 Weiwei Zheng1,2†
Weiwei Zheng1,2† Cheng Qian2†Yu Tang2Chunmei Yang1,2Yueke Zhou2
Cheng Qian2†Yu Tang2Chunmei Yang1,2Yueke Zhou2 Peiliang Shen1Wenxing Chen2,3Suyun Yu1,2
Peiliang Shen1Wenxing Chen2,3Suyun Yu1,2 Zhonghong Wei2
Zhonghong Wei2 Aiyun Wang2,3*
Aiyun Wang2,3* Yin Lu2,3*
Yin Lu2,3* Yang Zhao1,2*
Yang Zhao1,2*- 1Department of Biochemistry and Molecular Biology, School of Medicine & Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing, China
- 2Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, China
- 3Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine (TCM) Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing, China
Immunotherapy has been recognized as an effective and important therapeutic modality for multiple types of cancer. Nevertheless, it has been increasing recognized that clinical benefits of immunotherapy are less than expected as evidenced by the fact that only a small population of cancer patients respond favorably to immunotherapy. The structurally and functionally abnormal tumor vasculature is a hallmark of most solid tumors and contributes to an immunosuppressive microenvironment, which poses a major challenge to immunotherapy. In turn, multiple immune cell subsets have profound consequences on promoting neovascularization. Vascular normalization, a promising anti-angiogenic strategy, can enhance vascular perfusion and promote the infiltration of immune effector cells into tumors via correcting aberrant tumor blood vessels, resulting in the potentiation of immunotherapy. More interestingly, immunotherapies are prone to boost the efficacy of various anti-angiogenic therapies and/or promote the morphological and functional alterations in tumor vasculature. Therefore, immune reprograming and vascular normalization appear to be reciprocally regulated. In this review, we mainly summarize how tumor vasculature propels an immunosuppressive phenotype and how innate and adaptive immune cells modulate angiogenesis during tumor progression. We further highlight recent advances of anti-angiogenic immunotherapies in preclinical and clinical settings to solidify the concept that targeting both tumor blood vessels and immune suppressive cells provides an efficacious approach for the treatment of cancer.
1 Introduction
The last decade has undoubtedly visualized the striking rise of immunotherapy, in particular immune checkpoint blockade (ICB) therapy, for the treatment of cancer. In contrast to conventional cytotoxic agents, which directly target cancer cells, a major goal of cancer immunotherapy is to alleviate tumor-associated suppression of anticancer immune responses. A significant portion of cancer immunotherapy research has focused on heightening the functions of effector T cells, which play a direct role in recognizing tumor-associated antigens and in mediating tumoricidal responses (1, 2). The immune checkpoint mediators, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed death 1 (PD-1) as well as programmed death-ligand 1 (PD-L1) have been validated to be effectively targeted and their antibodies have been approved by the Food and Drug Administration (FDA) for treating various types of cancer. To date, the development of ICB-based immunotherapy has remarkably transformed the current therapeutic paradigm in oncology (3). However, ICB therapy commonly benefits <15% of cancer patients and leads to immune-associated adverse effects (4, 5). An alternative immunotherapy, utilizing engineered chimeric antigen receptor (CAR) T-cells to specifically target tumor-associated antigens, has opened the door to a promising novel treatment of numerous “liquid” cancers and achieved similar success in solid tumors (6). Among numerous crucial factors of immunity to cancer, the tumor microenvironment (TME) serves as a predominant challenge that overtly diminishes the effectiveness of ICB. The interactions between tumor endothelial cells (ECs) and immunosuppressive immune cells tend to form a vicious cycle that extensively distorts anti-tumor immune response and aggravates the development of tumors in the TME (7, 8). Notably, tumor blood vessels are prone to facilitate the infiltration of immunosuppressive immune cells into tumors, which in turn fuels tumor angiogenesis (9). This deteriorative crosstalk between immune suppression and angiogenesis not only produces the endothelium that brakes the penetration of T cells into tumors, but also curtails the functions of T cells and even results in elevated apoptosis of T cells. To this end, manipulation of tumor blood vessels is inclined to act as a reliable strategy to boost anti-tumor immune response and counteract the resistance to ICB (10).
In comparison to ICB, anti-angiogenesis therapy gained substantial attention at an earlier stage (11). It has been well established that neovascularization or angiogenesis plays a pivotal role in maintaining homeostasis due to the fact that blood vessels are able to transport nutrients to body’s tissues and organs and remove metabolic wastes (12). Of note, excessive growth of blood vessels tends to aggravate the progression of a variety of diseases especially tumors and intraocular vascular diseases (13). In light of therapy, conventional anti-angiogenic strategy aims to destroy tumor blood vessels (14). Nevertheless, the therapeutic outcomes for cancer patients received anti-angiogenesis therapy alone in clinic are less than what is expected. Excessive suppression of blood vessel formation in tumors confers diminished vascular perfusion, which sets up difficult hurdles for immune cell infiltration and drug delivery (9, 15). Vessel normalization theory, firstly proposed by Rakesh Jain, offers a novel and promising perspective in terms of anti-angiogenesis and shows potential synergistic effect in combination with other therapies (16). More intriguingly, vascular normalization and immune reprogramming can generate a positive feedback, which implies that reinforcement in one side has high propensity to strengthen the other’s effects (9).
In this review, we mainly discuss the latest advances on how abnormal tumor vasculature modulates the infiltration of various types of immune suppressive cells and incites an immunosuppressive phenotype, as well as outline how immunosuppressive TME influences tumor angiogenesis. We further gain insight into the latest knowledge of ICB in combination with anti-angiogenesis therapy and highlight the advances of relevant clinical trials.
2 Manipulation of angiogenesis has pivotal impacts on immunosuppressive tumor microenvironment
2.1 Description of blood vessel formation in tumors
It has been widely held that the hypoxic microenvironment in solid tumors results in rapid but abnormal blood vessel establishment. In fact, tumor angiogenesis is regarded as an extremely complicated process (17). This is owing to (a) the perturbed equilibrium between pro-angiogenic and anti-angiogenic factors, leading to the so-called “angiogenic switch” with excessive pro-angiogenic signaling (Table 1). For instance, vascular endothelial growth factor (VEGF) as the major factor to initiate angiogenesis is substantially up-regulated upon the stimulation of hypoxia. It plays a vital role in strengthening the formation of blood vessels mainly through leading to the activation of VEGF receptor-2 (VEGFR-2) that is predominantly expressed by ECs. The activation of VEGFR-2 is inclined to trigger the transduction of multiple critical signaling pathways, resulting in specific endothelial responses such as cell survival, proliferation, migration as well as vascular permeability (13). Similarly, fibroblast growth factor (FGF) and its receptor (FGFR) exert significant effects on propelling the proliferation, migration, and survival of ECs, thereby contributing to elevated angiogenesis (47). (b) angiogenic signaling cascades are usually located at the downstream of oncogene activation. Accordingly, tumor blood vessels are rather blind ended as well as leaky with disrupted endothelial junctions and disorganized endothelial lining, and display aberrant basement membrane and poor pericyte coverage (48). These characteristics seem eventually to be hallmarks of immature and dysfunctional tumor vasculature, inciting the consequence that tumor parenchyma maintains constantly hypoxic, which renders a negative feedback loop whereby pro-angiogenic signals never cease (49). Dysfunctional tumor blood vessels featured with reduced vascular perfusion end up producing swollen and thick vessel wall, where clotting events and hemostasis frequently occur. Thus, tumors with intensified vascular density are prone to be highly hypoxic and vice versa, depending on their vascular functionality and metabolic demand (50).
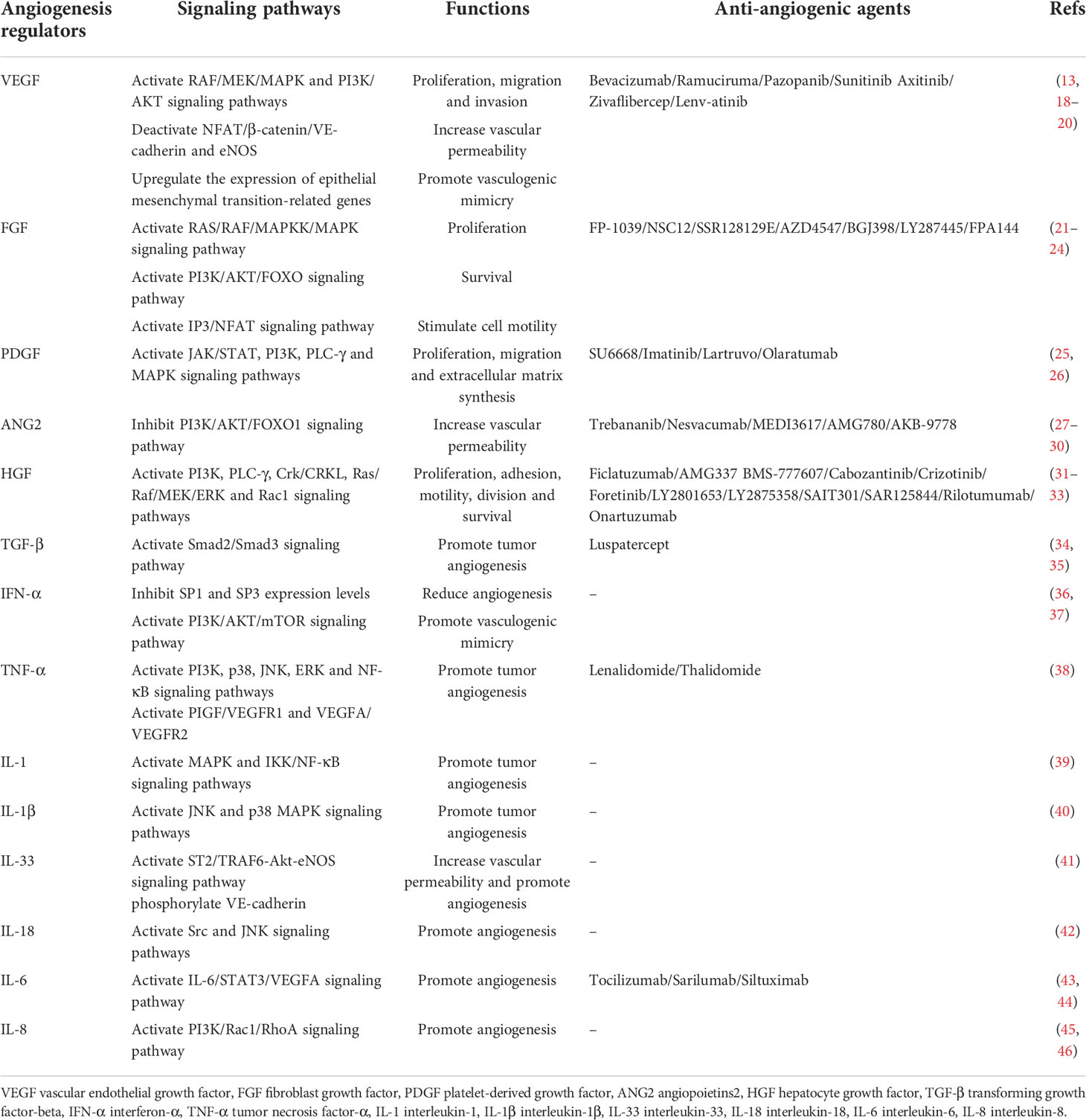
Table 2 The main angiogenic modulators involved in angiogenesis.
It has been well accepted that the formation of tumor blood vessels is able to occur in an array of manners, including sprouting angiogenesis, vasculogenesis, vessel co-option, vasculogenic mimicry, intussusceptive angiogenesis and trans-differentiation of cancer cells (Figure 1). In fact, angiogenesis and vasculogenesis are the predominant modes of tumor angiogenesis. Angiogenesis, the most widely investigated pattern of new vessel formation in tumors, refers to the initiation of tumor blood vessels from existing ECs and the production of new neoplastic capillaries in the format of sprouting (51). Indeed, sprouting angiogenesis is triggered by a panel of pro-angiogenic growth factors (eg. VEGF) produced by hypoxic and nutrient-deprived microenvironment, which allow quiescent ECs to exhibit activated phenotype. Further, activated ECs tend to release matrix metalloproteinases (MMPs) to degrade the basement membrane and turn to invasive profile (52), which enables tip cells to protrude and migrate towards the core of the angiogenic stimulus. Tip cells with minimal proliferative capability extend filopodia and lamellipodia to lead the nascent sprout towards oxygen-deprived regions. In addition, a wealth of molecules associated with extracellular matrix (ECM) degradation and basement membrane deposition have been observed to be highly expressed in tip cells (53). Tip cells are followed by another type of ECs named stalk cells, which proliferate to propel the elongation of sprouts and strengthen the formation of lumens. Specification in migratory tip versus proliferating stalk cell is overtly dynamic, resulting in that ECs persistently compete for the lead position. Upon the condition that two tip cells from adjacent sprouts meet, they are inclined to anastomose to generate a perfused new vessel (54). Vasculogenesis is achieved via the recruitment of endothelial progenitor cells (EPCs) that are capable of differentiating into ECs and penetrating into tumors to be directly involved in the generation of tumor blood vessels (55). Vasculogenic mimicry is a new tumor microcirculation mode that is different from the classical patterns of tumor angiogenesis as it relies on tumor cells rather than ECs (56). Intussusceptive angiogenesis is frequently observed in the lumen of existing blood vessels and is governed by the interstitial columnar structure, leading to the incision of the original vascular lumen and the production of new blood vessels. More specifically, it splits pre-existing vessels to induce new blood vessel formation (57). Vessel co-option has preference to appear in various malignancies, which means that tumor cells migrate along the existing or newly triggered blood vessels (hijacking the vasculature) to support tumor growth and metastasis (58). Vessel co-option can be visualized in a plethora of types of tumors in humans at multiple anatomic sites and influences the prognosis of cancer patients, but it still remains a frequently overlooked phenomenon (59). Trans-differentiation of cancer cells is recognized as the trans-differentiation of cancer stem cells (CSCs) to ECs and vascular smooth muscle-like cells, yielding the occurrence of neovascularization (60). Furthermore, intussusceptive angiogenesis is recognized as a faster phenomenon to orchestrate a plexus compared to sprouting angiogenesis. Intussusceptive angiogenesis has been deemed to be less energetically demanding, which allows it to minimally rely on the migration and proliferation of ECs (61).
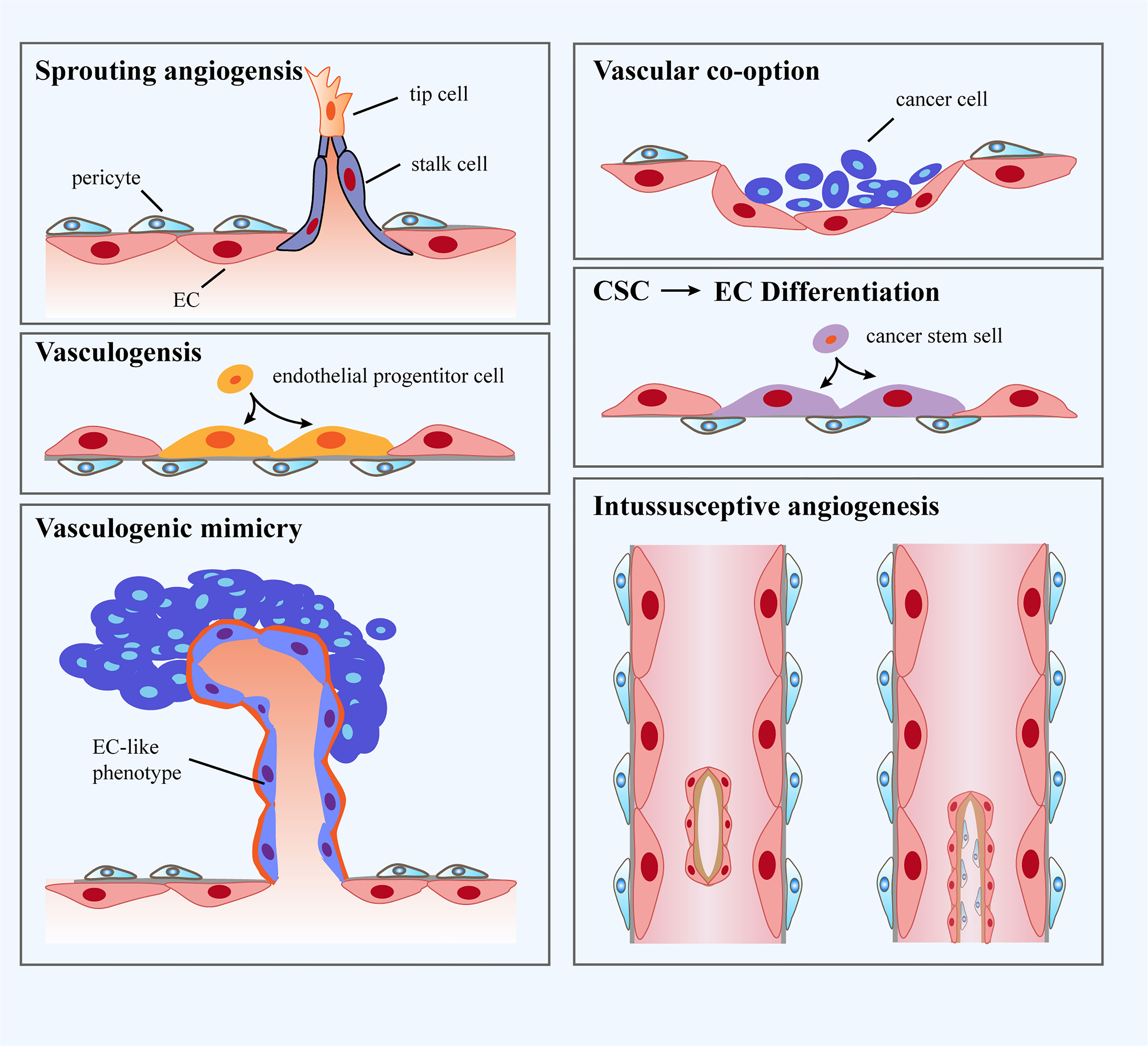
Figure 1 Classification of blood vessel formation. i: Sprouting angiogenesis refers to a process involving production and outgrowth of sprouts (tip cells), which ultimately integrate with an existing vessel or newly formed sprout. ii: Vasculogenesis is prenatal neo-vascularization occurred from endothelial progenitor cells (EPCs). The EPCs proliferate and generate lumens, ultimately gathering into new blood vessels. iii: Vasculogenic mimicry is regarded as a matrix-embedded blood vessel network produced by tumor cells rather than endothelial cells (ECs). iv: Vascular co-option represents that tumors grow by hijacking preexisting vessels in the peritumoral tissues. v: Trans-differentiation of cancer stem cells (CSCs) reflects neo-vascularization in tumors through differentiating CSCs to ECs. vi: Intussusceptive angiogenesis refers to the formation of new vasculature where a pre-existing vessel splits into two.
2.2 Tumor angiogenesis creates immunosuppressive microenvironment
In the majority of solid tumors, uncontrolled angiogenesis generates immunosuppressive microenvironment via influencing an array of immune-associated steps (Figure 2) (62–64). On one hand, hyperactive and aberrant angiogenesis impedes the number and tumor-killing activity of tumor-infiltrating lymphocytes (TILs). Firstly, leaky and dysfunctional blood vessels with poor pericyte coverage and abnormal basement membrane support underlines high interstitial fluid pressure (IFP), which indicates that there is elevated pressure difference to overcome for T cell trafficking into tumors. This undoubtedly leads to that simply fewer T cells are able to transmigrate across the physical barrier and infiltrate into tumor bed. Secondly, a bunch of critical adhesion molecules including vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) expressed on ECs are significantly down-regulated in newly formed tumor blood vessels, which hampers the adhesion and transmigration of T cells (65). Thirdly, neovascularization fails to compensate for enhanced oxygen consumption, and concurrent hypoxia potently declines the functions of TILs as a result of that hypoxia increases the expression of multiple inhibitory mediators for anti-tumor immune response, such as PD-L1, indoleamine 2, 3-dioxygenase (IDO), interleukin-6 (IL-6), and interleukin-10 (IL-10) (11). Hypoxia is also inclined to trigger the expression levels of immune checkpoint molecules including PD-L1 on tumor cells as well as PD-L1, T cell immunoglobulin and mucin-domain containing-3 (TIM-3) and CTLA-4 on tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) (66, 67). Moreover, upon the stimulation of VEGF, hypoxia tends to indirectly upregulate the expression of PD-1 on CD8+ T cells, thus further inhibiting immune effector cell activation and function Moreover, hypoxia together with tumor cell necrosis gives rise to the elevated the extracellular concentrations of the immune-suppressive metabolites such as adenosine and lactate. The enhanced level of lactate further results in metabolic lactic acidosis (a low pH in the blood due to accumulation of lactic acid) and leads to declined CD8+ T cell function by interfering with T cell receptor (TCR)-induced interferon-γ (IFN-γ) production (68, 69). Lastly, the high expression of Fas ligand (FasL) on tumor ECs selectively eliminates effector CD8+ T cells instead of Tregs, which is attributed to the augmented level of cellular FLICE-inhibitory protein (c-FLIP) on Tregs (70).
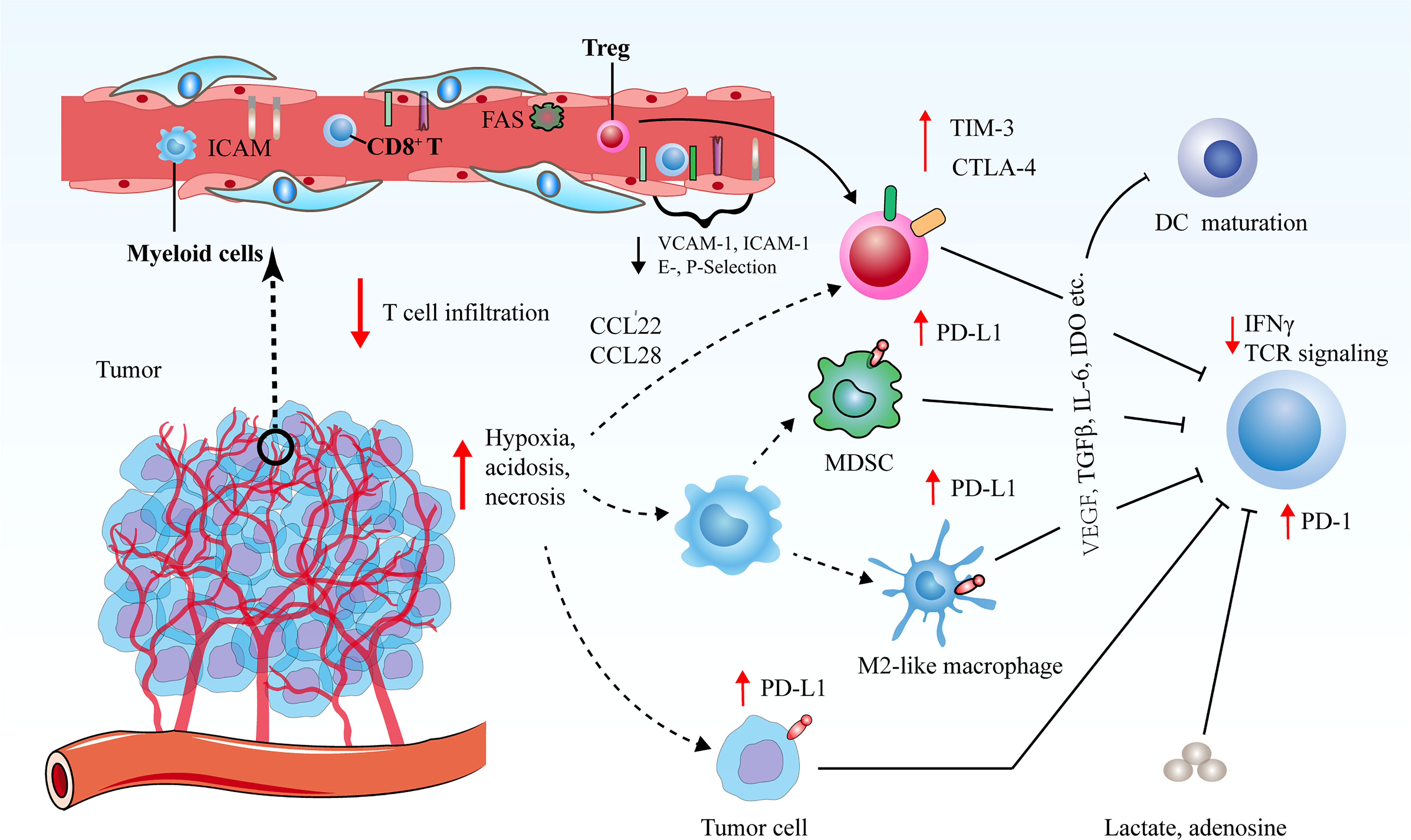
Figure 2 Tumor angiogenesis confers immunosuppressive tumor microenvironment via various routes. Decreased vascular perfusion and elevated vascular permeability augment tumor hypoxia, acidosis and necrosis, which initiate various immune suppressive processes to mitigate effector T cell functions. Hypoxia not only induces the secretion of cytokines and chemoattractants to promote the recruitment of immunosuppressive cells, but also boost the expression levels of CTLA-4 or TIM-3 on Tregs, and PD-L1 on MDSCs, TAMs and tumor cells to restrict the function of immune-supportive cells. The ECs of tumor vessels with lower levels of cell adhesion molecules are capable of triggering endothelial anergy, thereby inhibiting the infiltration of effector T cells into tumors.
On the other hand, excessive angiogenesis tends to promote the abundance of pro-tumor immune subsets that are crucial for immunosuppressive microenvironment. Upon expansion of abnormal tumor blood vessels, hypoxic microenvironment in tumors leads to the upregulation of chemokine (C-C motif) ligand-22 (CCL-22) and chemokine (C-C motif) ligand-28 (CCL-28), which are both involved in the recruitment of Tregs into tumors (71, 72). Additionally, hypoxia promotes the accumulation of MDSCs and facilitates the differentiation and polarization of TAMs into an immunosuppressive M2-like phenotype (73). In addition, circulating VEGF hinders the maturation and function of dendritic cells (DCs) to enable tumor cells to escape immune surveillance (74). These processes collaborate to hamper the maturation of effector immune cells and propel the anergy and exhaustion of T cells. Therefore, dysfunctional tumor vessels play an indispensable role in yielding the immunosuppressive nature of the TME and limiting the effects of cancer immunotherapies.
3 Different types of immune cells exert versatile effects on modulating tumor angiogenesis
3.1 Innate immune cells regulate tumor angiogenesis
It has been widely held that tumors, partially due to their hypoxic and acidic property, are able to recruit a large number of distinct innate immune cells that constitute approximately 30% of the total cell populations in tumors (75). Notably, tumor-associated neutrophils (TANs), TAMs, MDSCs, DCs and mast cells appear to be the most frequently observed cell populations and are correlated to intra-tumoral vascular density (8, 76–82). Indeed, in addition to cancer cells and cancer-associated fibroblasts, myeloid cells have been perceived as a crucial source of growth factors and chemokines to influence tumor vessels and determine the endothelial phenotype and function (83, 84), which has been substantiated in various mouse tumor models, such as breast, skin, brain, and cervical tumors (76). Additionally, certain classes of innate immune cells are capable of interfering with and polarizing other specific groups of immune cells to obtain either pro-angiogenic or anti-angiogenic properties, further influencing the amount and grade of angiogenesis in tumors (85). An increasing number of striking studies using murine tumor models have demonstrated the functional significance of innate immune cells in modulating tumor angiogenesis and subsequent tumor growth and metastasis, some of which are highlighted in detail as follows.
3.1.1 TANs
It has been well known that neutrophils are prone to give rise to the production of either pro-angiogenic or anti-angiogenic factors (86). Despite that neutrophils are featured with an eventually differentiated phenotype with short half-life property, these cells are still able to harbor a certain type of plasticity that enables them to differentiate into two different subpopulations: type 1 neutrophils (N1) with anti-microbial function, and TANs or N2 neutrophils with conspicuous pro-tumor and pro-angiogenic characteristics in the presence of TGF-β (87). It has been widely recognized that N1 neutrophils exert central effects on the front-line defense against harmful invading pathogens. They are able to either phagocytose micro-organisms, secrete soluble anti-microbial molecules from the granules or propel the formation of neutrophil extracellular traps (NETs), which are classic web-like fibers that consist of chromatin and serine proteases, playing a crucial role in trapping and destructing the extracellular microbes (88, 89). It has been reported that TANs/N2 neutrophils are able to be converted into N1 neutrophils in both mouse lung cancers and human melanomas following the stimulation of interferon-β (IFN-β) (90). In the context of malignant tumors, TANs tend to predominantly produce a list of pro-angiogenic factors including VEGF, FGF and MMPs as well as generate proteases (86, 91–93). Interestingly, neutrophils are known to contain VEGF-enriched granules that are released following the stimulation of TNF and create a reliable pathway of VEGF accessibility to improve the expansion of blood vessels (94). In the tumor microenvironment, TANs were also demonstrated to generate matrix metalloproteinase-9 (MMP-9) to orchestrate sequestered VEGF from the ECM in dysplastic pancreatic islet lesions of Rip1Tag2 transgenic mouse model. The produced VEGF initiated the onset of angiogenesis in these premalignant lesions and allowed the malignant transformation and progression (95–97). In line with this observation, the depletion of TANs with antibodies against Gr1 dramatically repressed the angiogenic switch in the pancreatic islet lesions (96). Moreover, neutrophils stimulated with colony-stimulating factor (G-CSF) promoted the expression levels of the angiogenic factor Bv8 (prokineticin-2) in various tumor models that boosted the proliferation, migration and survival of ECs, thereby generating a positive feedback loop for strengthening the recruitment and activation TANs (98).
3.1.2 TAMs
Macrophages are deemed to work as the professional phagocytes of innate immune cells with distinct specific roles, relying on the type of danger signals and endogenous molecules to which they are exposed. Macrophages are capable of triggering inflammatory responses and collaborating with other immune cells to activate adaptive T lymphocyte responses through antigen processing and presentation, thereby acting as the sentinels in all tissues of the body against invading pathogens. In tumors, it has been revealed that TAMs preferentially stay pro-tumorigenesis M2/T helper (Th) 2-like status, however, polarization of macrophages to a pro-inflammatory and anti-tumorigenesis (M1/Th1-like) phenotype tends to lead to impaired angiogenesis in multiple mouse and human tumor models (99–101).
Indeed, it was uncovered that M1-phenotypic macrophages mitigated sprouting angiogenesis and yielded the maturation of blood vessels through inducing the secretion of numerous anti-angiogenic cytokines including interleukin-12 (IL-12) and TNF-α (102, 103). Interestingly, IL-12 produced from M1 form of TAMs was capable of skewing TAMs to the M1 phenotype, therefore generating a positive feedback loop aiming at driving the anti-angiogenic M1 pattern. To this end, IL-12 administration not only curtailed microvascular density but also polarized TAMs from M2 towards M1 phenotype in tumors (102, 104). Moreover, M2-phenotypic TAMs are inclined to strengthen tumor angiogenesis via potentiating the secretion of multiple pro-angiogenic growth factors (e.g. VEGF, FGF, EGF, and PDGF-b), angiogenic chemokine (C-X-C motif) ligands (e.g. CXCL-8 and CXCL-12), as well as angiogenesis-associated elements (e.g. TGF-β, TNF-α, and thymidine phosphorylase) (105). These imperative factors not only have preference to reinforce EC proliferation and migration but also polarize TAMs away from tumor-suppressing M1 manner to tumor-promoting M2 manner.
Qiu and colleagues demonstrated that M2-like TAMs were positively correlated with the microvascular density in human pancreatic cancer. More specifically, the exosomes derived from M2-like TAMs were able to deliver miR-155-5p and miR-221-5p into ECs, which boosted the angiogenesis in pancreatic ductal adenocarcinoma (PDAC) through targeting E2F2 (106). Chen and co-workers highlighted that M2 TAMs-derived exosomal miR-942 tended to escalate angiogenesis via binding to FOXO1 to alleviate the suppression of β-catenin as well as harness the migration and invasion of lung adenocarcinoma (LUAD) cells (107). Pollard et al. also unveiled that M2-phenotypic TAMs had high propensity to propel the progression of tumor angiogenesis, they found that the infiltration of TAMs into the parenchyma of primary tumors was potently enhanced shortly prior to the development of a dense vascular network in the tumor area. As such, blockade of the infiltration of macrophages into tumors repressed the angiogenic switch (97). It has been well accepted that M2-phenotypic TAMs are the more predominant subset compared with M1-phenotypic TAMs in most solid tumors. In this regard, inactivation of TAMs via counteracting the CSF1/CSF1R signaling pathway, extensive antagonization of TAMs through clodronate liposomes, or genetic depletion of VEGF in TAMs was able to impede the development of angiogenic switch. In contrast, restoration of the macrophages through genetic approaches was capable of rescuing the angiogenic phenotype in tumors (97, 108–111).
3.1.3 MDSCs
As defined by the morphology and function, immature myeloid cells that can be classified into two main subtypes: granular MDSC (G-MDSC) and monocytic MDSC (M-MDSC). The studies focusing on the role of MDSCs in tumors showed that the main capabilities of these cells are to inhibit immunity by perturbing both innate and adaptive immune responses as well as to strengthen angiogenesis (76, 112). MDSCs have been gaining growing attention in curbing immune response in tumors owing to their ability to suppress the infiltration and activity of T cells (113, 114). In light of angiogenesis, MDSC-released VEGF was sufficient to prohibit the expression levels of various key adhesion molecules including ICAM-1 and VCAM-1 in tumor-associated ECs, thereby preventing the adhesion and extravasation of T cells (115, 116). MDSCs promote angiogenesis through heightening the levels of IL-10 but diminishing the leves of IL-12 in the TME. Furthermore, MDSCs are capable of enhancing angiogenesis by giving rise to the productions of Bv8 and MMP-9. MDSC-derived Bv8 is able to directly fuel the formation of neovessels through endocrine gland-derived VEGF1 (EG-VEGF1) and VEGF2 (EG-VEGF2) and can also further accumulate MDSCs within the tumors. Of note, a series of important inflammatory factors including granulocyte G-CSF, IL-1β and IL-6 are prone to confer the recruitment, activation and expansion of MDSCs in tumors through inducing the activation of STAT3 signaling pathway, thus further driving the pro-angiogenic property of MDSCs while hindering their differentiation potential into neutrophils or macrophages. Hence, it seems to imply that it is not surprising that the pro-angiogenic expression characteristics and functions of MDSCs remarkably overlap with those of mature macrophages (92, 117, 118).
3.1.4 DCs
DCs as another crucial innate immune subpopulation in the TME have also been revealed to play a pivotal role in modulating tumor angiogenesis on the basis of their maturation status (119). It has been increasingly recognized that DCs are prone to prime naiüve T cells and further lead to the activation of antigen-specific immunity. In fact, human DCs contain two types of predominant subsets, which are myeloid dendritic cells (MDCs) and plasmacytoid dendritic cells (PDCs). MDCs expressing tumor antigens have been employed in clinical trials to confer prominent therapeutic responses against various types of tumors. Zou et al. demonstrated that MDCs were observed to be absent from malignant ascites. Further, they showed that MDCs exerted striking effect on hampering tumor angiogenesis via promoting the secretion of IL-12 (120). On the contrary, a large number of PDCs and high level of stromal-derived factor including CXCL-12/SDF-1 were found to be present in their malignant ascites, further enabling PDCs to penetrating into the TME. It was shown that tumor-associated PDCs triggered tumor angiogenesis via provoking the secretion of TNF-α and IL-8 (120). Notably, the most predominant subpopulation of DCs in the TME is deemed to be immature DCs (iDCs) as cancer cells are able to preferentially recruit iDCs from peripheral blood vessels by producing a wealth of cytokines (e.g. VEGF, β-defensin, CXCL-12, HGF, and CXCL-8) (121). Moreover, iDCs are endowed with pro-angiogenic property and exert indispensable effect on bolstering tumor angiogenesis via secreting multiple angiogenic factors including VEGF-A and FGF (122).
3.1.5 Mast cells
MCs, a population of multifunctional immune cells that were originally identified in human tumors by Paul Ehrlich in the 1870s, are derived from bone marrow and are also thought to be involved in tumor angiogenesis (123). Indeed, there are considerable mast cells that are observed in tumors and peritumor tissues of cancer patients, and their functions in cancer insurgence and progression are dependent on tumor types. Intriguingly, mast cells releases a set of pro-angiogenic factors including FGF-2, VEGF-A, TNFs, CXCL-8 (123) and various proteases including MMPs (predominantly MMP-9), as well as chymase and tryptase that enable pro-MMPs to become their active forms (124, 125). Multiple lines of evidence highlighted that mast cell proteases serve as the pivotal regulators in rendering tumor angiogenesis (125). Furthermore, Li et al. showed that recruitment of mast cells into tumor parenchyma contributed to elevated angiogenesis in both in vitro and in vivo models. Mechanistic studies deciphered that infiltrated mast cells were validated to orchestrating tumor angiogenesis by activating PI3K/AKT/GSK3β/AM signaling cascade in the renal cell carcinoma (126).
3.2 Adaptive immune cells influence tumor angiogenesis
In fact, the function of lymphocytes in regulating tumor angiogenesis are still ambiguous. Nevertheless, a growing number of studies have documented that lymphocytes are able to govern the growth and maturation of tumor blood vessels in a direct or indirect manner.
3.2.1 B lymphocytes
Although the role of B lymphocytes in curbing tumor angiogenesis and the underlying mechanisms are still obscure, an increasing number of studies have gradually unveiled that the functions of B lymphocytes in tumor angiogenesis vary under distinct situations. For example, Yang and colleagues elucidated that tumor-associated B lymphocytes with activated STAT3 resulted in the accelerated tumor progression by virtue of augmenting tumor angiogenesis (127). Additionally, the amount of B cells in tumor tissues was positively correlated with the expression levels of a list of pro-angiogenic molecules that are located at the downstream of STAT3, as well as the extent of tumor angiogenesis. Indeed, it has also been documented that B lymphocytes are capable of potently strengthening tumor angiogenesis via promoting the secretion of an array of critical pro-angiogenic factors including VEGF, FGF-2, and MMP-9 (127), or indirectly via skewing the polarization of TAMs towards an pro-angiogenic and immunosuppressive M2-phenotypic TAMs in an IgG-dependent manner (128). Furthermore, Christofori and co-workers observed that the levels of a type of bone marrow-derived cell population, CD45dimVEGFR1-CD31low, were significantly elevated in the peripheral blood from multiple mouse models of tumor angiogenesis compared with that from healthy mice. More interestingly, gene expression profile analysis identified CD45dimVEGFR1-CD31low cells as an immature B lymphocyte population. Long-term treatment with potent compounds (BIBF-1120, nintedanib) that inhibiting various pro-angiogenic factors (VEGF, FGF and PDGF) resulted in a striking impairment in the number of immature B lymphocytes in the blood from tumor-bearing mice as well as prominent reductions in tumor volume and microvascular density. Hence, CD45dimVEGFR1-CD31low cell population may emerge as surrogate marker for tumor angiogenesis and be able to reflect the therapeutic response to anti-angiogenic strategies (129).
3.2.2 T lymphocytes
In comparison to B lymphocytes, T lymphocytes are prone to positively or negatively control tumor angiogenesis on the basis of the types of T cells. It has been shown that CD8+ T cells and CD4+ Th1 cells yield IFN-γ that limits the proliferation of ECs and triggers the generation of the angiostatic chemokines including CXCL-9, CXCL-10 and CXCL-11 in TAMs (91, 130). Another preponderant role of IFN-γ in influencing tumor angiogenesis is the polarization of TAMs from M2- to M1-phenotypic TAMs. It has been shown that hyperactive IFN-γ/STAT1 signaling cascade propels the reprograming of TAMs to M1-like profile, contributing to vascular remodeling and subsequent tumor destruction (83, 131). In contrast, Th2 cells as one type of CD4+ T cells play an important role in provoking profound tumor angiogenesis. Th2 cells expressing IL-4, IL-5, and IL-13 tend to promote the recruitment of M2-like TAMs via activating STAT6 and eventually instruct tumor angiogenesis (130). Th17 cells, another subtype of CD4+ T cells, have been demonstrated to be related to intensify angiogenesis in various types of human cancer. The expression of IL-17 in Th17 cells are associated with the infiltration of ECs and the formation of abnormal tumor blood vessels (132, 133). Tregs exert significant effects on inhibiting the function of IFN-γ-expressing CD4+ Th1 cells and producing the secretion of VEGF through hypoxia-mediated CCL-28, which both strikingly result in a pro-angiogenic tumor microenvironment (72) (Figure 3).
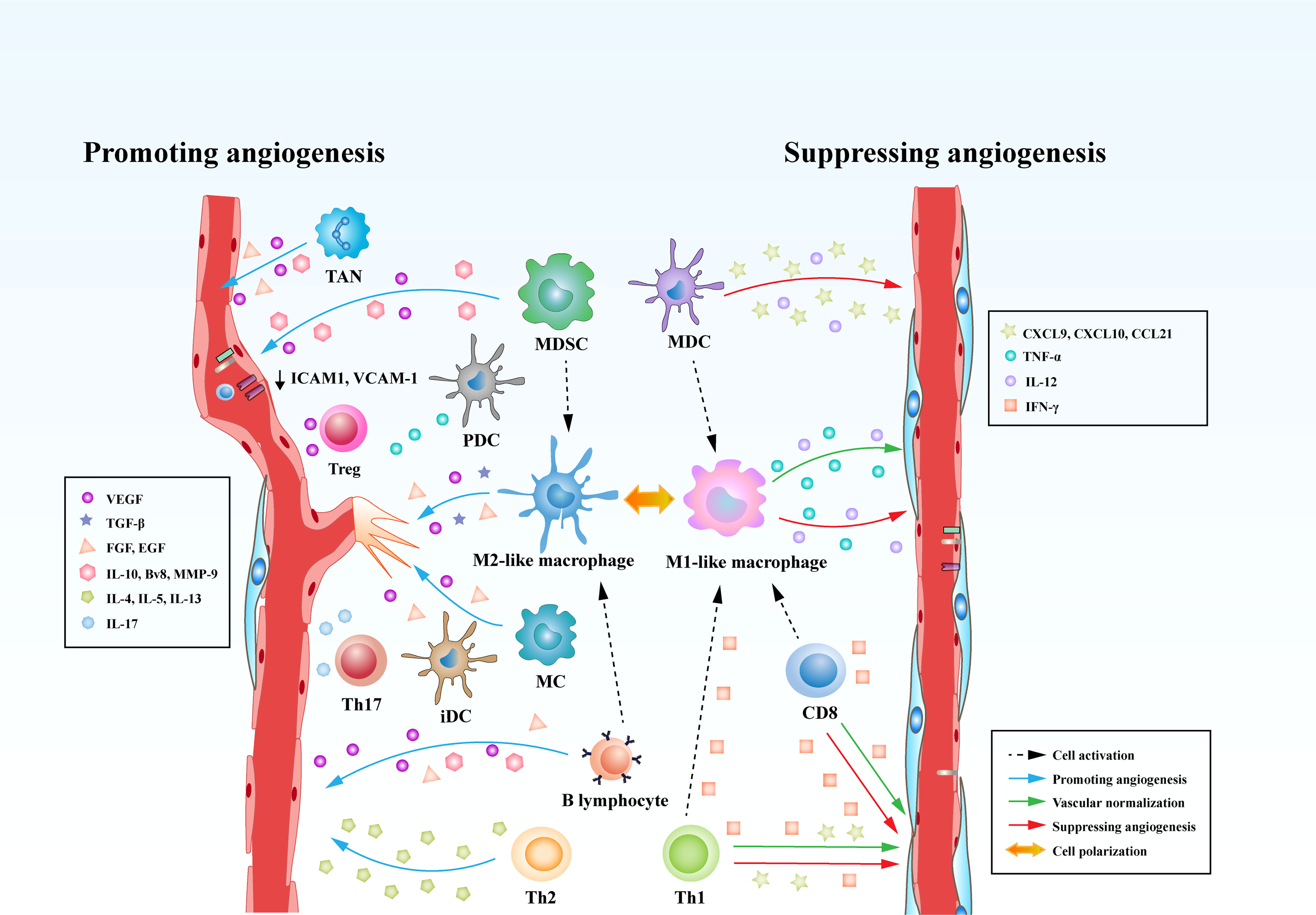
Figure 3 Immune cells play versatile roles in regulating tumor angiogenesis. Immune cells are able to directly curb the phenotypes and functions of tumor blood vessels through modulating the production of various cytokines. Some types of innate immune cells, such as MDCs and M1-like macrophages, produce cytokines (IL-12 or TNF) and chemokines (CXCL-9, CXCL-10, or CCL-21) that suppress tumor angiogenesis. Meanwhile, adaptive immune cells, such as CD8+ T cells and T helper 1 (Th1) cells, are capable of secreting IFN-γ, which is a potent cytokine that inhibits angiogenesis and induces vascular normalization in the TME. However, MDSCs, M2-like TAMs robustly augment tumor angiogenesis by secreting multiple factors including VEGF, IL-10, Bv8, and MMP-9. Moreover, Tregs, Th2, and Th17 cells can release pro-angiogenic factors such as VEGF, IL-4, IL-5, IL-13, and IL-17 to initiate the angiogenic switch. In addition to the direct effects on tumor vasculature, immune cells can also influence tumor vasculature indirectly through communicating and interfering with each other. CD8+ T cells and Th1 cells are able to skew macrophage polarization away from the M2 to the M1 phenotype. However, MDSCs and B lymphocytes reprogram TAMs from M1 to M2-like profile.
4 Intrinsic interactions between vascular normalization and immunotherapy
4.1 Advancements of anti-angiogenic strategies: From tumor starvation to vascular normalization
Systemic anti-angiogenic therapies have been well established with the rationale that suppressing the formation of tumor blood vessels is prone to confer conspicuous vascular eradication, consequently starving tumors to death due to disruption of delivery route for oxygen and nutrients or driving them to be “dormant”. In 1993, Napoleone Ferrara’s group uncovered a remarkable decrease in microvascular density and dramatic retardation of tumor growth in the nude mice harboring xenografts of glioblastoma multiforme, rhabdomyosarcoma and leiomyosarcoma following the treatment of an effective anti-VEGF monoclonal antibody (134). Moreover, single-agent treatment with bevacizumab contributed to striking tumor growth suppression of 20 diverse human tumor cell lines (13 different tumor types) implanted into nude mice regardless of the route of administration or the location of tumors (135). As such, it was believed that anti-angiogenesis therapeutic strategy was essential for repressing the growth of tumor and retarding metastasis by virtue of blocking the fuel supply and destroying the circulating principle routes for the tumor cells through prohibiting tumor angiogenesis. In spite of the promising preclinical data, the clinical outcomes of VEGF inhibition monotherapy for the treatment of solid tumors have been observed to be less than expected, with only limited objective response rates (ORRs) and a lack of meaningful survival benefits in phase 3 clinical trials (136). For example, in metastatic colorectal cancer, an ORR of 3.3% was calculated among chemotherapy-pretreated patients receiving bevacizumab, a well-known monoclonal antibody against human VEGF. On the contrary, a wide range of randomized phase 3 clinical trials of bevacizumab treatment in combination with systemic chemotherapy have shown profound enhancement in progression-free survival (PFS) and overall survival (OS) when in comparison to systemic chemotherapy alone (137–139). These clinical data seem to be counterintuitive. Anti-VEGF therapy is designed to induce the destruction of tumor blood vessels and thus tumor starvation, whereas the effectiveness of chemotherapeutic agents relies on adequate tumor blood flow and thereby efficient delivery of therapeutic drugs.
In 2005, “vascular normalization” theory was firstly proposed by Rakesh Jain, which potentially addresses this paradox (140). The theory postulates that instead of pruning blood vessels, the judicious employment of anti-angiogenic treatment reverses the structurally and functionally aberrant tumor blood vessels towards a more normal phenotype. The normalized tumor blood vessels tend to improve the treatment outcomes of chemotherapy, immunotherapy and radiotherapy since they offer reliable pathways for the anti-tumor elements penetrating into the tumor parenchyma efficiently (141, 142). Notably, the conception of vascular normalization possesses various limitations owing to the fact that it seems to be difficult to predict the accurate regimen (therapeutic window of dosage and time) that will confer vascular normalization rather than vessel pruning. In this regard, the same anti-angiogenic strategy (e. g. VEGF suppression), administrated at divergent doses or with the same dose in distinct tumor types, is able to harness vessel repression and further aggravate hypoxia or vascular normalization, and thereby reoxygenation in tumors (143, 144). For example, Huang and co-workers demonstrated that rational usage of anti-angiogenic treatment is crucial to ameliorate hypoxia in the tumor parenchyma and subsequently generate an immune-supportive TME (145). In their study, it was revealed that excessive treatment of anti-VEGFR2 tended to aggravate intratumoral hypoxia and restricted CD8+ T cell infiltration into the TME, thereby prohibiting anti-tumor immune response. Rivera and colleagues also unveiled that high-dose of anti-angiogenic treatment was able to result in the activation of PI3K signaling cascade in the myeloid cells that further attenuate immunity and bolster neovascularization (117). Hence, further preclinical and clinical studies are required to achieve the optimization of anti-angiogenic treatment in the era of cancer immunotherapy, aiming at opening the window of vascular normalization within the TME and fuel anti-cancer immune response.
4.2 Vascular normalization triggered by vasculature-targeting strategies potentiates cancer immunotherapy
Indeed, VEGF inhibition has been demonstrated to increase the number of tumor-infiltrating lymphocytes by virtue of resulting in tumor vascular normalization in both animal models and humans (146). In addition to repressing the classical VEGF-VEGFR signaling axis, targeting a plethora of alternative components to achieve tumor vascular normalization is also prone to exhibit striking therapeutic benefits. It has been increasingly recognized that fortifying vascular structure and enhancing vascular function can be fulfilled mainly through heightening pericyte coverage, restoring EC junctions, boosting tumor vascular perfusion and alleviating tumor hypoxia, thereby facilitating the infiltration of T cells and further provoking an immune-supportive TME. To this end, vascular normalization achieved by proper vasculature-targeting strategies has high propensity to improve cancer immunotherapy.
4.2.1 Regulating pericyte coverage
Pericytes have been regarded as an indispensable element of normal blood vessels, in which they result in vascular maturation and integrity, but inadequate pericyte coverage is frequently observed in tumor vasculature. As such, orchestrating pericyte coverage has been perceived as an effective and efficient therapeutic strategy to normalize tumor blood vessels (147, 148). A list of critical signaling pathways that are exploited to modulate pericyte coverage have been outlined in Figure 4.
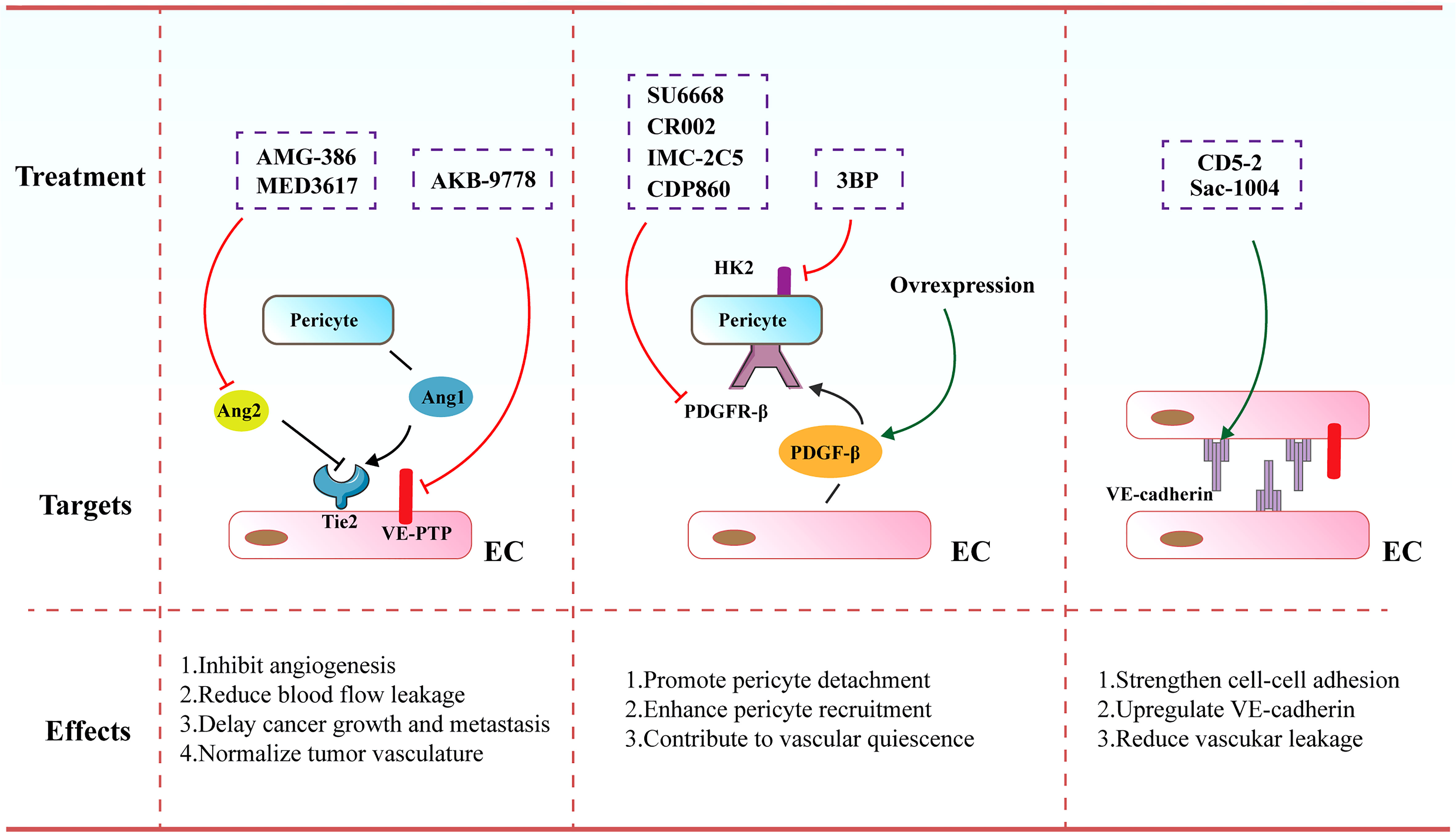
Figure 4 Therapeutic strategies and targets to normalize tumor blood vessels. Increased pericyte coverage, enhanced EC junctions, elevated tumor vascular perfusion as well as alleviated tumor hypoxia tend to result in the achievement of vascular normalization, further improving the distribution and efficiency of anti-cancer therapies.
Angiopoietin (Ang) signaling cascade has been reported to participate in governing the vascular permeability, vasodilation and vasoconstriction. The angiopoietins and their receptor Tie-2 (TEK) exert vital effects on controlling blood vessel formation and remodeling. Ang-1 that is secreted by perivascular cells plays an important role in driving maturation of blood vessels. In contrast, Ang-2 mainly secreted by ECs is involved in the disruption of EC-pericyte interaction ECs as it harbors antagonistic function to the Tie-2 receptor. Various inhibitors targeting the Ang/Tie signaling cascades have been formulated and are undergoing investigations in preclinical and clinical studies. For instance, MEDI3617 as an Ang-2 antibody was shown to lead to the reduction in tumor angiogenesis, a striking enhancement of chemotherapy, as well as impaired metastasis in multiple preclinical models including colorectal, renal, hepatocellular, and ovarian carcinoma (149). AMG-386 (trebananib), a peptide-Fc fusion protein, was observed to diminish tumor angiogenesis and further retard tumor progression via interfering with the binding of Ang-1 and Ang-2 to the Tie-2 receptor (150). Furthermore, Koh and colleagues conducted a comparison of Ang-2-Binding and Tie-2-Activating Antibody (ABTAA) and Ang-2-Blocking Antibody (ABA) in mice implanted with various types of tumors. ABTAA favorably resulted in immune reprogramming in tumors, implying that simultaneous Tie-2 activation and Ang-2 suppression potently confer a favorable TME to facilitate the delivery of chemotherapeutic drugs into tumors (151). Furthermore, Jain and co-workers treated glioblastoma-bearing mice with antibodies targeting Ang-2/VEGF (CrossMab, A2V), and found that A2V attenuated microvascular density and polarized M2-like TAMs toward the anti-tumoral M1-like TAMs, leading to the retardation of tumor progression (152).
Of note, vascular endothelial protein tyrosine phosphatase (VE-PTP) as a specific EC-phosphatase tends to dephosphorylate and further inactivate Tie2. AKB-9778 emerges as a selective and potent VE-PTP inhibitor that triggers the activation of Tie2 in ECs. The treatment of AKB-9778 mitigated the growth of breast cancer and diminished the size, number and progression of metastatic foci (153). Further, Vestweber et al. demonstrated that silence of VE-PTP with AKB-9778 and genetic strategy declined vascular permeability mediated by inflammatory factors. Additionally, leukocyte transmigration across the endothelial barrier was prevented. Mechanistical analysis implicated that suppression of VE-PTP restored endothelial junctions through activating Tie-2, which yielded the activation of Rap1 and further conferred the dissolution of radial stress fibers via Rac1 and inhibition of nonmuscle myosin II (154).
PDGF/PDGFR signaling cascade are also reported to be involved in cancer progression via autocrine stimulation of cancer cell proliferation and paracrine effect on stromal cells triggering tumor angiogenesis. Notably, it was demonstrated that anti-angiogenic resistance was partially owing to robust mural cell proliferation and elevated PDGF-B level. SU6668 serves as a PDGFR-β inhibitor that eliminates tumor vasculature through propelling the detachment of pericytes from ECs. SU6668 in combination with SU5416 as a VEGFR inhibitor exhibited a synergistic effect on tumor eradication (155, 156). Decrease in intratumoral pressure and alleviated hypoxia ascribed to increased pericyte coverage are the driving forces for augmented T cell infiltration. Likewise, vascular normalization contributes to an elevated and uniform distribution of adhesion molecules on the luminal surface of ECs lining the tumor blood vessels, thereby facilitating more efficient docking and rolling of T cells, both of which promote the infiltration of T cells into tumor parenchyma (157). Of note, PDGFR-β inhibitors contained various antibodies, such as CR002, IMC-2C5 and CDP860. They are engineered Fab’ fragment–polyethylene glycol conjugates that target PDGFR-β and repress its activation (158, 159). However, a number of studies demonstrated that depletion of the pericytes-associated blood vessels is not more effective than anti-VEGF antibody alone (160). Further, loss of pericyte coverage hampered vessel maturation and promoted metastasis. As such, an alternative efficient therapy to induce vascular normalization seems to activate PDGF/PDGFR-β signaling cascade. Ellis and colleagues highlighted that overexpression of PDGF-B in mouse models of colorectal and pancreatic cancer resulted in the suppression of tumor growth through boosting the recruitment of pericytes and limiting the proliferation of ECs. Further, treatment with imatinib mesylate (PDGFR-β inhibitor) led to impaired pericyte coverage, which was associated with the destabilization of tumor blood vessels (161).
Moreover, Wong and co-workers elucidated that increased hexokinase 2 (HK2)-mediated pericyte glycolysis was capable of strengthening ROCK2-MLC2 axis-mediated contractility, thereby resulting in disrupted tumor vascular function (162). Treatment of a HK2 inhibitor (3BP) conferred the recruitment of pericytes, thereby facilitating tumor vasculature remodeling and further enhancing drug delivery into tumors (162). Additionally, genetic elimination of regulator of G-protein signaling 5 (RGS5) enabled immature PDGFR-β+ pericytes to turn into mature αSMA+NG2+ pericytes without influencing their overall coverage onto the blood vessels (163, 164). This phenotypic alteration in tumors eventually diminished hypoxia and vascular permeability, resulting in significant penetration of immune effector cells into the tumor parenchyma.
4.2.2 Enhancement of endothelial junctions
It has been accepted that VE-cadherin acts as a central molecule for maintaining EC junctions and vascular integrity. Disruption of VE-cadherin expression or membrane location leads to augmented vascular permeability, reflecting its pivotal role in governing vascular barrier function (165, 166). Sac-1004 was a recently formulated molecule to build up appropriate IFP, ameliorate vascular permeability and enhance vascular perfusion. It intensified adherens junctions via modulating the cAMP/Rac/Cortactin signaling cascade to generate a cortical actin ring, which was able to boost the expression level of VE-cadherin (167). The dissociation of VE-cadherin from VE-PTP is a master regulatory factor of leukocyte migration across the endothelial barrier. The majority of leukocytes including lymphocytes transmigrate across the endothelium via a paracellular pathway, in comparison to the alternative transcellular route that occupies approximately 10% of transendothelial migration (168, 169). Vockel et al. demonstrated that upon α4β1 integrin in lymphocytes binding to endothelial VCAM-1, an important signaling pathway was initiated that resulted in the activation of Rac1. Rac1 subsequently triggered the activation of NOX, which transformed molecular oxygen to reactive oxygen species (ROS). These ROS activated the kinase Pyk2, which contributed to the phosphorylation of an undefined substrate for VE-PTP and further provoked VE-PTP to dissociate from VE-cadherin. This dissociation was correlated to elevated leukocyte transmigration and augmented vascular leakage (170).
Further, Zhao et al. uncovered an oligonucleotide-based inhibitor (171), which disrupted the interaction of microRNA-27α with its target VE-cadherin, leading to increased VE-cadherin expression (172, 173). Administration of CD5-2 in tumor-bearing mice boosted the expression of VE-cadherin in tumor endothelium, activating Tie-2 and tight junction pathways and normalizing vascular structure and function. CD5-2 treatment upregulated the levels of chemokines including CCL-2 and CXCL-10 that are involved in leukocyte transmigration, which facilitated the penetration of CD8+ T cells into tumor parenchyma (172). Qian et al. showed that Salvianolic acid B (SalB) elicited vascular normalization in the mouse models of breast cancer. More interestingly, SalB in combination with anti-PD-L1 inhibited the growth of tumors, which was mainly owing to increased infiltration of CD8+ T cells and enhanced delivery of anti-PD-L1 into tumors. Mechanistically, tumor cell enhancer of zeste homolog 2 (Ezh2)-driven cytokines perturbed the endothelial junctions with decreased VE-cadherin expression, which was reversed following the treatment of SalB (174).
4.2.3 Modulation of vascular perfusion and hypoxia
Intriguingly, multiple studies showed that radiotherapy or chemotherapy could modulate the tumor vasculature and further promote vascular perfusion. Recently, it was revealed that fractionated radiation therapy profoundly augmented tumor vascular perfusion following two weeks of treatment. It was correlated to how quality of tissue viability and diminished hypoxia, but the increased VEGF level was counteracted in the presence of sunitinib (175). Gold nanoparticles (AuNPs) have been regarded as a promising nanomaterial resulting from their efficient drug-delivery properties and robust anti-tumor function. Huang et al. unveiled the anti-neoplastic activity of an improved targeting polymer and folic acid-modified gold nanoparticles (AuNPP-FA). AuNPP-FA was capable of normalizing tumor blood vessels via boosted vascular perfusion, alleviated hypoxia as well as improved EC junctions that was mediated by increased VE-cadherin expression in ECs. In this sense, the immunotherapeutic outcomes were significantly potentiated owing to the escalated penetration of CD8+ T cells. Mechanistic analysis revealed that AuNPP-FA reinforced the expression and production of semaphorin 3A (SEMA3A) in cancer cells, which further led to the suppression of Smad2/3 signaling cascade in ECs (176). Chloroquine (CQ), commonly employed as an autophagy inhibitor, was also observed to regulate tumor vasculature (177). CQ influenced the TME through promoting vascular perfusion, diminishing hypoxic area, and restraining the dissemination and intravasation of cancer cells. The vascular normalization effect of CQ predominantly relied on changes in the trafficking of endosomal Notch1 and its downstream signaling activation in ECs, which could be abrogated upon the silence of Notch1 in ECs (177).
Further, it has been held that the oxygen-sensing prolyl hydroxylase domain proteins (PHD1-3) are capable of targeting hypoxia-inducible transcription factors (HIFs) for degradation. Under the condition that oxygen tension drops, PHDs tend to be less active, in which case stabilized HIFs induce an adaptive response including angiogenesis. Nevertheless, aggravated hypoxia in tumors leads to excessive secretion of pro-angiogenic factors and subsequently tumor angiogenesis. Carmeliet et al. showed that haplodeficiency of PHD-2 failed to influence tumor vascular density and lumen size, whereas enhanced the endothelial junctions and vascular maturation. This contributed to improved tumor vascular perfusion and oxygenation, further inhibiting tumor cell invasion and metastasis. These normalized tumor blood vessels phenotypes relied on HIF-mediated upregulation of (soluble) VEGFR-1 and VE-cadherin (178). Moreover, Mazzone et al. illustrated that the B55α/PP2A complex limited PHD-2 function and enhanced the survival of ECs in a HIF-dependent manner. Furthermore, it tended to dephosphorylate p38 to protect ECs from the occurrence of cell stress including the onset of blood flow. Their data emphasize a specific role of the B55α/PP2A phosphatase complex in tumor vascular remodeling and suggested the promising application values of PP2A-inhibitors as robust anti-angiogenic agents exclusively targeting nascent blood vessels with a mode-of-action complementary to anti-VEGFR therapies (179).
4.3 Immune reprograming-mediated tumor vascular normalization
Intriguingly, despite that immune cells have been proposed to encrypt the functions of ECs, thereby having profound consequences on curbing tumor angiogenesis, it has been recently demonstrated that the activation of a series of certain types of immune cells could also exert essential effects on improving tumor vascular normalization (Table 2) (180, 181, 187).

Table 2 Immunotherapies inducing vascular normalization.
It is noteworthy that, in addition to orchestrating immune-induced tumor cell elimination, ICB was also documented to provoke tumor vessel normalization at least in the orthotopic breast and ectopic colon cancer models. In both preclinical models, it was observed that repression of CTLA-4 or PD-1 resulted in diminished tumor vascular density, enhanced vascular perfusion, as well as alleviated hypoxia in the tumor bed, all of which are perceived as the central features of tumor vascular normalization (72, 180). In the both CD4−/− and CD8−/− mouse tumor models, it was shown that suppression of CTLA-4 and PD-1 was able to induce tumor vascular normalization through activating the Th1 cells. Nonetheless, it was deciphered that CD4+ T cells alone failed to sufficiently give rise to the remodeling of tumor blood vessels (180). Conversely, the deletion of CD4+ T cells contributed to the recruitment and accumulation of CD8+ T cells as well as elevated levels of IFN-γ, with a markedly tumor vascular normalization phenotype. In this regard, Zheng and co-workers drew the conclusion that the vascular normalization effect mediated by ICB therapy was potentially associated with the activation of CD8+ T cells induced by the IFN-γ signaling cascade (181). Additionally, Huang and colleagues highlighted that augmented release of Delta-like 1 (DLL1) in the TME gave rise to durable vascular normalization to diminish tumor hypoxia and facilitated the infiltration of CD8+ T cells as well as the polarization of M1-phenotypic TAMs. Mechanical analysis revealed that in vivo knockout of CD8+ T cells or silence of IFN-γ reversed the suppression of tumor growth and attenuated DLL1-mediated vascular normalization without influencing DLL1-induced macrophage polarization. Collectively, the data implicated that increased DLL1 levels in the TME conferred long-term tumor vascular normalization in a CD8+ T cell- and IFN-γ- dependent manner and enhanced the effectiveness of ICB (188).
Furthermore, Huang and co-workers also demonstrated that activated CD11b+Gr1loF4/80+Siglec-F+ eosinophils were capable of yielding tumor vascular normalization, which further provide robust support to provoke tumor elimination by virtue of CD8+ T cells. Of note, the underlying mechanism that eosinophils triggered tumor vascular normalization remained elusive, whereas it might be due to that eosinophils skewed polarization of TAMs away from M2-like into M1-like profile through activating IFN-γ and TNF derived from eosinophils, leading to impaired secretion of VEGF. The normalized tumor vasculature enabled T cells to penetrate into tumors, which contributed to the formation of a positive feedback loop that promoted further polarization of TAMs to M1-phenotypic profile, as well as vascular normalization to facilitate more efficient T cell infiltration (182).
In addition to the above-mentioned immune-associated signaling cascades, there are also other elements that may normalize tumor vasculature. Recently, Yang and colleagues identified the stimulator of IFN gene (STING) agonists (cGAMP and RR-CDA) that activated the expression of STING in tumors to normalize tumor vasculature (185). Also, IFN-β was observed to be a potent anti-angiogenic cytokine that limited EC proliferation and subsequently triggered the maturation of tumor blood vessels via upregulating Angpt1 (37). Administration of intratumoral STING agonist contributed to the activation of type I IFN signaling together with the boosted expression levels of vascular integrity-related genes, such as Ang1, PDGFR-β and collagen-α. The transcriptional alterations ultimately resulted in tumor vascular normalization with increased pericyte coverage and more intact basement membrane support, thereby improving the penetration of CD8+ T cells into tumor sites and diminishing hypoxic area in the TME. Nonetheless, prohibition of the type I IFN signaling cascade via IFN-α/β receptor blockade almost entirely abrogated STING-mediated transcriptional alterations and also phenotypic changes in blood vessels. These data implicated that the elevated expression levels of vascular integrity-associated molecules in the STING-induced vascular remodeling partially relied on the function of type I IFN.
5 Anti-angiogenic therapy meets immunotherapy
5.1 ICB plus anti-angiogenic therapy in preclinical studies
It has been well accepted that, on one hand, tumor immune evasion is tightly associated with angiogenesis. On the other hand, tumor angiogenesis is thought to rely substantially on immunosuppressive tumor microenvironment (18, 189, 190). The potential to combine anti-angiogenic therapy with immunotherapy, in particular with immune checkpoint inhibitors, has been corroborated by numerous combinations in various preclinical mouse models (Table 3). However, apart from decreased IFP and correspondingly improved T cell infiltration into tumor parenchyma, we are not able to rule out other mechanisms by which ICB and anti-angiogenesis synergistically kill tumor cell. Thus, further explorations should be conducted in expanding models. To date, multiple mechanisms have been found to be related to synergistic effects.
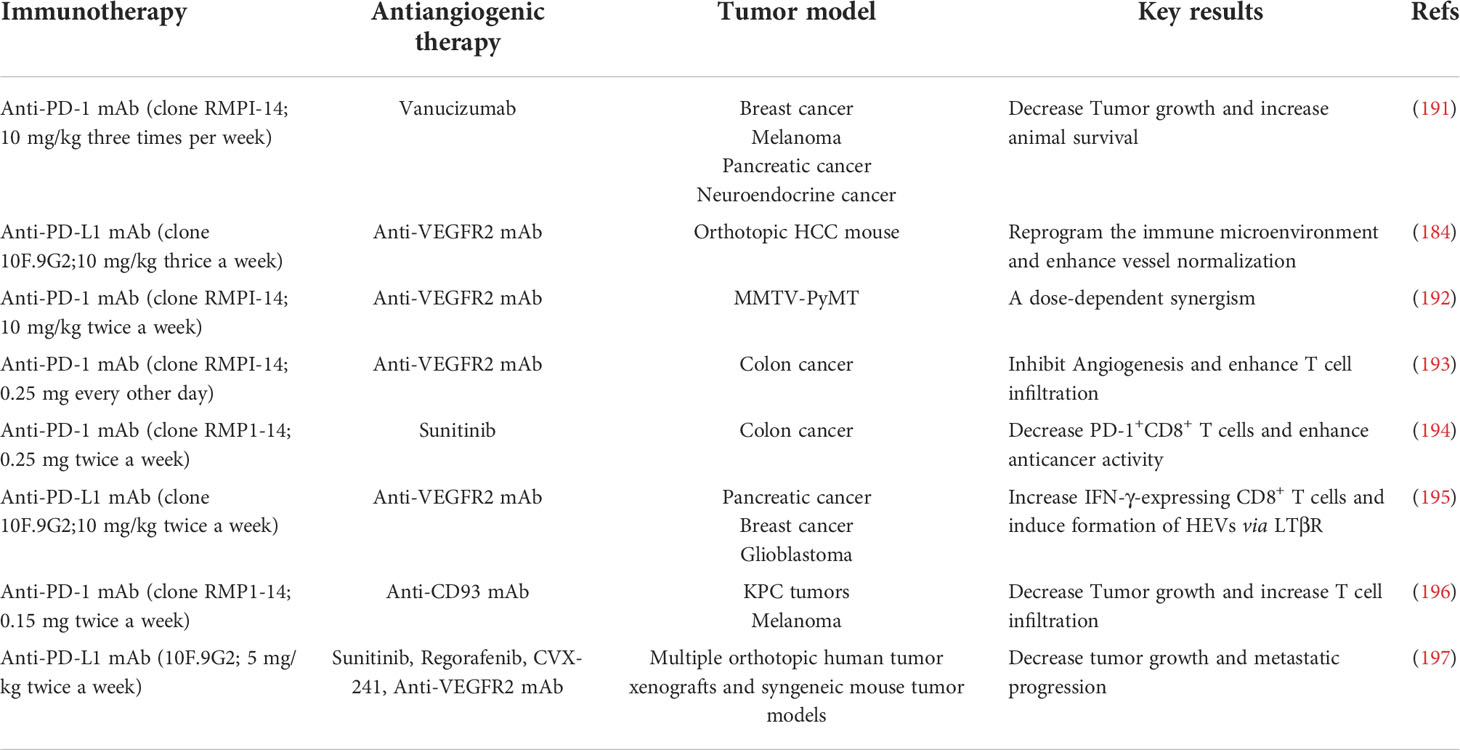
Table 3 Preclinical studies testing combinations of anti-angiogenic agents and immunotherapies.
5.1.1 Inducing formation of high endothelial venules
Allen and colleagues explored the effects of anti-PD-L1 (B20S) therapy in combination with anti-VEGFR2 (DC101) in mice bearing mammary carcinoma, glioblastoma and pancreatic neuroendocrine tumor. It was observed that the combination therapy exhibited robust synergistic effects on tumor eradication and OS compared with monotherapy in pancreatic neuroendocrine tumor and mammary carcinoma (195). After the combination therapy of anti-PD-L1 and anti-VEGFR2 for two weeks, the expression levels of IFN-γ+CD8+ and IFN-γ+CD4+ T cells were dramatically boosted in the pancreatic neuroendocrine tumor and mammary carcinoma, whereas the number of IFN-γ+CD8+ T cells was only modestly elevated for approximately 50% in glioblastomas (195). As the direct barrier for the extravasation T cells, abnormal blood vessels in tumors are recognized as the determinant resulting in the restricted T cell infiltration in glioblastomas. In addition to augmented pericyte coverage, the endothelium following the combination therapy was thickened with flat ECs instead of plump ECs, showing the specific property of the presence of HEVs. Immunohistochemical examination verified the phenotypic changes in ECs. Indeed, it has been commonly believed that HEVs are associated with lymphocyte homing (198, 199). Similarly, it was shown that intratumoral HEVs propelled the infiltration of T cells into tumor parenchyma. LTβR signaling cascade was found to be critical to give rise to the HEV phenotype. Activation of LTβR signaling cascade with its agonist accompanying to combination therapy could strikingly retard the progression of glioblastoma, suggesting the pivotal role of HEVs in the combination therapy (195).
5.1.2 Reciprocal regulation of VEGF/VEGFR2 signaling inhibition and ICB therapy
Voron et al. depicted that VEGF was able to augment the expression of PD-1 by leading to the activation of VEGFR2-PLCγ-calcineurin-NFAT signaling cascade (194). Consistently, anti-VEGF antibody profoundly down-regulated the expression of a bunch of preponderant immune checkpoint molecules (e. g. PD-1, CTLA-4, and TIM-3) on the CD8+ T cells in tumors (194). To this end, anti-PD-1 antibody in combination with anti-VEGF antibody was able to tremendously inhibit PD-1/PD-L1 signaling axis and synergistically delay the growth of tumor in particular for tumor with high VEGF levels in a synergistical way (194). Additionally, Ullrich and co-workers elucidated that, in a mouse model of small cell lung cancer (SCLC), anti-VEGF in combination with anti-PD-L1 synergistically enhanced the therapeutic outcomes in terms of the tumor progression compared with anti-PD-L1 or anti-VEGF monotherapy. Mechanistical studies revealed that VEGF-A was prone to elevate the co-expression of TIM-3 as an inhibitory receptor on T cells, suggesting the immunosuppressive role of VEGF in SCLC patients in the presence of anti-PD-1 therapy. This exhausted phenotype of T cells by PD-L1 blockade could be reversed following the supplement of anti-VEGF therapy (200).
5.1.3 Overcoming the resistance and negative feedback of anti-angiogenesis and ICB
In addition to VEGF/VEGFR2 signaling cascade, Ang-2/Tie-2 is recognized as an alternative pro-angiogenic pathway that emerges as a substitute when encountering resistance to anti-VEGF therapy (201–203). Schmittnaegel et al. illustrated that the dual inhibition of VEGF and Ang-2 by a bispecific antibody A2V displayed a more robust therapeutic function compared to monotherapy (191). Upon the treatment of A2V, the numbers of tumor-killing immune cell subsets including mature DCs, M1-like TAMs and IFN-γ+/CD69+ CD8+ T cell were significantly enhanced (191). In the meanwhile, elevated number of perivascular CD8+ T cells was found to be accompanied to the high level of PD-L1 on the tumor cells, which was responsible for the IFN-γ-induced negative feedback modulating principle (191). Accordingly, the combination therapy of anti-PD-1 and A2V destructed the negative feedback loop and amplified the immune response in tumors (191). Moreover, anti-angiogenic treatment was also capable of ameliorating resistance to anti-PD-1 therapy through precluding the TOX-induced T-cell exhaustion phenotype in the TME. Kim and colleagues elucidated that VEGF could conspicuously increase the expression of the transcription factor TOX, which further governed the characteristics and functions of CTLs (204). It was shown that the TOX-induced transcriptional event led to compelling T-cell exhaustion and boosted the expression levels of a series of immune checkpoints including PD-1, TIM-3, LAG-3, and TIGIT, thereby attenuating the secretion of killing cytokines from CTLs (204). Intriguingly, anti-VEGFR2 in combination with anti-PD-1 exerted striking effect on elevating the immunotherapeutic effectiveness and T-cell revitalization. Taken together, combination therapy of anti-angiogenic drugs and ICB has emerged as a promising therapeutic strategy in the anti-PD-1-resistant cancer.
5.2 ICB plus anti-angiogenic therapy in clinical studies
As discussed above, the interaction between immune suppression and angiogenesis renders tumor immune escape and treatment resistance. As summarized above, on the basis of the promising data of preclinical studies, considerable clinical studies have been performed to determine the synergistic effect of ICB and anti-angiogenic therapy in cancer patients (Table 4). The details of Table 4 can be obtained from http://clinicaltrials.gov/.

Table 4 Clinical trials examining the effects of ICB plus anti-angiogenic therapy.
5.2.1 Combination therapy of atezolizumab and bevacizumab
NCT02921269 is a phase II clinical trial for the combination therapy of atezolizumab (MPDL3280A) and bevacizumab in treating patients with recurrent, persistent or metastatic cervical cancer (205). Patients with advanced cervical cancer were intravenously injected with 15 mg/kg of bevacizumab and 1200 mg of atezolizumab every 3 weeks. In the entire evaluable patients (n=10), the disease control rate (DCR) by response evaluation criteria in solid tumors (RECIST) V.1.1 was 60% (0% complete response, 0% partial response, 60% stable disease). Median PFS was 2.9 months and median OS was 8.9 months (205). The combination of bevacizumab and atezolizumab did not meet the predefined efficacy endpoint, as addition of bevacizumab to PD-L1 blockade did not appear to enhance the ORR in cervical cancer.
However, The combination of atezolizumab and bevacizumab (NCT02715531) showed encouraging antitumor activity and safety in a phase 1b trial involving patients with unresectable hepatocellular carcinoma (206). Inspired by the encouraging outcome of NCT02715531, NCT03434379 is a phase III, open-label, randomized study of atezolizumab in combination with bevacizumab compared with sorafenib inpatients with untreated locally advanced or metastatic hepatocellular carcinoma (207). OS at 12 months was 67.2% (95% CI, 61.3 to 73.1) with atezolizumab-bevacizumab and 54.6% (95% CI, 45.2 to 64.0) with sorafenib. Median PFS was 6.8 months (95% CI, 5.7 to 8.3) and 4.3 months (95% CI, 4.0 to 5.6) in the respective groups (hazard ratio for disease progression or death, 0.59; 95% CI, 0.47 to 0.76; P<0.001). In short, atezolizumab combined with bevacizumab resulted in better overall and progression-free survival outcomes than sorafenib.
5.2.2 Combination therapy of avelumab and axitinib
In 2018, Choueiri et al. firstly demonstrated the efficacy of avelumab plus axitinib (VEGFR1-3 TKI) in the treatment of RCC. NCT02493751 is a phase Ib study with the purpose to assess the safety, pharmacokinetics, and pharmacodynamics of avelumab (anti-PD-L1) plus axitinib therapy (208). For a total of 55 patients recruited in the study, 54 patients received avelumab plus axitinib therapy except for one patient due to abnormally increased blood creatine phosphokinase (208). Within a follow-up period of appropriate one year, 58% (32 of 55) patients displayed complete response or partial response to the combination therapy while 20% (11 of 55) patients had stable disease (208). Notably, it was found that PD-L1 level fail to dramatically influence the treatment effectiveness. No matter choosing cut-off value as 1% or 5%, the ORRs of PD-L1 high expression group and PD-L1 low expression group were comparable (208).
Inspired by the encouraging outcome of NCT02493751, a phase III clinical trial (NCT02684006) was carried out to compare the efficacy of avelumab plus axitinib vs. sunitinib (a multitargeted tyrosine kinase inhibitor) monotherapy in the advanced RCC (209–212). Treatment-naive patients with RCC were divided (1:1) to receive avelumab (10 mg/kg) intravenously every 2 weeks plus axitinib (5 mg) orally twice daily or sunitinib (50 mg) orally once daily for 4 weeks (6-week cycle). Of the 886 patients, 442 were randomized to the avelumab plus axitinib treated group and 444 to the sunitinib treated group. After a minimum follow-up of 13 months (data cut-off 28 January 2019), the PFS was significantly longer in the avelumab plus axitinib treated group than in the sunitinib treated group, but the OS data were still immature (211).
5.2.3 Combination therapy alleviates adverse effects
ICB was shown to provoke the appearance of edema in patients with brain tumors, sometimes even leading to death (213). This phenomenon was different from the vessel normalization triggered by ICB therapy in various breast cancer models (180), which indicates that the vasculature-associated effects rely on the location and/or type of tumors. The possibility requires to be further explored. However, anti-VEGF drugs were able to alleviate the glioblastoma-related brain edema in both mice and patients (214, 215), offering a strong rationale for combining low-dose anti-VEGF therapy with immunotherapy in particular ICB therapy for potentially reducing the brain metastasis as well as ameliorating the side effects.
Of note, it has been observed that most adverse events of ICB therapy are reported to be closely associated with the hyperactive immune response, thereby displaying T cell mediated auto-immune like inflammatory disease (216, 217). Perturbed immune homeostasis by ICB therapy contributes to immune-mediated damage in normal tissues, including hepatic, gastrointestinal, and skin system (216). In general, it was demonstrated that the risk of adverse events of monoclonal antibodies that target either PD-1 or PD-L1 was lower than that of anti-CTLA-4 monoclonal antibody (grade 3-4 adverse event: 7-12% vs. 10-18%) (216). Interestingly, these adverse events could be mitigated through terminating the ICB therapy or diminishing the dosage of ICB (218). Theoretically, anti-angiogenic therapy leads to tumor vascular normalization, which generates a favorable microenvironment for T cell infiltration and drug delivery into tumors. However, in terms of the combination therapy of anti-angiogenesis and ICB, it was speculated that the positive feedback loop between the two therapies enabled reduced dose usage of ICB, which was sufficient to counteract immunosuppressive microenvironment with less adverse events (218).
6 Discussion
To date, it has been widely held that anti-angiogenic therapy is commonly used for the treatment of cancer. Nevertheless, various limitations including off-target effects, high effective doses and drug resistance have built up hurdles for the usage of anti-angiogenic therapies in patients with cancer. As such, novel vasculature-targeting therapeutic strategies with improved efficacy and safety are urgently required for preventing cancer progression in clinic (219, 220). With the burgeoning advancements in nanotechnology, smart nanotherapeutics have provided preponderant possibility to exclusively target tumor blood vessels. For instance, through specifically delivering thrombin loaded DNA nanorobots (Nanorobot-Th) into tumor blood vessels, an intratumoral thrombosis was initiated to trigger vascular infarction, and eventually tumor necrosis (221). Furthermore, anti-angiogenic drug resistance has been observed in clinic partially owing to the heterogeneity of tumor blood vessels. Tumor endothelial cells (TECs) are likely to be influenced by their parental tumors through the secreted tumor-derived factors, such as growth factors, chemokines, and microRNAs (222). TECs possibly differ on the basis of their TME, as well as tumor progression stage. To this end, it has the potential to develop the markers of TECs into biomarkers that characterize the heterogeneous tumor blood vessels, enabling the more rational selection of anti-angiogenic strategies. Multiple peptides that recognize TEC plasma membrane molecules have been identified. An asparagine-glycine-arginine (NGR) motif peptide ligand for CD13 has been shown to be overexpressed in TECs. NGR motif peptides have been successfully used to specifically deliver drugs as well as liposomes to the tumor blood vessels (223). Another peptide motif, named arginine-glycine-aspartic acid (RGD), has also been employed for drug delivery due to that it can recognize integrin αVβ3 that is highly expressed in TECs (224). Therefore, novel drug delivery systems to exclusively target heterogeneous tumor blood vessels have the potential to become promising anti-angiogenic strategies for avoiding side-effects and improving overall anti-cancer efficacy. Further in-depth studies on TEC heterogeneity may pave the way for facilitating the development of appropriate anti-angiogenic drug delivery systems.
It has been widely held that ICB has deemed to be a revolutionary milestone in light of cancer therapy. Nevertheless, a large number of cancer patients fail to respond to the ICB monotherapy, or even undergo relapse, sometimes harbor nonnegligible long-term toxicity, all of which have given rise to continuous attempts to address these issues by virtue of combination treatment of anti-angiogenesis and immunotherapy. As what we have outlined in this review, the interactions between tumor angiogenesis and immune suppression is very dynamic and a two-way process, facilitating the formation of positive feedback loop to propel the two processes reciprocally. To this end, modulation of either side is able to disrupt the vicious circle in the TME, further retarding the tumor progression. More specifically, Anti-angiogenic therapies in particular vascular normalization strategy are able to result in striking alterations in the tumor blood vessels of the TME, which tend to further potentiate the penetration and function of immune effector cells in tumors to fulfill efficient cancer immunotherapies. Moreover, the striking effects of ICB on the TME, in particular the tumor vascular alterations it triggers, have preference to offer new and realistic variables by which to examine and forecast the responses of tumor immunotherapy (225). There have been multiple lines of evidence verifying the effective and efficient clinical outcomes of the combination therapy of anti-angiogenesis treatment and ICB. Such promising data are required to be further confirmed in an array of currently ongoing randomized clinical studies with longer follow-up.
Of note, a bunch of points remain to be optimized aiming at orchestrating the effectiveness of combination therapy. Firstly, predictive clinical biomarkers are supposed to be discovered to lock in the subpopulation of cancer patients who are able to respond to the combination therapy. It is imperative to notice that the majority of the promising outcomes from the combination therapy of ICB and anti-angiogenic treatment have been predominantly observed in RCC, which is regarded as a high angiogenic and immunogenic tumor type with massive infiltrating immune cells (226). Secondly, it is also worthy of note that the core of anti-VEGF(R) agents as the predominant anti-angiogenesis strategies is needed to be diversified. The less expected clinical benefits propose the issue of resistance to anti-VEGF(R) therapy, which drives the proceedings in the strategies for modulating numerous pro-angiogenic signaling molecules, such as VEGFA-D, PDGF, and FGF. Thirdly, whether the functions of combination therapy of anti-angiogenic treatment and ICB are additive or synergistic is required to be investigated. Additionally, since the angiogenic profiles are distinctive depending on the types of organ, more solid evidence at the organ level should be obtained to increase the understanding on the responses to ICB. More importantly, a critical issue requires to be solved is the optimization of the dose and application arrangement of anti-angiogenesis during the combination therapy procedure. In fact, prolonging the therapeutic window of vascular normalization and controlling excessive vascular disruption tend to fulfill the maximal therapeutic benefit. Collectively, we believe that anti-angiogenesis treatment in combination with ICB is capable of working as an effective and efficient approach to brake the anti-cancer drug resistance, as well as increase the therapeutic outcomes and improve the prognosis of cancer patients.
Author contributions
YaZ, YL and AW: Conceptualization and design the review, and review final version approval. WZ and CQ: Bibliographic research. WC, WZ, SY, and PS: Writing - original draft preparation. YT, YuZ and CY: Table and figure design. ZW, AW and YaZ: Supervision. YaZ, AW and SY: Funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by National Natural Science Foundation of China (Grant No.: 82003991, 81973587 and 81973734), Jiangsu Province Traditional Chinese Medicine Leading Talents Program (Grant No.: SLJ0229), Natural Science Foundation of Higher School of Jiangsu Province (Grant No.: 18KJA360007), National TCM Innovation Backbone Training Program (Chinese medicine practitioners letter (2019) No.128), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica (Grant No.: JKLPSE201812), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX22_0791 and KYCX21_1743).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liu X, Hogg GD, DeNardo DG. Rethinking immune checkpoint blockade: 'Beyond the T cell'. J Immunother Cancer (2021) 9(1):e001460. doi: 10.1136/jitc-2020-001460
2. Watson RA, Tong O, Cooper R, Taylor CA, Sharma PK, de Los Aires AV, et al. Immune checkpoint blockade sensitivity and progression-free survival associates with baseline Cd8(+) T cell clone size and cytotoxicity. Sci Immunol (2021) 6(64):eabj8825. doi: 10.1126/sciimmunol.abj8825
3. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. Pd-1 and pd-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi: 10.3389/fphar.2017.00561
4. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (Keynote-224): A non-randomised, open-label phase 2 trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/S1470-2045(18)30351-6
5. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (Checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
6. Munn LL, Jain RK. Vascular regulation of antitumor immunity. Science (2019) 365(6453):544–5. doi: 10.1126/science.aaw7875
7. Cao H, Ni C, Han L, Wang R, Blasig R, Haseloff R, et al. Claudin-12 deficiency inhibits tumor growth by impairing transendothelial migration of myeloid-derived suppressor cells. Cancer Res (2022) 82(13):2472–84. doi: 10.1158/0008-5472.CAN-21-3896
8. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
9. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol (2018) 18(3):195–203. doi: 10.1038/nri.2017.145
10. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
11. Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis (2017) 20(2):185–204. doi: 10.1007/s10456-017-9552-y
12. Eelen G, Treps L, Li X, Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ Res (2020) 127(2):310–29. doi: 10.1161/CIRCRESAHA.120.316851
13. Apte RS, Chen DS, Ferrara N. Vegf in signaling and disease: Beyond discovery and development. Cell (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
14. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res (2013) 73(10):2943–8. doi: 10.1158/0008-5472.CAN-12-4354
15. Jain RK. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell (2014) 26(5):605–22. doi: 10.1016/j.ccell.2014.10.006
16. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer (2019) 18(1):60. doi: 10.1186/s12943-019-0974-6
17. Claesson-Welsh L, Welsh M. Vegfa and tumour angiogenesis. J Intern Med (2013) 273(2):114–27. doi: 10.1111/joim.12019
18. Pircher A, Wolf D, Heidenreich A, Hilbe W, Pichler R, Heidegger I. Synergies of targeting tumor angiogenesis and immune checkpoints in non-small cell lung cancer and renal cell cancer: From basic concepts to clinical reality. Int J Mol Sci (2017) 18(11):2291. doi: 10.3390/ijms18112291
19. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-vegf and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol (2018) 52(Pt 2):117–24. doi: 10.1016/j.semcancer.2017.12.002
20. Uemura A, Fruttiger M, D'Amore PA, De Falco S, Joussen AM, Sennlaub F, et al. Vegfr1 signaling in retinal angiogenesis and microinflammation. Prog Retin Eye Res (2021) 84:100954. doi: 10.1016/j.preteyeres.2021.100954
21. Liu G, Chen T, Ding Z, Wang Y, Wei Y, Wei X. Inhibition of fgf-fgfr and vegf-vegfr signalling in cancer treatment. Cell Prolif (2021) 54(4):e13009. doi: 10.1111/cpr.13009
22. Repetto M, Crimini E, Giugliano F, Morganti S, Belli C, Curigliano G. Selective Fgfr/Fgf pathway inhibitors: Inhibition strategies, clinical activities, resistance mutations, and future directions. Expert Rev Clin Pharmacol (2021) 14(10):1233–52. doi: 10.1080/17512433.2021.1947246
23. Yang L, Zhou F, Zheng D, Wang D, Li X, Zhao C, et al. Fgf/Fgfr signaling: From lung development to respiratory diseases. Cytokine Growth Factor Rev (2021) 62:94–104. doi: 10.1016/j.cytogfr.2021.09.002
24. Hui Q, Jin Z, Li X, Liu C, Wang X. Fgf family: From drug development to clinical application. Int J Mol Sci (2018) 19(7):1875. doi: 10.3390/ijms19071875
25. Zou X, Tang XY, Qu ZY, Sun ZW, Ji CF, Li YJ, et al. Targeting the Pdgf/Pdgfr signaling pathway for cancer therapy: A review. Int J Biol Macromol (2022) 202:539–57. doi: 10.1016/j.ijbiomac.2022.01.113
26. Jitariu AA, Raica M, Cimpean AM, Suciu SC. The role of pdgf-B/Pdgfr-Beta axis in the normal development and carcinogenesis of the breast. Crit Rev Oncol Hematol (2018) 131:46–52. doi: 10.1016/j.critrevonc.2018.08.002
27. Khan KA, Wu FT, Cruz-Munoz W, Kerbel RS. Ang2 inhibitors and Tie2 activators: Potential therapeutics in perioperative treatment of early stage cancer. EMBO Mol Med (2021) 13(7):e08253. doi: 10.15252/emmm.201708253
28. Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells (2019) 8(5):471. doi: 10.3390/cells8050471
29. Duran CL, Borriello L, Karagiannis GS, Entenberg D, Oktay MH, Condeelis JS. Targeting Tie2 in the tumor microenvironment: From angiogenesis to dissemination. Cancers (Basel) (2021) 13(22):5730. doi: 10.3390/cancers13225730
30. Leong A, Kim M. The angiopoietin-2 and tie pathway as a therapeutic target for enhancing antiangiogenic therapy and immunotherapy in patients with advanced cancer. Int J Mol Sci (2020) 21(22):8689. doi: 10.3390/ijms21228689
31. Zambelli A, Biamonti G, Amato A. Hgf/C-met signalling in the tumor microenvironment. Adv Exp Med Biol (2021) 1270:31–44. doi: 10.1007/978-3-030-47189-7_2
32. Raj S, Kesari KK, Kumar A, Rathi B, Sharma A, Gupta PK, et al. Molecular Mechanism(S) of Regulation(S) of c-Met/Hgf signaling in head and neck cancer. Mol Cancer (2022) 21(1):31. doi: 10.1186/s12943-022-01503-1
33. Mulcahy EQX, Colomicronn RR, Abounader R. Hgf/Met signaling in malignant brain tumors. Int J Mol Sci (2020) 21(20):7546. doi: 10.3390/ijms21207546
34. Chen Z, Chen Y, Li Y, Lian W, Zheng K, Zhang Y, et al. Prrx1 promotes stemness and angiogenesis Via activating tgf-Beta/Smad pathway and upregulating proangiogenic factors in glioma. Cell Death Dis (2021) 12(6):615. doi: 10.1038/s41419-021-03882-7
35. Aashaq S, Batool A, Mir SA, Beigh MA, Andrabi KI, Shah ZA. Tgf-beta signaling: A recap of smad-independent and smad-dependent pathways. J Cell Physiol (2022) 237(1):59–85. doi: 10.1002/jcp.30529
36. Ciccarese F, Grassi A, Pasqualini L, Rosano S, Noghero A, Montenegro F, et al. Genetic perturbation of ifn-alpha transcriptional modulators in human endothelial cells uncovers pivotal regulators of angiogenesis. Comput Struct Biotechnol J (2020) 18:3977–86. doi: 10.1016/j.csbj.2020.11.048
37. Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. Endothelial cell functions impaired by interferon in vitro: Insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res (2018) 163:105–16. doi: 10.1016/j.thromres.2018.01.039
38. Bakshi HA, Quinn GA, Nasef MM, Mishra V, Aljabali AAA, El-Tanani M, et al. Crocin inhibits angiogenesis and metastasis in colon cancer Via tnf-Alpha/Nf-Kb/Vegf pathways. Cells (2022) 11(9):1502. doi: 10.3390/cells11091502
39. Fahey E, Doyle SL. Il-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol (2019) 10:1426. doi: 10.3389/fimmu.2019.01426
40. Malkova AM, Gubal AR, Petrova AL, Voronov E, Apte RN, Semenov KN, et al. Pathogenetic role and clinical significance of interleukin-1beta in cancer. Immunology (2022) 1–14. doi: 10.1111/imm.13486
41. Dong Q, Tian J, Zheng W, Fan Q, Wu X, Tang Y, et al. Interleukin-33 protects mice against hindlimb ischemic injury by enhancing endothelial angiogenesis. Int Immunopharmacol (2022) 109:108850. doi: 10.1016/j.intimp.2022.108850
42. Kobori T, Hamasaki S, Kitaura A, Yamazaki Y, Nishinaka T, Niwa A, et al. Interleukin-18 amplifies macrophage polarization and morphological alteration, leading to excessive angiogenesis. Front Immunol (2018) 9:334. doi: 10.3389/fimmu.2018.00334
43. Cheng WX, Huang H, Chen JH, Zhang TT, Zhu GY, Zheng ZT, et al. Genistein inhibits angiogenesis developed during rheumatoid arthritis through the il-6/Jak2/Stat3/Vegf signalling pathway. J Orthop Translat (2020) 22:92–100. doi: 10.1016/j.jot.2019.07.007
44. El-Sherbiny M, El-Sayed RM, Helal MA, Ibrahiem AT, Elmahdi HS, Eladl MA, et al. Nifuroxazide mitigates angiogenesis in ehlrich's solid carcinoma: Molecular docking, bioinformatic and experimental studies on inhibition of il-6/Jak2/Stat3 signaling. Molecules (2021) 26(22):6858. doi: 10.3390/molecules26226858
45. Wu HX, Cheng X, Jing XQ, Ji XP, Chen XZ, Zhang YQ, et al. Lifr promotes tumor angiogenesis by up-regulating il-8 levels in colorectal cancer. Biochim Biophys Acta Mol Basis Dis (2018) 1864(9 Pt B):2769–84. doi: 10.1016/j.bbadis.2018.05.004
46. Fousek K, Horn LA, Palena C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol Ther (2021) 219:107692. doi: 10.1016/j.pharmthera.2020.107692
47. Ronca R, Giacomini A, Rusnati M, Presta M. The potential of fibroblast growth Factor/Fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin Ther Targets (2015) 19(10):1361–77. doi: 10.1517/14728222.2015.1062475
48. Takakura N. Vascular reconstitution in the tumor for more effective tumor immunotherapy. Cancer Sci (2021) 112(4):1348–56. doi: 10.1111/cas.14854
49. Schito L, Rey S. Hypoxia: Turning vessels into vassals of cancer immunotolerance. Cancer Lett (2020) 487:74–84. doi: 10.1016/j.canlet.2020.05.015
50. Mazzone M, Bergers G. Regulation of blood and lymphatic vessels by immune cells in tumors and metastasis. Annu Rev Physiol (2019) 81:535–60. doi: 10.1146/annurev-physiol-020518-114721
51. Kretschmer M, Rudiger D, Zahler S. Mechanical aspects of angiogenesis. Cancers (Basel) (2021) 13(19):4987. doi: 10.3390/cancers13194987
52. Arroyo AG, Iruela-Arispe ML. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc Res (2010) 86(2):226–35. doi: 10.1093/cvr/cvq049
53. del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood (2010) 116(19):4025–33. doi: 10.1182/blood-2010-02-270819
54. Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol (2010) 22(5):617–25. doi: 10.1016/j.ceb.2010.08.010
55. Zarychta E, Ruszkowska-Ciastek B. Cooperation between angiogenesis, vasculogenesis, chemotaxis, and coagulation in breast cancer metastases development: Pathophysiological point of view. Biomedicines (2022) 10(2):300. doi: 10.3390/biomedicines10020300
56. Chavoshi H, Poormolaie N, Vahedian V, Kazemzadeh H, Mir A, Nejabati HR, et al. Vascular mimicry: A potential therapeutic target in breast cancer. Pathol Res Pract (2022) 234:153922. doi: 10.1016/j.prp.2022.153922
57. Diaz-Flores L, Gutierrez R, Gayoso S, Garcia MP, Gonzalez-Gomez M, Diaz-Flores L Jr., et al. Intussusceptive angiogenesis and its counterpart intussusceptive lymphangiogenesis. Histol Histopathol (2020) 35(10):1083–103. doi: 10.14670/HH-18-222
58. Lugassy C, Vermeulen PB, Ribatti D, Pezzella F, Barnhill RL. Vessel Co-option and angiotropic extravascular migratory metastasis: A continuum of tumour growth and spread? Br J Cancer (2022) 126(7):973–80. doi: 10.1038/s41416-021-01686-2
59. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel Co-option in cancer. Nat Rev Clin Oncol (2019) 16(8):469–93. doi: 10.1038/s41571-019-0181-9
60. Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
61. De Spiegelaere W, Casteleyn C, Van den Broeck W, Plendl J, Bahramsoltani M, Simoens P, et al. Intussusceptive angiogenesis: A biologically relevant form of angiogenesis. J Vasc Res (2012) 49(5):390–404. doi: 10.1159/000338278
62. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and nk-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discovery (2014) 4(5):522–6. doi: 10.1158/2159-8290.CD-13-0985
63. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
64. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med (2020) 52(9):1475–85. doi: 10.1038/s12276-020-00500-y
65. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and pd-L1. Cancer Res (2015) 75(11):2139–45. doi: 10.1158/0008-5472.CAN-15-0255
66. Koh HS, Chang CY, Jeon SB, Yoon HJ, Ahn YH, Kim HS, et al. The hif-1/Glial Tim-3 axis controls inflammation-associated brain damage under hypoxia. Nat Commun (2015) 6:6340. doi: 10.1038/ncomms7340
67. Ruf M, Moch H, Schraml P. Pd-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer (2016) 139(2):396–403. doi: 10.1002/ijc.30077
68. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses ctl function by inhibition of P38 and Jnk/C-jun activation. Int J Cancer (2012) 131(3):633–40. doi: 10.1002/ijc.26410
69. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J, et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int J Mol Sci (2020) 21(21):8363. doi: 10.3390/ijms21218363
70. Kim S, Park J, Kim J, Jeon JS. Microfluidic tumor vasculature model to recapitulate an endothelial immune barrier expressing fasl. ACS Biomater Sci Eng (2021) 7(3):1230–41. doi: 10.1021/acsbiomaterials.0c01542
71. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
72. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis Via Ccl28 and T(Reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
73. He Z, Zhang S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol (2021) 12:741305. doi: 10.3389/fimmu.2021.741305
74. Han Z, Dong Y, Lu J, Yang F, Zheng Y, Yang H. Role of hypoxia in inhibiting dendritic cells by vegf signaling in tumor microenvironments: Mechanism and application. Am J Cancer Res (2021) 11(8):3777–93.
75. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
76. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8(8):618–31. doi: 10.1038/nrc2444
77. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13(10):739–52. doi: 10.1038/nrc3581
78. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res (1996) 56(20):4625–9.
79. Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab (2016) 24(5):701–15. doi: 10.1016/j.cmet.2016.09.008
80. Shu Y, Cheng P. Targeting tumor-associated macrophages for cancer immunotherapy. Biochim Biophys Acta Rev Cancer (2020) 1874(2):188434. doi: 10.1016/j.bbcan.2020.188434
81. Zhang X, Zhang W, Yuan X, Fu M, Qian H, Xu W. Neutrophils in cancer development and progression: Roles, mechanisms, and implications (Review). Int J Oncol (2016) 49(3):857–67. doi: 10.3892/ijo.2016.3616
82. Pylaeva E, Spyra I, Bordbari S, Lang S, Jablonska J. Transfer of manipulated tumor-associated neutrophils into tumor-bearing mice to study their angiogenic potential in vivo. J Vis Exp (2019) 149). doi: 10.3791/59807
83. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17(8):457–74. doi: 10.1038/nrc.2017.51
84. Lewis CE, Harney AS, Pollard JW. The multifaceted role of perivascular macrophages in tumors. Cancer Cell (2016) 30(1):18–25. doi: 10.1016/j.ccell.2016.05.017
85. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
86. Liang W, Ferrara N. The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol Res (2016) 4(2):83–91. doi: 10.1158/2326-6066.CIR-15-0313
87. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by tgf-beta: "N1" versus "N2" tan. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
88. Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, Sessa WC. Direct actions of angiopoietin-1 on human endothelium: Evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Invest (1999) 79(2):213–23.
89. Bird L. Tumour immunology: Neutrophils help tumours spread. Nat Rev Immunol (2016) 16(2):74–5. doi: 10.1038/nri.2016.17
90. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous ifn-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120(4):1151–64. doi: 10.1172/JCI37223
91. Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med (2010) 10(4):369–73. doi: 10.2174/156652410791316968
92. Coffelt SB, de Visser KE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol (2015) 36(4):198–216. doi: 10.1016/j.it.2015.02.006
93. Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res (2016) 39(11):1588–96. doi: 10.1007/s12272-016-0820-y
94. Gaudry M, Bregerie O, Andrieu V, El Benna J, Pocidalo MA, Hakim J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood (1997) 90(10):4153–61. doi: 10.1182/blood.V90.10.4153
95. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2(10):737–44. doi: 10.1038/35036374
96. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U.S.A. (2006) 103(33):12493–8. doi: 10.1073/pnas.0601807103
97. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi: 10.1158/0008-5472.CAN-06-1278
98. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-Cell-Dependent tumour angiogenesis. Nature (2007) 450(7171):825–31. doi: 10.1038/nature06348
99. Asiry S, Kim G, Filippou PS, Sanchez LR, Entenberg D, Marks DK, et al. The cancer cell dissemination machinery as an immunosuppressive niche: A new obstacle towards the era of cancer immunotherapy. Front Immunol (2021) 12:654877. doi: 10.3389/fimmu.2021.654877
100. Dallavalasa S, Beeraka NM, Basavaraju CG, Tulimilli SV, Sadhu SP, Rajesh K, et al. The role of tumor associated macrophages (Tams) in cancer progression, chemoresistance, angiogenesis and metastasis - current status. Curr Med Chem (2021) 28(39):8203–36. doi: 10.2174/0929867328666210720143721
101. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J BioMed Sci (2019) 26(1):78. doi: 10.1186/s12929-019-0568-z
102. Jarosz-Biej M, Kaminska N, Matuszczak S, Cichon T, Pamula-Pilat J, Czapla J, et al. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PloS One (2018) 13(1):e0191012. doi: 10.1371/journal.pone.0191012
103. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
104. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229(2):176–85. doi: 10.1002/path.4133
105. Larionova I, Kazakova E, Gerashchenko T, Kzhyshkowska J. New angiogenic regulators produced by tams: Perspective for targeting tumor angiogenesis. Cancers (Basel) (2021) 13(13):3253. doi: 10.3390/cancers13133253
106. Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X, et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2f2. Mol Ther (2021) 29(3):1226–38. doi: 10.1016/j.ymthe.2020.11.024
107. Wei K, Ma Z, Yang F, Zhao X, Jiang W, Pan C, et al. M2 macrophage-derived exosomes promote lung adenocarcinoma progression by delivering mir-942. Cancer Lett (2022) 526:205–16. doi: 10.1016/j.canlet.2021.10.045
108. Reusser NM, Dalton HJ, Pradeep S, Gonzalez-Villasana V, Jennings NB, Vasquez HG, et al. Clodronate inhibits tumor angiogenesis in mouse models of ovarian cancer. Cancer Biol Ther (2014) 15(8):1061–7. doi: 10.4161/cbt.29184
109. Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-Liposome-Mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br J Cancer (2006) 95(3):272–81. doi: 10.1038/sj.bjc.6603240
110. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting csf-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood (2010) 115(7):1461–71. doi: 10.1182/blood-2009-08-237412
111. Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature (2008) 456(7223):814–8. doi: 10.1038/nature07445
112. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat Rev Cancer (2010) 10(7):505–14. doi: 10.1038/nrc2868
113. Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol (2020) 21(5):555–66. doi: 10.1038/s41590-020-0666-9
114. Zhang Md J, Zhang Md L, Yang Md Y, Liu Md Q, Ma Md H, Huang Md A, et al. Polymorphonuclear-mdscs facilitate tumor regrowth after radiation by suppressing Cd8(+) T cells. Int J Radiat Oncol Biol Phys (2021) 109(5):1533–46. doi: 10.1016/j.ijrobp.2020.11.038
115. Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res (2003) 63(9):2322–9.
116. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol (2018) 15(5):310–24. doi: 10.1038/nrclinonc.2018.9
117. Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E, Bergers G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep (2015) 11(4):577–91. doi: 10.1016/j.celrep.2015.03.055
118. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of Cd8(+) T cells. Nat Immunol (2015) 16(6):609–17. doi: 10.1038/ni.3159
119. Sozzani S, Rusnati M, Riboldi E, Mitola S, Presta M. Dendritic cell-endothelial cell cross-talk in angiogenesis. Trends Immunol (2007) 28(9):385–92. doi: 10.1016/j.it.2007.07.006
120. Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res (2004) 64(16):5535–8. doi: 10.1158/0008-5472.CAN-04-1272
121. Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of vegf-a. Nat Med (2004) 10(9):950–8. doi: 10.1038/nm1097
122. Fainaru O, Almog N, Yung CW, Nakai K, Montoya-Zavala M, Abdollahi A, et al. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J (2010) 24(5):1411–8. doi: 10.1096/fj.09-147025
123. Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Marone G, et al. Are mast cells masters in cancer? Front Immunol (2017) 8:424. doi: 10.3389/fimmu.2017.00424
124. Bruno A, Pagani A, Pulze L, Albini A, Dallaglio K, Noonan DM, et al. Orchestration of angiogenesis by immune cells. Front Oncol (2014) 4:131. doi: 10.3389/fonc.2014.00131
125. de Souza Junior DA, Santana AC, da Silva EZ, Oliver C, Jamur MC. The role of mast cell specific chymases and tryptases in tumor angiogenesis. BioMed Res Int (2015) 2015:142359. doi: 10.1155/2015/142359
126. Chen Y, Li C, Xie H, Fan Y, Yang Z, Ma J, et al. Infiltrating mast cells promote renal cell carcinoma angiogenesis by modulating Pi3k–>Akt–>Gsk3beta–>Am signaling. Oncogene (2017) 36(20):2879–88. doi: 10.1038/onc.2016.442
127. Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, et al. B cells promote tumor progression Via Stat3 regulated-angiogenesis. PloS One (2013) 8(5):e64159. doi: 10.1371/journal.pone.0064159
128. Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. Fcrgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell (2010) 17(2):121–34. doi: 10.1016/j.ccr.2009.12.019
129. Fagiani E, Bill R, Pisarsky L, Ivanek R, Ruegg C, Christofori G. An immature b cell population from peripheral blood serves as surrogate marker for monitoring tumor angiogenesis and anti-angiogenic therapy in mouse models. Angiogenesis (2015) 18(3):327–45. doi: 10.1007/s10456-015-9470-9
130. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. Cd4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16(2):91–102. doi: 10.1016/j.ccr.2009.06.018
131. Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microrna activity amplifies ifn-Gamma-Induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol (2016) 18(7):790–802. doi: 10.1038/ncb3371
132. Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M, et al. Tumor-infiltrating Cd4+ Th17 cells produce il-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep (2011) 25(5):1271–7. doi: 10.3892/or.2011.1201
133. Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, et al. Il-17 is associated with poor prognosis and promotes angiogenesis Via stimulating vegf production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun (2011) 407(2):348–54. doi: 10.1016/j.bbrc.2011.03.021
134. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature (1993) 362(6423):841–4. doi: 10.1038/362841a0
135. Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res (2005) 65(3):671–80. doi: 10.1158/0008-5472.671.65.3
136. Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev (2020) 86:102017. doi: 10.1016/j.ctrv.2020.102017
137. Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, et al. Bevacizumab in combination with fluorouracil and leucovorin: An active regimen for first-line metastatic colorectal cancer. J Clin Oncol (2005) 23(15):3502–8. doi: 10.1200/JCO.2005.10.017
138. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med (2004) 350(23):2335–42. doi: 10.1056/NEJMoa032691
139. Salvatore L, Bria E, Sperduti I, Hinke A, Hegewisch-Becker S, Aparicio T, et al. Bevacizumab as maintenance therapy in patients with metastatic colorectal cancer: A meta-analysis of individual patients' data from 3 phase iii studies. Cancer Treat Rev (2021) 97:102202. doi: 10.1016/j.ctrv.2021.102202
140. Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science (2005) 307(5706):58–62. doi: 10.1126/science.1104819
141. Kim SJ, Jung KH, Son MK, Park JH, Yan HH, Fang Z, et al. Tumor vessel normalization by the Pi3k inhibitor hs-173 enhances drug delivery. Cancer Lett (2017) 403:339–53. doi: 10.1016/j.canlet.2017.06.035
142. Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: Progress, opportunities, and challenges. Annu Rev Physiol (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
143. Tolaney SM, Boucher Y, Duda DG, Martin JD, Seano G, Ancukiewicz M, et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci USA (2015) 112(46):14325–30. doi: 10.1073/pnas.1518808112
144. Garcia-Romero N, Palacin-Aliana I, Madurga R, Carrion-Navarro J, Esteban-Rubio S, Jimenez B, et al. Bevacizumab dose adjustment to improve clinical outcomes of glioblastoma. BMC Med (2020) 18(1):142. doi: 10.1186/s12916-020-01610-0
145. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
146. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
147. Xian X, Hakansson J, Stahlberg A, Lindblom P, Betsholtz C, Gerhardt H, et al. Pericytes limit tumor cell metastasis. J Clin Invest (2006) 116(3):642–51. doi: 10.1172/JCI25705
148. Procter TV, Williams A, Montagne A. Interplay between brain pericytes and endothelial cells in dementia. Am J Pathol (2021) 191(11):1917–31. doi: 10.1016/j.ajpath.2021.07.003
149. Leow CC, Coffman K, Inigo I, Breen S, Czapiga M, Soukharev S, et al. Medi3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int J Oncol (2012) 40(5):1321–30. doi: 10.3892/ijo.2012.1366
150. Coxon A, Bready J, Min H, Kaufman S, Leal J, Yu D, et al. Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: Implications for amg 386, an angiopoietin-1/2-Neutralizing peptibody. Mol Cancer Ther (2010) 9(10):2641–51. doi: 10.1158/1535-7163.MCT-10-0213
151. Park JS, Kim IK, Han S, Park I, Kim C, Bae J, et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell (2016) 30(6):953–67. doi: 10.1016/j.ccell.2016.10.018
152. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/Vegf bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci USA (2016) 113(16):4476–81. doi: 10.1073/pnas.1525360113
153. Goel S, Gupta N, Walcott BP, Snuderl M, Kesler CT, Kirkpatrick ND, et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J Natl Cancer Inst (2013) 105(16):1188–201. doi: 10.1093/jnci/djt164
154. Frye M, Dierkes M, Kuppers V, Vockel M, Tomm J, Zeuschner D, et al. Interfering with ve-ptp stabilizes endothelial junctions in vivo Via tie-2 in the absence of ve-cadherin. J Exp Med (2015) 212(13):2267–87. doi: 10.1084/jem.20150718
155. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest (2003) 111(9):1287–95. doi: 10.1172/JCI17929
156. Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, et al. Combined inhibition of vegf and pdgf signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J (2004) 18(2):338–40. doi: 10.1096/fj.03-0271fje
157. Liu Y, Shaw SK, Ma S, Yang L, Luscinskas FW, Parkos CA. Regulation of leukocyte transmigration: Cell surface interactions and signaling events. J Immunol (2004) 172(1):7–13. doi: 10.4049/jimmunol.172.1.7
158. Hawthorne T, Giot L, Blake L, Kuang B, Gerwien R, Smithson G, et al. A phase I study of Cr002, a fully-human monoclonal antibody against platelet-derived growth factor-d. Int J Clin Pharmacol Ther (2008) 46(5):236–44. doi: 10.5414/cpp46236
159. Jayson GC, Parker GJ, Mullamitha S, Valle JW, Saunders M, Broughton L, et al. Blockade of platelet-derived growth factor receptor-beta by Cdp860, a humanized, pegylated di-fab', leads to fluid accumulation and is associated with increased tumor vascularized volume. J Clin Oncol (2005) 23(5):973–81. doi: 10.1200/JCO.2005.01.032
160. Nisancioglu MH, Betsholtz C, Genove G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-a blockade. Cancer Res (2010) 70(12):5109–15. doi: 10.1158/0008-5472.CAN-09-4245
161. McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of pdgf-bb decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest (2007) 117(8):2114–22. doi: 10.1172/JCI31334
162. Meng YM, Jiang X, Zhao X, Meng Q, Wu S, Chen Y, et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat Commun (2021) 12(1):6011. doi: 10.1038/s41467-021-26259-y
163. Roth M, Gaceb A, Enstrom A, Padel T, Genove G, Ozen I, et al. Regulator of G-protein signaling 5 regulates the shift from perivascular to parenchymal pericytes in the chronic phase after stroke. FASEB J (2019) 33(8):8990–8. doi: 10.1096/fj.201900153R
164. Chang L, Scott MA, Meyers CA, James AW. Pericytes in sarcomas and other mesenchymal tumors. Adv Exp Med Biol (2019) 1147:109–24. doi: 10.1007/978-3-030-16908-4_4
165. Jannaway M, Scallan JP. Ve-cadherin and vesicles differentially regulate lymphatic vascular permeability to solutes of various sizes. Front Physiol (2021) 12:687563. doi: 10.3389/fphys.2021.687563
166. Grimsley-Myers CM, Isaacson RH, Cadwell CM, Campos J, Hernandes MS, Myers KR, et al. Ve-cadherin endocytosis controls vascular integrity and patterning during development. J Cell Biol (2020) 219(5):e201909081. doi: 10.1083/jcb.201909081
167. Maharjan S, Kim K, Agrawal V, Choi HJ, Kim NJ, Kim YM, et al. Sac-1004, a novel vascular leakage blocker, enhances endothelial barrier through the Camp/Rac/Cortactin pathway. Biochem Biophys Res Commun (2013) 435(3):420–7. doi: 10.1016/j.bbrc.2013.04.104
168. Zhao Y, Li J, Ting KK, Chen J, Coleman P, Liu K, et al. The ve-Cadherin/Beta-Catenin signalling axis regulates immune cell infiltration into tumours. Cancer Lett (2021) 496:1–15. doi: 10.1016/j.canlet.2020.09.026
169. Burn T, Alvarez JI. Reverse transendothelial cell migration in inflammation: To help or to hinder? Cell Mol Life Sci (2017) 74(10):1871–81. doi: 10.1007/s00018-016-2444-2
170. Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase ve-ptp from ve-cadherin. Blood (2013) 122(14):2512–22. doi: 10.1182/blood-2013-04-499228
171. Lei X, Chen M, Huang M, Li X, Shi C, Zhang D, et al. Desacetylvinblastine monohydrazide disrupts tumor vessels by promoting ve-cadherin internalization. Theranostics (2018) 8(2):384–98. doi: 10.7150/thno.22222
172. Zhao Y, Ting KK, Li J, Cogger VC, Chen J, Johansson-Percival A, et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes T-Cell-Mediated immunotherapy. Cancer Res (2017) 77(16):4434–47. doi: 10.1158/0008-5472.CAN-16-3129
173. Ting KK, Zhao Y, Shen W, Coleman P, Yam M, Chan-Ling T, et al. Therapeutic regulation of ve-cadherin with a novel oligonucleotide drug for diabetic eye complications using retinopathy mouse models. Diabetologia (2019) 62(2):322–34. doi: 10.1007/s00125-018-4770-4
174. Qian C, Yang C, Tang Y, Zheng W, Zhou Y, Zhang S, et al. Pharmacological manipulation of Ezh2 with salvianolic acid b results in tumor vascular normalization and synergizes with cisplatin and T cell-mediated immunotherapy. Pharmacol Res (2022) 182:106333. doi: 10.1016/j.phrs.2022.106333
175. Kleibeuker EA, Fokas E, Allen PD, Kersemans V, Griffioen AW, Beech J, et al. Low dose angiostatic treatment counteracts radiotherapy-induced tumor perfusion and enhances the anti-tumor effect. Oncotarget (2016) 7(47):76613–27. doi: 10.18632/oncotarget.12814
176. Huang N, Liu Y, Fang Y, Zheng S, Wu J, Wang M, et al. Gold nanoparticles induce tumor vessel normalization and impair metastasis by inhibiting endothelial Smad2/3 signaling. ACS Nano (2020) 14(7):7940–58. doi: 10.1021/acsnano.9b08460
177. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell (2014) 26(2):190–206. doi: 10.1016/j.ccr.2014.06.025
178. Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, et al. Heterozygous deficiency of Phd2 restores tumor oxygenation and inhibits metastasis Via endothelial normalization. Cell (2009) 136(5):839–51. doi: 10.1016/j.cell.2009.01.020
179. Ehling M, Celus W, Martin-Perez R, Alba-Rovira R, Willox S, Ponti D, et al. B55alpha/Pp2a limits endothelial cell apoptosis during vascular remodeling: A complementary approach to disrupt pathological vessels? Circ Res (2020) 127(6):707–23. doi: 10.1161/CIRCRESAHA.119.316071
180. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544(7649):250–4. doi: 10.1038/nature21724
181. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest (2018) 128(5):2104–15. doi: 10.1172/JCI96582
182. Zheng X, Zhang N, Qian L, Wang X, Fan P, Kuai J, et al. Ctla4 blockade promotes vessel normalization in breast tumors Via the accumulation of eosinophils. Int J Cancer (2020) 146(6):1730–40. doi: 10.1002/ijc.32829
183. Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-targeted tnfalpha stabilizes tumor vessels and enhances active immunotherapy. Proc Natl Acad Sci USA (2012) 109(20):7841–6. doi: 10.1073/pnas.1118296109
184. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (2020) 71(4):1247–61. doi: 10.1002/hep.30889
185. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. Sting activation reprograms tumor vasculatures and synergizes with Vegfr2 blockade. J Clin Invest (2019) 129(10):4350–64. doi: 10.1172/JCI125413
186. McGee MC, Hamner JB, Williams RF, Rosati SF, Sims TL, Ng CY, et al. Improved intratumoral oxygenation through vascular normalization increases glioma sensitivity to ionizing radiation. Int J Radiat Oncol Biol Phys (2010) 76(5):1537–45. doi: 10.1016/j.ijrobp.2009.12.010
187. Johansson-Percival A, He B, Ganss R. Immunomodulation of tumor vessels: It takes two to tango. Trends Immunol (2018) 39(10):801–14. doi: 10.1016/j.it.2018.08.001
188. Zhang N, Yin R, Zhou P, Liu X, Fan P, Qian L, et al. Dll1 orchestrates Cd8(+) T cells to induce long-term vascular normalization and tumor regression. Proc Natl Acad Sci USA (2021) 118(22):e2020057118. doi: 10.1073/pnas.2020057118
189. Ganss R. Tumour vessel normalization and immune checkpoint blockade: A new synergism. Immunol Cell Biol (2017) 95(6):497–8. doi: 10.1038/icb.2017.30
190. Gao F, Yang C. Anti-Vegf/Vegfr2 monoclonal antibodies and their combinations with pd-1/Pd-L1 inhibitors in clinic. Curr Cancer Drug Targets (2020) 20(1):3–18. doi: 10.2174/1568009619666191114110359
191. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and vegfa inhibition elicits antitumor immunity that is enhanced by pd-1 checkpoint blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
192. Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to pd-1 blockade. Clin Cancer Res (2020) 26(7):1712–24. doi: 10.1158/1078-0432.CCR-19-2179
193. Yasuda S, Sho M, Yamato I, Yoshiji H, Wakatsuki K, Nishiwada S, et al. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (Vegfr2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol (2013) 172(3):500–6. doi: 10.1111/cei.12069
194. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. Vegf-a modulates expression of inhibitory checkpoints on Cd8+ T cells in tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
195. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-Pd-L1 therapy stimulates tumor immunity through hev formation. Sci Transl Med (2017) 9(385):eaak9679. doi: 10.1126/scitranslmed.aak9679
196. Sun Y, Chen W, Torphy RJ, Yao S, Zhu G, Lin R, et al. Blockade of the Cd93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci Transl Med (2021) 13(604):eabc8922. doi: 10.1126/scitranslmed.abc8922
197. Wu FT, Man S, Xu P, Chow A, Paez-Ribes M, Lee CR, et al. Efficacy of cotargeting angiopoietin-2 and the vegf pathway in the adjuvant postsurgical setting for early breast, colorectal, and renal cancers. Cancer Res (2016) 76(23):6988–7000. doi: 10.1158/0008-5472.CAN-16-0888
198. Vella G, Guelfi S, Bergers G. High endothelial venules: A vascular perspective on tertiary lymphoid structures in cancer. Front Immunol (2021) 12:736670. doi: 10.3389/fimmu.2021.736670
199. Hussain B, Kasinath V, Ashton-Rickardt GP, Clancy T, Uchimura K, Tsokos G, et al. High endothelial venules as potential gateways for therapeutics. Trends Immunol (2022) 43(9):728–40. doi: 10.1016/j.it.2022.07.002
200. Meder L, Schuldt P, Thelen M, Schmitt A, Dietlein F, Klein S, et al. Combined vegf and pd-L1 blockade displays synergistic treatment effects in an autochthonous mouse model of small cell lung cancer. Cancer Res (2018) 78(15):4270–81. doi: 10.1158/0008-5472.CAN-17-2176
201. Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res (2017) 5(1):17–28. doi: 10.1158/2326-6066.CIR-16-0206
202. D'Amico G, Korhonen EA, Anisimov A, Zarkada G, Holopainen T, Hagerling R, et al. Tie1 deletion inhibits tumor growth and improves angiopoietin antagonist therapy. J Clin Invest (2014) 124(2):824–34. doi: 10.1172/JCI68897
203. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to vegf signaling blockade. Cell Rep (2014) 8(3):696–706. doi: 10.1016/j.celrep.2014.06.059
204. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. Vegf-a drives tox-dependent T cell exhaustion in anti-Pd-1-Resistant microsatellite stable colorectal cancers. Sci Immunol (2019) 4(41):eaay0555. doi: 10.1126/sciimmunol.aay0555
205. Friedman CF, Snyder Charen A, Zhou Q, Carducci MA, Buckley De Meritens A, Corr BR, et al. Phase ii study of atezolizumab in combination with bevacizumab in patients with advanced cervical cancer. J Immunother Cancer (2020) 8(2):e001126. doi: 10.1136/jitc-2020-001126
206. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (Go30140): An open-label, multicentre, phase 1b study. Lancet Oncol (2020) 21(6):808–20. doi: 10.1016/S1470-2045(20)30156-X
207. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
208. Choueiri TK, Larkin J, Oya M, Thistlethwaite F, Martignoni M, Nathan P, et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (Javelin renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol (2018) 19(4):451–60. doi: 10.1016/S1470-2045(18)30107-4
209. Rini BI, Atkins MB, Choueiri TK, Thomaidou D, Rosbrook B, Thakur M, et al. Time to resolution of axitinib-related adverse events after treatment interruption in patients with advanced renal cell carcinoma. Clin Genitourin Cancer (2021) 19(5):e306–12. doi: 10.1016/j.clgc.2021.03.019
210. Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 javelin renal 101 trial. Nat Med (2020) 26(11):1733–41. doi: 10.1038/s41591-020-1044-8
211. Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell MT, Venugopal B, et al. Updated efficacy results from the javelin renal 101 trial: First-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol (2020) 31(8):1030–9. doi: 10.1016/j.annonc.2020.04.010
212. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med (2019) 380(12):1103–15. doi: 10.1056/NEJMoa1816047
213. Zhu X, McDowell MM, Newman WC, Mason GE, Greene S, Tamber MS. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: Case report. J Neurosurg Pediatr (2017) 19(2):249–53. doi: 10.3171/2016.8.PEDS16326
214. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. Azd2171, a pan-vegf receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell (2007) 11(1):83–95. doi: 10.1016/j.ccr.2006.11.021
215. Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol (2009) 27(15):2542–52. doi: 10.1200/JCO.2008.19.9356
216. Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-Pd-1 and anti-Pd-L1 immune checkpoint antibodies. Ann Oncol (2015) 26(12):2375–91. doi: 10.1093/annonc/mdv383
217. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2021) 184(21):5309–37. doi: 10.1016/j.cell.2021.09.020
218. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
219. Ding Y, Li S, Nie G. Nanotechnological strategies for therapeutic targeting of tumor vasculature. Nanomedicine (Lond) (2013) 8(7):1209–22. doi: 10.2217/nnm.13.106
220. Zhao Y, Ji T, Wang H, Li S, Zhao Y, Nie G. Self-assembled peptide nanoparticles as tumor microenvironment activatable probes for tumor targeting and imaging. J Control Release (2014) 177:11–9. doi: 10.1016/j.jconrel.2013.12.037
221. Li Z, Di C, Li S, Yang X, Nie G. Smart nanotherapeutic targeting of tumor vasculature. Acc Chem Res (2019) 52(9):2703–12. doi: 10.1021/acs.accounts.9b00283
222. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell (2020) 37(1):21–36.e13. doi: 10.1016/j.ccell.2019.12.001
223. Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, Ribatti D, et al. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res (2003) 63(21):7400–9.
224. Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science (1998) 279(5349):377–80. doi: 10.1126/science.279.5349.377
225. Li SJ, Chen JX, Sun ZJ. Improving antitumor immunity using antiangiogenic agents: Mechanistic insights, current progress, and clinical challenges. Cancer Commun (Lond) (2021) 41(9):830–50. doi: 10.1002/cac2.12183
Keywords: immune checkpoint blockade, anti-angiogenesis, tumor immune microenvironment, vessel normalization, targeted therapy
Citation: Zheng W, Qian C, Tang Y, Yang C, Zhou Y, Shen P, Chen W, Yu S, Wei Z, Wang A, Lu Y and Zhao Y (2022) Manipulation of the crosstalk between tumor angiogenesis and immunosuppression in the tumor microenvironment: Insight into the combination therapy of anti-angiogenesis and immune checkpoint blockade. Front. Immunol. 13:1035323. doi: 10.3389/fimmu.2022.1035323
Received: 02 September 2022; Accepted: 26 October 2022;
Published: 10 November 2022.
Edited by:
Tong Xiang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Avishek Bhuniya, Wistar Institute, United StatesShuixing Zhang, First Affiliated Hospital of Jinan University, China
Copyright © 2022 Zheng, Qian, Tang, Yang, Zhou, Shen, Chen, Yu, Wei, Wang, Lu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiyun Wang, wangaiyun@njucm.edu.cn; Yin Lu, luyingreen@njucm.edu.cn; Yang Zhao, y.zhao@njucm.edu.cn
†These authors have contributed equally to this work