossart, University of Bonn, GermanyReviewed by:
Sam Basta, Queen’s University, CanadaDimitrios P. Kontoyiannis, University of Texas MD Anderson Cancer Center, United States
Copyright © 2023 Lazarus, Pitts, Wang, Lee, Buchbinder, Dougan, Armstrong, Paine, Ragsdale, Boyd, Rock and Gale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hillard M. Lazarus, hillard.lazarus@case.edu